Introduction
Endothelial-mesenchymal transition (EndMT) is a
cellular process whereby endothelial cells (ECs) undergo a change
in phenotype toward mesenchymal cells. EndMT results in the
upregulation of mesenchymal markers, including α-smooth muscle
actin (SMA) and vimentin, and the downregulation of EC markers,
including vascular endothelial (VE)-cadherin and cluster of
differentiation 31 (1). EndMT has
been demonstrated to be a key source of mesenchymal cells and an
important process in the development of fibrotic diseases (1-4).
Despite the indication of EndMT as an important process in numerous
animal models of fibrotic diseases, a number of studies have
suggested that EndMT may participate in the pathogenesis of
pulmonary arterial hypertension (PAH) (5-7).
Ranchoux et al (7)
obtained in situ evidence of the presence of EndMT in human
pulmonary arteries from patients with PAH. This study indicated
their findings in a well-established animal model of PAH
(monocrotaline and SuHx). A study by Hopper et al (6) also identified the occurrence of
EndMT in patients with PAH and in animal models. These findings
provided convincing experimental evidence that specific therapeutic
agents could be used to abrogate the process and may improve the
outcome of the patients with PAH.
Hydrogen sulfide (H2S) is a gaseous
mediator and is generated endogenously by cystathionine β-synthase
(EC4.2.1.22) and cystathionine γ-lyase (CSE; EC4.4.1.1) during
cysteine metabolism (8).
Endogenous H2S has previously been identified as a
gasotransmitter that is associated with physiological actions
during cardiovascular regulation, with carbon monoxide and nitric
oxide (9,10). For example, it has been suggested
that the abnormal generation and dysfunction of H2S
serves crucial roles in the development of pulmonary hypertension
(11-14). Treatment with exogenous
H2S has been indicated to promote apoptosis of pulmonary
artery smooth muscle cells (SMCs), inhibit proliferation of
pulmonary artery SMCs and reduce collagen deposition, leading to
improvement in pulmonary vascular remodeling (10-14). However, the link between
H2S and pulmonary vascular EndMT in PAH has not yet been
fully determined. Therefore, the current study aimed to explore the
regulatory role of H2S in EndMT during PAH. Due to the
fact monocrotaline (MCT) causes direct endothelial damage and
vascular remodeling, and can induce the occurrence of severe and
lethal PAH (15), an MCT-induced
rodent model was used in the present study. The results indicated
that endogenous H2S insufficiency accompanies the
process of pulmonary EndMT in MCT-induced PAH. The supplementation
of H2S inhibited the activation of the NF-κB-Snail
pathway in endothelial cells, resulting in the attenuation of EndMT
and PAH. These results revealed that H2S warrants
further study as a novel therapeutic target for EndMT in PAH.
Materials and methods
Animal model
The current study was carried out in accordance with
the recommendations in the Guide for the Care and Use of Laboratory
Animals of the Shanghai Jiaotong University School of Medicine. The
protocol was approved by the Committee on the Ethics of Animal
Experiments of the Shanghai Jiaotong University School of Medicine
[permit no. (2015)-130]. During the current study, 60 male
Sprague-Dawley rats (250±15 g; age, 10 weeks), obtained from the
Central Laboratory of Shanghai Ninth People's Hospital (Shanghai,
China), were kept in sterilized filter top cages with controlled
humidity and a 12-h day/night cycle at 22°C. Standard rat chow and
tap water were provided ad libitum. Rats received a single
intraperitoneal (i.p.) injection of MCT (60 mg/kg; Sigma-Aldrich;
Merck KGaA) to induce PAH. Rats were sacrificed at day 7, 14, 21
and 28 following MCT injection to observe the time-dependent
alterations of EndMT and H2S synthesis. Rats were
randomly administered NaHS (an H2S donor; i.p., 1
mg/kg/day), DL-propagylglycine (an inhibitor of H2S
synthesis; PAG; i.p., 10 mg/kg/day), or saline, 7 days after MCT
injection (16,17). After a period of 14 days, rats
were anesthetized with chloral hydrate (i.p., 400 mg/kg) and their
blood was collected via cardiac puncture. After observation for 10
min, the death of the animals was confirmed by the presence of
cardiac arrest and apnea, which was assessed using a stethoscope.
Subsequently, rat lungs were harvested and stored at -80°C for
subsequent use. Rat hearts were isolated, washed and weighed using
an analytical balance to calculate the weight ratio of the right
ventricle to the left ventricle plus septum [RV/(LV+S)]. At the
beginning and the end of the current study, systolic blood pressure
was monitored using a non-invasive computerized tail cuff system
(Blood Pressure Analysis System BP- 98AW monitor; Softron Co.,
Ltd.).
Cell culture
Human pulmonary artery endothelial cells (HPAECs;
ScienCell Research Laboratories, Inc.) cultured in endothelial cell
medium (ScienCell Research Laboratories, Inc.) containing 5% fetal
bovine serum (FBS; ScienCell Research Laboratories, Inc.), 100
µg/ml streptomycin, 100 U/ml penicillin and 1% endothelial
cell growth factor (ScienCell Research Laboratories, Inc.). Cells
were plated onto six-well plates or 100 mm tissue culture dishes 24
h prior to experimentation. Prior to treatment or stimulation,
near-confluent cultures were starved for ≥12 h in medium containing
0.5% FBS.
Cell treatment
HPAECs at ~80% confluence were rinsed twice with
serum-free culture medium prior to treatment. Cells were
pre-incubated with saline or NaHS (50, 100 or 200 µM) for 2
h. Cells were then incubated with transforming growth factor
(TGF)-β1 (10 ng/ml) for 24 h, in the continuous presence of NaHS,
to investigate the activation of the Snail-nuclear factor (NF)-κB
pathway. In an attempt to evaluate EndMT, HPAECs were subsequently
incubated with TGF-β1 (10 ng/ml) for an additional 9 days.
Measurement of pulmonary artery pressure
and right ventricular hypertrophy
Invasive closed-chest right heart catherization
(RHC) was performed to obtain the pulmonary artery pressure.
Briefly, animals were anesthetized via i.p., injection of 4%
chloral hydrate (360 mg/kg). Following the exposure of the right
external jugular vein, a polyethylene pipe catheter was inserted.
The catheter was then advanced through the superior vena cava,
right atrium and right ventricle, into the pulmonary artery.
Systolic pulmonary artery pressure, diastolic pulmonary artery
pressure and mean pulmonary artery pressure was continuously
monitored using ScisenseADV Pressure-Volume Measurement System and
LabScribe2 software (iWORX), using a pressure sensor, which was
connected to the extracorporal end of the catheter.
Measurement of right heart function using
ultrasound biomicroscopy
Right heart function was measured using an
ultrasound biomicroscopy system (Vevo 770; FUJIFILM VisualSonics,
Inc.) equipped with a 40 MHz transducer. An apical four-chamber
view was used to visualize the tricuspid annular plane systolic
excursion (TAPSE), which is a specific echocardiographic parameter
of RV function. All tests were performed twice and analyzed by a
sonographer who was blinded to the experiment.
Histological examination
After anesthetization, rats were subjected to left
ventricle perfusion with 10% neutralized formalin, at a stable
pressure of 100 mmHg. The right upper lung was subsequently
isolated and processed. Paraffin-embedded lung tissue sections (5
mm) were stained with hematoxylin for 5 min and eosin for 2 min at
room temperature. The histology of the muscular arteries (vessels
of ~150 µm diameter) in the lungs was investigated using a
light microscope with an objective lens magnification of x40 and an
eyepiece magnification of x10 (Nikon Corporation). Remodeling of
pulmonary arteries was quantified by ImageJ 1.47 software (National
Institutes of Health). Lumen area is defined as the area within the
lamina elastica interna. The medial area is defined as the area
between the lamina elastica interna and the lamina elastica
externa. Measurement of media and lumen area of pulmonary arteries
was acquired by tracing the border of the lumen, the internal and
external elastic laminae. A total of 10 arteries were measured per
animal. The average of 10 values obtained was used for
calculations. The ratio of media area (MA) to lumen area (LA) was
calculated.
Transmission electron microscopy
(TEM)
For TEM morphological analysis, freshly excised lung
tissues or cell pellets were fixed for 2 h in 2.5% glutaraldehyde
in 0.1 M sodium cacodylate buffer (pH 7.4) at room temperature.
These were then post-fixed with 1% osmium tetroxide, contrasted
with uranyl acetate 2% in water for 2 h at 4°C, dehydrated through
a graded ethanol series at 4°C and embedded in Epon medium for 4 h
at room temperature. Ultrathin sections (70 nm) were placed onto
200 mesh cooper grids, and counter stained with lead citrate for 10
min at room temperature prior to examination using an electron
microscope, which was operating at 80 kV. Finally, sections were
observed using electron microscopy (HT7700; Hitachi, Ltd.).
Western blot analysis
Lungs were homogenized at 4°C in
radioimmunoprecipitation lysis buffer (Beyotime Institute of
Biotechnology). Cultured cells (3x106) were lysed at 4°C
in radioimmunoprecipitation assay lysis buffer. Tissue homogenates
or cell lysates were extracted using centrifugation at 14,000 x g
for 10 min at 4°C. The protein concentration was assayed using the
Bradford method. Proteins (20 µg) were size-fractionated
using 10% SDS-PAGE and transferred onto nitrocellulose membranes.
The membranes were blocked for 2 h at room temperature with 5%
non-fat milk and then incubated overnight at 4°C with the following
primary antibodies: CSE rabbit monoclonal antibody (cat. no. 19689;
1:1,000; Cell Signaling Technology, Inc.), VE-cadherin rabbit
polyclonal antibody (cat. no. ab33168; 1:1,000; Abcam), α-SMA mouse
monoclonal antibody (cat. no. ab7817; 1:1,000; Abcam), IκBα
antibody (cat. no. 4814; 1:1,000; Cell Signaling Technology, Inc.),
phosphorylated (phospho)-IκBα (Ser32/36) (5A5) mouse monoclonal
antibody (cat. no. 9246; 1:1,000; Cell Signaling Technology, Inc.),
Snail rabbit monoclonal antibody (cat. no. 3879; 1:1,000; Cell
Signaling Technology, Inc.) and GAPDH rabbit monoclonal antibody
(cat. no. ab181602; 1:1,000; Abcam). Membranes were then incubated
with secondary antibodies for 2 h with Dylight™ 800
4XPEG-conjugated anti-rabbit IgG (H+L) (cat. no. 5151; 1:5,000;
Cell Signaling Technology, Inc.) or Dylight™ 8004XPEG-conjugated
anti-mouse IgG (H+L; cat. no. 5257; 1:5,000; Cell Signaling
Technology, Inc.) at room temperature. The results were visualized
using an Odyssey®Clx Imaging Infrared System and
analyzed by ImageJ 1.47 software (National Institutes of
Health).
Immunofluorescent staining
Lung sections were deparaffinized in xylene and
rehydrated in aqueous solutions with a decreasing alcohol series,
followed by washing with PBST (1xPBS with 0.5% Tween 20; pH 7.4).
Antigen retrieval was performed by heating the slides in Tris-EDTA
solution for 20 min at 95°C. Slides were then cooled for 15 min
before washing twice with PBS. Endogenous peroxidase activity and
nonspecific staining were blocked by incubation with 3%
H2O2 for 15 min and 5% goat serum (Beyotime
Institute of Biotechnology) for 1 h, respectively. Slides were
subsequently incubated with VE-cadherin rabbit polyclonal antibody
(cat. no. ab33168; Abcam) and α-SMA mouse monoclonal antibody (cat.
no. ab7817; Abcam) at a 1:100 dilution overnight at 4°C. For the
immunofluorescent staining of HPAECs, cells were rinsed with PBS,
fixed with 4% paraformaldehyde for 20 min at room temperature and
incubated with VE-cadherin rabbit polyclonal antibody (cat. no.
ab33168; Abcam; 1:100) and α-SMA mouse monoclonal antibody (cat.
no. ab7817; Abcam; 1:100) overnight at 4°C. Slides were washed
three times and incubated with Alexa Fluro®594
conjugated anti-rabbit IgG (cat. no. 8889; Cell Signaling
Technology, Inc.) and Alexa Fluro®488 conjugated
anti-mouse IgG (cat. no. 4408; Cell Signaling Technology, Inc.) for
1 h at room temperature, at a 1:1,000 dilution. Nuclei were stained
using 4,6-diamidino-2-phenylindole (DAPI) for 5 min at room
temperature. Stained cells were observed using light microscopy
(Nikon Corporation) and analyzed using NIS-Elements Imaging 4.50
software (Nikon Corporation).
Transcription activity of NF-κB
Nuclear extracts from lungs or HPAECs were obtained
using a nuclear extraction kit (Active Motif, Inc.). Nuclear
protein concentrations were assayed using the Bradford method
(Bio-Rad Laboratories, Inc.). The DNA binding activity of NF-κB p65
in nuclear extracts was determined, in duplicate, using TransAM™
NF-κB p65 Colorimetric kits (Active Motif, Inc.) as according to
the protocol described by the manufacturer. The absorbance was read
on a 96-well microplate reader (Tecan Group, Ltd.) at 450 nm with a
reference wavelength of 655 nm.
CSE cDNA cloning and transient
transfection
The open reading frame of CSE from the C57BL/6J
mouse liver (GenBank™ accession no. NM_145953) was amplified using
PCR and then subcloned into the mammalian expression vector
pcDNA3.1 (Invitrogen; Thermo Fisher Scientific, Inc.) (16). The CSE expression vectors or
control vectors were transiently transfected into HPAECs using
FuGENE HD Transfection reagent (Roche Diagnostics). A period of 48
h after transfection, cells were pretreated with PAG (2 mM) or
saline and then stimulated with TGF-β1 (10 ng/ml) for 24 h. A
number of cells were harvested to investigate the changes in the
NF-κB-Snail pathway. The remaining cells were subsequently
stimulated with TGF-β1 (10 ng/ml) for an additional 9 days to
estimate the process of EndMT.
Measurement of plasma H2S
Reactions containing 220 µl plasma, 60
µl 1% zinc acetate, 40 µl N,
N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl (20 µM)
and 40 µl FeCl3 in 1.2 M HCl (30 µM) were
prepared. After 10 min, reactions were terminated by adding 120
µl 10% trichloroacetic acid, which was used to precipitate
the proteins. The resulting solution was centrifuged at 14,000 x g
for 5 min at 4°C. The absorbance of the final solution was measured
at 670 nm using a 96-well microplate reader (Tecan Group, Ltd.).
The plasma concentration of H2S was calculated against a
calibration curve of NaHS.
H2S synthesizing activity
assay
H2S synthesizing activity in the lungs
was measured as previously described (16). The reaction mixture containing 100
mM potassium phosphate buffer (pH 7.4), 20 mM L-cysteine, 2 mM
pyridoxyal 5'-phosphate and tissue homogenate, was prepared in
tightly sealed tubes on ice. Tubes were subsequently transferred to
a water bath at 37°C to initiate the reactions. After 30 min, 1%
zinc acetate was added to entrap synthesized H2S and 10%
trichloroacetic acid was subsequently added to stop the reaction.
Additionally, 20 µM N,N-dimethyl-p-phenylenediamine sulfate
in 7.2 M HCl was added, followed by 30 µM FeCl3
in 1.2 M HCl. The absor-bance of the final solution was determined
at 670 nm using spectrophotometry (TecanGroup, Ltd.). The
concentration of H2S in each reaction mixture was
calculated against a calibration curve of NaHS. H2S
synthesizing activity in the lungs was expressed as nmol
H2S formed/mg DNA after being adjusted to the DNA
concentration in lung homogenates.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical analysis was performed using SPSS software 19
(IBM Corp.). Significant differences were determined using a
two-tailed Student's t test or using one-way analysis ovariance
with a Tukey's multiple comparison test. P<0.05 was considered
to indicate a statistically significant result. Each experiment was
repeated a minimum of three times.
Results
Time-dependent alterations of EndMT in
MCT-induced PAH
To assess the time-dependent alterations of EndMT in
MCT-induced PAH, pulmonary protein levels of VE-cadherin (an
endothelial marker), α-SMA (a mesenchymal marker) and Snail were
measured, as crucial regulators of EndMT (18,19). As presented in Fig. 1A and B, VE-cadherin expression
time-dependently reduced during the development of PAH, with a
considerable decrease 21 days after MCT injection. In contrast,
α-SMA expression was significantly upregulated at 14 days after MCT
injection (P<0.05) and remained highly expressed throughout the
experiment. Furthermore, the pulmonary expression of Snail was
increased following MCT injection, with a peak 21 days after the
initial MCT injection. The results also indicated that the plasma
concentration of H2S, lung H2S synthesizing
activity and lung CSE expression significantly reduced 14 days
after MCT injection and remained at a decreased level (P<0.05;
Fig. 1C-E). The time course
experiments demonstrated that an insufficiency of endogenous
H2S production in lungs accompanied the process of EndMT
in MCT-induced PAH, with a peak alteration 21 days after MCT
injection.
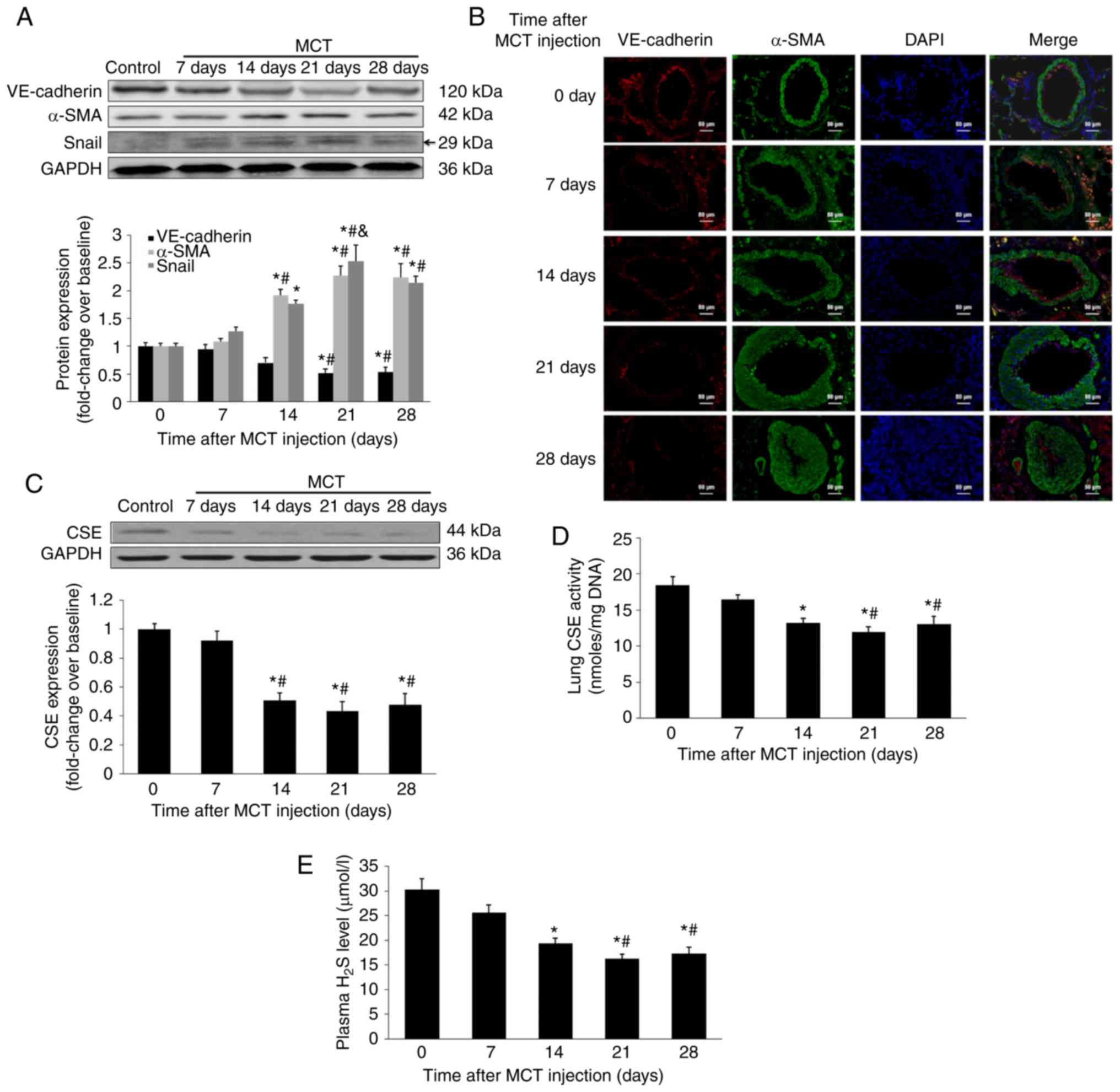 | Figure 1Time dependent alterations in protein
signatures of EndMT and the dynamic characteristics of endogenous
H2S synthesis in MCT-induced PAH. After injection with
MCT, rats were sacrificed at days 0, 7, 14, 21 and 28. Temporal
characteristics of VE-cadherin and α-SMA in lungs were measured
using (A) western blot analysis and (B) immunofluorescent staining.
Red fluorescence represents VE-cadherin, green fluorescence
represents α-SMA and blue fluorescence indicates DAPI nuclei
staining. Expression of (A) Snail and (C) CSE in the lungs was
measured using western blot analysis. (D) Lung CSE enzymatic
activity and (E) plasma H2S level were also measured.
Results are presented as the mean ± standard error of the mean (n=6
animals in each group). *P<0.05 vs. rats at baseline;
#P<0.05 vs. rats 7 days after MCT injection;
&P<0.05 vs. rats 14 days after MCT injection.
EndMT, endothelial-to-mesenchymal transition; H2S,
hydrogen sulfide; MCT, monocrotaline; PAH, pulmonary arterial
hypertension; CSE, cystathionine γ-lyase; SMA, smooth muscle actin;
VE, vascular endothelial; DAPI, 4,6-diamidino-2-phenylindole. |
Effect of H2S on EndMT in
MCT-induced PAH
A period of 7 days after MCT injection, rats
randomly received a daily administration of NaHS (1 mg/kg), PAG (an
inhibitor of H2S synthesis; 10 mg/kg) or saline. A
period of 3 weeks after MCT injection, pulmonary hemodynamic, right
ventricular hypertrophy and pulmonary structural changes were
assessed (Fig. 2). Hemodynamic
and echocardiographic measurements demonstrated that a preventive
application of NaHS significantly reduced PAH responses (pulmonary
arterial systolic and mean pressures), right ventricular
hypertrophy (RV/LV+S) and right ventricular systolic function,
while inhibition of H2S formation by PAG aggravated them
(Fig. 2A-D). Furthermore, NaHS
significantly alleviated pulmonary vascular structural remodeling,
as characterized by a reduced thickness of the medial layer of
muscular arteries (vessels of ~150 µm diameter) and
decreased narrowness in muscular arteries, whereas PAG aggravated
the severity of PAH (P<0.05; Fig.
2E and 2F). Additionally, the
therapeutic effect of NaHS was examined via administration 14 days
after MCT injection. Unfortunately, the therapeutic administration
of NaHS was unable to mitigate the severity of PAH (data not
shown).
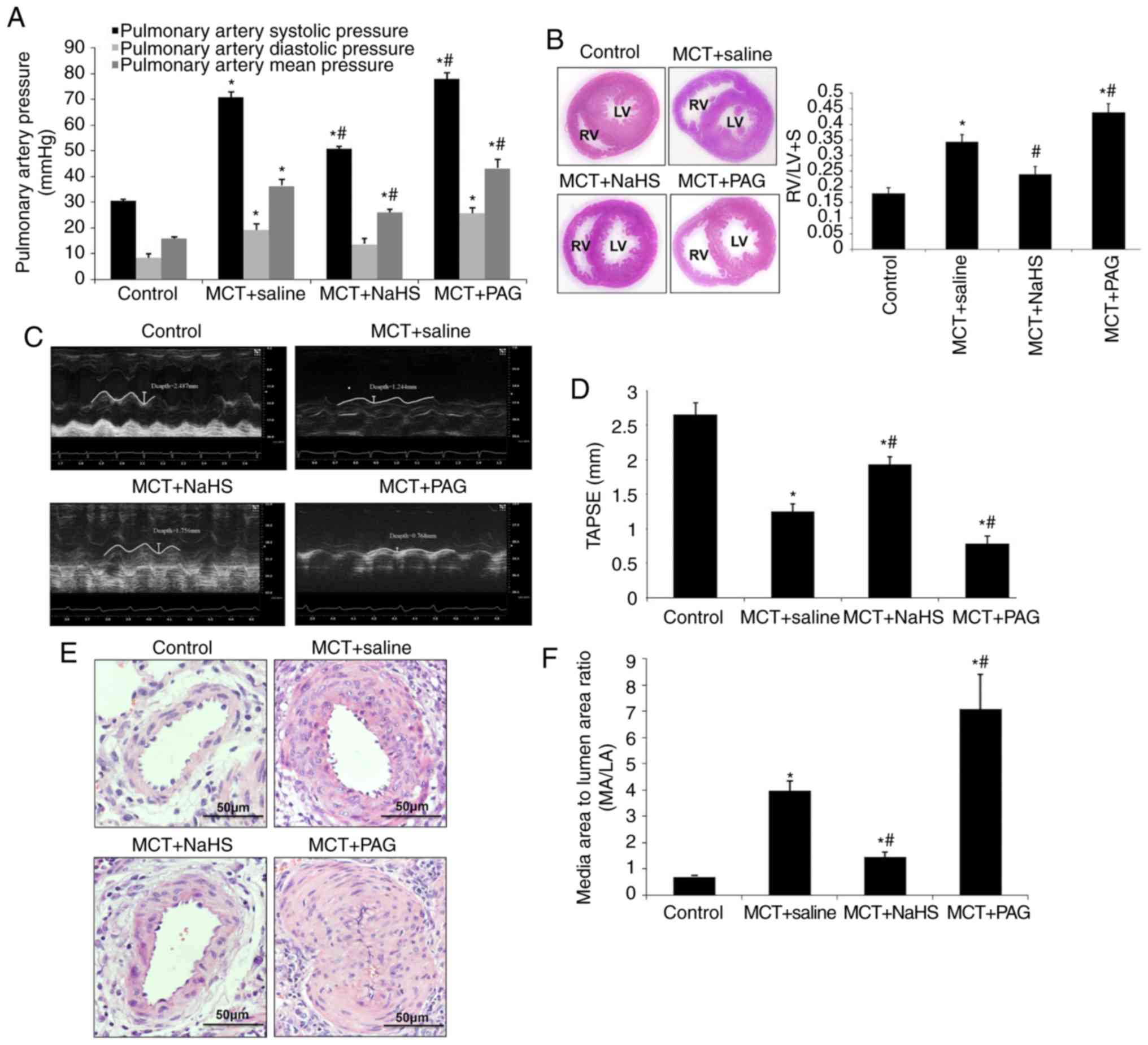 | Figure 2Effect of supplementation with
exogenous H2S (NaHS) or inhibition of H2S
formation by PAG on MCT-induced PAH. Rats received NaHS (i.p., 1
mg/kg/day) or PAG (i.p, 10 mg/kg/day) 7 days after MCT injection. A
period of 21 days after injection of MCT, rats were anesthetized
and underwent RHC. (A) Pulmonary artery systolic pressure,
pulmonary arterial diastolic pressure and pulmonary arterial mean
pressure were continuously recorded. (B) The RV/(LV+S) ratio was
calculated, as described in materials and methods, to estimate RV
hypertrophy. (C) Right ventricular ejection fraction was indicated
by TAPSE measured using ultrasound and (D) quantitative analysis.
(E) Pulmonary vascular remodeling was assessed using hematoxylin
and eosin staining. (F) Media to lumen ratio (MA/LA) was measured
as described in the materials and methods, to evaluate the
remodeling of pulmonary arteries. The results are presented as the
mean ± standard error of the mean (n=6 animals in each group).
*P<0.05 vs. the control group; #P<0.05
vs. rats with MCT+saline. H2S, hydrogen sulfide; PAG,
DL-propagylglycine; PAH, pulmonary arterial hypertension;
RV/(LV+S), weight ratio of the right ventricle to left ventricle
plus septum; MCT, monocrotaline. TAPSE, tricuspid annular plane
systolic excursion. MA, media area; LA, lumen area; i.p.,
intraperitoneal. |
Subsequently, whether NaHS exhibited an effect on
pulmonary EndMT in MCT-induced PAH was assessed. As presented in
Fig. 3A, NaHS significantly
upregulated the expression of VE-cadherin and reduced the
expression of α-SMA in the lungs, suggesting a partial reversion of
pulmonary EndMT (P<0.05). An additional loss of VE-cadherin and
an additional gain of α-SMA were observed in the lungs of PAH rats
treated with PAG. To further determine whether NaHS affected EndMT
in pulmonary ECs, double immuno-fluorescent staining was used for
VE-cadherin and α-SMA in the lung tissue sections (Fig. 3B). Red-stained VE-cadherin
positive ECs were detected in the lung tissue of control rats, or
in PAH rats treated with NaHS, whereas green-stained α-SMA positive
ECs were identified in small numbers. However, PAH rats that were
administered PAG indicated decreased expression of VE-cadherin and
increased expression of α-SMA when compared with PAH rats
administered saline.
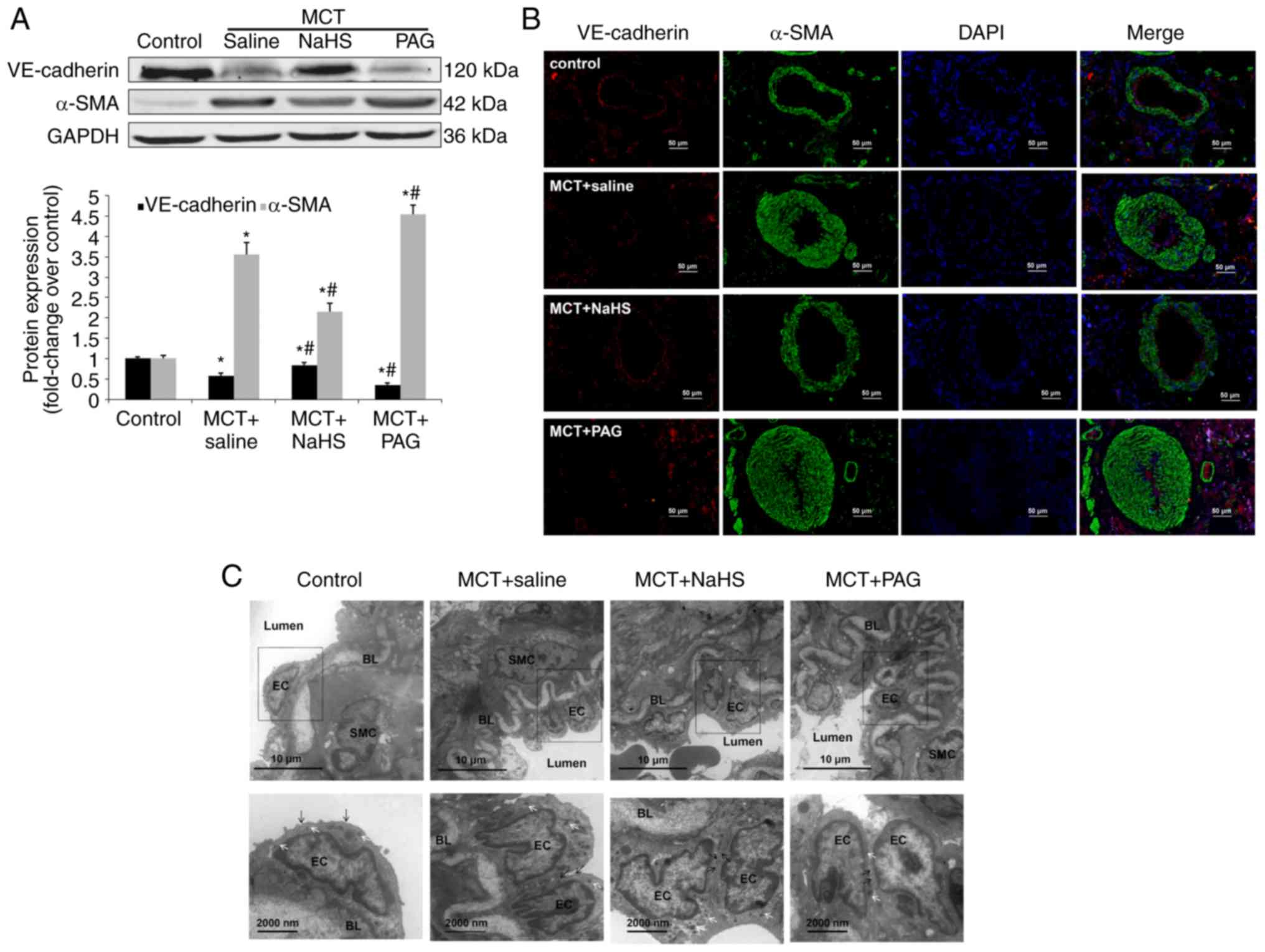 | Figure 3Effect of NaHS or PAG on EndMT in
MCT-induced PAH. Rats received NaHS (i.p., 1 mg/kg/day) or PAG
(i.p, 10 mg/kg/day) 7 days after MCT injection. A period of 21 days
after injection of MCT, rats were sacrificed (n=6 animals in each
group). The pulmonary expression levels of EndMT markers,
VE-cadherin and α-SMA were examined using (A) western blot analysis
and (B) immunofluorescent staining. Red fluorescence represents
VE-cadherin, green fluorescence represents α-SMA and blue
fluorescence represents 4,6-diamidino-2-phenylindole nuclei
staining. (C) The ultrastructure of the pulmonary artery was
assessed using transmission electron microscopy. In the control
group, endothelial cells are flat, elongated and separated from
smooth muscle cells via a thick basement membrane, the BL. In the
MCT group, endothelial cells display migration in the intima and
alter their nuclei orientation. Endothelial characteristics of
these cells are confirmed by the presence of caveolae (black arrow)
and Weibel-Palade bodies (white arrows). *P<0.05 vs.
the control group; #P<0.05 vs. rats with MCT+saline.
NaHS, exogenous H2S; PAG, DL-propagylglycine; PAH,
pulmonary arterial hypertension; EndMT, endothelial-to-mesenchymal
transition; MCT, monocrotaline; EC, endothelial cells; SMC, smooth
muscle cells; BL, basal lamina; i.p., intraperitoneal; SMA, smooth
muscle actin; VE, vascular endothelial. |
In accordance with the protein pattern, electron
microscopy observations revealed that NaHS affected the
ultrastructural characteristics of EndMT in MCT-induced PAH. As
presented in Fig. 3C, ECs were
flat, elongated and characterized by a high density of caveolae and
Weibel-Palade bodies (WPB) in the control pulmonary arteries. In
MCT-induced rats, luminal pulmonary ECs revealed a mixed
ultrastructural phenotype, as characterized by possessing
high-density caveolae and WPBs, exhibiting detectable
microfilaments and ongoing cell migration/invagination directed
toward the intima. In conclusion, NaHS treatment restored the
ultrastructural alterations of EndMT induced by MCT, while PAG
aggravated it (Fig. 3C).
Effect of H2S on NF-κB-Snail
signaling pathway in MCT-induced PAH
The activation of the NF-κB-Snail signaling pathway
is known to be required for the EndMT (18-20). Therefore, the role of
H2S in regulating the NF-κB-Snail pathway in MCT-induced
EndMT was assessed. The results revealed that treatment with NaHS
inhibited the activation of NF-κB and the subsequent transcription
of Snail, and this was demonstrated by a decreased level of
phospho-IκBα, an increased level of IκBα, impaired DNA binding
activity of p65 and the consequent downregulation of Snail
expression (Fig. 4A and B).
However, the inhibition of H2S synthesis, by PGA,
affected the NF-κB-Snail signaling pathway in the opposite manner
(Fig. 4A and B).
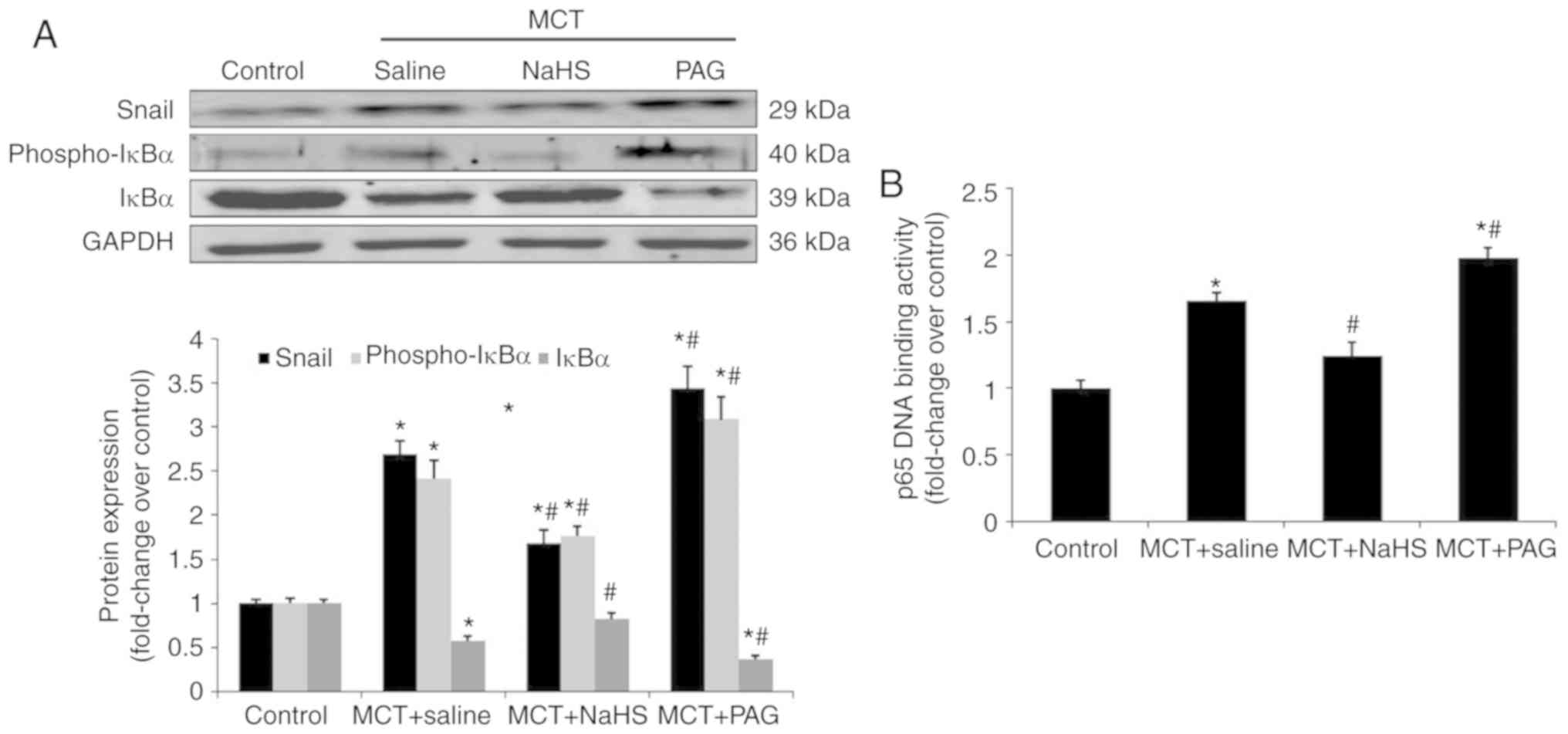 | Figure 4Effect of NaHS or PAG on NF-κB-Snail
pathway in MCT-induced PAH. Rats received NaHS (i.p., 1 mg/kg/day)
or PAG (i.p, 10 mg/kg/day) 7 days after MCT injection. A period of
21 days after injection of MCT, rats were sacrificed. Pulmonary
expression of Snail, phospho-IκBα and IκBα were measured using (A)
western blot analysis. (B) p65 DNA binding activity in lung nuclear
extracts were measured as described in the materials and methods.
The results are presented as the mean ± standard error of the mean
(n=6 animals in each group). *P<0.05 vs. the control
group; #P<0.05 vs. rats with MCT+saline. NaHS,
exogenous H2S; PAG, DL-propagylglycine; PAH, pulmonary
arterial hypertension; MCT, monocrotaline; phospho, phosphorylated;
i.p., intraperitoneal; NF, nuclear factor. |
Effect of H2S on
TGF-β1-induced EndMT in HPAECs
To further investigate the role of H2S in
endothelial cell EndMT, TGF-β1 (10 ng/ml) was used to stimulate
HPAECs. The results of western blot analysis and double
immunofluorescent staining indicated that TGF-β1 stimulation
induced the upregulation of α-SMA expression with a concomitant
downregulation of VE-cadherin in HPAECs (Fig. 5A and B). Treatment with NaHS
significantly inhibited TGF-β1 induced EndMT, as demonstrated by a
dose-dependent decrease in α-SMA expression and a dose-dependent
increase in VE-cadherin (Fig.
5A). A similar pattern was indicated using immunofluorescent
staining (Fig. 5B). Furthermore,
TEM was used to investigate the ultrastructural alterations of
HPAECs. After exposure to TGF-β1 for 10 days, HPAECs presented a
mixed ultrastructural phenotype, exhibiting the features of ECs
(caveolae and WPBs) and the characteristics of SMC (microfilaments;
Fig. 5C). Treatment with NaHS, at
a dose of 100 µM, partially reversed the ultrastructural
phenotypic shift in HPAECs that were induced by TGF-β1 (Fig. 5C). Consistent with this was the
alteration in cell morphology. HPAECs changed from a cobblestone
appearance into an elongated, spindle shape when stimulated with
TGF-β1 for 10 days (Fig. 5D).
Treatment with NaHS partially reversed the TGF-β1-induced
morphological change (Fig. 5D).
Ultrastructural and morphology observations further supported the
potential role of H2S in the process of EndMT.
Additionally, the impact of NaHS on the NF-κB-Snail pathway was
examined. In agreement with the aforementioned results, NaHS
dose-dependently inhibited the activation of NF-κB and subsequent
transcription of Snail in TGF-β1-stimulated HPAECs (Fig. 5E and F).
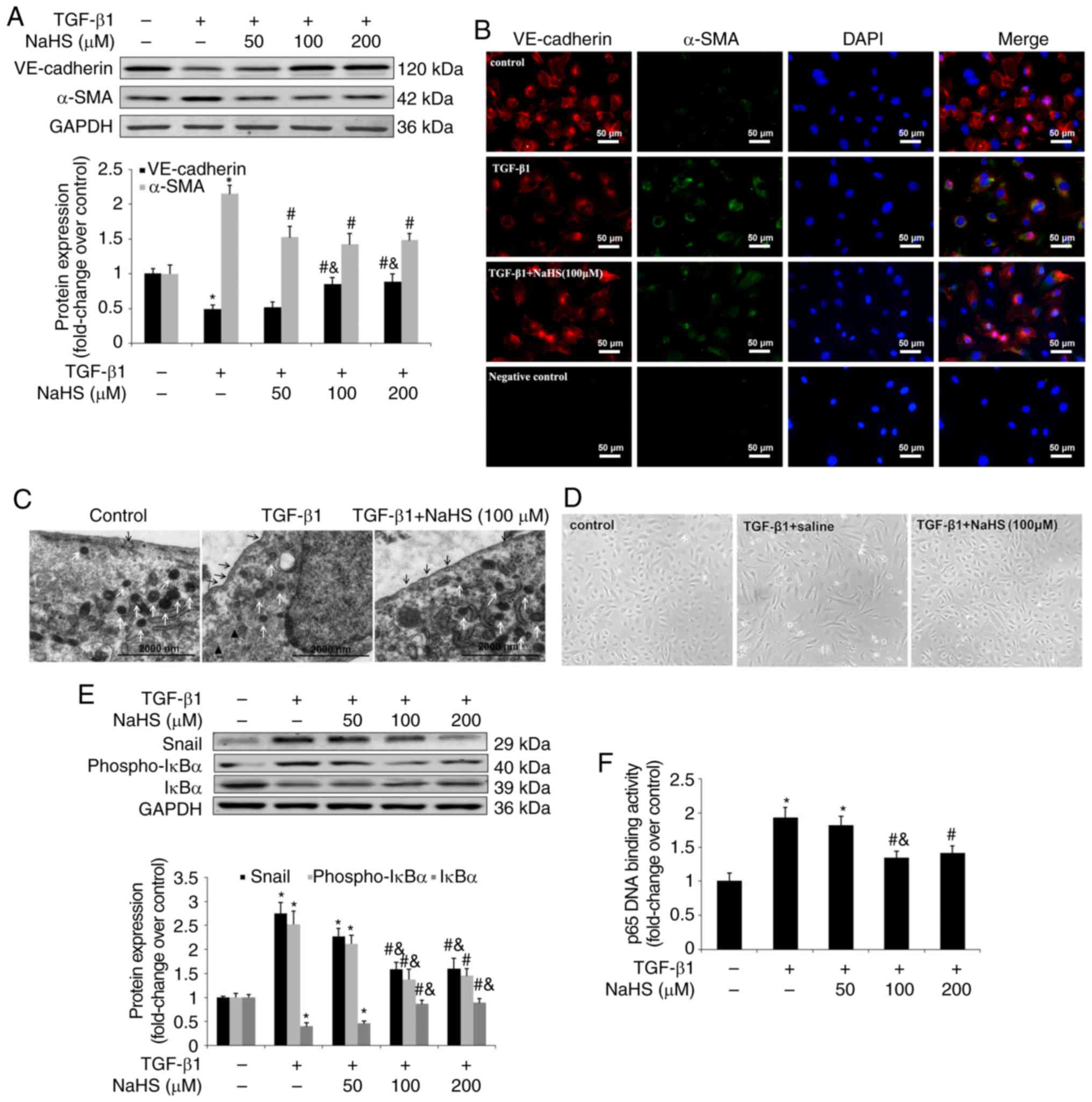 | Figure 5Effect of NaHS on TGF-β1-induced
EndMT in HPAECs. HPAECs were pre-incubated with saline or NaHS (50,
100 and 200 µM) for 2 h and stimulated with TGF-β1 (10
ng/ml) for 24 h in the continuous presence of NaHS (50, 100 and 200
µM) or saline. Some cells were harvested to investigate the
changes in NF-κB-Snail pathway. Remaining cells were subsequently
stimulated with TGF-β1 (10 ng/ml) for an additional 9 days in an
attempt to estimate the process of EndMT. Expression of EndMT
markers, VE-cadherin and α-SMA was examined using (A) western blot
analysis and (B) immunofluorescent staining. Red fluorescence
represents VE-cadherin, green fluorescence represents α-SMA and
blue fluorescence represents DAPI nuclei staining. The
ultrastructure of HPAECs was assessed using (C) TEM. Black arrows
indicate caveolae. White arrows indicate WPB. Black triangles
indicate microfilaments in the cytoplasm. Effect of NaHS on cell
morphology in TGF-β1-induced EndMT in HPAECs was investigated using
(D) phase-contrast light microscopy. Normal HPAECs exhibit a
cobblestone appearance and indicate caveolae and WPB, with few
microfilaments. Following exposure to TGF-β1, microfilamentation
appeared in the cytoplasm and cells are elongated and
spindle-shaped. The expression of Snail, phospho-IκBα and IκBα was
measured using (E) western blot analysis. (F) p65 DNA binding
activity in nuclear extracts was measured as described in the
materials and methods. The data are presented as the mean ±
standard error of the mean of at least three independent
experiments. *P<0.05 vs. the control group at
baseline; #P<0.05 vs. TGF-β1-stimulated HPAECs
treated with saline; &P<0.05 vs.
TGF-β1-stimulated HPAECs treated with NaHS at a concentration of 50
µM. NaHS, exogenous H2S; EndMT,
endothelial-to-mesenchymal transition; HPAEC, human pulmonary
artery endothelial cells; WPB, Weibel-Palade bodies; TEM,
transmission electron microscopy; VE, vascular endothelial; TGF,
transforming growth factor; DAPI, 4,6-diamidino-2-phenylindole. |
Effect of CSE overexpression on EndMT in
TGF-β1 stimulated HPAECs
HPAECs were transfected with a CSE plasmid to mimic
the supplementation of H2S (Fig. S1). In accordance with the
findings demonstrated in the NaHS treatment, the overexpression of
CSE considerably inhibited TGF-β1 induced EndMT. Western blot
analysis and immunofluorescent staining indicated that
significantly upregulated α-SMA expression and suppressed
VE-cadherin expression, which was caused by TGF-β1, were
significantly reversed by CSE overexpression (P<0.05; Fig. 6A and B). An examination of the
ultrastructure also revealed that CSE overexpression was associated
with the repression of the endothelial phenotype shift (Fig. 6C). Furthermore, CSE overexpression
resulted in an obvious reduction in the TGF-β1-induced
transactivation of NF-κB and transcription of Snail (Fig. 6D and E). However, the inhibitory
effect of CSE overexpression on TGF-β-induced EndMT and the
activation of NF-κB-Snail pathway, was abrogated by pretreatment
with PAG (Fig. 6). In conclusion,
CSE overexpression was unable to alter EndMT, cell morphology and
ultrastructure in TGF-β1-stimulated HPAECs when cells were
pretreated with PAG.
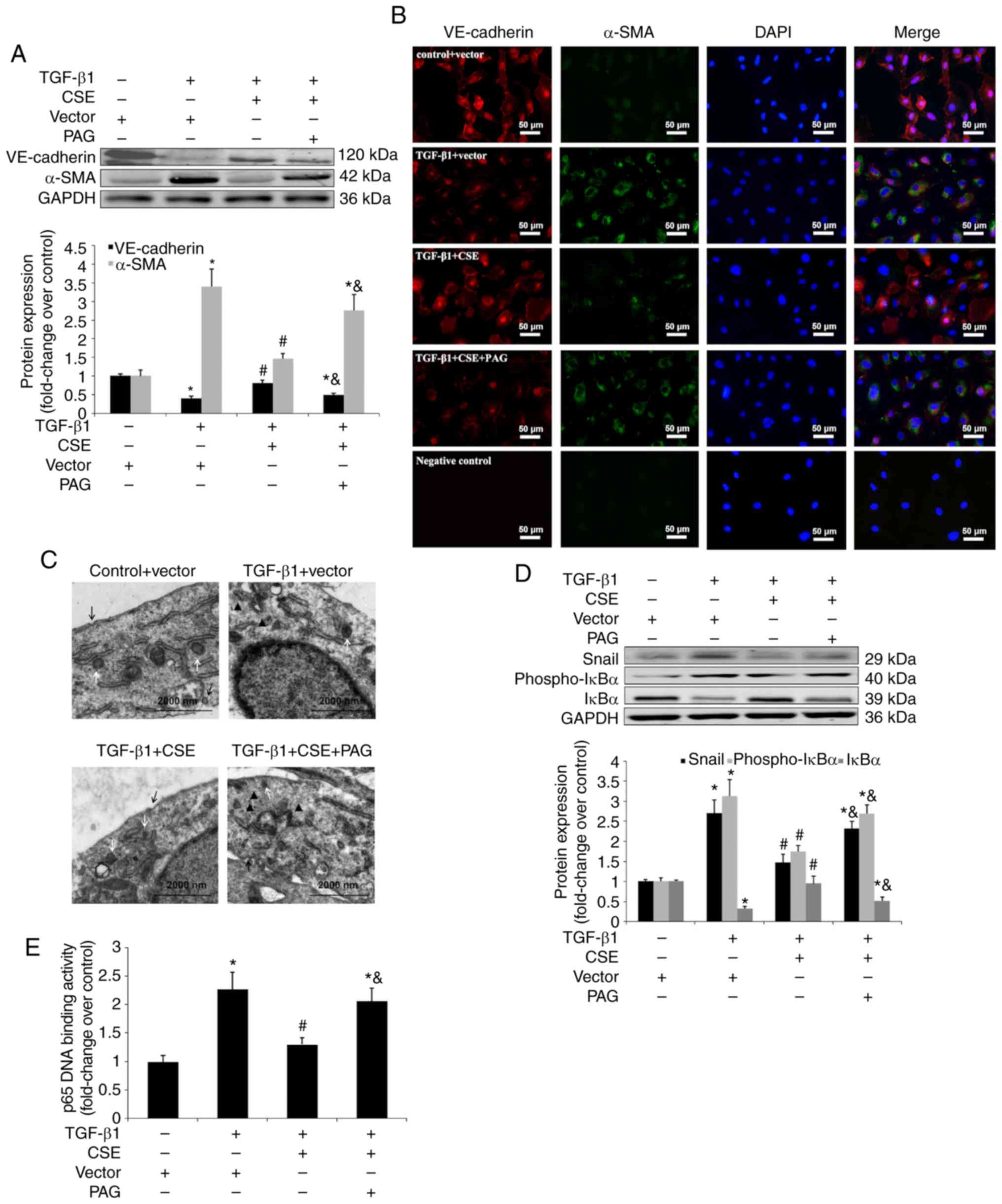 | Figure 6Effect of CSE overexpression on
TGF-β1-induced EndMT in HPAECs. HPAECs were transfected with a
vector or CSE plasmid and stimulated with TGF-β1 (10 ng/ml) for 24
h in the presence of PAG (2 mM) or saline. These cells were
harvested to investigate the changes in the NF-κB-Snail pathway.
Some cells were subsequently stimulated with TGF-β1 (10 ng/ml) for
an additional 9 days to estimate the process of EndMT. Expression
of EndMT markers, VE-cadherin and α-SMA and signal molecules were
examined using (A) western blotting and (B) immunofluorescent
staining. Red fluorescence represents VE-cadherin, green
fluorescence represents α-SMA and blue fluorescence represents DAPI
nuclei staining. Scale bar=50 µm. (C) The ultrastructure of
HPAECs was assessed using TEM. White arrows indicate caveolae.
Black arrows indicate WPB. Black triangles indicate microfilaments
in the cytoplasm. (D) Expression of Snail, phospho-IκBα and IκBα
were measured using western blot analysis. (E) p65 DNA binding
activity in nuclear extracts were measured as described in the
materials and methods. The data are presented as the mean ±
standard error of the mean of at least three independent
experiments. *P<0.05 vs. control group;
#P<0.05 vs. TGF-β1-stimulated HPAECs transfected with
vector; &P<0.05 vs. TGF-β1-stimulated HPAECs
transfected with CSE plasmid. CSE, cystathionine γ-lyase; EndMT,
endothelial-to-mesenchymal transition; DAPI,
4,6-diamidino-2-phenylindole; HPAEC, human pulmonary artery
endothelial cells; PAG, DL-propagylglycine; WPB, Weibel-Palade
bodies; TEM, transmission electron microscopy; TGF, transforming
growth factor; NF, nuclear factor; phospho, phosphorylated; VE,
vascular endothelial. |
Discussion
Epithelial to mesenchymal transition (EMT) is
defined as a phenotypic switch from an epithelial to mesenchymal
cell, which serves important roles in embryonic development and a
variety of adult conditions, including fibrosis, wound repair,
inflammation and malignancy (2).
The endothelium is a specialized form of squamous epithelial tissue
and as such, EndMT is a subcategory of EMT. EndMT has previously
been recognized as a new paradigm in PAH pathobiology (5-7).
Although the protective effect of H2S in PAH has been
well established, its role in EndMT has not yet been determined. In
the present study, the results demonstrated that EndMT is
accompanied by an endogenous deficiency of H2S in PAH.
The administration of exogenous H2S was indicated to
prevent MCT-induced PAH. The protective mechanism governing this is
likely to be associated with the reversal of the EndMT process by
inhibiting NF-κB-Snail axis molecules.
In the current study, a time-dependent feature of
the endothelial phenotype shift was identified in MCT-induced PAH.
The expression of α-SMA, a mesenchymal marker, was increased 14
days after MCT injection, while the expression of VE-cadherin, an
endothelial differentiation marker, was inhibited. EndMT continued
until 28 days after MCT injection. These findings are supported by
the temporal trend of EndMT obtained by a study performed by
Ranchoux et al (7), which
identified the overexpression of mesenchymal markers, Twist-1,
vimentin and P-vimentin, were associated with a strong repression
of VE-cadherin and p120-catenin protein expression. This
aforementioned study also observed pulmonary luminal cells
displaying a mixed phenotype endothelial/mesenchymal cells at 21
and 28 days after MCT injection (7). Furthermore, Nikitopoulou et
al (21) reported that
injection of rats with MCT produced a significant decline of
VE-cadherin at 24 h and 15 days, with recovery observed at 28 days.
The current study also indicated a slight rebound in the expression
of VE-cadherin and Snail 28 days after MCT injection. This may be
due to vascular remodeling by transformed endothelial cells
responding to angiogenic and proliferative stimuli at the late
stage of PAH rather than a true recovery. Conversely, CSE
expression in lungs was revealed to be significantly downregulated
14 days after MCT injection. CSE activity in the lungs and the
plasma level of H2S, consequently dropped and remained
at a low level. The temporal observations in EndMT and
H2S biosynthesis suggested that MCT triggered the
process of EndMT and inhibited the synthesis of endogenous
H2S.
In an attempt to further determine the association
between H2S and EndMT, an exogenous H2S
donor, NaHS, was used. NaHS treatment reversed the process of EndMT
and alleviated the severity of PAH. Using western blot analysis and
double immunofluorescent staining, the results demonstrated that
the decrease of endothelial cadherin and the increase of α-SMA were
partially ameliorated by NaHS. The ultrastructure characteristics
of EndMT, which were evaluated by TEM supported these previous
findings. In contrast, abolishing systemic H2S by PAG
promoted the endothelial phenotype shift and aggravated PAH and
pulmonary vascular remodeling. These results support the assumption
that H2S is important in the development and progression
of EndMT in MCT-induced PAH.
It is known that the TGF-β signaling pathway plays
important roles in the onset or development of MCT-induced PAH
(22-24). TGF-β1 induces pulmonary
endothelial cells to undergo EndMT (25). Then, the effect of H2S
on EndMT in TGF-β1 stimulated HPAECs was investigated. Cultured
HPAECs with TGF-β1 for 10 days resulted in EndMT, as characterized
by a decrease of VE-cadherin and an increase of α-SMA, with the
typical ultrastructure feature, elongated and spindle-like
appearance with microfilamentation in the cytoplasm.
Supplementation with exogenous H2S by NaHS or
transfection with CSE plasmid significantly reversed this phenotype
shift and these results are supported by findings in the MCT rat
model. Collectively, the in vivo and in vitro
findings of the current study supported the novel concept that
EndMT is regulated by H2S in PAH.
As H2S exhibits anti-fibrotic potential
in the pathogenesis of pulmonary, renal and hepatic fibrosis, in
which EMT participates, the effect of H2S on EMT was
initially investigated (26-28). H2S has been indicated
to attenuate TGF-β1-induced EMT in human alveolar epithelial cells
and renal tubular epithelial cells (27,29,30). H2S has also been
revealed to inhibit EMT in human breast cancer cells and
hepatocarcinoma cells, and exhibits an anti-cancer action (31,32). Recently, one study investigated
the role of H2S in EndMT and demonstrated that
H2S protected against endoplasmic reticulum
stress-induced EndMT via the Src pathway in human umbilical vein
ECs (33). Together with the
findings obtained in the present study, these results support the
import role of H2S in suppressing the
epithelial/endothelial phenotype shift, by which H2S
exhibits an anti-fibrotic, anti-oncogenic and anti-PAH effect in a
variety of conditions.
Snail is a family of transcription factors that
repress the expression of VE-cadherin to promote EMT/EndMT
(19). Snail is known to be
transcriptionally regulated by NF-κB (19,20). Therefore, the expression of signal
proteins associated with the NF-κB-Snail axis was measured in the
present study to investigate the detailed mechanisms underlying the
inhibitory effect of H2S on EndMT. The time-course study
indicated early augmentation of Snail expression 7 days after MCT
injection with a peak on day 21 and the maintenance of high
expression on day 28. The temporal pattern of Snail expression is
coincident with the chronological shift in endogenous
H2S synthesis in MCT-induced PAH, suggesting the
possible association between H2S and Snail. The addition
of NaHS downregulated the activation of NF-κB, as characterized by
the decreased phosphorylation of IκBα, reduced degradation of IκBα
and decreased DNA binding activity, leading to the repressed
transcription of Snail. However, the eradication of endogenous
H2S by PAG stimulated the transactivation of NF-κB and
the expression of Snail. Exogenous H2S or the
overexpression of CSE suppressed the activation of NF-κB and the
expression of downstream Snail induced by TGF-β1 in HPAECs. From
these results, it can be assumed that H2S induced NF-κB
inactivation and consequently caused the transcriptional inhibition
of Snail, resulting in the reversal of EndMT. The underlying
mechanism by which H2S modulates the activity of the
NF-κB-Snail signaling pathway remains undetermined. Recently, it
has been recognized that H2S functions via a variety of
signaling pathways through S-sulfhydration (34). A number of proteins, including
transcription factors, could be modified by H2S via
S-sulfhydration and change their original function, serving as
important switches or regulators (34). Therefore, future studies should
assess whether H2S regulates the epithelial/endothelial
phenotypic transition by sulfhydrating NF-kB, or via Snail
molecules at their cysteine residues.
In addition to the NF-κB-snail signaling pathway,
H2S may affect EndMT/EMT via a variety of mechanisms. In
tubular epithelial cells, H2S reduced the expression of
TGF-β receptor type I and TGF-β receptor type II, and attenuated
the TGF-β1-induced transduction of extracellular signal regulated
kinase and Wnt/catenin pathways, mitigating TGF-β1-induced EMT
(30). H2S also
counteracted TGF-β1-induced EMT and exhibited therapeutic potential
in renal sclerotic diseases via cleaving the disulfide bond in the
dimeric active TGF-β1 and generating an inactive TGF-β1 monomer
(29). Furthermore,
H2S has been suggested to reserve the epithelial
phenotype and inhibit EMT with partial dependence on the Smad and
p38 pathways (31,35,36). Collectively, these results
establish the important role of H2S in EndMT/EMT, which
contributes to numerous conditions, including fibrosis, wound
repair, inflammation and malignancy. However, the underlying
precise mechanisms and the potential application of H2S
in the treatment of these conditions require further study.
In conclusion, the results of the current study
demonstrated that NaHS, a H2S donor, exhibits a
protective effect on MCT-induced PAH, at least in part, via the
reversal of EndMT and impeding pulmonary vascular remodeling. The
underlying mechanisms of the inhibitory effect of H2S on
EndMT may be dependent on the inactivation of the NF-κB-Snail
signaling pathway. These results provide new evidence for the role
of H2S in PAH.
Supplementary Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81570037), Medical
Guiding Project of Shanghai Science and Technology Committee (grant
no. 19411963300) and Clinical Research Project of
Multi-Disciplinary Team, Shanghai Ninth People's Hospital, Shanghai
Jiaotong University School of Medicine (grant no. 2017-1-015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HuilZ and CW proposed the concept, designed the
experiment and revised the manuscript for important intellectual
content. HuiZ, YL and YM performed the major experiments. HuilZ
wrote the manuscript. JZ analyzed the data and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocol was approved by the Committee on the
Ethics of Animal Experiments of the Shanghai JiaoTong University
School of Medicine [permit no. (2015)-130].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goumans MJ, van Zonneveld AJ and ten Dijke
P: Transforming growth factor beta-induced
endothelial-to-mesenchymal transition: A switch to cardiacfibrosis?
Trends Cardiovasc Med. 18:293–298. 2008. View Article : Google Scholar
|
|
2
|
Willis BC and Borok Z: TGF-beta-induced
EMT: Mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeisberg EM, Potenta SE, Sugimoto H,
Zeisberg M and Kalluri R: Fibroblasts in kidney fibrosis emerge via
endothelial-to-mesenchymal transition. J Am Soc Nephrol.
19:2282–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arciniegas E, Frid MG, Douglas IS and
Stenmark KR: Perspectives onendothelial-to-mesenchymal transition:
Potential contribution to vascular remodeling in chronic pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 293:L1–L8. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hopper RK, Moonen JR, Diebold I, Cao A,
Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L and Rabinovitch M:
In pulmonary arterial hypertension, reduced BMPR2 promotes
endothelial-to-mesenchymal transition via HMGA1 and its target
slug. Circulation. 133:1783–1794. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ranchoux B, Antigny F, Rucker-Martin C,
Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F,
Planté S, et al: Endothelial-to-mesenchymal transition in pulmonary
hypertension. Circulation. 131:1006–1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lefer DJ: A new gaseous signaling molecule
emerges: Cardioprotective role of hydrogen sulfide. Proc Natl Acad
Sci USA. 104:17907–17908. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elsey DJ, Fowkes RC and Baxter GF:
Regulation of cardiovascular cell function by hydrogen sulfide
(H2S). Cell Biochem Funct. 28:95–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang C, Li X and Du J: Hydrogen sulfide as
a new endogenous gaseous transmitter in the cardiovascular system.
Curr Vasc Pharmacol. 4:17–22. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hongfang J, Cong Bailin, Bin Zhao, Zhang
Chunyu, Xinmin Liu, Weijin Zhou, Ying Shi, Tang Chaoshu and Junbao
D: Effects of hydrogen sulfide on hypoxic pulmonary vascular
structural remodeling. Life Sci. 78:1299–1309. 2006. View Article : Google Scholar
|
|
12
|
Li X, Du J, Jin H, Tang X, Bu D and Tang
C: The regulatory effect of endogenous hydrogen sulfide on
pulmonary vascular structure and gasotransmitters in rats with high
pulmonary blood flow. Life Sci. 81:841–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Du J, Jin H, Geng B and Tang C:
Sodium hydrosul-fide alleviates pulmonary artery collagen
remodeling in rats with high pulmonary blood flow. Heart Vessels.
23:409–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo L, Liu D, Tang C, Du J, Liu AD,
Holmberg L and Jin H: Sulfur dioxide upregulates the inhibited
endogenous hydrogen sulfide pathway in rats with pulmonary
hypertension induced by high pulmonary blood flow. Biochem Biophys
Res Commun. 433:519–525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stenmark KR, Meyrick B, Galie N, Mooi WJ
and McMurtry IF: Animal models of pulmonary arterial hypertension:
The hope for etiological discovery and pharmacological cure. Am J
Physiol Lung Cell Mol Physiol. 297:L1013–L1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang H, Guo C, Wu D, Zhang A, Gu T, Wang
L and Wang C: Hydrogen sulfide inhibits the development of
atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS
One. 7:e411472012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin Y, Zeng H, Gao L, Gu T, Wang C and
Zhang H: Hydrogen sulfide attenuates atherosclerosis in a partially
ligated carotid artery mouse model via regulating angiotensin
converting enzyme 2 expression. Front Physiol. 8:7822017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barberà MJ, Puig I, Domínguez D,
Julien-Grille S, Guaita-Esteruelas S, Peiró S, Baulida J, Francí C,
Dedhar S, Larue L and García de Herreros A: Regulation of Snail
transcription during epithelial to mesenchymal transition of tumor
cells. Oncogene. 23:7345–7354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J, García De and Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumor cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-kappaB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nikitopoulou I, Orfanos SE, Kotanidou A,
Maltabe V, Manitsopoulos N, Karras P, Kouklis P, Armaganidis A and
Maniatis NA: Vascular endothelial-cadherin downregulation as a
feature of endothelial transdifferentiation in
monocrota-line-induced pulmonary hypertension. Am J Physiol Lung
Cell Mol Physiol. 311:L352–L363. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Long L, Crosby A, Yang X, Southwood M,
Upton PD, Kim DK and Morrell NW: Altered bone morphogenetic protein
and transforming growth factor-beta signaling in rat models of
pulmonary hypertension: Potential for activin receptor-like
kinase-5 inhibition in prevention and progression of disease.
Circulation. 119:566–576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zakrzewicz A, Kouri FM, Nejman B,
Kwapiszewska G, Hecker M, Sandu R, Dony E, Seeger W, Schermuly RT,
Eickelberg O and Morty RE: The transforming growth
factor-beta/Smad2,3 signaling axis is impaired in experimental
pulmonary hypertension. Eur Respir J. 29:1094–1104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reynolds AM, Holmes MD, Danilov SM and
Reynolds PN: Targeted gene delivery of BMPR2 attenuates pulmonary
hypertension. Eur Respir J. 39:329–343. 2012. View Article : Google Scholar
|
|
25
|
Frid MG, Kale VA and Stenmark KR: Mature
vascular endothelium can give rise to smooth muscle cells via
endothe-lial-mesenchymal transdifferentiation: In vitro analysis.
Circ Res. 90:1189–1196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan HN, Wang HJ, Ren L, Ren B, Dan CR, Li
YF, Hou LZ and Deng Y: Decreased expression of p38 MAPK mediates
protective effects of hydrogen sulfide on hepatic fibrosis. Eur Rev
Med Pharmacol Sci. 17:644–652. 2013.PubMed/NCBI
|
|
27
|
Fang L, Li H, Tang C, Geng B, Qi Y and Liu
X: Hydrogen sulfide attenuates the pathogenesis of pulmonary
fibrosis induced by bleomycin in rats. Can J Physiol Pharmacol.
87:531–538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jung KJ, Jang HS, Kim JI, Han SJ, Park JW
and Park KM: Involvement of hydrogen sulfide and homocysteine
transsulfuration pathway in the progression of kidney fibrosis
after ureteral obstruction. Biochim Biophys Acta. 1832:1989–1997.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang Y, Zhang Z, Huang Y, Mao Z, Yang X,
Nakamura Y, Sawada N, Mitsui T, Takeda M and Yao J: Induction of
inactive TGF-β1 monomer formation by hydrogen sulfide contributes
to its suppressive effects on Ang II- and TGF-β1-induced EMT in
renal tubular epithelial cells. Biochem Biophys Res Commun.
501:534–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo L, Peng W, Tao J, Lan Z, Hei H, Tian
L, Pan W, Wang L and Zhang X: Hydrogen sulfide inhibits
transforming growth factor-β1-induced EMT via Wnt/Catenin pathway.
PLoS One. 11:e01470182016. View Article : Google Scholar
|
|
31
|
Lv M, Li Y, Ji MH, Zhuang M and Tang JH:
Inhibition of invasion and epithelial-mesenchymal transition of
human breast cancer cells by hydrogen sulfide through decreased
phospho-p38 expression. Mol Med Rep. 10:341–346. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan Y, Zhou C, Yuan D, Zhang J and Shao C:
Radiation exposure promotes hepatocarcinoma cell invasion through
epithelial mesenchymal transition mediated by H2S/CSE
pathway. Radiat Res. 185:96–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ying R, Wang XQ, Yang Y, Gu ZJ, Mai JT,
Qiu Q, Chen YX and Wang JF: Hydrogen sulfide suppresses endoplasmic
reticulum stress-induced endothelial-to-mesenchymal transition
through Src pathway. Life Sci. 144:208–217. 2016. View Article : Google Scholar
|
|
34
|
Zhang D, Du J, Tang C, Huang Y and Jin H:
H2S-induced sulf-hydration: Biological function and
detection methodology. Front Pharmacol. 8:6082017. View Article : Google Scholar
|
|
35
|
Cheng S, Lu Y, Li Y, Gao L, Shen H and
Song K: Hydrogen sulfide inhibits epithelial-mesenchymal transition
in peritoneal mesothelial cells. Sci Rep. 8:58632018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fang LP, Lin Q, Tang CS and Liu XM:
Hydrogen sulfide attenuates epithelial-mesenchymal transition of
human alveolar epithelial cells. Pharmacol Res. 61:298–305. 2010.
View Article : Google Scholar
|














