Introduction
Sepsis, also known as systemic inflammatory response
syndrome, arises as a result of an overwhelming immune response to
infection, which is highly associated with tissue injury, multiple
organ failure and even death (1,2).
In addition, sepsis is the most common cause of acute kidney injury
(AKI) (3), which is a type of
clinical syndrome characterized by a rapid decline in the
glomerular filtration rate, resulting in retention of nitrogenous
wastes, primary creatinine and blood urea nitrogen (4). The incidence of AKI increases with
the severity of sepsis and AKI occurs in ~19% of patients with
moderate sepsis, 23% with severe sepsis and 51% with septic shock
(5,6). Notably, the mortality rate for AKI
patients in the setting of sepsis is approximately twice as high as
that for sepsis alone (7). A
previous study showed that pathogenesis of sepsis-induced AKI was
distinct from AKI without sepsis and some studies demonstrated that
sepsis-induced renal ischemia and acute tubular necrosis (ATN) were
the primary pathophysiologies to AKI (8,9).
However, previous research involving postmortem observations in
septic animals and humans showed that sepsis-induced AKI was
characterized by obviously bland histology with focal areas of
tubular damage, but rare diffuse renal tubular cell death,
indicating that ATN could not completely explain this phenotype
(10,11). Meanwhile, three distinct
alterations [cellular bioenergetic responses to injury (10), diffused microcirculatory flow
abnormalities (12) and
inflammation (13,14)] were consistently observed
regardless of species, organ, disease stage or severity, indicating
that these three alterations played vital roles in the progression
and development of tubular injury and AKI. This study focused on
the mechanism of the inflammatory response in the sepsis-associated
AKI and aimed to improve the present status of treatment for
AKI.
CD39, alternatively known as ecto-nucleotide
triphosphate diphosphohydrolase-1, works as an inflammatory
suppressor that could catalyze the conversion of extracellular
adenosine triphosphate and diphosphate (ATP and ADP) into adenosine
monophosphate and promote the generation of adenosine (15). It is known that the extracellular
ATP triggers NLR family pyrin domain containing 3 (NLRP3)
inflammasome activation, which could lead to the generation of a
series of inflammatory cytokines, such as cleaved caspase-1,
interleukin (IL)-1β, IL-18, IL-6 and tumor necrosis factor-α
(TNF-α) (16,17). Meanwhile, adenosine was reported
to be anti-inflammatory, immunosuppressive and have protective
effects in sepsis-associated organ injury (18,19). Therefore, CD39 plays a suppressive
role in the NLRP3 inflammasome activation and its mediated
downstream pro-inflammatory cytokines. A study on sepsis-induced
liver injury showed that CD39 limited sepsis-caused systemic
inflammation and restored liver homeostasis by suppressing the
activation of NLRP3 through scavenging eATP and generating
adenosine (20). However, the
functional effects of CD39 in sepsis-induced AKI are less studied,
therefore the present study aimed to investigate whether CD39
effectively exerted its anti-inflammatory activity in
sepsis-associated AKI.
Materials and methods
Animal model of AKI
Adult male C57BL/6 mice (n=40, 6-8 weeks old, 22-26
g) were obtained from the Shanxi Dayi Hospital (Taiyuan, China).
All mice were allowed to have free access to food and water and
maintained in a specific pathogen-free animal facility under a 12-h
light/dark cycle at a controlled temperature (20-24°C) and in a
humid (50±5%) environment. Mice were randomly divided into 4
groups. In the control group, a total of 8 mice were
intraperitoneally injected with normal saline (Control). In the
lipopolysaccharide (LPS) group, a total of 32 mice were injected
with a single dose of LPS (20 mg/kg; Sigma-Aldrich; Merck KGaA).
Mice in the LPS and Control group were intraperitoneally
anesthetized with 50 mg/kg of 5% barbitone and subjected to a
thoracic incision 12, 24 and 48 h after the injection. The blood
samples (~0.3 ml per mouse) were collected by heart puncture and
maintained at 37°C for 30 min. After 10 min of centrifugation at
3,500 × g at 4°C, the serum was used for measurement of renal
function and ELISA. Kidney tissues were collected by bilateral
nephrectomy. After removing the kidney capsule and perirenal fat,
the left kidney tissues were fixed in 10% neutral formalin at 4°C
for 24 h for histopathology, while the right kidney tissues were
segmented for gene expression analyses. Animal experimental
procedures were approved by the Ethics Committee of Committee of
Shanxi Dayi Hospital and conducted in accordance with Guide for the
Care and Use of Laboratory Animal.
Hematoxylin and eosin staining
(H&E)
The kidney tissues were fixed with 10% neutral
formalin for <24 h and then washed with PBS. Mice kidneys were
dehydrated with a series of graded ethanol and then embedded in
paraffin and sliced into 3-μm-thick sections for H&E.
The paraffin-embedded sections were stained with hematoxylin and
eosin for 5 min at room temperature (Beijing Solarbio Science &
Technology Co., Ltd.) and visualized under a light microscope
(Leica Microsystems GmbH).
Biochemical index detection
The collected serum was analyzed on the Roche
molecular P800 autobiochemical analyzer (Roche Diagnostics GmbH).
Blood urea nitrogen (BUN) levels were determined using Urease GLDH
method, according to Urea Nitrogen Assay kits (Ningbo Purebio
Biotechnology Co., Ltd.) and the levels of serum creatinine (Cr)
were assayed following the protocol of the Creatinine Detection
kits (Whitman Biotechnology Co., Ltd.).
Cell culture and treatment
The renal tubular epithelial HK-2 cells (American
Type Culture Collection) were cultured in 5% CO2 at 37°C
in a humidified incubator with Dulbecco's modified Eagle's medium
(HyClone/Pierce; Thermo Fisher Scientific, Inc.), which was
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.) and 1% ampicillin. The culture medium was
refreshed every 2 days until cell confluence reached 70-80%. Then,
the cells were resuspended by digestion with 0.25% trypsin/EDTA.
The cells were exposed to LPS (Sigma-Aldrich; Merck KGaA) in
concentrations of 0, 0.1, 1, 10 and 100 μg/ml. A total of 24
h after LPS exposure, changes in cell viability and
inflammation-associated factor levels were assessed to determine
appropriate LPS concentration.
Cell transfection
In order to investigate the effects of CD39 on the
LPS-induced injury in HK-2 cells, the full-length mouse CD39 in
pcDNA3.1 overexpression vector, empty pcDNA3.1 vector (Mock), small
interfering RNA of CD39 (siCD39; 5′-GCG ATT GTC AGT GAA ACT T-3′)
and small interfering-negative control RNA (siNC; 5′-CCT ATC TGG
TCA ACA CGT ATT-3′) were obtained by GenePharma (Shanghai
GenePharma Co., Ltd). A total of ~4×105 HK-2 cells were
incubated in each well of 6-well plates with complete medium until
the cells reached 70% confluence. Cell transfection with 50 nmol/l
vector was conducted according to the manufacturer's protocol for
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The transfected cells were subjected to 10
μg/ml LPS (Sigma-Aldrich; Merck KGaA) treatment for
subsequent experiments. The activity was measured 24 h after the
transfection experiment.
Measurement of cell viability
The cell viability was investigated using Cell
Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology)
according to the manufacturer's protocol. All types of transfected
cells treated with or without LPS exposure were cultured in 96-well
plates (4×103 cells/well). Then, 10 μl CCK-8
solution was added to each well and the absorbance was determined
using a microplate reader (Bio-Rad Laboratories, Inc.) at a
wavelength of 450 nm.
ELISA
The levels of IL-1β, IL-18, IL-6 and TNF-α in blood
serum and HK-2 cell supernatant were determined using commercially
available ELISA kits (cat. nos. H002, H007, H015 and H052; Nanjing
Jiancheng Bioengineering Institute). HK-2 cell culture supernatant
was collected by a 5-min centrifugation at 10,000 × g at 4°C. In
all cases, the ELISA was performed according to the manufacturer's
protocols. In brief, the ELISA plates were coated with capture
antibodies (100 μl/well). The serum or supernatant
(serum/supernatant: 1:4) was incubated in the ELISA plate
supplemented with the prepared standard liquid. The horseradish
peroxidase-labeled antibodies were added into each well and
maintained at 37°C for 1 h. After the reaction was terminated by
phosphoric acid, optical density was determined by a microplate
reader at 450 nm (Biotek, Synergy HT).
Cell apoptosis
For apoptosis analysis, HK-2 cells from each
experimental group were stained with Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) using
Annexin-V-FITC cell apoptosis assay kit (Sigma-Aldrich; Merck KGaA)
in the dark at 25°C for 15 min. Then, apoptosis rate was analyzed
under the FACSCalibur flow cytometer (BD Biosciences) equipped with
Cell Quest software (version 3.3, BD Biosciences). The proportions
of early and late apoptotic cells were counted and compared.
Analysis of reactive oxygen species
(ROS)
After 24 h of exposure to LPS, the ROS levels of
each type of transfected HK-2 cells was assessed by the
2′,7′-dichlorofluorescein diace-tate (DCFDA) Cellular Reactive
Oxygen Species Detection Assay kit (Abcam). In brief, DCFDA was
dissolved in PBS at a final concentration of 20 μl/ml. The
cells were cultured with DCFDA solution for 30 min at 37°C. After
being washed with PBS, the levels of ROS were analyzed by flow
cytometry (BD Biosciences).
RNA extraction and real time-quantitative
PCR (RT-qPCR)
Total RNA was extracted from the collected kidney
tissues and transfected HK-2 cells using a high-purity total RNA
Rapid Extraction kit (BioTeke Corporation). The isolated RNAs (1
μg) were mixed with nuclease-free water for cDNA synthesis
using a Script cDNA Synthesis kit (Bio-Rad Laboratories, Inc.) at
37°C for 15 min and at 85°C for 5 sec. RT-qPCR was performed using
LightCycler technology (Roche Diagnostics GmbH) with FastStart DNA
MasterPLUS SYBR-Green I (Roche Diagnostics GmbH) detection. The
qPCR reaction volume was 20 μl and contained template cDNA,
250 nM of each primer, and 4 μl of 5X SYBR-Green Master Mix.
The PCR was performed by 50 cycles at 95°C for 10 sec, at 95°C for
10 sec, at 60°C for 20 sec and at 72°C for 30 sec. Relative gene
expression was normalized to GAPDH and calculated using the
2-ΔΔCq method (21).
All primers are listed in Table
I.
 | Table IPrimers for reverse
transcription-quantitative PCR. |
Table I
Primers for reverse
transcription-quantitative PCR.
| Gene name | Species | Primer
sequences |
|---|
| NLRP3 | Mouse | Forward:
5′-AGCTTCAGGTGTTGGAATTAGACA-3′ |
| Reverse:
5′-GCAGCAAACTGGAAAGGAAG-3′ |
| Human | Forward:
5′-CTTCTCTGATGAGGCCCAAG-3′ |
| Reverse:
5′-CGCCACAAAGATGGTCAC-3′ |
| CD39 | Mouse | Forward:
5′-AGCTGCCCCTTATGGAAGAT-3′ |
| Reverse:
5′-TCAGTCCCACAGCAATCAAA-3′ |
| Human | Forward:
5′-AGCAGCTGAAATATGCTGGC-3′ |
| Reverse:
5′-GAGACAGTATCTGCCGAAGTCC-3′ |
| GAPDH | Mouse | Forward:
5′-GAACATCATCCCTGCATCCA-3′ |
| Reverse:
5′-CCAGTGAGCTTCCCGTTCA-3′ |
| Human | Forward:
5′-CGGAGTCAACGGATTTGGTCGTAT-3′ |
| Reverse:
5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ |
Western blot assay
Following treatment, the protein expression in
kidney tissues and HK-2 cells was detected by a western blot assay.
Total protein was isolated using radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology), following the
protocols of the manufacturer. Protein concentrations were
quantified using a bicinchoninic protein assay kit (Beyotime
Institute of Biotechnology). The lysates (20 μg/lane) were
separated on 10% sodium dodecyl sulfate-polyacrylamide gels and
transferred onto polyvi-nylidene fluoride membranes. Blots were
blocked with non-fat milk (5%) at room temperature for 2 h and then
incubated with various primary antibodies at 4°C overnight. The
membranes were then washed and cultured with secondary antibodies
(1:2,000; cat. nos. ab205718 and ab205719; Abcam) for 2 h at room
temperature. The blots were detected using enhanced
chemiluminescence-plus reagents (GE Healthcare Life Sciences). The
relative proteins were normalized to GAPDH. The primary antibodies
used in this paper were NLRP3 (1:100; cat. no. ab214185; Abcam),
cleaved Caspase-1 (1:1,000; cat. no. ab10836; Abcam) and CD39
(1:1,000; cat. no. ab108248; Abcam).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6 (GraphPad Software, Inc.). Data were expressed as mean ±
standard error of mean. A Student's t-test was used for analyzing
the difference between two groups. One-way analysis of variance
followed by Dunnett's t-test was used to calculate the statistical
significance between multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Injection of LPS induces kidney function
and pathological changes in mice
To determine kidney function and pathological
changes after LPS exposure, kidney tissue sections were subjected
to H&E staining (Fig. 1A). In
the control group, the kidney tissues had clear structures without
degeneration, atrophy, swelling or necrosis of the renal tubular
epithelial cells or inflammatory infiltration. In the LPS group,
the kidney tissues showed obvious kidney injury at 12 h. A total of
24 h after LPS administration, kidney pathological changes were
more significant and were characterized by vacuolar degeneration,
luminal narrowing, loss of the brush border, tubule dilation and
infiltration of interstitial inflammatory cells. After 48 h, the
histopathology of the kidney was improved, however, inflammatory
cell infiltration was still observed in the renal interstitium. In
addition, as shown in Fig. 1B and
C, the Cr and BUN levels in mice were significantly increased
within 24 h after LPS administration (P<0.01), while both Cr and
BUN levels decreased at 48 h, indicating that the levels of Cr and
BUN were positively correlated with kidney pathological
severity.
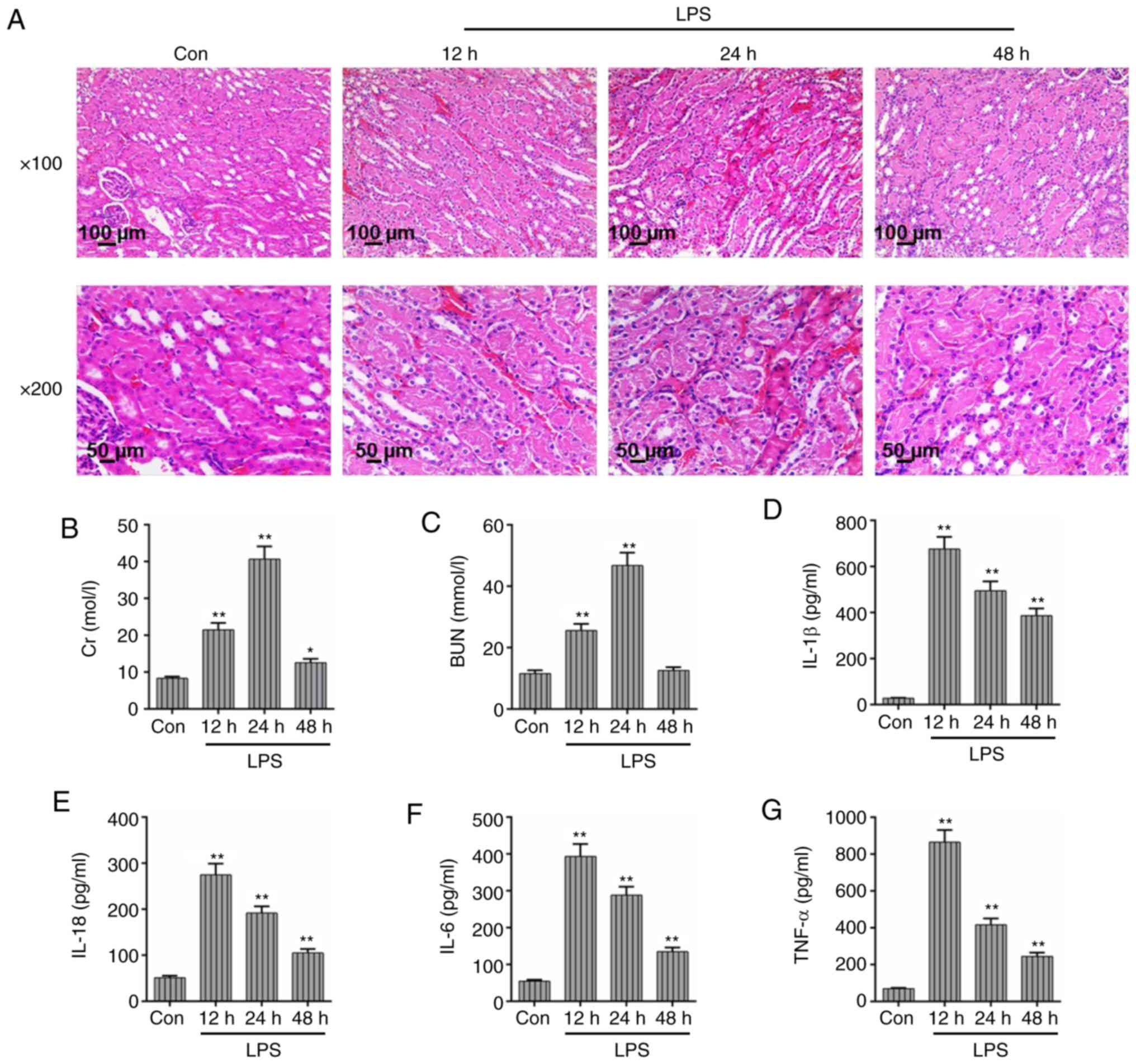 | Figure 1Injection of LPS induces kidney
function and pathological changes in mice. In order to determine
kidney function and pathological changes after LPS exposure, kidney
tissue sections were subjected to H&E staining and
histopathological observation. (A) After LPS treatment, the degree
of kidney injury and pathological changes were observed in mice at
different time points (12, 24 and 48 h). (Magnification: ×100 and
×200; Scale bar, 100 and 50 μm). The levels of (B) serum Cr
and (C) BUN in mice analyzed at different time points by the fully
automatic biochemical analyzer. The levels of (D) IL-1β, (E) IL-18,
(F) IL-6 and (G) TNF-α were assessed through the corresponding
ELISA kits. Each value represents the mean ± standard error of the
mean (n=3). *P<0.05 and **P<0.01 vs.
Con group. IL, interleukin; BUN, blood urea nitrogen; TNF-α, tumor
necrosis factor-α; Cr, creatinine; LPS, lipopolysaccharide;
H&E, hematoxylin and eosin; Con, control. |
Moreover, the levels of IL-1β, IL-18, IL-6 and TNF-α
peaked at 12 h and then reduced gradually as the time of LPS
administration increased (Fig.
1D-G). However, the levels of these inflammatory cytokines were
still much increased compared with the control group. The
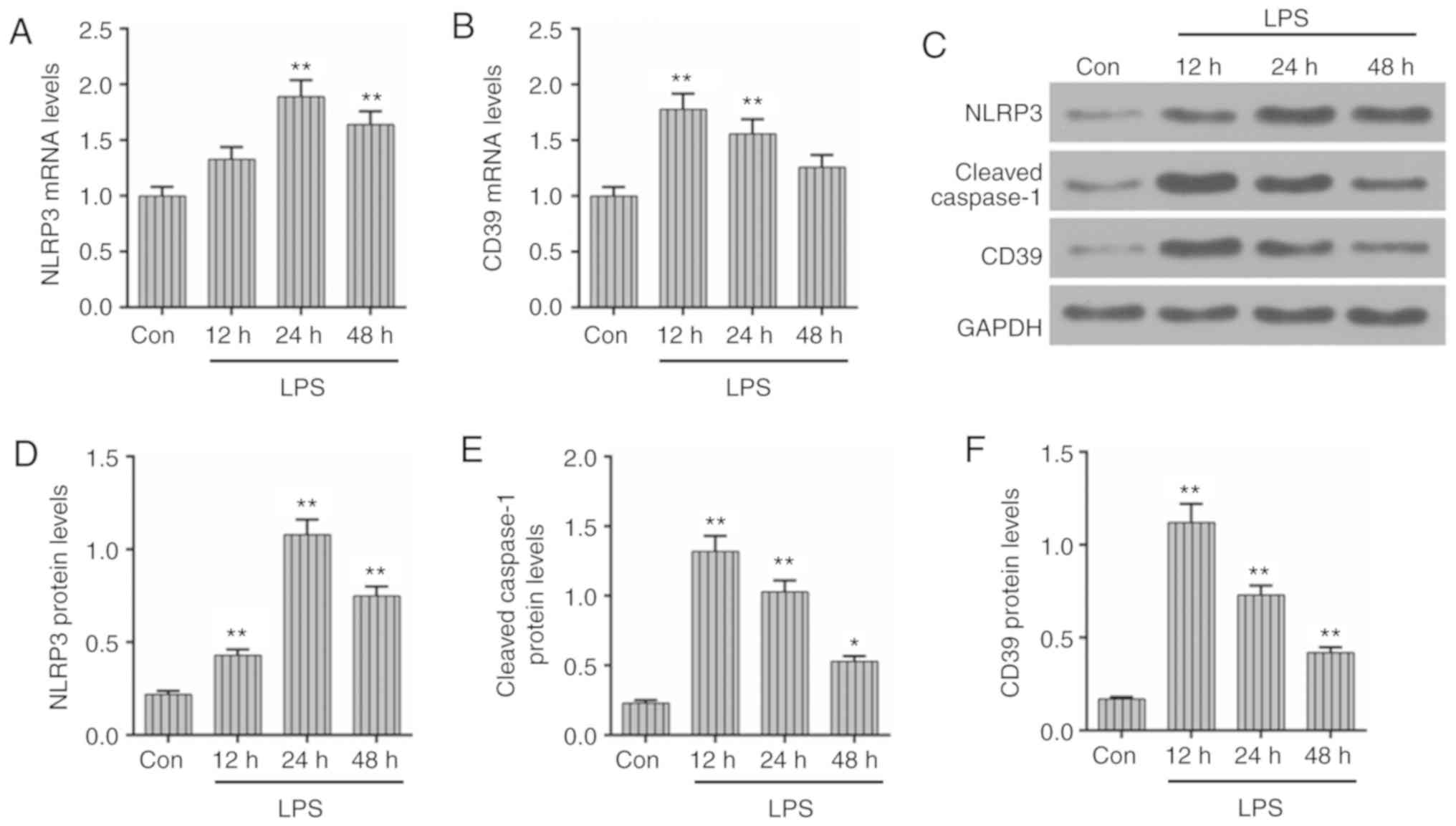
expression of CD39, inflammasome NLRP3 and cleaved caspase-1 were
also measured by RT-qPCR and western blotting. As presented in
Fig. 2A, C and D, both NLRP3 mRNA
and protein levels increased significantly at 24 h after LPS
injection (P<0.01) but reduced at 48 h. The expression of CD39
peaked at 12 h and then continuously reduced at 24 and 48 h
(Fig. 2B, C and F). The changes
in protein level of cleaved caspase-1 were consistent with the
expression of CD39 (Fig. 2C and
E). Taken together, the kidney injury and inflammation were the
most serious at 48 h of LPS administration.
Concentration of LPS shows a positive
association with the expression of inflammatory mediators in HK-2
cells
To determine an optimal LPS concentration for
subsequent experiment, HK-2 cells were treated with a series of
concentrations (0, 0.1, 1, 10 and 100 μg/ml) of LPS. As
shown in Fig. 3A, the HK-2 cell
viability was gradually decreased as the concentration of LPS
increased and reduced significantly when the cells were treated
with 10 μg/ml LPS (P<0.01). As listed in Fig. 3B-E, the inflammatory cytokines
(IL-1β, IL-18, IL-6 and TNF-α) showed a positive association with
the concentration of LPS. The treatment of LPS (1 μg/ml) was
enough to cause a significant inflammation in HK-2 cells
(P<0.01). Furthermore, the expression of NLRP3 and cleaved
caspase-1 was increased under the treatment of 10 μg/ml LPS
(Fig. 3F and H-J). Meanwhile, the
concentration of 0.01 μg/ml LPS slightly affected the
expression of CD39. However, 1 μg/ml LPS could induce an
increase of the CD39 expression levels and both its mRNA and
protein levels, which then decreased as the concentrations of LPS
increased (Fig. 3G, H and K).
Together, the concentration of LPS was positively correlated with
the level of inflammation, while the expression of CD39 increased
slightly under a low concentration of LPS but gradually decreased
as the LPS concentration increased.
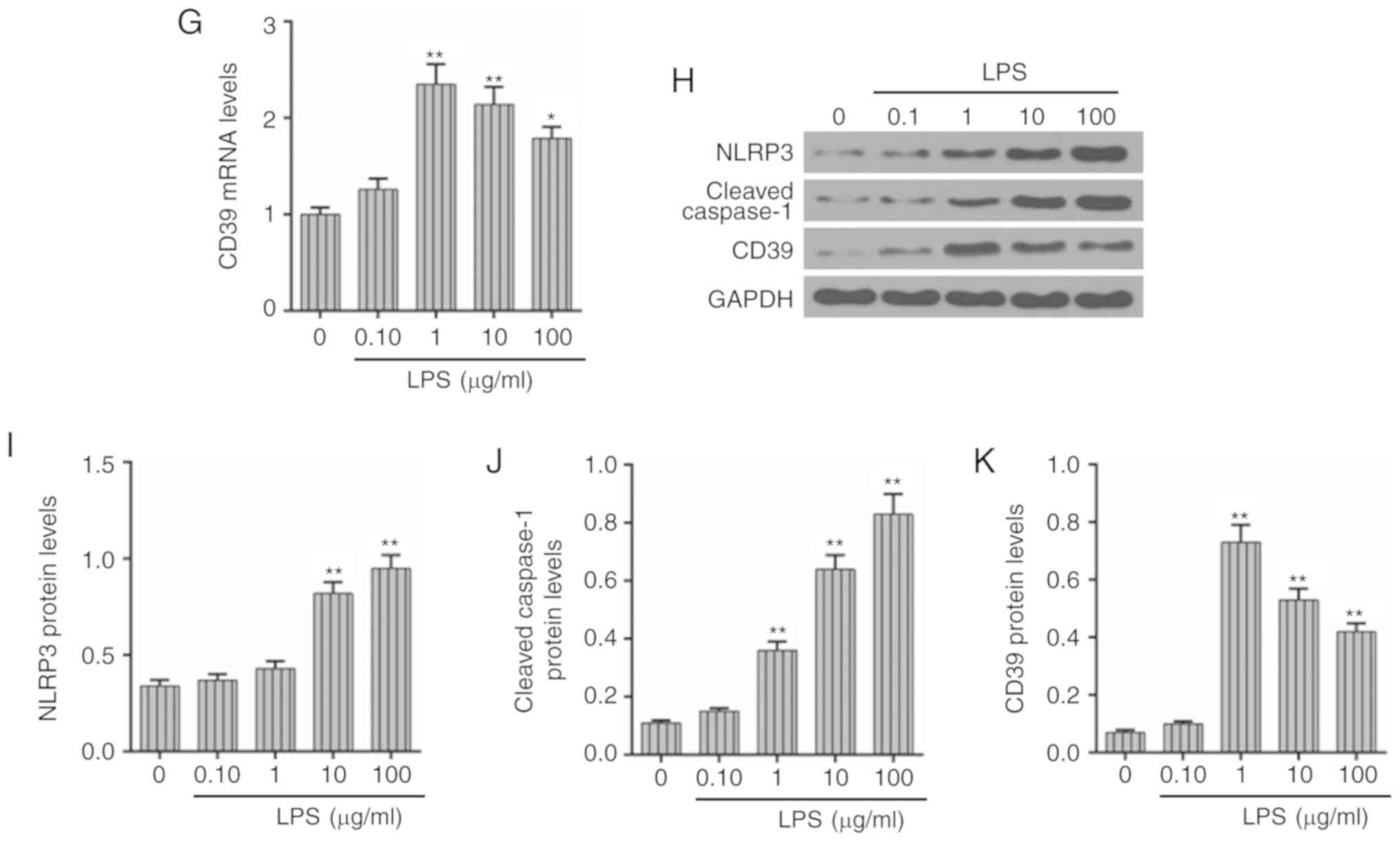 | Figure 3Concentration of LPS shows a positive
association with the expression of inflammatory mediators in HK-2
cells. To study the effects of LPS treatment at a series of
concentrations (0, 0.1, 1, 10 and 100 μg/ml) on HK-2 cells,
the cell viability, inflammation-associated factors and CD39
expression were measured after LPS treatment. (A) The cell
viability was examined using a Cell Counting Kit-8 assay. The
levels of (B) IL-1β, (C) IL-18, (D) IL-6 and (E) TNF-α were
measured under the different concentrations of LPS by corresponding
ELISA kits. The mRNA levels of (F) NLRP3 and (G) CD39 were
determined by RT-qPCR. (H) Western blot assay was used to assess
the protein levels of (I) NLRP3, (J) cleaved caspase-1 and (K)
CD39. Each value represents mean ± standard error of the mean
(n=3). GAPDH served as an internal control. *P<0.05
and **P<0.01 vs. Con group. RT-qPCR, reverse
transcription-quantitative PCR; NLRP3, NLR family pyrin domain
containing 3; IL, interleukin; TNF-α, tumor necrosis factor-α; LPS,
lipopolysaccharide; CD, cluster of differentiation; OD, optical
density; Con, control. |
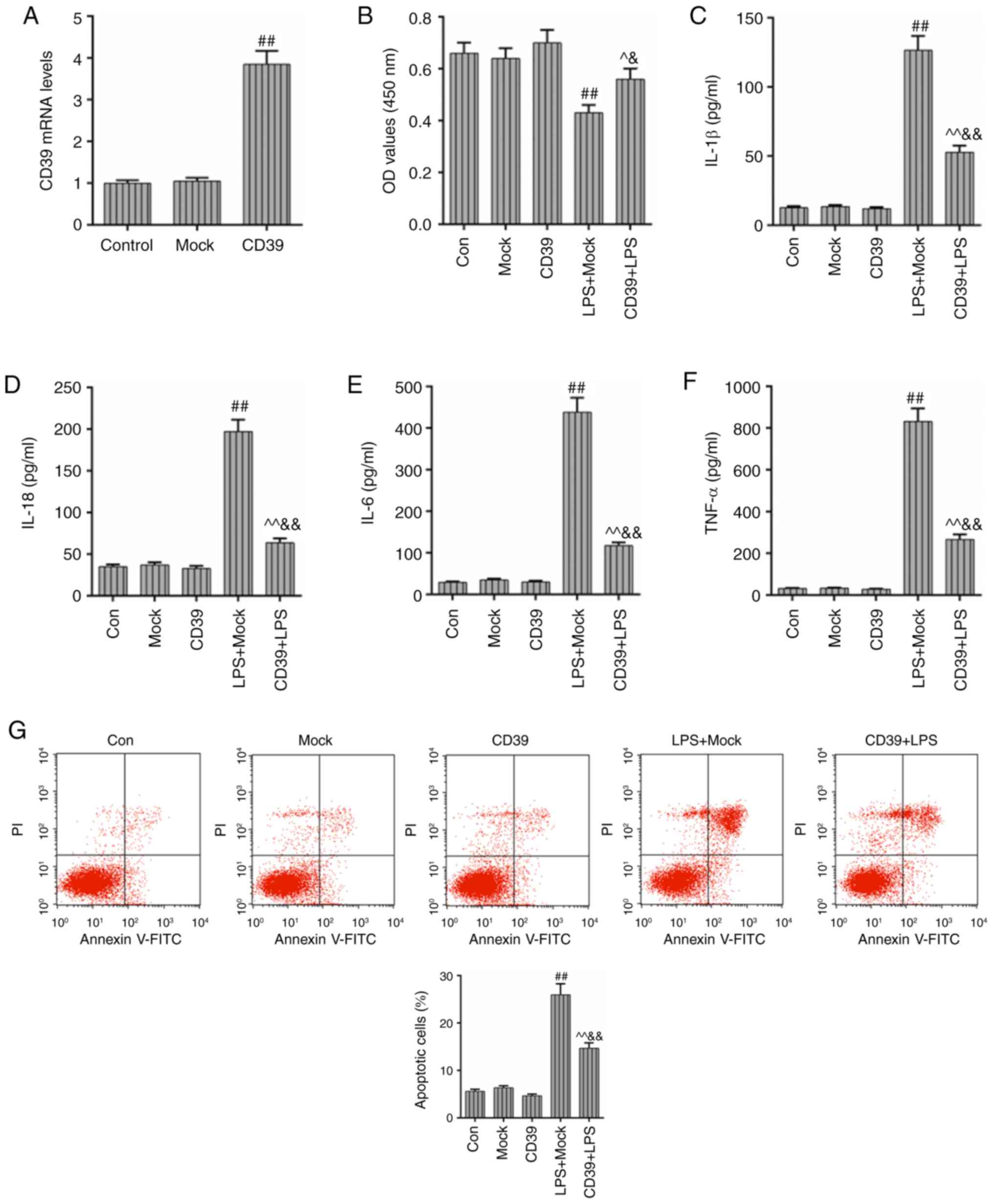
Overexpression of CD39 could mitigate
LPS-induced inflam- mation and apoptosis in HK-2 cells
In order to study the functional effects of CD39 on
the LPS-induced inflammation and apoptosis in HK-2 cells, the CD39
overexpression vector was constructed and transfected into HK-2
cells. As Fig. 4A shows, the CD39
vector was expressed stably in HK-2 cells. The CCK-8 assay showed
that the cell viability in LPS + Mock group was significantly
reduced in comparison with the Mock group, while the transfection
of CD39 could enhance the HK-2 cell viability (P<0.01; Fig. 4B). In addition, the treatment of
LPS could induce strong inflammation, as the levels of IL-1β,
IL-18, IL-6 and TNF-α were significantly increased in the LPS
group, however, increasing CD39 expression could effectively
inhibit the levels of these inflammatory cytokines (P<0.01;
Fig. 4C-F). The apoptosis and ROS
level was also measured by flow cytometry. As shown in Fig. 4G and H, the apoptosis rate and ROS
level significantly increased following LPS administration
(P<0.01), while transfection of CD39 had the ability to decrease
the apoptosis rate and ROS level. Therefore, the results of the
present study indicated that elevated CD39 may contribute to the
mitigation of the LPS-induced injury, inflammation and ROS
accumulation in HK-2 cells.
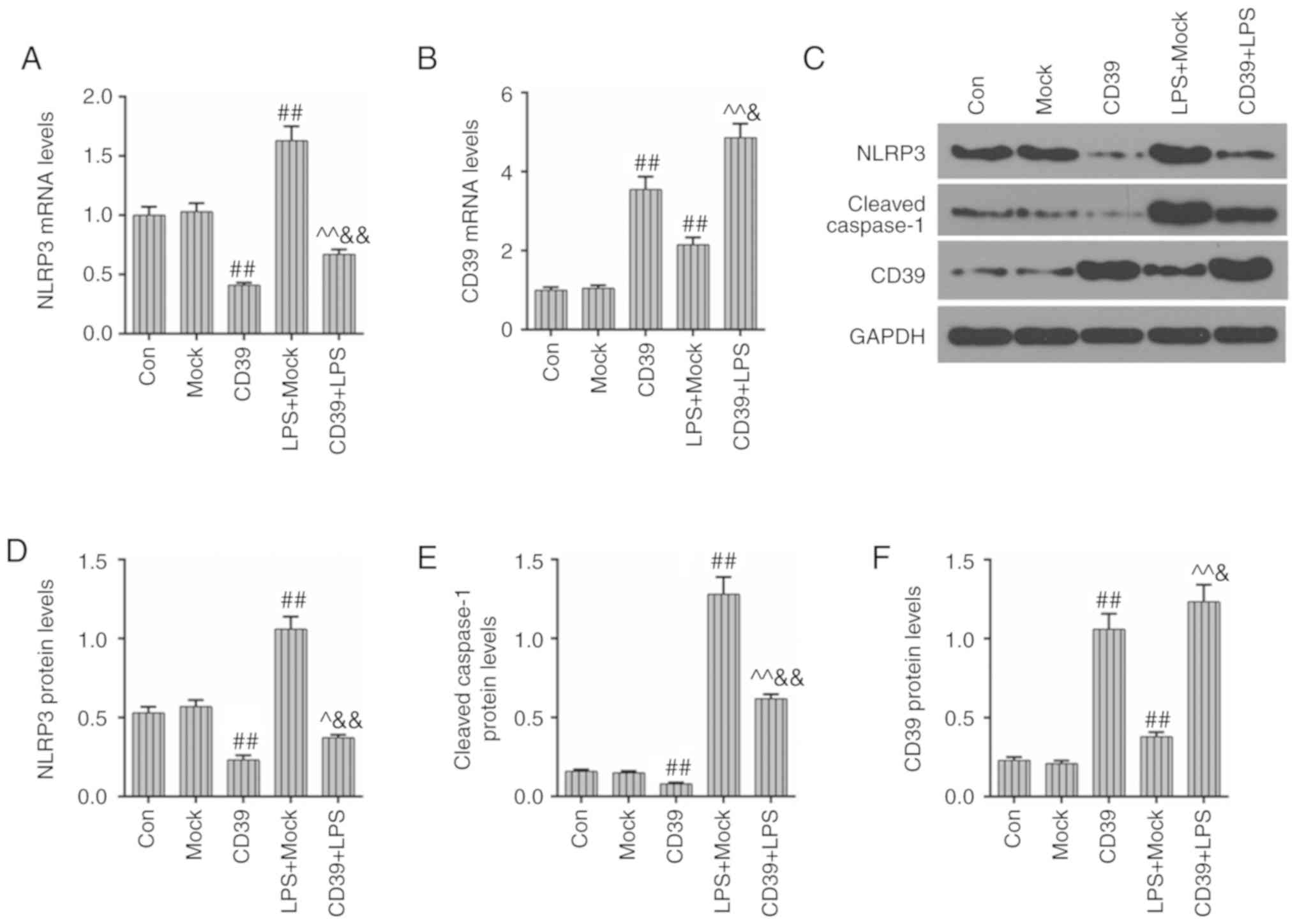
Overexpressed CD39 could inhibit the
activation of the inflammasome in HK-2 cells
As mentioned earlier, the overexpression of CD39
could significantly inhibit the LPS-induced inflammation in HK-2
cells. After transfection of CD39, the expression of NLRP3, which
works as an inflammasome and whose activation serves an essential
role in increasing inflammatory cytokines, was measured. As
indicated in Fig. 5A, the
expression of NLRP3 was increased under LPS administration, while
the transfection of CD39 overexpression could significantly inhibit
LPS-induced increased NLRP3 level. The transfection of CD39 induced
a significant upregulation of CD39 mRNA, compared with LPS + Mock
group (P<0.01; Fig. 5B). The
changes of their protein levels are shown in Fig. 5C-F. The protein levels of NLRP3
and CD39 were basically consistent with their mRNA levels. In
comparison to the LPS + Mock group, the protein level of cleaved
caspase-1 was significantly reduced after the transfection with
CD39 overexpression (P<0.01). Therefore, these results suggested
that the inhibitory effects of CD39 overexpression on LPS-induced
inflammation may rely on suppressing the activation of NLRP3.
CD39 inhibition aggravates LPS-induced
inflammation and cell apoptosis in HK-2 cells
In order to further verify the protective effects of
CD39 on the HK-2 cells under the treatment of LPS, the siCD39
vector was transfected into HK-2 cells and the levels of
inflammation and apoptosis rate were detected. Fig. 6A demonstrated that the expression
of CD39 was effectively inhibited by siRNA in HK-2 cells. LPS
treatment could induce the reduced cell viability, while the
transfection of siCD39 could further decrease cell viability,
compared with the LPS + NC group (P<0.05; Fig. 6B). The levels of those
inflammatory cytokines (IL-1β, IL-18, IL-6 and TNF-α) are shown in
Fig. 6C-F, and it was observed
that the transfection of siCD39 aggravated the inflammation caused
by LPS treatment. The levels of IL-1β, IL-18 and IL-6 in LPS +
siCD39 group were significantly increased compared with those in
LPS + NC group (P<0.05), while the TNF-α level was increased
slightly. In addition, compared with cells transfected with the NC
vector, the apoptosis rate and ROS level both had a slight increase
in the cells transfected with siCD39 after LPS treatment (Fig. 6G and H). Collectively, the results
of the present study indicated that in contrast to CD39
overexpression, the transfection of siCD39 could aggravate
LPS-induced inflammation, cell apoptosis and ROS accumulation in
HK-2 cells.
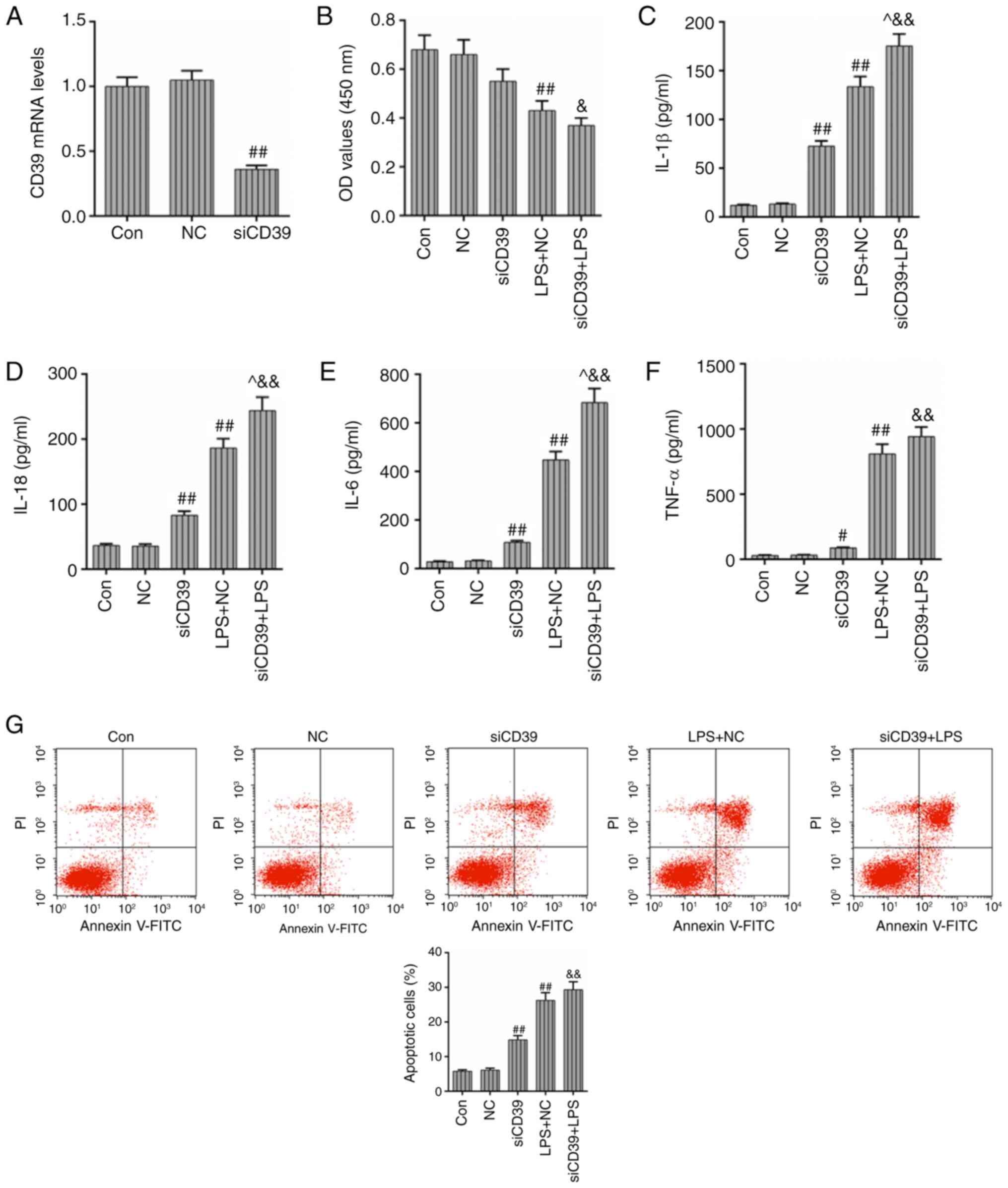 | Figure 6Transfection of siCD39 aggravates
LPS-induced inflammation and cell apoptosis in HK-2 cells. The
siCD39 vector was transfected into HK-2 cells and then, the levels
of inflammation and apoptosis rate were determined. (A) The
transfection efficiency of the siCD39 vector was assessed by
RT-qPCR. (B) The effects of siCD39 transfection on the HK-2 cell
viability were assessed by Cell Counting Kit-8 assay after the LPS
treatment. The effects of siCD39 on the levels of inflammatory
cytokines (C) IL-1β, (D) IL-18, (E) IL-6 and (F) TNF-α were
detected by the corresponding ELISA kits. (G) The effects of CD39
inhibition on the apoptosis rate were assessed by flow cytometry.
(H) The effects of CD39 inhibition on the ROS level were assessed
by flow cytometry. Each value represents the mean ± standard error
of the mean (n=3). GAPDH served as an internal control.
#P<0.05 and ##P<0.01 vs. NC group;
^P<0.05 vs. LPS + NC group; &P<0.05 and
&&P<0.01 vs. siCD39 group. ROS, reactive
oxygen species; CD39 siRNA, cluster of differentiation small
interfering RNA; IL, interleukin; TNF-α, tumor necrosis factor-α;
RT-qPCR, reverse transcription-quantitative PCR; LPS,
lipopolysaccharide; NC, negative control; CD, cluster of
differentiation. |
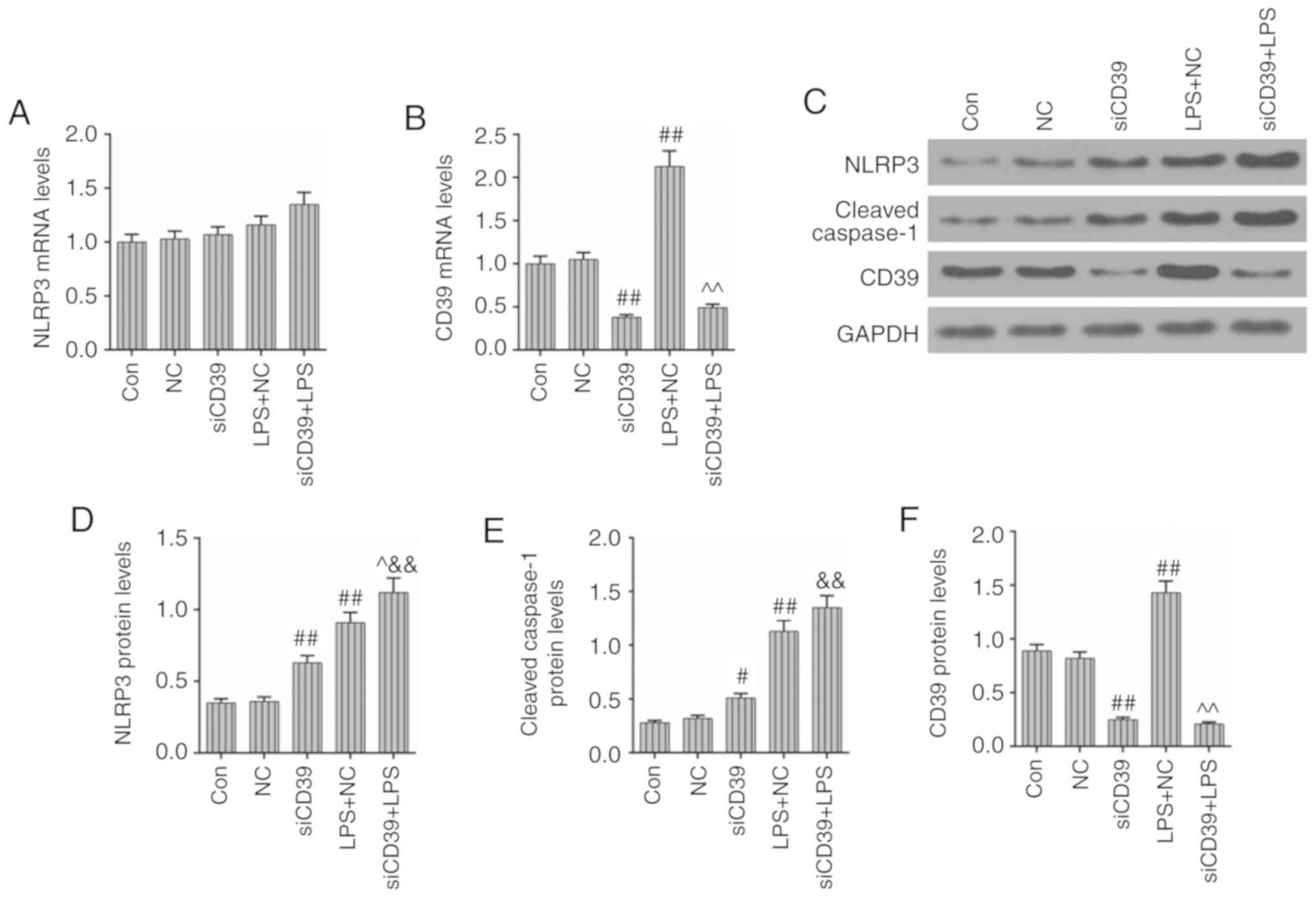
CD39 inhibition has a promoting effect on
the activation of the inflammasome in HK-2 cells
After transfection with siCD39, the NLRP3 mRNA
levels were increased again, compared with the LPS + NC group
(Fig. 7A). In the meantime, the
transfection of siCD39 obviously decreased the expression of CD39
and further weakened the stress reaction of CD39 to LPS treatment
(P<0.01; Fig. 7B). The protein
levels of NLRP3 and CD39 were basically consistent with their mRNA
levels, and the cleaved caspase-1 protein level had a slight
upregulation under the effects of CD39 inhibition and LPS treatment
(Fig. 7C-F). The results of the
present study suggested that the inhibition of CD39 may promote the
activation of NLRP3 and subsequent inflammation in HK-2 cells.
Discussion
Results obtained in this study revealed a protective
role for CD39 in LPS-induced inflammation and injury in HK-2 cells.
LPS stimulation could cause the activation of the inflammatory
response, which is an integral part of innate immunity. However, an
excessive inflammatory response can lead to severe tissue injury,
acute organ failure or chronic inflammatory conditions (22,23). The previous study has demonstrated
that the mechanism of CD39 in macrophages regulating adaptive
immunity involves the control of NLRP3 inflammasome activation
(24). This study investigated
changes of CD39 expression level in the murine kidney after LPS
intraperitoneal injection and found that CD39 had a stressed
upregulation at the beginning of LPS administration and then
decreased as the inflammation severity became greater, meanwhile,
the expression of NLRP3 was increased. In order to further study
the potential relationship between NLRP3 and CD39, the levels of
NLRP3 and inflammation-associated factors under the effects of CD39
overexpression and inhibition were detected in vitro. The
results of the present study indicated that overexpressed CD39
could negatively regulate the activation of NLRP3 and its
downstream signaling, therefore decreasing cell apoptosis and ROS
accumulation in HK-2 cells.
The NLRP3 inflammasomemediated inflammatory system
has been demonstrated to participate in a number of regulatory
mechanisms such as the secretion of pro-inflammatory cytokines,
pyroptosis, ROS accumulation and mitochondria damage and autophagy
(25,26). The overactive NLRP3 inflammasome
was reported to be closely associated with multiple inflammatory
diseases (27,28) including sepsis (29). A previous study showed that
upregulated CD39 during abdominal sepsis could inhibit the NLRP3
activation and diminish inflammation, ultimately improving the
survival of septic mice (30). In
the present study, the transfection of CD39 overexpression also
could inhibit the expression and activity of NLRP3, and
subsequently improve survival of HK-2 cells. In this study, it was
observed that the elevated CD39 could inhibit the cell apoptosis
and ROS accumulation in HK-2 cells. When cells were stimulated by
extracellular medium, the damaged mitochondria released
mitochondrial (mt)ROS into the cytoplasm, which then induced the
activation of NLRP3, resulting in the secretion of cleaved
caspase-1 and several pro-inflammatory cytokines (31). A recent study demonstrated that
mtROS was upstream from NLRP3 and that the treatment with an mtROS
inhibitor could effectively induce a reduction of NLRP3 expression
(32). Similarly, Celastrol was
reported to inhibit the activation of the NLRP3 inflammasome and
ameliorate inflammation by reducing ROS production (33). Combined with these studies, the
present study suggested that CD39 overexpression suppressing the
expression and activity of NLRP3 may also partially rely on the
inhibition of ROS accumulation.
Intracellular ATP works as the molecular unit of
currency of energy transfer (34), however, cell injury could induce
the secretion of ATP into the extracellular space where the
extracellular ATP (eATP) acts as a 'warning signal'. A study
revealed that eATP could contribute to the activation of the P2X
purinoceptor 7 and initiate inflammation (35). In 2017, Savio et al
(20) indicated that the CD39
protein impeded the NLRP3 activation and subsequent
pro-inflammatory cytokines through scavenging eATP, ultimately
inhibiting systemic inflammation and contributing to liver
homeostasis restoration. In general, the level of eATP was under
the careful control of CD39 (36), however, the present study
indicated that the functional effects of CD39 were gradually
decreased as the inflammation severity increased. The downregulated
CD39 may have less control over eATP level, which could cause a
vicious circle as the inflammation was aggravated, therefore
promoting cell injury. Moreover, a previous study demonstrated that
CD39 could effectively improve survival of sepsis via decreasing
systemic inflammation (30).
Compared with that, the results of the present study provided more
details about the underlying mechanisms of the positive effects of
CD39 on protecting the kidney from LPS-induced septic organ injury.
In the septic HK-2 cell injury induced by LPS, NLRP-3-related
inflammatory pathway was over-activated. Additionally CD39
overexpression/inhibition could affect the NLRP3-related
inflammatory pathway, cell apoptosis and cell cycle arrest in HK-2
cells. The present study revealed a high association between the
anti-septic ability of CD39 and the repression of the NLRP3-related
inflammatory pathway in septic AKI. Taken together, the present
study indicated that overexpressed CD39 could diminish the
expression and activity of NLRP3 and subsequent secretion of
pro-inflammatory cytokines, however, whether the protective ability
of CD39 involves the control of eATP remains to be further
investigated.
In conclusion, the present study indicated that CD39
had a promoting effect on the ability of renal tubular epithelial
cells to resist LPS-induced damage. The enhanced CD39 improved cell
viability and apoptosis, diminished the activation of NLRP3 and
mediated inflammatory system through impeding the overproduction of
ROS. In addition, considering the close relationship between CD39
and eATP, the authors also speculated that the protective ability
of CD39 in renal tubular epithelial cells was also mediated through
the control of eATP, however, further experiments are needed to
validate the hypothesis. Therefore, CD39 might be a potential
therapeutic target in sepsis-induced AKI.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
Substantial contributions to conception and design:
MY. Data acquisition, data analysis and data interpretation: ZK,
YW, LL and TM. Drafting the article or critically revising it for
important intellectual content: MY. Final approval of the version
to be published: All authors. Agreement to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of the work are appropriately investigated
and resolved: All authors.
Ethics approval and consent to
participate
Animal experimental procedures were approved by the
Ethics Committee of Committee of Shanxi Dayi Hospital (Taiyuan,
China) and conducted in accordance with Guide for the Care and Use
of Laboratory Animal.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of
interest.
References
|
1
|
Angus DC and van der Poll T: Severe sepsis
and septic shock. N Engl J Med. 369:20632013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aziz M, Jacob A, Yang WL, Matsuda A and
Wang P: Current trends in inflammatory and immunomodulatory
mediators in sepsis. J Leukoc Biol. 93:329–342. 2013. View Article : Google Scholar :
|
|
3
|
Hoste EA, Bagshaw SM, Bellomo R, Cely CM,
Colman R, Cruz DN, Edipidis K, Forni LG, Gomersall CD, Govil D, et
al: Epidemiology of acute kidney injury in critically ill patients:
The multinational AKI-EPI study. Intensive Care Med. 41:1411–1423.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khwaja A: KDIGO clinical practice
guidelines for acute kidney injury. Nephron Clin Pract. 120:pp.
c179–c184. 2012, PubMed/NCBI
|
|
5
|
Alobaidi R, Basu RK, Goldstein SL and
Bagshaw SM: Sepsis-associated acute kidney injury. Semin Nephrol.
35:2–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cruz DN, Bolgan I, Perazella MA, Bonello
M, de Cal M, Corradi V, Polanco N, Ocampo C, Nalesso F, Piccinni P,
et al: North east italian prospective hospital renal outcome survey
on acute kidney injury (NEiPHROS-AKI): Targeting the problem with
the RIFLE criteria. Clin J Am Soc Nephrol. 2:418–425. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mayeux PR and MacMillan-Crow LA:
Pharmacological targets in the renal peritubular microenvironment:
Implications for therapy for sepsis-induced acute kidney injury.
Pharmacol Ther. 134:139–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Langenberg C, Bellomo R, May C, Wan L, Egi
M and Morgera S: Renal blood flow in sepsis. Crit Care.
9:R363–R374. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schrier RW and Wang W: Acute renal failure
and sepsis. N Engl J Med. 351:159–169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takasu O, Gaut JP, Watanabe E, To K,
Fagley RE, Sato B, Jarman S, Efimov IR, Janks DL, Srivastava A, et
al: Mechanisms of cardiac and renal dysfunction in patients dying
of sepsis. Am J Respir Crit Care Med. 187:509–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong W, Li Z, Chen Y, Zhang L, Ye Z, Liang
H, Li R, Xu L, Zhang B, Liu S, et al: Necrostatin-1 attenuates
sepsis-associated acute kidney injury by promoting autophagosome
elimination in renal tubular epithelial cells. Mol Med Rep.
17:3194–3199. 2018.
|
|
12
|
De Backer D, Creteur J, Preiser JC, Dubois
MJ and Vincent JL: Microvascular blood flow is altered in patients
with sepsis. Am J Respir Crit Care Med. 166:98–104. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cen C, Aziz M, Yang WL, Zhou M, Nicastro
JM, Coppa GF and Wang P: Milk fat globule-epidermal growth
factor-factor VIII attenuates sepsis-induced acute kidney injury. J
Surg Res. 213:281–289. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mir SM, Ravuri HG, Pradhan RK, Narra S,
Kumar JM, Kuncha M, Kanjilal S and Sistla R: Ferulic acid protects
lipopolysaccha-ride-induced acute kidney injury by suppressing
inflammatory events and upregulating antioxidant defenses in Balb/c
mice. Biomed Pharmacother. 100:304–315. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eltzschig HK, Sitkovsky MV and Robson SC:
Purinergic signaling during inflammation. N Engl J Med.
367:2322–2333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan J, Li Y, Yang H, Zhang L, Yang B, Wang
M and Li Q: Interleukin-17A participates in podocyte injury by
inducing IL-1β secretion through ROS-NLRP3 inflammasome-caspase-1
pathway. Scand J Immunol. 87:pp. e126452018, View Article : Google Scholar
|
|
17
|
Yao ST, Cao F, Chen JL, Chen W, Fan RM, Li
G, Zeng YC, Jiao S, Xia XP, Han C and Ran QS: NLRP3 is required for
complement-mediated caspase-1 and IL-1beta activation in ICH. J Mol
Neurosci. 61:385–395. 2017. View Article : Google Scholar
|
|
18
|
Pérez-Cabeza de Vaca R, Dominguez-López M,
Guerrero-Celis N, Rodriguez-Aguilera JR and Chagoya de Sánchez V:
Inflammation is regulated by the adenosine derivative molecule,
IFC-305, during reversion of cirrhosis in a CCl4 rat
model. Int Immunopharmacol. 54:12–23. 2018. View Article : Google Scholar
|
|
19
|
Xu Y, Wang Y, Yan S, Yang Q, Zhou Y, Zeng
X, Liu Z, An X, Toque HA, Dong Z, et al: Regulation of endothelial
intracellular adenosine via adenosine kinase epigenetically
modulates vascular inflammation. Nat Commun. 8:9432017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Savio LEB, de Andrade Mello P, Figliuolo
VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, Silva CLM,
Feldbrügge L, Csizmadia E, Minshall RD, et al: CD39 limits P2X7
receptor inflammatory signaling and attenuates sepsis-induced liver
injury. J Hepatol. 67:716–726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Tabas I and Glass CK: Anti-inflammatory
therapy in chronic disease: Challenges and opportunities. Science.
339:166–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dinarello CA, Simon A and van der Meer JW:
Treating inflammation by blocking interleukin-1 in a broad spectrum
of diseases. Nat Rev Drug Discov. 11:633–652. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mascanfroni ID, Yeste A, Vieira SM, Burns
EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, et al:
IL-27 acts on DCs to suppress the T cell response and autoimmunity
by inducing expression of the immunoregulatory molecule CD39. Nat
Immunol. 14:1054–1063. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016. View Article : Google Scholar :
|
|
26
|
Zhong Z, Sanchez-Lopez E and Karin M:
Autophagy, NLRP3 inflammasome and auto-inflammatory/immune
diseases. Clin Exp Rheumatol. 34(4 Suppl 98): pp. S12–S16. 2016
|
|
27
|
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang
B, He Z, Zeng Y, Hu Y, Sun S, et al: Nitric oxide suppresses NLRP3
inflammasome activation and protects against LPS-induced septic
shock. Cell Res. 23:201–212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et
al: NLRP3 is activated in Alzheimer's disease and contributes to
pathology in APP/PS1 mice. Nature. 493:674–678. 2013. View Article : Google Scholar
|
|
29
|
Stearnskurosawa DJ, Osuchowski MF,
Valentine C, Kurosawa S and Remick DG: The pathogenesis of sepsis.
Annu Rev Pathol. 6:19–48. 2011. View Article : Google Scholar
|
|
30
|
Csóka B, Németh ZH, Törő G, Koscsó B,
Kókai E, Robson SC, Enjyoji K, Rolandelli RH, Erdélyi K, Pacher P
and Haskó G: CD39 improves survival in microbial sepsis by
attenuating systemic inflammation. FASEB J. 29:25–36. 2015.
View Article : Google Scholar :
|
|
31
|
Bhat M, Romagnuolo J, da Silveira E,
Reinhold C, Valois E, Martel M, Barkun JS and Barkun AN: Randomised
clinical trial: MRCP-first vs. ERCP-first approach in patients with
suspected biliary obstruction due to bile duct stones. Aliment
Pharmacol Ther. 38:1045–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li F, Xu M, Wang M, Wang L, Wang H, Zhang
H, Chen Y, Gong J, Zhang JJ, Adcock IM, et al: Roles of
mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced
lung inflammation and emphysema. Respir Res. 19:2302018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu X, Zhao Q, Zhang X and Zhang H, Liu Y,
Wu X, Li M, Li X, Zhang J, Ruan X and Zhang H: Celastrol
ameliorates inflammation through inhibition of NLRP3 inflammasome
activation. Oncotarget. 8:67300–67314. 2017.PubMed/NCBI
|
|
34
|
Knowles JR: Enzyme-catalyzed phosphoryl
transfer reactions. Annu Rev Biochem. 49:877–919. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morandini AC, Savio LE and Coutinho-Silva
R: The role of P2X7 receptor in infectious inflammatory diseases
and the influence of ectonucleotidases. Biomed J. 37:169–177. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Robson SC, Sévigny J and Zimmermann H: The
E-NTPDase family of ectonucleotidases: Structure function
relationships and pathophysiological significance. Purinergic
Signal. 2:409–430. 2006. View Article : Google Scholar
|