Introduction
Asthma represents a chronic respiratory disease that
affects more than 300 million individuals worldwide (1). It is characterized by airway
inflammation, hyperresponsiveness and remodeling (2). As the first line of contact with the
external environment, airway epithelial (AE) cells form a barrier
to the outside world comprising airway surface fluids, mucus and
apical junctional complexes (AJCs) between neighboring cells.
Previous studies have focused on AE barrier injury in asthma
(3-5), reporting dysfunctional epithelial
AJCs in asthmatic airways, without describing the precise
mechanisms and/or consequences for airway inflammation (6,7).
Impaired epithelium could increase airway vulnerability to insults
from viruses, allergens and toxic substances, aggravating airway
inflammation and remodeling. Due to ethical constraints, animal
models serve as a golden tool for understanding the pathology of
asthma in vivo. However, most animal studies have used acute
sensitization and exposure to allergens. In addition, the available
models often lack features of chronic remodeling. Therefore, the
potential mechanisms obtained in mouse studies are merely used to
understand the effects of particular therapies in acute allergic
inflammation and do not comprehensively explain the chronic phase
of the disease.
In the past decades, increasing attention has been
paid to the management of childhood asthma. The orosomucoid-like
protein isoform 3 (ORMDL3) gene is strongly and significantly
associated with childhood-onset asthma (8). Several in vitro and in
vivo studies have suggested that ORMDL3 contributes to airway
remodeling and inflammation by selectively activating the unfolded
protein response in the endoplasmic reticulum (9,10),
regulating chemokine expression (11) and altering Ca2+ influx
for T-lymphocyte activation (12). Previous findings indicated that
intranasal administration of cytokines significantly induces ORMDL3
mRNA expression in the bronchial epithelium of mice (9). Previously, ORMDL3 was shown to
regulate the metabolism of the cell membrane component sphingolipid
in A549 cells (13,14). Sphingolipid has attracted
increasing attention in recent years. Indeed, several studies have
established its role in cell growth, survival and migration
(15-17). ORMDL3 is involved in sphingolipid
metabolism and de novo sphingolipid synthesis (9). An in vitro study revealed
ORMDL3 overexpression at mid-levels inhibits serine
palmitoyltransferase (SPT) activity, while a more pronounced ORMDL3
overexpression results in increased SPT levels (18).
As a key lipid kinase in sphingolipid metabolism,
sphingosine kinase 1 (SPHK1) regulates sphingosine 1-phosphateas
well as the SPT balance in the lung tissue. SPHK1 mRNA levels are
significantly increased in airway diseases such as lung cancer,
rhinitis and asthma (19-21). Previous studies have proposed that
SPHK1 is associated with airway inflammation, goblet cell
hyperplasia and hyperresponsiveness (19,22,23). The possible mechanisms include
calcium flux control, arachidonic acid release and ERK
phosphorylation induction (24-26). In vivo studies have
demonstrated that treatment with an SPHK1 inhibitor or SPHK1
knockout could ameliorate OVA-induced airway hyperreactivity (AHR)
and airway inflammation in mice (22,26). Meanwhile, SPHK1 suppression
upregulates E-cadherin in A549 cells (17). Based on these data, it was
hypothesized that ORMDL3 overexpression causes AE barrier injury by
activating SPHK1.
Materials and methods
Animal sensitization and challenge
A total of 12 female Balb/c mice (4 weeks old, 18-22
g) were obtained from Beijing Vital River Laboratory Animal
Technology Co., Ltd. Animals were housed in the experimental animal
center of the Nanjing University of Chinese Medicine, with a 12 h
light/dark cycle at a constant temperature of 22±2°C and a relative
humidity of 50%. Cages, bedding, food and water were sterilized
before use and animals received ad libitum access to food
and water. The animals were acclimatized for 7 days prior to
initiating the experiments. The animals were immunized with 200
µl of 2.5% ovalbumin (OVA; Sigma-Aldrich; Merck KGaA) in
saline by intraperitoneal injection on days 1 and 8 (sensitization
stage). On days 15-28, mice were exposed to 2.5% OVA by inhalation
for 30 min per day (acute challenge stage). On days 32-85, the
animals were further exposed to aerosolized 2.5% OVA for 30 min
once every three days (chronic challenge stage). On days 29, 42 and
55, mice were anesthetized by inhaled ether; when the mouse's
breathing became slow and deep, and loss of righting reflex, it
ensured the mouse was fully anesthetized, and then 50 µl
respiratory syncytial virus was administered (RSV;
103.015 TCID50/50 µl) by the
intranasal route(virus infection stage). Control mice were
administered normal saline instead of OVA and RSV in the
sensitization, challenge and virus infection stages of the
protocol. Human RSV (A2 strain, purchased from the Type Culture
Collection Center of Wuhan University) was amplified as previously
described (27). Titers were
determined by the TCID50 method in Hep-2 cells
(CBP60246; Type Culture Collection of the Chinese Academy of
Sciences). The protocols for sensitization, challenge and virus
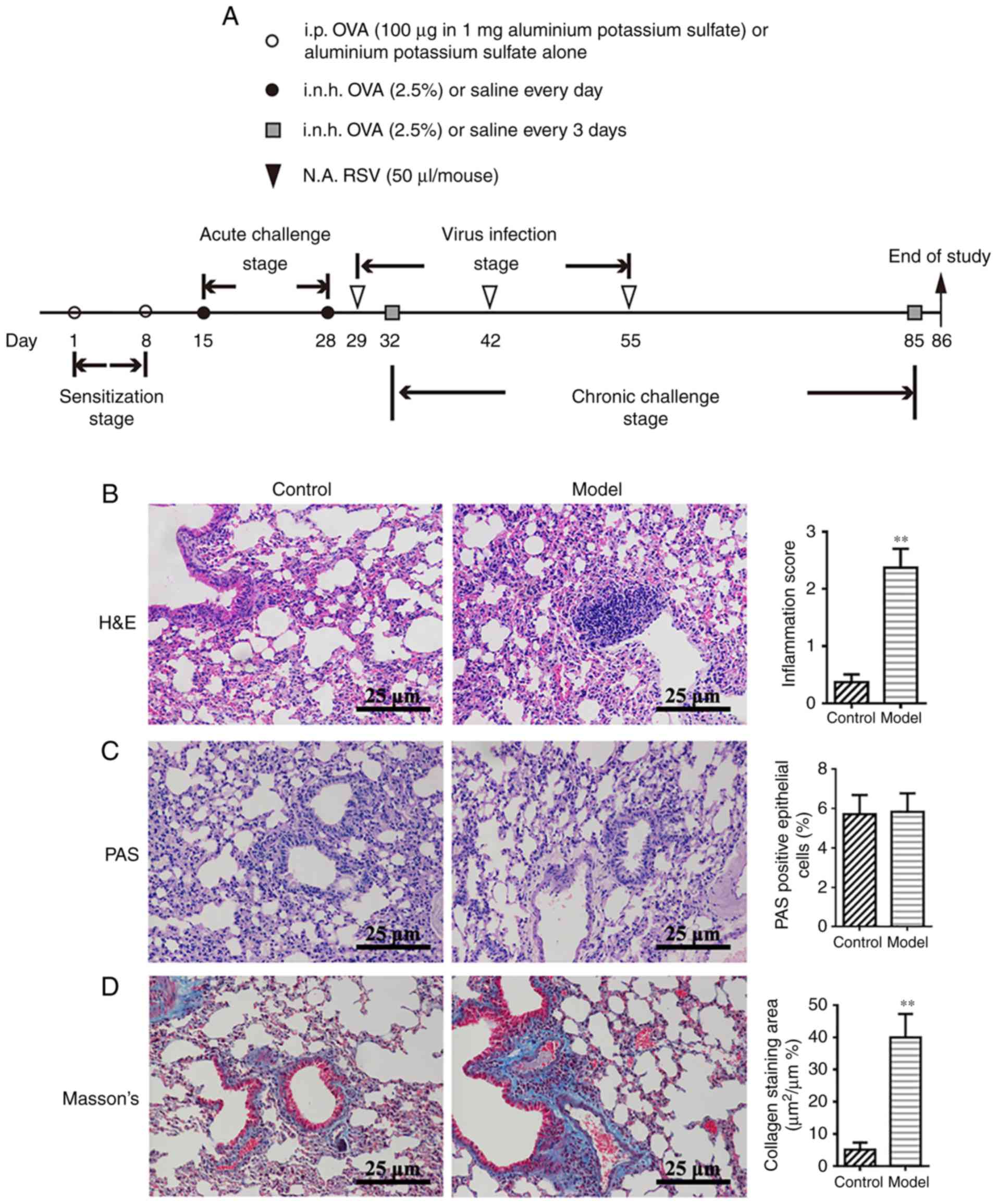
infection are summarized in Fig.
1A. The animal experiments were performed according to the
National Institutes of Health Guidelines for Laboratory Animals and
approved by the Animal Ethics Committee of Nanjing University of
Chinese Medicine.
Lung tissue collection and
histopathology
On day 86, mice were anesthetized by chloral hydrate
(400 mg/kg), the right eyeball was removed to collect blood (~1.0
ml) and sacrificed by cervical dislocation. When the mice no longer
moved, exhibited no response to external stimuli and breathing and
cardiac arrest, the mice were confirmed as dead. Then, the lungs
were removed from the thoracic cavity by careful dissection,
inflated with 1 ml of 10% neutral-buffered formalin and fixed with
10% neutral-buffered formalin at room temperature for 72 h. After
fixation, the left lung was dissected, embedded in paraffin and
sectioned at 6 µm. The resulting sections were stained with
hematoxylin-eosin (H&E) at room temperature for 3 min. Total
lung inflammation was defined as the sum of peri-bronchial and
peri-vascular scores (Table SI).
Mucus production and goblet cell hyperplasia were examined by
periodic acid-Schiff (PAS) staining as described previously
(28). Sections stained (at room
temperature for 15 min) with the Masson's trichromedye were
required to include the extracellular matrix (ECM) and contractile
elements associated with the airway. Peri-bronchiolar areas
positive for Masson's trichrome staining were determined by light
microscopy with an Image-Pro Plus V6.0 image analysis system (Media
Cybernetics, Inc.).
Quantification of cytokine levels
Lung tissue and serum cytokine levels were assessed
with ELISA kits specific for mouse interleukin (IL)-4(MM-0173M2),
IL-13 (MM-0165M2), and tumor necrosis factor (TNF)-α (MM-0132M2;
all, Feiya Biotechnology). The assays were performed according to
the manufacturer's protocol.
Cell Counting Kit (CCK)-8 assay for TNF-a
concentration selection
To quantitate any cytotoxic effects of TNF-α, a
CCK-8 cell viability detection assay was performed on the
conditioned media from 16HBE cells according to the manufacturer's
protocol (Dojindo Molecular Technologies, Inc.). Briefly, CCK-8 (10
µl) was added to each well and the plates were incubated for
2 h at 37°C. Optical density was measured using a Microplate Reader
at 450 nm.
Cell culture and treatment
Human bronchial epithelial 16HBE cells (CBP60550,
purchased from the Cell Bank Center, Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences) cultured in
RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
All cells were maintained in a humidified chamber containing 5%
CO2 at 37°C. 16HBE cells transfected with small
interfering (si)-SPHK1/si-control (ctrl) were exposed to
10 ng/ml TNF-α (purchased from PeproTech, Inc.); 16HBE cells
over-expressing ORMDL3 and the corresponding empty control
(Vector; purchased from Applied Biological Materials Inc.)
were exposed to 10 nM N,N-dimethyl-D-erythro-sphingosine (DMS;
purchased from Sigma-Aldrich; Merck KGaA) for 5 days, followed by
the evaluation of epithelial TEER and mRNA and protein expression
levels.
siRNA preparation and transfection
For si-RNA transfection, 16HBE cells were seeded at
a density of 2×105 cells/well in 6-well plates until
50-70% confluence. The si-RNA targeting ORMDL3 (si-ORMDL3)
and its negative control siRNA (si-control), and the si-RNA
targeting SPHK1 (si-SPHK1) and its negative control si-RNA
(si-ctrl; Synthesized by GenePharma Co., Ltd.). si-RNA was
transfected into 16HBE cells with si-RNA-Mate (50 pmol si-RNA; 16
µl si-RNA-Mate; GenePharma Co., Ltd.) in RPMI-1640-low serum
medium in accordance with the manufacturer's protocols. The
sequences of the siRNAs used are listed in Table I and activity was measured via
reverse transcription-quantitative PCR (RT-qPCR) 24 h after
transfection. Thereafter, the transfected cells were trypsinized
and seeded at a density of 0.33×105 cells per
cm2 in Transwell inserts with a 0.4 µm pore size
(Corning, Inc.). The medium was changed the following day and
subsequently refreshed every other day throughout the experiment.
The silencing effects of si-RNAs were confirmed by RT-qPCR.
 | Table ISequences of si-RNAs. |
Table I
Sequences of si-RNAs.
| Gene | Sense (5′-3′) | Anti-sense
(5′-3′) |
|---|
| ORMDL3 |
CCAACCUCAUUCACAACAUTT |
AUGUUGUGAAUGAGGUUGGTT |
| SPHK1 |
GAGGCUGAAAUCUCCUUCATT |
UGAAGGAGAUUUCAGCCUCTT |
| GAPDH |
UGACCUCAACUACAUGGUUTT |
AACCAUGUAUUGAGGUCATT |
TEER and epithelial barrier integrity
assessments
ORMDL3/Vector and si-ORMDL3/si-control
cells were seeded in Transwell inserts as mentioned above, and
incubated for 72 h to yield a cell monolayer. Chemicals at proper
concentrations were added to the inserts (TNF-α, 10 ng/ml; DMS, 10
nM). Cells were serum-starved overnight before addition of TNF-α or
DMS. TEER was monitored before (day 0) and at days 1 (24 h) to 5
daily with a Millicell-ERS2 Volt-Ohm Meter (EMD Millipore)
according to the manufacturer's protocol. TEER was derived as
(R sample-R blank) x surface area (cm2).
The surface area was 0.33 cm2.
At day 5, epithelial permeability in
ORMDL3/Vector and si-ORMDL3/si-control cells was measured by
fluorescein isothiocyante (FITC)-conjugated dextran (4-kDa; 1
mg/ml; Sigma-Aldrich; Merck KGaA) fluorescence strength. In this
assay, 4-kDa-FITC-dextran was added to the apical side of the
inserts while RPMI-1640 containing 2% FBS was placed in lower
wells. After 3 h, 100 µl of fluid was collected from the
basolateral compartment of each insert and transferred to 96-well
plates (Corning, Inc.). Fluorescence was measured at 490 nm,
reflecting the amounts of fluoresce in sodium diffused from the
apical side of the insert to the basal one. The values of
FITC-dextran signals were normalized to the corresponding control
groups (Vector or si-control) and presented as Pa/Pc%.
SPHK1 activity assay
ORMDL3/Vector and si-ORMDL3/si-control cells
were cultured in 24-well Transwell plates. At 5 days of culture,
the cells were harvested and processed with SPHK1 Activity Assay
kit I (Shanghai Haling Biotechnology Co., Ltd. http://www.halingbio.com). SPHK1 activity and
percentage inhibition were calculated based on the manufacturer's
protocol.
Western blot analysis
Western blot analysis was performed as described
previously by the authors' team (15). The following antibodies were used
and incubated overnight at 4°C: Anti-ORMDL3 (1:2,000; ab107639;
Abcam), anti-SPHK1 (1:1,000; ab71700; Abcam) and anti-Tubulin
(1:2,000; ab179513; Abcam), anti-GAPDH (1:5,000; ab181602; Abcam),
anti-Claudin-18 (1:2,000; 21126-1-AP; ProteinTech Group, Inc.),
anti-E-cadherin (1:2,000; YT1454; ImmunoWay Biotechnology),
anti-phospho-ERK1/2 (1:1,000; ab201015; Abcam; kindly provided by
Dr Xiao-Fei, Jiang) and anti-ERK1/2 (1:1,000; ab17942; Abcam;
kindly provided by Dr Xiao-Fei, Jiang). Goat anti-rabbit
horseradish peroxidase-conjugated IgG (1:5,000; ab97051; Abcam) was
used to detect antibody binding at room temperature for 2 h. Target
proteins were visualized using a ChemiDoc™ MP Imaging system
(Bio-Rad Laboratories, Inc.) and analyzed using ImageLab software
V5.1 (Bio-Rad Laboratories, Inc.).
RNA extraction and RT-qPCR
Total RNA was isolated with TRIzol reagent according
to the manufacturer's protocol (Takara Biotechnology, Co., Ltd.).
cDNA was synthesized using the Prime Script® RT reagent
kit with gDNA Eraser (Takara Biotechnology Co., Ltd.) according to
the manufacturer's protocol. RT-qPCR was performed with cDNA
samples using SYBR® Premix ExTapTMII (Takara
Biotechnology Co., Ltd.). The primers are listed in Tables II and III. The reaction conditions were as
follows: 30 sec at 95°C, followed by 40 cycles at 95°C for 5 sec,
60°C for 30 sec and 72°C for 30 sec. Relative changes in mRNA
levels were measured by the 2−∆∆Cq method (29) and normalized to endogenous
GAPDH.
| Table IIPrimers used in quantitative PCR for
mouse lung assessment. |
Table II
Primers used in quantitative PCR for
mouse lung assessment.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| IL-4 |
TCTCGAATGTACCAGGAGCCATAT |
AAGCACCTTGGAAGCCCTACAGA |
| IL-13 |
GCAGCATGGTATGGAGTGTG |
CCTCTGGGTCCTGTAGATGG |
| TNF-α |
CTGGATGTCAATCAACAATGGGA |
ACTAGGGTGTGAGTGTTTTCTGT |
| Claudin-18 |
GACCGTTCAGACCAGGTACA |
GCGATGCACATCATCACTC |
| E-cadherin |
CAGGCTGGCTGAAAGTGACA |
ACGGATCCCTCAAACACCTC |
| SPHK1 |
CATCACGGCCTGTAAAAAGGT |
ATCTTCCACAAACCCAATCTGG |
| GAPDH |
AATGGATTTGGACGCATTGGT |
TTTGCACTGGTACGTGTTGAT |
| Table IIIPrimers used in quantitative PCR for
cell assessment. |
Table III
Primers used in quantitative PCR for
cell assessment.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Claudin-18 |
CCTGATGATCGTAGGCATCG |
TGCATTTCAGGGCAAAGATG |
| E-cadherin |
TCGTCACCACAAATCCAGTG |
CATTCACATCAAGCACATCC |
| ORMDL3 |
TCACCAACCTCATTCACAACAT |
GACCCCATAATCCATATGCTC |
| SPHK1 |
ATCACGGATGTATGACGTTTTGG |
CAGGCTATTGCTGCGAAGAAC |
| GAPDH |
TGTGGGCATCAATGGATTTGG |
ACACCATGTATTCCGGGTCAAT |
Immunofluorescence (IF)
IF staining of SPHK1, E-cadherin and Claudin-18
proteins in mouse lung samples, ORMDL3/Vector cells, and
si-ORMDL3/si-control cells was performed. Cells were seeded
on slides for 24 h, fixed with 4% paraformaldehyde for 15 min at
room temperature and washed 3 times with PBS. The samples were then
blocked with goat serum (Gibco; Thermo Fisher Scientific, Inc.) in
5% bovine serum albumin/PBS for 1 h at room temperature and
incubated with Alexa Fluor 647 (Abcam)-conjugated SPHK1, E-cadherin
and Claudin-18, respectively, overnight at 4°C. After two PBS
washes, the cells were mounted with ProLong Gold containing DAPI
(Life Technologies; Thermo Fisher Scientific, Inc.) and visualized
under a Leica TCS SP5 confocal microscope (Leica Microsystems,
Inc.).
Statistical analysis
Data are mean ± standard deviation from six
independent experiments with GraphPad Prism 6.0 (GraphPad Software,
Inc.). Statistical analysis was performed with unpaired Student's
t-test (for two experimental groups) or one-way analysis of
variance (for multiple experimental groups), followed by Dunnett's
post hoc test to determine the differences among multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Successful establishment of a model of
chronic asthma in mice by OVA-RSV induction
The authors' previous studies demonstrated that
OVA-RSV induces mild airway inflammation in remittent asthmatic
mice (30,31). To ensure successful establishment
of an animal model of chronic asthma, histopathological features
were compared between the control and experimental groups.
Consistent with the authors' previous findings (30), the amounts of inflammatory cells
around the respiratory tract and vessels in the OVA-RSV group, as
well as bronchial wall thickness, were significantly increased
compared with control values (P<0.01; Fig. 1B). PAS staining showed notable
mucus secretion (Fig. 1C), while
Masson's trichrome staining of lung sections from asthmatic mice
revealed increased collagen deposition around the airway (Fig. 1D). Taken together, these findings
indicated that the chronic asthmatic model was successfully
established with OVA-RSV induction in mice.
OVA-RSV induction increases IL-4, IL-13
and TNF-a levels in serum and lung homogenates
High secretion levels of Th2 and pro-inflammatory
cytokines is a well-established feature of asthma. To determine
whether OVA-RSV treatment affects cytokine levels in serum, IL-4,
IL-13 and TNF-α were detected by ELISA. Compared with the control
values, serum IL-4, IL-13 and TNF-α levels were increased
significantly by the OVA-RSV airway challenge (P<0.01; Fig. 2A-C). In addition, IL-4, IL-13 and
TNF-α levels in lung homogenates were assessed by RT-qPCR and
ELISA. Compared with the control group, the OVA-RSV group showed
significantly increased expression levels of IL-4, IL-13 and TNF-α,
at the mRNA, and ELISA levels (P<0.01; Fig. 2A-D). These findings indicated that
OVA-RSV promoted allergic airway reactions by altering airway
inflammation.
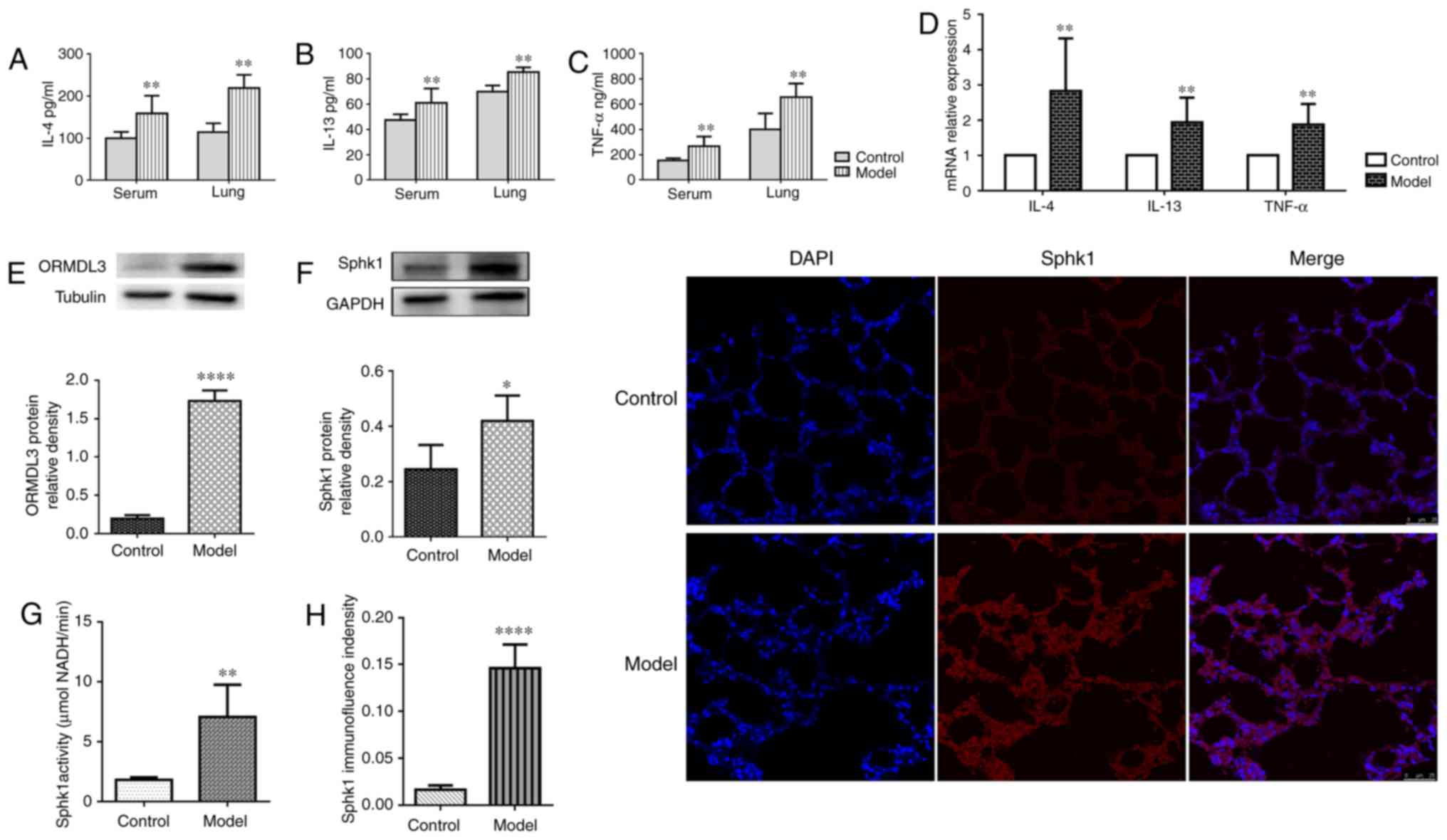 | Figure 2OVA-RSV upregulates pro-inflammatory
cytokines, ORMDL3 and SPHK1 in asthmatic mice. (A-C) OVA-RSV
treatment resulted in elevated (A) IL-4, (B) IL-13 and (C) TNF-α
levels in asthmatic mouse serum and lung tissue samples. (D)
OVA-RSV induced IL-4, IL-13 and TNF-α mRNA expression in the lung
tissue of asthmatic mice. (E) ORMDL3 and (F) SPHK1 proteins were
upregulated in the mouse lung of the OVA-RSV group. (G) SPHK1
activity was detected with SPHK1 Activity Assay kit I in the mouse
lung tissue. (H) Immunofluorescent staining was used to assess the
SPHK1 localization in the mouse lung tissue. Values are mean ±
standard deviation (n=6 per group). *P<0.05,
**P<0.01 and ****P<0.001 vs. the
control group. E, epithelial; SPHK1, sphin-gosine kinase 1; ORMDL3,
orosomucoid-like protein isoform 3; si, small interfering; Ctrl,
control; TNF, tumor necrosis factor; IL, interleukin; OVA-RSV,
ovalbumin-respiratory syncytial virus. |
OVA-RSV induction increases ORMDL3 and
SPHK1 expression levels in the mouse lung
Given that ORMDL3 is upregulated by OVA challenge
(32), the effects of OVA-RSV on
ORMDL3 expression were examined in vivo. OVA-RSV
administration in mice resulted in significantly increased ORMDL3
protein levels in the lung tissue compared with control values
(Fig. 2E).
It was reported that SPHK1 is involved in the
pathological process of asthma (17). Meanwhile, a previous study
revealed that ORMDL3 mediates sphingolipid metabolism in yeasts
(33) and dysregulates ceramide
homeostasis in A549 cells (18).
SPHK1 represents a key enzyme in the regulation of sphingolipid
metabolism and plays an essential role in plasma membrane formation
(8,15). Increased SPHK1 protein expression
levels were observed by western blotting (Fig. 2F) in OVA-RSV stimulated mice vs.
control animals. Furthermore, increased SPHK1 activity (Fig. 2G) and IF (Fig. 2H) density were found in the lung
tissue of the model group compared with the control animals. These
results implied that both ORMDL3 and SPHK1 were upregulated in
OVA-RSV associated asthma in mice.
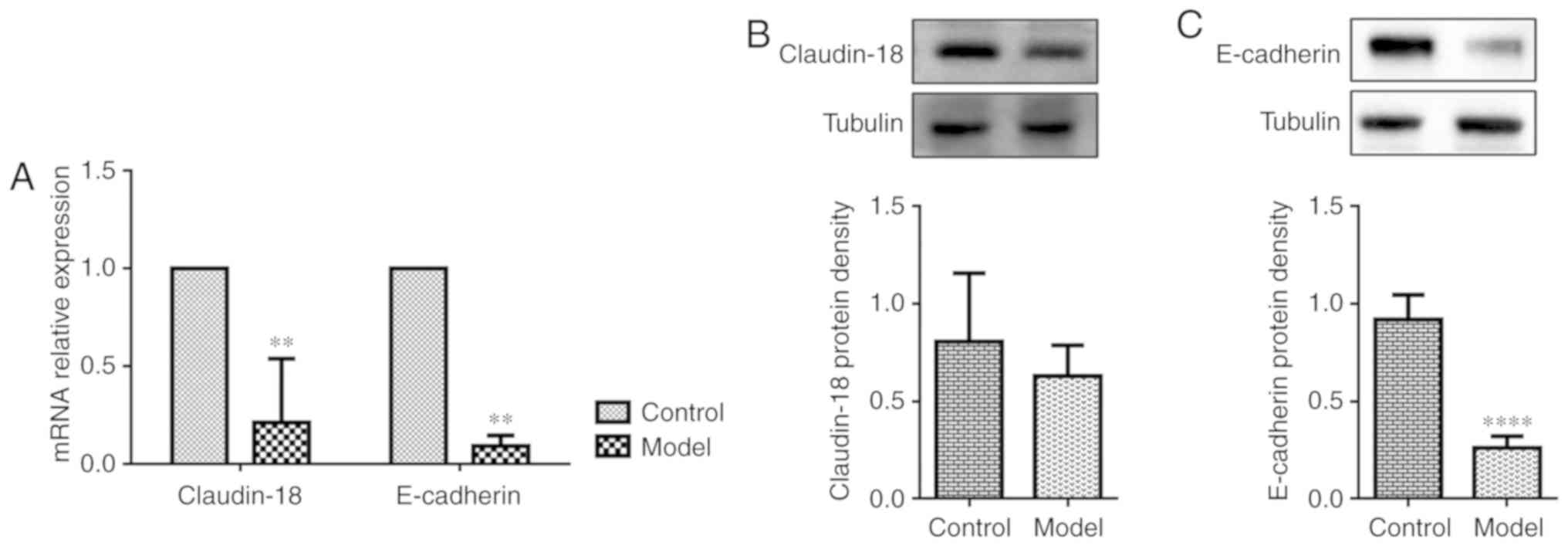
OVA-RSV induces loss of AE junction
proteins in the lung tissue of asthmatic mice
Since asthmatic patients show dysfunctional AE
barrier properties, whether OVA-RSV treatment affects the
expression of epithelial junction proteins that maintain barrier
integrity was examined. Claudin-18 and E-cadherin play important
roles in maintaining the epithelial barrier integrity. Exposure to
allergens or cytokines decreases E-cadherin expression in primary
cells from asthmatic mice (34).
Recent findings demonstrated that Claudin-18 expression is
associated with the degree of lung injury (35). Consistent with previously reported
data in allergen-exposed mice, OVA-RSV challenged mice exhibited
loss of the AE junction molecule E-cadherin compared with control
mice as shown by RT-qPCR (Fig.
3A) and western blotting (Fig.
3C), while OVA-RSV only decreased Claudin-18 mRNA expression
(Fig. 3A) and not protein levels
(Fig. 3B). To explore the
detailed mechanisms behind ORMDL3's association with airway barrier
dysfunction, subsequent studies in 16HBE cells were performed since
in vitro cell models allow the use of a large number of
purified cells for molecular and biochemical analyses; in addition,
using human cells could overcome species discrepancies which are
common in experimental studies.
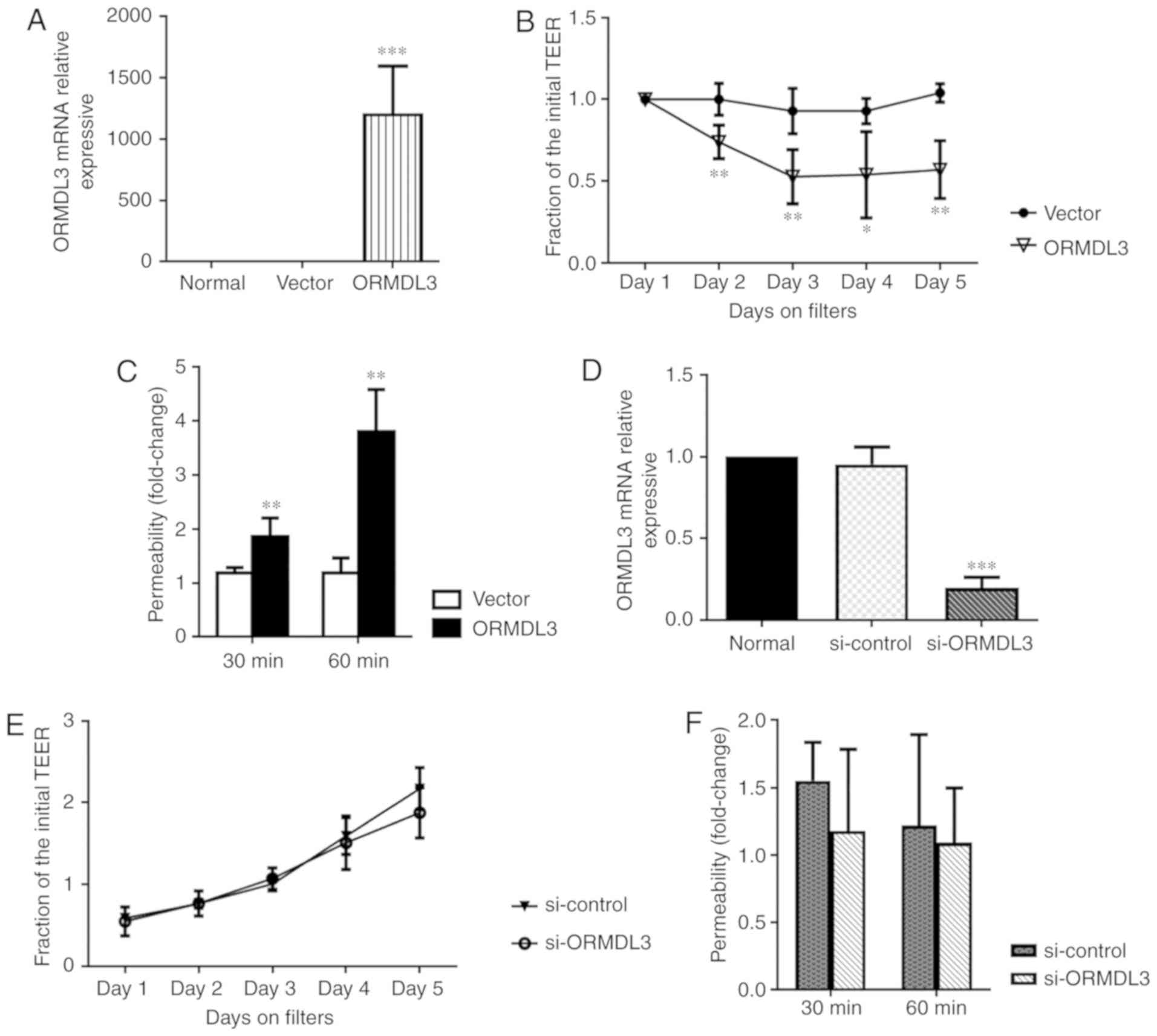
ORMDL3 overexpression decreases AE
barrier integrity
To assess the effects of ORMDL3 on AE barrier
integrity, ORMDL3 in 16HBE cells was overexpressed or knocked down
and the resulting cells were analyzed by RT-qPCR (Fig. 4A and B). TEER was measured at days
2-5 after ORMDL3/Vector or si-ORMDL3/si-control cells
were seeded in Transwell plates. TEER was significantly decreased
in cells overexpressing ORMDL3 compared with Vector
transfected cells, while there was no difference between
si-ORMDL3 and si-control cells (P<0.01; Fig. 4C and D). Epithelial membrane
permeability assessment is an important method for evaluating AE
barrier function. In the present study, epithelial permeability was
measured by treatment with 4-kDa-FITC-dextran. As shown in Fig. 4E and F, ORMDL3 overexpression
resulted in increased cell permeability compared with Vector
trans-fected cells, while ORMDL3 knockdown did not affect cell
permeability compared with the si-ctrl group. These data
suggested that ORMDL3 overexpression resulted in barrier function
damage in 16HBE cells.
ORMDL3 overexpression downregulates
Claudin-18 and E-cadherin
Whether ORMDL3 overexpression downregulates
Claudin-18 and E-cadherin in 16HBE cells was assessed. The results
showed that in 16HBE cells overexpressing ORMDL3, Claudin-18 and
E-cadherin mRNA levels were roughly 50 and 70%, respectively,
compared with those of the controls (Fig. 5A), while ORMDL3 knockdown had
opposite effects (Fig. 5B and D).
However, only Claudin-18 protein expression was significantly
decreased by ORMDL3 overexpression (P<0.05; Fig. 5C), while ORMDL3 knockdown had
opposite effects (Fig. 5F).
However, E-cadherin protein expression was not influenced by ORMDL3
(Fig. 5E and H). Moreover, IF
data showed that ORMDL3 overexpression resulted in decreased
Claudin-18 and E-cadherin fluorescence intensities (Fig. 5I). However, ORMDL3 knockdown did
not influence Claudin-18 and E-cadherin fluorescence intensities
and distribution (Fig. 5J). These
data suggested that ORMDL3 overexpression induced bronchial
epithelial barrier dysfunction by decreasing Claudin-18 protein
levels, possibly disrupting the localization of Claudin-18 and
E-cadherin in cell-cell contact.
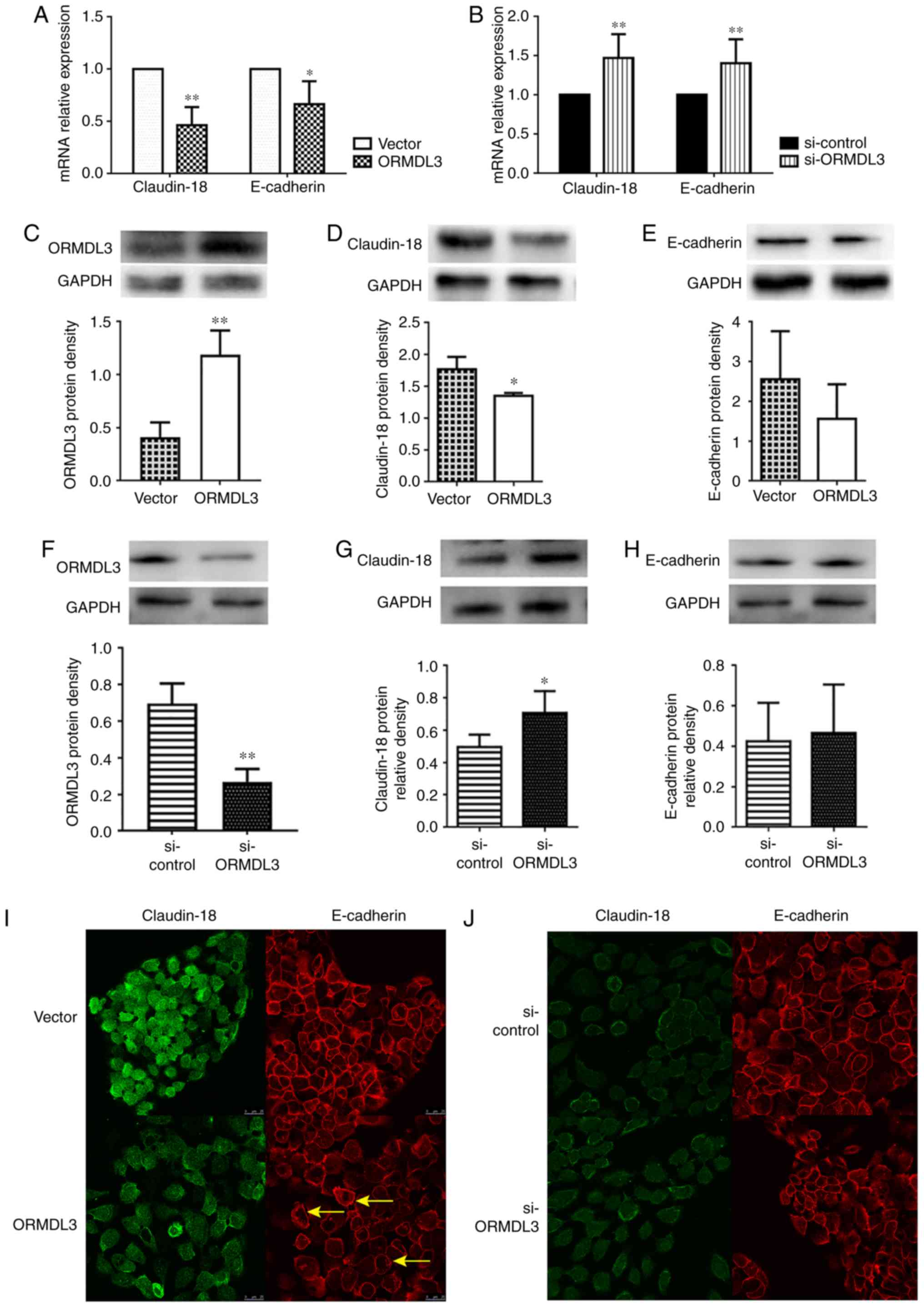 | Figure 5ORMDL3 overexpression downregulates
TJ proteins. Assessment of Claudin-18 and E-cadherin mRNA
expression levels in (A) ORMDL3/Vector and (B)
si-ORMDL3/si-control 16HBE cells showing that ORMDL3
overexpression resulted in increased ORMDL3, reduced Claudin-18 and
E-cadherin amounts, whereas ORMDL3 silencing led to reduced ORMDL3,
elevated Claudin-18 and E-cadherin levels. The protein expression
levels of (C) ORMDL3, (D) Claudin-18 and (E) E-cadherin in
ORMDL3/Vector and the protein expression levels of (F)
ORMDL3, (G) Claudin-18 and (H) E-cadherin in
si-ORMDL3/si-control 16HBE cells were consistent with mRNA
analysis. Immunofluorescence staining showing the localizations and
signal intensities of Claudin-18 and E-cadherin in (I)
ORMDL3/Vector and (J) si-ORMDL3/si-control 16HBE
cells. Values are mean ± standard deviation (n=6 per group in
duplicate). *P<0.05 and **P<0.01 vs.
the corresponding control group. Arrows show the stratification of
TJs. E, epithelial; SPHK1, sphingosine kinase 1; ORMDL3,
orosomucoid-like protein isoform 3; si, small interfering; Ctrl,
control; TNF, tumor necrosis factor; ERK, extracellular signal
regulated kinase; TJ, tight junction. |
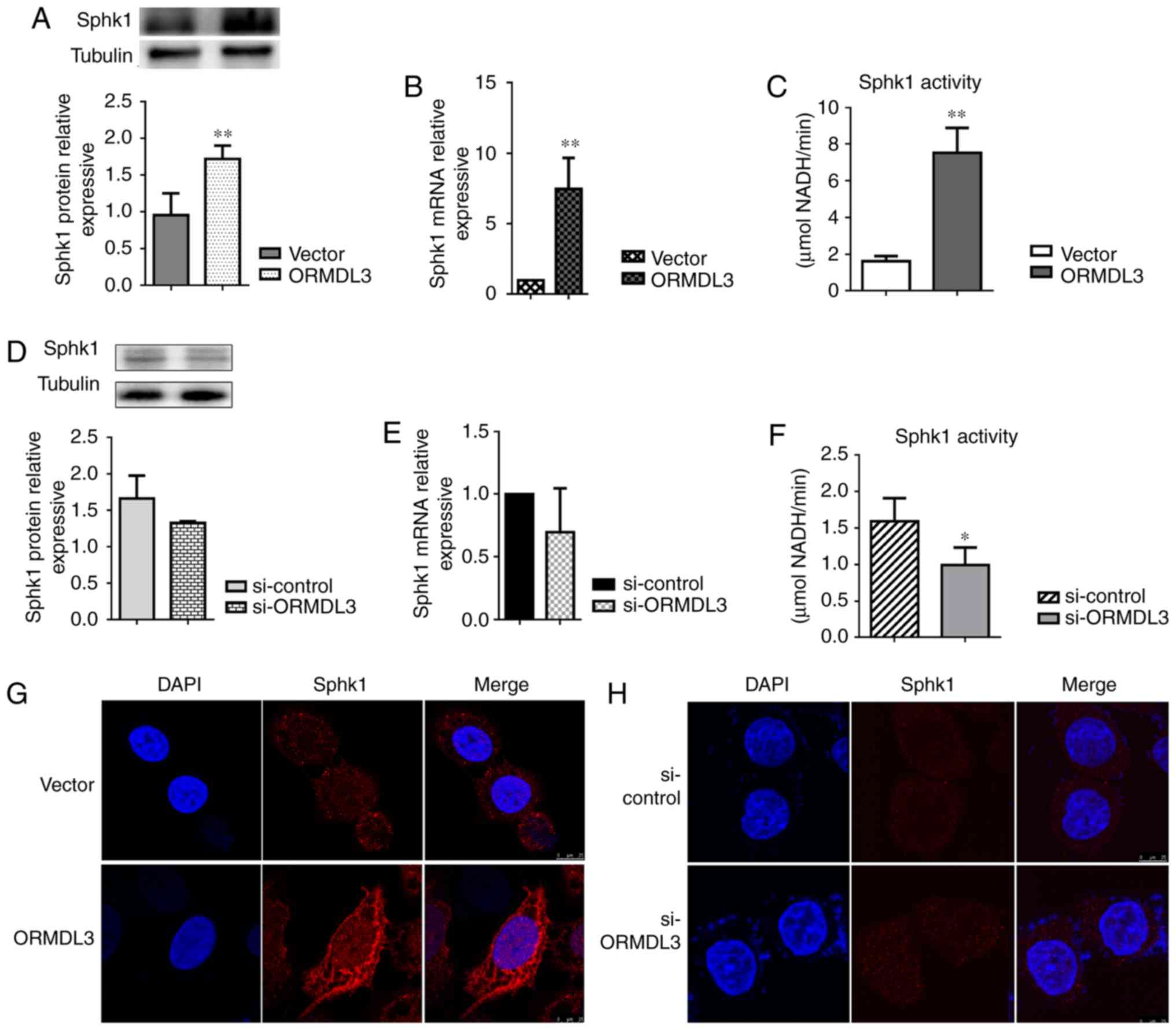
ORMDL3 promotes SPHK1 activation and
changes its distribution in 16HBE cells
To assess whether ORMDL3 induces changes in plasma
membrane composition, SPHK1 activity was determined and
distribution in ORMDL3 overexpressing and silenced 16HBE cells,
respectively. Consistent with the findings in OVA-RSV mice, SPHK1
expression in 16HBE cells showed altered patterns in response to
ORMDL3 overexpression, with mRNA and protein expression
levels significantly increased in 16HBE ORMDL3 overexpressing cell
monolayers (P<0.01; Fig. 6A and
B). Meanwhile, the opposite effects were observed after ORMDL3
knockdown although there were no statistically significant
differences (Fig. 6C and D). To
evaluate whether SPHK1 is activated more after upregulation, SPHK1
activities in ORMDL3 overexpressing and silenced cells were
assessed, respectively. ORMDL3 overexpressing cells displayed
increased SPHK1 activity compared with the Vector group
(Fig. 6E) and ORMDL3 silencing
resulted in significantly decreased SPHK1 activity compared with
si-ctrl cells (P<0.05; Fig.
6F). Moreover, cellular localization of SPHK1 was assessed in
ORMDL3/Vector and si-ORMDL3/si-control cells. The
results showed that different from the cytoplasmic localization of
SPHK1 in Vector and si-ctrl cells, ORMDL3
overexpression resulted in SPHK1 localizing to the plasma membrane,
thereby indicating SPHK1 activation (Fig. 6G); ORMDL3 suppression caused no
change of SPHK1 localization (Fig.
6H). These data indicated that ORMDL3 overexpression resulted
in SPHK1 relocation and activation.
SPHK1 silencing alleviates AE dysfunction
induced by ORMDL3 overexpression
TNF-α is a powerful epithelial barrier (23,36,37), damage inducer and has been
confirmed as a SPHK1 agonist. Moreover, a recent study found TNF-α
increases ORMDL3 protein expression in 293 cells (38). The cell viability assay (Fig. S1) shows that 10 ng/ml TNF-α is
the most suitable concentrate for 16HEB cells. TNF-α (10 ng/ml) was
used in the following assays to investigate whether inhibiting
SPHK1 by si-RNA could rescue the phenotypes associated with TNF-α,
similar to ORMDL3 overexpression. To evaluate the relationship
between ORMDL3 and SPHK1, was first transfected with
si-SPHK1 in normal 16HBE cells for 48 h before treatment
with TNF-α for another 5 days. The knockdown efficiency of
si-SPHK1 was determined by western blotting. As shown in
Fig. 7A, SPHK1 protein levels in
cells transfected with si-SPHK1 were only 5% of those found
in cells transfected with si-ctrl. It was found that reduced
TEER in 16HBE cell monolayers due to TNF-α treatment could be
largely prevented by SPHK1 knockdown (Fig. 7B). The mRNA and protein levels of
Claudin-18 and E-cadherin were significantly increased in the
si-SPHK1 and si-SPHK1+TNF-α groups compared with the
si-ctrl+TNF-α group (P<0.01; Fig. 7C and D, panels c and d). These
results indicated a negative correlation between SPHK1 and ORMDL3
proteins (Fig. 7D, panels a and
b). Since SPHK1 induces ERK1/2 trans-activation (31,39), ERK phosphorylation was evaluated
and it was found that SPHK1 knockdown alleviated TNF-α induced ERK
phosphorylation (Fig. 7D, panel
e).
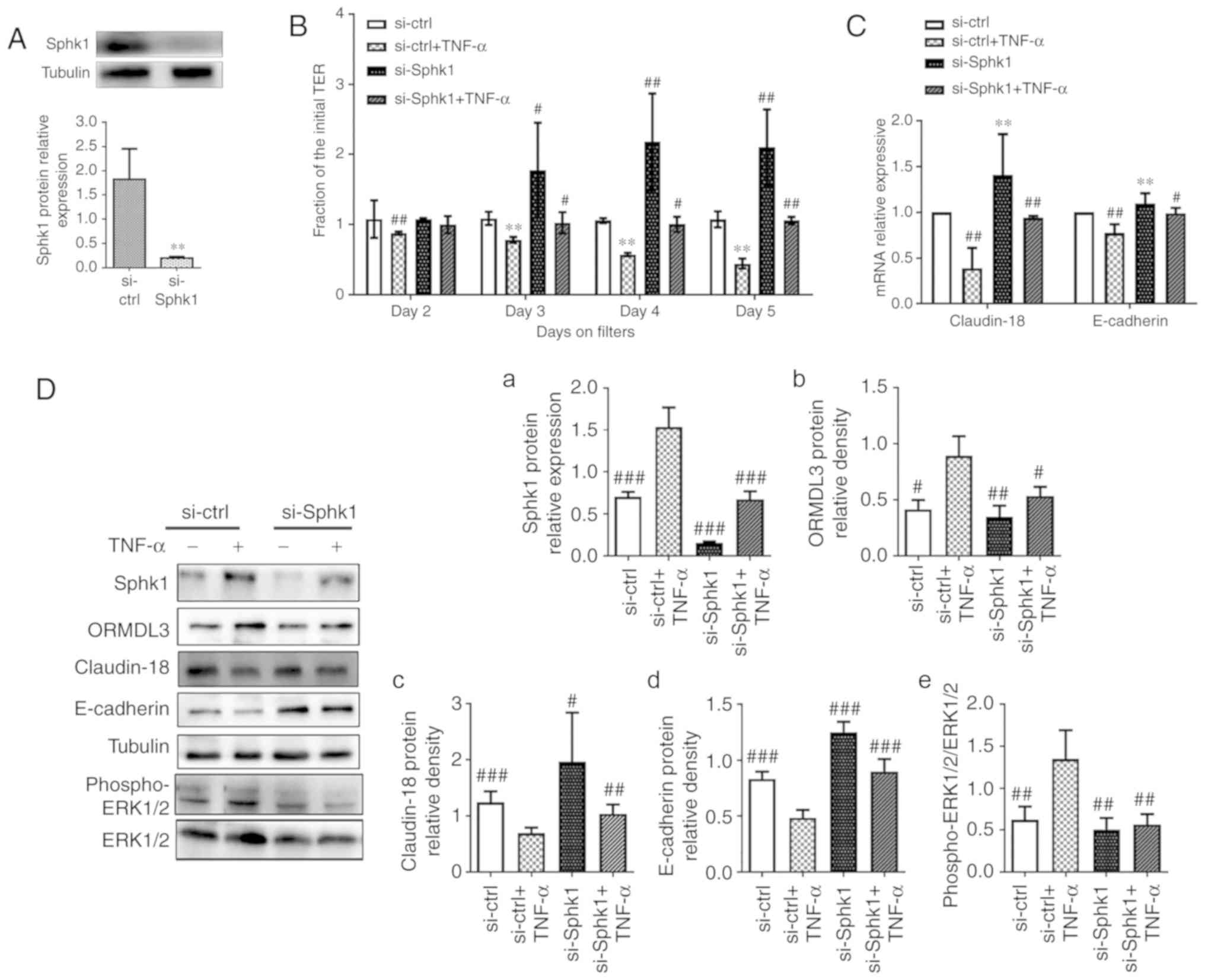 | Figure 7SPHK1 silencing alleviates ORMDL3
induced airway barrier dysfunction. (A) SPHK1 protein expression in
si-SPHK1/si-ctrl 16HBE cells was detected by western
blotting. (B) si-SPHK1/si-ctrl cells were treated with TNF-α
(10 ng/ml) for 5 days and TEER was measured daily. (C) Claudin-18
and E-cadherin mRNA expression levels in si-SPHK1/si-ctrl
and si-SPHK1+TNF-α/si-ctrl+TNF-α cells were detected
by qRT-PCR. (D-a) Sphk1, (b) ORMDL3, (c) Claudin-18, (d) E-cadherin
and (e) ERK1/2 protein expression levels in si-SPHK1/si-ctrl
and si-SPHK1+TNF-α/si-ctrl+TNF-α cells were detected
by western blotting. (E) ORMDL3/Vector cells were treated
with DMS (10 nM) for 5 days and TEER was measured daily. (F)
Claudin-18 and E-cadherin mRNA expression levels in
ORMDL3/Vector and ORMDL3+DMS/Vector+DMS cells were
detected by qRT-PCR. (G-a) Sphk1, (b) ORMDL3, (c) Claudin-18, (d)
E-cadherin and (e) ERK1/2 protein expression levels in
ORMDL3+DMS/Vector+DMS cells were detected by western
blotting. Data are presented as the mean ± standard deviation of
six independent experiments. **P<0.01 vs.
si-ctrl group. #P<0.05, ##P<0.01
and ###P<0.001 vs. si-ctrl+TNF-a group.
★P<0.05 vs. the Vector group. ∆P<0.05,
∆∆P<0.01 and ∆∆∆P<0.001 vs. ORMDL3
group. E, epithelial; SPHK1, sphingosine kinase 1; ORMDL3,
orosomucoid-like protein isoform 3; si, small interfering; ctrl,
control; TNF, tumor necrosis factor; ERK, extracellular signal
regulated kinase. |
To further assess the role of SPHK1 in ORMDL3
induced airway barrier dysfunction, ORMDL3 overexpressing cells
were treated with the specific SPHK1 inhibitor DMS (10 nM) for 5
days. The results showed that DMS restored TEER decrease associated
with ORMDL3 overexpression (Fig.
7E). ORMDL3 overexpression induced SPHK1 and ORMDL3 protein
expression, but this was decreased by SPHK1 inhibitor (Fig. 7G, panels a and b). RT-qPCR and
western blotting demonstrated that ORMDL3 overexpression induced
SPHK1 protein expression, and decreased Claudin-18 and E-cadherin
levels; these effects were restored by the above SPHK1 inhibitor
(Fig. 7F and G, panels c and d).
Furthermore, ORMDL3 overexpression induced robust ERK1/2
phosphorylation and was partly blocked by DMS (Fig. 7G, panel e). Collectively, these
data demonstrated that SPHK1/ERK1/2 signaling may be downstream of
ORMDL3 in promoting bronchial epithelial barrier dysfunction.
Discussion
Asthma is considered a heterogeneous disorder
resulting from a complex interplay between genetic susceptibility,
allergenic triggers and viral infection. Substantial efforts have
been directed toward its management. Rational therapy of chronic
asthma is important for maintaining the asthmatic patient's daily
life. However, current studies mainly focus on acute or severe
asthma rather than chronic disease. Thus, it is urgent to develop
proper animal models to investigate the potential molecular
mechanisms of chronic asthma.
Challenge allergens, methods, times and periods vary
in different studies. OVA is a conventional allergen used in
asthmatic animal studies; it induces airway inflammation in mice
but does not affect most structural changes associated with chronic
asthma. Airway infection always induces asthmatic attack. As the
most common respiratory pathogen in childhood, RSV is a cause of
morbidity and mortality in elderly and high-risk adults (40). RSV infection alters the lung
microenvironment, impairs tolerance to inhaled allergens and
increases the patient's susceptibility to asthma (41). Emerging evidence points out that
RSV contributes to AE barrier disruption via AJC disassembly and
increased paracellular permeability (42). A short-term study compared airway
hyperresponsiveness and pathological changes of the lung tissue in
RSV infected mice, OVA challenged mice, and OVA-sensitized and RSV
infected (OVA/RSV) mice. The results indicated RSV infection during
OVA sensitization prolongs AHR and increases lung lymphocyte
amounts as well as airway mucus production (43). To the best of our knowledge, no
studies have combined viral infection with allergen challenge in
long-term asthmatic animal models. In a previous study, it was
demonstrated that OVA-RSV significantly increases inflammation
scores, goblet cell hyperplasia and collagen deposition following
the last RSV infection (56 days). A total of 30 days after the last
infection, respiratory mucus hypersecretion and airway remodeling
were absent, but airway inflammation was still observed (30). In the present study, the chronic
asthmatic mouse model was modified and the expected characteristics
were obtained, including airway inflammation and remodeling, with
no mucus hypersecretion. These findings indicate that chronic
changes associated with the characteristic features of inflammation
and remodeling have been generated successfully in the present
animal model.
In the pathogenesis of asthma, inflammatory cells
initiate and maintain key pathological features through production
of cytokines (42). In
preliminary experiments, increased CXCL1, CXCL2 and CCL11 levels in
serum, elevated interferon-γ amounts in the lung tissue, and
significantly increased IL-6 and TGF-β levels in the
bronchoalveolar lavage fluid 30 days after the last OVA-RSV
challenge were observed (23,31). In the present study, the levels of
cytokines in the modified chronic model of murine allergic asthma
induced by OVA-RSV were evaluated and higher serum and lung tissue
IL-4, IL-13 and TNF-α amounts were found. Previous studies have
indicated that the above three cytokines affect TJs in vivo
and in vitro (23,44-46). In addition, administration of
IL-4, IL-13 and TNF-α significantly induces ORMDL3 expression
(9,38). These findings prompted the present
study to investigate the relationship between ORMDL3 expression and
bronchial epithelial barrier dysfunction. Previous data have
suggested that ORMDL3 plays a role in airway remodeling (including
increased airway smooth muscle, sub-epithelial fibrosis and mucus
formation), airway responsiveness, and lung elasticity (47,48); however, its effects on AE cells
are not well understood. The results of the present study indicated
that OVA-RSV challenge increased ORMDL3 expression in the lung
tissue of chronic asthmatic mice.
Bronchial epithelial cells play an important role in
asthma pathology. Increased permeability to external substances,
reduced TEER and disruption of junctional proteins are used to
assess epithelial barrier dysfunction. Hyperpermeability of
bronchial epithelial cells results in greater penetration of
inhaled allergens and particles into the sub-epithelial space,
facilitating antigen sampling and inducing innate and adaptive
immune responses (49).
Junctional proteins are important for initiating and maintaining
cell-cell adhersion, and participate in numerous signal
transduction cascades. E-cadherin is important in maintaining
tissue morphogenesis and polarity, and necessary for adhesion
junction formation (50,51). Claudin-18.1 is localized at TJs
and represents the only known lung-specific tight junction gene
product (52-54). Claudin-18 levels are decreased in
subjects with asthma and Claudin-18-deficient mice show exacerbated
serum IgE increase and enhanced airway hyperresponsiveness
following intranasal antigen sensitization (55). Meanwhile, decreased Claudin-18 and
E-cadherin protein levels in epithelial cells contribute to
defective AE barrier in patients with atopic asthma (35,56). It was demonstrated that OVA-RSV
decreased Claudin-18 and E-cadherin mRNA levels, with no effect on
Claudin-18 protein expression in the lung tissue of mouse models.
To further assess the role of ORMDL3 in bronchial epithelial
barrier dysfunction, ORMDL3 expression was manipulated in 16HBE
cells. The results suggested that ORMDL3 overexpression
significantly increased TEER, decreased permeability, downregulated
Claudin-18 at the mRNA and protein levels, and altered E-cadherin
distribution in 16HBE cells, while si-RNA-mediated ORMDL3 silencing
had opposite effects. This implied that ORMDL3 overexpression could
impair bronchial epithelial barrier function.
The cell membrane component sphingolipid has
attracted substantial attention in recent years. Several studies
have established its role in cell growth, survival and migration
(15-17). ORMDL3 is involved in sphingolipid
metabolism and de novo sphingolipid synthesis (9). An In vitro study revealed
ORMDL3 overexpression at mid-levels inhibits SPT activity; however,
more pronounced ORMDL3 overexpression results in increased
sphingolipid levels (18). SPHK1
is a key lipid kinase involved in the regulation of sphingolipid
metabolism. Interestingly, SPHK1 mRNA levels are significantly
increased in airway diseases (19-21). Previous studies have proposed that
SPHK1 is associated with airway inflammation, goblet cell
hyperplasia, and hyperresponsiveness (19,22,23). The possible mechanisms include
calcium flux control, arachidonic acid release, and induced ERK
phosphorylation (24-26). In vivo studies have
demonstrated that treatment with a SPHK1 inhibitor or SPHK1
knockout ameliorates OVA-induced AHR and airway inflammation in
mice (22,26). The present findings indicated that
OVA-RSV challenge could boost SPHK1 activity in the lung tissue of
asthmatic mice, and SPHK1 levels paralleled those of ORMDL3.
IF revealed SPHK1 distribution in the cytoplasmic
compartment of normal 16HBE cells, whereas ORMDL3 over-expression
caused SPHK1 to translocate to the cell membrane; however, ORMDL3
knockdown did not affect SPHK1 location. A study of head and neck
carcinoma indicated that SPHK1 expression correlates with
E-cadherin downregulation (15).
To investigate whether SPHK1 is involved in ORMDL3 induced airway
barrier dysfunction, ORMDL3 overexpressing cells were treated with
DMS, which resulted in decreased ORMDL3 levels in these cells,
although not reaching the normal levels. Meanwhile, DMS alleviated
TEER and junctional protein expression changes associated with
ORMDL3 overexpression. These results demonstrated that SPHK1 is
involved in ORMDL3 induced bronchial epithelial barrier dysfunction
as a downstream effector.
The mitogen-activated protein kinase (MAPK) family
is a group of serine/threonine kinases, which include p38 MAPK and
ERK protein kinase pathways. Increasing evidence indicates that
activation of the MAPK/ERK pathway promotes the proliferation,
migration and differentiation of peripheral cells around airway
epithelial damage (57,58). Impaired alveolar barrier function
in septic rats is associated with ERK-mediated downregulation of
several tight junction components (59). Furthermore, the ERK pathway is
implicated in barrier function dysregulation attributed to reduced
levels of junctional proteins in AE cells (60). Previous studies reported that the
SPHK1/ERK1/2 signaling pathway is involved in mucus production in
human AE cells. In the present study, the effects of ORMDL3 on the
ERK1/2 signaling cascade were assessed (19,24,61). The results showed that the SPHK1
inhibitor DMS alleviated AE barrier dysfunction and ERK signaling
pathway activation associated with ORMDL3 overexpression.
However, there were certain limitations in this
study. AHR and de novo sphingolipid metabolism were not
evaluated in the current mouse model, which requires further
assessment using ORMDL3 transgenic mice.
In conclusion, this study provided novel findings
that ORMDL3 overexpression results in damaged epithelial barrier
integrity, reflected by higher epithelial permeability, increased
inflammatory cytokine secretion, decreased TEER and hampered
expression of AE molecules. In long-term chronic asthma mouse
models, induced ORMDL3overexpression was observed alongside
Claudin-18 and E-cadherin down-regulation. The in vitro
study suggested that ORMDL3 overexpression promotes ERK1/2
phosphorylation, and via SPHK1 activation and inhibition, reverses
ORMDL3 induced AE barrier dysfunction by alleviating ERK1/2
phosphorylation. Taken together, these findings reveal a novel role
for ORMDL3 in the pathogenesis of asthma.
Supplementary Data
Acknowledgments
The authors would like to thank Dr Xiaofei Jiang
from Jiangsu Key Laboratory of Pediatric Respiratory Disease for
providing the anti-phospho-ERK1/2 and anti-ERK1/2 antibodies.
Funding
The present study was partly supported by grants
from the the National Natural Science Foundation of China, China
(grant no. 81473723) and the Innovation Program for Graduate
Students of Jiangsu Province, China (grant no. KYZZ17_1316).
Availability of data and materials
The authors declare that all supporting data are
available within the article and its online supplementary
files.
Authors' contributions
RY and MT performed the experiments. RY, MT and JX
analyzed and interpreted the data. XZ made substantial
contributions to the design supervision of the present study. RY
wrote the manuscript. All authors reviewed the results and approved
the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were performed according to
procedures approved by the Animal Ethics Committee of Nanjing
University of Chinese Medicine.
Patient consent for publication
Not applicable.
Compenting interests
All authors declare that they have no any conflict
of interests.
References
|
1
|
Global Initiative for Asthma: Global
strategy for asthma management and prevention. 2017.
|
|
2
|
Martinez FD and Vercelli D: Asthma Lancet.
382:1360–1372. 2013. View Article : Google Scholar
|
|
3
|
McGarvey LP, Butler CA, Stokesberry S,
Polley L, McQuaid S, Abdullah H, Ashraf S, McGahon MK, Curtis TM,
Arron J, et al: Increased expression of bronchial epithelial
transient receptor potential vanilloid 1 channels in patients with
severe asthma. J Allergy Clin Immunol. 133:704–712.e4. 2014.
View Article : Google Scholar
|
|
4
|
Hackett TL and Knight DA: The role of
epithelial injury and repair in the origins of asthma. Curr Opin
Allergy Clin Immunol. 7:63–68. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung KF: Intrinsic differences of the
airway epithelium in childhood allergic asthma. Am J Respir Crit
Care Med. 174:1066–1067. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao C, Puddicombe SM, Field S, Haywood J,
Broughton-Head V, Puxeddu I, Haitchi HM, Vernon-Wilson E, Sammut D,
Bedke N, et al: Defective epithelial barrier function in asthma. J
Allergy Clin Immunol. 128:549–556. e1–e12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kicic A, Sutanto EN, Stevens PT, Knight DA
and Stick SM: Intrinsic biochemical and functional differences in
bronchial epithelial cells of children with asthma. Am J Respir
Crit Care Med. 174:1110–1118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moffatt MF: Genes in asthma: New genes and
new ways. Curr Opin Allergy Clin Immunol. 8:411–417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller M, Rosenthal P, Beppu A, Mueller
JL, Hoffman HM, Tam AB, Doherty TA, McGeough MD, Pena CA, Suzukawa
M, et al: ORMDL3 transgenic mice have increased airway remodeling
and airway responsiveness characteristic of asthma. J Immunol.
192:3475–3487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hjelmqvist L, Tuson M, Marfany G, Herrero
E, Balcells S and Gonzàlez-Duarte R: ORMDL proteins are a conserved
new family of endoplasmic reticulum membrane proteins. Genome Biol.
3:RESEARCH00272002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller M, Tam AB, Cho JY, Doherty TA, Pham
A, Khorram N, Rosenthal P, Mueller JL, Hoffman HM, Suzukawa M, et
al: ORMDL3 is an inducible lung epithelial gene regulating
metal-loproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci
USA. 109:16648–16653. 2012. View Article : Google Scholar
|
|
12
|
Carreras-Sureda A, Cantero-Recasens G,
Rubio-Moscardo F, Kiefer K, Peinelt C, Niemeyer BA, Valverde MA and
Vicente R: ORMDL3 modulates store-operated calcium entry and
lymphocyte activation. Hum Mol Genet. 22:519–530. 2013. View Article : Google Scholar
|
|
13
|
Worgall TS, Veerappan A, Sung B, Kim BI,
Weiner E, Bholah R, Silver RB, Jiang XC and Worgall S: Impaired
sphingolipid synthesis in the respiratory tract induces airway
hyperreactivity. Sci Transl Med. 5:186ra672013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levy BD: Sphingolipids and susceptibility
to asthma. N Engl J Med. 369:976–978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young MM, Kester M and Wang HG:
Sphingolipids: Regulators of crosstalk between apoptosis and
autophagy. J Lipid Res. 54:5–19. 2013. View Article : Google Scholar :
|
|
16
|
Schiefler C, Piontek G, Doescher J,
Schuettler D, Mißlbeck M, Rudelius M, Haug A, Reiter R, Brockhoff G
and Pickhard A: Inhibition of SphK1 reduces radiation-induced
migration and enhances sensitivity to cetuximab treatment by
affecting the EGFR/SphK1 crosstalk. Oncotarget. 5:9877–9888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tamashiro PM, Furuya H, Shimizu Y and
Kawamori T: Sphingosine kinase 1 mediates head & neck squamous
cell carcinoma invasion through sphingosine 1-phosphate receptor 1.
Cancer Cell Int. 14:762014. View Article : Google Scholar :
|
|
18
|
Oyeniran C, Sturgill JL, Hait NC, Huang
WC, Avni D, Maceyka M, Newton J, Allegood JC, Montpetit A, Conrad
DH, et al: Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3)
expression dysregulates ceramide homeostasis in cells and ceramide
exacerbates allergic asthma in mice. J Allergy Clin Immunol.
136:1035–1046.e6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Natarajan V, Ha AW, Dong Y, Reddy NM,
Ebenezer DL, Kanteti P, Reddy SP, Usha Raj J, Lei Z,
Maienschein-Cline M, et al: Expression profiling of genes regulated
by sphingosine kinase1 signaling in a murine model of hyperoxia
induced neonatal bronchopulmonary dysplasia. BMC Genomics.
18:6642017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nishiuma T, Nishimura Y, Okada T, Kuramoto
E, Kotani Y, Jahangeer S and Nakamura S: Inhalation of sphingosine
kinase inhibitor attenuates airway inflammation in asthmatic mouse
model. Am J Physiol Lung Cell Mol Physiol. 294:L1085–L1093. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu CY, Liu SQ, Qin MB, Zhuge CF, Qin L,
Qin N, Lai MY and Huang JA: SphK1 modulates cell migration and
EMT-related marker expression by regulating the expression of p-FAK
in colorectal cancer cells. Int J Mol Med. 39:1277–1284. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haberberger RV, Tabeling C, Runciman S,
Gutbier B, König P, Andratsch M, Schütte H, Suttorp N, Gibbins I
and Witzenrath M: Role of sphingosine kinase 1 in allergen-induced
pulmonary vascular remodeling and hyperresponsiveness. J Allergy
Clin Immunol. 124:933–941. e1–e9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sim TY, Harith HH, Tham CL, Md Hashim NF,
Shaari K, Sulaiman MR and Israf DA: The protective effects of a
synthetic geranyl acetophenone in a cellular model of TNF-α-induced
pulmonary epithelial barrier dysfunction. Molecules. 23:pii: E1355.
2018. View Article : Google Scholar
|
|
24
|
Kono Y, Nishiuma T, Okada T, Kobayashi K,
Funada Y, Kotani Y, Jahangeer S, Nakamura S and Nishimura Y:
Sphingosine kinase 1 regulates mucin production via ERK
phosphorylation. Pulm Pharmacol Ther. 23:36–42. 2010. View Article : Google Scholar
|
|
25
|
Oskeritzian CA, Alvarez SE, Hait NC, Price
MM, Milstien S and Spiegel S: Distinct roles of sphingosine kinases
1 and 2 in human mast-cell functions. Blood. 111:4193–4200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Price MM, Oskeritzian CA, Falanga YT,
Harikumar KB, Allegood JC, Alvarez SE, Conrad D, Ryan JJ, Milstien
S and Spiegel S: A specific sphingosine kinase 1 inhibitor
attenuates airway hyperresponsiveness and inflammation in a mast
cell-dependent murine model of allergic asthma. J Allergy Clin
Immunol. 131:501–511.e1. 2013. View Article : Google Scholar
|
|
27
|
Huang Z, Gao L, Zhao X, Ling H and Chen W:
Effect of Gubenfangxiao decoction on respiratory syncytial
virus-induced asthma and expression of asthma susceptibility gene
orosomucoid 1-like protein 3 in mice. J Tradit Chin Med.
36:101–106. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Page K, Lierl KM, Herman N and Wills-Karp
M: Differences in susceptibility to German cockroach frass and its
associated proteases in induced allergic inflammation in mice.
Respir Res. 8:912007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Lu Y, Xu JY, Zhang XH and Zhao X:
Gu-Ben-Fang-Xiao decoction attenuates sustained airway inflammation
by suppressing ER stress response in a murine asthma remission
model of respiratory syncytial virus infection. J Ethnopharmacol.
192:496–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xia P, Gamble JR, Wang L, Pitson SM,
Moretti PA, Wattenberg BW, D'Andrea RJ and Vadas MA: An oncogenic
role of sphingosine kinase. Curr Biol. 10:1527–1530. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng Q and Shang Y: ORMDL3 may
participate in the pathogenesis of bronchial epithelial-mesenchymal
transition in asthmatic mice with airway remodeling. Mol Med Rep.
17:995–1005. 2018.
|
|
33
|
Breslow DK, Collins SR, Bodenmiller B,
Aebersold R, Simons K, Shevchenko A, Ejsing CS and Weissman JS: Orm
family proteins mediate sphingolipid homeostasis. Nature.
463:1048–1053. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nawijn MC, Hackett TL, Postma DS, van
Oosterhout AJ and Heijink IH: E-cadherin: Gatekeeper of airway
mucosa and allergic sensitization. Trends Immunol. 32:248–255.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sweerus K, Lachowicz-Scroggins M, Gordon
E, LaFemina M, Huang X, Parikh M, Kanegai C, Fahy JV and Frank JA:
Claudin-18 deficiency is associated with airway epithelial barrier
dysfunction and asthma. J Allergy Clin Immunol. 139:72–81.e1. 2017.
View Article : Google Scholar
|
|
36
|
Hardyman MA, Wilkinson E, Martin E,
Jayasekera NP, Blume C, Swindle EJ, Gozzard N, Holgate ST, Howarth
PH, Davies DE and Collins JE: TNF-α-mediated bronchial barrier
disruption and regulation by src-family kinase activation. J
Allergy Clin Immunol. 132:665–675.e8. 2013. View Article : Google Scholar
|
|
37
|
Ma TY, Iwamoto GK, Hoa NT, Akotia V,
Pedram A, Boivin MA and Said HM: TNF-alpha-induced increase in
intestinal epithelial tight junction permeability requires NF-kappa
B activation. Am J Physiol Gastrointest Liver Physiol.
286:G367–G376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qiu L: Effect of TNF-α on ORMDL3 gene
promoter regulation and its possible mechanism. Dissertation.
Nanjing Medical University; 2014
|
|
39
|
Pitson SM, Moretti PA, Zebol JR, Xia P,
Gamble JR, Vadas MA, D'Andrea RJ and Wattenberg BW: Expression of a
catalytically inactive sphingosine kinase mutant blocks
agonist-induced sphingosine kinase activation. A dominant-negative
sphingosine kinase. J Biol Chem. 275:33945–33950. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Falsey AR, Hennessey PA, Formica MA, Cox C
and Walsh EE: Respiratory syncytial virus infection in elderly and
high-risk adults. N Engl J Med. 352:1749–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Krishnamoorthy N, Khare A, Oriss TB,
Raundhal M, Morse C, Yarlagadda M, Wenzel SE, Moore ML, Peebles RS
Jr, Ray A and Ray P: Early infection with respiratory syncytial
virus impairs regulatory T cell function and increases
susceptibility to allergic asthma. Nat Med. 18:1525–1530. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rezaee F, DeSando SA, Ivanov AI, Chapman
TJ, Knowlden SA, Beck LA and Georas SN: Sustained protein kinase D
activation mediates respiratory syncytial virus-induced airway
barrier disruption. J Virol. 87:11088–11095. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Peebles RS Jr, Sheller JR, Johnson JE,
Mitchell DB and Graham BS: Respiratory syncytial virus infection
prolongs methacholine-induced airway hyperresponsiveness in
oval-bumin-sensitized mice. J Med Virol. 57:186–192. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wawrzyniak P, Wawrzyniak M, Wanke K,
Sokolowska M, Bendelja K, Ruckert B, Globinska A, Jakiela B, Kast
JI, Idzko M, et al: Regulation of bronchial epithelial barrier
integrity by type 2 cytokines and histone deacetylases in asthmatic
patients. J Allergy Clin Immunol. 139:93–103. 2017. View Article : Google Scholar
|
|
45
|
Sugita K, Steer CA, Martinez-Gonzalez I,
Altunbulakli C, Morita H, Castro-Giner F, Kubo T, Wawrzyniak P,
Rückert B, Sudo K, et al: Type 2 innate lymphoid cells disrupt
bronchial epithelial barrier integrity by targeting tight junctions
through IL-13 in asthmatic patients. J Allergy Clin Immunol.
141:300–310.e11. 2018. View Article : Google Scholar
|
|
46
|
Shang VC, Kendall DA and Roberts RE:
Δ9-Tetrahydrocannabinol reverses TNFα-induced increase
in airway epithelial cell permeability through CB2
receptors. Biochem Pharmacol. 120:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hsu KJ and Turvey SE: Functional analysis
of the impact of ORMDL3 expression on inflammation and activation
of the unfolded protein response in human airway epithelial cells.
Allergy Asthma Clin Immunol. 9:42013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ha SG, Ge XN, Bahaie NS, Kang BN, Rao A,
Rao SP and Sriramarao P: ORMDL3 promotes eosinophil trafficking and
activation via regulation of integrins and CD48. Nat Commun.
4:24792013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Georas SN and Rezaee F: Epithelial barrier
function: At the front line of asthma immunology and allergic
airway inflammation. J Allergy Clin Immunol. 134:509–520. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Boussadia O, Kutsch S, Hierholzer A,
Delmas V and Kemler R: E-cadherin is a survival factor for the
lactating mouse mammary gland. Mech Dev. 115:53–62. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Van den Bossche J and Van Ginderachter JA:
E-cadherin: From epithelial glue to immunological regulator. Eur J
Immunol. 43:34–37. 2013. View Article : Google Scholar
|
|
52
|
Soini Y: Claudins in lung diseases. Respir
Res. 12:702011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Niimi T, Nagashima K, Ward JM, Minoo P,
Zimonjic DB, Popescu NC and Kimura S: Claudin-18, a novel
downstream target gene for the T/EBP/NKX2.1 homeodomain
transcription factor, encodes lung- and stomach-specific isoforms
through alternative splicing. Mol Cell Biol. 21:7380–7390. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ohta H, Chiba S, Ebina M, Furuse M and
Nukiwa T: Altered expression of tight junction molecules in
alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell
Mol Physiol. 302:L193–L205. 2012. View Article : Google Scholar
|
|
55
|
Li G, Flodby P, Luo J, Kage H, Sipos A,
Gao D, Ji Y, Beard LL, Marconett CN, DeMaio L, et al: Knockout mice
reveal key roles for claudin 18 in alveolar barrier properties and
fluid homeo-stasis. Am J Respir Cell Mol Biol. 51:210–222.
2014.PubMed/NCBI
|
|
56
|
Abe-Yutori M, Chikazawa T, Shibasaki K and
Murakami S: Decreased expression of E-cadherin by Porphyromonas
gingivalis-lipopolysaccharide attenuates epithelial barrier
function. J Periodontal Res. 52:42–50. 2017. View Article : Google Scholar
|
|
57
|
Montiel M, Quesada J and Jiménez E:
Activation of calcium-dependent kinases and epidermal growth factor
receptor regulate muscarinic acetylcholine receptor-mediated
MAPK/ERK activation in thyroid epithelial cells. Cell Signal.
19:2138–2146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cohen TS, Gray Lawrence G and Margulies
SS: Cultured alveolar epithelial cells from septic rats mimic in
vivo septic lung. PLoS One. 5:e113222010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Petecchia L, Sabatini F, Usai C, Caci E,
Varesio L and Rossi GA: Cytokines induce tight junction disassembly
in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab
Invest. 92:1140–1148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lai WQ, Goh HH, Bao Z, Wong WS, Melendez
AJ and Leung BP: The role of sphingosine kinase in a murine model
of allergic asthma. J Immunol. 180:4323–4329. 2008. View Article : Google Scholar : PubMed/NCBI
|