Introduction
Bladder cancer, the ninth most common type of cancer
worldwide, is associated with a high recurrence rate (1). Considering that the tumor
microenvironment (TME) plays an important role in bladder cancer
growth, research on the TME has been prevalent. The richly diverse
TME implies that tumor cells need adequate nutrients for growth
through energy metabolism. Glutamine (Gln), one of the most
abundant amino acids in the TME, is mainly involved in critical
physiological processes, such as energy synthesis, biosynthesis,
antioxidant defense and cell signaling regulation (2,3).
In fact, Gln can serve as an alternative metabolite
to drive the tricarboxylic acid (TCA) cycle for energy generation,
exerting a 'replenishment effect' (4). After being transferred into a cell
via a transporter on the cell membrane, Gln participates in the TCA
cycle with glutamate (Glu) via glutaminase (GLS), which produces
2-oxoglutarate (αKG) via Glu dehydrogenase (GLUD1) (5).
In addition to being metabolic fuel, Gln can help
neutralize reactive oxygen species (ROS) (6,7).
ROS, such as superoxide anions and hydrogen peroxide, can act as
intracellular second messengers and activate apoptosis (8). Glu, the product of glutaminolysis,
is also directly used for the synthesis of the antioxidant,
glutathione (9). A significant
fraction of Gln-derived carbon can be used to produce NADPH for
redox balance (10). Thus, Gln
becomes an essential amino acid for Gln-dependent cancer cells, and
this process is related to cancer cell viability (2,11,12).
However, in contrast to the above description,
Cacace et al (13)
proposed that Gln activates signal transducer and activator of
transcription 3 (STAT3) to control cancer cell proliferation,
independently of its activity as a metabolic fuel or ROS scavenger.
The overactivation of STAT3, a protein present in the cytoplasm
that is coupled with the tyrosine phosphorylation signaling
pathway, results in aberrant cell proliferation and apoptosis, and
promotes tumor formation and development (14,15). It is well known that STAT3 is
activated through phosphorylation on Y705 or S727, after which it
binds to extracellular signaling proteins. The activated proteins
can be translocated to the nucleus, where they bind to the
promoters of genes involved in cell survival, cell cycling,
invasion, migration and angiogenesis (16).
Therefore, we sought to determine whether the
characteristics of Gln metabolism in the bladder cancer cell line,
T24, are consistent with the mechanisms proposed by Cacace et
al (13). Existing research
on the mechanisms through which Gln promotes the proliferation of
bladder cancer cells remains inadequate.
Materials and methods
Cells and reagents
The bladder cancer cell line, T24, purchased from
the Cell Bank of the Chinese Academy of Sciences, was routinely
cultured in RPMI-1640 medium (BI) containing 2 g/l glucose and 300
mg/l Gln. The assay medium was modified Eagle's medium (BI) without
glucose or Gln reconstituted with 2 g/l of glucose. Both media were
supplemented with 10% fetal bovine serum and 1% penicillin and
streptomycin. The cells were grown at 37°C in a humidified 5%
CO2 atmosphere. L-Gln (Sigma-Aldrich), D-(+)-glucose
(Sigma-Aldrich), 0-100 µM 6-diazo-5-oxo-L-norleucine (Don)
(Sigma-Aldrich), 0-10 mM N-acetylcysteine (NAC) (MCE), 2 mM
L-glutamic acid dimethyl ester hydrochloride (Glu) (Sigma-Aldrich)
and 2 mM dimethyl αKG (Sigma-Aldrich) were used during the
experiment. The reagents were dissolved either in ultrapure water
or directly in modified Eagle's medium (BI) according to the
manufacturer's indications. All drugs and reagents were
administered to adherent cells in fresh assay medium.
Cell proliferation assay
Cell proliferation was assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, the T24 cells were plated into 96-well plates at a
density of 5x103 cells/well. Following 24 h of
incubation at 37°C, the medium was replaced with 200 µl of
specific medium for 24, 48 or 72 h. A total of 10 µl of MTT
was added to the wells, and the cells were incubated for a further
4 h at 37°C. Finally, 100 µl of dimethyl sulfoxide (DMSO)
were added to each well, and the wells were gently mixed for 1 min.
Finally, the cell numbers were measured spectrophoto-metrically by
an absorbance microplate reader (Thermo Fisher Scientific) at 490
nm.
Cell cycle assays
The T24 cells were seeded in 6-well plates, grown
for 24 h, and then incubated in Gln(+) or Gln(-) medium. After 24
or 48 h, a cell cycle assay was conducted as previously described
(17). Following the instructions
of the kit, the cells were fixed with 75% cold ethanol overnight
and then washed with phosphate-buffered saline (PBS). Subsequently,
propidium iodide (50 µg/ml) containing RNase was added to
the cells for DNA staining before flow cytometric analysis.
Measurement of intracellular adenosine
triphosphate (ATP) levels
The T24 cells were cultured in a 6-well plate at a
density of 2x105 cells/well for 24 h and then cultured
under various Gln conditions for 48 h. The supernatant was added to
a 96-well plate with a black border according to the protocol
provided by the manufacturer for a commercial ATP assay kit
(Beyotime Institute of Biotechnology) and as previously described
by Castaneda et al (18).
The assay buffer was gently mixed with the substrate at room
temperature, and the mixed reagent (100 µl) was added to
each well. The plate was then incubated with shaking for 15 min at
room temperature. Following incubation, luminescence was measured
using a microplate reader (Beckman Coulter).
Measurement of intracellular ROS
levels
ROS levels were determined using
2',7'-dichlorodihydrofluorescein diacetate (H2DCF-DA)
with a Reactive Oxygen Species Assay kit (Beyotime Institute of
Biotechnology). The T24 cells were pre-seeded into 6-well plates at
2x105/well and then cultured with Gln(+) or Gln(-)
medium for 48 h. The cells were then treated with 10 mM
H2DCF-DA dissolved in PBS (1 ml) at 37°C for 20 min. The
assay was conducted as previously described (19). The fluorescence intensity was
monitored at an excitation wavelength of 488 nm and an emission
wavelength of 530 nm. In addition, Applied 1.0-2.0 ml of 5
µM MitoSOX™ reagent working solution was added to cover the
cells adhering to the coverslip. The cells were incubated for 10
min at 37°C, protected from light. The cells were then washed
gently 3 times with warm buffer as described in the MitoSOX™ Red
mitochondrial superoxide indicator *for live-cell imaging*
(Molecular Probes). Finally, we observed the intracellular red
fluorescence under a confocal fluorescence microscope (BD
Biosciences). ROSUP was added to the positive control well as a
positive control, which can significantly increase the level of ROS
following stimulation of the cells for 20-30 min.
Apoptosis assay
The T24 cells were pre-seeded in a 6-well plate at a
density of 3x105 cells/well with RPMI-1640 medium for 24
h and were then cultured with Gln(+), Gln(-), Gln(-)+αKG, or
Gln(-)+Glu medium for 48 h. Cell morphology was assessed as
previously described (20) with
an Apoptosis and Necrosis Assay kit (Beyotime Institute of
Biotechnology). Briefly, cells in all groups were washed 3 times
with cold PBS, and the cells in the supernatant and those that
adhered to the plate were then collected. Following incubation with
Annexin V-FITC for 15 min in the dark at 37°C, the cells were
washed 3 more times with cold PBS. The results were analyzed by
flow cytometry (BD Biosciences).
Western blot analysis
The cells were lysed in cell lysis buffer (Beyotime
Institute of Biotechnology) containing 1 mM phenylmethylsulfonyl
fluoride. Following determination with the BCA method, the protein
samples (30 µg protein/lane) were then subjected to SDS-PAGE
and transferred to a nitrocellulose membrane. The membrane was
blocked with 5% bovine serum albumin and subsequently incubated
overnight at 4°C with the appropriate primary antibodies. The
membrane was then incubated with horseradish peroxidase-conjugated
goat anti-rabbit (111-035-045)/mouse (111-035-062) IgG (1:10,000,
Jackson ImmunoResearch Laboratories, Inc.) at 4°C for 2 h. To
verify equal loading of the samples, the same membrane was
incubated with a monoclonal β-actin antibody followed by
horseradish peroxidase-conjugated goat anti-rabbit IgG. The protein
bands were visualized with a FluorChem Q instrument (ProteinSimple,
type: AlphaImager). The data were analyzed using ImageJ software
(NIH). Primary antibodies against total-STAT3 (9139T), p-Y705-STAT3
(4113S), c-Myc (13987S), Bcl2 (15071S) and β-actin (3700S) were
purchased from Cell Signaling Technology, while primary antibodies
against proliferating cell nuclear antigen (PCNA, ab18197), Gls
(ab156876) and Glud1 (ab168352), were purchased from Abcam.
Statistical analysis
Each experiment was performed at least 3 times. All
values are presented as the means ± SD in Prism 7 statistical
software. Differences between 3 or more groups were determined by
one-way analysis of variance (ANOVA) followed by a post hoc test
with Sidak's multiple comparisons test in Prism 7. In addition,
t-tests was performed for comparisons between 2 groups. Differences
were considered statistically significant at P<0.05.
Results
Effects of Gln on T24 bladder cancer cell
proliferation
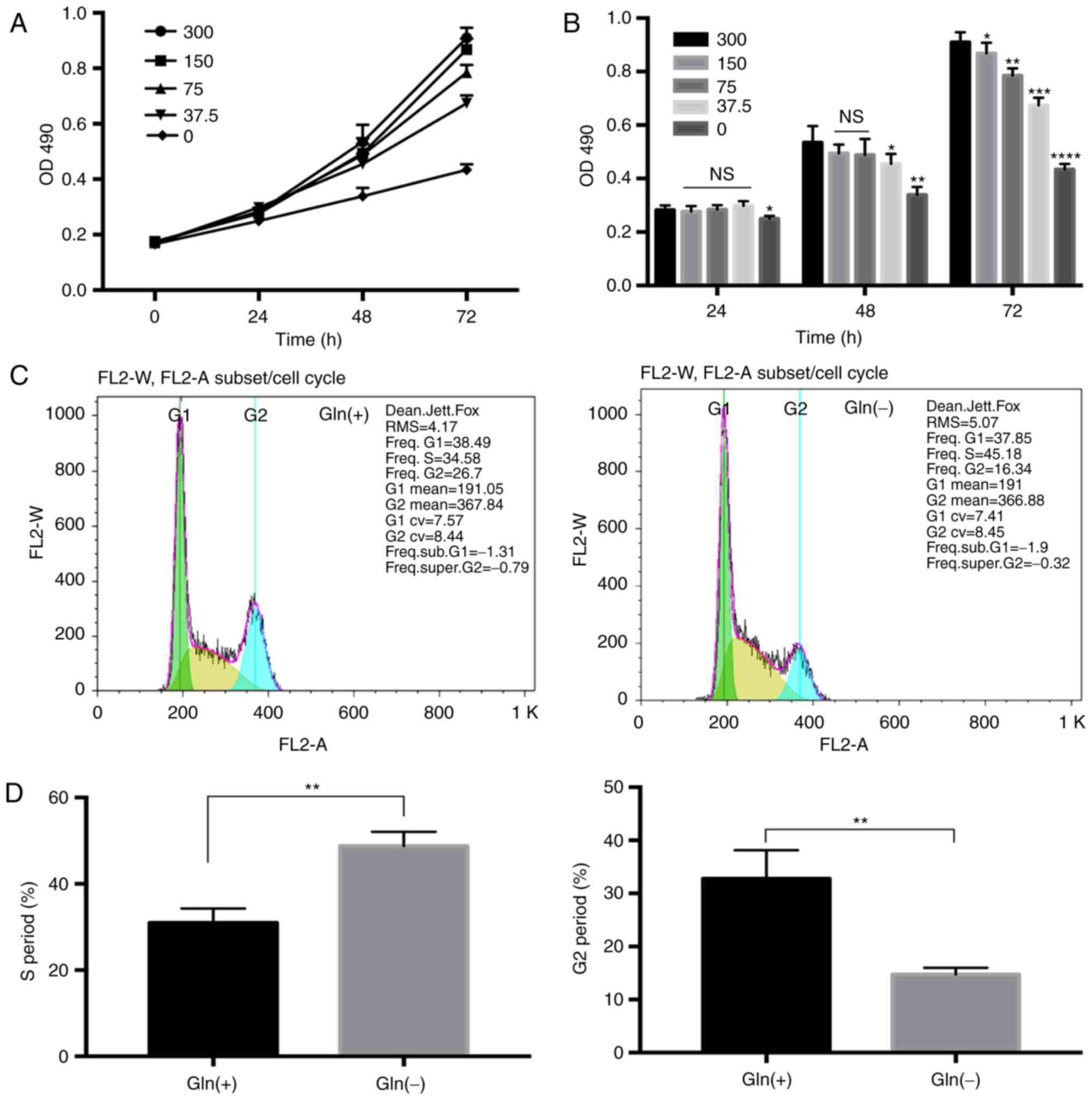
The results of MTT assay revealed that the
proliferation rate of the T24 cells was indeed positively
associated with the Gln concentration. No apparent differences were
observed in the bladder cancer cell proliferation rate among the
groups cultured with the different Gln concentrations (0, 37.5, 75,
150 and 300 mg/l) after 24 h. However, the proliferation rates of
the T24 cells decreased with the decreasing Gln concentrations at
48 and 72 h (P<0.05). The proliferation rate of the Gln
deprivation group was lower than that of the other Gln
concentration groups (Fig. 1A).
As the differences in the proliferation rate were most evident at
72 h (Fig. 1B), we selected 72 h
as the time point for use in further experiments.
Furthermore, compared with that in the Gln(+) group
(300 mg/l), the proportion of cells in the S phase of the cell
cycle in the Gln(-) group (0 mg/l) was approximately 11% higher on
average, while the proportion of cells in the G2 phase was almost
11% lower; cell cycle analysis thus revealed that Gln deprivation
caused T24 growth arrest in the S phase (Fig. 1C and D).
Effect of the inhibition of Gln uptake on
the proliferation of T24 bladder cancer cells
To further identify the critical role of Gln in T24
cell proliferation, we used a Gln analog, Don, to inhibit T24 Gln
utilization, as Don competes for Gln uptake. The effects of various
Don concentrations (0, 12.5, 25, 50 and 100 µM) on T24
proliferation were evaluated. The results revealed that the
proliferation rates of the T24 cells gradually decreased with the
increasing concentrations of Don. Additionally, Don most
effectively inhibited T24 cell proliferation in the presence of
extracellular Gln at the 25 µM concentration (Fig. 2A), contributing to our selection
of 25 µM as the optimal concentration of Don at 72 h
(Fig. 2B). In fact, compared with
the Gln(+) group, the different Gln(+)-Don groups exhibited
significantly decreased proliferation rates similar to those in the
Gln(-) group (Fig. 2C). In
addition, the levels of proteins associated with both Gln
metabolism (GLS and GLUD1) and proliferation (PCNA) were
significantly decreased in the Gln(-) group and the Gln(+)+Don
groups compared with the Gln(+) group (Fig. 2D).
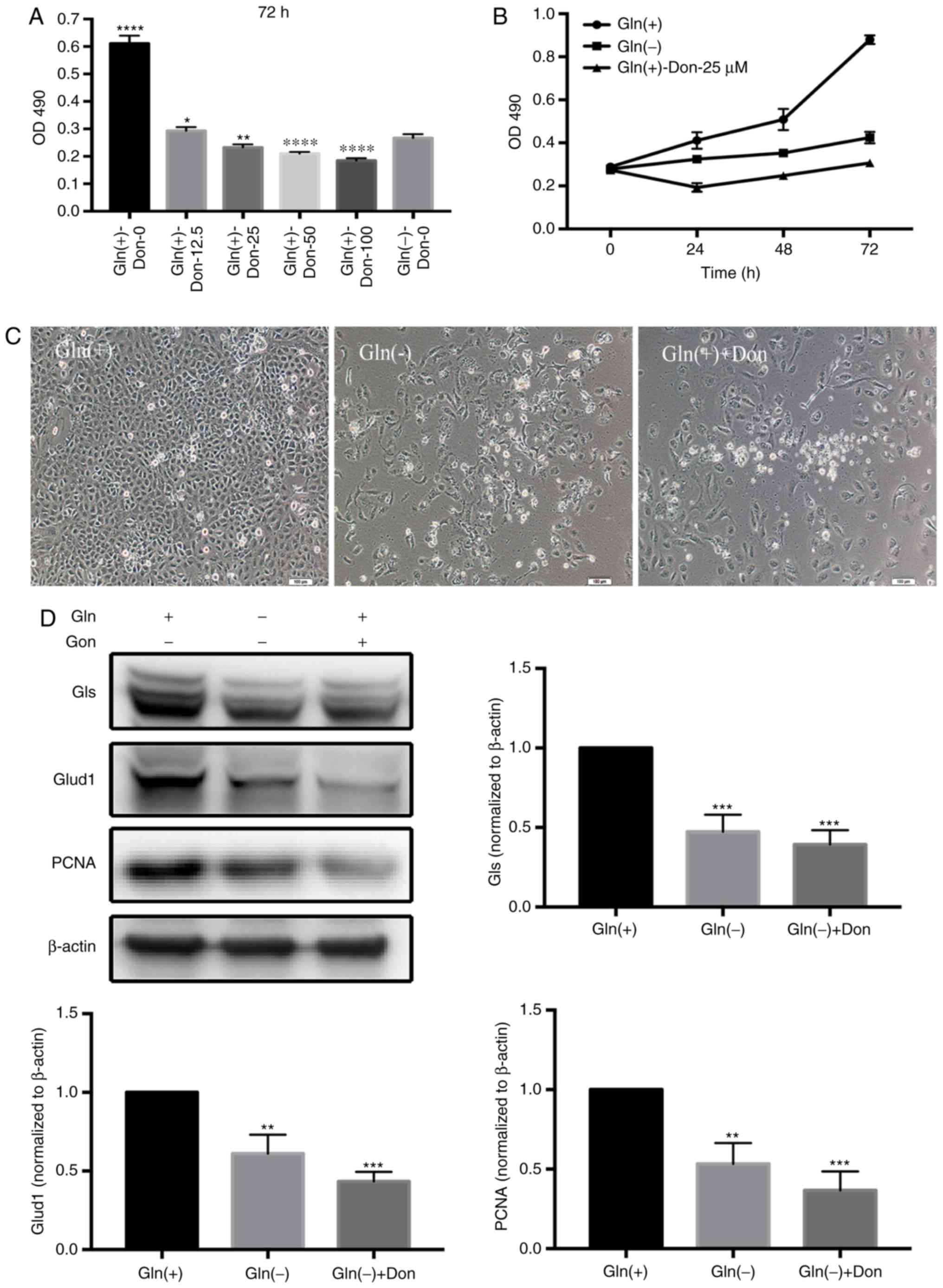 | Figure 2Gln analogs affect T24 bladder cancer
cell proliferation similarly to Gln starvation. (A) Analysis of the
effects of a range of Don concentrations on T24 proliferation
rates. (B) Gln deprivation affected the proliferation rates of T24
bladder cancer cells incubated with the optimum Don concentration
(25 µM) for 24, 48 and 72 h. (C) The growth status of T24
bladder cancer under Gln(+), Gln(-), and Gln(+)+Don conditions.
Scale bars, 100 µm. (D) Western blot analysis of proteins
associated with Gln metabolism (Gls and Glud1) and proliferation
(PCNA) using Gln(+) as the control. One-way ANOVA with a test for
homogeneity of variance was used to compare the cell lines.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. Gln,
glutamine; Don, 6-diazo-5-oxo-L-norleucine; Gls, glutaminase;
Glud1, Glu dehydrogenase. |
Glutaminolysis-dependent ATP production
in T24 cells
As shown in Fig.
3A, Gln serves as a crucial carbon source for cellular
bioenergetic and biosynthetic needs. Cell proliferation is
associated with a high influx of Gln-derived carbon into the TCA
cycle (4). In this study, we
found that the ATP levels in the T24 bladder cancer cells decreased
gradually with the reduction in Gln (Fig. 3B). We divided the cells into 4
groups and treated two of these with the cell-permeable precursors,
dimethyl-Glu and dimethyl αKG, creating the Gln(+), Gln(-),
Gln(-)+αKG, and Gln(-)+Glu groups. When used at a 2 mM
concentration (13), both Glu and
αKG were capable of restoring the proliferation of the Gln-deprived
T24 cells (Fig. 3C). No obvious
differences were observed among the groups after 24 h; however, the
restoring effects of Glu and αKG on cell proliferation were evident
at 48 and 72 h (Fig. 3D). In
addition, both Glu and αKG were able to restore ATP production in
the Gln-deprived cells (Fig. 3E).
In summary, these data indicate that Gln can promote T24 cell
proliferation through ATP supplementation via glutaminolysis.
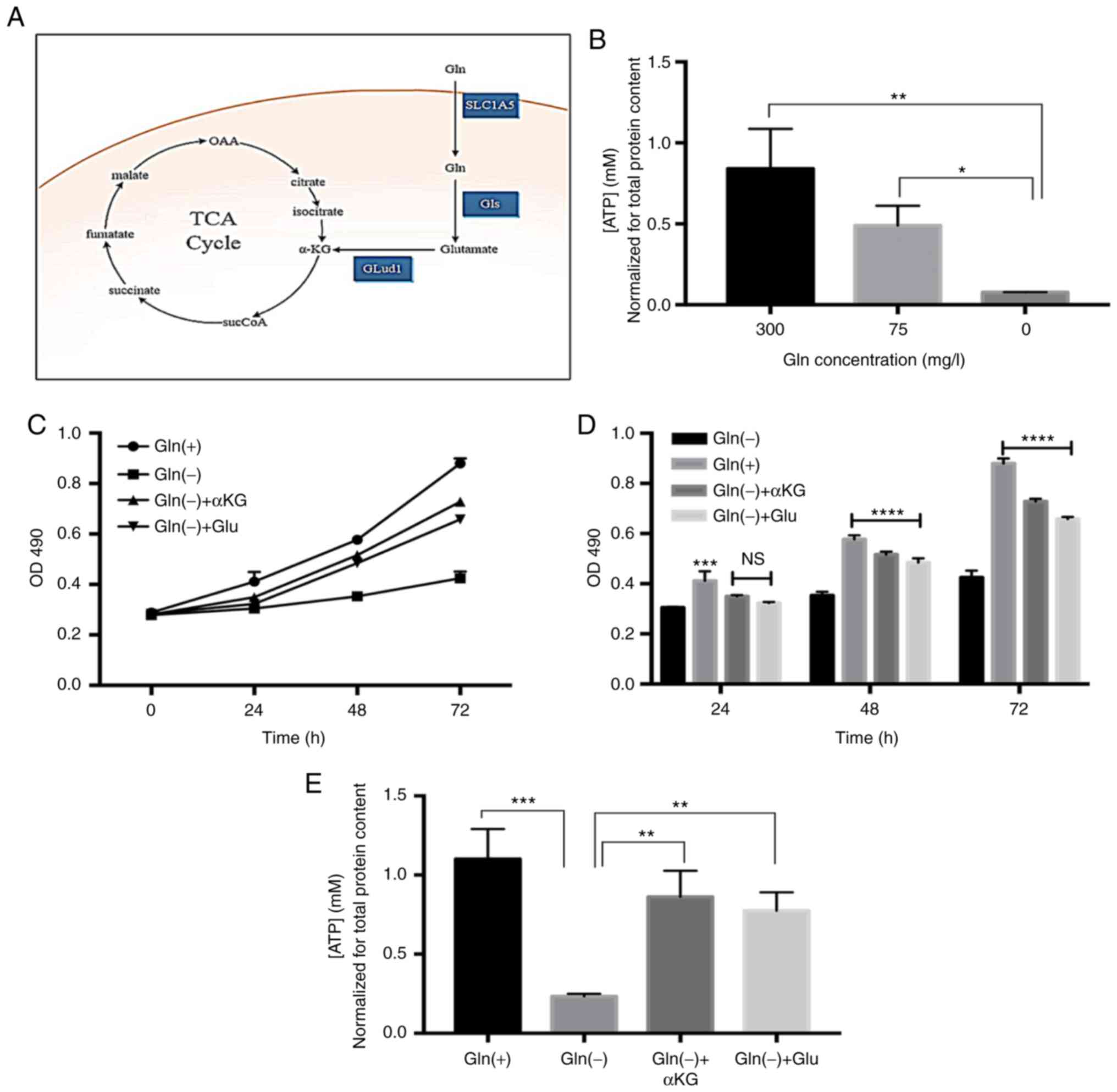 | Figure 3Gln glutaminolysis dependently
promotes ATP production. (A) Gln contributes to the TCA cycle
metabolite pool. Enzymes involved in the metabolism of Gln, Gls and
Glud1. (B) Gln modified ATP content in T24 cells. (C and D)
Supplementation with Gln catabolism intermediates partially
restored cellular proliferation at 24, 48 and 72 h; Gln(-) was used
as the control condition. (E) Supplementation with Gln catabolism
intermediates significantly restored cellular ATP levels. One-way
ANOVA with a test for homogeneity of variance was used to compare
the cell lines. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001; ns, no
significance. Gln, glutamine; Gls, glutaminase; Glud1, Glu
dehydrogenase; SLC1A5, recombinant solute carrier family 1, member
5. |
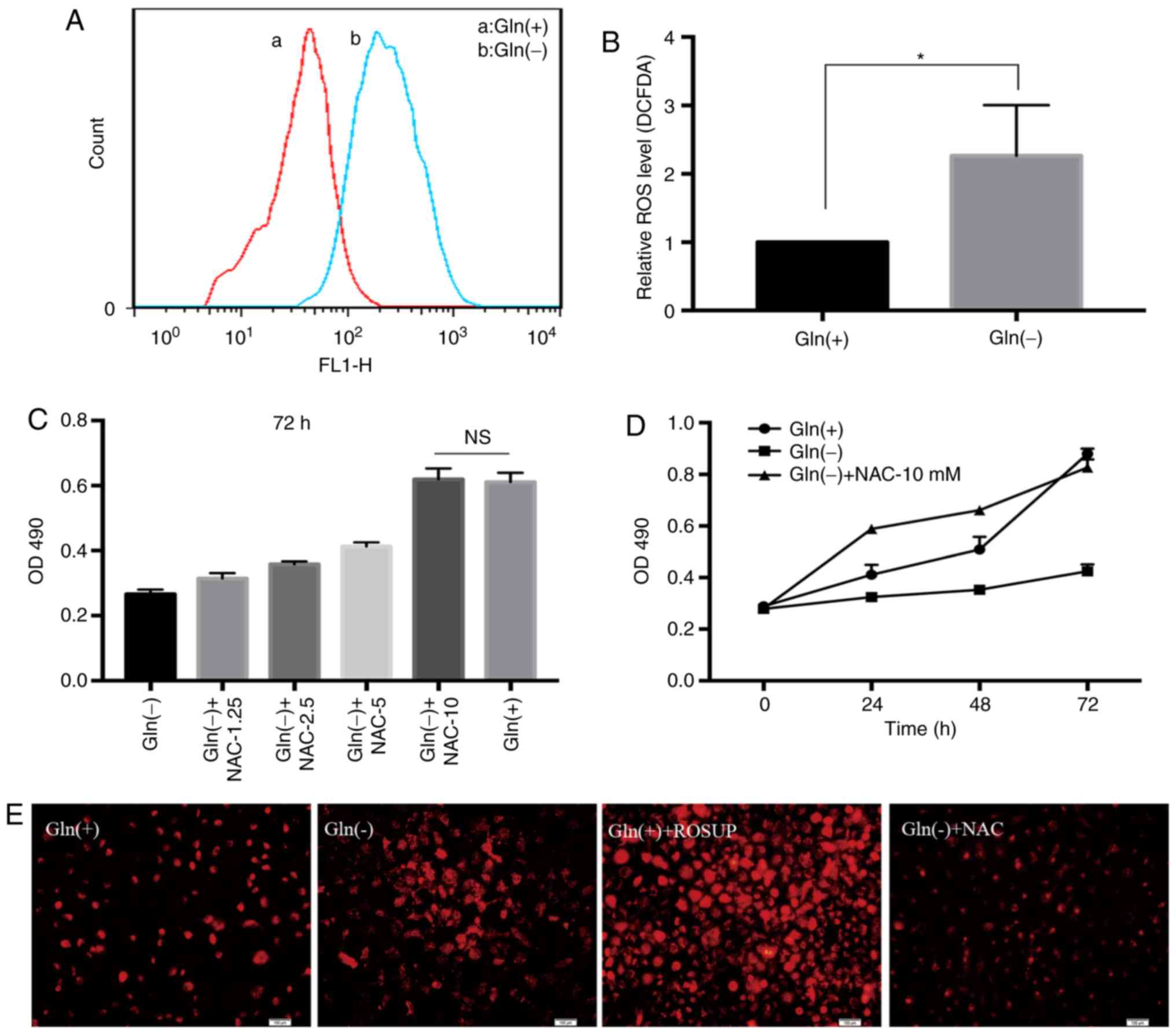
Effect of Gln on ROS production in T24
cells
The levels of ROS in the Gln(-) group were markedly
higher than those in the Gln(+) group (Fig. 4A and B). NAC, a classic ROS
inhibitor, was used to create Gln(-)+NAC groups in addition to the
Gln(+) and Gln(-) groups. NAC restored the proliferation rate of
the Gln-deprived T24 cells. As the concentration of NAC increased,
the proliferation rate of the T24 cells also gradually increased in
the Gln(-)+NAC groups compared with the Gln(-) group. At the 10 mM
optimal concentration, the proliferation rate of the Gln(-)+NAC
group was equivalent to that of the Gln(+) group (Fig. 4C). We compared the viability of
the cells in the Gln(+), Gln(-), and Gln(-)+NAC groups at 24, 48
and 72 h by MTT assays. The results revealed that the proliferation
rate was lower in the Gln(-) group than in the other 2 groups, and
at 24 and 48 h, the proliferation rate was much greater in the
Gln(-)+NAC group than that in the other groups. However, at 72 h,
there was no obvious difference between the Gln(+) group and the
Gln(-)+NAC group (Fig. 4D). In
addition, the analysis of the fluorescence intensities in the
different treatment groups by intracellular ROS fluorescence
staining confirmed that Gln deprivation increased ROS
concentrations in the T24 bladder cancer cells (Fig. 4E).
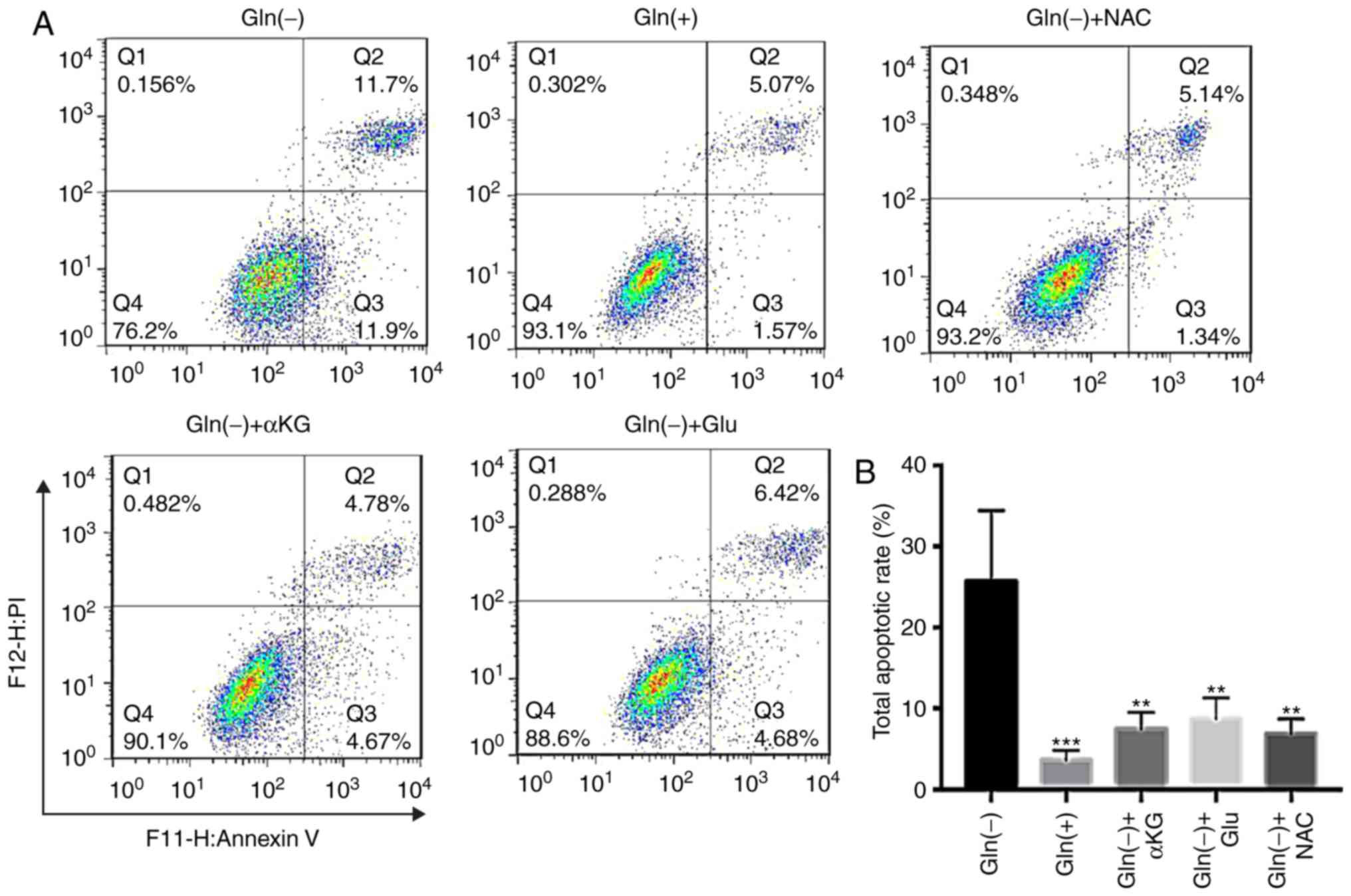
Gln promotes cell viability via its
effects on glutaminolysis and ROS production
The analysis of the apoptotic effects of different
conditions, including Gln(+), Gln(-), Gln(-)+αKG, Gln(-)+Glu, and
Gln(-)+NAC, revealed that the Gln(-) group exhibited the highest
apoptotic rate among the 5 groups. The groups exposed to the
glutaminolysis intermediates, αKG and Glu, exhibited lower
apoptotic rates than those in the Gln(-) group. In addition, the
ROS scavenger, NAC, restored cell viability in the absence of Gln
(Fig. 5). Thus, increasing
glutaminolysis intermediate levels and reducing intracellular ROS
levels decreased the apoptotic rates of the T24 cells under Gln
deprivation.
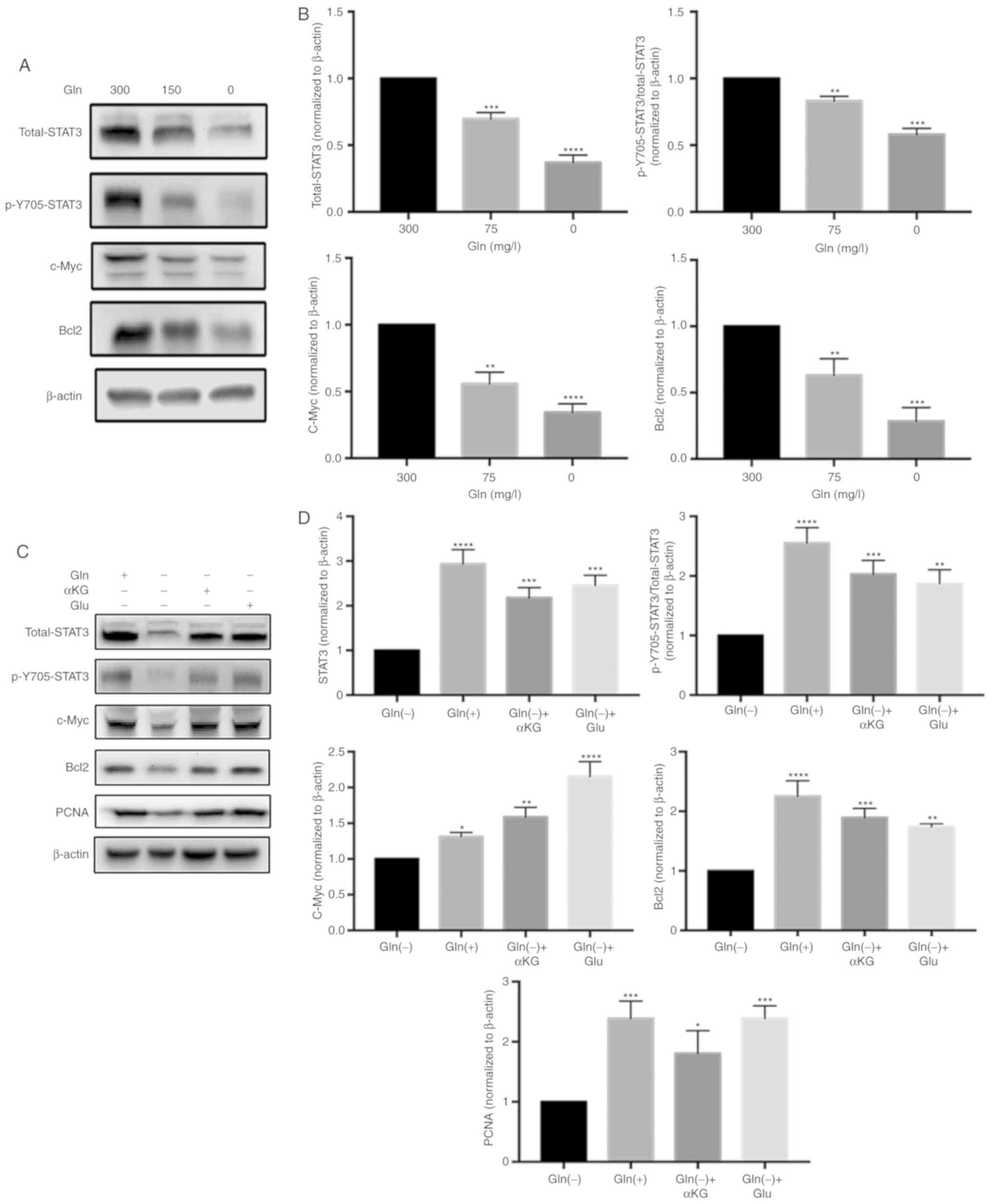
Effect of Gln on STAT3 expression in T24
cells
To verify the association between Gln and STAT3, the
groups of cells were treatd with 0, 150 or 300 mg/l Gln and we then
examined STAT3 expression at the protein level (Fig. 6A and B). The results of western
blot analysis revealed that the expression of c-Myc, Bcl2, PCNA and
STAT3 (including total-STAT3, and p-Y705-STAT3/total-STAT3)
decreased gradually with the decreasing Gln concentrations at 48 h.
This findings clearly suggests that Gln promotes the proliferation
and decreases the apoptosis of T24 bladder cancer cells by
promoting STAT3 expression.
Glutaminolysis and ROS levels modulate
STAT3 activation
We then sought to determine whether glutaminolysis
and ROS levels regulate STAT3 expression. The expression levels of
c-Myc, Bcl2, PCNA and STAT3 (including total-STAT3 and
p-Y705-STAT3) in the Gln(+), Gln(-), Gln(-)+αKG, Gln(-)+Glu, and
Gln(-)+NAC groups (Fig. 6C and
6E) were analyzed. The results of
western blot analysis revealed that proteins associated with
proliferation (c-Myc, Bcl2 and PCNA) were expressed at higher
levels in the Gln(-)+αKG, Gln(-)+Glu, and Gln(-)+NAC groups than in
the Gln(-) group. The expression of total-STAT3 and
p-Y705-STAT3/total-STAT3 was efficiently restored in the Gln(-)+αKG
and Gln(-)+Glu groups compared with the Gln(-) group (Fig. 6D). In addition, the expression of
total-STAT3 and p-Y705-STAT3/total-STAT3 was higher in the
Gln(-)+NAC group than in the Gln(-) group (Fig. 6F).
Discussion
Bladder cancer ranks 13th in terms of mortality,
although mortality rates are increasing in most countries (1,21).
The majority of cases are transitional cell carcinoma (TCC), a
neoplasm originating from transitional urothelial cells. The T24
cell line was derived from malignant bladder cancer undergoing
progression to the muscle layer of the bladder wall, in contrast to
cell lines derived from non-muscle-invasive bladder cancer, an
early-stage type of bladder cancer (22). Gln metabolism contributes to T24
cancer cell proliferation. Indeed, Lea et al (23) found that Gln deprivation affected
the proliferation rates of several bladder cancer cell lines,
including the T24 and UM-UC-3 lines.
In this study, the T24 cell proliferation rates were
positively associated with the Gln concentrations. Compared with
that in the Gln(+) group, the proportion of cells in the S phase
was much higher in the Gln(-) group. In response to Gln
deprivation, K-Ras-driven cancer cells can arrest in either the S
or G2/M phase due to insufficient nucleotide biosynthesis (24-26). Aspartate, which is essential for
nucleotide biosynthesis, is produced in a transamination reaction
catalyzed by GOT2. Therefore, in the absence of Gln, a lack of
aspartate for the GOT2 catalytic reaction leads to replication
stress due to insufficient nucleotides, which may be the cause of
the S phase arrest observed in this study. Consistent with this
hypothesis, S phase arrest can be overcome by providing cells with
α-ketoglutarate and aspartic acid (24). To confirm the direct association
between Gln and bladder cancer, T24 cell proliferation was further
examined by using the Gln analog, Don. Compared to Gln alone [in
the Gln(+) group], Don markedly inhibited the proliferation of the
T24 cells and significantly decreased the protein expression of the
key enzymes, GLS and GLUD1, which participate in Gln
metabolism.
Cancer cells undergo metabolic transformation to
meet their increased anabolic demand for glycolytic and TCA cycle
intermediates to synthesize important biomolecules required for
cell growth. The key to this metabolic transformation is the
mitochondrial excretion of citric acid, which is used in the TCA
cycle to produce the cytosolic acetyl-CoA necessary for lipid
biosynthesis. Upon the loss of mitochondrial citrate, cancer cells
become dependent on the 'conditionally essential' amino acid Gln,
which serves as a supplemental carbon source for the TCA cycle.
We further explored the mechanism through which Gln
affects T24 proliferation from the perspectives of ATP production,
ROS neutralization and direct stimulation of the production of
STAT3 as a signaling factor. The supplementation of Gln-deprived
T24 cells with Gln catabolism intermediates (Glu and αKG) restored
both the ATP levels and proliferation rates to a certain extent,
which illustrated that Gln also affects the proliferation of T24
bladder cancer cells through the catabolic pathway. Gln-derived
carbon enters the TCA cycle following the conversion of Glu to αKG.
Gln-derived carbon also contributes to ATP production through its
oxidation in the TCA cycle (27).
Accordingly, cell-permeable forms of Glu and αKG, which have
previously been reported to effectively supply Glu and αKG
intracellularly, can be substituted for Gln to support cancer cell
metabolism and ATP production (10,27).
In addition, Gln is a regulator of intracellular
redox balance associated with glutathione synthesis and metabolic
NADPH production. These functions of Gln require its metabolic
processing and account for its effects on the viability of T24
cells. The results of this study revealed that Gln starvation
induced cell death by augmenting the ROS levels in T24 bladder
cancer cells. We investigated the possibility that Gln promotes the
survival of T24 cells by mitigating the toxic effects of increased
ROS production. Notably, the addition of a ROS scavenger to the
culture medium evidently increased the proliferation of the
Gln-deprived T24 cells, suggesting that Gln is required for the
alleviation of the effects of enhanced ROS production. Therefore,
Gln controls the proliferation of T24 cells, which are
Gln-dependent bladder cancer cells, in a manner dependent on its
metabolic use. These findings are inconsistent with the hypothesis
described in the study by Cacace et al (13), namely that Gln controls cancer
cell proliferation independently of ROS production modulation.
Subsequently, we wished to determine whether Gln
affects T24 proliferation by modulating STAT3 activation. Yang
et al (28) proposed that
Gln regulates the activation of STAT3, a signaling pathway mediator
that regulates cancer hallmarks in invasive OVCA cells. The
inhibition of Gln utilization in these cancer cells through GLS
inactivation significantly reduced phosphorylated STAT3 expression
(29). However, whether this
phenomenon is applicable to bladder cancer has remained unknown.
Activated STAT3 has been detected in bladder cancer (30), and the inhibition of STAT3
activity with antisense oligonucleotides, decoy oligonucleotides
and siRNA has been shown to result in the apoptosis and reduced
growth of tumor cells (31).
Notably, STAT3 expression is closely related to T24 bladder cancer
cell proliferation. As mentioned previously, STAT3 pathway
activation and inactivation clearly affect T24 cell proliferation.
For example, it has been identified that the inhibition of STAT3
efficiently decreases T24 cell proliferation (32,33). The rapid activation of STAT3 has
also been found to modulate the proliferation of the T24 cells
(34). In this study, we did not
aim to repeat such experiments. Instead, we explored the
association between Gln and the STAT3 pathway. We found that STAT3
was an essential mediator of Gln-dependent T24 cancer cell
proliferation and that Gln promoted total-STAT3 and
p-Y705-STAT3/total-STAT3 expression. To ascertain whether Gln
affects STAT3 expression to modulate cancer cell proliferation and
apoptosis, we also detected c-Myc and Bcl2, which have previously
been reported to be influenced by Gln and STAT3. The oncogene c-Myc
coordinates the expression of genes necessary for cells to engage
in Gln catabolism that exceeds the cellular requirements for
protein and nucleotide biosynthesis (35). STAT3 mostly mediates the rapid
activation of the c-Myc gene (36). Information on the metabolic
regulation of Bcl2 proteins may provide insight into alternative
routes of apoptosis activation. For example, cells surviving Gln
withdrawal exhibit a particularly enhanced expression of BIM and
the binding of BIM to Bcl2 (37).
Huang et al (38) found
that Roxyl-zhc-84 reduced JAK1 and STAT3 phosphorylation and
markedly downregulated the expression of the anti-apoptotic
protein, Bcl2, which is known as a STAT3 target gene.
STAT3 is constitutively phosphorylated on tyrosine
in various tumor types (39), and
this phosphorylation is associated with a reduced ROS production,
delayed senescence and protection from apoptosis. Both nuclear
p-Y705-STAT3 and mitochondrial p-S727-STAT3 can promote cell
survival and reduce ROS production (40). Previous studies have demonstrated
that the inactivation of STAT3 promotes apoptosis by decreasing
Bcl2 expression (41,42) and that it is feasible to induce
cancer cell apop-tosis by regulating ROS-modulated STAT3 expression
(43,44). In addition, Cetinbas et al
(45) hypothesized that increases
in ROS production compensate for the increases in Gln ingestion and
utilization. As described above, Gln can promote T24 cell
proliferation through glutaminolysis, regulating ROS production and
promoting STAT3 expression. It was previously unclear whether Gln
affects bladder cancer cell T24 proliferation by activating STAT3
through ROS and glutaminolysis. The results of this study revealed
that Gln can act intracellularly to activate STAT3 through
glutaminolysis and mediate the proliferative effects of STAT3,
indicating that Gln affects STAT3 expression via its catabolism.
Under Gln deprivation, the replenishment of Gln catabolism
intermediates (αKG and Glu) significantly restored the STAT3
expression levels. Similarly, the elimination of intracellular ROS
with NAC initially reduced the use of Gln for ROS scavenging,
causing Gln to accumulate in cells; this effect was directly
reflected by the total STAT3 expression. These findings identify
the critical role of ROS in regulating STAT3 expression. These
experiments reveal that STAT3 is modulated by glutaminolysis and
ROS levels. In other words, Gln can induce T24 bladder cancer cell
proliferation mainly through STAT3 by affecting ATP and ROS
generation.
Above all, this study highlights the essential role
of Gln in the proliferation of Gln-dependent T24 bladder cancer
cells. In addition, our findings regarding the cause-effect
relationship between Gln metabolism and proliferation in the T24
bladder cancer cell line show the importance of Gln-dependent
cancer therapy in clinical practice. The observation that
KRas-driven Gln-dependent cancer cells arrest in the S phase due to
a lack of aspartic acid (46)
provides a compelling treatment opportunity for KRas-driven late
metastatic bladder cancer. In fact, Ras oncogene mutations
cooperate with β-catenin activation to drive bladder tumourigenesis
(47). It has also been reported
that the inhibition of STAT3 partially reverses gemcitabine-induced
migration, chemoresistance, and tumor relapse (48).
Radical cystectomy, regarded for decades as the
standard treatment for muscle-invasive bladder cancer, is
associated with significant morbidity and high complication rates
after surgery. Interest in alternative treatment options that
protect the bladder has increased (49-51). Three methods have been widely
recognized as bladder-protecting therapies: Transurethral resection
of bladder tumors (TURBT), chemotherapy and radiation therapy
(51-53). However, radiation therapy is often
associated with recurrence and distant metastasis, as several
tumors are radioresistant; thus, its implementation can result in
complications (54,55). Furthermore, the search for
strategies with which to prevent the recurrence of bladder cancer
after using chemotherapeutics, such as gemcitabine, a frequently
used drug in the treatment of bladder cancer, is a vital part of
clinical chemotherapy. The identification of prognostic and
predictive biomarkers may provide other strategies through which
patients can avoid radical cystectomy (56,57).
Understanding the role of Gln in STAT3 activation
may aid in the prevention and treatment of Gln-dependent bladder
cancer in clinical practice from the perspective of metabolism.
Thus far, several GLS inhibitors have been developed in the search
for selective inhibitors of Gln catabolism; these agents, including
968, BPTES and CB-389, have exhibited tumor-suppressive activities
in preclinical models (58-61). In addition, radiolabeled Gln can
be used to locate tumors, and currently popular carrier materials
can be used with Gln analogs to block Gln metabolism in tumor cells
at the gene level for tumor starvation.
In conclusion, the findings of this study indicated
that Gln affects the proliferation of T24 bladder cancer cells
mainly by modifying STAT3 expression. This effect is dependent on
the metabolic use of Gln and on the participation of Gln in the
regulation of ROS levels. Gln deprivation results in K-Ras-driven
bladder cancer cell arrest in S phase. Notably, S phase-arrested
cells are vulnerable to the cytotoxic drugs capecitabine,
paclitaxel and rapamycin (24,26). Thus, Gln deprivation mediates the
'synthetic lethality' of KRas-driven cancer cells upon
capecitabine, paclitaxel and rapamycin treatment. The findings of
this study further establish the potential for exploiting metabolic
changes in cancer cells that confer novel opportunities for
therapeutic intervention.
Acknowledgements
The authors would like to thank the members of the
Key Laboratory of Urinary System Diseases (Affiliated Hospital of
Qingdao University). The authors are also grateful to the
Department of Urology, Affiliated Hospital of Qingdao University
for providing helpful discussions with regards to this
manuscript.
Funding
This study was financially supported by the National
Natural Science Foundation of China (grant nos. 81772713, 81472411,
81372752 and 81401899); the Taishan Scholar Program of Shandong
Province (tsqn20161077); the Natural Science Foundation
(ZR2014HM088) and the Key Research and Development Program
(2018GSF118197) of Shandong Province; the China Postdoctoral
Science Foundation (2017M622144) and Qingdao Postdoctoral
Application Research Project; and the Qingdao Young Scientist
Applied Basic Research Fund (15-9-1-105-jch. 15-9-1-51-jch).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
NS, YL, YC LS, YW and HN conceived and designed the
study. NS, LW, DL and ZL performed the experiments and analyzed the
data. NS and LS wrote the manuscript. YW and HN reviewed and edited
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Antoni S, Ferlay J, Soerjomataram I, Znaor
A, Jemal A and Bray F: Bladder cancer incidence and mortality: A
global overview and recent trends. Eur Urol. 71:96–108. 2017.
View Article : Google Scholar
|
|
2
|
Zhang J, Pavlova NN and Thompson C: Cancer
cell metabolism: The essential role of the nonessential amino acid,
glutamine. EMBO J. 36:1302–1315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Daye D and Wellen KE: Metabolic
reprogramming in cancer: Unraveling the role of glutamine in
tumorigenesis. Semin Cell Dev Biol. 23:362–369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DeBerardinis RJ, Mancuso A, Daikhin E,
Nissim I, Yudkoff M, Wehrli S and Thompson CB: Beyond aerobic
glycolysis: Transformed cells can engage in glutamine metabolism
that exceeds the requirement for protein and nucleotide synthesis.
Proc Natl Acad Sci USA. 104:19345–19350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rajagopalan KN and DeBerardinis RJ: Role
of glutamine in cancer: Therapeutic and imaging implications. J
Nucl Med. 52:1005–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matés JM, Pérez-Gómez C, Núñez de Castro
I, Asenjo M and Márquez J: Glutamine and its relationship with
intracellular redox status, oxidative stress and cell
proliferation/death. Int J Biochem Cell Biol. 34:439–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ratnikov B, Aza-Blanc P, Ronai ZA, Smith
JW, Osterman AL and Scott DA: Glutamate and asparagine cataplerosis
underlie glutamine addiction in melanoma. Oncotarget. 6:7379–7389.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shanware NP, Bray K, Eng CH, Wang F,
Follettie M, Myers J, Fantin VR and Abraham RT: Glutamine
deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat
Commun. 5:49002014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wise DR and Thompson CB: Glutamine
addiction: A new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Le A, Lane AN, Hamaker M, Bose S, Gouw A,
Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al:
Glucose-independent gluta-mine metabolism via TCA cycling for
proliferation and survival in B cells. Cell Metab. 15:110–121.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cacace A, Sboarina M, Vazeille T and
Sonveaux P: Glutamine activates STAT3 to control cancer cell
proliferation independently of glutamine metabolism. Oncogene.
36:2074–2084. 2017. View Article : Google Scholar :
|
|
14
|
Fukada T, Hibi M, Yamanaka Y,
Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K and Hirano
T: Two signals are necessary for cell proliferation induced by a
cytokine receptor gp130: Involvement of STAT3 in anti-apoptosis.
Immunity. 5:449–460. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Du XL, Wang CJ, Lin DC, Ruan X,
Feng YB, Huo YQ, Peng H, Cui JL, Zhang TT, et al: Reciprocal
activation between PLK1 and Stat3 contributes to survival and
proliferation of esophageal cancer cells. Gastroenterology.
142:521–530. 2012. View Article : Google Scholar
|
|
16
|
Mandal PK, Ren Z, Chen X, Xiong C and
McMurray JS: Structure-affinity relationships of glutamine mimics
incorporated into phosphopeptides targeted to the SH2 domain of
signal transducer and activator of transcription 3. J Med Chem.
52:6126–6141. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fabrício F, de Oliveira CP, Rockenbach L,
Mendes FB, Bergamin L, Jandrey EH, Edelweiss MI, Guterres SS,
Pohlmann AR and Battastini AM: Pharmacological improvement and
preclinical evaluation of methotrexate-loaded lipid-core
nanocapsules in a glioblastoma model. J Biomed Nanotechnol.
11:1808–1818. 2015. View Article : Google Scholar
|
|
18
|
Castaneda JM, Hua R, Miyata H, Oji A, Guo
Y, Cheng Y, Zhou T, Guo X, Cui Y, Shen B, et al: TCTE1 is a
conserved component of the dynein regulatory complex and is
required for motility and metabolism in mouse spermatozoa. Proc
Natl Acad Sci USA. 114:E5370–E5378. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen W, Shen X, Hu Y, Xu K, Ran Q, Yu Y,
Dai L, Yuan Z, Huang L, Shen T and Cai K: Surface functionalization
of titanium implants with chitosan-catechol conjugate for
suppression of ROS-induced cells damage and improvement of
osteogenesis. Biomaterials. 114:82–96. 2017. View Article : Google Scholar
|
|
20
|
Dietrich F, Figueiró F, Filippi-Chiela EC,
Cappellari AR, Rockenbach L, Tremblay L, de Paula PB, Roesler R,
Filho AB, Sévigny J, et al: Ecto-5'-nucleotidase/CD73 contributes
to the radiosensitivity of T24 human bladder cancer cell line. J
Cancer Res Clin Oncol. 144:469–482. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
22
|
Rockenbach L, Bavaresco L, Fernandes
Farias P, Cappellari AR, Barrios CH, Bueno Morrone F and Oliveira
Battastini AM: Alterations in the extracellular catabolism of
nucleotides are involved in the antiproliferative effect of
quercetin in human bladder cancer T24 cells. Urol Oncol.
31:1204–1211. 2013. View Article : Google Scholar
|
|
23
|
Lea MA, Altayyar M and desBordes C:
Inhibition of growth of bladder cancer cells by
3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one in combination with
other compounds affecting glucose metabolism. Anticancer Res.
35:5889–5899. 2015.PubMed/NCBI
|
|
24
|
Saqcena M, Mukhopadhyay S, Hosny C,
Alhamed A, Chatterjee A and Foster DA: Blocking anaplerotic entry
of glutamine into the TCA cycle sensitizes K-Ras mutant cancer
cells to cytotoxic drugs. Oncogene. 34:2672–2680. 2015. View Article : Google Scholar
|
|
25
|
Gaglio D, Soldati C, Vanoni M, Alberghina
L and Chiaradonna F: Glutamine deprivation induces abortive s-phase
rescued by deoxyribonucleotides in k-ras transformed fibroblasts.
PLoS One. 4:e47152009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saqcena M, Patel D, Menon D, Mukhopadhyay
S and Foster DA: Apoptotic effects of high-dose rapamycin occur in
S-phase of the cell cycle. Cell Cycle. 14:2285–2292. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan J, Kamphorst JJ, Mathew R, Chung MK,
White E, Shlomi T and Rabinowitz JD: Glutamine-driven oxidative
phosphorylation is a major ATP source in transformed mammalian
cells in both normoxia and hypoxia. Mol Syst Biol. 9:7122013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang L, Moss T, Mangala LS, Marini J, Zhao
H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, et al:
Metabolic shifts toward glutamine regulate tumor growth, invasion
and bioenergetics in ovarian cancer. Mol Syst Biol. 10:7282014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo L, Zhou B, Liu Z, Xu Y, Lu H, Xia M,
Guo E, Shan W, Chen G and Wang C: Blockage of glutaminolysis
enhances the sensitivity of ovarian cancer cells to PI3K/mTOR
inhibition involvement of STAT3 signaling. Tumour Biol.
37:11007–11015. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Santoni M, Conti A, Piva F, Massari F,
Ciccarese C, Burattini L, Cheng L, Lopez-Beltran A, Scarpelli M,
Santini D, et al: Role of STAT3 pathway in genitourinary tumors.
Future Sci. 1:FSO152015.
|
|
31
|
Zhang B, Lu Z, Hou Y, Hu J and Wang C: The
effects of STAT3 and survivin silencing on the growth of human
bladder carcinoma cells. Tumour Biol. 35:5401–5407. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu F, Chen Y, Li Y, Ju J, Wang Z and Yan
D: RNA-interference-mediated Cdc42 silencing down-regulates
phosphorylation of STAT3 and suppresses growth in human
bladder-cancer cells. Biotechnol Appl Biochem. 49:121–128. 2008.
View Article : Google Scholar
|
|
33
|
Tsujita Y, Horiguchi A, Tasaki S, Isono M,
Asano T, Ito K, Asano T, Mayumi Y and Kushibiki T: STAT3 inhibition
by WP1066 suppresses the growth and invasiveness of bladder cancer
cells. Oncol Rep. 38:2197–2204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen RJ, Ho YS, Guo HR and Wang YJ: Rapid
activation of Stat3 and ERK1/2 by nicotine modulates cell
proliferation in human bladder cancer cells. Toxicol Sci.
104:283–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kiuchi N, Nakajima K, Ichiba M, Fukada T,
Narimatsu M, Mizuno K, Hibi M and Hirano T: STAT3 is required for
the gp130-mediated full activation of the c-myc gene. J Exp Med.
189:63–73. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bajpai R, Matulis SM, Wei C, Nooka AK, Von
Hollen HE, Lonial S, Boise LH and Shanmugam M: Targeting glutamine
metabolism in multiple myeloma enhances BIM binding to BCL-2
eliciting synthetic lethality to venetoclax. Oncogene.
35:3955–3964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang Z, Zhou W, Li Y, Cao M, Wang T, Ma
Y, Guo Q, Wang X, Zhang C, Zhang C, et al: Novel hybrid molecule
overcomes the limited response of solid tumours to HDAC inhibitors
via suppressing JAK1-STAT3-BCL2 signalling. Theranostics.
8:4995–5011. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Demaria M, Giorgi C, Lebiedzinska M,
Esposito G, D'Angeli L, Bartoli A, Gough DJ, Turkson J, Levy DE,
Watson CJ, et al: A STAT3-mediated metabolic switch is involved in
tumour transformation and STAT3 addiction. Aging (Albany NY).
2:823–842. 2010. View Article : Google Scholar
|
|
40
|
Poli V and Camporeale A: STAT3-mediated
metabolic repro-graming in cellular transformation and implications
for drug resistance. Front Oncol. 5:1212015. View Article : Google Scholar
|
|
41
|
Tan Y, Huang N, Zhang X, Hu J, Cheng S, Pi
L and Cheng Y: KIAA0247 suppresses the proliferation, angiogenesis
and promote apoptosis of human glioma through inactivation of the
AKT and Stat3 signaling pathway. Oncotarget. 7:87100–87113. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang X, Qiu W, Zhang G, Xu S, Gao Q and
Yang Z: MicroRNA-204 targets JAK2 in breast cancer and induces cell
apoptosis through the STAT3/BCl-2/survivin pathway. Int J Clin Exp
Pathol. 8:5017–5025. 2015.PubMed/NCBI
|
|
43
|
Sun Q, Lu NN and Feng L: Apigetrin
inhibits gastric cancer progression through inducing apoptosis and
regulating ROS-modulated STAT3/JAK2 pathway. Biochem Biophys Res
Commun. 498:164–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cai N, Zhou W, Ye LL, Chen J, Liang QN,
Chang G and Chen JJ: The STAT3 inhibitor pimozide impedes cell
proliferation and induces ROS generation in human osteosarcoma by
suppressing catalase expression. Am J Transl Res. 9:3853–3866.
2017.PubMed/NCBI
|
|
45
|
Cetinbas N, Daugaard M, Mullen AR, Hajee
S, Rotblat B, Lopez A, Li A, DeBerardinis RJ and Sorensen PH: Loss
of the tumor suppressor Hace1 leads to ROS-dependent glutamine
addiction. Oncogene. 34:4005–4010. 2015. View Article : Google Scholar :
|
|
46
|
Mukhopadhyay S, Saqcena M and Foster DA:
Synthetic lethality in KRas-driven cancer cells created by
glutamine deprivation. Oncoscience. 2:807–808. 2015.PubMed/NCBI
|
|
47
|
Ahmad I, Patel R, Liu Y, Singh LB, Taketo
MM, Wu XR, Leung HY and Sansom OJ: Ras mutation cooperates with
β-catenin activation to drive bladder tumourigenesis. Cell Death
Dis. 2:e1242011. View Article : Google Scholar
|
|
48
|
Zhang Z, Duan Q, Zhao H, Liu T, Wu H, Shen
Q, Wang C and Yin T: Gemcitabine treatment promotes pancreatic
cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade.
Cancer Lett. 382:53–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jacobs BL, Lee CT and Montie JE: Bladder
cancer in 2010: How far have we come? CA Cancer J Clin. 60:244–272.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kotwal S, Choudhury A, Johnston C, Paul
AB, Whelan P and Kiltie AE: Similar treatment outcomes for radical
cystectomy and radical radiotherapy in invasive bladder cancer
treated at a United Kingdom specialist treatment center. Int J
Radiat Oncol Biol Phys. 70:456–463. 2008. View Article : Google Scholar
|
|
51
|
Russell CM, Lebastchi AH, Borza T, Spratt
DE and Morgan TM: The role of transurethral resection in trimodal
therapy for muscle-invasive bladder cancer. Bladder Cancer.
2:381–394. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kulkarni GS, Hermanns T, Wei Y, Bhindi B,
Satkunasivam R, Athanasopoulos P, Bostrom PJ, Kuk C, Li K,
Templeton AJ, et al: Propensity score analysis of radical
cystectomy versus bladder-sparing tri-modal therapy in the setting
of a multidisciplinary bladder cancer clinic. J Clin Oncol.
35:2299–2305. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rödel C, Grabenbauer GG, Kühn R,
Papadopoulos T, Dunst J, Meyer M, Schrott KM and Sauer R:
Combined-modality treatment and selective organ preservation in
invasive bladder cancer: Long-term results. J Clin Oncol.
20:3061–3071. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
James ND, Hussain SA, Hall E, Jenkins P,
Tremlett J, Rawlings C, Crundwell M, Sizer B, Sreenivasan T,
Hendron C, et al: Radiotherapy with or without chemotherapy in
muscle-invasive bladder cancer. N Engl J Med. 366:1477–1488. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Park JC, Citrin DE, Agarwal PK and Apolo
AB: Multi-modal management of muscle-invasive bladder cancer. Curr
Probl Cancer. 38:80–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen RC, Shipley WU, Efstathiou JA and
Zietman AL: Trimodality bladder preservation therapy for
muscle-invasive bladder cancer. J Natl Compr Cancer Netw.
11:952–960. 2013. View Article : Google Scholar
|
|
57
|
Shrivastava S, Mansure JJ, Almajed W, Cury
F, Ferbeyre G, Popovic M, Seuntjens J and Kassouf W: The role of
HMGB1 in radioresistance of bladder cancer. Mol Cancer Ther.
15:471–479. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang JB, Erickson JW, Fuji R, Ramachandran
S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV and
Cerione RA: Targeting mitochondrial glutaminase activity inhibits
oncogenic transformation. Cancer Cell. 18:207–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gross MI, Demo SD, Dennison JB, Chen L,
Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et
al: Antitumor activity of the glutaminase inhibitor CB-839 in
triple-negative breast cancer. Mol Cancer Ther. 13:890–901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor
RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, et al: Targeted
inhibition of tumor-specific glutaminase diminishes cell-autonomous
tumorigenesis. J Clin Invest. 125:2293–2306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stalnecker CA, Ulrich SM, Li Y,
Ramachandran S, McBrayer MK, DeBerardinis RJ, Cerione RA and
Erickson JW: Mechanism by which a recently discovered allosteric
inhibitor blocks glutamine metabolism in transformed cells. Proc
Natl Acad Sci USA. 112:394–399. 2015. View Article : Google Scholar
|