Introduction
Myocardial infarction (MI) constitutes one of the
most serious cardiac events with high morbidity and mortality
worldwide (1,2). Myocardial remodeling occurs after MI
as a compensatory mechanism to decrease wall stress which may lead
to inevitable cardiac dilatation, followed by heart failure
(3). At present, pharmacological
intervention remains the most effective treatment for preventing
post-infarction cardiac remodeling and dysfunction (4). The one year mortality rate of
patients hospitalized for heart failure after MI was as high as
43.2% (5). Therefore, it has been
crucial to exploit effective intervention targets for attenuating
cardiac remodeling and dysfunction post MI, thereby preventing the
development of heart failure in post-infarction patients (6).
Apoptosis and autophagy are reported to participate
in physiological processes that occur constitutively in the
myocardium (7,8). The roles of apoptosis and autophagy
in ischemic myocardial injury have been extensively demonstrated in
ischemic induced myocardial injury. Increasing evidence suggests
that apoptosis and autophagy disruption results in the development
of severe left ventricular dysfunction (9,10).
As a result, developing a strategy aimed at regulating apoptosis
and autophagy is necessary to protect against post-MI cardiac
dysfunction. Therefore, the accurate molecular mechanisms that
regulate apoptotic and autophagic responses are pivotal for
preventing the development of heart failure post MI.
The mammalian target of rapamycin (mTOR) is a highly
conserved serine/threonine kinase, belonging to the
phosphoinositide kinase-related kinase family (11). mTOR has been reported to activated
via the protein kinase B/phosphoinositide 3 kinase pathway
(12). This signaling mechanism
is centrally involved in physiological hypertrophy but also takes
part in pathological remodeling of the heart (12). It is well established that mTOR
plays an important role in regulating cellular physiology,
metabolism and stress responses (11). Previous studies have confirmed
that mTOR can concurrently modulate apoptosis and autophagy related
pathogenesis in numerous heart diseases (13-15). Furthermore, inhibiting mTOR
induced apoptosis and autophagy may exert a protective influence
against MI injury (16,17). The exact mechanisms explaining how
this signaling pathway regulates apoptosis and autophagy during MI
remain to be clarified.
Regulated in development and DNA damage response-1
(Redd1), located on chromosome 10 (10q22.1), is a 232 amino acids
protein which is transcriptionally induced by DNA damage in various
types of cells (18,19). Redd1 plays a vital role in
biological, physiological functions and cellular functions, such as
cell growth, inflammation and autophagy (20-22). Despite its relationship with mTOR
and extensive research in multiple diseases, such as
neurodegenerative disorders (23)
and cancer (24), little is known
about the role of Redd1 in the heart. Few previous studies have
evaluated the hypothesis that Redd1 may have relevance to cardiac
physiopathology. Studies have shown that Redd1 inhibition protected
cardiomyocytes from ischemia-reperfusion (I/R) injury (25) or methamphetamine-induced
myocardial damage (26).
Paradoxically, Liu et al (27) demonstrated that Redd1 attenuated
cardiac hypertrophy induced by phenylephrine via enhancing
autophagy. These observations imply that Redd1 is possibly
associated with cardiac dysfunction. However, there was no study on
whether Redd1 could ameliorate the prognosis of cardiac dysfunction
post MI. At present, the role of Redd1 in the heart remains
unknown. Nonetheless, extrapolating experimental data from other
cell types, Redd1 appears to play a pivotal role in inhibiting mTOR
activation (28-30).
In this context, the present study aimed to explore
the potential contribution of Redd1 during the development of heart
failure after MI. The study presented here demonstrates the
critical role of Redd1 overexpression in cardiomyocytes during the
chronic phases of MI. A single intravenous injection of an
adeno-associated virus 9 (AAV9) vector expressing Redd1 reduced
left ventricular dysfunction. In addition, Redd1 improved cardiac
function after myocardial infarction through apoptosis inhibition
and autophagy enhancement mediated by mTOR inactivation. The
results of the present study suggest the critical importance of
Redd1 in the development of heart failure post MI.
Materials and methods
Animals
A total of 30 C57BL/6 male mice weighing 14-16 g
(4-5 weeks) were purchased from the Beijing HFK Bioscience Co.,
Ltd. Mice were kept in cages at 22±2°C with 40±5% humidity under a
12 h light/dark cycle in the Tongji Medical School Experimental
Animal Center, and fed a chow diet and water. Animal experiments
were carried out in accordance with the Guide for the Care and Use
of Laboratory Animals published by the National Institute of Health
and were approved by the Institutional Animal Care and Use
Committee at Tongji Medical College, Huazhong University of Science
and Technology.
Injection of AAV9 vectors
The AAV9 vectors carrying enhanced green fluorescent
protein (AAV9-GFP) or mouse Redd1 (AAV9-Redd1) were purchased from
Weizhen Biotechnology Company. The sequence of the Redd1 vector was
consistent with the coding sequence of mouse Redd1, shown as
follows,
ATGCCTAGCCTCTGGGATCGTTTCTCGTCCTCCTCTTCCTCTTCGTCCTCGTCTCGAACTCCGGCCGCTGATCGGCCGCCGCGCTCCGCCTGGGGGTCTGCAGCCAGAGAAGAGGGCCTTGACCGCTGCGCGAGCCTGGAGAGCTCGGACTGCGAGTCCCTGGACAGCAGCAACAGTGGCTTCGGGCCGGAGGAAGACTCCTCATACCTGGATGGGGTGTCCCTGCCCGACTTTGAGCTGCTCAGTGACCCCGAGGATGAGCACCTGTGTGCCAACCTGATGCAGCTGCTGCAGGAGAGCCTGTCCCAGGCGCGATTGGGCTCGCGGCGCCCTGCGCGTTTGCTCATGCCGAGCCAGCTGGTGAGCCAGGTGGGCAAGGAACTCCTGCGCCTGGCATACAGTGAGCCGTGCGGCCTGCGGGGGGCACTGCTGGACGTGTGTGTGGAGCAAGGCAAGAGCTGCCATAGCGTGGCTCAGCTGGCCCTCGACCCCAGCCTGGTGCCCACCTTTCAGTTGACCCTGGTGCTGCGTCTGGACTCTCGCCTCTGGCCCAAGATCCAGGGGCTGTTAAGTTCTGCCAACTCTTCCTTGGTCCCTGGTTACAGCCAGTCCCTGACGCTAAGTACCGGCTTCAGAGTCATCAAGAAGAAACTCTACAGCTCCGAGCAGCTGCTCATTGAAGAGTGTTGA.
Mice were injected with viral solution (2.8×1011 vector
genomes per mouse) via the tail vein 4 weeks before MI surgery
(31).
MI surgery and experimental groups
MI was induced by permanent ligation of the
left-anterior descending coronary artery (LAD) as previous reported
(32). Briefly, mice were
anesthetized with 3% pentobarbital sodium (50 mg/kg) by
intraperitoneal injection. Mice were mechanically ventilated. A
thoracotomy was conducted between the left third and fourth ribs.
The thymus was retracted upwards and the auricular appendix was
exposed. The LAD was ligated by a 6-0 silk suture. The sham group
mice underwent the same process except for ligating the LAD. Mice
were randomly divided into four groups: Sham with AAV9-GFP
(Sham+GFP; n=6), Sham with AAV9-Redd1 (Sham+Redd1; n=6), MI with
AAV9-GFP (MI+GFP; n=8) and MI with AAV9-Redd1 (MI+Redd1; n=8). Mice
were treated with AAV9-Redd1 or AAV9-GFP for 4 weeks before MI or
sham operation. Mice were sacrificed at 4 weeks post MI or sham
surgery.
Echocardiography
A total of 4 weeks following MI, mice were
anesthetized with 1.5% isoflurane via inhalation (33). The depth of anesthesia was
determined by immobility and assessing the absence of the
withdrawal reflex of the right paw. Subsequently, cardiac function
was measured by transthoracic echocardiography with a Vevo 2100
high-resolution micro imaging system (VisualSonics, Inc.). The
echocardiography images were acquired from the long axes and the
short axis. The following parameters were measured in M-mode: Left
ventricular end-diastolic diameter (LVEDd) and left ventricular
end-systolic diameter (LVESd). The percentage of left ventricular
fractional shortening (LVFS, %) and left ventricular ejection
fraction (LVEF, %) were automatically calculated. The parameters
were acquired and averaged from six cardiac cycles.
Masson's trichrome staining and
picrosirius red staining
Heart tissues were fixed with 4% paraformaldehyde
for 24 h at room temperature, subsequently paraffin embedded and
sectioned into 5 µm thick slices. Masson's trichrome and
picrosirius red staining were carried out in accordance with
standard procedures (34). The
stained sections were used to estimate scar thickness, infarct size
and expansion index as previously stated (35,36). Picrosirius red staining was
applied to calculate the percentage of collagen content in the
border zone of the infarcted heart. The images were acquired at
×200 magnification in the border zone of the infarct area, using a
light microscope (Olympus Corporation), and analyzed with Image-Pro
Plus software 6.0 (Media Cybernetics, Inc.).
Immunofluorescent assay
Paraffin slides were deparaffinized for
immunofluorescent staining. All sections were incubated with
primary antibodies for Redd1 (1:50; cat. no. ab106356; Abcam) and
LC3B (1:50; cat. no. Arg55799; Arigo Biolaboratories, Corps.) at
4°C overnight. Fluorescein tetramethylrhodamine-conjugated
secondary antibody affinipure goat anti-rabbit Immunoglobulin G
(H+L; 1:50; cat no. 111-025-144; Jackson ImmunoResearch Europe,
Ltd.,) was incubated for 1 h at room temperature. Then, all slides
were stained with 300 nM DAPI (D1306, Invitrogen; Thermo Fisher
Scientific, Inc.) for 5 min at room temperature. In each sample,
three random fields were observed by fluorescence microscopy at
×200 magnification. The relative fluorescence intensity was
estimated with ImageJ software (version 1.51, National Institutes
of Health).
Terminal deoxynucleotidyl-transferase
mediated dUTP nick-end labeling (TUNEL) assay
The cardiac cell apoptosis level in the infarct zone
of the heart was determined by TUNEL staining. In accordance with
the manufacturer's protocol supplied by the TUNEL detection kit
(Roche Diagnostics), the heart sections were treated with TUNEL
reagent for 1 h at 37°C. Subsequent to washing with twice PBS, the
sections were counterstained with 300 nM DAPI (D1306, Invitrogen;
Thermo Fisher Scientific, Inc.) for 5 min at room temperature. In
each sample, images of three random fields were captured with a
100X lens by fluorescence microscopy. The percentage of positive
apoptotic cells was estimated with ImageJ software.
Tissue separation
The atrial tissue was removed and the left ventricle
was cut open. The infarct tissue and the border tissue were
separated from the heart for western blotting and PCR analysis. As
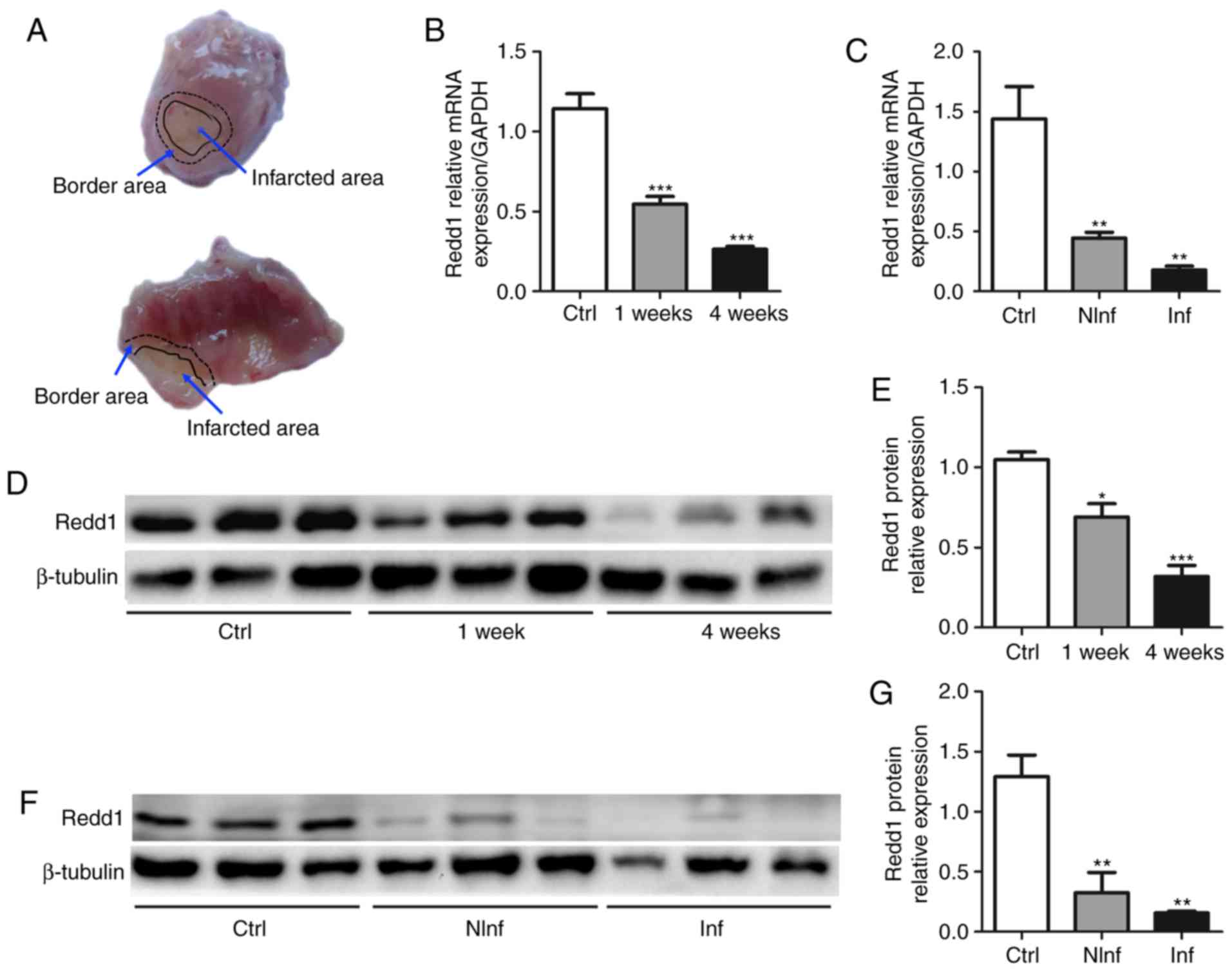
shown in Fig. 1A, the infarcted
area was thin and pale, located within the black solid line. The
border zone was a transitional pink area, thinner than normal
myocardial tissue, located between the black solid line and the
black dotted line (37).
Western blotting
The myocardial total protein was extracted using
with RIPA lysis buffer (Beyotime Institute of Biotechnology) at 4°C
for 30 min, then quantified with a bicin-choninic acid kit
(Beyotime Institute of Biotechnology). Equal amounts of denatured
protein samples (100 µg) were separated by 12% SDS-PAGE and
were transferred to nitrocellulose membranes. The nitrocellulose
membranes were blocked in 5% milk diluted with TBS/0.1% Tween-20
(TBST) for 2 h at room temperature, following by primary antibody
incubation overnight at 4°C. The primary antibodies were as
follows: Anti-β tubulin (1:1,000; cat. no. 66240-1-Ig, ProteinTech
Group, Inc.); anti-Redd1 (1:1,000; cat. no. ab106356; Abcam);
anti-Bcl2 (1:1,000; cat. no. A11025, Abclonal Biotech Co., Ltd.);
anti-Bax (1:1,000; cat. no. A12009; Abclonal Biotech Co., Ltd.);
anti-LC3B (1:1,000; cat. no. Arg55799; Arigo Biolaboratories,
Corps.); and anti-Beclin1 (1:1,000; cat. no. A7353; Abclonal
Biotech Co., Ltd.); anti-total-mTOR (1:1,000; cat. no. A2445;
Abclonal Biotech Co., Ltd.); anti-phosphorylated (phospho)-mTOR
(1:1,000; cat. no. AP0094; Abclonal Biotech Co., Ltd.),
anti-total-P70/S6 kinase [1:1,000; cat. no. 2708t; Cell Signaling
Technology, Inc., (CST)]; anti-phospho-P70/S6 kinase (1:1,000; cat.
no. 9208t; CST); anti-total-4EBP1 (1:1,000; cat. no. 9644t; CST);
anti-phospho-4EBP1 (1:1,000; cat. no. 9451t; CST). The membranes
were washed with TBST 3 times, following by incubation with
HRP-conjugated secondary antibodies anti-rabbit IgG (H+L; 1:2,000;
cat. no. ANT020) and anti-mouse IgG (H+L; 1:2,000; cat. no. ANT019;
both Antgene Biotechnology co., Ltd.) for 2 h at room temperature.
Bands were detected with a chemiluminescence detection System (ECL;
Thermo Fisher Scientific, Inc.) and quantified by densitometry
using ImageJ software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Myocardial total RNA was extracted with TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Complementary
DNA was synthesized using the PrimeScript RT Master Mix kit (Takara
Bio, Inc.) and then used for qPCR with a SYBR-Green Master Mixture
(Takara Bio, Inc.) on the ABI StepOnePlus RT-PCR system (Applied
Biosystems, Thermo Fisher Scientific, Inc.). The reverse
transcription conditions were as follows: 37°C for 15 min and 85°C
for 5 sec. The qPCR cycling conditions were as follows: 95°C for 10
min and 40 cycles of 95°C for 15 sec and 60°C for 15 sec. qPCR was
performed with 3 replicates of each sample. The relative expression
quantity was analyzed by normalizing to the GAPDH level and
calculated with the 2−ΔΔCq method (38). The primers were as follows: 5′-AGG
TCG GTG TGA ACG GAT TTG-3′ and 5′-AGG TCG GTG TGA ACG GAT TTG-3′
for GAPDH, 5′-GCT TCC AGG CCA TAT TGG AG-3′ and 5′-GGG GGC ATG ACC
TCA TCT T-3′ for natriuretic peptide type A (ANP), 5′-GAG GTC ACT
CCT ATC CTC TGG-3′ and 5′-GCC ATT TCC TCC GAC TTT TCT C-3′ for
natriuretic peptide type B (BNP), 5′-ACT GTC AAC ACT AAG AGG GTC
A-3′ and 5′-TTG GAT GAT TTG ATC TTC CAG GG-3′ for myosin heavy
polypeptide 7 (β-MHC), 5′-GCT CCT CTT AGG GGC CAC T-3′ and 5′-CCA
CGT CTC ACC ATT GGG G-3′ for collagen I, 5′-ACG TAG ATG AAT TGG GAT
GCA G-3′ and 5′-GGG TTG GGG CAG TCT AGT G-3′ for collagen III.
Statistical analysis
Each experiment was repeated ≥3 times. All data were
analyzed by Prism 5 (GraphPad Software Inc.) and presented as the
mean ± standard error of the mean. The differences in the data were
analyzed. Unpaired, two-tailed Student's t-test was used for two
groups. One-way analysis of variance was used for multiple
comparisons. Post hoc differences were determined by Newman Keuls
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Redd1 is downregulated in the heart
during the myocardial infarction process
To investigate whether Redd1 is involved in MI,
LAD-induced MI models were used (Fig.
1A). Redd1 mRNA and protein levels in the infarct zone declined
significantly at different time points (1 and 4 weeks) after MI
compared with sham-operation animals (P<0.001; Fig. 1B, D and E). Meanwhile, Redd1 mRNA
and protein expression declined significantly in different regions
(border area and infarct area) at 4 weeks compared with the sham
group (P<0.05; Fig. 1C, F and
G). These data indicate that Redd1 expression in the heart is
downregulated during MI, suggesting that Redd1 may participate in
the process of MI.
Redd1 overexpression protects against
cardiac dysfunction after MI
To investigate the influence of Redd1 on cardiac
dysfunction and remodeling post-MI, AAV9-Redd1 or control AAV9-GFP
was injected via the tail vein 4 weeks prior to permanent LAD
ligation. A total of 8 weeks after injection, AAV9-Redd1 group
displayed increased fluorescence intensity of Redd1 compared with
the AAV9-GFP group (Fig. 2A and
B). In addition, heart Redd1 protein level was increased nearly
three times in the sham operation mice injected with AAV9-Redd1
compared with the sham operation mice injected with AAV9-GFP.
Notably, the heart Redd1 protein level was also increased in MI
mice injected with AAV9-Redd1 compared with MI mice injected with
AAV9-GFP (Fig. 2C and D). In line
with preceding studies, the present study discovered that mice
undergoing MI surgery exhibited decreased LVEF and LVFS, as well as
increased LVEDd and LVESd (39,40), suggesting that MI surgery
successfully leads to cardiac insufficiency and ventricular
dilatation (Fig. 2E). Parameters
of echocardiography in mice at 4 weeks after sham or MI surgery are
listed in Table I. Redd1
overexpression significantly increased LVEF and LVFS (P<0.001;
Fig. 2F and G), while LVEDd and
LVESd were attenuated (Fig. 2H and
I). Sham+Redd1 group mice displayed no significant changes
compared with Sham+GFP group mice. Consistently, the MI+Redd1 group
mice also displayed decreased mRNA levels of ANP and BNP compared
with the MI+GFP group mice in the border zone (Fig. 2J and K). The β-MHC mRNA level was
also slightly decreased after the AAV-Redd1 injection but not
significantly (Fig. 2L).
Collectively, Redd1 overexpression protected against cardiac
dysfunction and cardiac dilatation at 4 weeks after MI after
MI.
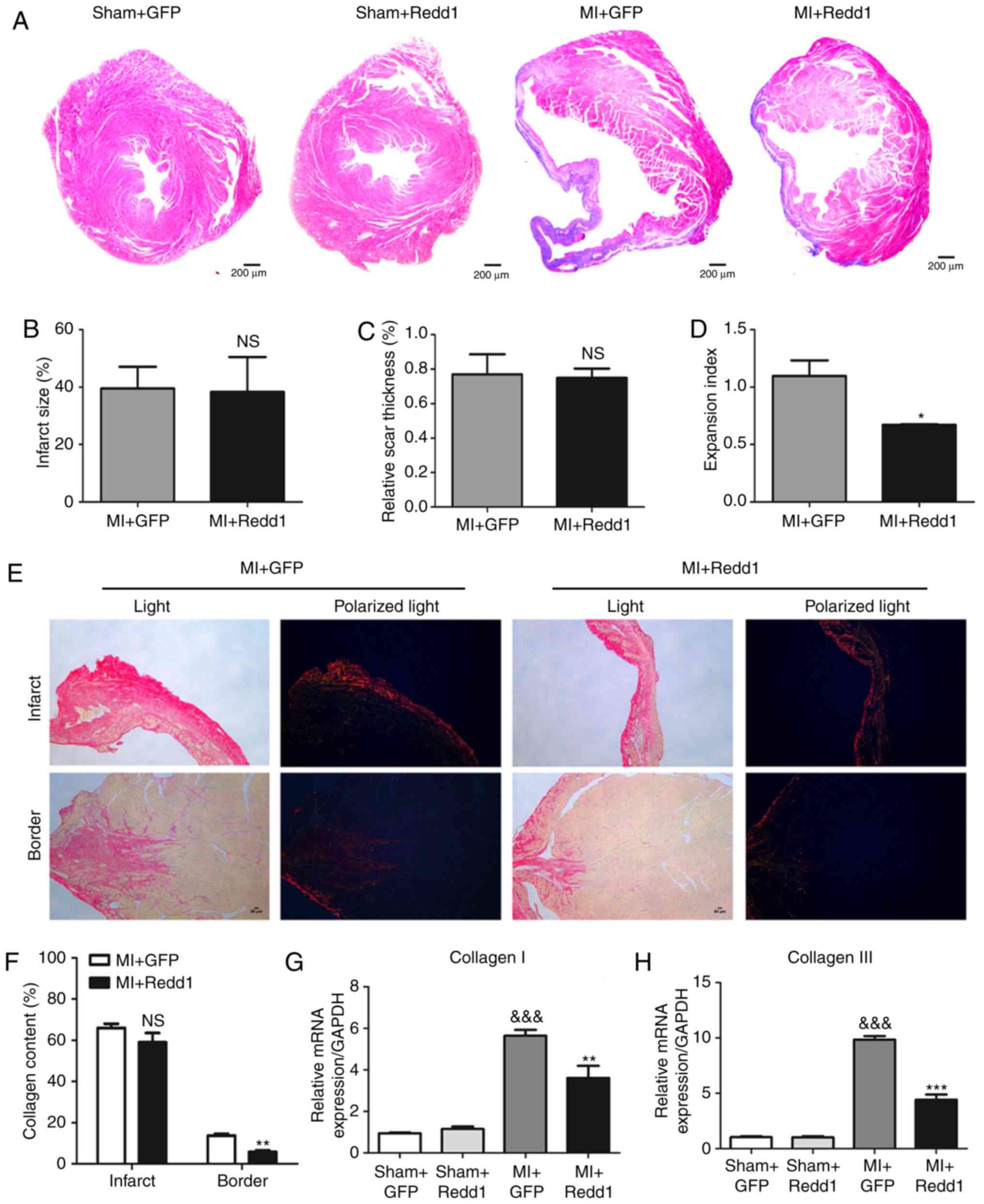
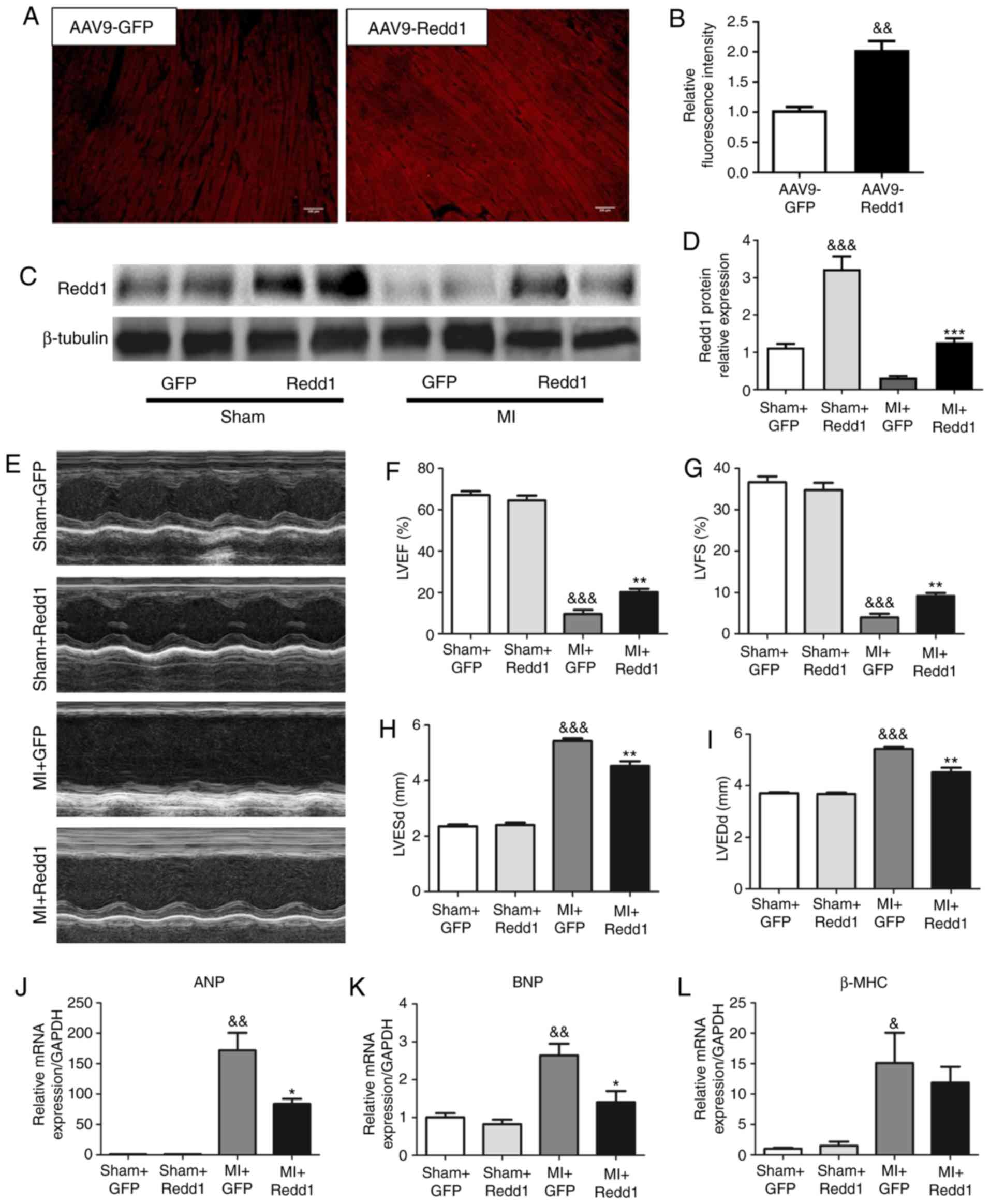 | Figure 2Redd1 overexpression protects against
cardiac dysfunction after MI. (A) The temporal expression pattern
of Redd1 tested in fluorescence in the heart of AAV9-GFP and
AAV9-Redd1 mice. (B) Quantitative results of Redd1 fluorescence
intensity (n=6). (C) Protein expression of Redd1 at 4 weeks
following MI in the myocardium of mice treated with AAV9-GFP or
AAV9-Redd1 and (D) the quantitative analysis (n=6-8). (E)
Representative M-mode echocardiography image of the LV from the
four groups at 4 weeks after MI. (F-I) Quantitative results of (F)
LVEF, (G) LVFS, (H) LVESd and (I) LVEDd of mice from the four
groups at 4 weeks after MI (n=6-8). (J) ANP, (K) BNP and (L) β-MHC
determined by reverse transcription-quantitative PCR assay (n=6-8).
Data presented as the mean ± standard error of the mean.
&P<0.05, &&P<0.01 and
&&&P<0.001 vs. Sham+GFP group;
*P<0.05, **P<0.01 and
***P<0.001 vs. the MI+GFP group. Redd1, regulated in
development and DNA damage response-1; MI, myocardial infarction;
AAV9, adeno-associated virus 9; GFP, green fluorescent protein; LV,
left ventricular; LVEF, left ventricular ejection fraction; LVFS,
left ventricular fractional shortening; LVEDd, left ventricular
end-diastolic diameter; LVESd, left ventricular end-systolic
diameter; ANP, natriuretic peptide type A; BNP, natriuretic peptide
type B; β-MHC, myosin heavy polypeptide 7. |
 | Table IParameters in mice at 4 weeks after
sham or MI surgery. |
Table I
Parameters in mice at 4 weeks after
sham or MI surgery.
| Parameters | Sham+GFP (n=6) | Sham+Redd1
(n=6) | MI+GFP (n=8) | MI+Redd1 (n=8) |
|---|
| LVEF (%) | 67.10±1.84 | 64.58±2.35 | 9.55±2.01a | 20.23±1.59b |
| LVFS (%) | 36.63±1.44 | 34.76±1.73 | 4.00±0.89a | 9.17±0.76b |
| LVEDd (mm) | 3.71±0.04 | 3.68±0.06 | 5.54±0.10a | 4.85±0.19b |
| LVESd (mm) | 2.35±0.04 | 2.40±0.08 | 5.42±0.06a | 4.52±0.12b |
Redd1 overexpression protects against
cardiac expansion and fibrosis after MI
To determine cardiac dilatation post MI, heart
tissues were stained with Masson's trichrome 4 weeks after MI
(Fig. 3A). In the present study,
the MI+Redd1 group displayed a reduced heart expansion index, as
well as unaltered scar size and wall thickness compared with the
MI+GFP group (Fig. 3B-D).
Meanwhile, to analyze cardiac fibrosis, heart tissues were stained
with picrosirius red staining in the border zone. The MI+Redd1
group displayed less collagen content compared with the MI+GFP
group (Fig. 3E and F).
Furthermore, MI+Redd1 group mice also exhibited lower mRNA levels
of collagen I and collagen III compared with MI+GFP group mice
(Fig. 3G and H). Collectively,
Redd1 over-expression protected against cardiac expansion and
fibrosis after MI.
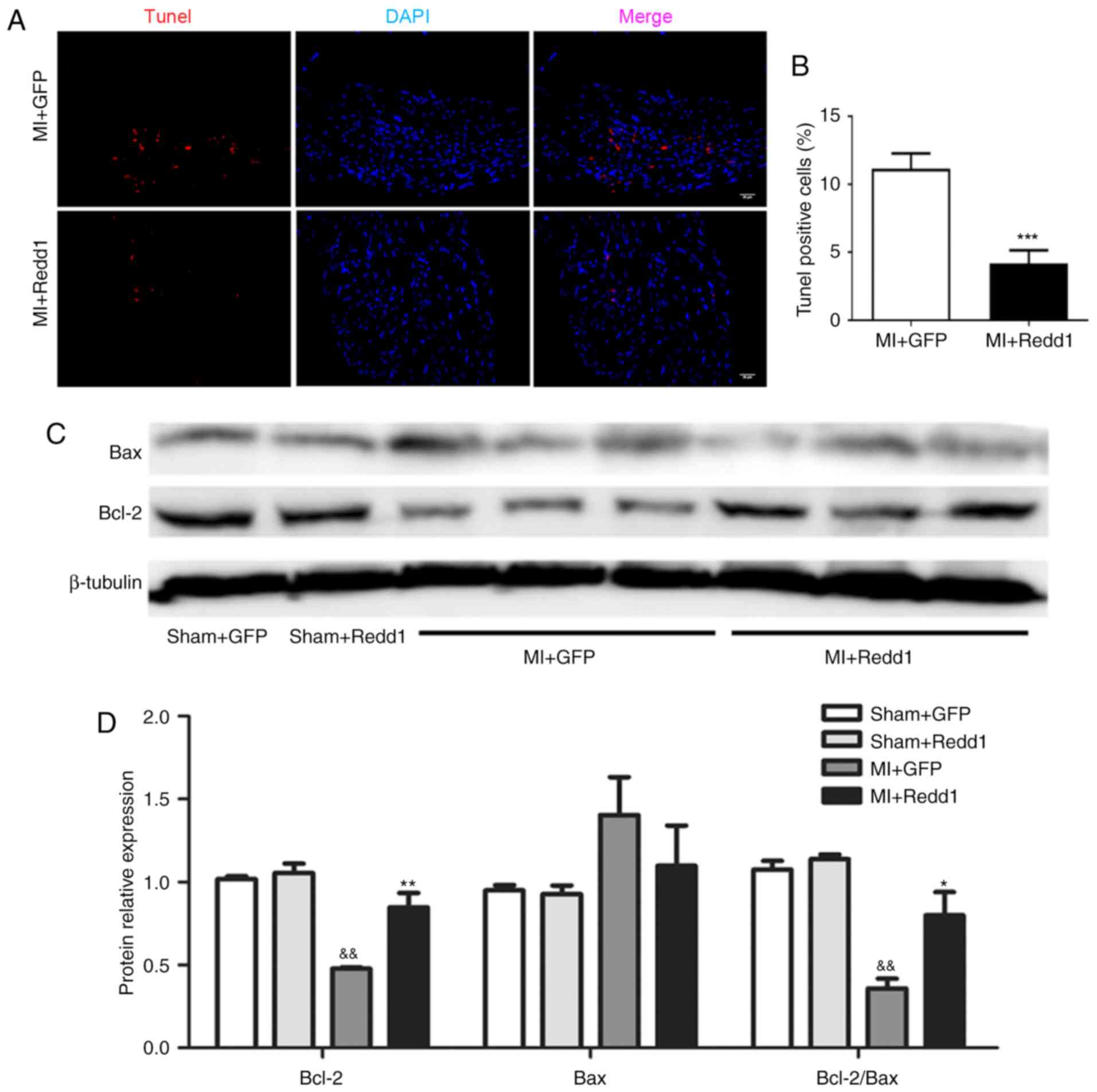
Redd1 overexpression inhibits myocardial
apoptosis after MI
In order to explore the effect of Redd1 on cardiac
cell apoptosis in the infarct zone, TUNEL staining was conducted. A
total of 4 weeks after MI, it was found that the MI+Redd1 group
mice displayed decreased TUNEL-positive cells compared with the
MI+GFP group (Fig. 4A and B). In
addition, the expression of Bcl-2 family members in the infarct
zone was examined through western blot assay. The MI+Redd1 group
mice exhibited a significant increase in the expression of Bcl-2
and the ratio of Bcl-2/Bax compared with MI+GFP group mice
(P<0.01; Fig. 4C and D). These
results showed that Redd1 could attenuate cardiac cell apoptosis in
response to ischemia injury.
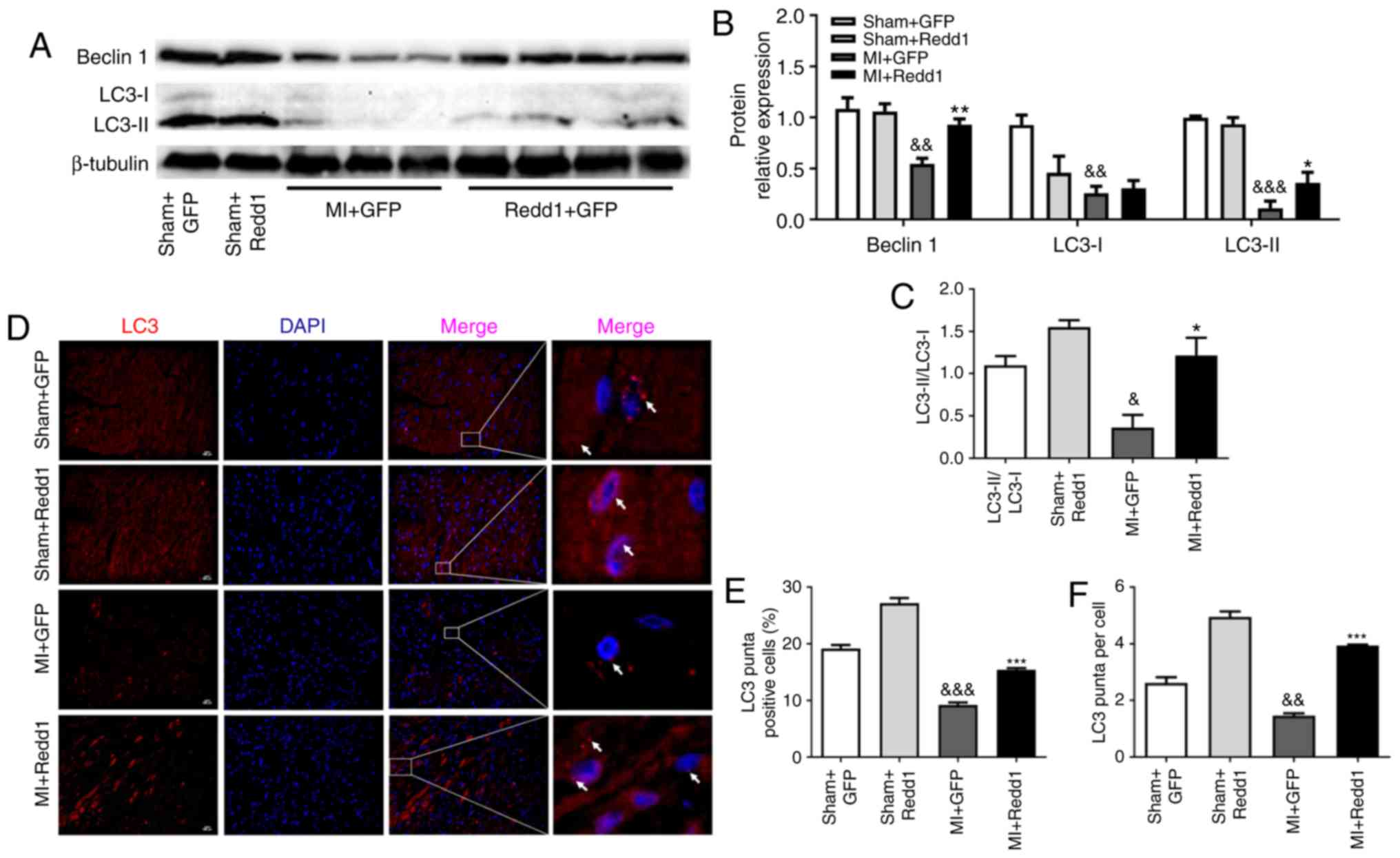
Redd1 overexpression enhances myocardial
autophagy after MI
To investigate the effect of Redd1 on myocardial
autophagy in the border zone of the infarction, LC3 and Beclin1
levels were measured through western blotting. A total of 4 weeks
following LAD ligation, Beclin1 expression and the LC3-II/LC3-I
ratio were increased in the MI+Redd1 group mice compared with in
the MI+GFP group mice (Fig.
5A-C). Similarly, the ratio of LC3 puncta positive cells to the
total cells in the border zone was increased in the MI+Redd1 group
mice compared with in the MI+GFP group mice, as well as LC3 puncta
per cell (Fig. 5D-F). These
results demonstrated that overexpression of Redd1 could improve
cardiac cell autophagy at 4 weeks after MI.
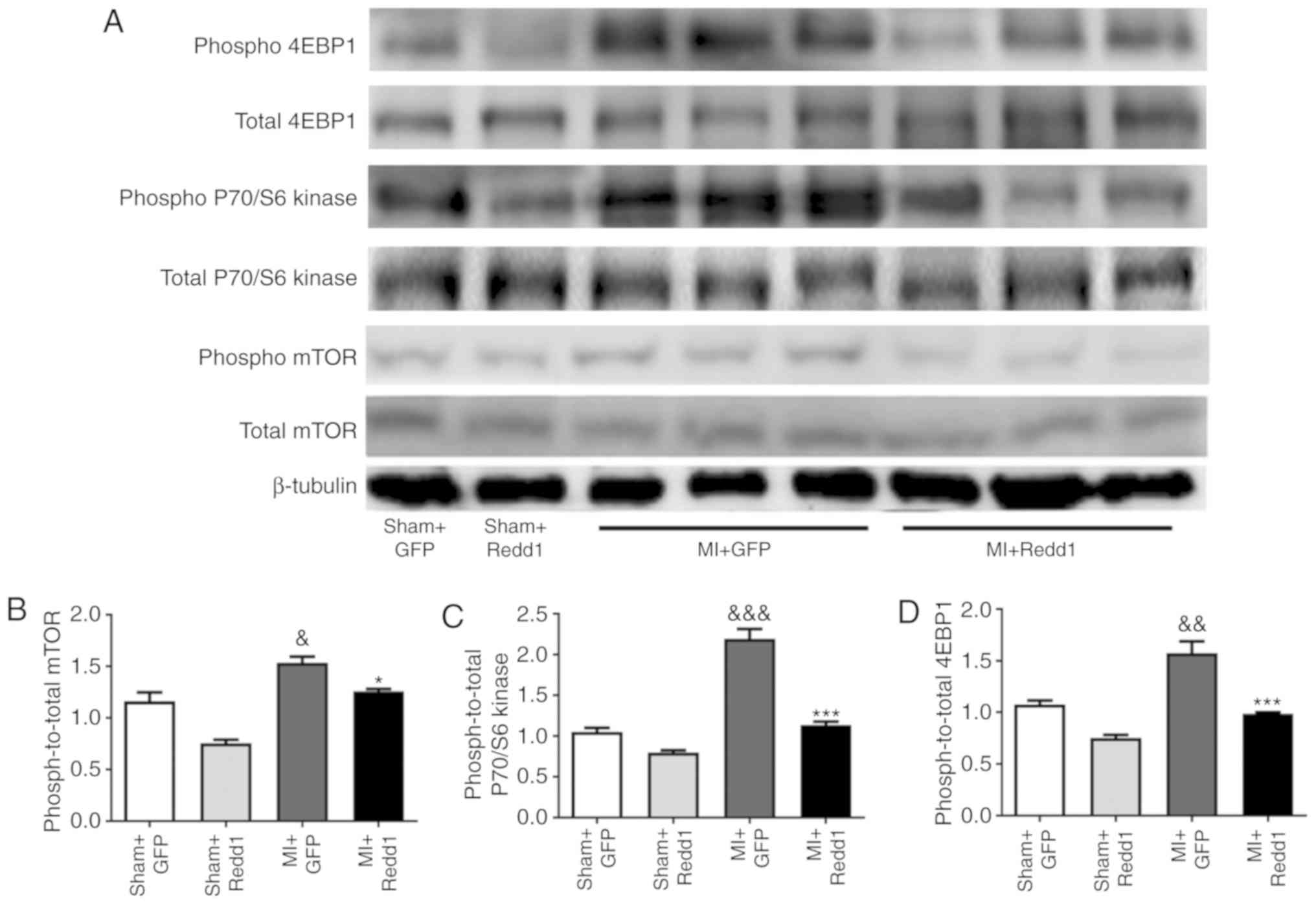
Redd1 overexpression downregulates the
mTOR/P70/S6 kinase/4EBP1 pathway after MI
To further study whether the mTOR signaling pathway
is involved in the regulation of MI mediated by Redd1, the
influence of Redd1 overexpression on mTOR phosphorylation level in
the border zone of the infarction was measured. MI surgery resulted
in increased mTOR phosphorylation, while Redd1 overexpression
partially inhibited this effect (Fig.
6A and B). The phosphorylation state of P70/S6 kinase and 4EBP1
was examined and targets of the mTOR1 complex were established. As
shown in Fig. 6, Redd1 also
significantly decreased the phosphorylation levels of P70/S6 kinase
and 4EBP1 (P<0.001; Fig. 6C and
D). These results suggested that Redd1 could alleviate ischemia
injury induced ventricular dysfunction partly through the mTOR
signaling pathway.
Discussion
MI is a major cause of morbidity and mortality
worldwide (1,2). Although significant progress has
been made in minimizing myocardial loss after MI, large numbers of
patients still develop heart failure due to myocardial remodeling
(41). The present study
demonstrated a critical role for Redd1 in post-MI cardiac
dysfunction. The Redd1 expression level decreased in different
regions (border area and infarct area) of the infarcted myocardium
at 4 weeks. Cardiac-specific overexpression of Redd1 improved
cardiac function and decreased cardiac dilatation. This protective
effect was produced via inhibiting apoptosis and enhancing
autophagy. This pattern indicates that Redd1 plays a protective
role in myocardial ischemia injury.
Redd1 has been identified as a critical
stress-regulated protein whose expression is transcriptionally
induced in various types of cells by DNA damage involved in a
variety of physiological and pathological processes (18). Most studies indicate that Redd1
serves an important role in inflammation and autophagy (20-22). In addition, Liu et al
(27) explored whether Redd1
attenuated cardiac hypertrophy induced by phenylephrine via
enhancing autophagy. Consistent with these results, the present
study also demonstrated the protective effect of Redd1 in post-MI
cardiac dysfunction. Paradoxically, previous studies have
illustrated that inhibition of Redd1 could protect cardiac against
I/R injury (42,43). Recently, Gao et al
(44) demonstrated that increased
Redd1 expression contributed to I/R injury by exaggerating
excessive autophagy during reperfusion. The present study supposed
that the contradictory conclusion was due to the different models,
as Hashmi and Al-Salam (45) has
declared that the processes of cardiomyocyte injury in MI and I/R
were indeed distinct. In addition, the local microenvironment of
the myocardium determined the particular roles of molecules and
enzymes that were part of their pathogenesis. Actually, the
pathological changes, such as inflammation, apoptosis and autophagy
in MI vary along with time (46).
It makes more sense for further experiments to explore the
functions of Redd1 in different periods of MI.
To inspect the underlying mechanism that Redd1
protects against cardiac remodeling and dysfunction, the present
study investigated the effect of Redd1 on cardiac fibrosis and
apoptosis. The occurrence of cardiac fibrosis has been identified
as a definitive characteristic of pathological remodeling and
results in cardiac dysfunction post MI (47). Previous research has demonstrated
that cardiac fibrosis not only causes systolic dysfunction, but
also interrupts the coordination of myocardial
excitation-contraction coupling (48). The results of the present study
showed that overexpression of Redd1 alleviated heart failure and
cardiac fibrosis induced by MI in vivo, as indicated by
increased cardiac function and decreased collagen content.
Apoptosis, a process of programmed cell death, has been identified
in the pathogenesis of myocardial ischemia injury (7,49)
and could determine the fate of the heart (50). In the present study, Redd1
overexpression could inhibit myocardial apoptosis in post-MI hearts
in vivo. The results indicate that Redd1 can inhibit
apoptosis by upregulating the expression of anti-apoptotic
molecules Bcl-2 and the ratio of Bcl-2/Bax. However, the exact
mechanism underlying Redd1-mediated apoptosis inhibition post MI is
not well understood.
Autophagy is a crucial intracellular process which
can increase protein turnover and may be involved in preventing the
accumulation of aberrant proteins or impaired organelles (51). Autophagy deficiency may be
responsible for polyubiquitinated protein accumulation and
endoplasmic reticulum stress elevation (52). Autophagy seems to regulate cardiac
homeostasis in response to various stresses and has a protective
role in the cardiac response to ischemia by removing damaged
mitochondria (8,53). A previous study demonstrated that
impaired autophagy contributes to adverse cardiac remodeling post
MI (54). It has been reported
that enhancing autophagy could decrease infarction size and
alleviate adverse cardiac remodeling (55), while suppressing autophagy led to
adverse cardiac remodeling following MI. The results revealed that
autophagy plays a dynamic role in the determination of infarct size
and cardiac function. In line with the previous studies, the
present study observed that the cardiac cells of MI mice exhibited
a lower rate of autophagy (46,56). This was indicated by 2
well-defined markers of Beclin1 (an indicator of autophagy
induction) and conversion of LC3-I to lipidated LC3-II (an index of
autophagosome abundance), which suggested that impaired autophagy
was related to the pathological mechanism of cardiac remodeling in
response to MI. However, Redd1 over-expression improved cardiac
cell autophagy, accompanied by elevated heart function and reduced
cardiac expansion. Based on these results, it can be concluded that
Redd1 serves a protective role in enhancing autophagy, therefore
ameliorating cardiac remodeling post MI.
mTOR is recognized as a highly conserved
serine/threonine kinase that controls cellular metabolism and
growth in response to diverse stimuli (11,57,58). Studies have demonstrated that mTOR
is necessary for embryonic cardiovascular development (59,60). However, selective pharmacological
and genetic inhibition of mTOR was shown to reduce myocardial
damage after acute and chronic MI (17). Previous studies have demonstrated
that mTOR and its downstream signaling pathways can concurrently
regulate apoptosis and autophagy (13-15). Furthermore, inhibiting mTOR
induced apoptosis and autophagy may exert a protective influence
against MI injury (16,17). Consistent with a previous study,
the current results revealed that the phosphorylation of mTOR and
its downstream signaling pathways were induced in response to MI,
along with increased apoptosis and decreased autophagy.
Additionally, Redd1 overexpression could inhibit apoptosis and
enhance autophagy, as well as suppress the phosphorylation of mTOR.
Accordingly, the results of the present study suggest that the
inhibition of mTOR signaling pathway contributes to the protective
effects of Redd1 on reducing apoptosis and improving autophagy in
the myocardium in response to MI surgery. Therefore, the authors
speculated that Redd1 protected against cardiac dysfunction and
remodeling through inhibiting the mTOR pathway.
In conclusion, the results of the present study have
provided the new insight that Redd1 exerts a beneficial influence
in improving cardiac dysfunction post-infarction. The cardiac
protective effects of Redd1 post MI are associated with the
inhibition of apoptosis and the improvement of autophagy. Redd1
exerts its effect, at least in part, by suppressing the mTOR
signaling pathway. However, the precise mechanism by which Redd1
regulates apoptosis and autophagy remains to be elucidated, and
further investigations are underway. From the clinical point of
view, Redd1 could potentially be a therapeutic candidate for the
prevention and treatment of MI-induced irreversible myocardial
remodeling and heart failure, reducing the risk of mortality. From
a clinical point of view, Redd1 could potentially be a promising
therapeutic candidate for patients with MI and other cardiovascular
diseases.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Nature Science Foundation of China (grant no. 8157051059), the
National Nature Science Foundation of China (grant no. 81601217)
and the Natural Science Foundation of Hubei Province (grant no.
2017CFB627).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
PPH designed and performed the study, analyzed the
data and wrote the manuscript. JF contributed to conducting the
experiments, analyzing data and writing the manuscript. LC, CHJ,
KFW and HXL were involved in performing the study. YL and BMQ
contributed to data analysis and interpretation. BLQ and LHL
conceived the study, participated in its design and helped to draft
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Ethics
committee of Tongji Medical college, Huazhong University of Science
and Technology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Michaud CM: Murray CJ and Bloom BR: Burden
of disease-implications for future research. JAMA. 285:535–539.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deedwania PC: The key to unraveling the
mystery of mortality in heart failure: An integrated approach.
Circulation. 107:1719–1721. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White HD, Norris RM, Brown MA, Brandt PW,
Whitlock RM and Wild CJ: Left ventricular end-systolic volume as
the major determinant of survival after recovery from myocardial
infarction. Circulation. 76:44–51. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gajarsa JJ and Kloner RA: Left ventricular
remodeling in the post-infarction heart: A review of cellular,
molecular mechanisms, and therapeutic modalities. Heart Fail Rev.
16:13–21. 2011. View Article : Google Scholar
|
|
5
|
Chen J, Hsieh AF, Dharmarajan K, Masoudi
FA and Krumholz HM: National trends in heart failure
hospitalization after acute myocardial infarction for medicare
beneficiaries: 1998-2010. Circulation. 128:2577–2584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maejima Y, Kyoi S, Zhai P, Liu T, Li H,
Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al: Mst1
inhibits autophagy by promoting the interaction between Beclin1 and
Bcl-2. Nat Med. 19:1478–1488. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Konstantinidis K, Whelan RS and Kitsis RN:
Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc
Biol. 32:1552–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan L, Sadoshima J, Vatner DE and Vatner
SF: Autophagy: A novel protective mechanism in chronic ischemia.
Cell Cycle. 5:1175–1177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou L, Guo J, Xu F, Weng X, Yue W and Ge
J: Cardiomyocyte dimethylarginine dimethylaminohydrolase1
attenuates left-ventricular remodeling after acute myocardial
infarction: Involvement in oxdative stress and apoptosis. Basic Res
Cardiol. 113:282018. View Article : Google Scholar
|
|
10
|
Liu CY, Zhang YH, Li RB, Zhou LY, An T,
Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, et al: LncRNA CAIF
inhibits autophagy and attenuates myocardial infarction by blocking
p53-mediated myocardin transcription. Nat Commun. 9:292018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Betz C and Hall MN: Where is mTOR and what
is it doing there? J Cell Biol. 203:563–574. 2013. View Article : Google Scholar :
|
|
12
|
McMullen JR, Shioi T, Zhang L, Tarnavski
O, Sherwood MC, Kang PM and Izumo S: Phosphoinositide
3-kinase(p110alpha) plays a critical role for the induction of
physiological, but not pathological, cardiac hypertrophy. Proc Natl
Acad Sci USA. 100:12355–12360. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boluyt MO, Zheng JS, Younes A, Long X,
O'Neill L, Silverman H, Lakatta EG and Crow MT: Rapamycin inhibits
alpha 1-adren-ergic receptor-stimulated cardiac myocyte hypertrophy
but not activation of hypertrophy-associated genes. Evidence for
involvement of p70 S6 kinase Circ Res. 81:176–186. 1997.
|
|
14
|
Mazelin L, Panthu B, Nicot AS, Belotti E,
Tintignac L, Teixeira G, Zhang Q, Risson V, Baas D, Delaune E, et
al: mTOR inactivation in myocardium from infant mice rapidly leads
to dilated cardio-myopathy due to translation defects and
p53/JNK-mediated apoptosis. J Mol Cell Cardiol. 97:213–225. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McMullen JR, Sherwood MC, Tarnavski O,
Zhang L, Dorfman AL, Shioi T and Izumo S: Inhibition of mTOR
signaling with rapamycin regresses established cardiac hypertrophy
induced by pressure overload. Circulation. 109:3050–3055. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan L, Guo N, Cao Y, Zeng S, Wang J, Lv F,
Wang Y and Cao X: miRNA145 inhibits myocardial infarctioninduced
apoptosis through autophagy via Akt3/mTOR signaling pathway in
vitro and in vivo. Int J Mol Med. 42:1537–1547. 2018.PubMed/NCBI
|
|
17
|
Buss SJ, Muenz S, Riffel JH, Malekar P,
Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M,
Izumo S, et al: Beneficial effects of Mammalian target of rapamycin
inhibition on left ventricular remodeling after myocardial
infarction. J Am Coll Cardiol. 54:2435–2446. 2009. View Article : Google Scholar
|
|
18
|
Ellisen LW, Ramsayer KD, Johannessen CM,
Yang A, Beppu H, Minda K, Oliner JD, McKeon F and Haber DA: REDD1,
a developmentally regulated transcriptional target of p63 and p53,
links p63 to regulation of reactive oxygen species. Mol Cell.
10:995–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tirado-Hurtado I, Fajardo W and Pinto JA:
DNA damage inducible transcript 4 Gene: The switch of the
metabolism as potential target in cancer. Front Oncol. 8:1062018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brugarolas J, Lei K, Hurley RL, Manning
BD, Reiling JH, Hafen E, Witters LA, Ellisen LW and Kaelin WG Jr:
Regulation of mTOR function in response to hypoxia by REDD1 and the
TSC1/TSC2 tumor suppressor complex. Genes Dev. 18:2893–2904. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee DK, Kim JH, Kim J, Choi S, Park M,
Park W, Kim S, Lee KS, Kim T, Jung J, et al: REDD-1 aggravates
endotoxin-induced inflammation via atypical NF-κB activation. FASEB
J. 32:4585–4599. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qiao S, Dennis M, Song X, Vadysirisack DD,
Salunke D, Nash Z, Yang Z, Liesa M, Yoshioka J, Matsuzawa S, et al:
A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to
control stress-induced autophagy and sustain exercise capacity. Nat
Commun. 6:70142015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Canal M, Romani-Aumedes J, Martin-Flores
N, Pérez-Fernández V and Malagelada C: RTP801/REDD1: A stress
coping regulator that turns into a troublemaker in
neurodegen-erative disorders. Front Cell Neurosci. 8:3132014.
View Article : Google Scholar
|
|
24
|
Ben Sahra I, Regazzetti C, Robert G,
Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF,
Giorgetti-Peraldi S and Bost F: Metformin, independent of AMPK,
induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer
Res. 71:4366–4372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hernandez G, Lal H, Fidalgo M, Guerrero A,
Zalvide J, Force T and Pombo CM: A novel cardioprotective
p38-MAPK/mTOR pathway. Exp Cell Res. 317:2938–2949. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen R, Wang B, Chen L, Cai D, Li B, Chen
C, Huang E, Liu C, Lin Z, Xie WB and Wang H: DNA damage-inducible
transcript 4 (DDIT4) mediates methamphetamine-induced autophagy and
apoptosis through mTOR signaling pathway in cardiomyocytes. Toxicol
Appl Pharmacol. 295:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu C, Xue R, Wu D, Wu L, Chen C, Tan W,
Chen Y and Dong Y: REDD1 attenuates cardiac hypertrophy via
enhancing autophagy. Biochem Biophys Res Commun. 454:215–220. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sciarretta S, Volpe M and Sadoshima J:
Mammalian target of rapamycin signaling in cardiac physiology and
disease. Circ Res. 114:549–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Volkers M, Konstandin MH, Doroudgar S,
Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ,
et al: Mechanistic target of rapamycin complex 2 protects the heart
from ischemic damage. Circulation. 128:2132–2144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao Y, Xiong X, Jia L and Sun Y:
Targeting Cullin-RING ligases by MLN4924 induces autophagy via
modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis.
3:e3862012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prasad KM, Xu Y, Yang Z, Acton ST and
French BA: Robust cardiomyocyte-specific gene expression following
systemic injection of AAV: In vivo gene delivery follows a poisson
distribution. Gene Ther. 18:43–52. 2011. View Article : Google Scholar
|
|
32
|
Wang X, Meng H, Chen P, Yang N, Lu X, Wang
ZM, Gao W, Zhou N, Zhang M, Xu Z, et al: Beneficial effects of
muscone on cardiac remodeling in a mouse model of myocardial
infarction. Int J Mol Med. 34:103–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oliveira AC, Melo MB, Motta-Santos D,
Peluso AA, Souza-Neto F, da Silva RF, Almeida JFQ, Canta G, Reis
AM, Goncalves G, et al: Genetic deletion of the alamandine receptor
MRGD leads to dilated cardiomyopathy in mice. Am J Physiol Heart
Circ Physiol. 316:H123–H133. 2019. View Article : Google Scholar
|
|
34
|
Jia LX, Qi GM, Liu O, Li TT, Yang M, Cui
W, Zhang WM, Qi YF and Du J: Inhibition of platelet activation by
clopidogrel prevents hypertension-induced cardiac inflammation and
fibrosis. Cardiovasc Drugs Ther. 27:521–530. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dai W, Hale SL, Martin BJ, Kuang JQ, Dow
JS, Wold LE and Kloner RA: Allogeneic mesenchymal stem cell
transplantation in postinfarcted rat myocardium: Short- and
long-term effects. Circulation. 112:214–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hochman JS and Choo H: Limitation of
myocardial infarct expansion by reperfusion independent of
myocardial salvage. Circulation. 75:299–306. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lubbe WF, Peisach M, Pretorius R, Bruyneel
KJ and Opie LH: Distribution of myocardial blood flow before and
after coronary artery ligation in the baboon. Relation to early
ventricular fibrillation. Cardiovasc Res. 8:478–487. 1974.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
39
|
Lee EJ, Park SJ, Kang SK, Kim GH, Kang HJ,
Lee SW, Jeon HB and Kim HS: Spherical bullet formation via
E-cadherin promotes therapeutic potency of mesenchymal stem cells
derived from human umbilical cord blood for myocardial infarction.
Mol Ther. 20:1424–1433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Yang R, Guo B, Zhang H and Liu S:
Exosomal miR-301 derived from mesenchymal stem cells protects
myocardial infarction by inhibiting myocardial autophagy. Biochem
Biophys Res Commun. 514:323–328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hochman JS, Reynolds HR, Dzavik V, Buller
CE, Ruzyllo W, Sadowski ZP, Maggioni AP, Carvalho AC, Rankin JM,
White HD, et al: Long-term effects of percutaneous coronary
intervention of the totally occluded infarct-related artery in the
subacute phase after myocardial infarction. Circulation.
124:2320–2328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park KM, Teoh JP, Wang Y, Broskova Z,
Bayoumi AS, Tang Y, Su H, Weintraub NL and Kim IM:
Carvedilol-responsive microRNAs, miR-199a-3p and -214 protect
cardiomyocytes from simulated ischemia-reperfusion injury. Am J
Physiol Heart Circ Physiol. 311:H371–H383. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou Y, Chen Q, Lew KS, Richards AM and
Wang P: Discovery of potential therapeutic miRNA targets in cardiac
ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther.
21:296–309. 2016. View Article : Google Scholar
|
|
44
|
Gao C, Wang R, Li B, Guo Y, Yin T, Xia Y,
Zhang F, Lian K, Liu Y, Wang H, et al: TXNIP/Redd1 signalling and
excessive autophagy: A novel mechanism of myocardial
ischaemia/reper-fusion injury in mice. Cardiovasc Res. Jun
26–2019.Epub ahead of print. View Article : Google Scholar
|
|
45
|
Hashmi S and Al-Salam S: Acute myocardial
infarction and myocardial ischemia-reperfusion injury: A
comparison. Int J Clin Exp Pathol. 8:8786–8796. 2015.PubMed/NCBI
|
|
46
|
Wang X, Guo Z, Ding Z and Mehta JL:
Inflammation, autophagy, and apoptosis after myocardial infarction.
J Am Heart Assoc. 7:pii: e008024. 2018. View Article : Google Scholar
|
|
47
|
Creemers EE and Pinto YM: Molecular
mechanisms that control interstitial fibrosis in the
pressure-overloaded heart. Cardiovasc Res. 89:265–272. 2011.
View Article : Google Scholar
|
|
48
|
Berk BC, Fujiwara K and Lehoux S: ECM
remodeling in hypertensive heart disease. J Clin Invest.
117:568–575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ren J, Zhang S, Kovacs A, Wang Y and
Muslin AJ: Role of p38alpha MAPK in cardiac apoptosis and
remodeling after myocardial infarction. J Mol Cell Cardiol.
38:617–623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Krijnen PA, Nijmeijer R, Meijer CJ, Visser
CA, Hack CE and Niessen HW: Apoptosis in myocardial ischaemia and
infarction. J Clin Pathol. 55:801–811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lekli I, Haines DD, Balla G and Tosaki A:
Autophagy: An adaptive physiological countermeasure to cellular
senescence and ischaemia/reperfusion-associated cardiac
arrhythmias. J Cell Mol Med. 21:1058–1072. 2017. View Article : Google Scholar
|
|
53
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu X, He L, Chen F, He X, Cai Y, Zhang G,
Yi Q, He M and Luo J: Impaired autophagy contributes to adverse
cardiac remodeling in acute myocardial infarction. PLoS One.
9:e1128912014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wu P, Yuan X, Li F, Zhang J, Zhu W, Wei M,
Li J and Wang X: Myocardial upregulation of cathepsin D by ischemic
heart disease promotes autophagic flux and protects against cardiac
remodeling and heart failure. Circ Heart Fail. 10:pii: e004044.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jewell JL and Guan KL: Nutrient signaling
to mTOR and cell growth. Trends Biochem Sci. 38:233–242. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Johnson SC, Rabinovitch PS and Kaeberlein
M: MTOR is a key modulator of ageing and age-related disease.
Nature. 493:338–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Land SC, Scott CL and Walker D: MTOR
signalling, embryo-genesis and the control of lung development.
Semin Cell Dev Biol. 36:68–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sciarretta S, Forte M, Frati G and
Sadoshima J: New insights into the role of mTOR signaling in the
cardiovascular system. Circ Res. 122:489–505. 2018. View Article : Google Scholar : PubMed/NCBI
|