Introduction
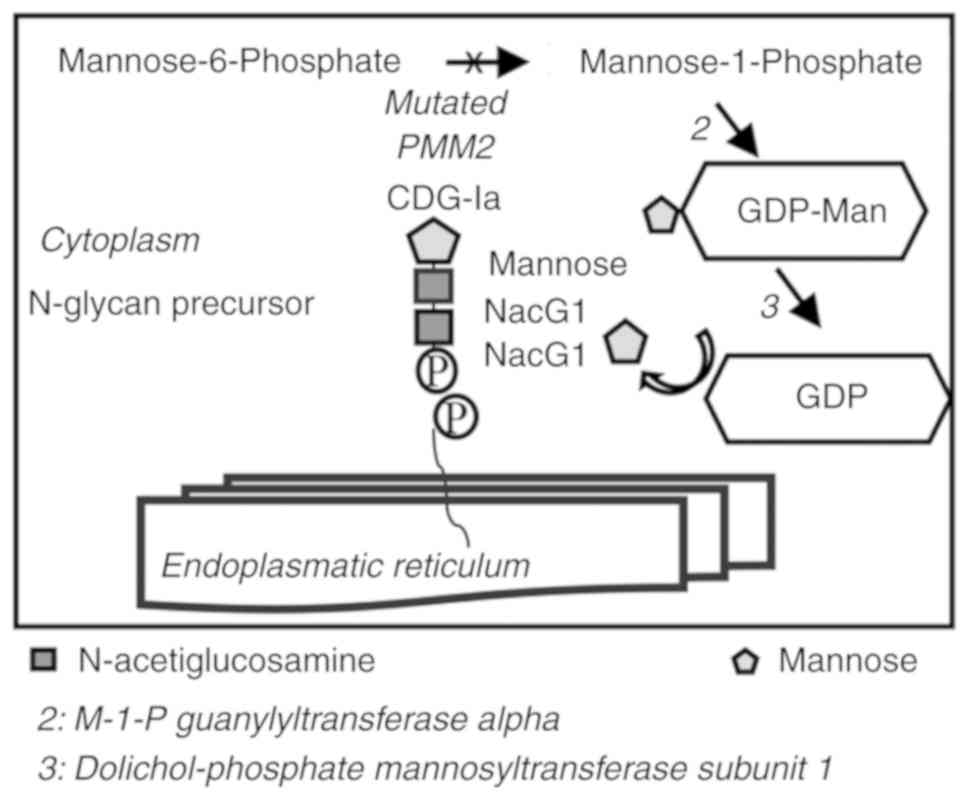
Congenital disorder of glycosylation type Ia
(CDG-Ia) is a rare autosomal recessive multisystem disorder with
severe neurological disabilities (encephalopathy and psychomotor
retardation) due to mutations in the phosphomannomutase 2
(PMM2) gene, which encodes for an enzyme involved in the
isomerization of mannose-6-phosphate (M6P) to mannose-1-phosphate
(M1P). In this metabolic pathway, M1P is the precursor of guanosine
5′-diphospho-D-mannose (GDP-Man). GDP-Man is the substrate of the
enzyme mannose-1-phosphate guanylyltransferase α, which is crucial
for protein N-glycosylation (Fig.
1). The mutated PMM2 gene leads to a decreased
conversion of M6P to M1P, corresponding to limited synthesis of
GDP-Man in the cytoplasm. GDP-Man is a key substrate in
glycoprotein formation and is involved in several reactions
catalysed by the N-glycosylation pathway. Therefore, it is used
directly or indirectly as a mannose donor for all mannosylation
reactions. Decreased GDP-Man results in the hypoglycosylation of
newly synthesised proteins with a limited number of branched
oligosaccharide chains linked to asparagine amino groups.
N-glycosylation is a post-translational modification
that is important in determining protein structure and function.
Several proteins, including lysosomal enzymes, serum proteins and
membrane glycoproteins, are sensitive to hypoglycosylation
(1,2). Therefore, the lack of mannosylation
disrupts the lysosomal enzyme cycle, resulting in low enzyme
concentration levels within the cells and abnormally increased
levels in the extracellular fluids and serum. Previous studies have
examined the activity of several lysosomal enzymes in the serum and
leukocytes of patients with CDG-Ia (1). Barone et al demonstrated that
the enzymatic activities of β-galactosidase, β-glucuronidase,
β-hexosaminidase and arylsulfatase A in the serum of patients were
increased between 2- and 4-fold compared with those of normal
subjects. However, in CDG-Ia leukocytes, the activities of other
enzymes, including α-fucosidase, β-glucuronidase and α-mannosidase,
were reduced (1). These findings
demonstrated an alteration of the intra- and extracellular
lysosomal enzyme activity levels in patients with CDG-Ia, as a
consequence of the disturbed isomerization of M6P to M1P (1-3).
The current treatment options used for the symptoms
caused by CDG-Ia are inefficient. Enzyme replacement therapy (ERT)
can be an available treatment for patients with CDG-Ia by the
modification of PMM2 and the attachment of a transduction domain
which allows the nanoparticle to cross biological membranes
(4,5). Alternatively, it can utilise
molecules to improve PMM2 uptake (6). Unfortunately, the low stability of
compounds in serum and the presence of the blood-brain barrier
(BBB) render ERT a therapeutic strategy with limited success.
Additional attempts to treat PMM2 deficiency have been reported
(7), including gene therapy,
dietary mannose supplementation, and phosphomannose isomerase
inhibitors that increase the availability of M6P. The addition of 1
mM Man to the fibroblasts of patients with CDG-Ia is able to
ameliorate the glycosylation defect and restore the deficient
levels of GDP-Man (8).
Patients with CDG-Ia do not respond well to
treatment (9-11); this is due to the fact that
Man-1-P is a compound that cannot cross the cell membrane.
Therefore, the use of a derivative that can pass via the cell
membrane may re-establish the level of GDP-Man that is necessary
for correct protein glycosylation (8-12).
Therefore, the present study examined the possibility of direct
treatment with GDP-Man, by bypassing the reaction catalysed by
PMM2. This approach was used by in vitro administration of
poly (D,L-lactide-co-glycolide) (PLGA) nanoparticles (NPs), which
can overcome the drawbacks associated with the low stability of the
compound. It is widely recognised that PLGA nanoparticle vectors
exhibit optimal physicochemical properties, including high
biodegradability and biocompatibility with tissues and cells and
low toxicity (13). The use of
the PLGA polymers was approved by the Food and Drug administration
and the European Medicine Agency in different clinical protocols of
drug delivery in humans (14). It
has been shown that these compounds can cross the BBB (15,16). In the present study, a therapeutic
strategy was proposed to treat CDG-Ia in fibroblast cultures using
PLGA NPs loaded with GDP-Man, which is a nucleotide-activated sugar
essential for the production of oligosaccharide chains. The effects
of this approach were assessed in human deficient fibroblasts,
prior to and following treatment with GDP-Man-NPs, by measuring the
activities of several lysosomal enzymes that were impaired in this
pathway.
Materials and methods
PLGA NP preparation
PLGA was used according to the protocol provided by
the manufacturer (Boehringer-Ingelheim, Ingelheim am Rhein,
Germany). PLGA conjugated with Rhodamine B piperazine amide
(R-PLGA; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was
prepared as previously described (17). Poly vinyl alcohol (PVA; 13-15,000
Da, degree of hydrolysation 86-89 Mol%, viscosity at 4% w/v in
water 3.5-4.5 cps) and the solvents (HPLC grade) used for analyses
were purchased from Sigma-Aldrich; Merck KGaA.
Lyophilised guanosine 5′-diphospho-D-mannose sodium
salt (GDP-Man, Sigma-Aldrich; Merck KGaA) was re-suspended in water
for injectable preparations (Galenica Senese, Monteromi d'Arbia,
Italy). The final concentration of the solution was 10 mg/ml of
GDP-Man. The GDP-Man-loaded PLGA NPs were prepared using the W/O/W
(double emulsion) solvent evaporation method (18,19). A total of 500 μl of GDP-Man
solution were emulsified in 2 ml of dichloromethane (DCM;
Sigma-Aldrich; Merck KGaA) solution containing polymer (47.5 mg of
PLGA and 2.5 mg of R-PLGA at 5°C) using a probe sonicator
(MicrosonUltrasonic cell disruptor, Misonix, Inc. Farmingdale, NY,
USA) at 100 W for 30 sec to obtain the W1/O emulsion
(first inner emulsion). The first inner emulsion was rapidly added
to 8.5 ml of 1% (w:v) PVA aqueous solution and the
W1/O/W2 emulsion was formed under sonication
(100 W for 60 sec) at 5°C. The preparation was mechanically stirred
(1,400 rpm) for at least 3 h (RW20DZM, Janke & Kunkel
IKA-Labortechnik, Staufen, Germany) at room temperature until
complete solvent evaporation. The GDP-Man-loaded PLGA NPs (sample
B1) were recovered and purified, to remove the un-encapsulated
GDP-Man and PVA, by hi-speed centrifugation (Beckman Coulter, Inc.,
Brea, CA, USA) at 15,000 rpm for 10 min at 5°C. They were
subsequently washed several times with sterile water, re-suspended
in physiological solution and used for in vitro experiments.
The preparations were stored at −20°C and restored at room
temperature by sonication prior to the treatments. Empty NPs
(sample B2) were used and were prepared in 500 ml of water, instead
of GDP-Man solution, in the presence of the same preparative
conditions used for sample B1.
Physico-chemical and technical
characterization of NPs
The morphology and the structure/architecture of the
samples (B1 and B2) were analysed by scanning transmission electron
microscopy (STEM). A drop of sample suspension was placed on a
200-mesh copper grid (TABB Laboratories Equipment, Berkshire, UK)
and allowed to adsorb. The suspension surplus was removed using
filter paper. All grids were analysed using a Nova NanoSEM 450
(FEI; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
transmission electron microscope operating at 30 kV. A TEM grid
holder was used and the STEM II detector was set in the
bright-field mode to analyse the transmitted electrons.
Size and zeta potential (ζ-pot)
The mean particle size (Z-Average) and the
polydispersitivity index (PDI) of the samples following
purification and re-suspension in physiological solution, were
determined at room temperature by photon correlation spectroscopy
using a Zetasizer Nano ZS (laser 4 mW He-Ne, 633 nm, laser
attenuator automatic, transmission 100-0.0003%, Avalanche
photodiode detector, Q.E. >50% at 633 nm) from Malvern
Instruments, Ltd. (Malvern UK). The results were also expressed as
the intensity distribution, i.e. the size 50% [Di(50)] and 90%
[Di(90)], below which all the NPs were placed. The ζ-pot was
measured using the same equipment with a combination of laser
Doppler velocimetry and phase analysis light scattering. All data
are expressed as the mean of at least three determinations (three
for each sample).
Residual surfactant
The residual quantity of surfactants (PVA) was
determined using a colorimetric method based on the formation of
the coloured complex between two adjacent hydroxyl groups of the
surfactant and of an iodine molecule (20). The protocol used was reported in a
previous study (21).
GDP-Man content
To evaluate the content of GDP-Man, 2 ml of NP
suspension was freeze-dried at −60°C and at 1×10−3 mm/Hg
for 48 h (LyoLab 300, Heto-Holten, Allerod, Denmark). The
freeze-dried NPs (10 mg) were dissolved in 1.5 ml of
dichloromethane. Subsequently, 3 ml of water were added to the
extract of the GDP-Man and the organic solvent was evaporated at
room temperature under stirring (1,500 rpm for at least 1 h;
RW20DZM, Janke & Kunkel IKA-Labortechnik). The aqueous solution
was filtered (cellulose acetate filter, porosity 0.2-μm,
Sartorius AG, Göttingen, Germany) to remove the polymer residues
and was analysed by HPLC in order to evaluate the drug content.
The HPLC apparatus (PerkinElmer, Inc., Waltham, MA,
USA) comprised a model series 200 pump with an injection valve and
a 200 μl sample loop. A UV detector (series 200) was used
for the determination of the NP concentration. Chromatography
separation was performed on a sunfire C 18 (4.6×150 mm; porosity 5
μm; Waters Corporation, Milford, MA, USA) column at room
temperature and at a flow rate of 1 ml/min. The pump operated in an
isocratic mode using a mobile phase of 43:23:36:1 v/v acetonitrile:
methanol: water: acetic acid. The chromatographic peak-areas of the
standard solutions were collected and used for the generation of
the calibration curve. The linearity of the method was achieved in
the range of 0.2-10 μg/ml (r2=0.9982). All data
are expressed as the mean of at least three determinations. Drug
loading is expressed as mg of GDP-Man encapsulated/100 mg of
NPs.
Cell culture
A total of three CDG-Ia fibroblast cultures
(CDG-Ia_F1, CDG-Ia_F2, CDG-Ia_F3) namely F1, F2 and F3, were
obtained from skin biopsies provided by the Cell Line and DNA
Biobank from Patients affected by Genetic Diseases (Centro di
Diagnostica Genetica e Biochimica delle Malattie Metaboliche,
Istituto G. Gaslini, Genova, Italy). The human fibroblasts were
maintained in RPMI 1640 supplemented with 10% fetal bovine serum,
1% L-glutamine and 1% penicillin/streptomycin (all Euroclone,
Milano, Italy) at 37°C in humidified air containing 5%
CO2. Healthy fibroblasts (HFs) were obtained from skin
specimens derived from the circumcision of healthy subjects.
Informed consent forms were provided from their parents or legal
representatives.
Mutational analysis of the three skin
specimens
Genomic DNA was isolated from the fibroblasts of
patients with CDG-Ia. Direct DNA sequencing of the fibroblast
cultures F1, F2 and F3 demonstrated that patients were compound
heterozygous with different percentages of PMM2 residual activity
(22-26).
In the F1 fibroblast cultures, the L32R/R123Q
mutation was identified. This PMM2 genotype has been demonstrated
to reduce PMM2 activity to ~2.2 mU/mg compared with normal activity
levels, which is estimated to be 5.34±1.74 mU/mg of protein
(23,24). The fibroblasts of the F2 genotype
revealed R14H/C241S mutations with a PMM2 residual activity of 1.7
mU/mg. These two genotypes have been frequently associated with a
mild phenotype, which is characterized by cerebellar ataxia,
dysmetria and dysarthria (23,24).
In the F3 fibroblasts, R141H/D223N mutations were
identified, which have been associated with low PMM2 residual
activity (undetectable) causing a severe phenotype of fibroblasts.
The majority of patients with the severe phenotype have severe
intellectual disability and microcephaly, and they are unable to
walk independently (23,24).
3-(4,5-Dimethylthiazol-2-yl) 2,5-diphenyl
tetrazolium bromide (MTT) toxicity assay of PLGA GDP-Man
An MTT assay is a colorimetric assay based on the
ability of viable cells to reduce the soluble yellow tetrazolium
salt, MTT, in order to form a blue formazan crystal by
mitochondrial succinate dehydrogenase. The MTT test (Sigma-Aldrich;
Merck KGaA) was performed in order to determine the cellular
toxicity on fibroblasts following treatment with GDP-Man PLGA NPs.
The cell viability was measured using the GLOMAX multi detection
system and the UV optical kit (Promega Corporation, Madison, WI,
USA) following a 4-h incubation period (27).
Glycoprotein isolation
A glycoprotein isolation kit using concanavalin A
(ConA; Thermo Fisher Scientific, Inc.) was used to separate
N-linked glycoproteins from complex protein mixtures, following the
manufacturer's protocol. The mix was passed through a column of
lectin ConA that was immobilised on agarose. Following washing, the
glycoprotein fraction was eluted using buffer (2% SDS and 50 mM
Tris-HCl, pH 7.4; Sigma-Aldrich; Merck KGaA).
Two-dimensional electrophoresis
(2-DE)
A total of 46 μg of glycoproteins from each
sample were denatured in 150 μl of dissolution buffer (7 M
urea, 2 M thiourea, 4% CHAPS, 40 mM Tris, 65 mM DTT and 0.24%
Bio-Lyte 3-10) in the presence of bromophenol blue, as previously
described (28). Subsequently,
7-cm pH 3.0-10.0 NL (IPG) readystrips (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) were rehydrated at 50 V for 12 h at 20°C.
Isoelectric focusing (IEF) was performed in a protein IEF Cell
(Proteane IEF Cells; Bio-Rad Laboratories, Inc.). Following IEF,
the IPG strips were equili-brated by serial incubations (15 min) in
equilibration buffer (6 M urea, 2% SDS, 50 mM Tris-HCl pH 8.8, 30%
glycerol, and 1% DTT) and in equilibration buffer containing 4%
iodo-acetamide instead of DTT. The equilibrated IPG strips were
transferred to a 12% polyacrylamide gel for electrophoresis. The
2-DE was run on a Mini-Protean 3 Cell (200 V constant voltage)
until the bromophenol blue reached the bottom of the gel. Following
protein fixation for 5 h in 40% methanol and 10% acetic acid, the
gels were stained for 15 h with Sypro Ruby (Bio-Rad Laboratories,
Inc.). The molecular masses were determined using precision protein
standard markers (Bio-Rad Laboratories, Inc.), covering a range of
10-250 kDa. The 2-DE gels were scanned with a Molecular Imager
PharosFX system and analysed using the Proteomeweaver 4 program
(Bio-Rad Laboratories, Inc.).
Lysosomal enzyme activity assays
The proteins were extracted from cells using
mammalian protein extraction reagent solution, following the
protocol recommended by the manufacturer (Thermo Fisher Scientific
Inc.). The specific activities of β-galactosidase, α-mannosidase,
β-mannosidase and β-glucuronidase were measured using
4-methyl-umbelliferyl β-D-galactopyranoside, 4-methyl-umbelliferyl
α-d-man-nopyranoside, 4-methyl-umbelliferyl β-d-mannopyranoside and
4-methyl-umbelliferyl-β-d-glucuronidase substrates (Sigma-Aldrich;
Merck KGaA). The 4-methyl-umbelliferyl-β-D-galactopyranoside and
4-methyl-umbelliferyl-α-d-mannopi ranoside substrates were used at
final concentrations of 1.5 and 3 mM in a buffer containing 0.1 M
citric acid and 0.20 M sodium-phosphate (pH 4.5). The
4-methyl-umbellifery l-β-d-mannopyranoside substrate was used at
the final concentration of 2.5 mM in a buffer containing 0.1 M
citric acid and 0.20 M sodium-phosphate buffer at a pH of 4.7.
The reactions were performed in triplicate in
96-well black multiplates (Corning 96-Well Black) provided from
Euroclone (Milan, Italy) with a final volume of 60 μl
(substrate 40 μl and sample 20 μl) for 30 min at
37°C, with the exception of
4-methyl-umbelliferyl-β-d-mannopyranoside and 4-methyl
-umbelliferyl-β-d-glucuronidase substrates. In these cases, the
reactions were performed for 60 min at 37°C. Each reaction mixture
was terminated by the addition of 0.290 ml of 0.4 M glycine-NaOH
buffer at pH 10.4. The fluorescence of the released
4-methyl-umbelliferyl was measured using a GLOMAX Multi Detection
system and a fluorescence optical kit (Promega Corporation), which
was set at an excitation wavelength of 360 nm and an emission
wavelength of 450 nm. The protein concentration of the cellular
extracts was determined with the Lowry assay using a PerkinElmer
Lamda 40 UV/VIS spectrophotometer. The enzymatic activity in the
fibroblast cultures is expressed as nmol of substrate hydrolysed
per/min and normalised per mg of protein.
Statistical analysis
All values of the in vitro experiments are
reported as the mean ± standard deviation. Three experimental
replicates were performed per sample. Student's unpaired t-test was
used to compare the activity of α-mannosidase, β-glucuronidase and
β-galactosidase between untreated and treated CDG-Ia fibroblasts.
P<0.01 was considered to indicate a statistically significant
difference. The statistical analysis was performed using the PRISM6
program (GraphPad Software, Inc., La Jolla, CA, USA).
Results
GDP-Man NPs characterization
In the present study, PLGA NPs were produced that
contained GDP-Man (GDP-Man NPs). Several technological conditions,
including the volume of inner, the organic and external phases, the
concentration of the polymers, the surfactant and drug
concentration, and the time and potency of sonication were
optimised in order to achieve the optimal preparative results in
terms of size, polidispersivity and drug content (data not shown).
B1 GDP-Man loaded NPs) and B2 (unloaded NPs) samples were selected
and assessed, which were prepared as described above. The results
of the physicochemical characterization are shown in Fig. 2A and B and Table I. The GDP-Man NPs (B1 sample) had
a higher PDI value (0.18, vs. 0.05) compared with that of the empty
NPs (B2 sample). In addition, the Z-Average values and the wide
size distribution (D90) increased from 200 to 300 nm and from 285
to 440 nm, respectively, indicating a major heterogeneity of B1
that typically occurred during drug encapsulation. These data
confirmed the presence of well-formed spheric-like structures in
the B1 samples, which exhibited higher heterogeneity in terms of
shape and size in comparison with the B2 samples. In addition, the
ζ-pot of B1 (~7.5 mV) was decreased following drug loading compared
with that of B2 (~2 mV), possibly due to a different reorganization
of the polymer with the drug into the matrix-like structure of NPs,
and due to the possible absorption of GDP-Man on the NP
surface.
 | Table IPhysico-chemical characterization of
GDP-Man PLGA NPs and PLGA NPs. |
Table I
Physico-chemical characterization of
GDP-Man PLGA NPs and PLGA NPs.
| Sample | W1/O (sonication
parameter) | W1/OAV2 (sonication
parameter) | Z-Average (nm) | PDI | Di(50) (nm) | Di(90) (nm) | ζ-pot
(mV) | GDP-Man content
(μg/100 mg NPs) | Surfactant content
(PVA %) |
|---|
| B1 | 100W/30 sec | 100 W/6 sec | 326±10 | 0.1S±0.4 | 257±55 | 43S±8 | −7.5±1 | 27.7±3 | 6±4 |
| B2 | 100W/30 sec | 100 W/6 sec | 197±4 | 0.05±0.03 | 202±2 | 285±5 | −1.9±2 | - | 7±2 |
GDP-Man PLGA NPs do not reduce the
viability of human skin fibroblasts
The B1 parameters were optimal with regard to the
cellular uptake, as described previously (29). In addition, NPs that are suitable
for in vivo application exhibit a particle size ranging
between 100 and 350 nm (30).
These structures are favourable in terms of the circulation
stability, the interaction with cells and the reduced elimination
by the kidneys (30). The present
study produced an NP content of ~27.7 μg of GDP-Man/100 mg
(Table I).
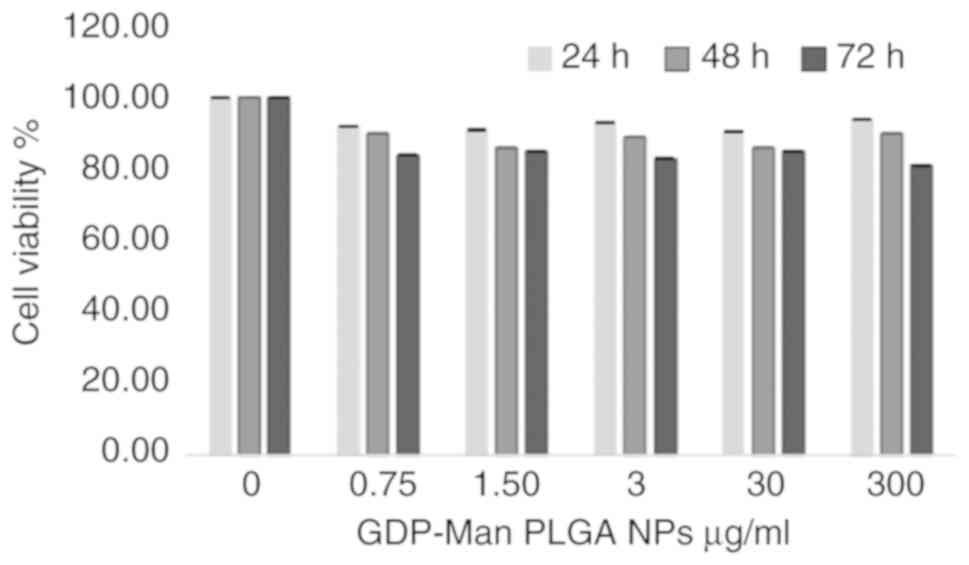
Subsequently, the in vitro toxicity of the
GDP-Man NPs was assayed by the addition of different concentrations
of B1 NPs to F3 (severe phenotype) fibroblasts. The NP content
ranged between 0.75 and 300 μg/ml, corresponding to 0.4-150
ng/well of GDP-Man in the culture medium, as determined by HPLC
analysis conducted on NP samples. Following 48 h of incubation with
five different concentrations of B1 preparation, there was no
significant decrease in the viability of the fibroblasts compared
with that of the untreated cultures (Fig. 3).
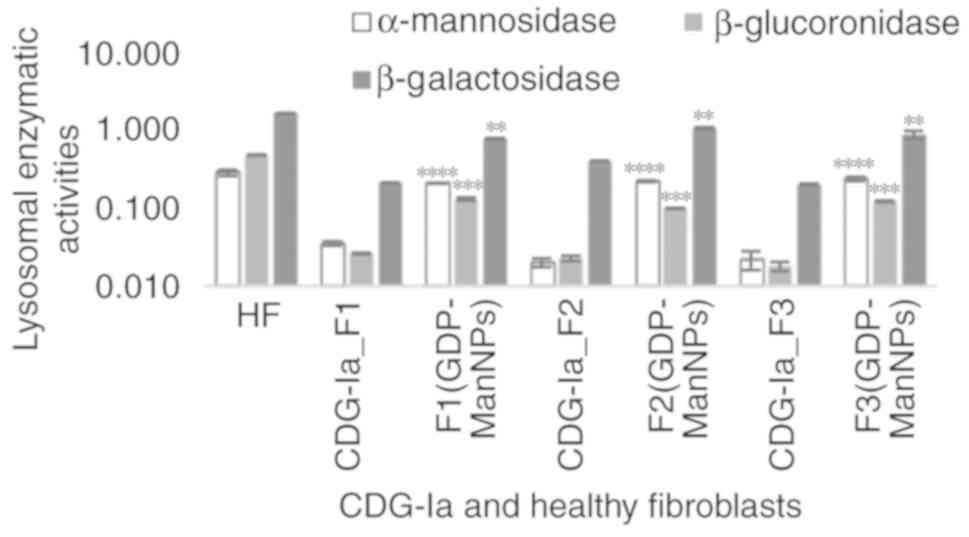
Identification of lysosomal enzyme
activity reductions in CDG-Ia fibroblasts
The ability of the GDP-Man NPs to restore the
N-glycosylation of glycoproteins, was further examined. The three
deficient cultures, namely F1, F2 and F3, were assayed and the
pattern of lysosomal enzymes deficits was assessed by comparing the
specific activity of several lysosomal enzymes with that of healthy
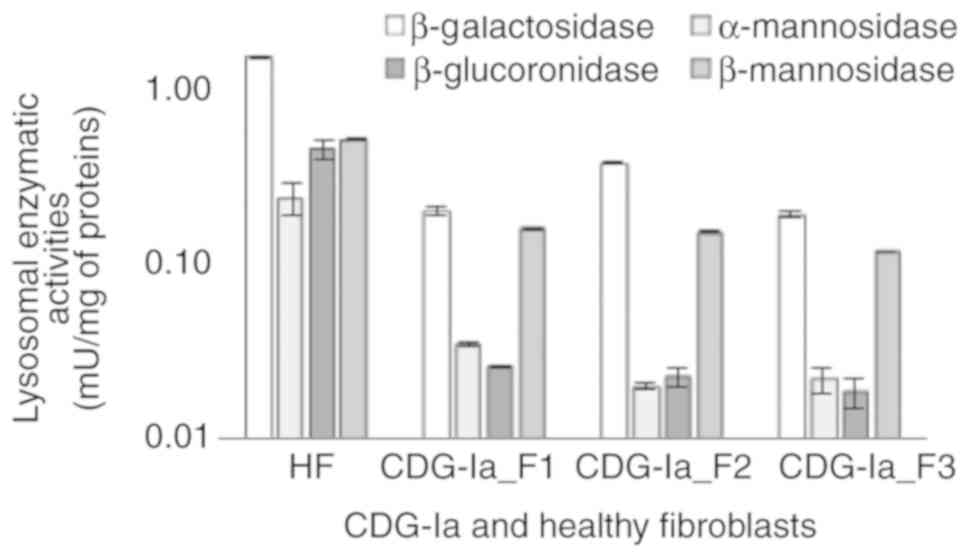
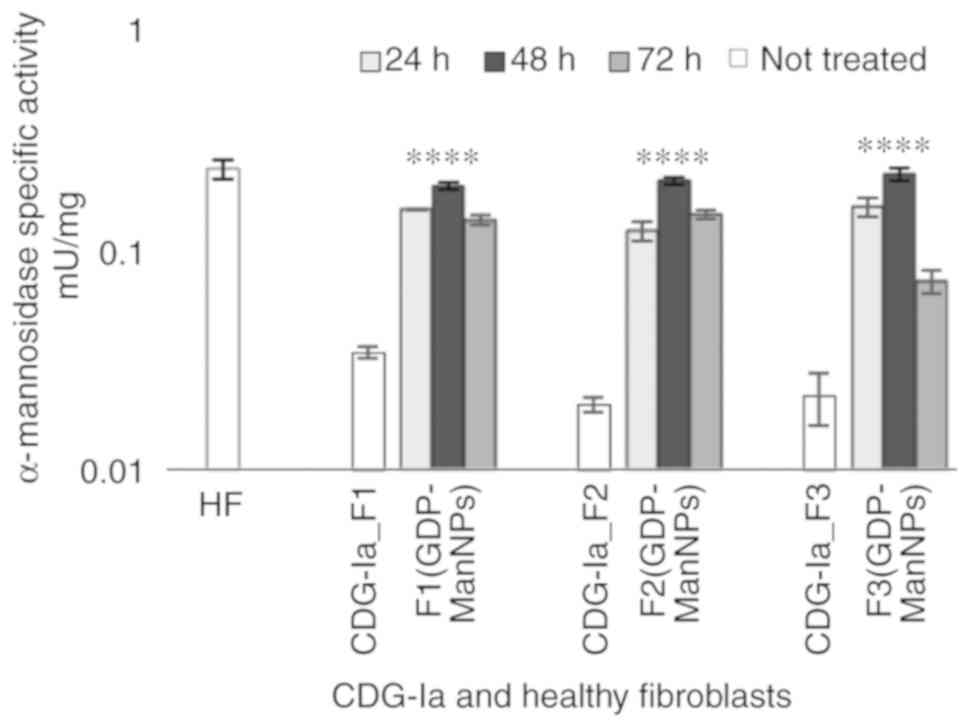
fibroblasts. As shown in Fig. 4,
it was found that the β-galactosidase, α-mannosidase,
β-glucuronidase, and β-mannosidase activities were significantly
lower in the CDG-Ia fibroblasts than in the control samples in all
three cultures investigated. The normal mean level of α-mannosidase
specific activity was 0.25±0.051 mU/mg, whereas levels in the F1,
F2 and F3 cultures were estimated to be 0.035-0.020±0.001 mU/mg.
The normal mean level of the β-glucuronidase activity was
0.476±0.064 mU/mg, whereas the levels were estimated to be
0.026-0.018±0.004 mU/mg in the F1, F2 and F3 cultures. The normal
mean level of β-mannosidase activity was 0.540±0.005 mU/mg, which
was reduced to 0.164-0.120±0.002 mU/mg in the F1, F2 and F3
cultures.
A previous study (1) determined that the activity of the
β-galactosidase enzyme was not reduced in the sera and leukocytes
of patients with CDG-Ia. Therefore, this activity of this enzyme
was not initially considered to determine the in vitro
correction of glycosylation. However, all three CDG-Ia fibroblast
cultures indicated a significant decrease in β-galactosidase
activity, which was estimated to be 7-10 times lower than that of
the HFs. The normal range of β-galactosidase activity in HFs was
estimated to be 1.62±0.021 mU/mg, whereas this was decreased to
0.40-0.20±0.013 mU/mg in the F1, F2 and F3 cultures (Fig. 4). Therefore, three fibroblast
cultures, F1, F2 and F3 were established; F1 and F2 were associated
with the mild phenotype, whereas F3 was associated with the severe
phenotype.
Identification of optimal dose and
incubation time
The capacity of the GDP-Man NPs to ameliorate
glycosylation deficits was examined by treating the three F1, F2
and F3 cultures with the B1 preparation. Prior to conducting the
assays, the optimum concentration of GDP-Man NPs and the optimum
incubation time period were determined. Finally, the time period
required to restore the lysosomal activity was determined. These
parameters were used in the 'in vitro therapeutic
protocol'.
Rush et al (31) reported that the average GDP-Man
contents in normal and CDG-Ia fibroblasts were 23 and 2.3
pmol/106 cells, respectively, which corresponded to 14
and 1.4 ng of GDP-Man, respectively. According to these results,
2×105 cells of F1, F2 and F3 were cultured in
6-multiwell plates and were used for the experiments below. The
three cultures were treated with GDP-Man NPs containing either 150
ng (300 μg/ml of B1 NPs) or 15 ng (30 μg/ml of B1
NPs) of GDP-Man. Furthermore, the CDG-Ia fibroblasts were treated
with a quantity of GDP-Man five times higher than that in the
1×106 HFs (32).
Incubation durations of 3 and 6 h were assessed.
Following overnight post-incubation with conditioned medium, the F1
(GDP-Man NPs), F2 (GDP-Man NPs), F3 (GDP-Man NPs) and HF cultures
were harvested and total proteins were extracted. The specific
activities of several lysosomal enzymes were assayed. The
α-mannosidase activity of the HFs was estimated to be 0.25±0.051
mU/mg, whereas those of the F1, F2 and F3 fibroblasts were
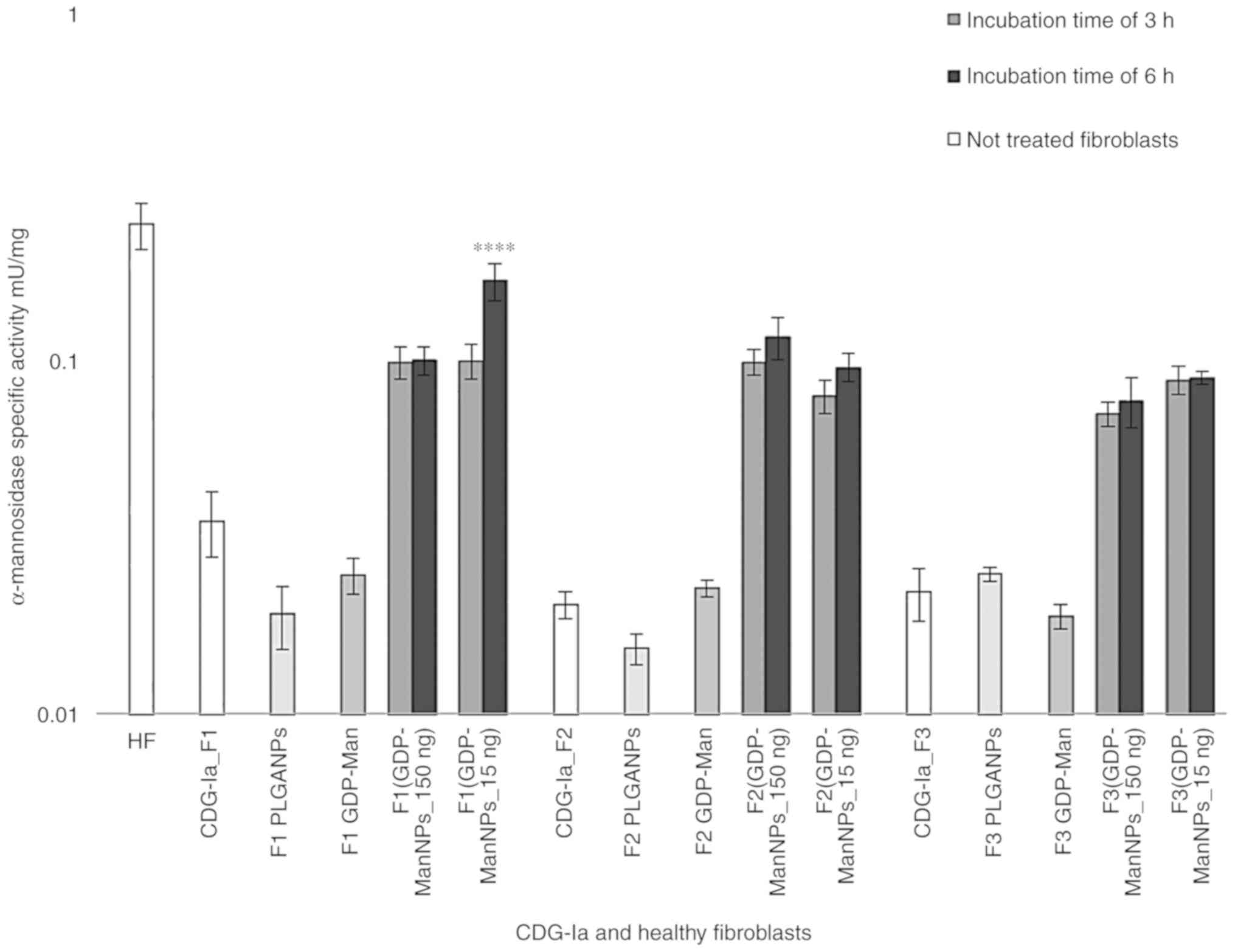
estimated to be 0.026±0.001 mU/mg. As shown in Fig. 5, the α-mannosidase activity
increased between 3- and 4-fold in all three F1 (GDP-Man NPs), F2
(GDP-Man NPs), F3 (GDP-Man NPs) cultures. Notably, the
α-mannosidase activity value of the F1 (GDP-Man NPs) culture was
estimated to be 0.172±0.021 mU/mg, which corresponded to 69% of
normal activity. Therefore, the F1 (GDP-Man NPs_15 ng) culture
indicated a significant increase of α-mannosidase activity
following 6 h treatment of 15 ng/well of GDP-Man (30 μg/ml
GDP-Man-PLGA NPs) in conditioned medium and 24 h post-incubation.
Incubation of the fibroblasts with 150 ng/well of GDP-Man (300
μg/ml GDP-Man-PLGA NPs) did not cause a linear improvements
in α-mannosidase activity. Therefore, 15 ng/well of GDP-Man was
selected as the optimal concentration used in the subsequent
assays. In contrast to these findings, incubation of the
fibroblasts for 6 h resulted in higher activity levels than those
noted at 3 h (33). Therefore, 6
h was selected as the optimal time period for the following
assessments.
In order to ensure that the effect observed was not
obtained by simple supplementation of free GDP-Man to the medium,
150 ng of free GDP-Man was added to the medium of the F1, F2 and F3
cultures under the same experimental conditions. No significant
increase in α-mannosidase activity was observed for the F1 GDP-Man,
F2 GDP-Man and F3 GDP-Man fibroblasts, which confirmed that free
GDP-Man did not affect glycosylation without the use of the PLGA
carrier. When adding PLGA NPs to F1, F2 and F3, no increase in the
enzymatic activity of α-mannosidase was observed (Fig. 5). Therefore, the loading of
GDP-Man in PLGA NPs was required for efficient delivery and
increase in the protein glycosylation levels. No lysosomal enzyme
activity was detected in the media of the treated or untreated
fibroblasts.
Evaluation of post-incubation time
necessary to correct the lysosomal enzyme activities
According to the aforementioned findings, the time
period required to correct the glycosylation of proteins was
determined by assaying the specific activity of α-mannosidase at
the three time points (24, 48 and 72 h) following 6 h of incubation
with 15 ng/well of GDP-Man loaded in PLGA NPs. The activities of
the α-mannosidase enzyme in the F1 (GDP-Man NPs), F2 (GDP-Man NPs)
and F3 (GDP-Man NPs) cultures were similar to those of the HFs at
48 h post-incubation and were marginally decreased following 72 h
of incubation (Fig. 6). Therefore
the 48-h period was selected as the standard post-incubation time
period used in the experimental assays.
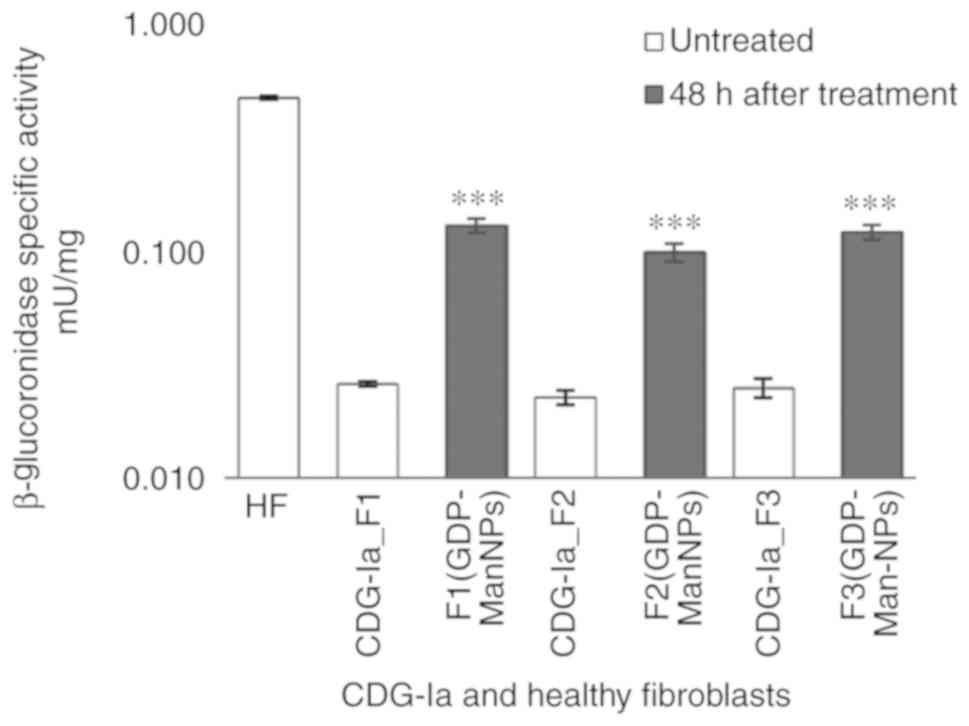
The activity of the lysosomal enzyme,
β-glucuronidase, was also detected; low activity was observed in
the F3 fibroblasts (0.0246±0.002 mU/mg) compared with that noted in
the HFs (0.476±0.006 mU/mg). Following treatment with NPs,
β-glucuronidase activities in the F1 (GDP-Man NPs), F2 (GDP-Man
NPs) and F3 (GDP-Man NPs) fibroblasts were higher than those in the
F1, F2 and F3 groups (Fig. 7).
The F1, F2 and F3 groups were incubated for 48 h in the presence of
15 ng/well of GDP-Man (30 μg/ml of GDP-Man-PLGA NPs).
Subsequently, the F1 (GDP-Man NPs) and F3 (GDP-Man NPs)
β-glucuronidase activity reached 0.130 mU/mg, which corresponded to
32.5% of the normal activity compared with that noted in the F1, F2
and F3 groups.
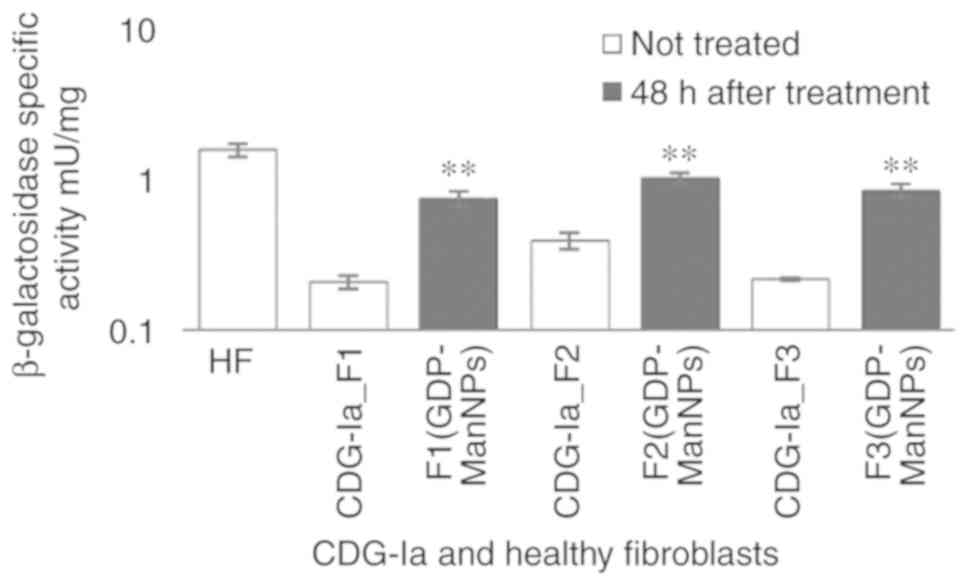
Finally the F1 (GDP-Man NPs), F2 (GDP-Man NPs) and
F3 (GDP-Man NPs) were incubated at 48 h with 15 ng/well of GDP-Man
(30 μg/ml of GDP-Man-PLGA NPs) and their β-galactosidase
activities were increased 5-7-fold compared with those noted in F1,
F2 and F3 (Fig. 8). Their levels
were comparable to those of the HFs.
To rule out that an increase in enzymatic activity
corresponds to higher production of protein, the lysosomal mRNA
levels were determined. Ichisaka et al demonstrated an
increase in the mRNA expression levels of β-hexosaminidase, which
corresponded to an increase in the β-hexosaminidase α-chain subunit
(3). The present study aimed to
quantify the mRNA expression levels of α-mannosidase,
β-gluocoronidase and β-galactosidase (34). No significant variations in mRNA
levels were observed among the F3 cells treated with GDP-Man PLGA
NPs and the F3 cells treated with single PLGA NPs. Based on these
results, it was confirmed that an improvement of lysosomal specific
activity does not depend on an increase in protein quantity, as the
mRNA production and expression levels did not alter following 48 h
of treatment with GDP-Man PLGA NPs. The details regarding the exact
methodological approach are available upon request.
The results reported in the present study confirmed
that GDP-Man NPs are internalised by CDG-Ia fibroblasts and
metabolised accordingly. It is noteworthy that the selected
quantity of GDP-Man was sufficient to partially restore the in
vitro N-glycosylation. The protein glycosylation pattern was
improved and several lysosomal enzymes increased in activity
following a single treatment (Fig.
9).
 | Figure 9Specific activities of α-mannosidase,
β-glucuronidase and β-galactosidase are expressed as mU/mg of
protein and were analysed 48 h following incubation. The increase
in the specific activity levels of these lysosomal enzymes in the
treated fibroblasts F1_GDP-Man NPs, F2_GDP-Man NPs and F3_GDP-Man
NPs was significant (P<0.01, P<0.001 and P<0.0001)
compared with the levels noted in the defective fibroblasts
(untreated CDG-Ia_F1, CDG-Ia_F2 and CDG-Ia_F3) following 54 h of
incubation. **P<0.01, ***P<0.001 and
****P<0.0001 among untreated and treated fibroblast
cultures. HF, healthy fibroblasts; CDG-Ia, congenital disorder of
glycosylation type Ia; GDP-Man, guanosine 5′-diphospho-D-mannose;
PLGA, poly (D,L-lactide-co-glycolide); NPs, nanoparticles. |
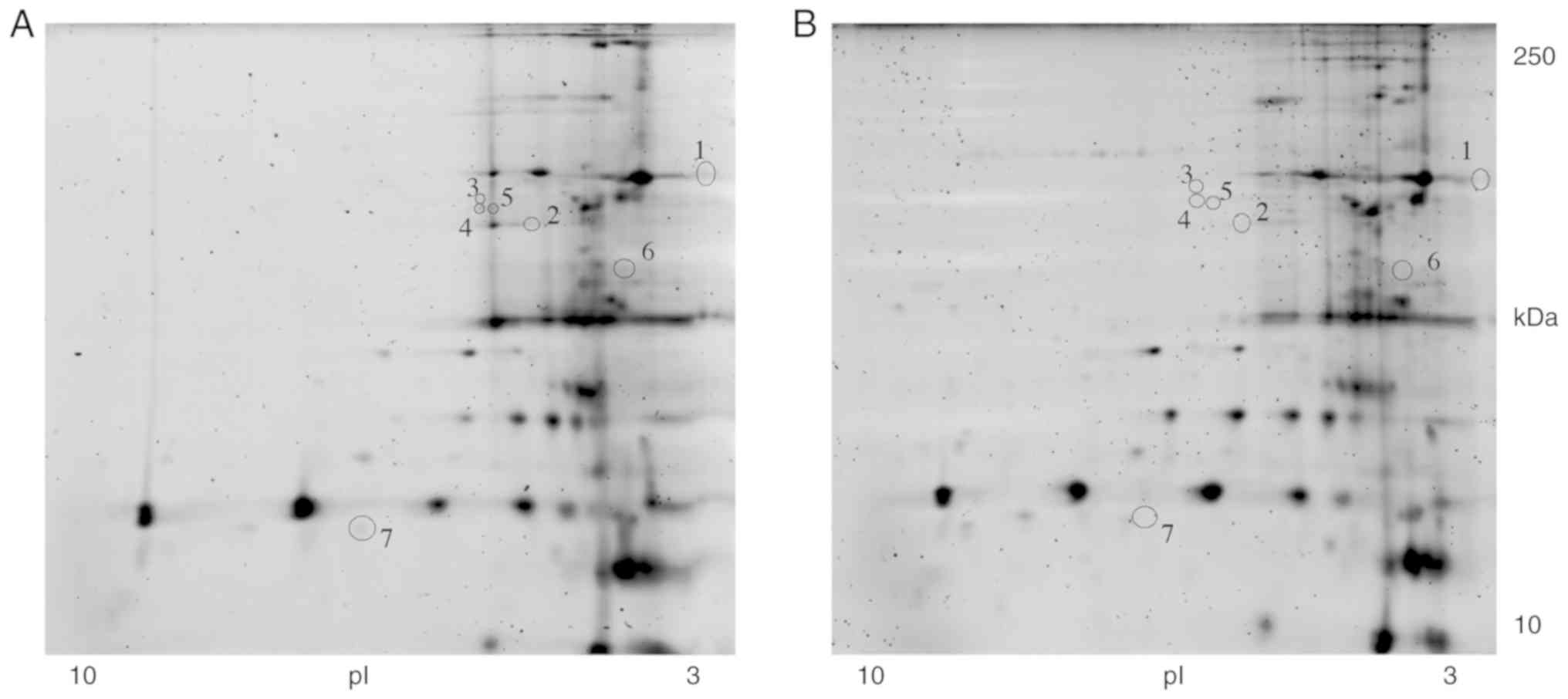
2-DE map of glycoproteins prior to and
following treatment
In order to confirm the restoration of
N-hypoglycosylation following treatment, the bulk of the extracted
proteins were used to analyse the glycosylation pattern of the
CDG-Ia fibroblasts. Total proteins were extracted from the F3 and
HF cultures, and the glycosylated protein fraction was separated as
described above. Subsequently, the glycosylation patterns between
F3 and HF were examined (32).
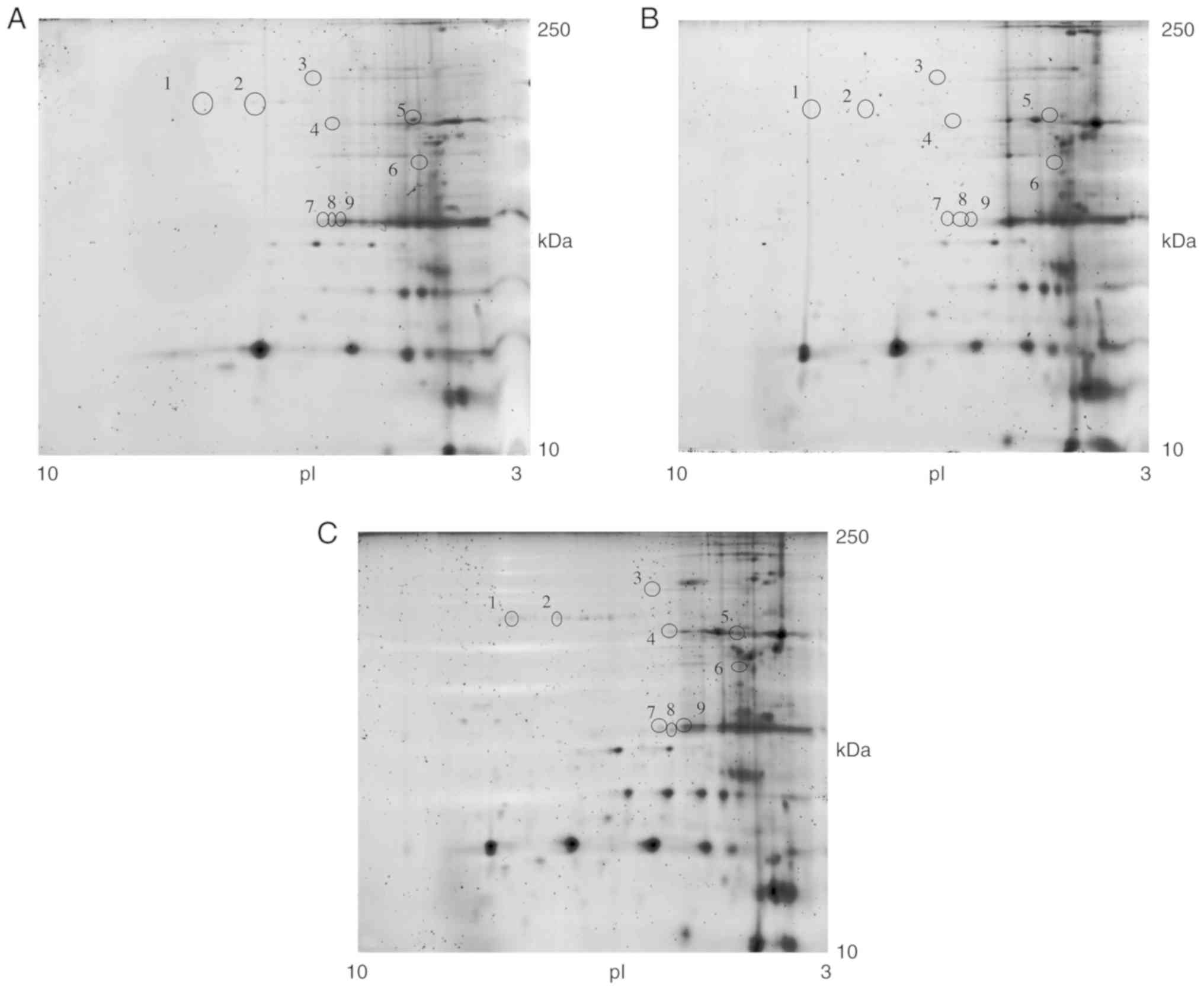
Approximately 450 glycoprotein spots were detected in the 2-DE gels
and the correlation of gel-pairs performed allowed an average
matching efficiency of ~81%. The 2-DE map analysis revealed nine
glycoprotein spots present in the HF culture (Fig. 10A) that were absent in the F3
culture (Fig. 10B).
The F3 culture was incubated for 6 h with 15 ng/well
of GDP-Man (30 μg/ml of GDP-Man-PLGA NPs). The conditioned
medium was subsequently replaced with fresh medium. At 48 h
post-incubation, the cells were harvested and the glycosylated
proteins were separated. Eelectrophoretic analysis of the
glycoprotein extracts of HFs (Fig.
10A), F3 prior to treatment (Fig. 10B) and F3 (GDP-Man NPs) following
treatment (Fig. 10C) were
analysed. The results indicated that, following treatment, the F3
(GDP-Man NPs) retained their normal glycosylation pattern. The
results in Fig. 10A, B and C,
indicate the spots that were present in the control samples, that
the deficient cells did not have spots, and the spots that
reappeared following treatment, respectively. Furthermore, seven
proteins present in the F3 fibroblasts were absent in the F3
(GDP-Man NPs) fibroblasts (Fig. 11A
and B). These results suggest that the treatment may improve
the post-translational protein glycosylation in vitro.
Discussion
Despite several attempts (35), no effective therapy has been
developed to alleviate CDG-Ia symptoms. The present study aimed to
demonstrate the efficacy of the administration of GDP-Man. The
current hypothesis was that the introduction of GDP-Man to the cell
restores the N-glycosylation pathway so that branched
oligosaccharide chains can be mounted on proteins. However, the
addition of free GDP-Man in the medium exerted no effects on the
activities of the enzymes examined. PLGA nanoparticles were used to
deliver GDP-Man into deficient cells. It was possible to produce
two batches, which exhibited optimal features, suggesting that
GDP-Man is a molecule that can be loaded on PLGA NPs.
Fibroblasts were considered an optimal marker to
assess the efficacy of the delivery method. This was achieved by
measuring the activity of lysosomal enzymes. Lysosomal enzymes
represent a class of proteins whose function mainly depends on
oligosaccharide chains that are linked to aspara-gine amino groups.
Assessment of the lysosomal enzymes allowed determination of the
dose of administration, the incubation time and the time required
for the successful delivery of GDP-Man. Of note, the most effective
dose was 15 ng/well, instead of 150 ng/well, whereas the optimal
incubation time was 6 h. The production of lysosomal enzymes is a
process that requires a long period of time, whereas the maximal
specific activity was recorded 48 h following incubation.
Lysosomal enzymes, including α-mannodisase and
β-glucoronidase, were used as reference enzymes to determine
glycosylation in vitro, as previous evidence has
demonstrated that their activity was reduced significantly in
CDG-Ia leukocytes (1). The in
vitro results of the present study demonstrated that GDP-Man
PLGA nanoparticles comprise a successful treatment for CDG-Ia with
optimal efficacy. To date, low levels of glycosylation have been
determined in fibroblast culture medium and in the serum of
patients with xCDG-Ia.
Increases in the serum activity of the α-fucosidase
and β-hexosaminidase enzymes were also noted in these patients. The
lack of GDP-Man impaired the processing of glycoproteins, and
affected the cell metabolism.
CDG-Ia can cause neurological disorders. Therefore,
PLGA nanoparticles are particularly useful due to their ability to
cross the BBB when coated with a short glycopeptide that is linked
to the endothelial μ opioid receptor (g7) (15,16). Recently, Chan et al
reported the generation of the first viable PMM2 hypomorphic mouse
model, which recapitulates several features of the CDG-Ia disease
(36). To the best of our
knowledge, the present study is the first to describe successful
in vitro treatment of a metabolic disorder using a
nanoparticle-mediated therapeutic approach, which is based on the
delivery of GDP-Man.
In conclusion, this model allows the assessment of
glyco-sylation-targeted therapies, and these strategies may be used
to treat CDG-Ia and other glycosylation genetic disorders with
similar aetiology.
Funding
The present study was supported by a grant from the
Institute for Maternal and Child Health IRCCS 'Burlo Garofolo'
(grant no. RC 37/09). DME PhD fellowship was supported by Azzurra
Malattie Rare ONLUS and the University of Trieste (grant no.
30/2013).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BB was involved in the conception and design of the
study, performed the majority of the cellular experiments and
contributed to the drafting and revision of the article. EDM, SB
and RA contributed to the experimental work, analysis, acquisition
and interpretation of data regarding the biochemical experiments.
AT contributed to the experimental work, analysis, acquisition and
interpretation of data regarding the biochemical characterization
of deficient fibroblasts. BU contributed to the experimental work,
analysis, acquisition and interpretation of data regarding the
2-DE. BR contributed to the experimental work regarding the
characterization of GDP-loaded NPs and contributed to the drafting
and revision of the article. MA contributed to the experimental
work, analysis, acquisition and interpretation of data regarding
the genetic characterization of the deficient fibroblasts. GT
contributed to the experimental work regarding the GDP-loaded NP
preparation and characterization, and contributed to the production
of the draft and revision of the article. DD was involved in the
conception and design of the study, analysis and interpretation of
the data, and made a fundamental scientific contribution to the
production of the draft and the revision of the article. GMS
coordinated the activities of all the groups, contributed to the
conception and design of the study, analysis and interpretation of
the data, and made a fundamental scientific contribution for the
production of the draft and the revision of the article. All
authors approved the submission of the present article.
Ethics approval and consent to
participate
Approval was granted by the Human Subjects
Protection Review Board of the Institute for Maternal and Child
Health IRCCS 'Burlo Garofolo' (Technical Scientific Committee of
the Scientific Directorate).
Patient consent for publication
Informed consent was provided by the parents or
legal representatives of subjects involved.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the 'Cell Line and
DNA Biobank from Patients affected by Genetic Diseases' and the
Centro di Diagnostica Genetica e Biochimica delle Malattie
Metaboliche, Istituto G. Gaslini Genova for providing patient
fibroblasts. The abstract was presented at the International
Conference on Nanomedicine and Nanobiotechnology, Sep 28-Sep 30
2016 in Paris, France.
References
|
1
|
Barone R, Carchon H, Jansen E, Pavone L,
Fiumara A, Bosshard NU, Gitzelmann R and Jaeken J: Lysosomal enzyme
activities in serum and leukocytes from patients with
carbohydrate-deficient glycoprotein syndrome type IA
(phosphomannomutase deficiency). J Inherit Metab Dis. 21:167–172.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaeken J: Congenital disorders of
glycosylation. Ann NY Acad Sci. 1214:190–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ichisaka S, Ohno K, Yuasa I, Nanba E,
Sakuraba H and Suzuki Y: Increased expression of
beta-hexosaminidase alpha chain in cultured skin fibroblasts from
patients with carbohydrate-deficient glycoprotein syndrome type I.
Brain Dev. 20:302–306. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schwarze SR, Ho A, Vocero-Akbani A and
Dowdy SF: In vivo protein transduction: Delivery of a biologically
active protein into the mouse. Science. 285:1569–1572. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elson-Schwab L, Garner OB, Schuksz M,
Crawford BE, Esko JD and Tor Y: Guanidinylated neomycin delivers
large, bioactive cargo into cells through a heparan
sulfate-dependent pathway. J Biol Chem. 282:13585–13591. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Snyder EL and Dowdy SF: Cell penetrating
peptides in drug delivery. Pharm Res. 21:389–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Freeze HH: Towards a therapy for
phosphomannomutase 2 deficiency, the defect in CDG-Ia patients.
Biochim Biophys Acta. 1792:835–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eklund EA, Merbouh N, Ichikawa M,
Nishikawa A, Clima JM, Dorman JA, Norberg T and Freeze HH:
Hydrophobic Man-1-P derivatives correct abnormal glycosylation in
type I congenital disorder of glycosylation fibroblasts.
Glycobiology. 15:1084–1093. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mayatepek E, Schröder M, Kohlmüller D,
Bieger WP and Nützenadel W: Continuous mannose infusion in
carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr.
86:1138–1140. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kjaergaard S, Kristiansson B, Stibler H,
Freeze HH, Schwartz M, Martinsson T and Skovby F: Failure of
short-term mannose therapy of patients with carbohydrate-deficient
glycoprotein syndrome type 1A. Acta Paediatr. 87:884–888. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mayatepek E and Kohlmüller D: Mannose
supplementation in carbohydrate-deficient glycoprotein syndrome
type I and phosphomannomutase deficiency. Eur J Pediatr.
157:605–606. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brasil S, Pascoal C, Francisco R,
Marques-da-Silva D, Andreotti G, Videira PA, Morava E, Jaeken J and
Dos Reis Ferreira V: CDG therapies: From bench to bedside. Int J
Mol Sci. 19:E13042018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Parveen S, Misra R and Sahoo SK:
Nanoparticles: A boon to drug delivery, therapeutics, diagnostics
and imaging. Nanomedicine. 8:147–166. 2012. View Article : Google Scholar
|
|
14
|
Tancini B, Tosi G, Bortot B, Dolcetta D,
Magini A, De Martino E, Urbanelli L, Ruozi B, Forni F, Emiliani C,
et al: Use of poly-lactide-co-glycolide-nanoparticles for lysosomal
delivery of a therapeutic enzyme in glycogenosis type II
fibroblasts. J Nanosci Nanotechnol. 15:2657–2666. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Costantino L, Gandolfi F, Tosi G, Rivasi
F, Vandelli MA and Forni F: Peptide-derivatized biodegradable
nanoparticles able to cross the blood-brain barrier. J Control
Release. 108:84–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tosi G, Bortot B, Ruozi B, Dolcetta D,
Vandelli MA, Forni F and Severini GM: Potential use of polymeric
nanoparticles for drug delivery across the blood-brain barrier.
Curr Med Chem. 20:2212–2225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bondioli L, Costantino L, Ballestrazzi A,
Lucchesi D, Boraschi D, Pellati F, Benvenuti S, Tosi G and Vandelli
MA: PLGA nanopar-ticles surface decorated with the sialic acid,
N-acetylneuraminic acid. Biomaterials. 31:3395–3403. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosca ID, Watari F and Uo M: Microparticle
formation and its mechanism in single and double emulsion solvent
evaporation. J Control. 99:271–280. 2004.
|
|
19
|
Ruozi B, Belletti D, Forni F, Sharma A,
Muresanu D, Mössler H, Vandelli MA, Tosi G and Sharma HS: Poly
(D,L-lactide-co-glycolide) nanoparticles loaded with cerebrolysin
display neuroprotective activity in a rat model of concussive head
injury. CNS Neurol Disord Drug Targets. 13:1475–1482. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Joshi DP, Lan-Chun-Fung YL and Pritchard
JG: Determination of poly(vinyl alcohol) via its complex with boric
acid and iodine. Anal Chim Acta. 104:153–160. 1979. View Article : Google Scholar
|
|
21
|
Belletti D, Grabrucker AM, Pederzoli F,
Menrah I, Vandelli MA, Tosi G, Duskey TJ, Forni F and Ruozi B:
Hybrid nanoparticles as a new technological approach to enhance the
delivery of cholesterol into the brain. Int J Pharm. 543:300–310.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pirard M, Achouri Y, Collet JF, Schollen
E, Matthijs G and Van Schaftingen E: Kinetic properties and
tissular distribution of mammalian phosphomannomutase isozymes.
Biochem J. 339:201–207. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barone R, Carrozzi M, Parini R, Battini R,
Martinelli D, Elia M, Spada M, Lilliu F, Ciana G, Burlina A, et al:
A nationwide survey of PMM2-CDG in Italy: High frequency of a mild
neurological variant associated with the L32R mutation. J Neurol.
262:154–164. 2015. View Article : Google Scholar
|
|
24
|
Vega AI, Pérez-Cerdá C, Abia D, Gámez A,
Briones P, Artuch R, Desviat LR, Ugarte M and Pérez B: Expression
analysis revealing destabilizing mutations in phosphomannomutase 2
deficiency (PMM2-CDG): Expression analysis of PMM2-CDG mutations. J
Inherit Metab Dis. 34:929–939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grünewald S, Schollen E, Van Schaftingen
E, Jaeken J and Matthijs G: High residual activity of PMM2 in
patients' fibroblasts: Possible pitfall in the diagnosis of CDG-Ia
(phosphomannomutase deficiency). Am J Hum Genet. 68:347–354. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bortot B, Cosentini D, Faletra F, Biffi S,
De Martino E, Carrozzi M and Severini GM: PMM2-CDG: Phenotype and
genotype in four affected family members. Gene. 531:506–509. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Monaco I, Arena F, Biffi S, Locatelli E,
Bortot B, La Cava F, Marini GM, Severini GM, Terreno E and Comes
Franchini M: Synthesis of lipophilic core-shell
Fe3O4 @SiO2 @Au nanoparticles and
polymeric entrapment into nanomicelles: A novel nanosystem for in
vivo active targeting and magnetic resonance-photoacoustic dual
imaging. Bioconjug Chem. 28:1382–1390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carcoforo P, Ura B, Mischiati C,
Squerzanti M, Lanzara V, Cervellati C, Calza R, De Laureto PP,
Frare E, Portinari M, et al: Comparative proteomic analysis of
ductal breast carcinoma demonstrates an altered expression of
chaperonins and cytoskeletal proteins. Mol Med Rep. 7:1700–1704.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sahay G, Alakhova DY and Kabanov AV:
Endocytosis of nano-medicines. J Control Release. 145:182–195.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu Q, Gu G, Liu Z, Jiang M, Kang T, Miao
D, Tu Y, Pang Z, Song Q, Yao L, et al: F3 peptide-functionalized
PEG-PLA nanoparticles co-administrated with tLyp-1 peptide for
anti-glioma drug delivery. Biomaterials. 34:1135–1145. 2013.
View Article : Google Scholar
|
|
31
|
Rush JS, Panneerselvam K, Waechter CJ and
Freeze HH: Mannose supplementation corrects GDP-mannose deficiency
in cultured fibroblasts from some patients with Congenital
Disorders of Glycosylation (CDG). Glycobiology. 10:829–835. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kleinert P, Kuster T, Arnold D, Jaeken J,
Heizmann CW and Troxler H: Effect of glycosylation on the protein
pattern in 2-D-gel electrophoresis. Proteomics. 7:15–22. 2007.
View Article : Google Scholar
|
|
33
|
Cartiera MS, Johnson KM, Rajendran V,
Caplan MJ and Saltzman WM: The uptake and intracellular fate of
PLGA nano-particles in epithelial cells. Biomaterials.
30:2790–2798. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Van Scherpenzeel M, Willems E and Lefeber
DJ: Clinical diagnostics and therapy monitoring in the congenital
disorders of glycosylation. Glycoconj J. 33:345–358. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chan B, Clasquin M, Smolen GA, Histen G,
Powe J, Chen Y, Lin Z, Lu C, Liu Y, Cang Y, et al: A mouse model of
a human congenital disorder of glycosylation caused by loss of
PMM2. Hum Mol Genet. 25:2182–2193. 2016. View Article : Google Scholar : PubMed/NCBI
|