Introduction
Hepatocellular carcinoma (HCC), the most prevalent
subtype of liver cancer, has become the third leading cause of
cancer-related death worldwide, accounting for nearly one-third of
malignancies (1,2). Hepatitis B and C viral infection,
aflatoxin B1 exposure, excessive alcohol consumption, obesity and
some inherited metabolic disorders are generally considered to be
major risk factors in the development of HCC (3). While current understanding of
molecular mechanisms underlying HCC development has significantly
improved, knowledge is limited in protein-coding genes that
constitute the part of the genome. Recently, high-throughput
studies of the human transcriptome have revealed that there is far
more genomic transcription than previously anticipated, with the
vast majority of the genome being transcribed into an astounding
number of non-coding RNAs (ncRNAs) (4-6). A
subset of these molecules displays distinct cancer-specific
expression patterns, suggesting that they may be potential drivers
of cancer biology and have increasing value as clinical
biomarkers.
These ncRNAs are classified according to their
sequence length into long ncRNA (lncRNA; >200 bp) and small
ncRNA (<200 bp), such as microRNA (miRNA/miR), and they do not
serve as a template for protein synthesis but nonetheless are
potent regulators of gene expression at epigenetic (7), transcriptional (8) and post-transcriptional levels
(9). For the past two decades,
miRNAs have been recognized as controlling gene expression by
binding to specific sites at the 3′ untranslated region of target
mRNAs, causing translational repression or degradation (10), while the lesser-studied lncRNAs
are implicated in almost every epigenetic regulation event
(11). In addition, both miRNAs
and lncRNAs have been implicated to modulate diverse cellular
pathways that lead to oncogenesis, metastasis and progression
(12,13). Despite growing evidence expanding
the understanding of the complex role of individual lncRNAs or
miRNAs in various steps of cancer development and progression,
co-expression profiles, regulatory networks and interaction of
lncRNA-mRNA-miRNA remain largely unexplored in cancer.
In the authors' previous study, the microarray
profiles of miRNAs in HCC were reported (14). In the present study,
high-throughput technologies were employed to simultaneously
establish expression profiles of lncRNAs and mRNAs in the same 7
pairs of HCC and adjacent tumor-free specimens. An integrative
method was then applied to comprehensively analyze the
relationships between aberrantly expressed lncRNAs, mRNAs and
miRNAs. The significant involvement of one novel lncRNA in the
regulation of cell cycle was highlighted. The comprehensive
analysis by the present study provided novel insights into the
lncRNA-mRNA-miRNA co-expression network at the transcriptional and
post-transcriptional levels in modulating HCC growth. These
findings may serve as the foundation for future studies of
lncRNA-mRNA-miRNA interactions in HCC and contribute to further
systematic studies of tumorigenesis in the future.
Materials and methods
Sample collection
A total of 37 paired HCC specimens and matched
adjacent tumor-free tissue counterparts were collected from
patients (average age: 56.92 years; male/female =6.4) admitted to
the Zhongshan Hospital of Fudan University between January 2008 and
December 2009. The diagnosis was based on imaging presentation with
a typical pattern of HCC and clinicopathological examination after
surgery (Fig. S1). Among these
specimens, seven paired HCC specimens and matched adjacent
tumor-free tissue counterparts were randomly selected for
microarray analysis. The clinical information of the seven patients
were provided in Table SI.
The present study was approved by the Institutional
Ethics Committee of Zhongshan Hospital of Fudan University and
conformed to the provisions of the Declaration of Helsinki. Written
informed consent was obtained from all patients who participated in
this study.
RNA preparation
Total RNAs were extracted and purified from 7 pairs
of specimens for lncRNA, mRNA and miRNA, respectively, using an
RNeasy mini kit for total RNA (Qiagen, GmBH) and mirVana™ miRNA
Isolation kit without phenol (Ambion; Thermo Fisher Scientific,
Inc.). The whole process was conducted according to the
manufacturer's protocols. NanoDrop ND-2000 (Thermo Fisher
Scientific, Inc.) was applied for quantification of total RNAs and
RNA Integrity Number was detected using Agilent Bioanalyzer 2100
(Agilent technologies, Inc.).
Microarray analysis
Two microarrays, including human lncRNA plus mRNA
microarray v5.0 (4x180 K) and human miRNA microarray v21.0 (Agilent
Technologies, Inc.), were used for detection of differential
expression of lncRNAs plus mRNAs, and miRNAs, respectively. The
threshold set for differentially expressed genes (DEGs) was
P<0.05, fold-change ≥2.0 and false discovery rate (FDR) value
<0.05. Vioplot was conducted to explore signal intensity of
DEGs. Hierarchical clustering was performed to display the
distinguishable gene expression pattern among samples. The data
discussed in this publication have been deposited in NCBI's Gene
Expression Omnibus and are accessible through GEO Series accession
number GSE101728 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101728)
and GSE108724 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108724).
Gene ontology (GO) enrichment and Kyoto
Encyclopedia of Genes and Genomes pathway (KEGG) pathway
analysis
GO enrichment analysis was evaluated by Database for
Annotation, Visualization and Integrated Discovery bioinformatics
tool (https://david.ncifcrf.gov) to display
the potential biological functions of DEGs. Enrichment map by
Cytoscape Desktop program (http://baderlab.org/Software/EnrichmentMap) was
applied to visualize the results of gene-set enrichment as a
network in the aspects of biological process, cellular component
and molecular function. KEGG pathway was used to identify
significantly enriched signaling transduction pathways. The
significance of the pathway was measured by P-value.
Furthermore, Gene Set Enrichment Analysis (GSEA) for all detected
mRNAs was performed in order to validate the consistence of the
findings from GO and KEGG pathway enrichment analysis.
Construction of the lncRNA-mRNA-miRNA
interaction network
To establish a co-expression network of lncRNAs,
mRNAs and miRNAs, differential expressed mRNAs involved in the most
significant pathway from the analysis above were recruited. During
this procedure, the interactions of miRNA-mRNA were predicted by
miRanda database tool (http://www.microrna.org). Furthermore, LncTar
(http://www.cuilab.cn/lnctar) was
employed to predict the candidate miRNAs and mRNAs that may have
underlying relationships with lncRNAs (free energy cutoff <-20).
Pearson correlation coefficient was calculated to measure the
intensity between lncRNA, mRNA and miRNA. Cytoscape was applied to
construct and visualize the potential networks between the three
types of RNAs.
Overall survival data collection
To estimate the potential roles of the targeted mRNA
in the prognosis of HCC patients, the RNA sequencing data of 365
HCC samples and related clinical data were retrieved from the
Cancer Genome Atlas (TCGA) database for later Kaplan-Meier
tests.
Quantitative PCR (qPCR)
In the validation step, total RNAs were extracted
from 30 pairs of HCC and matched tumor-free specimens by
TRIzol® reagent (Thermo Fisher Scientific, Inc.). The
reverse transcription and the detection of qualification were
conducted by the assistance of the PrimeScript™ RT Master Mix kit
and the SYBR® Premix Ex Taq™ kit (Takara
Biotechnology Co., Ltd.), respectively. Reverse transcription was
conducted at 37°C for 15 min, and then at 85°C for 5 sec for heat
inactivation. The conditions for qPCR were as follows: Stage 1:
Initial denaturation, 1 cycle, 95°C for 30 sec; stage 2: PCR, 40
cycles, 95°C for 5 sec, 60°C for 30 sec. Data were analyzed using
the 2−ΔΔCq method (15). Primers for the lncRNAs were
designed and specialized by Primer Premier version 5.0 (Premier
Biosoft International; Table SII). The expression level of the
candidate lncRNAs was measured by the ABI 7900HT Fast Real-Time PCR
system, with GAPDH as the internal control. qPCR was repeated three
times.
Statistical analysis
All statistical analysis of microarray data was
conducted using the R package, including vioplot (CRAN, v0.2),
pcaMethods (Bioconductor, v1.76.0), pheatmap (CRAN, v1.0.8) and
limma (Bioconductor, v3.40.0). Fold-change ≥2.0, P<0.05 and FDR
<0.05 were set up for further analysis. Expression of the
candidate lncRNAs was presented as the scatter plot with mean ±
standard deviation and the range of the log-transformed expression
levels was estimated by Wilcoxon rank-sum test. Kaplan-Meier
estimator was applied to analyze the overall survival data from
TCGA database. P<0.05 was considered to indicate a statistically
significant difference.
Results
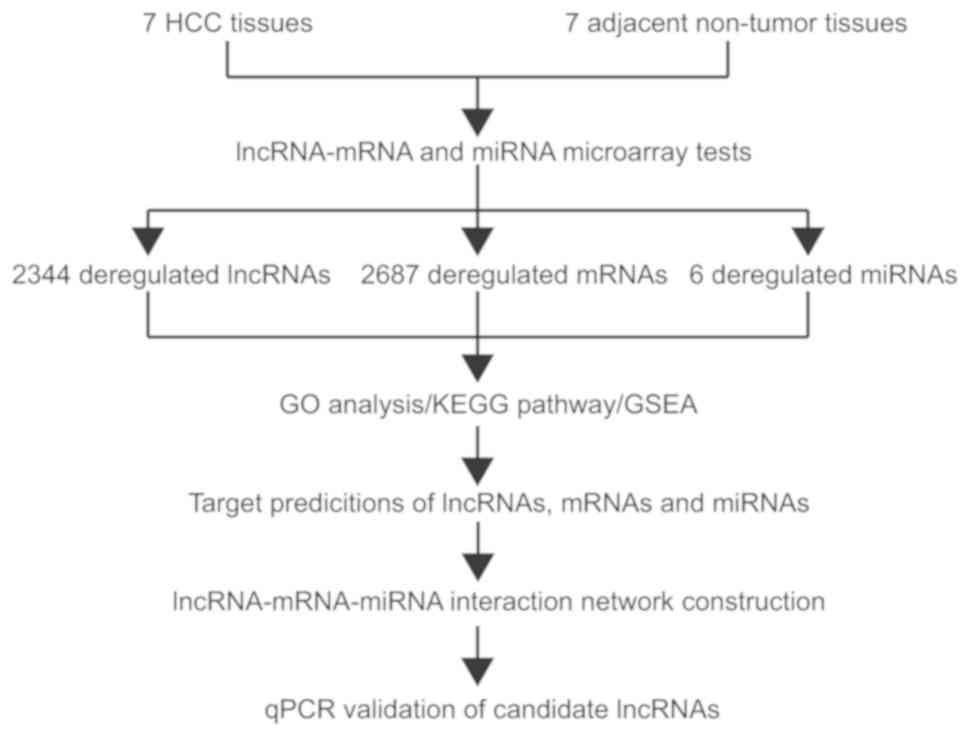
Strategy
To gain insights into possible interactions between
lncRNAs, mRNAs and miRNAs in HCC, the present study first sought to
create a high-throughput map of such an expression profile for use
in subsequent analyses. A five-step approach was applied to
identify targets of HCC-specific lncRNAs, mRNAs and miRNAs
(Fig. 1). Firstly, expression
profiles of lncRNAs, miRNAs and mRNAs in HCC and matched tumor-free
specimens were obtained using high-throughput microarray assays.
Secondly, GO enrichment, KEGG pathway analysis and GSEA were used
to enrich and analyze differentially expressed gene sets. Thirdly,
the significant correlation networks between lncRNAs, mRNAs and
miRNAs, were explored using online analysis tools (miRanda and
LncTar). Fourthly, the differentially expressed mRNAs involved in
the most significant pathways were retained to construct the
lncRNA-mRNA-miRNA interaction networks. Finally, some of the
predicted direct target-related lncRNAs were selected to be
validated in thirty paired HCC specimens and matched adjacent
tumor-free tissues.
Evaluation of microarray raw data
Vioplot was applied for visualization of the
distribution of the signal intensity of lncRNA-mRNA microarray from
seven paired HCC specimens and matched adjacent tumor-free tissue
counterparts. Specifically, lncRNAs from the microarray data were
divided into six types, including bidirectional, exonic antisense,
exonic sense, intergenic, intronic antisense and intronic sense
lncRNAs. The mean expression level of these six types of lncRNAs
was slightly lower than that of mRNAs, while the standard error
expression between lncRNA and mRNA was almost the same (Fig. S2).
In the lncRNA-mRNA microarray, a total of 91,007
lncRNA and 29,859 mRNA species were detected in the HCC and matched
tumor-free specimens. Meanwhile, 2,570 miRNA species were
identified in the miRNA microarray. After filtering out the low
signal intensity, 32,793 lncRNA, 22,224 mRNA and 369 miRNA species
were retained (Table SIII). By the application of PCA, top 1,000
lncRNA and mRNA species that showed the greatest variation and 369
miRNAs were visualized in a heatmap. The findings revealed that the
distribution of lncRNAs and mRNAs in HCC group was separated
remarkably from the controls, implicating that these abnormally
expressed lncRNAs and mRNAs may play an important role in HCC
(Fig. S3A-C). However, for the
miRNA microarray, the distribution did not present well (Fig. S3D).
Differentially expressed lncRNAs, mRNAs
and miRNAs in HCC specimens
With the inclusion criteria of fold-change ≥2.0 and
FDR <0.05, 2,344 lncRNA species (1,056 being upregulated and
1,288 being downregulated), 2,687 mRNA (1,171 being upregulated and
1,516 being downregulated) and 6 miRNA (5 being upregulated and 1
being downregulated) were confirmed to be differentially expressed
in the present study (Table SIV).
GO enrichment and KEGG pathway
analysis
To uncover the involvement of target genes for
lncRNAs and miRNAs in various biological processes, GO enrichment
and KEGG pathway analysis were used for differentially expressed
mRNAs. During this procedure, GO enrichment covers three domains,
including molecular functions (MF), biological processes (BP) and
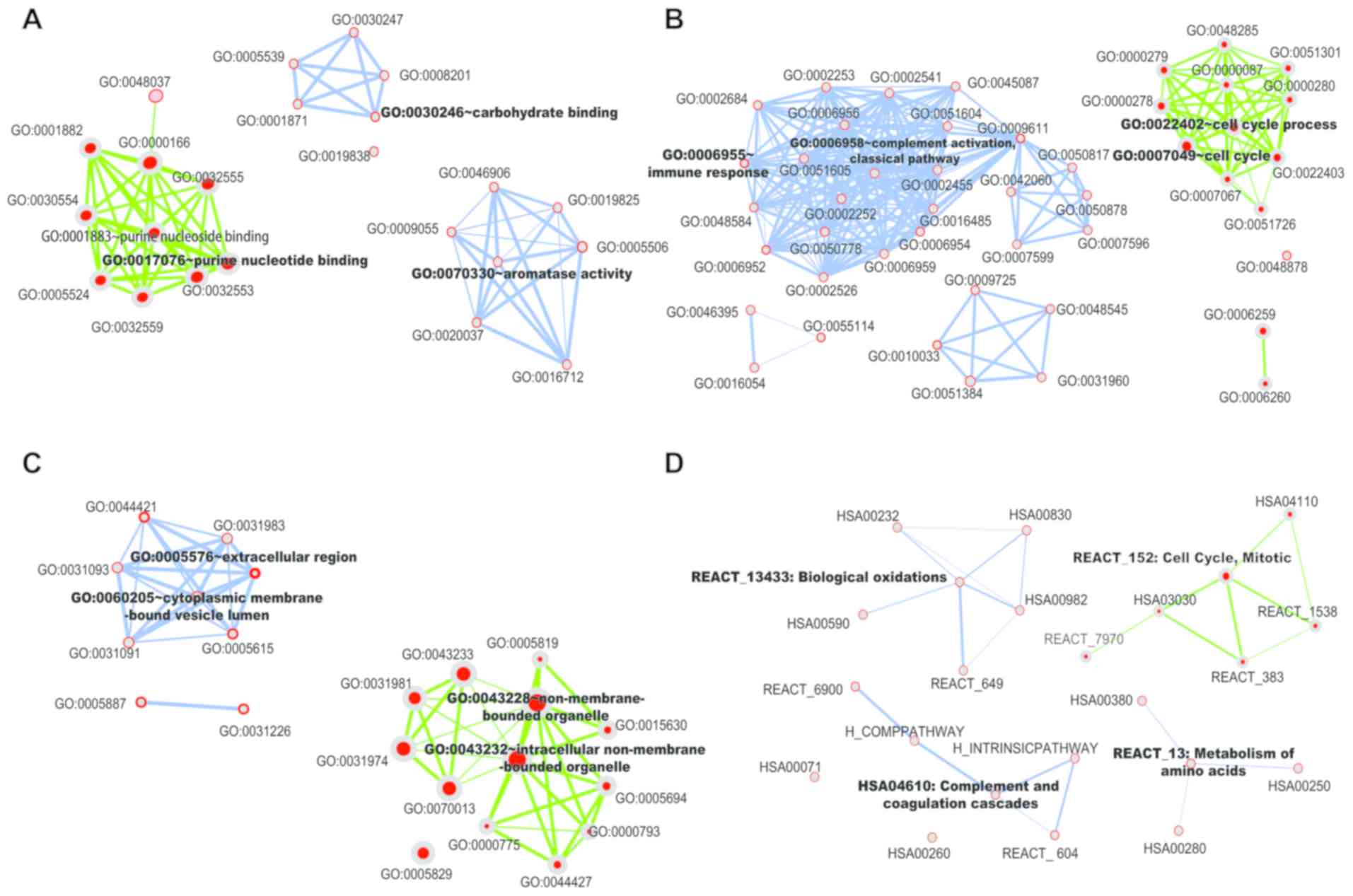
cellular components (CC). As shown in Fig. 2A-C and Table SV, predicted target
genes of upregulated mRNAs fall into purine nucleotide binding in
MF, cell cycle in BP and intracellular non-membrane-bound organelle
in CC, such as cell cycle-related genes CDKN2A (cyclin-dependent
kinase inhibitor 2A), cyclin-dependent kinase inhibitor 2B and
Aurora kinase A in BP. Furthermore, predicted target genes of
down-regulated mRNAs involved in carbohydrate binding in MF, immune
response in BP and extracellular region in CC, such as immune
response-related gene Mannan-binding lectin serine protease 1 and
Mannan-binding lectin serine protease 2 in BP.
In the KEGG pathway analysis, upregulated mRNAs were
predicted to modulate the cell cycle, such as cyclin-dependent
kinase 1 (CDK1), CCNE1, cyclin E2 (CCNE2), proliferating cell
nuclear antigen (PCNA) and ubiquitin-conjugating enzyme E2C; while
the downregulated mRNAs are associated with metabolic pathways,
such as formimidoyltransferase cyclodeaminase, glutamic-pyruvic
transaminase, glutamic-oxaloacetic transaminase 1,
glutamic-oxaloacetic transaminase 2 and spermidine
N1-acetyltransferase 1 (Fig. 2D
and Table SVI). All significant pathways for mRNAs that may exert
modulating functions are shown in Tables SVII and SVIII. Notably,
among these related pathways, target genes in controlling cell
cycle were validated as the most significant data set
(P=8.75x10-21).
To validate the results of GO enrichment and KEGG
pathway analysis, all of the mRNAs detected by microarray were
recruited into the GSEA based on fold-change value (16). The findings demonstrated that
mRNAs involved in the cell cycle were markedly upregulated, which
is consistent with the results of GO enrichment and KEGG pathway
analysis. Meanwhile, when the R language package was applied to
visualize the GSEA of top three significant pathways in modulating
cell cycle, G2-M checkpoint and cell cycle mitotic, are prominent,
which further confirms that the genes controlling cell cycle
pathways are very active in HCC development and progression
(Fig. S4).
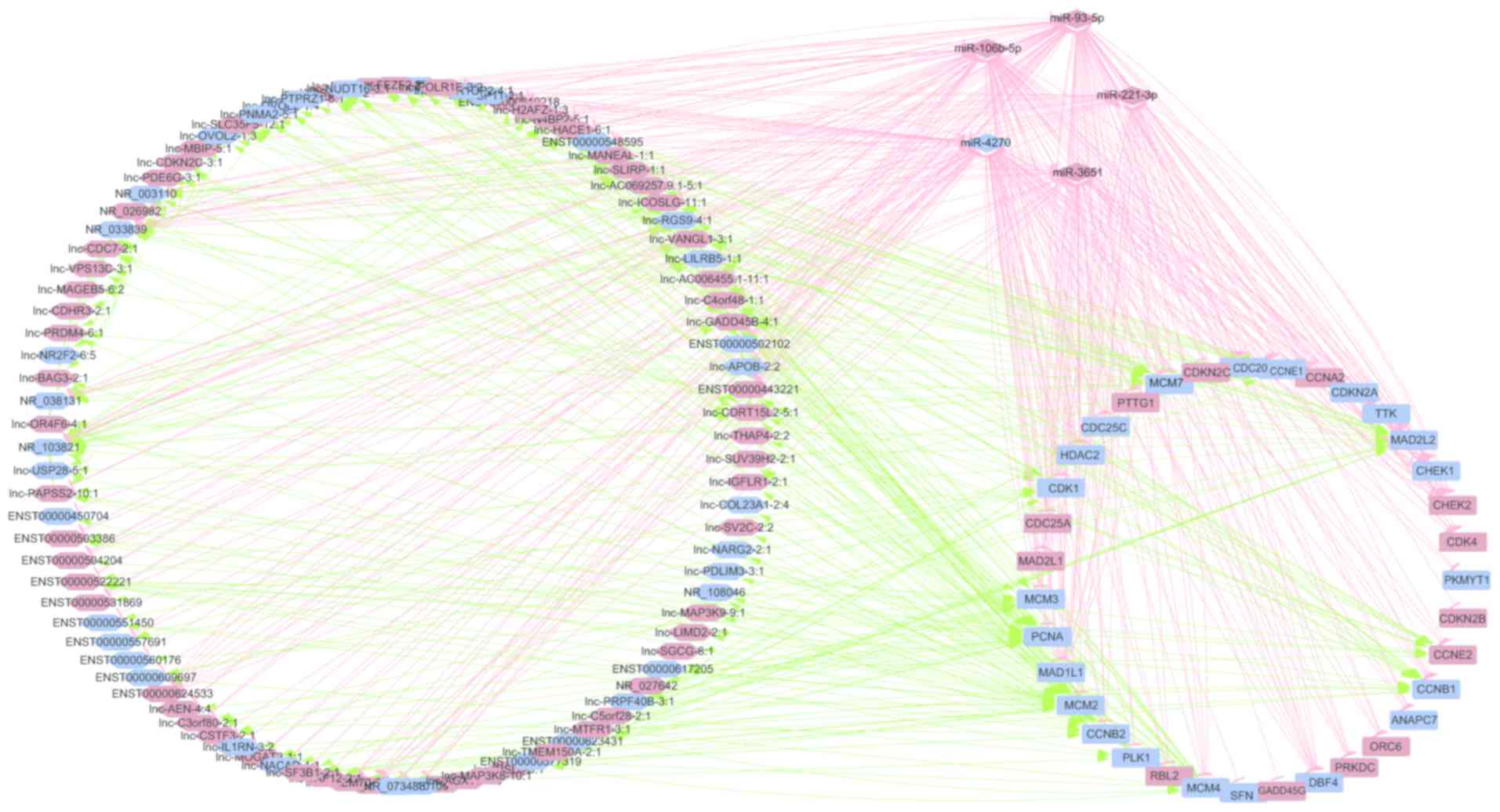
Construction of lncRNA-mRNA-miRNA
co-expression network
Based on the above network, it was shown that
differentially expressed mRNAs involved in the cell cycle pathway
played a crucial role in the pathogenesis of HCC and they were then
selected as basic elements for further exploration of the
coordination and orchestra of lncRNAs, mRNAs and miRNAs in specific
cell functions, such as cell cycle control. With the assistance of
miRanda and Lnctar, the significantly expressed miRNAs and lncRNAs
binding to the cell cycle-related genes were screened out. After
the calculation of Pearson correlation coefficients, the screening
results showed that a co-expression network consists of 193 nodes
and 655 connections between 95 lncRNAs, 36 mRNAs and 5 miRNAs.
Within this co-expression network, 187 pairs presented as positive
and 468 pairs presented as negative. This co-expression network
indicated that one lncRNA could target 10 mRNA at most in a given
process and that one mRNA could be under control by three lncRNAs
at most; while one miRNA could target 31 coding genes at most, and
one mRNA could correlate with three lncRNAs at most in a specific
biological activity. This provides a snapshot for gene expression
control for a particular gene in normal or malignant processes.
This multiple-layer modulation at epigenetic, transcriptional
(mRNA), post-translational levels (miRNA or lncRNAs) constitutes a
very comprehensive signaling network governing the cell cycle in a
fine-tuned fashion. Taken together, a total of 95 lncRNAs, 36 mRNAs
and 5 miRNAs are involved in the predicted lncRNA-mRNA-miRNA
interaction network after combining the proposed binding targets
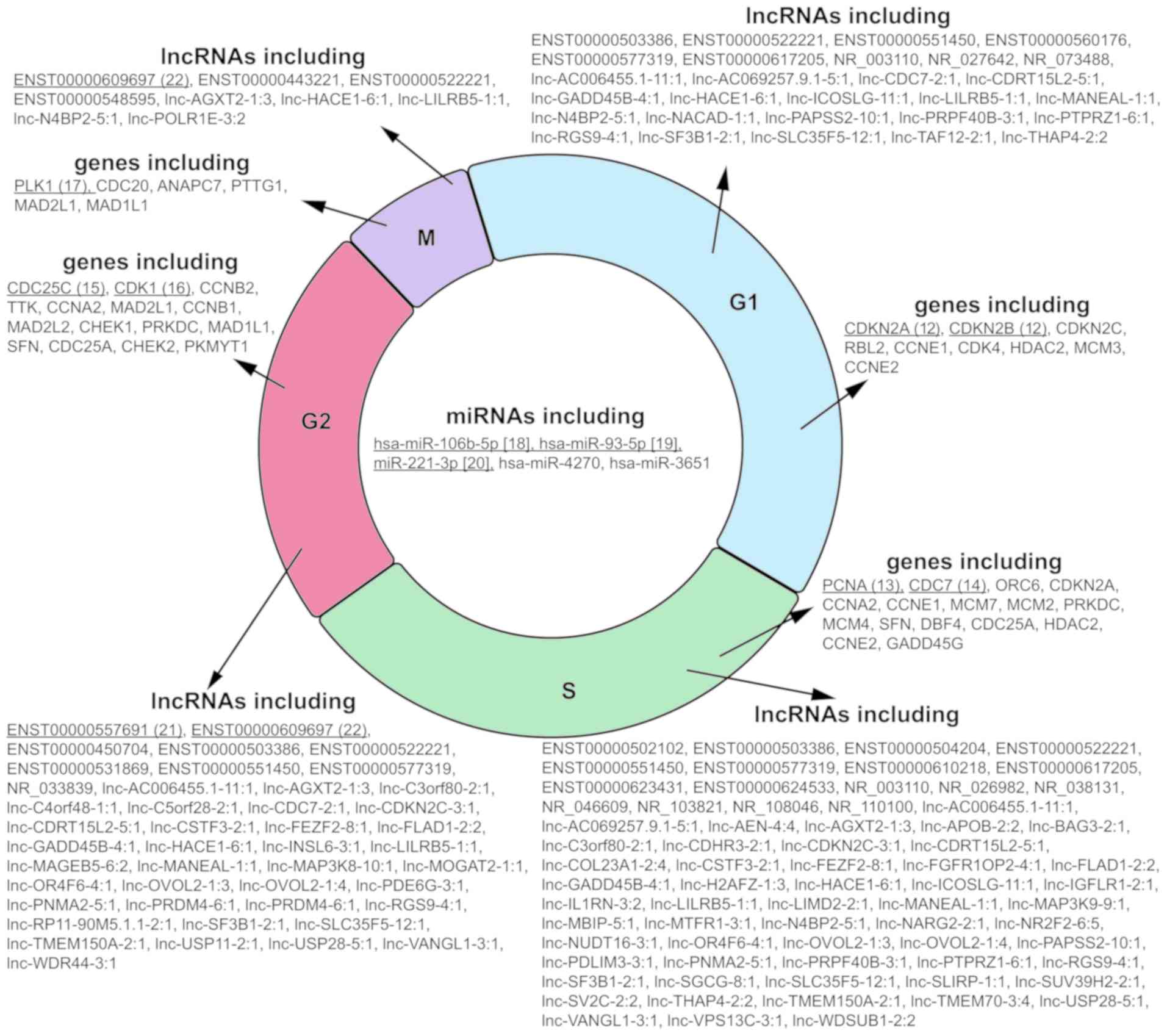
(Fig. 3). Next, these lncRNAs,
miRNAs and mRNAs were categorized into different phases of the cell
cycle based on the predicted target molecules (Fig. 4). Among these 95 lncRNAs, the top
five lncRNAs associated with cell cycle modulation, including
ENST00000522221, ENST00000577319, lnc-GADD45B-4:1, lnc-HACE1-6:1
and lnc-ICOSLG-11:1, were selected for validation analysis, and all
of targeted mRNAs, miRNAs and lncRNAs for these five lncRNAs are
also shown (Table I).
Kaplan-Meier curves of overall survival of targeted mRNAs,
including cyclin B1 (CCNB1), CCNB2, CCNE1, CCNE2, cell division
cycle 7 (CDC7), CDK1, mitotic arrest deficient 2 like 2,
minichromosome maintenance complex component 2 (MCM2),
minichromosome maintenance complex component 3 (MCM3),
minichromosome maintenance complex component 4 (MCM4),
minichromosome maintenance complex component 7 (MCM7), PCNA, polo
like kinase 1 (PLK1), are also shown using TCGA database (Fig. 5).
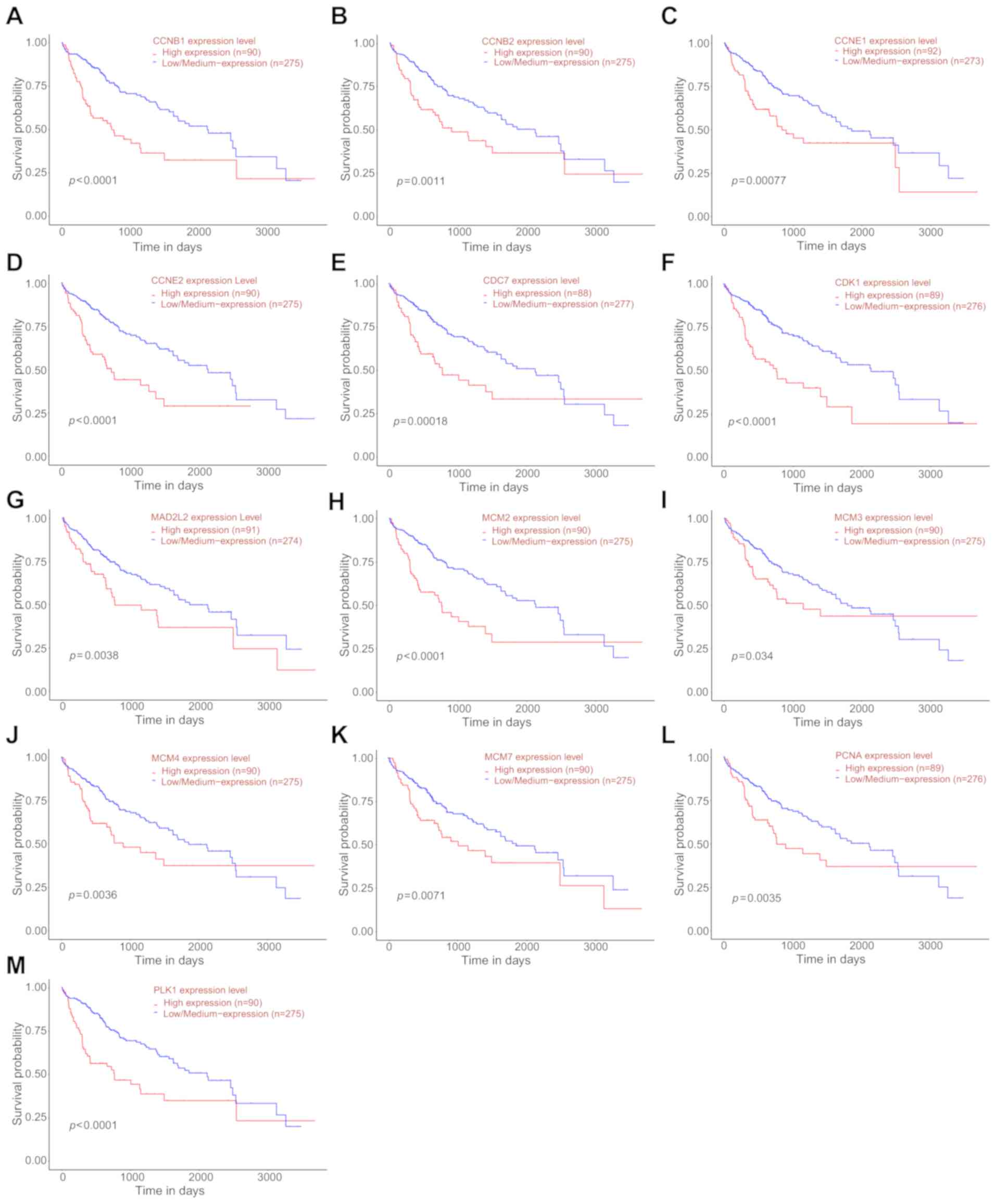 | Figure 5Kaplan-Meier survival analysis of
targeted mRNAs based on the data from TCGA database. Overall
survival rate of targeted mRNAs, including (A) CCNB1, (B) CCNB2,
(C) CCNE1, (D) CCNE2, (E) CDC7, (F) CDK1, (G) MAD2L2, (H) MCM2, (I)
MCM3, (J) MCM4, (K) MCM7, (L) PCNA and (M) PLK1. Adapted from
UALCAN: http://ualcan.path.uab.edu/index.html. |
 | Table IThe network of the interactions
between lncRNAs and miRNAs and mRNAs. |
Table I
The network of the interactions
between lncRNAs and miRNAs and mRNAs.
| Gene ID
(lncRNA) | Regulation | Target lncRNA | Target miRNA | Target mRNA |
|
ENST00000522221 | Up | - | hsa-miR-106b-5p,
hsa-miR-93-5p | CDC7, CCNE1, MCM3,
PCNA, MCM2, CCNB2, PLK1, CCNB1, CCNE2 |
|
ENST00000577319 | Down | - | - | MAD2L2, CDC7, CDK1,
MCM3, PCNA, MCM2, MCM4 |
|
lnc-GADD45B-4:1 | Up | ENST00000502102,
lnc-POLR1E-3:2, lnc- WDSUB1-2:2, NR_103821 | - | MAD2L2, CCNE1,
MCM7, MCM3, CCNB2, MCM4 |
| lnc-HACE1-6:1 | Up | ENST00000548595,
NR_038131, NR_103821, | hsa-miR-106b-5p,
hsa-miR-93-5p | CCNE1, MCM7, MCM2,
CCNB2, PLK1, CCNB1 |
|
lnc-ICOSLG-11:1 | Up | ENST00000502102,
lnc-POLR1E3:2, NR_038131 | hsa-miR-106b-5p,
hsa-miR-93-5p | CDC7, CCNE1, MCM7,
MCM3, MCM2, CCNE2 |
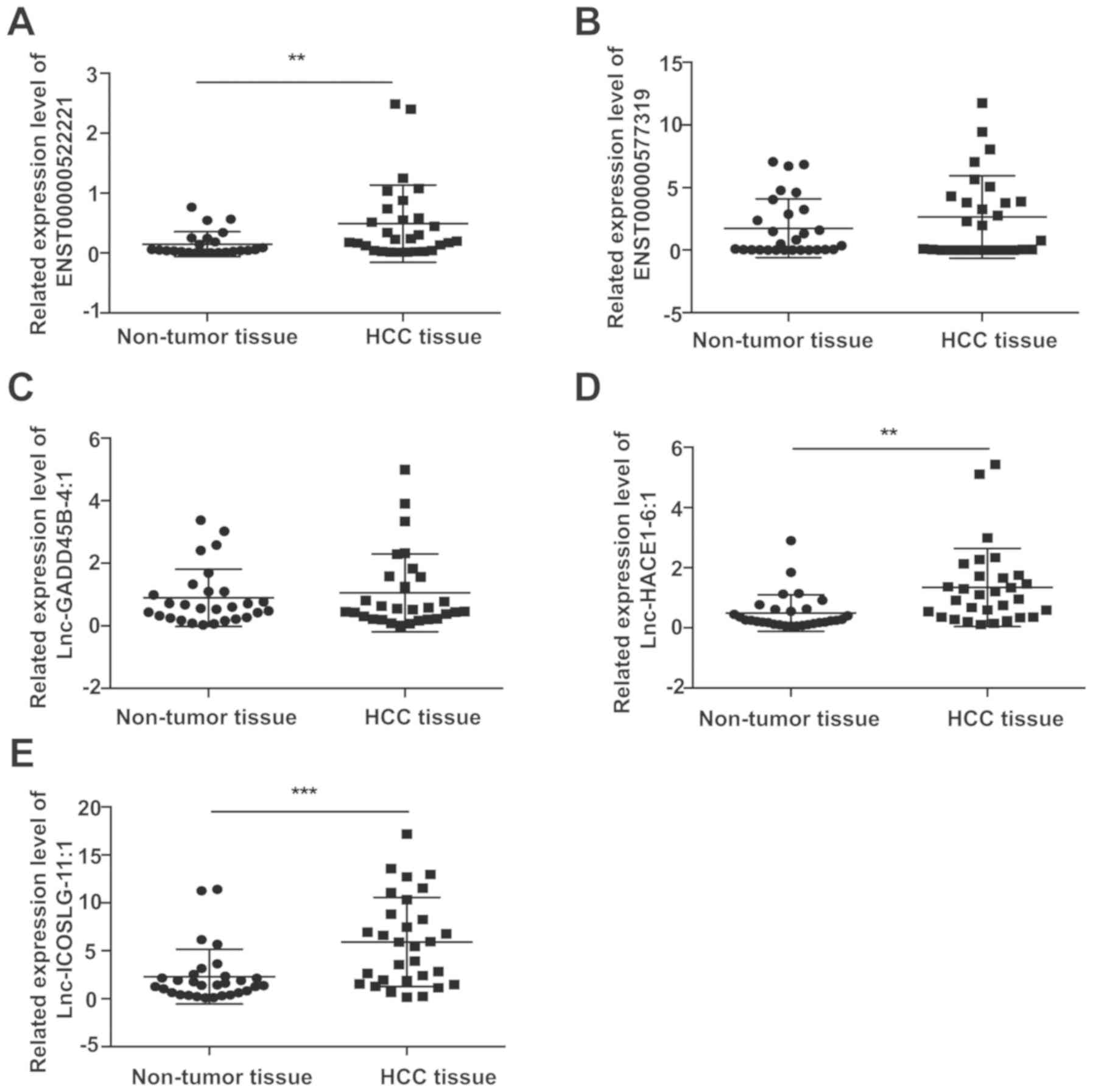
Validation of candidate lncRNAs
To validate the reliability of the results obtained
from the microarray, top five differentially expressed lncRNAs was
selected for qPCR validation in 30 pairs of HCC and matched
tumor-free specimens. The results found that ENST00000522221,
lnc-HACE1-6:1 and lnc-ICOSLG-11:1 are significantly upregulated in
HCC tissues compared with that in adjacent tumor-free tissues
(P=0.0080, P=0.0034 and P=0.0003, respectively; Fig. 6).
Discussion
Aberrant expression of ncRNAs, including lncRNAs and
miRNAs, as well as protein-coding genes have been widely used to
uncover underlying molecular mechanisms contributing to
tumorigenesis, progression, metastasis and drug resistance or
relapse. A comprehensive framework of orchestrated expression and
interaction of lncRNAs, mRNAs, miRNAs in HCC, however, remains to
be elucidated. In this study, an integrated lncRNA-mRNA-miRNA
signature that consists of co-expressed lncRNA, mRNA and miRNA
datasets was examined. Furthermore, GO enrichment and KEGG pathway
analyses of differentially expressed mRNAs point to potential
functions and pathways critical for HCC cell cycle, and a
co-expression network that was composed of 193 network nodes and
655 connections between 95 lncRNAs, 36 mRNAs and 5 miRNAs was
constructed to reflect possible interactions of these
molecules.
Involved mRNAs and ncRNAs in the interaction
network, which potentially modulate critical genes in four phases
of the cell cycle, including G1, S, G2 and M were listed. The mRNAs
that engaged in the four parts of the cycle belong to prominent and
classic cell cycle-related genes, such as CDKN2A/B (17), PCNA (18) and CDC7 (19) in G1/S phase and cell division
cycle 25C (20), CDK1 (21) and PLK1 (22) in G2/M phase. The miRNAs and
lncRNAs were categorized based on their predicted targeting mRNA
encoding genes involved in the cell cycle. It turned out that all
the five enriched miRNAs (including miR-4270, miR-3651, miR-93-5p,
hsa-miR-106b-5p and miR-221-3p) have potential target molecules
critical for the entire cell cycle. Notably, no prior studies have
proved that these miRNAs are associated with the cell cycle in HCC
specimens. In 2012, a study demonstrated that downregulated
miR-106b induces G1/S arrest in HCC cell lines (23). Another study showed that
overexpression of miR-93-5p promotes G1 or S arrest in ovarian
carcinoma (24). Additionally,
upregulated miR-221-3p promotes accumulation of human fibroblasts
in the G1/S phase (25). It is
noteworthy that none of the 95 lncRNAs have been reported to
participate in the modulation of the cell cycle in HCC. Only
ENST00000557691 and ENST00000609697 have been reported in studies
of microarray expression profiles in glioblastoma multiforme
(26) and multiple sclerosis
(27), respectively. Finally,
ENST00000522221, lnc-HACE1-6:1 and lnc-ICOSLG-11:1 were validated
as cell cycle-related regulators in an extended 30 pairs of HCC
specimens. Therefore, the present study provides a snapshot of how
mRNAs, miRNAs and lncRNAs govern a given process, such as the cell
cycle, at epigenetic, transcriptional and post-translational levels
in an orchestrated fashion in HCC.
Although the study of lncRNAs is still in its
infancy, it has become increasing apparent that lncRNAs are
critical regulators for cellular and physiological processes by
modulating gene and other ncRNA expression. Previous studies have
separately demonstrated the involvement of miRNAs (28) and lncRNAs (29) in the development of cancer. Since
the first cancer-targeted MRX34, a liposome-based miR-34 mimic,
entered Phase I clinical trials in patients with advanced HCC in
2013, understanding the biology of lncRNAs and its connections with
mRNAs and miRNAs may lead to promising clinical applications in
HCC. Moreover, several studies are available that focus on
individual lncRNA expression profiles and regulatory cascades in
the tumorigenesis of HCC, such as the HCC upregulated long
non-coding RNA (30), H19
(31), metastasis associated lung
adenocarcinoma transcript 1 (32)
and HCC upregulated EZH2-associated long non-coding RNA (33). However, no studies provide a
snapshot of multilayer modulation and interaction networks of
lncRNAs, mRNAs and miRNAs in hepatic carcinogenesis, although
dissecting each network or interactions involved is far beyond the
scope of this manuscript, and requires more advanced approaches and
extensive resources.
In the present study lnc-HACE1-6:1, lnc-ICOSLG-11:1
and ENST00000522221 in 30 pairs of HCC specimens were validated.
Located on chr6, lnc-HACE1-6:1 (865 bp) was identified as a
transcript of nucleophosmin 1 pseudogene 10 (ENST00000398310) from
the Ensemble database (http://asia.ensembl.org/index.html). Evidence exists
that NPM1 is associated with various cancers, such as prostate
cancer and acute myeloid leukemia (34-36). Especially, it was found that
overex-pression of NPM1 suppresses p53 via blocking cell
cycle-related proteins, such as ARF/MDM2 in colorectal carcinoma
(37). Similarly, lnc-ICOSLG-11:1
(395 bp), located in chr21, was identified as a transcript of H2A
histone family member Z pseudogene 1 (ENST00000416034) from the
Ensemble database. The reports revealed that H2AFZ was
overexpressed in HCC patients and promoted the cell growth by
affecting cell cycle-related proteins (38). In addition, there is a growing
belief that lncRNAs can function as endogenous miRNA sponges as a
part of competing endogenous RNA (ceRNA) network. For example, the
PTEN pseudogene 1, a lncRNA sharing a high degree of sequence
homology with tumor suppressor gene PTEN, acts as a decoy for
PTEN-targeting miRNAs (39). In
the present study, both lnc-HACE1-6:1 and lnc-ICOSLG-11:1 were
predicted to bind with miR-106b-5p and miR-93-5p, and to interact
with cell cycle-related genes, such as CCNE1 (40), MCM7 (41) and MCM2 (42). It was postulated that high levels
of lnc-HACE1-6:1 and lnc-ICOSLG-11:1 affect cell cycle-related
genes by acting as the ceRNA via mediating NPM1 and H2AFZ
expression, respectively. ENST00000522221, known as a transcript of
THUMP domain containing 3 antisense RNA 1, has never been reported
to associate with HCC. In the present study, it was predicted that
ENST00000522221 was associated with miRNAs, including miR-106b-5p
and miR-93-5p, and cell cycle-related genes. Nevertheless, more
studies are warranted to gain better understanding of the role of
ENST00000522221, lnc-HACE1-6:1 and lnc-ICOSLG-11:1 in hepatic
carcinogenesis.
In conclusion, the present study has laid a
framework to understand miRNA-lncRNA-mRNA co-expression profiles,
interaction and orchestrated modulation of cellular processes, such
as the cell cycle in hepatic carcinogenesis. Probing complicated
interplays between lncRNAs and mRNAs/miRNAs holds promise to
identify new diagnostic and prognostic markers for HCC and, at
least in part, novel targets for antitumor therapies.
Supplementary Data
Acknowledgements
The authors gratefully acknowledge TCGA and UALCAN
for providing data to analyze.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
mRNA
|
messenger RNAs
|
|
lncRNAs
|
long non-coding RNAs
|
|
miRNA
|
microRNA
|
|
3′ UTR
|
3′ untranslated region
|
|
FDR
|
false discovery rate
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DEGs
|
differential expressed genes
|
|
GSEA
|
Gene Set Enrichment Analysis
|
|
TCGA
|
The Cancer Genome Atlas
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
PCA
|
principle component analysis
|
|
MF
|
molecular functions
|
|
BP
|
biological processes
|
|
CC
|
cellular components
|
|
CCNE1
|
cyclin E1
|
|
CCNE2
|
cyclin E2
|
|
CCNB1
|
cyclin B1
|
|
CCNB2
|
cyclin B2
|
|
CDKN2A
|
cyclin-dependent kinase inhibitor
2A
|
|
CDK1
|
cyclin-dependent kinase 1
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
CDC7
|
cell division cycle 7
|
|
MCM2
|
minichromosome maintenance complex
component 2
|
|
MCM3
|
minichromosome maintenance complex
component 3
|
|
MCM4
|
minichromosome maintenance complex
component 4
|
|
MCM7
|
minichromosome maintenance complex
component 7
|
|
PLK1
|
polo like kinase 1
|
|
CDC25C
|
cell division cycle 25C
|
|
ceRNA
|
competing endogenous RNA
|
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81472673, 81672720
and 81672334), the Shanghai Science and Technology Commission
(grant no. 16ZR1406100), and the National Clinical Key Special
Subject of China.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
All the authors contributed to this manuscript,
including the conception and design (SQW, HLAJ, LD, XZS, JW and
JMZ), the acquisition of data (HRZ and XNY), the analysis and
interpretation of the data (HRZ, XNY, GCZ, XS, EB, HYG and GQS),
material support (TTL, LLL and YC), study supervision (XZS, JW and
JMZ), and the writing, review and revision of the manuscript (HRZ,
XNY, SQW, HLAJ, LD and JMZ). All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The project protocol was approved by Institutional
Ethics Committee of Zhongshan Hospital of Fudan University. All
patients recruited in the present study provided written informed
consent for the use of their tissue samples for clinical
research.
Patient consent for publication
Written informed consent was obtained from all
participants for the publication of their data.
Competing interests
The authors declare that there are no conflicts of
interest.
References
|
1
|
Cervello M, McCubrey JA, Cusimano A,
Lampiasi N, Azzolina A and Montalto G: Targeted therapy for
hepatocellular carcinoma: Novel agents on the horizon. Oncotarget.
3:236–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schlesinger S, Aleksandrova K, Pischon T,
Fedirko V, Jenab M, Trepo E, Boffetta P, Dahm CC, Overvad K,
Tjønneland A, et al: Abdominal obesity, weight gain during
adulthood and risk of liver and biliary tract cancer in a European
cohort. Int J Cancer. 132:645–657. 2013. View Article : Google Scholar
|
|
4
|
Kretz M, Siprashvili Z, Chu C, Webster DE,
Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, et al:
Control of somatic tissue differentiation by the long non-coding
RNA TINCR. Nature. 493:231–235. 2013. View Article : Google Scholar :
|
|
5
|
Lagarde J, Uszczynska-Ratajczak B,
Carbonell S, Pérez-Lluch S, Abad A, Davis C, Gingeras TR, Frankish
A, Harrow J, Guigo R and Johnson R: High-throughput annotation of
full-length long noncoding RNAs with capture long-read sequencing.
Nat Genet. 49:1731–1740. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lagarde J, Uszczynska-Ratajczak B,
Santoyo-Lopez J, Gonzalez JM, Tapanari E, Mudge JM, Steward CA,
Wilming L, Tanzer A, Howald C, et al: Extension of human lncRNA
transcripts by RACE coupled with long-read high-throughput
sequencing (RACE-Seq). Nat Commun. 7:123392016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gatto S, Della Ragione F, Cimmino A,
Strazzullo M, Fabbri M, Mutarelli M, Ferraro L, Weisz A, D'Esposito
M and Matarazzo MR: Epigenetic alteration of microRNAs in
DNMT3B-mutated patients of ICF syndrome. Epigenetics. 5:427–443.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huarte M, Guttman M, Feldser D, Garber M,
Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M,
et al: A large intergenic noncoding RNA induced by p53 mediates
global gene repression in the p53 response. Cell. 142:409–419.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao X, Patton JR, Davis SL, Florence B,
Ames SJ and Spanjaard RA: Regulation of nuclear receptor activity
by a pseu-douridine synthase through posttranscriptional
modification of steroid receptor RNA activator. Mol Cell.
15:549–558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu HR, Huang RZ, Yu XN, Shi X,
Bilegsaikhan E, Guo HY, Song GQ, Weng SQ, Dong L, Janssen HLA, et
al: Microarray expression profiling of microRNAs reveals potential
biomarkers for hepatocellular carcinoma. Tohoku J Exp Med.
245:89–98. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Subramanian A, Kuehn H, Gould J, Tamayo P
and Mesirov JP: GSEA-P: A desktop application for Gene Set
Enrichment Analysis. Bioinformatics. 23:3251–3253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soto JL, Cabrera CM, Serrano S and
López-Nevot MA: Mutation analysis of genes that control the G1/S
cell cycle in melanoma: TP53, CDKN1A, CDKN2A, and CDKN2B. BMC
Cancer. 5:362005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moldovan GL, Pfander B and Jentsch S: PCNA
controls establishment of sister chromatid cohesion during S phase.
Mol Cell. 23:723–732. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hess GF, Drong RF, Weiland KL, Slightom
JL, Sclafani RA and Hollingsworth RE: A human homolog of the yeast
CDC7 gene is overexpressed in some tumors and transformed cell
lines. Gene. 211:133–140. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang H, Hu M, Zhao R, Li P and Li M:
Dihydromyricetin suppresses the proliferation of hepatocellular
carcinoma cells by inducing G2/M arrest through the
Chk1/Chk2/Cdc25C pathway. Oncol Rep. 30:2467–2475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng P, Li Y, Yang L, Wen Y, Shi W, Mao
Y, Chen P, Lv H, Tang Q and Wei Y: Hepatitis B virus X protein
(HBx) induces G2/M arrest and apoptosis through sustained
activation of cyclin B1-CDK1 kinase. Oncol Rep. 22:1101–1107.
2009.PubMed/NCBI
|
|
22
|
Pellegrino R, Calvisi DF, Ladu S, Ehemann
V, Staniscia T, Evert M, Dombrowski F, Schirmacher P and Longerich
T: Oncogenic and tumor suppressive roles of polo-like kinases in
human hepatocellular carcinoma. Hepatology. 51:857–868.
2010.PubMed/NCBI
|
|
23
|
Shen G, Jia H, Tai Q, Li Y and Chen D:
miR-106b downregulates adenomatous polyposis coli and promotes cell
proliferation in human hepatocellular carcinoma. Carcinogenesis.
34:211–219. 2013. View Article : Google Scholar
|
|
24
|
Chen X, Chen S, Xiu YL, Sun KX, Zong ZH
and Zhao Y: RhoC is a major target of microRNA-93-5P in epithelial
ovarian carcinoma tumorigenesis and progression. Mol Cancer.
14:312015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Markopoulos GS, Roupakia E, Tokamani M,
Vartholomatos G, Tzavaras T, Hatziapostolou M, Fackelmayer FO,
Sandaltzopoulos R, Polytarchou C and Kolettas E:
Senescence-associated microRNAs target cell cycle regulatory genes
in normal human lung fibroblasts. Exp Gerontol. 96:110–122. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Q, Jia H, Li H, Dong C, Wang Y and Zou
Z: LncRNA and mRNA expression profiles of glioblastoma multiforme
(GBM) reveal the potential roles of lncRNAs in GBM pathogenesis.
Tumour Biol. 37:14537–14552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang F, Gao C, Ma XF, Peng XL, Zhang RX,
Kong DX, Simard AR and Hao JW: Expression profile of long noncoding
RNAs in peripheral blood mononuclear cells from multiple sclerosis
patients. CNS Neurosci Ther. 22:298–305. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar
|
|
29
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li D, Liu X, Zhou J, Hu J, Zhang D, Liu J,
Qiao Y and Zhan Q: Long noncoding RNA HULC modulates the
phosphorylation of YB-1 through serving as a scaffold of
extracellular signal-regulated kinase and YB-1 to enhance
hepatocarcinogenesis. Hepatology. 65:1612–1627. 2017. View Article : Google Scholar
|
|
31
|
Zhang L, Yang F, Yuan JH, Yuan SX, Zhou
WP, Huo XS, Xu D, Bi HS, Wang F and Sun SH: Epigenetic activation
of the MiR-200 family contributes to H19-mediated metastasis
suppression in hepatocellular carcinoma. Carcinogenesis.
34:577–586. 2013. View Article : Google Scholar
|
|
32
|
Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY,
Zhang F, Wu LM, Chen LM and Zheng SS: Long non-coding RNA MALAT-1
over-expression predicts tumor recurrence of hepatocellular
carcinoma after liver transplantation. Med Oncol. 29:1810–1816.
2012. View Article : Google Scholar
|
|
33
|
Yang F, Zhang L, Huo XS, Yuan JH, Xu D,
Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, et al: Long noncoding
RNA high expression in hepatocellular carcinoma facilitates tumor
growth through enhancer of zeste homolog 2 in humans. Hepatology.
54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dai L, Li J, Xing M, Sanchez TW, Casiano
CA and Zhang JY: Using serological proteome analysis to identify
serum anti-nucleophosmin 1 autoantibody as a potential biomarker in
European-American and African-American patients with prostate
cancer. Prostate. 76:1375–1386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jo SY, Park SH, Kim IS, Yi J, Kim HH,
Chang CL, Lee EY, Cho YU, Jang S, Park CJ and Chi HS: Correlation
of NPM1 type A mutation burden with clinical status and outcomes in
acute myeloid leukemia patients with mutated NPM1 type A. Ann Lab
Med. 36:399–404. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Versluis J, In 't Hout FE, Devillier R,
van Putten WL, Manz MG, Vekemans MC, Legdeur MC, Passweg JR,
Maertens J, Kuball J, et al: Comparative value of post-remission
treatment in cytoge-netically normal AML subclassified by NPM1 and
FLT3-ITD allelic ratio. Leukemia. 31:26–33. 2017. View Article : Google Scholar
|
|
37
|
Wong JC, Hasan MR, Rahman M, Yu AC, Chan
SK, Schaeffer DF, Kennecke HF, Lim HJ, Owen D and Tai IT:
Nucleophosmin 1, upreg-ulated in adenomas and cancers of the colon,
inhibits p53-mediated cellular senescence. Int J Cancer.
133:1567–1577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang HD, Kim PJ, Eun JW, Shen Q, Kim HS,
Shin WC, Ahn YM, Park WS, Lee JY and Nam SW: Oncogenic potential of
histone-variant H2A.Z.1 and its regulatory role in cell cycle and
epithelial-mesenchymal transition in liver cancer. Oncotarget.
7:11412–11423. 2016.PubMed/NCBI
|
|
39
|
Poliseno L, Salmena L, Zhang J, Carver B,
Haveman WJ and Pandolfi PP: A coding-independent function of gene
and pseudo-gene mRNAs regulates tumour biology. Nature.
465:1033–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nault JC, Datta S, Imbeaud S, Franconi A,
Mallet M, Couchy G, Letouzé E, Pilati C, Verret B, Blanc JF, et al:
Recurrent AAV2-related insertional mutagenesis in human
hepatocellular carcinomas. Nat Genet. 47:1187–1193. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou YM, Zhang XF, Cao L, Li B, Sui CJ, Li
YM and Yin ZF: MCM7 expression predicts post-operative prognosis
for hepato-cellular carcinoma. Liver Int. 32:1505–1509. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu F, Yuan JH, Huang JF, Yang F, Wang TT,
Ma JZ, Zhang L, Zhou CC, Wang F, Yu J, et al: Long noncoding RNA
FTX inhibits hepatocellular carcinoma proliferation and metastasis
by binding MCM2 and miR-374a. Oncogene. 35:5422–5434. 2016.
View Article : Google Scholar : PubMed/NCBI
|