Introduction
Non-alcoholic fatty liver disease (NAFLD) has become
a major public health concern, affecting over half a billion people
worldwide, and has been associated with a variety of metabolic
comorbidities (1). Hepatic
steatosis, as a key metabolic hallmark of NAFLD, is mainly caused
by excessive fat accumulation. An overload of free fatty acids,
particularly saturated free fatty acids, may induce the endoplasmic
reticulum (ER) stress response (2,3).
Additionally, the presence of non-functional unfolded or misfolded
proteins and subsequent ER stress may result in reduced lipid
droplet stability and increased lipogenesis, aggravating NAFLD
(4). Importantly, in the livers
of patients with NAFLD and animal models of diet-induced obesity,
chronic ER stress and prolonged activation of the unfolded protein
response (UPR) was observed; these factors play a key role in the
development of hepatic steatosis (5-7).
Erythropoietin (EPO), a glycoprotein hormone, has
been traditionally considered as a key regulator of erythropoiesis
(8,9); however, its protective role has
recently been extended to ameliorating metabolic disorders
(10). Specifically, EPO was
demonstrated to promote brown fat-like characteristics to increase
fatty acid oxidation in white adipose tissue from obese mice
(11); EPO was reported to
decrease adipose tissue mass by increasing fat utilization in
skeletal muscles from obese mice or humans during aerobic exercise
(12,13). In liver tissues, EPO was found to
reduce insulin resistance via peroxisome proliferator-activated
receptor γ (PPARγ)-dependent protein kinase B (AKT) activation, and
alleviate steatosis, partially via lipophagy (14,15); however, the mechanism underlying
the role of EPO in hepatic lipid metabolism, ER stress in
particular, remains unknown.
Sirtuin 1 (SIRT1), a member of the sirtuin family of
nicotinamide adenine dinucleotide (NAD+)-dependent
protein deacetylases, regulates key aspects of lipid metabolism
(16,17). SIRT1 can deacetylate protein
kinase-like ER kinase (PERK) and inhibit the PERK-eukaryotic
initiation factor 2α (eIF2α) axis of the UPR pathway, thereby
suppressing hepatic steatosis in mice (18-20). Our previous study revealed that
the NAD+/NADH ratio and SIRT1 activity increased in
response to EPO in HepG2 cells in vitro (15). However, the role of SIRT1 in
association with EPO in ER stress remains unclear. In addition,
SIRT1-induced fibroblast growth factor 21 (FGF21) expression was
found to play a critical role in controlling obesity and energy
balance (21,22).
The aim of the present study was to investigate the
effects of EPO on hepatic ER stress and FGF21 expression, and
determine whether this mechanism involves SIRT1.
Materials and methods
Animal model
All animal studies were performed in accordance with
the National Institutes of Health guidelines and were approved by
the Nanjing University Medical School Institutional Animal Care and
Use Committee. Male hepatocyte-specific SIRT1-deleted (SIRT1-LKO)
mice and wild-type (WT, C57BL/6J) littermates (8 weeks old) were
purchased from the Model Animal Research Center of Nanjing
University (Nanjing, China). The mice were housed under controlled
temperature (20-22°C) and humidity (50%) conditions, with a 12-h
light/dark cycle. All mice were given free access to high-fat diet
(HFD; 60% calories from fat, 25% calories from carbohydrates and
15% calories from protein; Yangzhou University, Yangzhou, China) or
standard chow (NC; 10% calories from fat, 75% calories from
carbohydrates and 15% calories from protein; Yangzhou University),
as described below. The mice were intraperitoneally injected with
3,000 U/kg of recombinant human EPO (Sunshine Pharmaceutical) or an
equivalent volume of PBS every other day (n=6 per group). For the
first experiment, SIRT1-LKO mice and WT littermates were fed HFD
for 12 weeks, and then divided into two groups prior to treatment
with EPO or PBS for 5 weeks. For the second experiment, C57BL/6J
mice were treated with EPO or PBS for 7 or 14 days; the mice were
given free access to NC and water. Finally, for the third
experiment, C57BL/6J mice were fed HFD or NC for 12 weeks; the HFD
group was intraperitoneally injected with EPO or PBS for 5 weeks.
Lean littermate control mice were intraperitoneally injected with
an equivalent volume of PBS. Body weight and fat were measured
weekly (AccuFat-1050, MAG-MED). After 5 weeks, the mice were fasted
for 8 h and underwent an intraperitoneal glucose tolerance test
(IPGTT), as previously described (14). Glucose levels were determined from
blood samples obtained via the tail veil at 0, 30, 60 and 120 min
following glucose administration (1.5 g/kg). Subsequently, all the
mice were deprived of food for 8 h prior to sacrifice for the
collection of blood samples and liver tissues for further
examination. After being weighed, fresh liver tissues were
subjected to hematoxylin and eosin (H&E; Goodbio) and Oil Red O
staining (Goodbio) (15). For
immunofluorescence, the sections were incubated with rabbit
anti-protein disulfide isomerase (PDI) antibody (1:500; Cell
Signaling Technology, Inc.; cat. no. 3501). For
immunohistochemistry (IHC), the sections were incubated with rabbit
anti-FGF21 antibody (1:200; Abcam; cat. no. ab66564) and
quantitated by ImageJ plugin IHC profiler (National Institutes of
Health), as previously described (23-25). Liver triglyceride (TG) content was
measured using an ELISA kit according to the manufacturer's
instructions (BioVision, Inc.). All samples were stored at −80°C
for further analysis.
Cell culture
Primary hepatocytes were isolated from 8-12-week-old
C57BL6/J mice (20-25 g) using a two-step perfusion method, and
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (FBS), as described previously
(26). For pharmacological
studies, cells were treated with 40 U/ml EPO, 0.3 mmol/l palmitate
(PA; Sigma-Aldrich; Merck KGaA) or 1 µmol/l thapsigargin
(Thp; Sigma-Aldrich; Merck KGaA) for 24 h. Where indicated, the
cells were pre-treated with 40 µmol/l resveratrol
(Sigma-Aldrich; Merck KGaA) or 2 µmol/l EX-527 (MedChem
Express, LLC) for 1 h prior to EPO treatment. Primary hepatocytes
isolated from EPO-treated and control mice were incubated with 1, 2
and 10 nmol/l human recombinant FGF21 (Abcam) for 4 h; vehicle
treatment served as control.
Small interfering RNA (siRNA)
transfection
Primary hepatocytes were transfected with siRNA
against SIRT1 (GenePharma Co., Ltd.) using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.). PA
and EPO were added to the cells at 36 h post-transfection. The
siRNA sequences were as follows: SIRT1 siRNA,
5′-GAUGAAGUUGACCUCCUCA-3′; and negative control,
5′-UUCUCCGAACGUGUCACGUTT-3′.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qRCR)
analysis
Total RNA was extracted from mouse liver tissue
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). RT of RNA into cDNA was performed with the RT
reagent kit (Takara Bio, Inc.), followed by qPCR analysis on an ABI
StepOne Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using SYBR Green (Roche Diagnostics) at a final
volume of 20 µl, according to the manufacturer's protocol.
The primers used were as follows: FGF21, forward
5′-CCTCCAGTTTGGGGGTCAAG-3′ and reverse 5′-CTGGTTTGGGGAGTCCTTCT-3′;
peroxisome proliferator-activated receptor-γ coactivator-1α
(PGC-1α), forward 5′-GTCCTTCCTCCATGCCTGAC-3′ and reverse
5′-TAGCTGAGCTGAGTGTTGGC-3′; FGF receptor 1 (FGFR1), forward
5′-GTGGAGAATGAGTATGGGAGC-3′ and reverse 5′-GGATCTGGACATACGGCAAG-3′;
single-pass transmembrane protein βKlotho, forward
5′-ACGAGGGCTGTTTTATGTGG-3′ and reverse 5′-CAGGTGAGGATCGGTAAACTG-3′;
acyl-CoA oxidase 1 (Acox1), forward 5′-TGCCATTCGATACAGTGCTG-3′ and
reverse 5′-CAGGAGCGGGAAGAGTTTATAC-3′; pyruvate dehydrogenase kinase
4, isoenzyme 4 (Pdk4), forward 5′-GGATTACTGACCGCCTCTTTAG-3′ and
reverse 5′-GGGAGCTTTTCTACAGACTCAG-3′; enoyl-CoA hydratase and
3-hydroxyacyl CoA dehydrogenase (Ehhadh), forward
5′-AGCTGTTTATGTACCTTCGGG-3′ and reverse
5′-CTGCTTTGGGTCTGACTCTAC-3′; glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), forward 5′-TGGCCTTCCGTGTTTCCTAC-3′ and
reverse 5′-GAGTTGCTGTTGAAGTCGCA-3′. Expression was normalized to
GAPDH and determined via the 2−ΔΔCq method (27).
Western blot analysis
Liver tissues or cultured hepatocytes were lysed
with radioimmunoprecipitation assay lysis buffer (Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with protease
inhibitor cocktail (Roche Diagnostics). Total protein concentration
was measured using the bicinchoninic acid method (BCA protein assay
kit; Thermo Fisher Scientific, Inc.). Subsequently, 20 µg of
each sample was separated via 8-15% Bis-Tris NuPAGE gels (Goodbio)
and transferred to polyvinylidene difluoride membranes (Invitrogen;
Thermo Fisher Scientific, Inc.). The membranes were blocked with 5%
non-fat milk or bovine serum albumin, and incubated overnight at
4°C with specific antibodies. Antibodies against SIRT1 (rabbit
monoclonal, cat. no. 9475, 1:1,000), PERK (rabbit monoclonal, cat.
no. 3192, 1:1,000), phosphorylated (p)-eIF2α (Ser51, rabbit
monoclonal, cat. no. 3398, 1:1,000), eIF2α (rabbit monoclonal, cat.
no. 5324, 1:1,000) and GAPDH (rabbit monoclonal, cat. no. 5174,
1:2,000) were obtained from Cell Signaling Technology, Inc. The
anti-FGF21 antibody (rabbit polyclonal, cat. no. ab66564, 1:1,000)
and glucose-regulated protein 78 (GRP78; rabbit polyclonal, cat.
no. ab21685, 1:1,000) were purchased from Abcam. The anti-p-PERK
antibody (Thr981, rabbit polyclonal, cat. no. YP1055, 1:1,000) was
obtained from ImmunoWay Biotechnology. The membranes were then
washed and incubated with a secondary antibody (goat anti-rabbit
IgG, ZSGB-BIO; OriGene Technologies, Inc.) at room temperature for
1 h. The antibody-antigen complexes were visualized by enhanced
chemiluminescence (ECL; EMD Millipore). Band intensities were
quantified using ImageJ software (National Institutes of Health).
All antibodies were validated by the manufacturer.
FGF21 levels
According to the manufacturer's instructions, the
levels of FGF21 in mouse serum and cell culture supernatant were
determined using a FGF21 ELISA kit (cat. no. ab212160, Abcam).
Briefly, after equilibrating all reagents at room temperature, 50
µl of samples or standards were added to the wells, followed
by the antibody cocktail. After 1 h of incubation, the wells were
washed three times to remove unbound material. Subsequently, 100
µl 3,3′,5,5′-tetramethylbenzidene substrate was added to
each well and incubated for 15 min at room temperature in the dark.
The reaction was terminated by adding 50 µl stop solution
and the optical density was recorded at 450 nm.
Statistical analysis
All analyses were performed using SPSS software
(version 22.0, IBM Corp.). All data are expressed as the mean ±
standard error of the mean. Differences between multiple groups
were determined by performing one-way ANOVA followed by the least
significant difference or Dunnett's T3 post hoc test. Student
t-tests were used to assess differences between two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
EPO-induced suppression of hepatic
steatosis and reductions in body weight are compromised by
hepatic-specific SIRT1 deletion in HFD-fed mice
After 5 weeks of treatment, EPO-treated HFD-fed mice
exhibited weight loss and reduced body fat compared with control
mice (Fig. 1A and B).
Additionally, IPGTTs revealed that the blood glucose levels were
lower in EPO-treated mice compared with those in the PBS-treated
group (Fig. 1C). Furthermore,
histological analysis, including H&E and Oil Red O staining,
demonstrated that EPO markedly alleviated hepatic steatosis in
HFD-fed WT mice (Fig. 1D).
Consistently, liver TG levels and liver weight were decreased in
response to EPO treatment in the WT group (Fig. 1E and F). Compared with WT mice,
these beneficial effects on body weight and hepatic steatosis were
not observed in SIRT1-LKO mice. These results suggested a critical
role for hepatic SIRT1 in mediating the protective effects of EPO
in attenuating obesity and hepatic lipid accumulation.
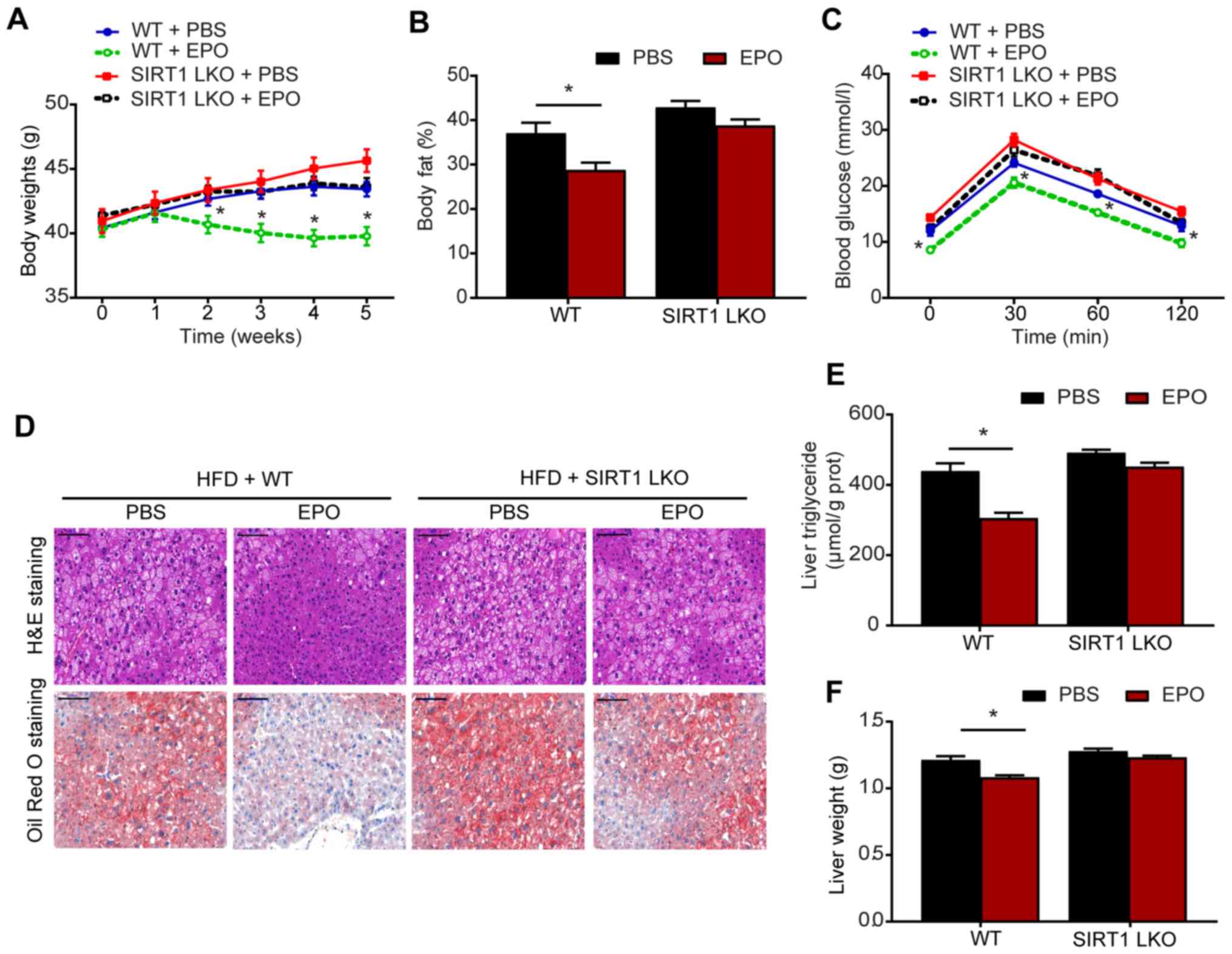 | Figure 1EPO-induced reductions in hepatic
steatosis and body weight were compromised by hepatic
specific-deletion of SIRT1 in HFD-fed mice. (A) Body weight, (B)
body fat ratio and (C) IPGTTs were conducted following treatment.
(D) H&E and Oil Red O staining (scale bar, 100 μm), and
analysis of (E) triglyceride (TG) content and (F) liver weight were
performed on the livers of WT and SIRT1-LKO mice. Data are
presented as the mean ± standard error of the mean (n=6/group,
*P<0.05 vs. WT + PBS group). EPO, erythropoietin; SIRT1, sirtuin
1; HFD, high-fat diet; IPGTTs, intraperitoneal glucose tolerance
tests; H&E, hematoxylin and eosin; WT, wild-type; SIRT1-LKO,
hepatocyte-specific SIRT1-deleted. |
EPO suppresses hepatic ER stress in a
SIRT1-dependent manner
ER stress plays an important role in the development
of hepatic steatosis (6). To
investigate whether EPO alleviates metabolic ER stress, we measured
the levels of the specific ER stress markers PDI and GRP78, and
investigated PERK/eIF2α signaling in the UPR pathway. In HFD-fed
mice, EPO intervention notably inhibited the expression of PDI,
GRP78, p-PERK and p-eIF2α (Fig. 2A
and B). In vitro, EPO-treated primary hepatocytes were
incubated with PA or the ER stress inducer Thp. Treatment with PA
or the ER stress agonist promoted the expression of GRP78, p-PERK
and p-eIF-2α, in parallel with an increase in TG content,
suggesting that activation of ER stress may induce hepatic
steatosis. Conversely, the upregulation of these ER stress markers
and increased TG content induced by Thp and PA were suppressed by
EPO (Figs. 3A and S1).
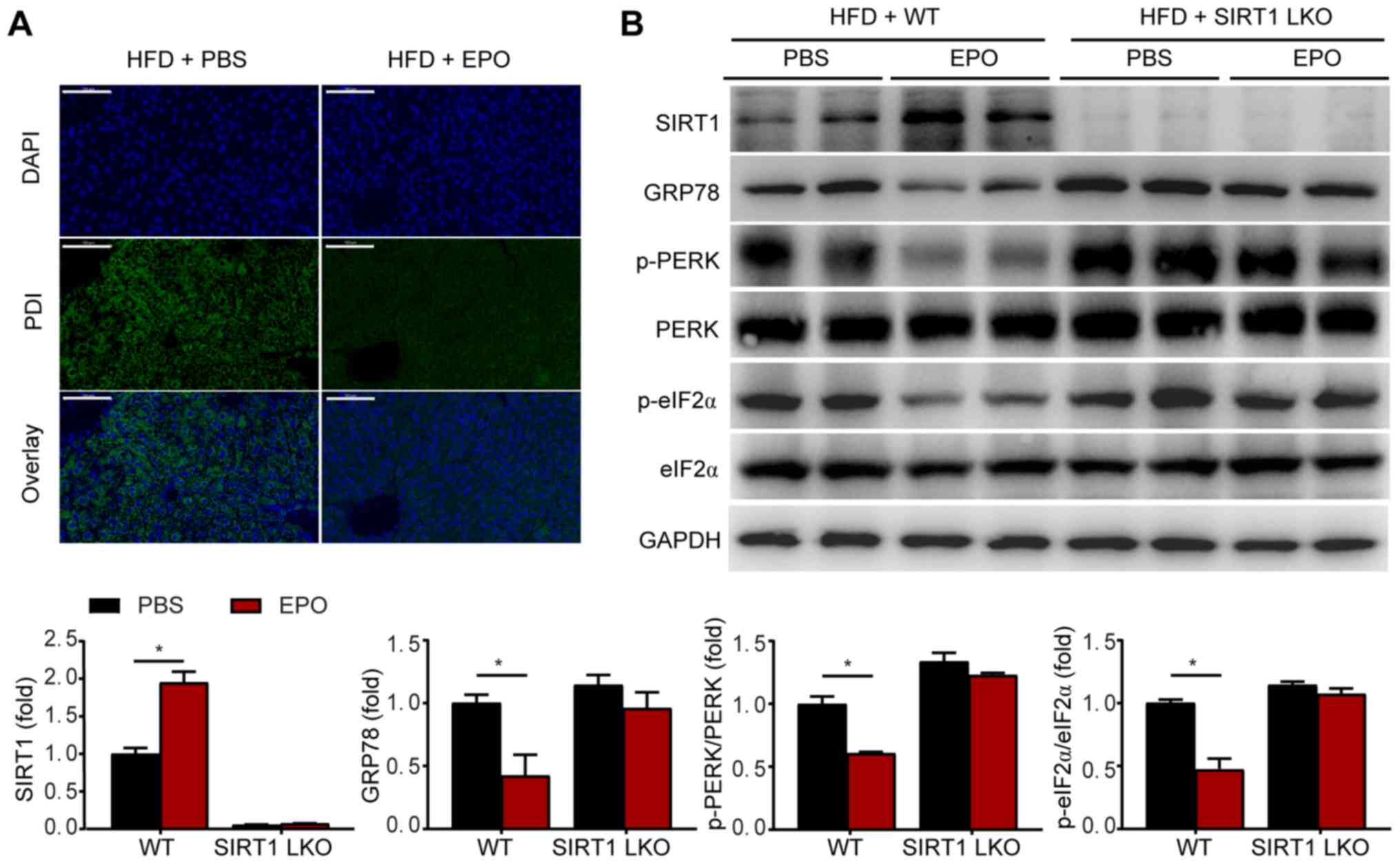 | Figure 2The effects of EPO on inhibiting
hepatic ER stress were eliminated in SIRT1-LKO mice. (A) The ER
stress marker PDI was detected by immunofluorescence staining in
liver tissues of HFD-fed WT mice with or without EPO intervention.
Green dots indicate PDI expression. Scale bar, 50 µm. (B)
Following EPO treatment, hepatic SIRT1, GRP78, p-PERK/PERK and
p-eIF2α/eIF2α protein expression were determined by western
blotting in the livers of EPO-treated WT and SIRT1-LKO mice. Data
are presented as the mean ± standard error of the mean (n=6/group,
*P<0.05 vs. WT + PBS group). EPO, erythropoietin; ER,
endoplasmic reticulum; SIRT1-LKO, hepatocyte-specific
SIRT1-deleted; WT, wild-type; HFD, high-fat diet; PDI, protein
disulfide isomerase; GRP78, glucose-regulated protein 78; p-PERK,
phosphorylation of PRKR-like endoplasmic reticulum kinase; p-eIF2α,
phosphorylation of eukaryotic translation initiation factor 2α. |
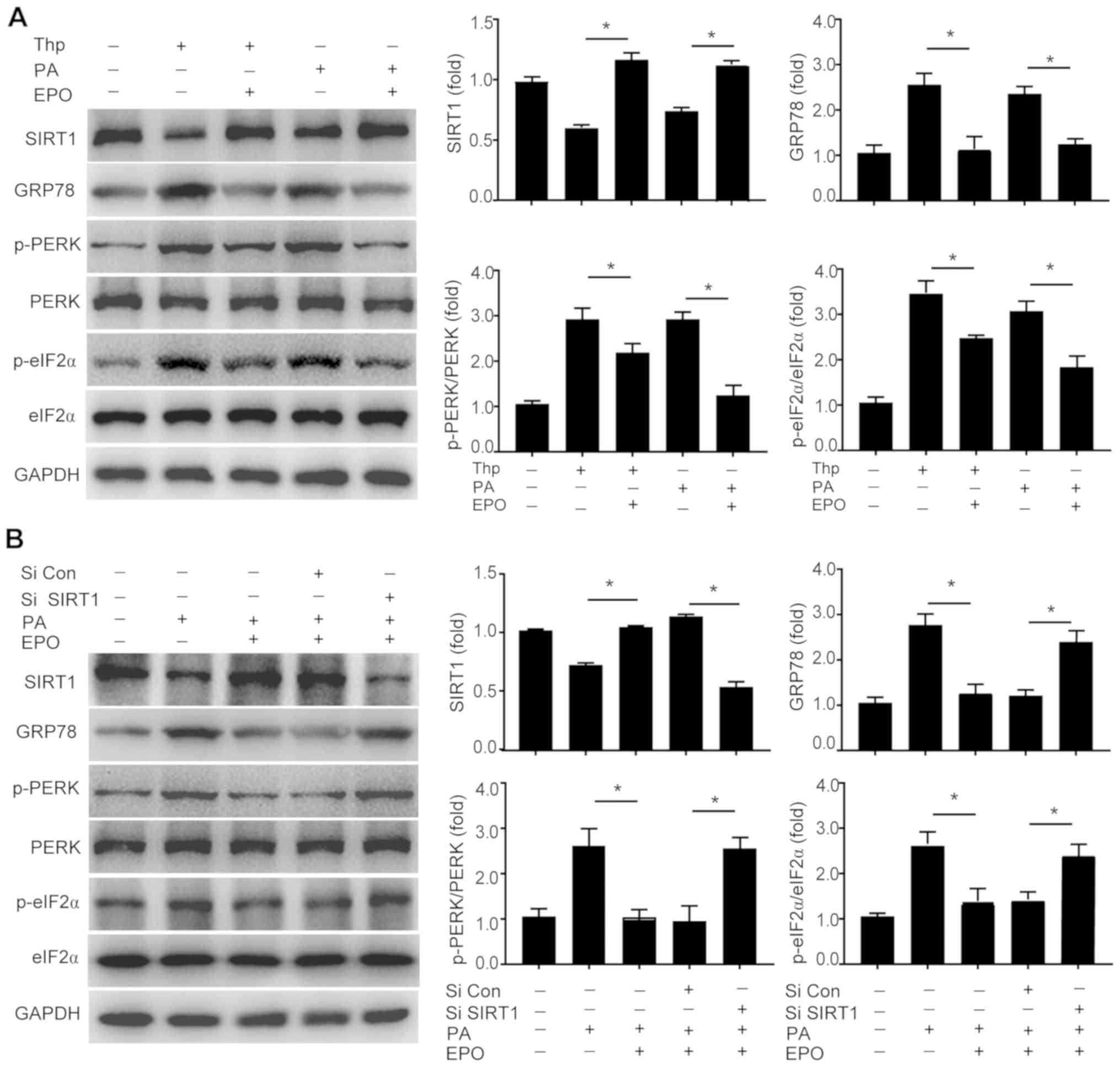 | Figure 3EPO inhibits lipid-induced hepatic ER
stress in a SIRT1-dependent manner. The protein expression levels
of SIRT1, GRP78, p-PERK/PERK and p-eIF2α/eIF2α protein expression
were determined (A) in Thp- or PA-treated primary hepatocytes, and
(B) in hepatocytes treated with SIRT1-targeting siRNA. Data are
presented as the mean ± standard error of the mean (n=3 independent
experiments, *P<0.05). EPO, erythropoietin; ER,
endoplasmic reticulum; SIRT1, sirtuin 1; GRP78, glucose-regulated
protein 78; p-PERK, phosphorylation of PRKR-like endoplasmic
reticulum kinase; p-eIF2α, phosphorylation of eukaryotic
translation initiation factor 2α; Thp, thapsigargin; PA, palmitate;
siRNA, small interfering RNA. |
SIRT1, a potent regulator of energy metabolism and
stress response, may play an important role in fatty liver and
obesity (18,28). The present study investigated
whether SIRT1 mediates the effects of EPO on hepatic ER stress and
steatosis. As shown in Fig. 2B,
no notable alterations in the expression of GRP78, p-PERK or
p-eIF2α were observed in SIRT1-LKO mice following EPO treatment. In
addition, the effects of EPO on decreasing the protein expression
levels of GRP78, p-PERK and p-eIF2α were attenuated following SIRT1
inhibition by siRNA-mediated silencing in PA-treated primary
hepatocytes (Figs. 3B and
S2). Collectively, these
findings indicate that SIRT1 may mediate the effects of EPO on
alleviating hepatic ER stress and lipid accumulation.
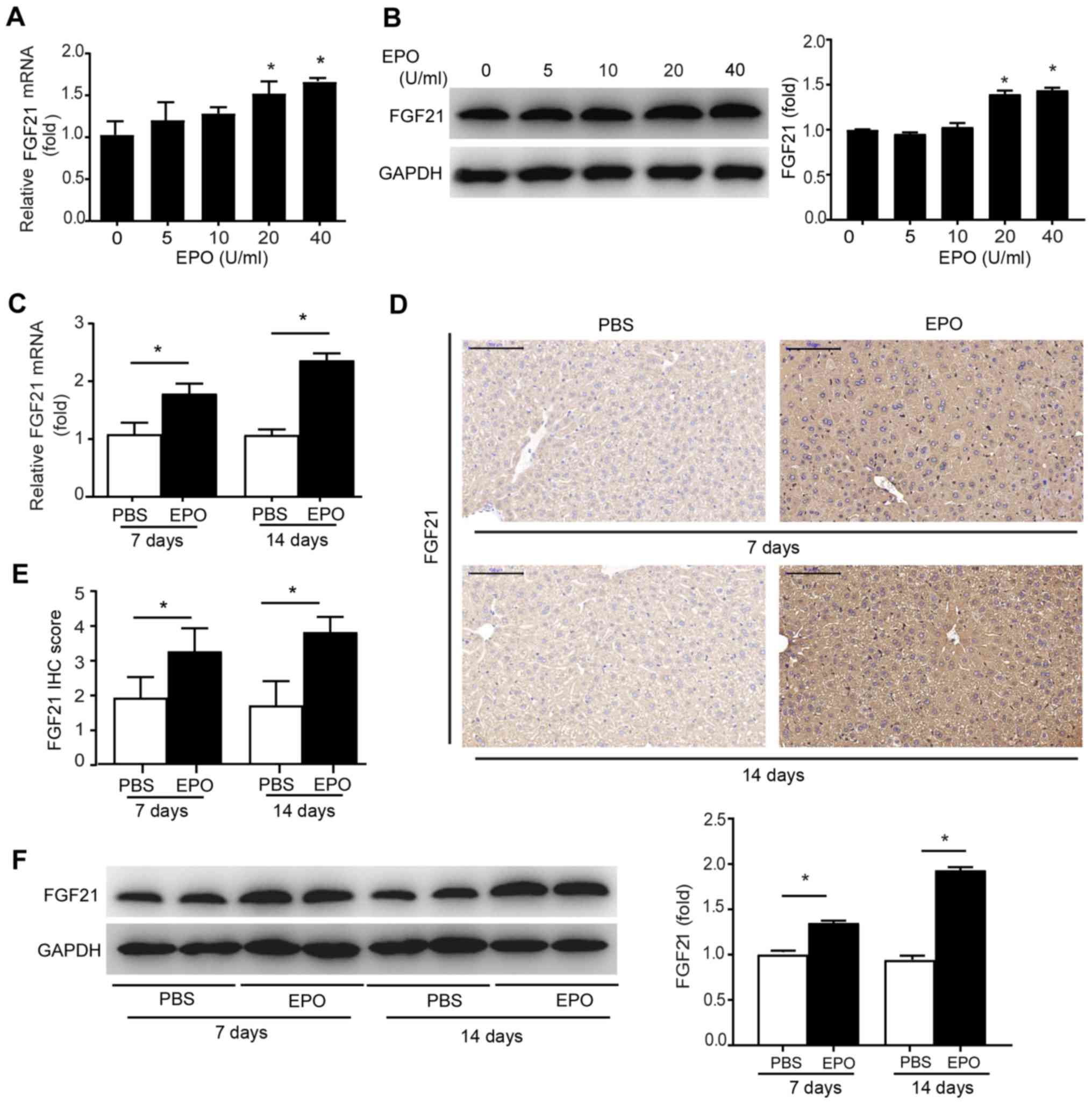
EPO increases hepatic FGF21
expression
In view of the notable effects of EPO treatment on
the liver and whole-body metabolism, it was hypothesized that a
hepatokine may mediate the effects of EPO on alleviating hepatic
lipid accumulation and obesity. Thus, the expression of FGF21, a
hormone secreted by the liver that acts as a potent regulator of
lipid metabolism, was analyzed (29). The results revealed that the mRNA
and protein expression levels of FGF21 were increased in primary
hepatocytes following treatment with 20-40 U/ml EPO (Fig. 4A and B), which was consistent with
the alterations in FGF21 expression in cell culture (Fig. S3A). The results of PCR, western
blotting and IHC revealed that short-term EPO treatment for 7 or 14
days in mice fed NC promoted FGF21 mRNA and protein expression in
liver tissues (Fig. 4C-F), which
was accompanied by increased levels of circulating FGF21 (Fig. S3B). Moreover, previous evidence
indicated that SIRT1-mediated activation of FGF21 prevented liver
steatosis and obesity (21).
Therefore, whether SIRT1 is required for EPO-induced FGF21
expression was investigated. As shown in Fig. 5A, EPO induced the expression of
SIRT1 and FGF21 in hepatocytes, mimicking the effects of the SIRT1
agonist resveratrol. Conversely, the SIRT1 inhibitor EX-527
suppressed the upregulated expression of FGF21 mediated by EPO. In
addition, EPO treatment increased the expression levels of FGF21 in
the circulation and liver tissues of HFD-fed WT mice, but this
effect was mitigated in mice harboring a hepatic-specific deletion
of SIRT1 (Figs. 5B and S3C). Collectively, these results
suggest that SIRT1 is required for EPO-stimulated FGF21 production
in hepatocytes.
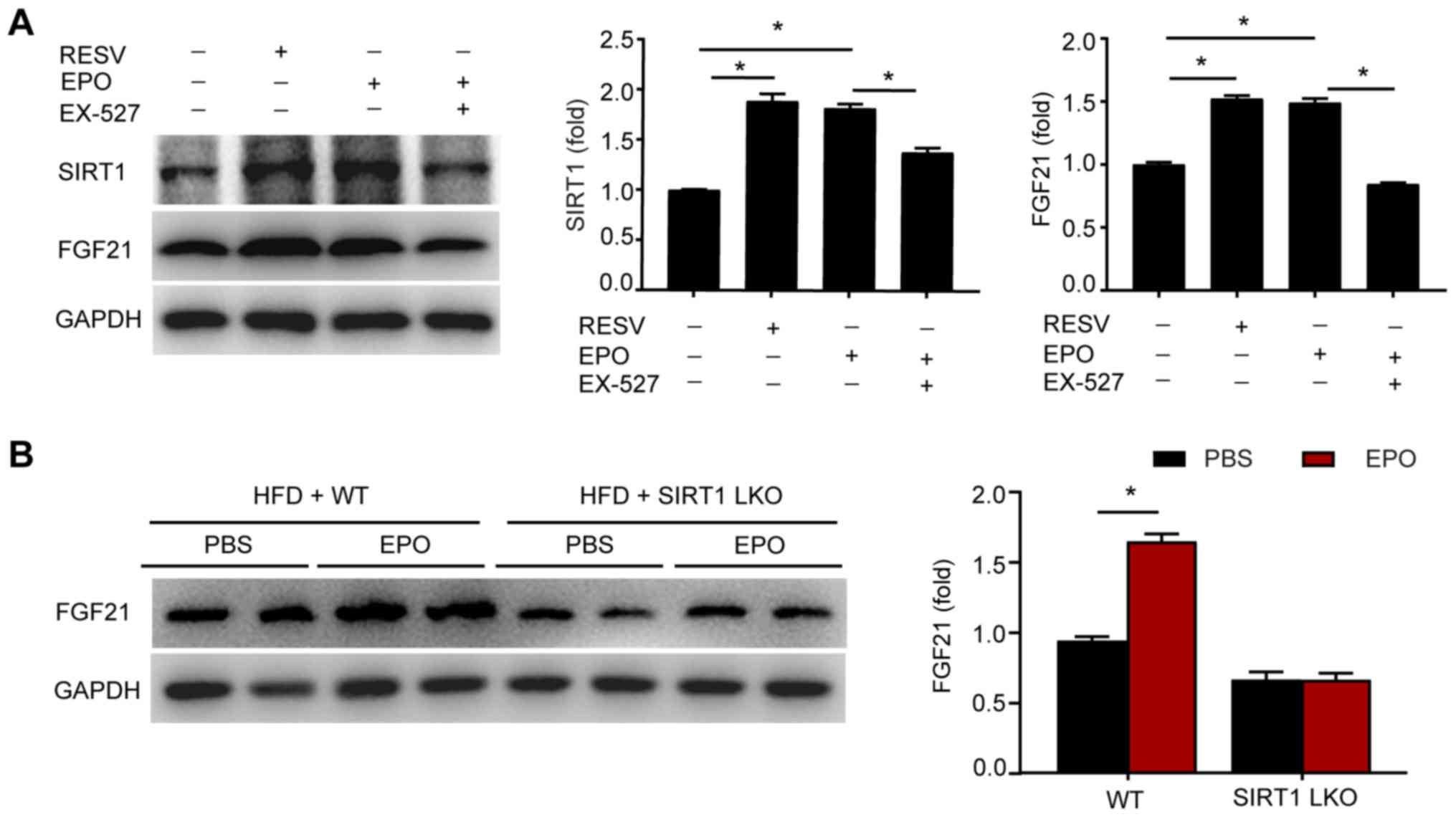 | Figure 5SIRT1 is required for EPO-induced
FGF21 expression. (A) The protein expression levels of SIRT1 and
FGF21 were determined by western blotting in primary hepatocytes
exposed to resveratrol, EPO or EX527. Data are shown as the mean ±
standard error of the mean (n=3 independent experiments,
*P<0.05). (B) The protein expression levels of FGF21
were determined in the liver tissues of HFD-fed WT and SIRT1-LKO
mice. Data are shown as the mean ± standard error of the mean
(n=6/group, *P<0.05 vs. WT + PBS group). EPO,
erythropoietin; SIRT1, sirtuin 1; FGF21, fibroblast growth factor
21; HFD, high-fat diet; WT, wild type; SIRT1-LKO,
hepatocyte-specific SIRT1-deleted. RESV, resveratrol. |
EPO partially restores sensitivity of
hepatocytes to FGF21 in HFD-fed mice
An HFD diet was reported to suppress the expression
of hepatic FGF21 receptors, including FGFR1 and βKlotho, and
suggested that obesity occurs under FGF21-resistant conditions
(30). In the livers of HFD-fed
mice, EPO treatment partially restored the expression of the
aforementioned receptors (Fig. 6A and
B). Additionally, Acox1, Pdk4 and Ehhadh have been proposed as
downstream target genes of the FGF21/PGC-1α axis associated with
hepatic lipid β-oxidation (30,31). The mRNA expression levels of these
genes were decreased in the livers of mice administered an HFD-fed
challenge. Conversely, EPO intervention restored the expression of
PGC-1α, Acox1 and Ehhadh, but not Pdk4 (Fig. 6C and D). In addition,
FGF21-induced phosphorylation of eIF2α was inhibited in mouse
hepatocytes isolated from HFD mice; however, FGF21 intervention
enhanced eIF2α phosphorylation (Fig.
6E). Collectively, these results demonstrated that EPO
treatment enhanced FGF21 sensitivity in response to alterations in
metabolism.
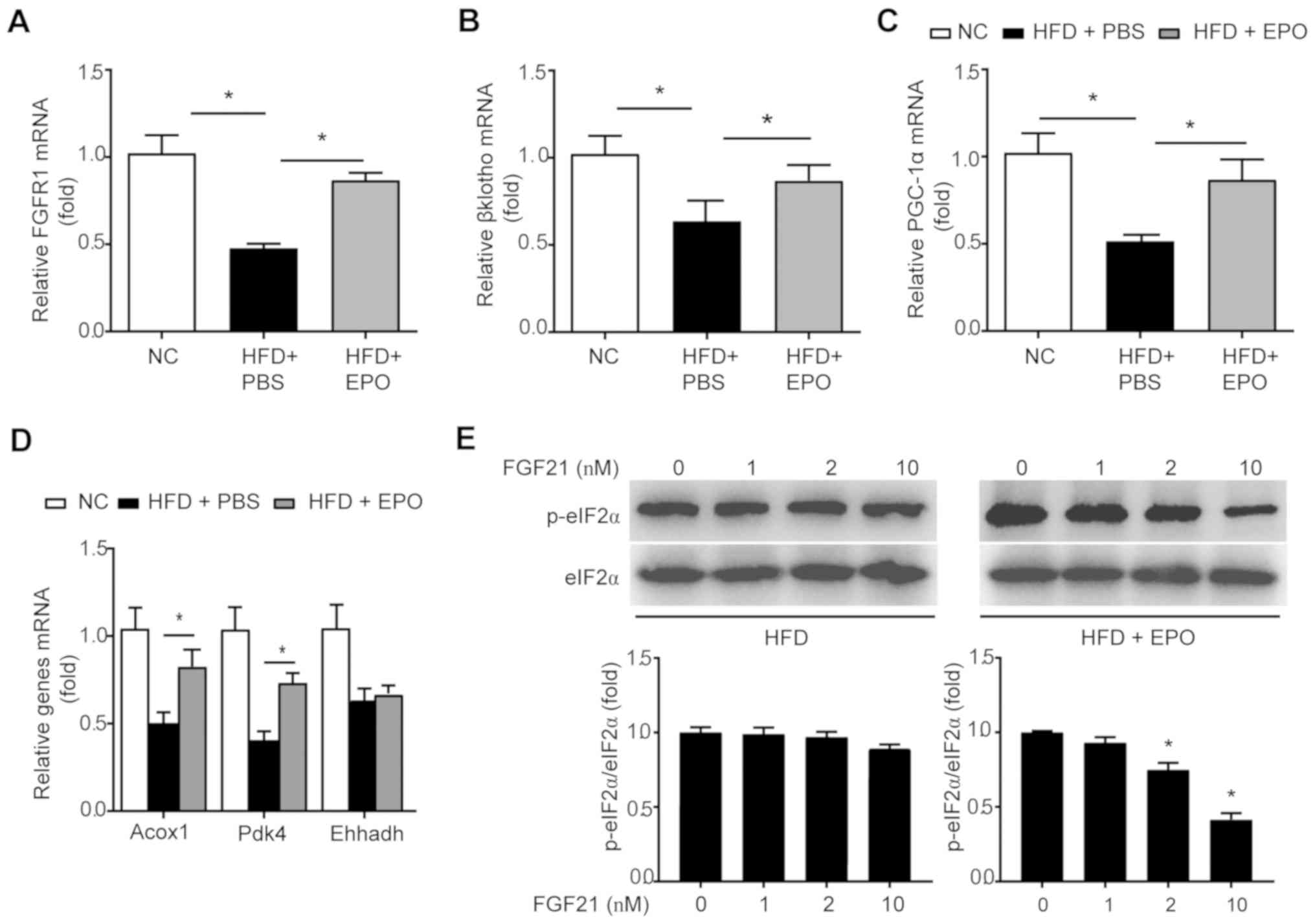 | Figure 6EPO partially restored FGF21
sensitivity in the hepatocytes of HFD-fed mice. The mRNA expression
levels of FGF21 co-receptors (A) FGFR1, (B) βKlotho, (C) PGC-1αand
its target genes, including (D) Acox1, Pdk4 and Ehhadh were
measured in liver tissues of HFD-fed mice following EPO treatment.
Data are presented as the mean ± standard error of the mean
(n=6/group, *P<0.05). (E) Primary hepatocytes,
isolated from HFD-fed mice treated with PBS or EPO, were incubated
with indicated dosage of recombinant FGF21, followed by western
blotting to analyze p-eIF2α/eIF2α expression. Data are shown as the
mean ± standard error of the mean (n=3 independent experiments,
*P<0.05). EPO, erythropoietin; SIRT1, sirtuin 1;
FGF21, fibroblast growth factor 21; HFD, high-fat diet; PGC-1α,
peroxisome proliferator-activated receptor-γ coactivator α; Acox1,
acyl-CoA oxidase 1; Pdk4, pyruvate dehydrogenase kinase 4,
isoenzyme 4; Ehhadh, enoyl-CoA hydratase and 3-hydroxyacyl CoA
dehydrogenase; p-eIF2α, phosphorylation of eukaryotic translation
initiation factor 2α. |
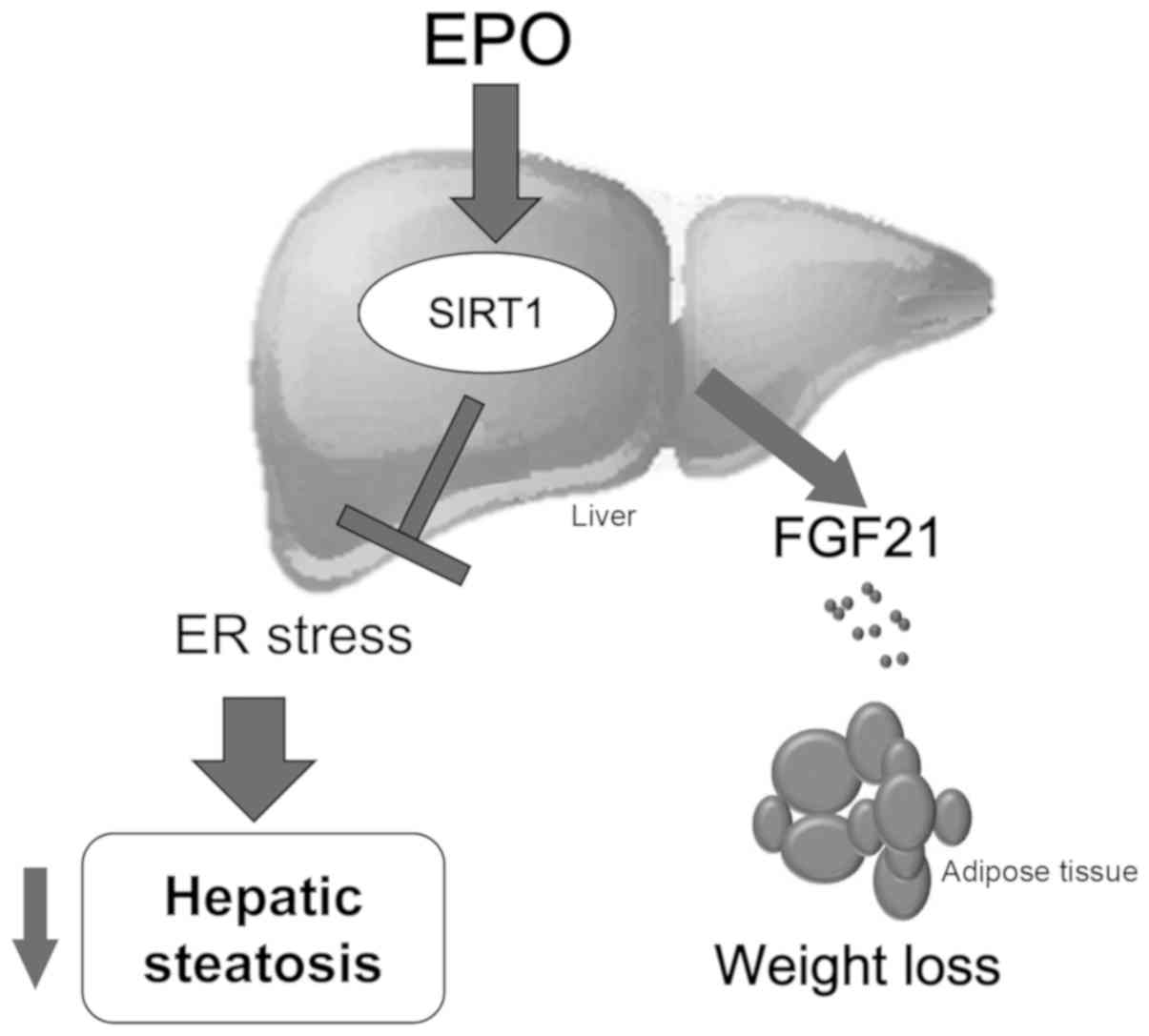
Discussion
EPO has been attracting increasing attention for its
potential beneficial effects against the progression of obesity,
diabetes and fatty liver disease. However, the underlying mechanism
mediating these effects remains unclear. In the present study, a
novel mechanism by which EPO alleviates obesity-induced ER stress
and hepatic steatosis in a SIRT1-dependent manner was proposed.
Additionally, EPO was reported to serve as a positive regulator of
hepatic FGF21 expression, further supporting the hypothesis that
EPO may be a promising agent for the treatment of hepatic steatosis
and obesity.
Hepatic ER stress, induced by pharmacological agents
or metabolic dysregulation, promotes hepatic lipid accumulation by
increasing lipogenesis (32).
Genetic ablation studies revealed that the phosphorylation of
eIF2α, a key downstream target of PERK, exacerbates the progression
of hepatic steatosis in mice subjected to pharmacologically induced
ER stress (33). Conversely,
transgenic mice with inactivated eIF2α via dephosphorylation were
protected from HFD-induced hepatic steatosis (34). In the present study, in addition
to the protective effects of EPO against weight gain and fat
accumulation, EPO was observed to decrease lipid content and
alleviate ER stress in the livers of HFD-fed mice and PA-induced
hepatocytes. These observations were associated with decreases in
the expression levels of ER stress markers, including PDI, GRP78,
p-PERK and p-eIF2α. Consistently, previous studies have reported
the ability of EPO to protect rats against cardiac dysfunction and
nephrotoxicity by attenuating intracellular ER stress (35,36). Furthermore, SIRT1 overexpression
was proposed to attenuate lipid-induced ER stress and hepatic
accumulation by decreasing mammalian target of rapamycin complex 1
activity (18,37). Our previous study reported that
EPO was able to increase hepatic SIRT1 activity in vitro
(15). The results of the present
study suggested the presence of in vivo cross-talk between
EPO and SIRT1 in liver tissue. As the alleviating effect of EPO on
ER stress and fatty liver was abrogated in SIRT1-LKO mice, SIRT1
may regulate the effects of EPO in the liver. Additionally, SIRT1
loss-of-function approaches in hepatocytes via siRNA further
indicated that SIRT1 is required for EPO to attenuate hepatic ER
stress and lipid deposition. Collectively, these findings revealed
a novel mechanism through which EPO alleviates ER stress in hepatic
steatosis in a SIRT1-dependent manner.
Furthermore, the effects of EPO on alleviating
metabolic dysregulation were eliminated in SIRT1-LKO mice. Previous
studies reported that hepatic SIRT1 markedly induced FGF21
expression and secretion to control whole-body energy metabolism
(21,38). The hepatokine FGF21 has been
considered as a promising therapeutic agent that acts by promoting
hepatic fatty acid oxidation, the browning of white adipose tissue
and the thermogenesis of brown adipose tissue (39-41). The present study suggested that
EPO could increase the expression of hepatic FGF21 in vivo
and in vitro. However, SIRT1 deficiency (genetic or
pharmacologically induced) inhibited the effects of EPO on the
induction of FGF21, which was accompanied by diminished effect on
increased expression of FGF21-targeted PGC-1α. These results
suggested that SIRT1-induced FGF21 expression may contribute to the
effects of EPO on alleviating hepatic steatosis and obesity. Of
note, obese animals or human subjects, or those with fatty liver,
had elevated FGF21 levels, indicating that the function of FGF21 is
impaired under conditions of fat accumulation (30,42). The expression of FGFR1 and
βKlotho, which serve as co-receptors of FGF21, was down-regulated
by HFD feeding (43).
Additionally, EPO treatment restored the expression of these
receptors, which was accompanied by the upregulation of
FGF21/PGC-1α-axis target genes, including Acox1 and Pdk4. In
addition, eIF2α is a molecular target of FGF21 in the regulation of
ER stress in the liver (44); the
present study demonstrated that eIF2α phosphorylation in response
to FGF21 in mouse hepatocytes was restored by EPO treatment,
compared with PBS-treated obese mice. As regards the feedback
mechanism underlying the effects of FGF21 on lipid metabolism
related to ER stress (45),
further investigation is required to determine the role of FGF21 in
association with the effects of EPO on alleviating ER stress.
Collectively, the results of the present study indicated that EPO
treatment induced FGF21 expression and restored its sensitivity
under conditions of obesity, contributing to improved systemic
metabolism.
In summary, the results of the present study
demonstrated that EPO ameliorated hepatic steatosis and obesity by
inducing SIRT1-mediated inhibition of ER stress and promoting FGF21
expression (Fig. 7). These
findings may improve our understanding of this mechanism and
provide more experimental evidence on the therapeutic value of EPO
in fatty liver and obesity.
Supplementary Data
Abbreviations:
|
EPO
|
erythropoietin
|
|
ER
|
endoplasmic reticulum
|
|
eIF2α
|
eukaryotic initiation factor 2α
|
|
FGF21
|
fibroblast growth factor 21
|
|
HFD
|
high-fat diet
|
|
IPGTT
|
intraperitoneal glucose tolerance
test
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
PA
|
palmitate
|
|
PDI
|
protein disulfide isomerase
|
|
GRP78
|
glucose-regulated protein 78
|
|
PERK
|
protein kinase-like ER kinase
|
|
PGC-1α
|
peroxisome proliferator-activated
receptor-γ coactivator α
|
|
PPARγ
|
proliferator-activated receptor γ
|
|
AKT
|
protein kinase B
|
|
siRNA
|
small interfering RNA SIRT1, sirtuin
1
|
|
TG
|
triglyceride
|
|
UPR
|
unfolded protein response
|
|
NAD
|
nicotinamide adenine dinucleotide
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
BCA
|
bicinchoninic acid
|
|
Pdk4
|
pyruvate dehydrogenase kinase 4,
isoenzyme 4
|
|
Acox1
|
acyl-CoA oxidase 1
|
|
Ehhadh
|
enoyl-CoA hydratase and 3-hydroxyacyl
CoA dehydrogenase
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China Grant Awards (grant nos.
81600637, 81770819, 81570736, 81570737, 81370947, 81500612,
81400832, 81600632 and 81703294), the National Key Research and
Development Program of China (grant no. 2016YFC1304804), the
Natural Science Foundation of Jiangsu Province of China (grant no.
BK20160116), the Jiangsu Provincial Key Medical Discipline (grant
no. ZDXKB2016012), the Key Project of Nanjing Clinical Medical
Science, the Key Research and Development Program of Jiangsu
Province of China (grant nos. BE2015604 and BE2016606), the Jiangsu
Provincial Medical Talent (grant no. ZDRCA2016062), and the Nanjing
Science and Technology Development Project (grant no.
201605019).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
TH contributed to the study design, data acquisition
and interpretation, drafting of the article, and revision of the
manuscript. ZG contributed to the study design, data acquisition,
data analysis, and the drafting and revision of the manuscript. BZ
and RM contributed to the acquisition and analysis of the data and
the drafting of the manuscript. YB contributed to the study design,
data interpretation, and the drafting and revision of the
manuscript. DZ contributed to the study design, data
interpretation, and the revision of the manuscript. All authors
have read and approved the final version of this manuscript for
publication.
Ethics approval and consent to
participate
All animal experiments were approved by the Nanjing
University Medical School Institutional Animal Care and Use
Committee, and were performed in accordance with the National
Institutes of Health guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests associated with this publication.
References
|
1
|
Younossi Z, Anstee QM, Marietti M, Hardy
T, Henry L, Eslam M, George J and Bugianesi E: Global burden of
NAFLD and NASH: Trends, predictions, risk factors and prevention.
Nat Rev Gastroenterol Hepatol. 15:11–20. 2018. View Article : Google Scholar
|
|
2
|
Deng X, Pan X, Cheng C, Liu B, Zhang H,
Zhang Y and Xu K: Regulation of SREBP-2 intracellular trafficking
improves impaired autophagic flux and alleviates endoplasmic
reticulum stress in NAFLD. Biochim Biophys Acta Mol Cell Biol
Lipids. 1862:337–350. 2017. View Article : Google Scholar
|
|
3
|
Leamy AK, Egnatchik RA and Young JD:
Molecular mechanisms and the role of saturated fatty acids in the
progression of non-alcoholic fatty liver disease. Prog Lipid Res.
52:165–174. 2013. View Article : Google Scholar
|
|
4
|
Fu S, Watkins SM and Hotamisligil GS: The
role of endoplasmic reticulum in hepatic lipid homeostasis and
stress signaling. Cell Metab. 15:623–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Basseri S and Austin RC: ER stress and
lipogenesis: A slippery slope toward hepatic steatosis. Dev Cell.
15:795–796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashraf NU and Sheikh TA: Endoplasmic
reticulum stress and Oxidative stress in the pathogenesis of
non-alcoholic fatty liver disease. Free Radic Res. 49:1405–1418.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jang MK, Nam JS, Kim JH, Yun YR, Han CW,
Kim BJ, Jeong HS, Ha KT and Jung MH: Schisandra chinensis extract
ameliorates nonalcoholic fatty liver via inhibition of endoplasmic
reticulum stress. J Ethnopharmacol. 185:96–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yien YY, Shi J, Chen C, Cheung JTM, Grillo
AS, Shrestha R, Li L, Zhang X, Kafina MD, Kingsley PD, et al:
FAM210B is an erythropoietin target and regulates erythroid heme
synthesis by controlling mitochondrial iron import and
ferrochelatase activity. J Biol Chem. 293:19797–19811. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jelkmann W: Erythropoietin: Back to
basics. Blood. 115:4151–4152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
She J, Yuan Z, Wu Y, Chen J and Kroll J:
Targeting erythropoietin protects against proteinuria in type 2
diabetic patients and in zebrafish. Mol Metab. 8:189–202. 2018.
View Article : Google Scholar :
|
|
11
|
Kodo K, Sugimoto S, Nakajima H, Mori J,
Itoh I, Fukuhara S, Shigehara K, Nishikawa T, Kosaka K and Hosoi H:
Erythropoietin (EPO) ameliorates obesity and glucose homeostasis by
promoting thermogenesis and endocrine function of classical brown
adipose tissue (BAT) in diet-induced obese mice. PLoS One.
12:e01736612017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caillaud C, Connes P, Ben Saad H and
Mercier J: Erythropoietin enhances whole body lipid oxidation
during prolonged exercise in humans. J Physiol Biochem. 71:9–16.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hojman P, Brolin C, Gissel H, Brandt C,
Zerahn B, Pedersen BK and Gehl J: Erythropoietin over-expression
protects against diet-induced obesity in mice through increased fat
oxidation in muscles. PLoS One. 4:e58942009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ge Z, Zhang P, Hong T, Tang S, Meng R, Bi
Y and Zhu D: Erythropoietin alleviates hepatic insulin resistance
via PPARγ-dependent AKT activation. Sci Rep. 5:178782015.
View Article : Google Scholar
|
|
15
|
Hong T, Ge Z, Meng R, Wang H, Zhang P,
Tang S, Lu J, Gu T, Zhu D and Bi Y: Erythropoietin alleviates
hepatic steatosis by activating SIRT1-mediated autophagy. Biochim
Biophys Acta Mol Cell Biol Lipids. 1863:595–603. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sathyanarayan A, Mashek MT and Mashek DG:
ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic
lipid droplet catabolism. Cell Rep. 19:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kong Q, Zhang H, Zhao T, Zhang W, Yan M,
Dong X and Li P: Tangshen formula attenuates hepatic steatosis by
inhibiting hepatic lipogenesis and augmenting fatty acid oxidation
in db/db mice. Int J Mol Med. 38:1715–1726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Xu S, Giles A, Nakamura K, Lee JW,
Hou X, Donmez G, Li J, Luo Z, Walsh K, et al: Hepatic
overexpression of SIRT1 in mice attenuates endoplasmic reticulum
stress and insulin resistance in the liver. FASEB J. 25:1664–1679.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan Q, Ren Y, Liu W, Hu Y, Zheng J, Xu Y
and Wang G: Resveratrol prevents hepatic steatosis and endoplasmic
reticulum stress and regulates the expression of genes involved in
lipid metabolism, insulin resistance, and inflammation in rats.
Nutr Res. 35:576–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang X, Yang W, Wang R, Xie T, Li H, Feng
D, Jin X, Sun H and Wu S: Sirtuin-1 (SIRT1) stimulates growth-plate
chondrogenesis by attenuating the PERK-eIF-2α-CHOP pathway in the
unfolded protein response. J Biol Chem. 293:8614–8625. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Wong K, Giles A, Jiang J, Lee JW,
Adams AC, Kharitonenkov A, Yang Q, Gao B, Guarente L and Zang M:
Hepatic SIRT1 attenuates hepatic steatosis and controls energy
balance in mice by inducing fibroblast growth factor 21.
Gastroenterology. 146:539–549.e7. 2014. View Article : Google Scholar :
|
|
22
|
Han HS, Choi BH, Kim JS, Kang G and Koo
SH: Hepatic Crtc2 controls whole body energy metabolism via a
miR-34a-Fgf21 axis. Nat Commun. 8:18782017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng Q, Tong M, Ou B, Liu C, Hu C and
Yang Y: Isorhamnetin protects against bleomycin-induced pulmonary
fibrosis by inhibiting endoplasmic reticulum stress and
epithelial-mesenchymal transition. Int J Mol Med. 43:117–126.
2019.
|
|
24
|
Varghese F, Bukhari AB, Malhotra R and De
A: IHC Profiler: An open source plugin for the quantitative
evaluation and automated scoring of immunohistochemistry images of
human tissue samples. PLoS One. 9:e968012014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shah KN, Bhatt R, Rotow J, Rohrberg J,
Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al:
Aurora kinase A drives the evolution of resistance to
third-generation EGFR inhibitors in lung cancer. Nat Med.
25:111–118. 2019. View Article : Google Scholar
|
|
26
|
Seglen PO: Hepatocyte suspensions and
cultures as tools in experimental carcinogenesis. J Toxicol Environ
Health. 5:551–560. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Zhang W, Sun Y, Liu W, Dong J and Chen J:
SIRT1 mediates the role of RNA-binding protein QKI 5 in the
synthesis of triglycerides in non-alcoholic fatty liver disease
mice via the PPARα/FoxO1 signaling pathway. Int J Mol Med.
43:1271–1280. 2019.PubMed/NCBI
|
|
29
|
Babaknejad N, Nayeri H, Hemmati R, Bahrami
S and Esmaillzadeh A: An overview of FGF19 and FGF21: The
therapeutic role in the treatment of the metabolic disorders and
obesity. Horm Metab Res. 50:441–452. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fisher FM, Chui PC, Antonellis PJ, Bina
HA, Kharitonenkov A, Flier JS and Maratos-Flier E: Obesity is a
fibroblast growth factor 21 (FGF21)-resistant state. Diabetes.
59:2781–2789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gasparin FRS, Carreño FO, Mewes JM,
Gilglioni EH, Pagadigorria CLS, Natali MRM, Utsunomiya KS,
Constantin RP, Ouchida AT, Curti C, et al: Sex differences in the
development of hepatic steatosis in cafeteria diet-induced obesity
in young mice. Biochim Biophys Acta Mol Basis Dis. 1864:2495–2509.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Henkel AS: Unfolded protein response
sensors in hepatic lipid metabolism and nonalcoholic fatty liver
disease. Semin Liver Dis. 38:320–332. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rutkowski DT, Wu J, Back SH, Callaghan MU,
Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, et al:
UPR pathways combine to prevent hepatic steatosis caused by ER
stress-mediated suppression of transcriptional master regulators.
Dev Cell. 15:829–840. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oyadomari S, Harding HP, Zhang Y,
Oyadomari M and Ron D: Dephosphorylation of translation initiation
factor 2alpha enhances glucose tolerance and attenuates
hepatosteatosis in mice. Cell Metab. 7:520–532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kong D, Zhuo L, Gao C, Shi S, Wang N,
Huang Z, Li W and Hao L: Erythropoietin protects against
cisplatin-induced nephrotoxicity by attenuating endoplasmic
reticulum stress-induced apoptosis. J Nephrol. 26:219–227. 2013.
View Article : Google Scholar
|
|
36
|
Lu J, Dai Q, Ma G, Zhu Y, Chen B, Li B and
Yao Y: Erythropoietin attenuates cardiac dysfunction in rats by
inhibiting endoplasmic reticulum stress-induced diabetic
cardiomyopathy. Cardiovasc Drugs Ther. 31:367–379. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jung TW, Lee KT, Lee MW and Ka KH: SIRT1
attenuates palmitate-induced endoplasmic reticulum stress and
insulin resistance in HepG2 cells via induction of oxygen-regulated
protein 150. Biochem Biophys Res Commun. 422:229–232. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsui S, Sasaki T, Kohno D, Yaku K,
Inutsuka A, Yokota- Hashimoto H, Kikuchi O, Suga T, Kobayashi M,
Yamanaka A, et al: Neuronal SIRT1 regulates macronutrient-based
diet selection through FGF21 and oxytocin signalling in mice. Nat
Commun. 9:46042018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jimenez V, Jambrina C, Casana E, Sacristan
V, Muñoz S, Darriba S, Rodó J, Mallol C, Garcia M, Leó X, et al:
FGF21 gene therapy as treatment for obesity and insulin resistance.
EMBO Mol Med. 10:pii: e8791. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu M, Cao H, Hou Y, Sun G, Li D and Wang
W: Liver plays a major role in FGF-21 mediated glucose homeostasis.
Cell Physiol Biochem. 45:1423–1433. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fisher FM, Kleiner S, Douris N, Fox EC,
Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS,
Maratos-Flier E and Spiegelman BM: FGF21 regulates PGC-1α and
browning of white adipose tissues in adaptive thermogenesis. Genes
Dev. 26:271–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dushay J, Chui PC, Gopalakrishnan GS,
Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML
and Maratos-Flier E: Increased fibroblast growth factor 21 in
obesity and nonalcoholic fatty liver disease. Gastroenterology.
139:456–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rusli F, Deelen J, Andriyani E,
Boekschoten MV, Lute C, van den Akker EB, Müller M, Beekman M and
Steegenga WT: Fibroblast growth factor 21 reflects liver fat
accumulation and dysregulation of signalling pathways in the liver
of C57BL/6J mice. Sci Rep. 6:304842016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiang S, Yan C, Fang QC, Shao ML, Zhang
YL, Liu Y, Deng YP, Shan B, Liu JQ, Li HT, et al: Fibroblast growth
factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded
protein response and counteracts endoplasmic reticulum
stress-induced hepatic steatosis. J Biol Chem. 289:29751–29765.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ye M, Lu W, Wang X, Wang C, Abbruzzese JL,
Liang G, Li X and Luo Y: FGF21-FGFR1 coordinates phospholipid
homeostasis, lipid droplet function, and ER stress in obesity.
Endocrinology. 157:4754–4769. 2016. View Article : Google Scholar : PubMed/NCBI
|