Introduction
Cardiac dysfunction is a major contributor to the
significantly increased mortality rate in patients with sepsis
compared with in septic patients without cardiac dysfunction
(1,2). Previous studies have demonstrated
that the mechanisms underlying sepsis-induced myocardial
dysfunction include inflammatory mediators, structural alterations,
dysfunctional cardiomyocyte contractility, reduced energy
metabolism and cell death (3-5).
However, the precise cause and molecular mechanism underlying the
pathogenesis of septic cardiomyopathy are not completely
understood. Lipopolysaccharide (LPS) from Gram-negative bacteria
uses the Toll-like receptor 4 (TLR4) signaling pathway to mediate
pro-inflammatory and pro-apoptotic activities and is recognized as
the best characterized activator of sepsis; it has been reported
that cardiomyocytes express TLR4 (6). Tumor necrosis factor (TNF)-α is an
inflammatory cytokine present in the circulation that can be
produced locally by cardiomyocytes (7). Evidence has indicated that
modulation of TNF-α levels, either directly (8,9) or
by downregulating TNF-α (10),
has therapeutic significance in sepsis-induced myocardial
dysfunction. Additionally, TNF-α, as a death ligand, is responsible
for death receptor-induced apoptosis, which also contributes to
sepsis-induced cardiac dysfunction. Previous studies have reported
that endotoxin injection activates apoptotic and survival pathways
in the hearts of rats. Apoptotic regulatory factors are activated
in the hearts of rats treated with endotoxin, and caspase
inhibition (11) or Bcl-2
overexpression (12) prevents
myocardial dysfunction and cardiac apoptosis in rodent models of
sepsis. It is well established that nuclear factor (NF)-κB and
mitogen-activated protein kinase (MAPK) serve critical roles in the
inflammatory response and apoptosis regulation, and participate in
the pathogenesis of sepsis-induced cardiac dysfunction.
The Tyro3, Axl and Mer (TAM) family comprises
receptor tyrosine kinases, which initiate intracellular signaling
cascades by binding to ligands, such as growth arrest-specific 6
(Gas6) and protein S (13). Among
the TAM family, Axl is expressed ubiquitously and has the highest
affinity to Gas6 (14,15). Numerous studies have reported that
the Gas6/Axl pathway exerts anti-inflammatory and anti-apoptotic
effects (16,17). Specifically, Axl is expressed in
the heart (18) and Gas6/Axl
signaling exerts a protective effect against myocardial ischemia
(19). However, the role of
Gas6/Axl in cardiac myocytes treated with endotoxin remains
unknown. Furthermore, the Gas6/Axl/PI3K/Akt pathway is well known
for its anti-apoptotic effects (20,21). Previous studies have demonstrated
that the PI3K/Akt-dependent pathway, which regulates cell
proliferation and survival, is protective against sepsis-induced
myocardial injury in vitro and in vivo (22,23). In addition, PI3K/Akt signaling
activation ameliorates sepsis-induced cardiac dysfunction and
improves survival rates (24,25). Therefore, it is reasonable to
speculate that modulation of the Gas6/Axl/PI3K/Akt pathway may
benefit sepsis-induced myocardial dysfunction.
Gas6, which is structurally similar to protein S, is
a vitamin K-dependent protein. The levels of plasma Gas6 are
increased in patients with sepsis (26,27); however, its biological effects may
be hindered by increased Axl (28). Recent studies have demonstrated
that exogenous administration of Gas6 arrests TNF-α and
interleukin-1 in monocytes/macrophages stimulated with LPS
(29), and exerts protective
effects on the lung (30) and
kidney (31) in rodent models of
sepsis, which indicates a practical role for the exogenous use of
Gas6.
This study hypothesized that Gas6 may protect
against sepsis-induced myocardial dysfunction via Axl/PI3K/Akt
signaling, which is associated with the MAPK and NF-κB pathways.
H9C2 cells were stimulated with LPS to mimic sepsis-induced cardiac
dysfunction and Gas6 was applied exogenously. MAPK and NF-κB
activation, TNF-α expression and apoptosis were then evaluated in
the presence or absence of TP-0903 (an Axl inhibitor) and
Wortmannin (a PI3K inhibitor).
Materials and methods
Cell culture and treatments
Cardiomyoblasts (H9C2 cells) obtained from the
Chinese Academy of Sciences Cell Bank were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), 100 IU/ml penicillin and
100 mg/ml streptomycin at 37°C in an atmosphere containing in 5%
CO2. Cells were seeded onto 6-well plates at a density
of 1.5×105 cells/well. Recombinant mouse Gas6 (100
ng/ml) (R&D Systems, Inc.) was added 2 h prior to stimulation
with LPS (10 µg/ml; from Escherichia coli;
Sigma-Aldrich; Merck KGaA). Inhibitors TP-0903 (15 nM) and
Wortmannin (3 nM) (Selleck Chemicals, LLC) were administered 15 min
prior to Gas6 administration. Cells were incubated for 15 min-24 h
with LPS at 37°C and were then harvested for analysis.
Cell counting kit 8 (CCK8) cell viability
assay
Cells were seeded in a 96-well plate
(5×103 cells/well) and exposed to various treatments
after reaching 50% confluence. CCK8 reagent (10 µl; Dojindo
Molecular Technologies, Inc.) and DMEM (100 µl) were then
added and the plates were incubated at 37°C for 3 h. The absorbance
was determined using a microplate reader (Tecan Group, Ltd.) at 450
nm.
Lactate dehydrogenase (LDH) assay
Cells were seeded in a 96-well plate
(5×103 cells/well) and were exposed to various
treatments after reaching 50% confluence. LDH release measurements
were analyzed using the relative LDH assay kit (cat. no. A020-2;
Nanjing Jiancheng Bioengineering Institute) according to the
manufacturer's protocol. Culture medium was retained to perform
TNF-α analysis.
Evaluation of TNF-α release
The levels of TNF-α in the culture medium were
evaluated using a TNF-α enzyme-linked immunosorbent assay kit (cat.
no. BPE10374-09R; Shanghai Boyun Biotech Co., Ltd.) in accordance
with the manufacturer's protocol.
Measurement of caspase-3 activity
Caspase-3 activity was determined using a commercial
Caspase Activity kit (cat. no. BC3830-50T; Beijing Solarbio Science
& Technology Co., Ltd.) according to the manufacturer's
protocol.
Annexin V-FITC/propidium iodide (PI)
staining for phosphatidylserine translocation
Annexin V-FITC/PI staining kit (BD Biosciences) was
used to detect apoptosis, according to the manufacturer's protocol.
Cells were harvested and resuspended in 150 ml binding buffer,
followed by staining with 5 µl FITC-conjugated Annexin V and
5 µl PI in the dark for 15 min at room temperature. The
mixture was detected by flow cytometry (FACSCalibur BD
Biosciences). In the early stage of apoptosis, cell membranes were
stained with FITC-conjugated Annexin V, whereas nuclei were not
stained with PI. In the late stage of apoptosis, cells were stained
with both FITC-conjugated Annexin V and PI. The results were
analyzed by FlowJo vX.0.7 (FlowJo LLC).
TUNEL assay
Cell apoptosis was also detected using the TUNEL
method. The assay was carried out with a commercial Cell Death
Detection kit (cat. no. 11684817910; Roche Molecular Diagnostics)
in accordance with the manufacturer's protocols. Images of the
cells were captured using a fluorescence microscope.
Hoechst staining for nuclear
morphology
Cells were fixed with 4% paraformaldehyde for 30 min
at room temperature and washed twice with PBS. Cells were incubated
with Hoechst 33342 (Beijing Solarbio Science & Technology Co.,
Ltd.) at room temperature for 5 min and washed with PBS three
times. Images of the cells were captured using a fluorescence
microscope.
Immunofluorescence analysis
Cells from the various treatment groups were fixed
with 4% paraformaldehyde at 4°C for 1 h and were permeabilized with
0.5% Triton X-100 for 10 min. After blocking with 1% bovine serum
albumin (BSA; Beyotime Institute of Biotechnology) at 4°C for 30
min, the cells were incubated with anti-P65 (cat. no. 8242S; 1:100;
Cell Signaling Technology, Inc.) at 4°C overnight. Subsequently,
samples were incubated with DyLightFluor® 488-conjugated
donkey anti-rabbit IgG secondary antibodies (1:400; cat. no.
BYE026; Shanghai Boyun Biotech Co., Ltd.) for 1 h at room
temperature and were stained with DAPI for 7 min in the dark. The
representative images were captured using an Olympus BX51
microscope (Olympus Corporation).
Western blotting
Cells were harvested with RIPA lysis buffer
(Beyotime Institute of Biotechnology) on ice once cell treatment
was completed. The protein concentration was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). Equal amounts of protein (5 µg/µl, 10
µl per lane) were separated by 8-12% SDS-PAGE and were
transferred onto PVDF membranes using Bio-Rad western blot analysis
apparatus (Bio-Rad Laboratories, Inc.). The membranes were then
blocked with 5% BSA at room temperature for 1 h and incubated at
4°C overnight with antibodies against Axl (cat. no. BZ12291),
phosphorylated (p)-Axl (cat. no. BS64021), Akt (cat. no. BS1810),
p-Akt (cat. no. BS4006), IκB-α (BS3601; Bioworld Technology, Inc.),
p-IκB-a (cat. no. 2859T), P65 (cat. no. 8242S), p-P65 (cat. no.
3033S), extracellular signal-regulated kinase 1/2 (ERK1/2) (cat.
no. 4695T), p-ERK1/2 (cat. no. 4370T), c-Jun N-terminal protein
kinase (JNK) (cat. no. 9252T), p-JNK (cat. no. 4668T), p38 MAPK
(cat. no. 8690T), p-p38 MAPK (cat. no. 4511T), Bcl-2 (cat. no.
2870T) (Cell Signaling Technology, Inc.), Bax (cat. no. ab32503;
Abcam) (dilutions, 1:1,000) or GAPDH (cat. no. BS60630; 1:8,000;
Bioworld Technology, Inc.), followed by incubation at room
temperature for 1 h with corresponding secondary antibodies
(1:5,000; cat. no. BL003A; Biosharp Life Science, Inc.). Protein
bands were detected with an enhanced chemiluminescence kit (Thermo
Fisher Scientific, Inc.) and were semi-quantified using Quantity
One v4.6.6 software (Bio-Rad Laboratories, Inc.).
Statistical analysis
The results are presented as the mean ± standard
deviation from at least three independent experiments. Statistical
comparisons were analyzed by one-way analysis of variance and
Tukey's test using GraphPad Prism 5 software (GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Gas6 attenuates LPS-induced cytotoxicity
in H9C2 cardiomyocytes
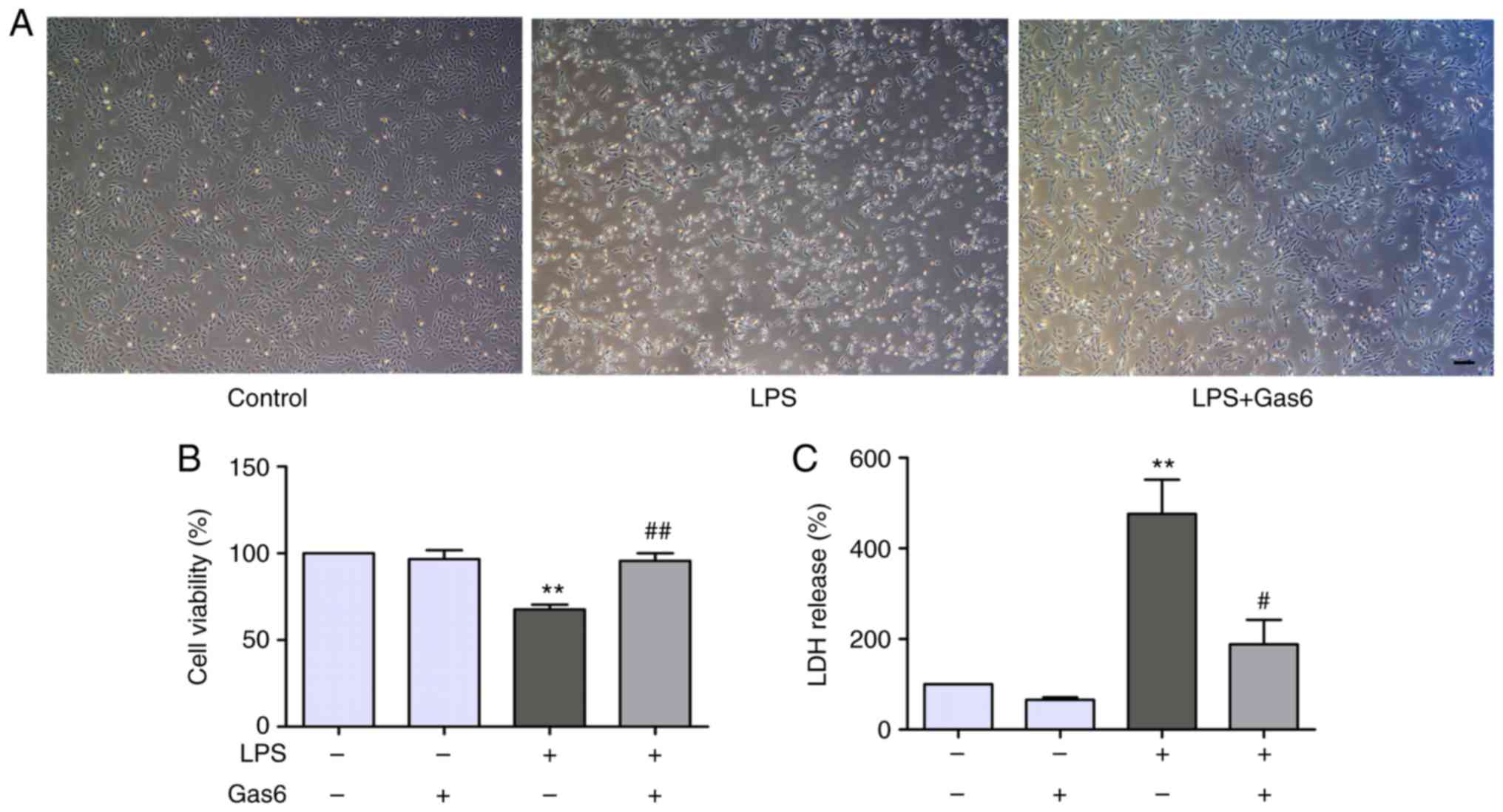
The present study determined the effects of Gas6 on
LPS-stimulated H9C2 cells using phase-contrast microscopy. Notably,
H9C2 cells treated with LPS for 24 h were markedly shrunk in size
and decreased in number compared with untreated cells (Fig. 1A). Treatment with Gas6 (100 ng/ml)
induced a significant improvement in cell morphology and decreased
cell death compared with in the LPS-treated group. To further
investigate the role of Gas6 in H9C2 cells challenged with LPS,
CCK8 and LDH assays were performed as indicators of cytotoxicity.
Treatment with LPS (10 µg/ml) decreased cell viability by
32.3% compared with the control group (P<0.01). Pretreatment
with Gas6 induced a marked increase (41.3%) in the viability of
cells compared with the LPS group (P<0.01; Fig. 1B). Furthermore, treatment of H9C2
cells with LPS increased LDH release by 476.1% compared with the
control cells (P<0.01), which was reduced by 60.4% with
co-treatment with Gas6 (P<0.05; Fig. 1C).
Gas6 activates the Axl/PI3K/Akt pathway
in LPS-challenged H9C2 cardiomyocytes
The association between Gas6/Axl and PI3K activation
in is well known various cell types (20,21). To identify the signaling pathway
associated with the protective effects of Gas6 on LPS-treated H9C2
cells, this study investigated whether Gas6 activated the
Axl/PI3K/Akt pathway. Western blotting results demonstrated that
Gas6 alone had no effect on the phosphorylation and expression of
Axl and Akt. However, Gas6 enhanced the phosphorylation and
expression of Axl and Akt in the presence of LPS. To determine
whether Gas6-activated PI3K/Akt signaling was mediated by Axl,
TP-0903, an Axl inhibitor, was administered. Pretreatment with
TP-0903 abolished the enhanced phosphorylation and expression of
Axl and Akt induced by Gas6 (Fig. 2A
and C-H). These results suggested that Axl may mediate
Gas6-induced activation of the PI3K/Akt signaling pathway. To
determine the effects of Wortmannin (PI3K inhibitor) on the
phosphorylation and expression of Akt, cells were treated with
Wortmannin prior to Gas6. Wortmannin decreased the phosphorylation
and expression of Akt induced by Gas6 treatment (Fig. 2B and F-H).
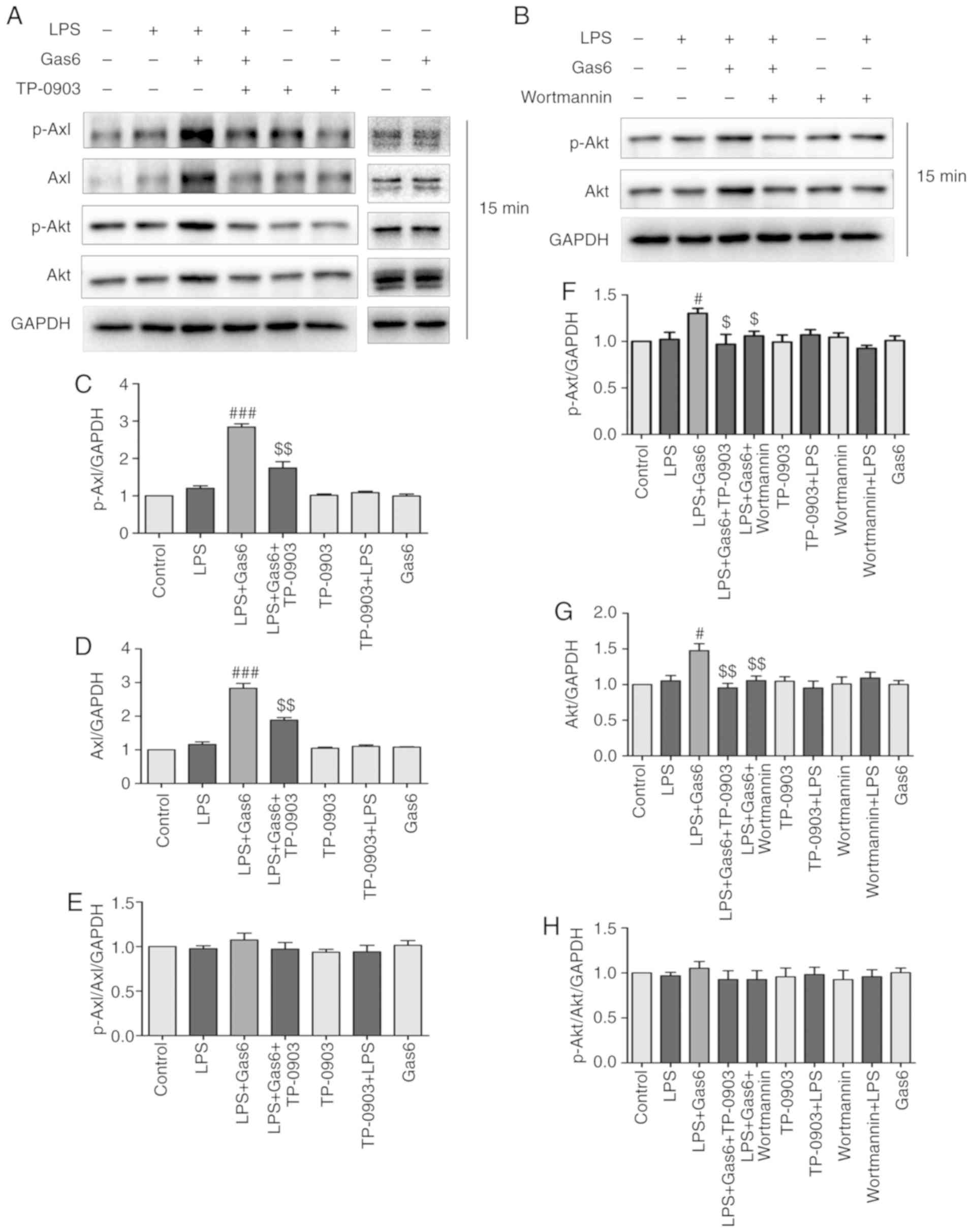 | Figure 2Gas6 activates the Axl/PI3K/Akt
signaling pathway in LPS-stimulated H9C2 cells. After pretreatment
with or without TP-0903 or Wortmannin for 15 min, the cells were
incubated with Gas6 for 2 h, followed by treatment with LPS for 15
min. After LPS administration, H9C2 cells were harvested for
analysis. (A and B) Representative western blots and (C-H)
semi-quantification of p-Axl, Axl, p-Akt and Akt in each group.
Data are presented as the mean ± standard deviation.
#P<0.05, ###P<0.001 vs. the LPS group;
$P<0.05, $$P<0.01 vs. the LPS + Gas6
group. Gas6, growth arrest-specific 6; LPS, lipopoly-saccharide;
p-, phosphorylated. |
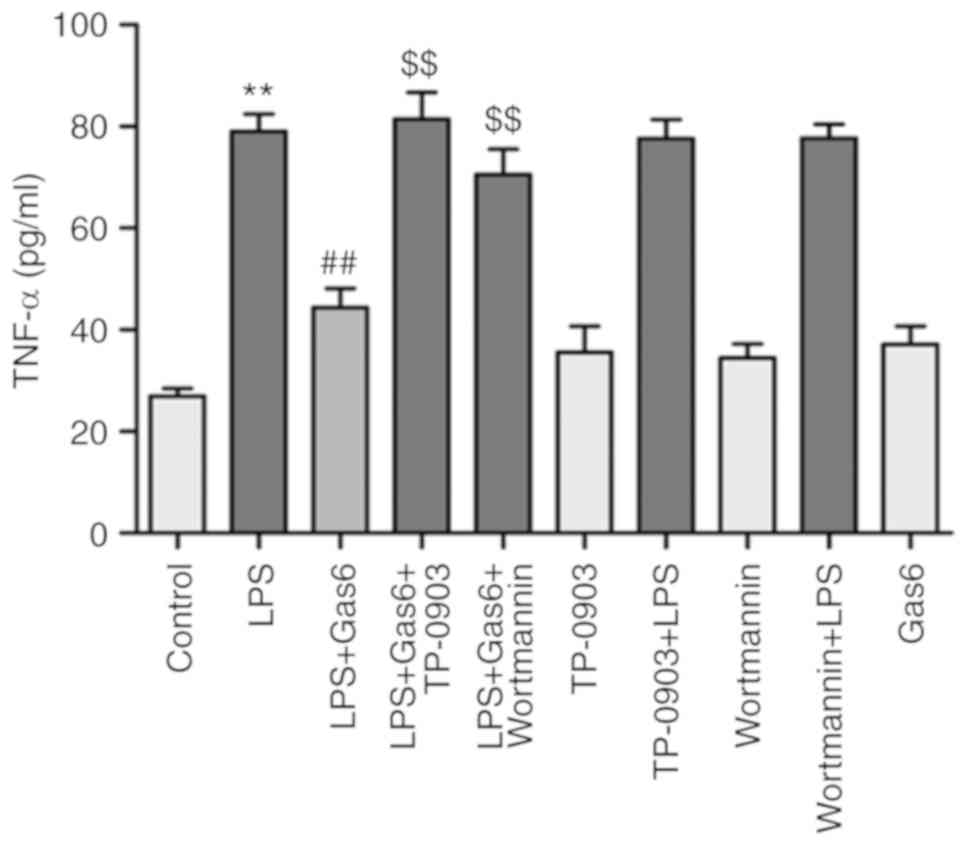
Gas6 suppresses the release of TNF-α via
the Axl/PI3K/Akt pathway in LPS-challenged H9C2 cells
TNF-α (death receptor ligand) binds to TNF-receptor
1 (TNF-R1; membrane-bound death receptor) and triggers the death
receptor-mediated apoptotic pathway, which has been reported to be
involved in the pathogenesis of sepsis-induced myocardial
dysfunction (32). As shown in
Fig. 3, LPS significantly
increased the release of TNF-α by 193.4% compared with in the
control group (P<0.01). Conversely, Gas6 treatment decreased the
release of TNF-α by 43.8% in H9C2 cells stimulated with LPS
(P<0.01). To confirm the relevance of the Axl/PI3K/Akt pathway
on the ability of Gas6 to decrease the production of TNF-α in
response to LPS, H9C2 cells were treated with TP-0903 or Wortmannin
in the presence of Gas6. TP-0903 and Wortmannin eliminated the
attenuating effects of Gas6 on TNF-α release by 83.6 and 58.9%,
respectively (P<0.01).
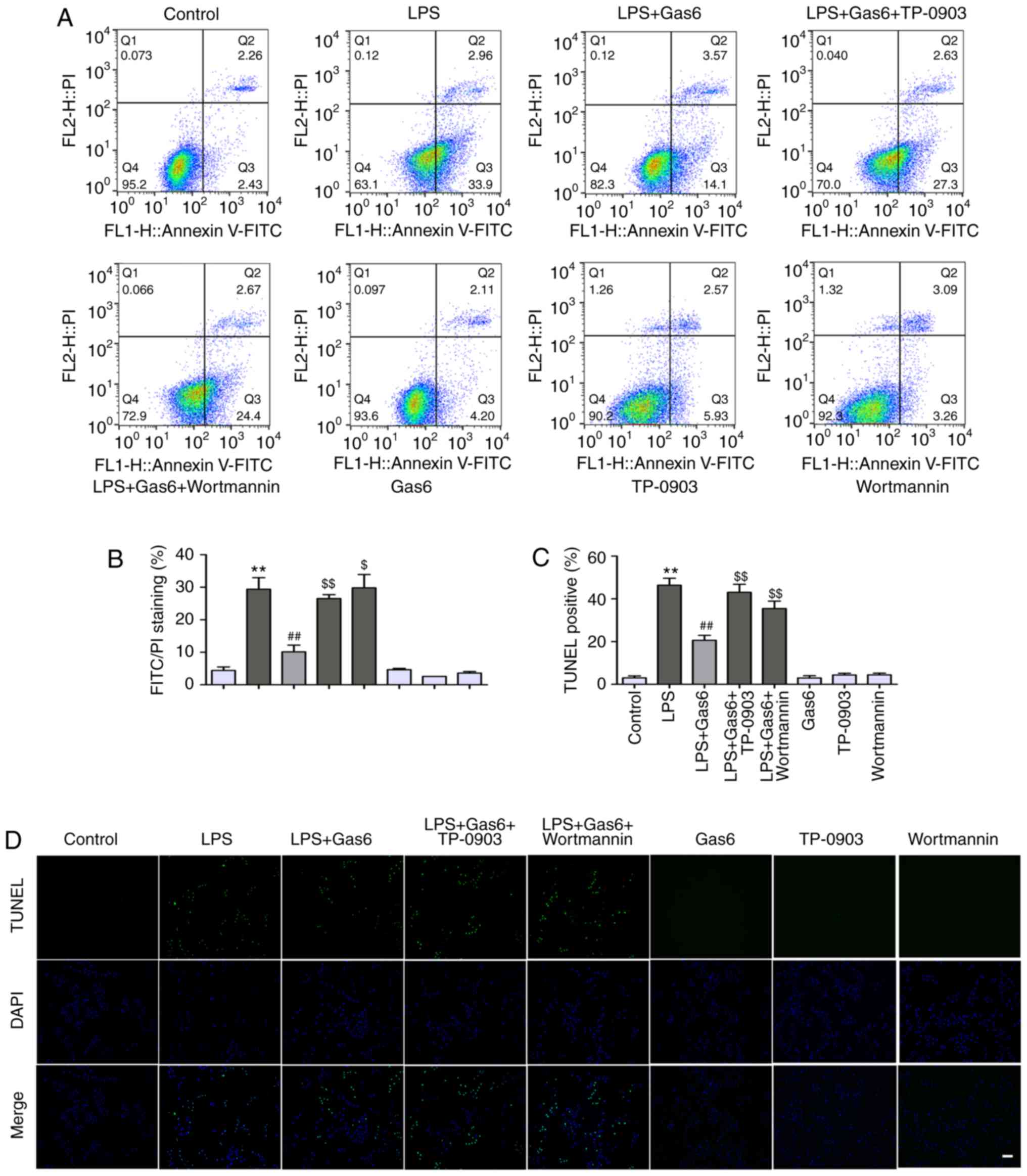
Gas6 suppresses apoptosis via the
Axl/PI3K/Akt pathway in LPS-challenged H9C2 cells
The present study examined the protective effects of
Gas6 on H9C2 cells exposed to LPS. Annexin V-PI staining and TUNEL
assay were applied to assess the early and late stages of apoptosis
in H9C2 cells, respectively. LPS exposure led to an incremental
increase in the percentage of early stage apoptotic cells from 4.4
to 29.4% compared with in the control group (P<0.01); Gas6
attenuated the percentage of early stage apoptotic cells to 10.1%
(P<0.01; Fig. 4A and B).
Similar results were observed using the TUNEL assay. As shown in
Fig. 4C and D, LPS stimulation
significantly increased the number of apoptotic H9C2 cells to 46.4%
(P<0.01), whereas pretreatment with Gas6 decreased the
percentage of apoptotic cells to 20.6% (P<0.01). To further
assess the role of Axl/PI3K/Akt signaling in the protective effect
of Gas6 on H9C2 cells stimulated with LPS, Hoechst staining,
Annexin V-PI staining, and TUNEL assay were performed in
LPS-treated cells incubated with Gas6 in the presence or absence of
TP-0903 and Wortmannin. As shown in Fig. 5A, TP-0903 and Wortmannin reversed
the amelioration of nuclear morphological alterations by Gas6 in
H9C2 cells challenged with LPS. Axl and PI3K inhibitors also
abolished the protective effect of Gas6 and increased the
percentage of apoptotic cells to 29.8% (P<0.01) and 32.4%
(P<0.05), respectively, as assessed by Annexin V-PI staining
(Fig. 4A and B). TUNEL analysis
also revealed that the protective effects of Gas6 on H9C2 cells
treated with LPS were abrogated by treatment with Axl and PI3K
inhibitors; these inhibitors enhanced the percentage of apoptotic
cells to 43.1 and 37.7%, respectively (P<0.01; Fig. 4C and D). Hoechst staining, which
detects nuclear condensation and fragmentation, revealed that
apoptosis was markedly increased following treatment with LPS,
which was ameliorated by pretreatment with Gas6 (Fig. 5A).
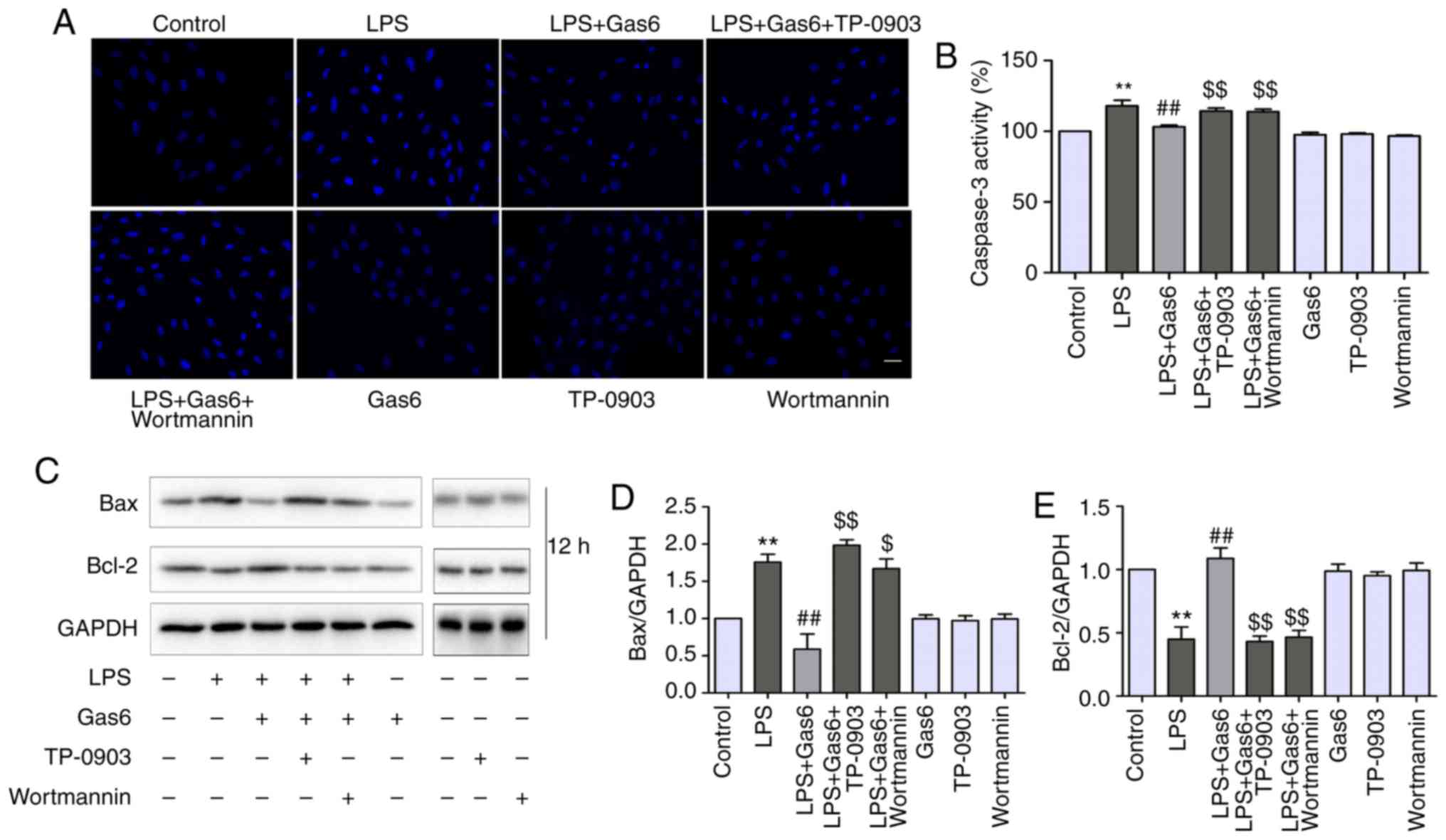
Axl/PI3K/Akt inhibition reverses the
effects of Gas6 on caspase-3 activity, and Bax and Bcl-2
expression
Since caspase-3 is considered a central regulator of
apoptosis (33), caspase-3
activity was examined to confirm apoptosis. In addition, Bax and
Bcl-2 expression levels were determined, as these are Bcl-2 family
members that are involved in the intrinsic apoptotic pathway
(34). As shown in Fig. 5B-D, caspase-3 activity and Bax
expression were increased by LPS treatment compared with the
control, whereas they were decreased by Gas6 pretreatment when
compared with cells stimulated with LPS. Conversely, Gas6 elevated
the expression levels of the anti-apoptotic protein Bcl-2, which
were decreased by LPS. To determine the role of Axl/PI3K/Akt
inhibition and the effects of Gas6 on caspase-3 activity, and Bax
and Bcl-2 expression, TP-0903 and Wortmannin were used to treat
cells. Caspase-3 activity was attenuated by Gas6 from 117.9 to
103.2% (P<0.01); this effect was reversed with the addition of
TP-0903 and Wortmannin, and caspase-3 activity was increased to
114.3 and 113.8%, respectively (both P<0.01). TP-0903 and
Wortmannin also reversed the effects of Gas6 on the proapoptotic
protein Bax and the anti-apoptotic protein Bcl-2 (Fig. 5C-E).
Axl/PI3K/Akt inhibition abolishes the
inhibitory effects of Gas6 on activation of NF-κB in LPS-challenged
H9C2 cells
Activation of NF-κB signaling is thought to be a key
event in the pathogenesis of sepsis and sepsis-induced myocardial
dysfunction (35). Therefore,
this study investigated the role of Gas6 in activation of the NF-κB
pathway in the presence of LPS. Pretreatment of cells with Gas6
resulted in inhibition of the phosphorylation and degradation of
IκB-α at 15 min (Fig. 6A-D).
Additionally, Gas6 suppressed the phosphorylation and expression of
P65 at 2.5 h (Fig. 6A, B and
E-H). Furthermore, the prominently enhanced nuclear
translocation of P65 caused by LPS was inhibited by Gas6 at 1 h
(Fig. 6I). Gas6 alone did not
affect the phosphorylation and expression of P65 or IκB-α, or the
localization of P65. In addition, to examine whether Gas6 blocked
the activation of NF-κB signaling via the Axl/PI3K/Akt pathway,
H9C2 cells were pretreated with TP-0903 or Wortmannin in the
presence or absence of Gas6. The effects of Gas6 on LPS-induced
phosphorylation and degradation of IκB-α were blocked by TP-0903
and Wortmannin (Fig. 6A-D).
Similarly, the effects of Gas6 on the phosphorylation, expression
and translocation of P65 were reversed by TP-0903 and Wortmannin
treatment (Fig. 6A, B and
E-I).
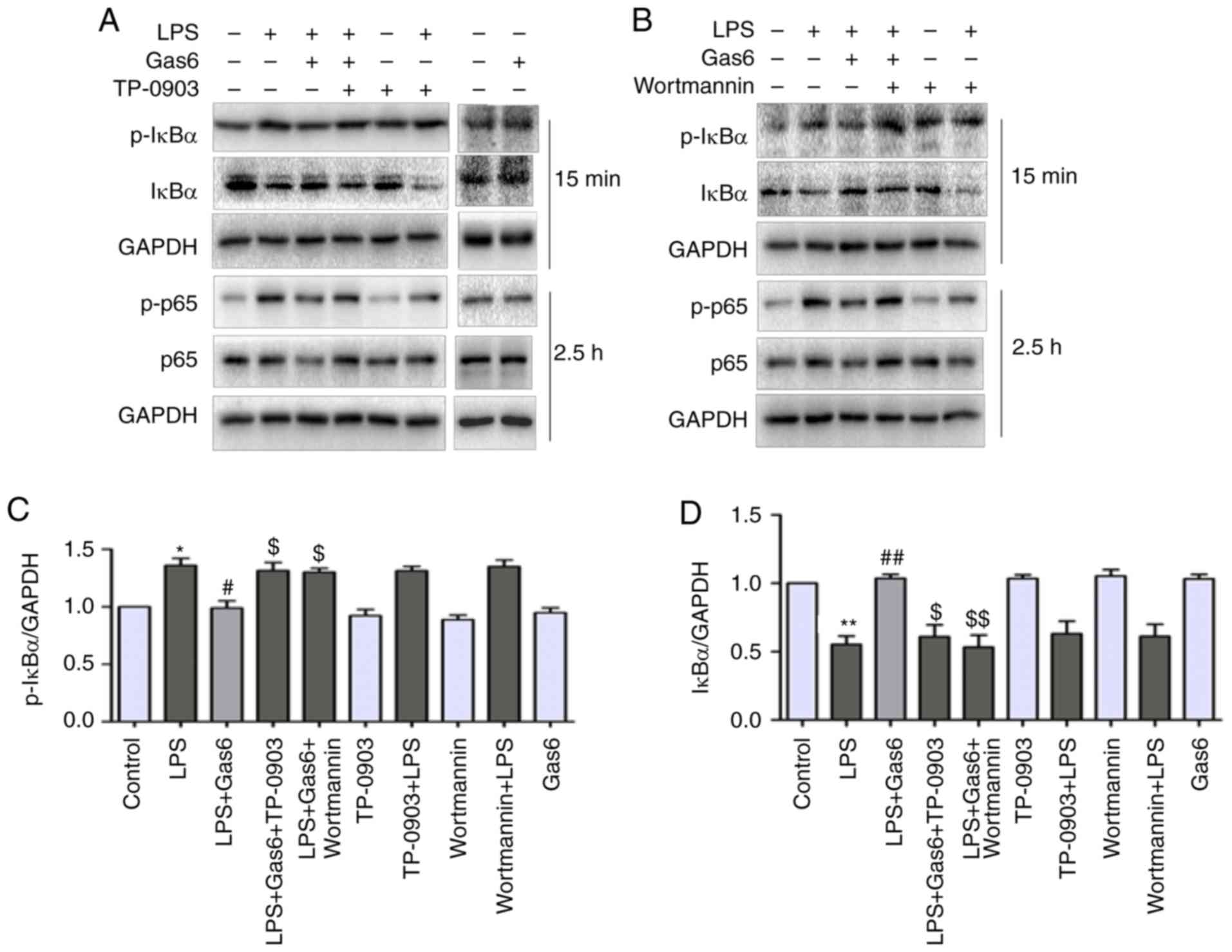 | Figure 6Inhibition of NF-κB signaling by Gas6
is Axl/PI3K/Akt-dependent in LPS-treated H9C2 cells. After
pretreatment with TP-0903 or Wortmannin for 15 min, the cells were
incubated with Gas6 for 2 h, followed by treatment with LPS for the
appropriate times. Following LPS administration, H9C2 cells were
harvested for analysis. (A and B) Representative western blots and
(C and D) semi-quantification of p-IκBα, IκBα, p-P65 and P65. (E-H)
Semi-quantification of p-IκBα, IκBα, p-P65 and P65. (I)
Translocation of NF-κB P65 was visualized by immunofluorescence
analysis (scale bar, 50 µm). Data are presented as the mean
± standard deviation. *P<0.05, **P<0.01
vs. the control group; #P<0.05,
##P<0.01 vs. the LPS group; $P<0.05,
$$P<0.01 vs. the LPS + Gas6 group. Gas6, growth
arrest-specific 6; LPS, lipopolysaccharide; NF-κB, nuclear
factor-κB; p-, phosphorylated. |
Axl/PI3K/Akt inhibition eliminates the
suppressive effects of Gas6 on activation of MAPKs in
LPS-challenged H9C2 cells
To further investigate the cardioprotective
mechanism underlying the effects of Gas6, the role of MAPKs in H9C2
cells pretreated with Gas6 in the presence of LPS was evaluated.
Western blotting results demonstrated that Gas6 significantly
decreased the phosphorylation of JNK and p38 MAPK induced by LPS
stimulation at 15 min and 2 h, respectively, whereas it did not
affect the expression of total JNK and p38 MAPK (Fig. 7A-D). Conversely, Gas6 increased
the phosphorylation and expression of ERK at 15 min (Fig. 7A, B and E-G). Gas6 alone had no
effect on the phosphorylation of JNK, ERK or p38 MAPK. To determine
whether Gas6 inhibited activation of MAPKs via the Axl/PI3K/Akt
pathway, LPS-stimulated cells were incubated with TP-0903 and
Wortmannin in the presence or absence of Gas6. The results revealed
that inhibition of Gas6/Axl signaling by TP-0903 and inhibition of
PI3K/Akt signaling by Wortmannin modified Gas6-induced suppression
of JNK and p38 MAPK phosphorylation in cells stimulated with LPS,
and reversed the effect of Gas6 on ERK phosphorylation and
expression.
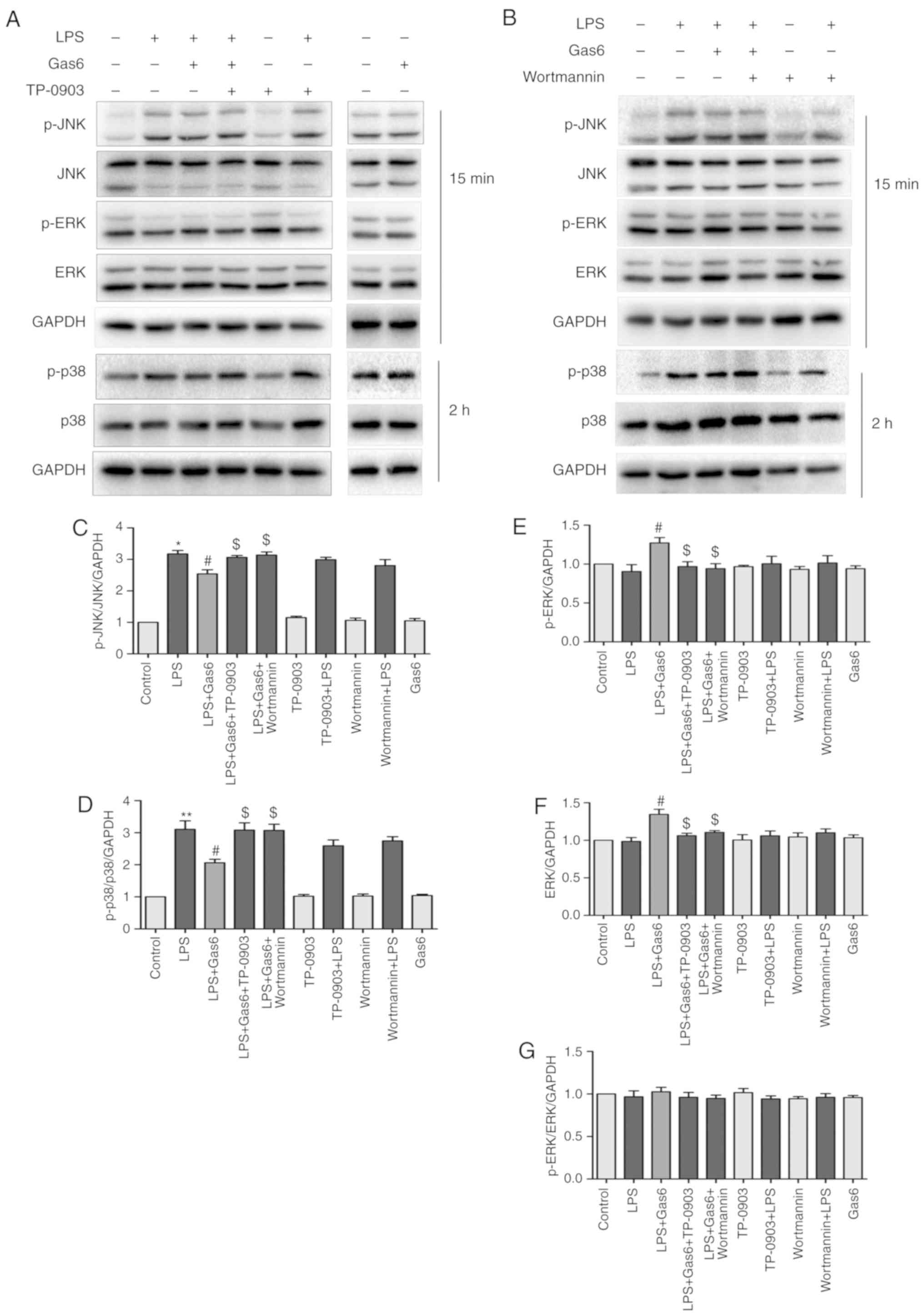 | Figure 7Inhibition of MAPK signaling by Gas6
is Axl/PI3K/Akt-dependent in LPS-treated H9C2 cells. After
pretreatment with TP-0903 or Wortmannin for 15 min, the cells were
incubated with Gas6 for 2 h, followed by LPS for the appropriate
times. Following LPS administration, H9C2 cells were harvested for
analysis. (A and B) Representative western blots and (C-G)
semi-quantification of p-JNK, JNK, p-ERK, ERK, p38 and p-p38. Data
are presented as the mean ± standard deviation.
*P<0.05, **P<0.01 vs. the control
group; #P<0.05 vs. the LPS group;
$P<0.05 vs. the LPS + Gas6 group. ERK, extracellular
signal-regulated kinase; Gas6, growth arrest-specific 6; JNK, c-Jun
N-terminal protein kinase; LPS, lipopolysaccharide; p-,
phosphorylated. |
Discussion
The present study demonstrated that Gas6 suppressed
TNF-α release and apoptosis in LPS-treated H9C2 cells, and
inhibited their related regulators NF-κB and MAPKs. TP-0903 and
Wortmannin abrogated the inhibitory effects of Gas6 on
LPS-challenged H9C2 cells. Therefore, these findings indicated that
Gas6-attenuated LPS-induced TNF-α release and apoptosis in H9C2
cells may involve inhibition of NF-κB and MAPK activation via the
Axl/PI3K/Akt signaling pathway.
Previous studies have demonstrated that apoptosis
(36) and TNF-α (37) produced by cardiomyocytes
contribute to septic cardiomyopathy, and an increased Gas6
concentration is observed in the circulation of patients with
sepsis (27). TNF-α, as a death
receptor ligand, participates in the extrinsic apoptotic pathway.
After binding with the death receptor TNF-R1, TNF-α increases the
expression of caspase-8 and TNF receptor-related death domain
protein, which leads to formation of the death-inducing signaling
complex, followed by activation of the apoptotic cascade (32). In the intrinsic pathway, Bcl-2
family proteins (Bax, Bak and Bid) are first activated and combine
with each other, and are inserted into the outer membrane, leading
to the release of key enzymes (such as cytochrome c),
followed by the formation of apoptotic bodies, amplification of the
death signal, and apoptosis (38). Additionally, previous studies have
demonstrated that exogenous administration of Gas6 prevents the
release of inflammatory cytokines and exerts anti-apoptotic effects
on models of sepsis (29-31). Therefore, this study investigated
the effects of Gas6 on TNF-α release and apoptosis, and analyzed
the underlying mechanism, in order to further improve existing
therapies and treatments for septic cardiomyopathy. In the human
circulatory system, the concentration of Gas6 is subnanomolar
(20-50 ng/ml or 0.25 nM) and increases ~2-fold in patients with
sepsis (28,39). This study confirmed the effects of
100 ng/ml Gas6 on TNF-α release and apoptosis in LPS-challenged
H9C2 cells. The present study provided evidence to suggest that
Gas6 may attenuate production of the proinflammatory cytokine TNF-α
as well as apoptosis.
The Axl/PI3K/Akt pathway is known to be involved in
the pro-survival and anti-apoptotic effects of Gas6 (21,40). Axl is a TAM receptor, which is
mainly expressed on the cell surface. After binding with its
ligands (e.g. Gas6), Axl is activated and autophosphorylated to
initiate signaling (41). Among
the signaling cascades, the PI3K/Akt pathway is necessary for the
Gas6/Axl-dependent anti-apoptotic effect (21). The PI3K/Akt pathway serves a
central role in cell growth and survival. Activation of Akt
directly regulates apoptotic regulatory factors, such as Bax, Bad
and caspase-9 (42,43), or facilitates its interaction with
transcription factors such as NF-κB. To determine whether
Axl/PI3K/Akt participated in the action of Gas6 in LPS-induced H9C2
cells, an Axl selective inhibitor (TP-0903) and a PI3K inhibitor
(Wortmannin) were used; the results demonstrated that these
inhibitors abrogated the inhibitory effects of Gas6 on TNF-α
production and apoptosis in LPS-stimulated H9C2 cells. The present
study revealed that Gas6 enhanced the phosphorylation and
expression of Axl and Akt, whereas TP-0903 and Wortmannin reversed
the phosphorylation and expression of Axl and Akt, respectively. In
other studies, only the phosphorylation of Axl and Akt were
affected, whereas the expression of Axl and Akt remained unaffected
by inflammatory injury or drugs. However, Gas6 increased both the
phosphorylation and expression of Axl and downstream signaling
molecules in a model of neuroinflammation (17). TP-0903 has also been shown to
inhibit both the phosphorylation and expression of Axl and Akt in
lymphocytic leukemia B cells (44). Various cell origins and
stimulations may explain the distinctions in the expression of Axl
and Akt in different studies. In the present study, the ratios of
p-Axl/Axl, p-Akt/Akt, p-p65/p65 and p-ERK/ERK were almost the same
in the different groups, which indicated that alterations in the
expression of phosphorylated proteins were due to alterations in
the expression of the total proteins. Taken together, the findings
indicated that Gas6 exerted its protective effect via the
Axl/PI3K/Akt pathway in LPS-challenged H9C2 cells.
Accumulating evidence has indicated that activation
of NF-κB (45) and MAPK (22) contributes to enhanced
cardiomyocyte TNF-α production and apoptosis in the presence of
LPS. Generally, the transcription factor NF-κB is inactive due to
its binding with the inhibitory protein IκB, which exists in the
cytoplasm. Once activated by LPS, NF-κB translocates to the
nucleus, subsequently triggering the transcription of various
inflammatory- and apoptosis-associated genes (46). In the present study, NF-κB was
activated in the presence of LPS, whereas Gas6 reversed the
phosphorylation and expression of NF-κB in response to LPS
treatment in cardiomyocytes. The MAPK family consists of at least
three members: ERK1/2, JNK1/2 and p38 MAPK. JNK and p38 MAPK
contribute to endotoxin-induced enhanced cardiomyocyte TNF-α
production and apoptosis. A previous study reported that ERK has
the opposite effect of JNK and p38 MAPK (47). The present findings indicated that
LPS induced an increase in JNK and p38 MAPK phosphorylation, but
marginally decreased the phosphorylation and expression of ERK.
Administration of Gas6 reversed the effects of LPS on H9C2 cells at
different time-points. However, other studies have demonstrated
that LPS activates ERK, JNK and p38 MAPK phosphorylation (48). The cell origin and time-point may
explain this discrepancy.
Numerous studies have revealed that Gas6/Axl
activation suppresses inflammation by inhibiting NF-κB (17,49). Gas6 also promotes cardiac
hypertrophy via ERK1/2, which indicates a relationship between
Gas6/Axl and MAPK (50).
Additionally, compelling evidence has revealed cross talk between
PI3K/Akt and NF-κB and MAPK in LPS-challenged cardiomyocytes
(25,48). To further investigate the
mechanism underlying the protective effects of Gas6 on LPS-treated
H9C2 cells, Axl and PI3K inhibitors were applied. TP-0903 and
Wortmannin reversed the effects of Gas6 on MAPK and NF-κB in
LPS-stimulated cardiomyocytes, suggesting that Gas6 may attenuate
MAPK and NF-κB in LPS-stimulated cardiomyocytes via the
Axl/PI3K/Akt pathway.
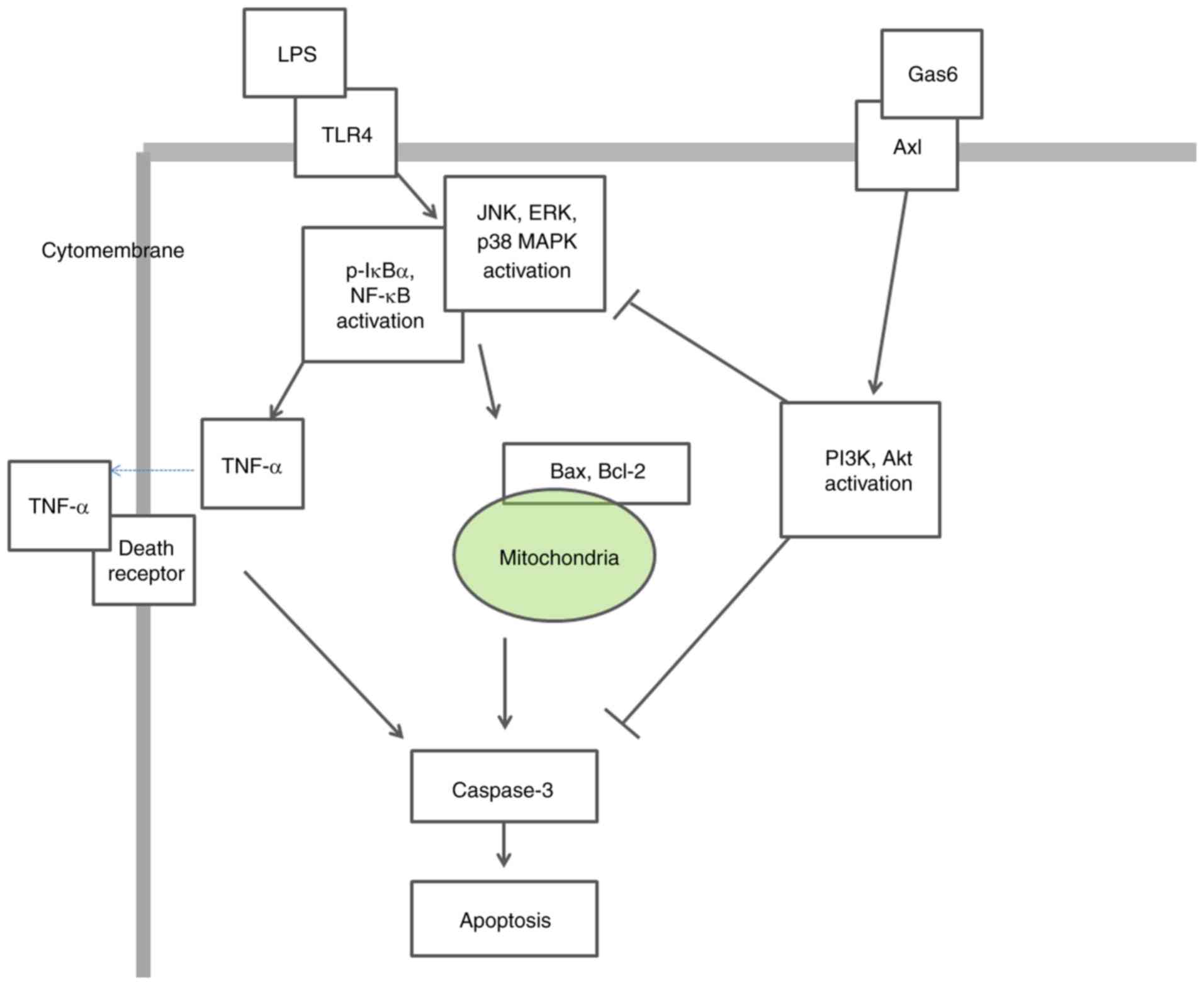
In conclusion, the present findings demonstrated
that the Axl/PI3K/Akt pathway may be essential for Gas6-mediated
suppression of MAPK and NF-κB activation, as well as the
attenuation of TNF-α production and apoptosis, in response to LPS
stimulation in H9C2 cardiomyoblasts (Fig. 8). These findings provide evidence
regarding the specific molecular signaling events that participate
in the protective effect of Gas6 on LPS-challenged H9C2
cardiomyocytes and support the hypothesis that Gas6 may emerge as a
pharmacological therapy for the treatment of sepsis-induced
myocardial dysfunction. Further studies are required to determine
the protective effects of Gas6 against sepsis-induced myocardial
injury in vivo and to identify its potential clinical
applications.
Funding
This study was funded by the Zhejiang Provincial
Natural Science Foundation (grant no. LQ15H150003), the Zhejiang
Provincial Medical and Health Science and Technology Project (grant
no. 2015KYA152), the Wenzhou Municipal Science and Technology
Project (grant no. Y20150182) and the Zhejiang Provincial
Administration of Traditional Chinese Medicine Project (grant no.
2015ZA120). This study was supported, in part, by grants from the
National Natural Science Foundation (grant nos. 81571937, 81772112
and 81871583) and the Medical Health Science and Technology Major
Project of the Zhejiang Provincial Health Commission (grant no.
WKJ-Z-J-1724).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL, ML and JY conceived of and designed the study.
ML, JY and XH performed the study. JY, KC, YW GZ and GH analyzed
the data. ML, JY, GZ and GH wrote the manuscript. All authors
reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Parrillo JE, Parker MM, Natanson C,
Suffredini AF, Danner RL, Cunnion RE and Ognibene FP: Septic shock
in humans. Advances in the understanding of pathogenesis,
cardiovascular dysfunction, and therapy. Ann Intern Med.
113:227–242. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landesberg G, Gilon D, Meroz Y, Georgieva
M, Levin PD, Goodman S, Avidan A, Beeri R, Weissman C, Jaffe AS and
Sprung CL: Diastolic dysfunction and mortality in severe sepsis and
septic shock. Eur Heart J. 33:895–903. 2012. View Article : Google Scholar :
|
|
3
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bayeva M, Sawicki KT, Butler J,
Gheorghiade M and Ardehali H: Molecular and cellular basis of
viable dysfunctional myocardium. Circ Heart Fail. 7:680–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frantz S, Kobzik L, Kim YD, Fukazawa R,
Medzhitov R, Lee RT and Kelly RA: Toll4 (TLR4) expression in
cardiac myocytes in normal and failing myocardium. J Clin Invest.
104:271–280. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grandel U, Fink L, Blum A, Heep M, Buerke
M, Kraemer HJ, Mayer K, Bohle RM, Seeger W, Grimminger F and
Sibelius U: Endotoxin-induced myocardial tumor necrosis
factor-alpha synthesis depresses contractility of isolated rat
hearts: Evidence for a role of sphingosine and
cyclooxygenase-2-derived thromboxane production. Circulation.
102:2758–2764. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vincent JL, Bakker J, Marécaux G,
Schandene L, Kahn RJ and Dupont E: Administration of anti-TNF
antibody improves left ventricular function in septic shock
patients. Results of a pilot study Chest. 101:810–815. 1992.
|
|
9
|
Herbertson MJ, Werner HA, Goddard CM,
Russell JA, Wheeler A, Coxon R and Walley KR: Anti-tumor necrosis
factor-alpha prevents decreased ventricular contractility in
endotoxemic pigs. Am J Respir Crit Care Med. 152:480–488. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng T, Lu X, Lei M, Moe GW and Feng Q:
Inhibition of p38 MAPK decreases myocardial TNF-alpha expression
and improves myocardial function and survival in endotoxemia.
Cardiovasc Res. 59:893–900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nevière R, Fauvel H, Chopin C, Formstecher
P and Marchetti P: Caspase inhibition prevents cardiac dysfunction
and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care
Med. 163:218–225. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lancel S, Petillot P, Favory R, Stebach N,
Lahorte C, Danze PM, Vallet B, Marchetti P and Neviere R:
Expression of apoptosis regulatory factors during myocardial
dysfunction in endotoxemic rats. Crit Care Med. 33:492–496. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stitt TN, Conn G, Gore M, Lai C, Bruno J,
Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al: The
anticoagulation factor protein S and its relative, Gas6, are
ligands for the Tyro 3/Axl family of receptor tyrosine kinases.
Cell. 80:661–670. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sasaki T, Knyazev PG, Clout NJ, Cheburkin
Y, Göhring W, Ullrich A, Timpl R and Hohenester E: Structural basis
for Gas6-Axl signalling. EMBO J. 25:80–87. 2006. View Article : Google Scholar
|
|
15
|
Zhao GJ, Zheng JY, Bian JL, Chen LW, Dong
N, Yu Y, Hong GL, Chandoo A, Yao YM and Lu ZQ: Growth
arrest-specific 6 enhances the suppressive function of
CD4+CD25+ regulatory T cells mainly through
Axl receptor. Mediators Inflamm. 2017:68484302017.
|
|
16
|
Goruppi S, Ruaro E and Schneider C: Gas6,
the ligand of Axl tyrosine kinase receptor, has mitogenic and
survival activities for serum starved NIH3T3 fibroblasts. Oncogene.
12:471–480. 1996.PubMed/NCBI
|
|
17
|
Wu G, McBride DW and Zhang JH: Axl
activation attenuates neuroinflammation by inhibiting the
TLR/TRAF/NF-κB pathway after MCAO in rats. Neurobiol Dis.
110:59–67. 2018. View Article : Google Scholar
|
|
18
|
Graham DK, Bowman GW, Dawson TL, Stanford
WL, Earp HS and Snodgrass HR: Cloning and developmental expression
analysis of the murine c-mer tyrosine kinase. Oncogene.
10:2349–2359. 1995.PubMed/NCBI
|
|
19
|
Wu M, Lu S, Zhong J, Huang K and Zhang S:
Protective effects of pterostilbene against myocardial
ischemia/reperfusion injury in rats. Inflammation. 40:578–588.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goruppi S, Ruaro E, Varnum B and Schneider
C: Requirement of phosphatidylinositol 3-kinase-dependent pathway
and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3
fibroblasts. Mol Cell Biol. 17:4442–4453. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee WP, Wen Y, Varnum B and Hung MC: Akt
is required for Axl-Gas6 signaling to protect cells from
E1A-mediated apoptosis. Oncogene. 21:329–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu CJ, Lo JF, Kuo CH, Chu CH, Chen LM,
Tsai FJ, Tsai CH, Tzang BS, Kuo WW and Huang CY: Akt mediates
17beta-estra-diol and/or estrogen receptor-alpha inhibition of
LPS-induced tumor necresis factor-alpha expression and myocardial
cell apoptosis by suppressing the JNK1/2-NFkappaB pathway. J Cell
Mol Med. 13:3655–3667. 2009. View Article : Google Scholar
|
|
23
|
An R, Zhao L, Xi C, Li H, Shen G, Liu H,
Zhang S and Sun L: Melatonin attenuates sepsis-induced cardiac
dysfunction via a PI3K/Akt-dependent mechanism. Basic Res Cardiol.
111:82016. View Article : Google Scholar
|
|
24
|
Gao M, Ha T, Zhang X, Wang X, Liu L,
Kalbfleisch J, Singh K, Williams D and Li C: The Toll-like receptor
9 ligand, CpG oligodeoxynucleotide, attenuates cardiac dysfunction
in poly-microbial sepsis, involving activation of both
phosphoinositide 3 kinase/Akt and extracellular-signal-related
kinase signaling. J Infect Dis. 207:1471–1479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun B, Xiao J, Sun XB and Wu Y:
Notoginsenoside R1 attenuates cardiac dysfunction in endotoxemic
mice: An insight into oestrogen receptor activation and PI3K/Akt
signalling. Br J Pharmacol. 168:1758–1770. 2013. View Article : Google Scholar :
|
|
26
|
Manfioletti G, Brancolini C, Avanzi G and
Schneider C: The protein encoded by a growth arrest-specific gene
(gas6) is a new member of the vitamin K-dependent proteins related
to protein S, a negative coregulator in the blood coagulation
cascade. Mol Cell Biol. 13:4976–4985. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gibot S, Massin F, Cravoisy A, Dupays R,
Barraud D, Nace L and Bollaert PE: Growth arrest-specific protein 6
plasma concentrations during septic shock. Crit Care. 11:R82007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ekman C, Linder A, Akesson P and Dahlbäck
B: Plasma concentrations of Gas6 (growth arrest specific protein 6)
and its soluble tyrosine kinase receptor sAxl in sepsis and
systemic inflammatory response syndromes. Crit Care. 14:R1582010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alciato F, Sainaghi PP, Sola D, Castello L
and Avanzi GC: TNF-alpha, IL-6, and IL-1 expression is inhibited by
GAS6 in monocytes/macrophages. J Leukoc Biol. 87:869–875. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giangola MD, Yang WL, Rajayer SR, Nicastro
J, Coppa GF and Wang P: Growth arrest-specific protein 6 attenuates
neutrophil migration and acute lung injury in sepsis. Shock.
40:485–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen LW, Chen W, Hu ZQ, Bian JL, Ying L,
Hong GL, Qiu QM, Zhao GJ and Lu ZQ: Protective effects of growth
arrest-specific protein 6 (Gas6) on sepsis-induced acute kidney
injury. Inflammation. 39:575–582. 2016. View Article : Google Scholar
|
|
32
|
Carlson DL, Willis MS, White DJ, Horton JW
and Giroir BP: Tumor necrosis factor-alpha-induced caspase
activation mediates endotoxin-related cardiac dysfunction. Crit
Care Med. 33:1021–1028. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lakhani SA, Masud A, Kuida K, Porter GA
Jr, Booth CJ, Mehal WZ, Inayat I and Flavell RA: Caspases 3 and 7:
Key mediators of mitochondrial events of apoptosis. Science.
311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li S, Zhao H, Wang Y, Shao Y, Li J, Liu J
and Xing M: The inflammatory responses in Cu-mediated elemental
imbalance is associated with mitochondrial fission and intrinsic
apoptosis in Gallus gallus heart. Chemosphere. 189:489–497. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma H, Wang X, Ha T, Gao M, Liu L, Wang R,
Yu K, Kalbfleisch JH, Kao RL, Williams DL and Li C: MicroRNA-125b
prevents cardiac dysfunction in polymicrobial sepsis by targeting
TRAF6-mediated nuclear factor κB activation and p53-mediated
apoptotic signaling. J Infect Dis. 214:1773–1783. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suzuki J, Bayna E, Li HL, Molle ED and Lew
WY: Lipopolysaccharide activates calcineurin in ventricular
myocytes. J Am Coll Cardiol. 49:491–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baumgarten G, Knuefermann P, Nozaki N,
Sivasubramanian N, Mann DL and Vallejo JG: In vivo expression of
proinflammatory mediators in the adult heart after endotoxin
administration: The role of toll-like receptor-4. J Infect Dis.
183:1617–1624. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Balogh I, Hafizi S, Stenhoff J, Hansson K
and Dahlbäck B: Analysis of Gas6 in human platelets and plasma.
Arterioscler Thromb Vasc Biol. 25:1280–1286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ming Cao W, Murao K, Imachi H, Sato M,
Nakano T, Kodama T, Sasaguri Y, Wong NC, Takahara J and Ishida T:
Phosphatidylinositol 3-OH kinase-Akt/protein kinase B pathway
mediates Gas6 induction of scavenger receptor a in immortalized
human vascular smooth muscle cell line. Arterioscler Thromb Vasc
Biol. 21:1592–1597. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mark MR, Chen J, Hammonds RG, Sadick M and
Godowsk PJ: Characterization of Gas6, a member of the superfamily
of G domain-containing proteins, as a ligand for Rse and Axl. J
Biol Chem. 271:9785–9789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kamada H, Nito C, Endo H and Chan PH: Bad
as a converging signaling molecule between survival PI3-K/Akt and
death JNK in neurons after transient focal cerebral ischemia in
rats. J Cereb Blood Flow Metab. 27:521–533. 2007. View Article : Google Scholar
|
|
43
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sinha S, Boysen J, Nelson M, Secreto C,
Warner SL, Bearss DJ, Lesnick C, Shanafelt TD, Kay NE and Ghosh AK:
Targeted Axl inhibition primes chronic lymphocytic leukemia B cells
to apop-tosis and shows synergistic/additive effects in combination
with BTK inhibitors. Clin Cancer Res. 21:2115–2126. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Y, Wang Y, Yang D, Yu X, Li H, Lv X,
Lu D and Wang H: β1-adrenoceptor stimulation promotes LPS-induced
cardiomyocyte apoptosis through activating PKA and enhancing CaMKII
and IκBα phosphorylation. Crit Care. 19:762015. View Article : Google Scholar
|
|
46
|
Pham CG, Bubici C, Zazzeroni F, Papa S,
Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C,
et al: Ferritin heavy chain upregulation by NF-kappaB inhibits
TNFalpha-induced apoptosis by suppressing reactive oxygen species.
Cell. 119:529–542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu X, Jia B, Wang F, Lv X, Peng X, Wang Y,
Li H, Wang Y, Lu D and Wang H: α1 adrenoceptor activation by
norepinephrine inhibits LPS-induced cardiomyocyte TNF-α production
via modulating ERK1/2 and NF-κB pathway. J Cell Mol Med.
18:263–273. 2014. View Article : Google Scholar
|
|
48
|
Yang P, Han Y, Gui L, Sun J, Chen YL, Song
R, Guo JZ, Xie YN, Lu D and Sun L: Gastrodin attenuation of the
inflammatory response in H9c2 cardiomyocytes involves inhibition of
NF-κB and MAPKs activation via the phosphatidylinositol 3-kinase
signaling. Biochem Pharmacol. 85:1124–1133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hasanbasic I, Cuerquis J, Varnum B and
Blostein MD: Intracellular signaling pathways involved in
Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart
Circ Physiol. 287:H1207–H1213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao YF, Xu DC, Zhu GF, Zhu MY, Tang K, Li
WM and Xu YW: Growth arrest-specific 6 exacerbates pressure
overload-induced cardiac hypertrophy. Hypertension. 67:118–129.
2016. View Article : Google Scholar
|