Introduction
Lung cancer is the most prevalent type of cancer
worldwide, leading annually to the deaths of more than 1.76 million
individuals (1). Non-small-cell
lung cancer (NSCLC) is the major type of lung adenocarcinoma found
in approximately 85% of all cases of lung cancer, with the majority
of patients presenting with advanced tumor stages in the United
States (2). Even though numerous
types of treatment strategies including immunotherapy,
chemotherapy, radiotherapy and combination therapy have been
developed for NSCLC, the 5-year survival rate for patients with
advanced stages of the disease (with metastasis) has only
marginally improved over the past 4 decades, remaining at
approximately 4% (3). Therefore,
the development of novel therapeutic strategies for NSCLC is of
utmost importance.
TNF-related apoptosis-inducing ligand (TRAIL) is
often referred to as a ʻmagic bulletʼ as it can specifically target
a broad range of tumors, including lung cancer, while avoiding
detrimental effects on normal cells. TRAIL is a potent apoptotic
inducer capable of binding to the death receptors, death receptor
(DR)4 and DR5, which can subsequently activate an apoptotic signal
pathway known as the extrinsic apoptotic pathway (4). However, a number of types of tumors
evade TRAIL-mediated apoptosis, such as via an increase in the
expression of anti-apoptotic molecules, by the downregulation of
apoptotic proteins, or with the inhibition of apoptotic signaling
pathways, leading to TRAIL resistance (5,6).
To overcome this resistance, several methods have used, such as the
modulation of apoptosis-related proteins or treatment with
chemotherapeutic drugs, leading to an increase in TRAIL
sensitivity.
CCR4-NOT transcription complex (CNOT), composed of
11 subunits, participates in ribonucleic acid (RNA) regulation,
including messenger RNA (mRNA) stability and export. In particular,
CNOT2, one of the CCR4-NOT subunits, plays a critical role in
deadenylase activity and the structural integrity of the complex
(7). In addition, this protein
participates in embryonic development and transcriptional
regulation via the recruitment of histone modifiers (8,9).
Recent studies have suggested that CNOT2 plays important roles in
tumor progression, such as in metastasis, proliferation and
angiogenesis via the regulation of vascular endothelial growth
factor signaling in breast cancer cells (10,11). However, to the best of our
knowledge, CNOT2 has not been evaluated as a molecular target for
tumor treatment to date. Hence, in the current study, the
therapeutic potential through which the suppression of CNOT2
enhances TRAIL sensitivity in TRAIL-resistant NSCLC cells through
the STAT3 signaling pathway was examined.
Materials and methods
Cells and cell culture
All cell culture experiments adhered to standard
biosafety level 2 guidelines. All cell lines used in this study
were tested for mycoplasma contamination using a mycoplasma
detection kit (JCBIO). The NSCLC cell lines, A549, H1299, H596 and
H460, were purchased from the Korean Cell Line Bank. The NSCLC
cells were cultured in Roswell Park Memorial Institute 1640
(RPMI-1640; Corning Inc.), supplemented with 10% fetal bovine serum
(Corning Inc.) and 1% penicillin-streptomycin (Corning Inc.). All
cells were cultured in a 37°C humidified atmosphere containing 5%
CO2.
Reagents and antibodies
TRAIL/Apo2L (ABIN2973530) was purchased from Atgen.
The antibodies for cleaved caspase-3 (sc-56053), caspase-3
(sc-7148), Src homology region 2 domain containing phosphatase-1
(SHP1) (sc-8425), DR4 (sc-8411), BCL2 associated X protein (Bax)
(sc-7480) and PARP[poly(ADP-ribose) polymerase] (sc-7150) were
purchased from Santa Cruz Biotechnology, while phospho-signal
transducer and activator of transcription 3 (STAT3) (#9131), STAT3
(#12640), phospho-Akt(#9271), phosphor-mitogen-activated protein
kinase (MAPK) (#9101), phospho-Src (#2105), Bcl-2 (#15071), DR5
(#3696), C/EBP homologous protein (CHOP) (#2895), glucose-regulated
protein 78 (GRP78) (#3177) and β-actin (#4967) antibodies were
purchased from Cell Signaling Technology. Finally, the antibodies
for CNOT2 (ab55679) and cytochrome c (ab13575) were
purchased from Abcam.
Cytotoxicity assay
Cell cytotoxicity was measured by way of the
3-(4,5-dimethylthialzol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) assay. MTT was purchased from Molecular Probes. The NSCLC
cells were seeded in 96-well plates at a density of
0.7×104 cells per well and incubated with various
concentrations (0, 3.12, 6.25, 12.5, 25, 50, 100, 200 and 400
ng/ml) of TRAIL for 24 h at 37°C. The viability of the cells was
analyzed as previously described (12,13). Optical density was measured using
a microplate reader (VersaMax; Molecular Devices) at 570 nm and
data were analyzed using the Softmax Pro software program
(Molecular Devices). The results are expressed as the means and
standard deviations after at least 3 independent experiments
performed in triplicate.
Apoptosis assay
Apoptosis was measured by flow cytometry after
staining with Annexin V-FITC and propidium iodide (PI). The cells
(2×105) were incubated with 0, 25 and 100 ng/ml of TRAIL
for 24 h at 37°C prior to analysis. The cells were stained using an
Annexin V-FITC apoptosis detection kit (Biovision) according to the
manufacturer's instructions. Apoptotic cells were measured using
the FACSCalibur platform (BD Biosciences). Data were analyzed using
the Cell Quest program (BD Biosciences). The experiment was
repeated in triplicate.
Stable cell lines
To establish a stable CNOT2-overexpressing H460 cell
line, the cells were seeded in the manner of 1.5×105
cells in 6-well plates at 1 day prior to transfection. Transfection
was performed using pcDNA3.1-CNOT2 and vector with X-treme GENE HP
DNA transfection reagent (Roche Holding AG), following the
manufacturer's instructions. Following 72 h of transfection, the
cells were treated with 500 µg/ml of G418 (Sigma-Aldrich)
every 2 days for 10 days total to establish a stable cell line.
To ensure the stable knockdown of CNOT2 in the NSCLC
cells, the A549, H1299 and H596 cells were transfected with pRS
vector containing CNOT2 shRNA (cat. no. TF81001; OriGene
Technologies, Inc.) or control shRNA (Cat. no. TF81001; OriGene
Technologies, Inc.). After transfection, the cells were incubated
for 2 days followed by selection with 1.5 to 2 µg/ml
puromycin for 3-5 days. Thereafter, cells were cultured with normal
media.
Transfection with siRNA
SHP1 siRNA and control siRNA were designed and
purchased from Santa Cruz Biotechnology. At 1 day prior to
transfection, the cells were seeded in the manner of
1×105 cells in a 6-well plate and incubated at 37°C. The
cells were transfected using the INTERFERin in vitro siRNA
transfection reagent (Polyplus-transfection SA) according to the
manufacturer's instructions. The mixture was applied to the cells
and incubated for 48 h at 37°C in the presence of 5%
CO2.
Colony-forming assay
The cells were seeded in the manner of
5×102 cells in 6-well plates and incubated for 7 days at
37°C in the presence of 5% CO2. Subsequently, 10%
buffered formaldehyde (Sigma-Aldrich) was added to the cells
followed by incubation for 10 min at room temperature, while 1 ml
of 0.1% crystal violet (Sigma-Aldrich) was also added to the cells
and shaken for 30 min at room temperature. The cells were washed 3
times with distilled water and dried.
Cell count
The cells were seeded in the manner of
1×105 cells in six6well plates and incubated for 7 days
at 37°C in the presence of 5% CO2. The numbers of cells
were counted using a light microscope (Olympus) and a hemocytometer
(Hausser Scientific Co.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells using TRIzol
reagent. Cells were mixed with 1 ml of TRIzol (Invitrogen; Thermo
Fisher Scientific) and incubated at room temperature for 10 min
following vortexing. Subsequently, 300 µl of chloroform was
mixed gently by inverting. Centrifugation was performed at 12,000 ×
g for 15 min at 4°C and the resultant supernatant was collected. At
this point, an equal amount of isopropanol was added, mixed gently,
and incubated at room temperature for 10 min. Centrifugation was
performed for 20 min at 10,000 × g at 4°C to remove the
supernatant. After washing with 70% ethanol, the RNA pellet was
dissolved in diethyl pyrocarbonate (DEPC) water. The RNA
concentration and purity of each sample were determined using a
NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific).
Complementary DNA (cDNA) was synthesized using the PrimeScript
first-strand cDNA synthesis kit (Takara Korea Biomedical Inc.)
according to the manufacturer's instructions. The amplification of
each cDNA was monitored using the Sensi FAST SYBR No-ROX kit
(Bioline) on a StepOnePlus instrument (Thermo Fisher Scientific).
All specific forward and reverse primers used were as follows:
Cardiotrophin-like cytokines factor 1 (CLCF1), forward, 5′-TAT GAC
CTC ACC CGC TAC CT-3′ and reverse, 5-′GGG GCC CAG GTA GTT CAG-3′;
interleukin (IL)6 forward, 5′-CCA CCG GGA ACG AAA GAG AA-3′ and
reverse, 5′-GAG AAG GCA ACT GGA CCG AA-3′; IL6 receptor (IL6R)
forward, 5′-GGG TCT CTA CCA TCC CCT GT-3′ and reverse, 5′-AGA AAT
GGC AGA AGC CCT CC-3′; IL6 signal transducer (IL6ST) forward,
5′-CAG TGG TCA CCT CAC ACT CC-3′ and reverse, 5′-TGA CAT GCA TGA
AGA CCC CC-3′; Janus kinase 2 (JAK2) forward, 5′-TGG GGT TTT CTG
GTG CCT TT-3′ and reverse, 5′-TAG AGG GTC ATA CCG GCA CA-3′;
leukemia inhibitory factor (LIF) forward, 5′-CTC GCC CAT CAC CTC
ATC TC-3′ and reverse, 5′-GCA GAG CTG TTT CAC GCA AA-3′; Erb-B2
receptor tyrosine kinase 4 (ERBB4) forward, 5′-CCT GGA AGA AAG ACG
ACT CGT TC-3′ and reverse, 5′-CGT CAC TCT GAT GGG TGA ATT TCC-3′;
and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward,
5′-CTG CAC CAC CAA CTG CTT AG-3′ and reverse, 5′-AGG TCC ACC ACT
GAC ACG TT-3′. GAPDH was used as an internal control. The relative
mRNA expression change was expressed as a fold change in comparison
with the GAPDH control. Data are presented as the means ± standard
deviations from at least 3 independent experiments in
triplicate.
RNA isolation and gene expression
profiling
In the present study, we performed global gene
expression analyses using Affymetrix GeneChip® Human
Gene 2.0 ST oligonucleotide arrays. Total RNA was isolated from the
CNOT2-depleted or control sh-RNA transfected-A549 cells using
TRIzol reagent (Qiagen). RNA quality was assessed with the Agilent
2100 Bioanalyser (Agilent Technologies) and quantity was determined
with the ND-2000 spectrophotometer (Thermo Fisher Scientific). For
each RNA, the synthesis of target cRNA probes and hybridization
were performed using Agilent's Low-input QuickAmp Labeling kit
(Agilent Technologies) according to the manufacturer's
instructions. The gene expression data were analyzed using
GeneSpringGX 7.3.1 software (Agilent Technologies). A
hierarchically clustered heatmap was generated using MeV version
4.9.0 software. The genes related to cell proliferation [Gene
Ontology (GO):0008283) and STAT3 GO:0042509] were classified and
visualized in a Venn diagram. Gene array hybridization raw data
were deposited in the National Center for Biotechnology Information
Gene Expression Omnibus database under accession no. GSE131518.
Western blot analysis
The cells were rinsed with ice-cold PBS and
harvested with a cell scraper followed by centrifugation at 300 × g
for 5 min at 4°C. The cell pellets were lysed as previously
described (13,14). Protein samples were quantified by
using a Bio-Rad DC protein assay kit II (Bio-Rad) and 10-20
µg of protein lysates were separated on 8 to 15% sodium
dodecyl sulfate-polyacrylamide gels and transferred to
polyvinylidene difluoride membranes (Millipore). The membranes were
blocked in 5% nonfat skim milk (in TBST buffer) for 1 h at RT and
then probed with primary antibodies at 1/1,000-2,000 diluted in
3-5% non-fat skim milk for 1 h at room temperature. After washing 3
times for 5 min with TBST, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (#7076 for
anti-mouse, #7074 for anti-rabbit from Cell Signaling Technology),
1/5,000 diluted in 5% non-fat skim milk, for 1 h at room
temperature. The protein expression levels were detected using an
enhanced chemiluminescence (ECL) system (Amersham Pharmacia),
according to the manufacturer's instructions.
Statistical analysis
All experiments were repeated 3 times. Data are
presented as the means ± standard deviation (SD). Differences
between the means of each group were analyzed using a t-test, while
P-values <0.05 were considered significant to indicate
statistically significant differences. The statistical software
package Excel (Microsoft Corp.) was used for the analysis. The
Prism software package (Graph Pad Prism 5.0 for Windows) was used
for data collection and presentation for Fig. 3B. The data ranged from 3 to 12
separate experiments and are presented as the means ± SD. Two-way
ANOVA followed by a Bonferroni post hoc test were used to determine
the significant differences between the various experimental and
control groups.
Results
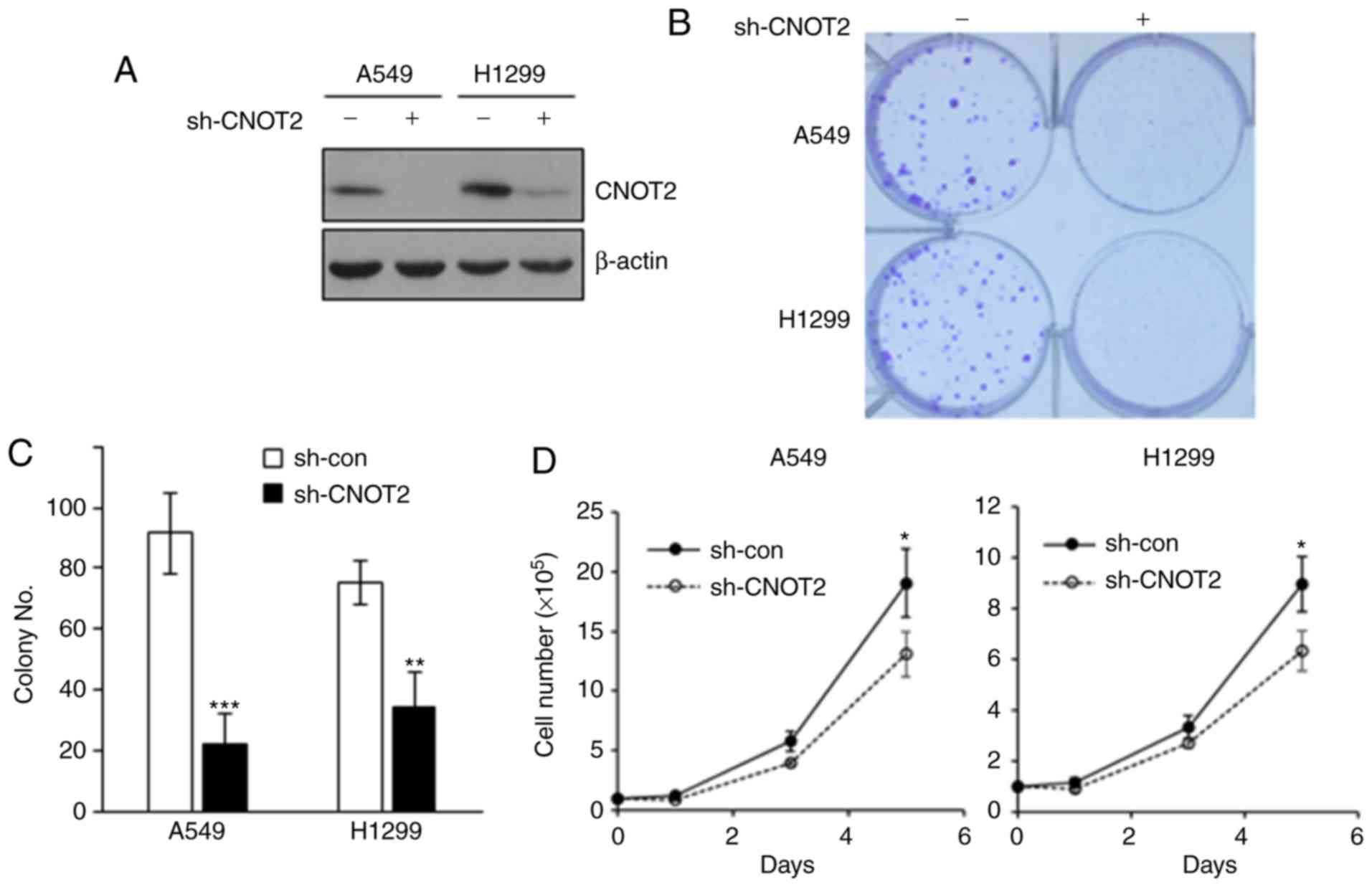
Depletion of CNOT2 affects the
proliferation and growth of NSCLC cells
To determine whether CNOT2 has an oncogenic function
in NSCLC similar to that in other cancer types as we previously
demonstrated (15), we depleted
CNOT2 using specific shRNA in the NSCLC cell lines, A549 and H1299
(Fig. 1A). We found that the
knockdown of CNOT2 resulted in a 50 to 75% decrease in colony
number (Fig. 1B and C) and an
approximately 40% decrease in the growth rates of NSCLC cells
(Fig. 1D). These results indicate
that CNOT2 affects the proliferation and growth of NSCLC cells.
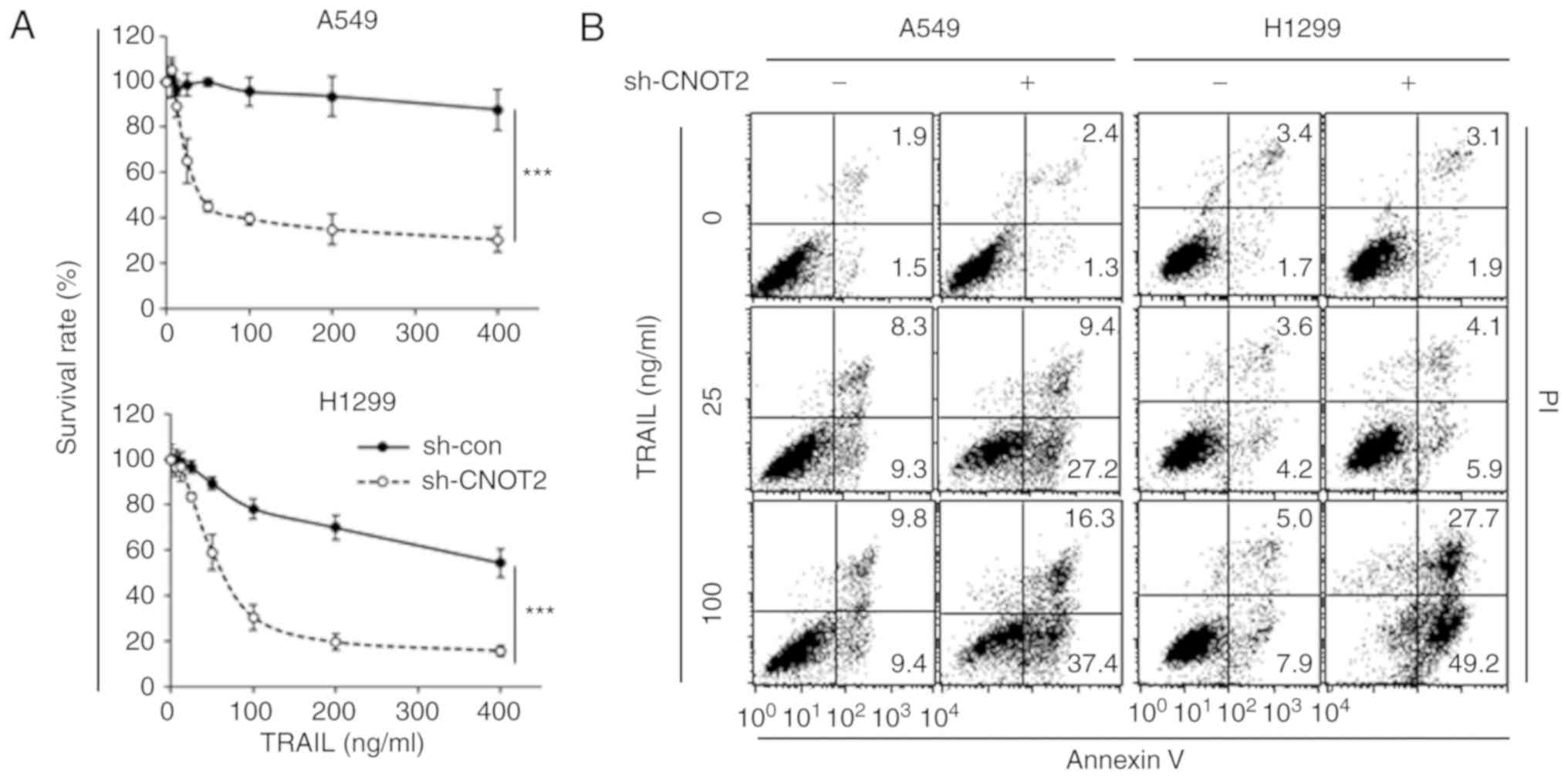
Knockdown of CNOT2 markedly increases
TRAIL sensitivity in A549 and H1299 cells
To explore whether CNOT2 participates in the
regulation of TRAIL sensitivity, the cells in which CNOT2 was
downregulated were treated with TRAIL (Figs. 2A and S1). As shown in Figs. 2A and S1B, treatment with TRAIL markedly
decreased the survival rates of cells when compared with the
control in the A549, H1299 and H596 cells. TRAIL treatment enhanced
apoptosis, the main death mechanism promoted by TRAIL, in a
dose-dependent manner in the CNOT2-depleted cells (Fig. 2B). These results indicate that
CNOT2 participates in the TRAIL-mediated apoptosis of NSCLC
cells.
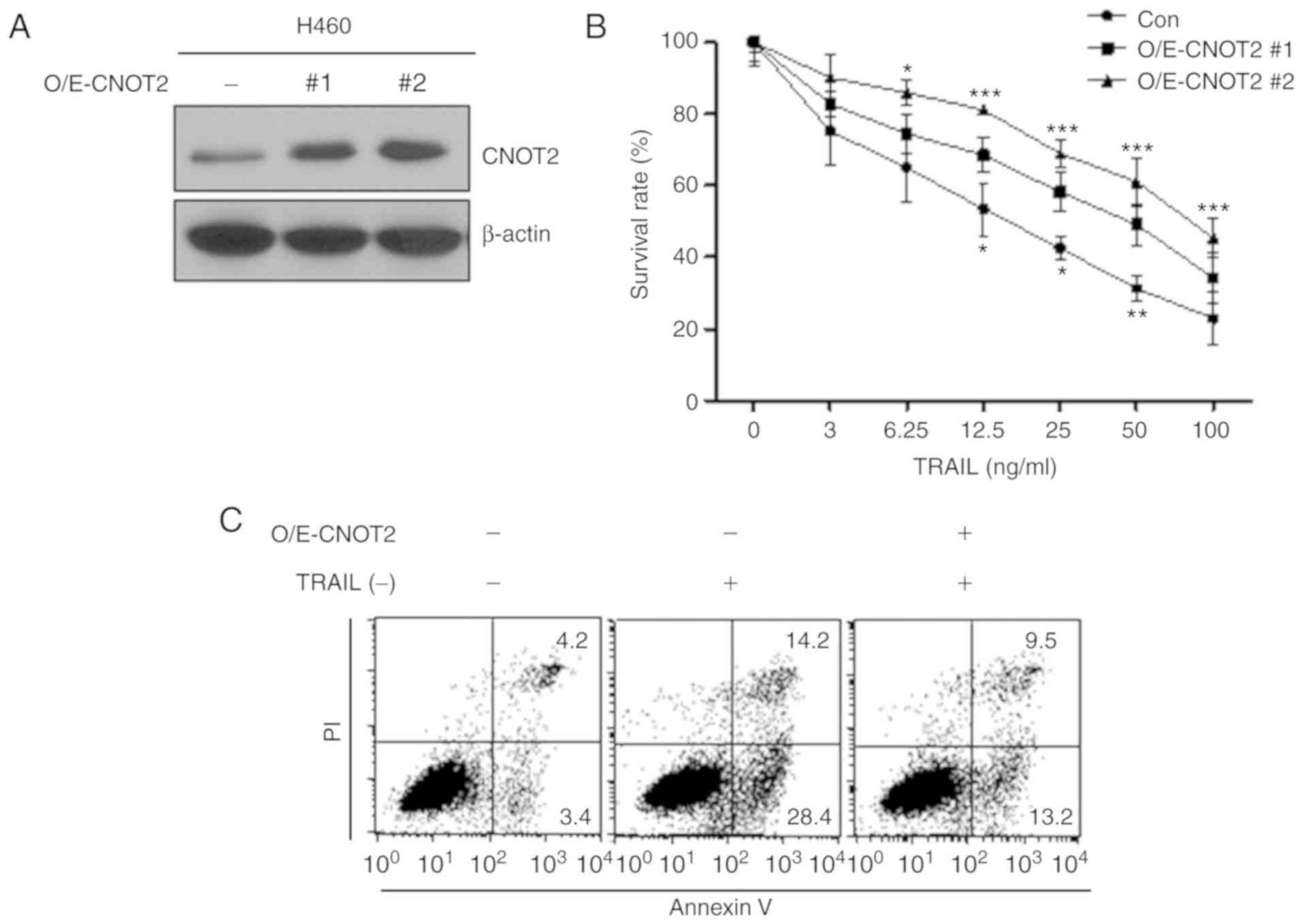
CNOT2 overexpression increases TRAIL
resistance in the TRAIL-sensitive cell line, H460
To confirm that CNOT2 participates in TRAIL
sensitivity in NSCLC, we established a CNOT2-overexpressing H460
line that is TRAIL-sensitive (Fig.
3A). Treatment of the CNOT2-overexpressing cells with TRAIL
increased the cell survival rates by 1.2- to 1.7-fold, as compared
with the control cells (Fig. 3B).
In addition, the CNOT2-overexpressing cells exhibited decreased
apoptosis following treatment with TRAIL (Fig. 3C). These data suggest that the
increase in CNOT expression imparts TRAIL resistance on
TRAIL-sensitive cells.
CNOT2 depletion downregulates the
SHP1/STAT3 signaling pathway
To validate the gene expression profiles and
molecular mechanisms involved in CNOT2-mediated TRAIL sensitivity,
we analyzed GO pathways in CNOT2-silenced A549 cells using a cDNA
microarray. CNOT2 depletion induced dominant changes in the
pathways of cell migration, angiogenesis, RNA splicing and cell
proliferation (Figs. 4A and
S2). In previous studies, it was
demonstrated that CNOT2 is highly associated with angiogenesis,
migration and metastasis in various cancer types (11,16-18). In this study, we focused on
proliferation, which was also altered in CNOT2-depleted lung cancer
cells (Fig. 4B). Sixteen genes
regulated by CNOT2 depletion were commonly STAT3-related genes,
involved in cell proliferation and CNOT2-related genes (Fig. 4C). Consistent with these findings,
RT-qPCR analysis confirmed that the inhibition of CNOT2
downregulated the gene expression levels of IL6 and CLCF1 as tumor
progression markers and STAT3-related genes (19,20) as well as LIF and ERBB4 as
proliferation markers (21,22) in A549 and H1299 cells (Fig. 4D). In the same analysis, IL6ST,
IL6R and JAK2, as STAT3-related genes without differences in
CNOT2-depleted cells, were confirmed (Fig. 4C and D). Subsequently, STAT3
activity was also verified by measuring the phosphorylation level
of STAT3 in CNOT2-depleted cells. As shown in Fig. 4E, the phosphorylation of STAT3 was
significantly decreased, while STAT3 protein expression was not
altered in the A549 or H1299 cells in which CNOT2 expression was
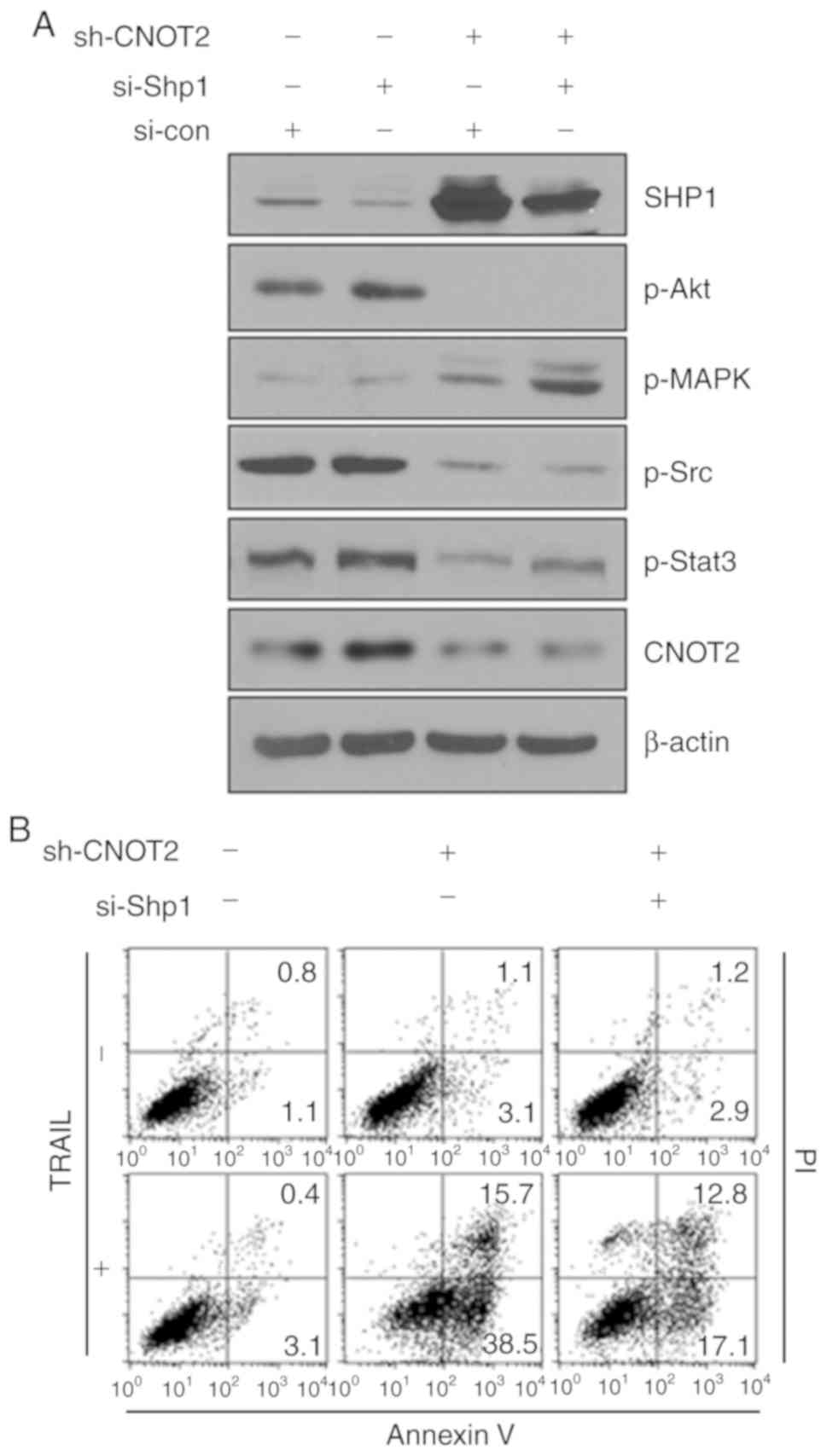
silenced. In addition, we found that SHP1 was dramatically
upregulated in the same sample (Fig.
4E). To verify whether SHP1 plays an important role in
CNOT2-mediated gene regulation, we inhibited the SHP1 gene in
CNOT2-depleted cells. The inhibition of SHP1 increased the
phosphorylation of STAT3 as compared with the control (Fig. 5A) and recovered the survival rates
of CNOT2-depleted cells (Fig.
5B). These results indicate that STAT3 is a main regulator of
the CNOT2-mediated signaling pathway.
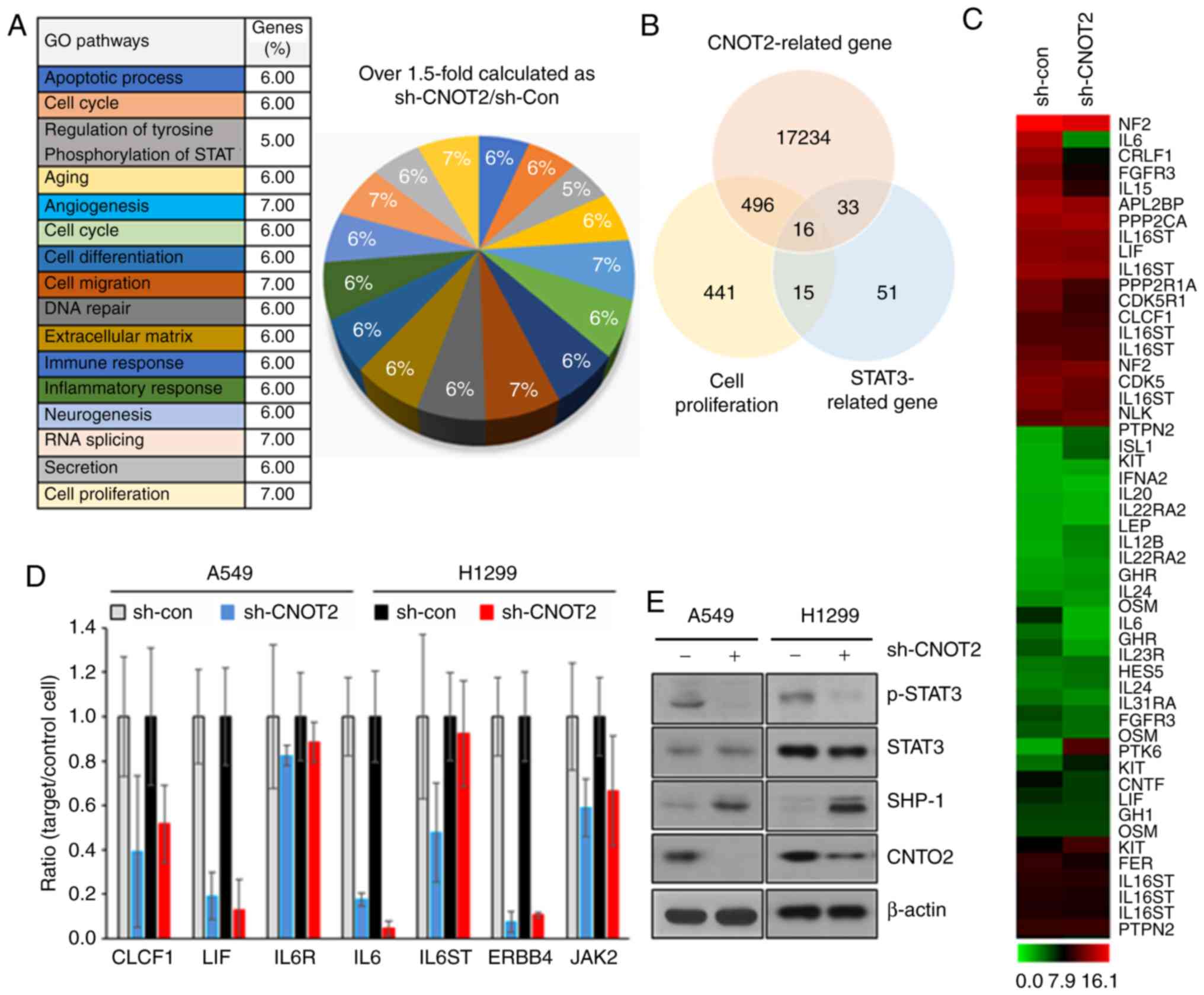 | Figure 4Gene profile in CNOT2-depleted cells
and verification by RT-qPCR and western blot analysis. (A) Gene
ontology (GO) pathway gene sets were compared to untreated controls
in CNOT2-depleted A549 cells. (B) Associations between cell
proliferation genes, STAT3-related genes and CNOT2-related genes in
CNOT2-depleted A549 cells. (C) Heatmap of top-ranked upregulated or
downregulated genes in CNOT2-depleted A549 cells. (D) Effect of
CNOT2 depletion on the mRNA levels of CLCF1, LIF, IL6R, IL6, IL6ST,
ERBB4 and JAK2 by RT-qPCR. GAPDH was used as a loading control. (E)
STAT3 activity in CNOT2-depleted A549 and H1299 cells was analyzed
by western blot analysis. Cell lysates were used for detecting
phospho-STAT3, STAT, SHP1 and CNOT2. β-actin was used as an
internal loading control. CNOT2, CCR4-NOT transcription complex
subunit 2; STAT3, signal transducer and activator of transcription
3; CLCF1, cardiotrophin-like cytokines factor 1; LIF, leukemia
inhibitory factor; IL6, interleukin 6; IL6R, interleukin 6
receptor; IL6ST, interleukin 6 signal transducer; ERBB4, Erb-B2
receptor tyrosine kinase 4; JAK2, Janus kinase 2; SHP1, Src
homology region 2 domain containing phosphatase-1. |
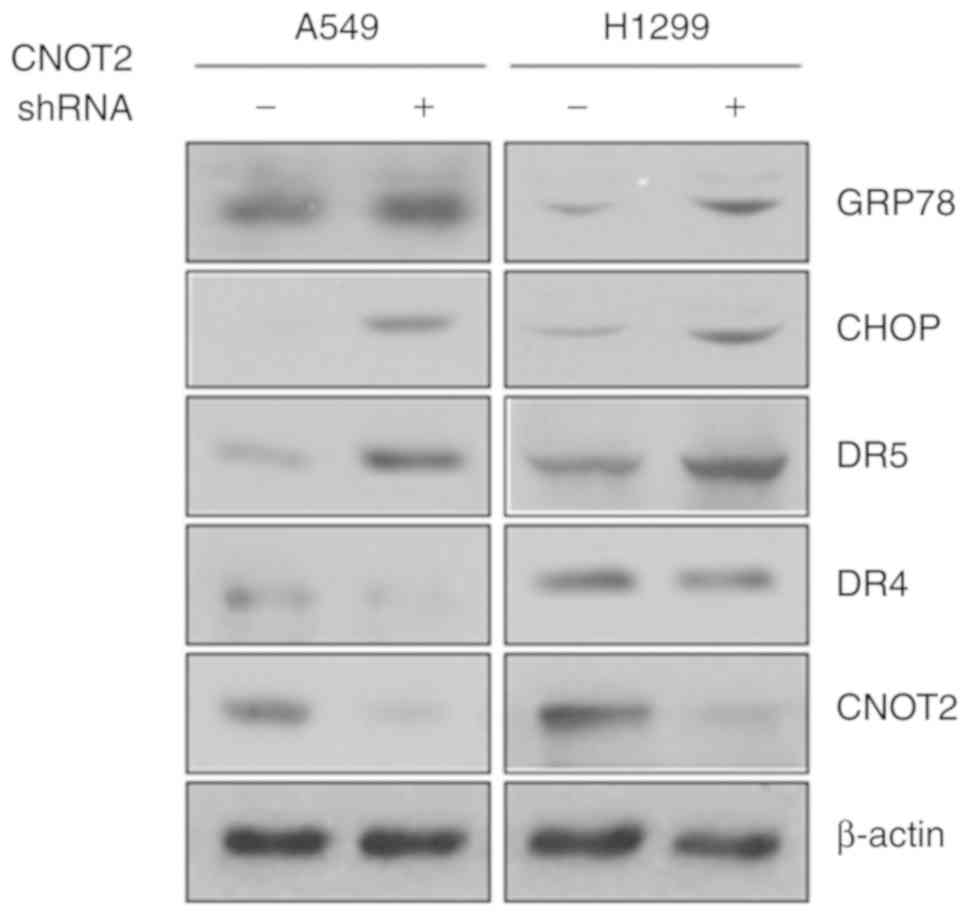
Inhibition of CNOT2 increases endoplasmic
reticulum (ER) stress
Recent studies have indicated that STAT3 and ER
stress signaling pathways interact with each other through diverse
mechanisms, leading to the control of cellular fate (23,24). Thus, it was hypothesized that
STAT3 inactivation by the depletion of CNOT2 may affect the ER
stress signaling pathway. To examine this hypothesis, we validated
the expression level of ER stress signaling molecules. As shown in
Fig. 6, CNOT2 depletion induced
the expression of GRP78, CHOP and DR5, ER stress-related molecules,
but not that of DR4 in the A549 and H1299 cells. These results
suggest that the CNOT2-mediated STAT3 signaling pathway is closely
associated with ER stress, but not apoptosis.
Discussion
In the current study, the findings demonstrated that
the depletion of CNOT2 induced the TRAIL-mediated apoptosis of
various TRAIL-resistant NSCLC cells. These increases in TRAIL
sensitivity by CNOT2 depletion were mediated by the induction of ER
stress, leading to the upregulation of DR5. In addition, it was
demonstrated that CNOT2 may participate in STAT3 signaling via the
regulation of SHP1 expression.
TRAIL is a promising molecule which can be used to
kill tumors, even though the majority of tumors present acquired
resistance against TRAIL-mediated cell death. Following the binding
of TRAIL to death receptors, DR4 and DR5 receptors are clustered by
the recruitment of Fas-associated protein with death domain (FADD),
which initiates the extrinsic apoptotic signaling pathway (25). Subsequently, FADD recruits
caspase-8 to form the death-inducing signaling complex (DISC),
leading to the cleavage of caspase-3, -6 and -7 (25-27). The activation of these caspases
induces apoptotic phenotypes, such as membrane blebbing, cleavage
of proteins and the cytoskeleton, and DNA fragmentation. In
addition, recruited caspase-8 activates the intrinsic apoptotic
pathway in certain instances, such as for example, the Bax/Bcl-2
pathway (28).
However, tumors, including lung cancer tumors
usually have a number of evasive tactics against TRAIL-induced
apoptosis. One typical evasive strategy of various tumors is the
upregulation of the anti-apoptotic proteins, Bcl-2, Bcl-xL, induced
myeloid leukemia cell differentiation protein (Mcl-1), BHRF1 and
E1b-19K (5). More than 50% of
cancer types possess overexpressed Bcl-2 (29) and XIAP is upregulated in various
types of cancer (6). The other
evasion mechanism involves the downregulation of pro-apoptotic or
apoptosis-inducing proteins, such as p53, caspases, PARP, Bax and
the tumor necrosis factor receptor family, including DR4 and DR5
(30-33). For example, the expression levels
of caspases, including caspase-8 are decreased in a diverse range
of tumor cells (6,34). Mutations in the p53 gene have been
reported in up to 25% of tumors (35) and the upregulation of p53 isoforms
leading to the inactivation of the canonical p53 function has also
been found in tumors (36).
To overcome these evasion strategies against
apoptosis, diverse combination therapies with TRAIL and agents to
either increase TRAIL activity or sensitize TRAIL-resistant cells
have been considered. A number of phytochemicals can modulate the
expression of various proteins to increase TRAIL efficiency. For
example, decursin enhances TRAIL-induced apoptosis through the
upregulation of ER stress-mediated DR5 in lung cancer cells
(12). Another approach is to
modulate genes regulating apoptosis-related proteins, leading to an
increased susceptibility to TRAIL efficiency. The depletion of
calcium and integrin-binding protein 1 (CIB1), regulators of
oncogenic PI3K/AKT and MET/ERK signaling, with TRAIL enhances the
apoptosis of triple-negative breast cancer cells (37).
Similar to CIB1, in the case of proteins with an
oncogenic or tumor suppressor function, the modulation of the
expression may affect TRAIL-mediated cell death. As previously
demonstrated, CNOT2 participates in the regulation of angiogenesis,
mobility autophagy and proliferation in diverse tumor types
(11,15). Based on these oncogenic functions
of CNOT2, it was hypothesized that CNOT2 may participate in
TRAIL-resistant mechanisms. Consistently, the depletion of CNOT2
with TRAIL also markedly increased the apoptotic rate of
TRAIL-resistant lung cancer cells.
STAT3 is a well-documented oncogenic transcription
factor that participates in diverse cellular signaling pathways,
including proliferation, apoptosis and cell growth (38,39). In various malignant tumors, STAT3
signaling is constitutively activated, enhancing tumorigenesis
(40,41). SHP1, a non-receptor protein
tyrosine phosphatase, is a major negative regulator by the
dephosphorylation of STAT3 (42).
A number of studies have indicated that the constitutive activation
of STAT3 in various tumor types is prompted by diminished or
abolished SHP1 expression (43,44). Therefore, the induction of SHP1
expression in cancer may be an effective method with which to
diminish tumors via the inactivation of STAT3. In this aspect,
CNOT2 may be an attractive target which may be used to boost SHP1
expression. Nakase et al reported that Sp1, Oct-1, NF-κB and
CREB-1 interacted with the core SHP1 P2 promoter in CD4+
T-cells and Jurkat cells (45).
Previously, Warner et al suggested that CNOT2 depletion
affected NF-κB expression (46).
These two results indicate that CNOT2 may regulate SHP1 expression
through the NF-κB signaling pathway. However, further detailed
studies are required to better elucidate the mechanisms through
which CNOT2 regulates SHP1.
Recent studies have suggested that the STAT
signaling pathway may be connected with ER stress (23,24,47). Based on the findings of this
study, under a CNOT2-depleted condition in NSCLC cells, SHP1 and ER
stress molecules were concurrently increased. In addition, the
inhibition of SHP1 suppressed apoptosis induced by TRAIL and CNOT2
depletion in NLCLC cells. However, this study was not able to
obtain any direct evidence to connect STAT3 and ER stress in
CNOT2-depleted NSCLC cells (data not shown). Further studies are
warranted to address this issue.
Taken together, the depletion of CNOT2 highly
induces the TRAIL-mediated apoptosis of various TRAIL-resistant
NSCLC cells. These increases in TRAIL sensitivity by CNOT2
depletion are mediated by the induction of ER stress, leading to
the upregulation of DR5. In addition, this study demonstrated that
CNOT2 may participate in STAT3 signaling via the regulation of
SHIP1 expression.
Supplementary Data
Abbreviations:
|
TRAIL
|
TNF-related apoptosis-inducing
ligand
|
|
CNOT2
|
CCR4-NOT transcription complex subunit
2
|
|
NSCLC
|
non-small cell lung cancer
|
|
DR4
|
death receptor 4
|
|
DR5
|
death receptor 5
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
FBS
|
fetal bovine serum
|
|
SHP1
|
Src homology region 2 domain
containing phosphatase-1
|
|
Bax
|
BCL2 associated X protein
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
CHOP
|
C/EBP homologous protein
|
|
GRP78
|
glucose-regulated protein 78
|
|
MTT
|
3-(4,5-dimethylthialzol-2-yl)-2,5-diphenyl-2H-tetrazolium
bromide
|
|
PI
|
propidium iodide
|
|
RNA
|
ribonucleic acid
|
|
DEPC
|
diethyl pyrocarbonate
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
ECL
|
enhanced chemiluminescence
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
IL6
|
interleukin 6
|
|
IL6R
|
interleukin 6 receptor
|
|
CLCF1
|
cardiotrophin-like cytokines factor
1
|
|
LIF
|
leukemia inhibitory factor
|
|
ERBB4
|
Erb-B2 receptor tyrosine kinase 4
|
|
IL6ST
|
interleukin 6 signal transducer
|
|
JAK2
|
Janus kinase 2
|
|
ER
|
endoplasmic reticulum
|
|
FADD
|
Fas-associated protein with death
domain
|
|
DISC
|
death-inducing signalling complex
|
Acknowledgments
Not applicable.
Funding
This study was supported by the National Research
Foundation of Korea (NRF) Grant funded by the Korea Government
(Ministry of Science and ICT) (NRF-2018R1D1A1B07048377 and
NRF-2016R1D1A1B03933853).
Availability of data and materials
The datasets supporting the conclusions of this
article are included within the article and its additional
files.
Authors' contributions
EOK was involved in acquisition, analysis and
interpretation of the data and development of the methodology. SEK
was involved in the acquisition of data. MC contributed to the
acquisition and analysis of the data during revision. KJR was
involved in the analysis of the data and revised critically for
important intellectual content. MY was involved in the analysis and
interpretation of the data and the writing of the manuscript,
conception and design of the study, and funding. All authors
discussed the results, commented on the manuscript, and approved
the final submitted and published versions. In addition, all
authors agree to be accountable for all aspects of the work in
ensuring that questions related to the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no that they have no competing
interests.
References
|
1
|
World Health Organization: Cancer.
https://www.who.int/news-room/fact-sheets/detail/cancer.
Accessed September 12, 2018.
|
|
2
|
Warth A, Muley T, Herpel E, Meister M,
Herth FJ, Schirmacher P, Weichert W, Hoffmann H and Schnabel PA:
Large-scale comparative analyses of immunomarkers for diagnostic
subtyping of non-small-cell lung cancer biopsies. Histopathology.
61:1017–1025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Noone AM, Howlader N, Krapcho M, Miller D,
Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, et al:
SEER Cancer Statistics Review, 1975-2015. National Cancer Institute
Bethesda; MD: 2018
|
|
4
|
Schneider P, Thome M, Burns K, Bodmer JL,
Hofmann K, Kataoka T, Holler N and Tschopp J: TRAIL receptors 1
(DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate
NF-kappaB. Immunity. 7:831–836. 1997. View Article : Google Scholar
|
|
5
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elkholi R, Renault TT, Serasinghe MN and
Chipuk JE: Putting the pieces together: How is the mitochondrial
pathway of apoptosis regulated in cancer and chemotherapy? Cancer
Metab. 2:162014. View Article : Google Scholar
|
|
7
|
Russell P, Benson JD and Denis CL:
Characterization of mutations in NOT2 indicates that it plays an
important role in maintaining the integrity of the CCR4-NOT
complex. J Mol Biol. 322:27–39. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zwartjes CG, Jayne S, van den Berg DL and
Timmers HT: Repression of promoter activity by CNOT2, a subunit of
the transcription regulatory Ccr4-not complex. J Biol Chem.
279:10848–10854. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jayne S, Zwartjes CG, van Schaik FM and
Timmers HT: Involvement of the SMRT/NCoR-HDAC3 complex in
transcriptional repression by the CNOT2 subunit of the human
Ccr4-Not complex. Biochem J. 398:461–467. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ito K, Inoue T, Yokoyama K, Morita M,
Suzuki T and Yamamoto T: CNOT2 depletion disrupts and inhibits the
CCR4-NOT deadenylase complex and induces apoptotic cell death.
Genes Cells. 16:368–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sohn EJ, Jung DB, Lee H, Han I, Lee J, Lee
H and Kim SH: CNOT2 promotes proliferation and angiogenesis via
VEGF signaling in MDA-MB-231 breast cancer cells. Cancer Lett.
412:88–98. 2018. View Article : Google Scholar
|
|
12
|
Kim J, Yun M, Kim EO, Jung DB, Won G, Kim
B, Jung JH and Kim SH: Decursin enhances TRAIL-induced apoptosis
through oxidative stress mediated- endoplasmic reticulum stress
signalling in non-small cell lung cancers. Br J Pharmacol.
173:1033–1044. 2016. View Article : Google Scholar
|
|
13
|
Jung JH, Kwon TR, Jeong SJ, Kim EO, Sohn
EJ, Yun M and Kim SH: Apoptosis induced by tanshinone IIA and
crypto-tanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2
signaling in chronic myeloid leukemia K562 cells. Evid Based
Complement Alternat Med. 2013:8056392013. View Article : Google Scholar
|
|
14
|
Jung JH, Jung DB, Kim H, Lee H, Kang SE,
Srivastava SK, Yun M and Kim SH: Zinc finger protein 746 promotes
colorectal cancer progression via c-Myc stability mediated by
glycogen synthase kinase 3β and F-box and WD repeat
domain-containing 7. Oncogene. 37:3715–3728. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jeong K, Kwon HY, Jeong MS, Sohn EJ and
Kim SH: CNOT2 promotes degradation of p62/SQSTM1 as a negative
regulator in ATG5 dependent autophagy. Oncotarget. 8:46034–46046.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Faraji F, Hu Y, Yang HH, Lee MP, Winkler
GS, Hafner M and Hunter KW: Post-transcriptional control of tumor
cell autonomous metastatic potential by CCR4-NOT deadenylase CNOT7.
PLoS Genet. 12:e10058202016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee J, Jung JH, Hwang J, Park JE, Kim JH,
Park WY, Suh JY and Kim SH: CNOT2 is critically involved in
atorvastatin induced apoptotic and autophagic cell death in
non-small cell lung cancers. Cancers (Basel). 11:pii: E1470. 2019.
View Article : Google Scholar
|
|
18
|
Vicente C, Stirparo R, Demeyer S, de Bock
CE, Gielen O, Atkins M, Yan J, Halder G, Hassan BA and Cools J: The
CCR4-NOT complex is a tumor suppressor in Drosophila mela-nogaster
eye cancer models. J Hematol Oncol. 11:1082018. View Article : Google Scholar
|
|
19
|
Grivennikov SI and Karin M: Inflammatory
cytokines in cancer: Tumour necrosis factor and interleukin 6 take
the stage. Ann Rheum Dis. 70(Suppl 1): i104–i108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vicent S, Sayles LC, Vaka D, Khatri P,
Gevaert O, Chen R, Zheng Y, Gillespie AK, Clarke N, Xu Y, et al:
Cross-species functional analysis of cancer-associated fibroblasts
identifies a critical role for CLCF1 and IL-6 in non-small cell
lung cancer in vivo. Cancer Res. 72:5744–5756. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haskins JW, Nguyen DX and Stern DF:
Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo
pathway target genes and promote cell migration. Sci Signal.
7:ra1162014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu H, Yue X, Zhao Y, Li X, Wu L, Zhang C,
Liu Z, Lin K, Xu-Monette ZY, Young KH, et al: LIF negatively
regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in
colorectal cancers. Nat Commun. 5:52182014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meares GP, Liu Y, Rajbhandari R, Qin H,
Nozell SE, Mobley JA, Corbett JA and Benveniste EN: PERK-dependent
activation of JAK1 and STAT3 contributes to endoplasmic reticulum
stress-induced inflammation. Mol Cell Biol. 34:3911–3925. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Banerjee K, Keasey MP, Razskazovskiy V,
Visavadiya NP, Jia C and Hagg T: Reduced FAK-STAT3 signaling
contributes to ER stress-induced mitochondrial dysfunction and
death in endothelial cells. Cell Signal. 36:154–162. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dickens LS, Boyd RS, Jukes-Jones R, Hughes
MA, Robinson GL, Fairall L, Schwabe JW, Cain K and Macfarlane M: A
death effector domain chain DISC model reveals a crucial role for
caspase-8 chain assembly in mediating apoptotic cell death. Mol
Cell. 47:291–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu H, Su D, Zhang J, Ge S, Li Y, Wang F,
Gravel M, Roulston A, Song Q, Xu W, et al: Improvement of
pharmacokinetic profile of TRAIL via trimertag enhances its
antitumor activity in vivo. Sci Rep. 7:89532017. View Article : Google Scholar
|
|
27
|
Xu W, Jing L, Wang Q, Lin CC, Chen X, Diao
J, Liu Y and Sun X: Bax-PGAM5L-Drp1 complex is required for
intrinsic apoptosis execution. Oncotarget. 6:30017–30034. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y and Zhang B: TRAIL resistance of
breast cancer cells is associated with constitutive endocytosis of
death receptors 4 and 5. Mol Cancer Res. 6:1861–1871. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di X, Zhang G, Zhang Y, Takeda K, Rivera
Rosado LA and Zhang B: Accumulation of autophagosomes in breast
cancer cells induces TRAIL resistance through downregulation of
surface expression of death receptors 4 and 5. Oncotarget.
4:1349–1364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fernald K and Kurokawa M: Evading
apoptosis in cancer. Trends Cell Biol. 23:620–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stupack DG: Caspase-8 as a therapeutic
target in cancer. Cancer Lett. 332:133–140. 2013. View Article : Google Scholar
|
|
35
|
Khoury MP and Bourdon JC: The isoforms of
the p53 protein. Cold Spring Harb Perspect Biol. 2:a0009272010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bourdon JC, Fernandes K, Murray-Zmijewski
F, Liu G, Diot A, Xirodimas DP, Saville MK and Lane DP: p53
isoforms can regulate p53 transcriptional activity. Genes Dev.
19:2122–2137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chung AH, Leisner TM, Dardis GJ, Bivins
MM, Keller AL and Parise LV: CIB1 depletion with docetaxel or TRAIL
enhances triple-negative breast cancer cell death. Cancer Cell Int.
19:262019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fagard R, Metelev V, Souissi I and
Baran-Marszak F: STAT3 inhibitors for cancer therapy: Have all
roads been explored? JAKSTAT. 2:e228822013.PubMed/NCBI
|
|
39
|
Xiong A, Yang Z, Shen Y, Zhou J and Shen
Q: Transcription factor STAT3 as a novel molecular target for
cancer prevention. Cancers (Basel). 6:926–957. 2014. View Article : Google Scholar
|
|
40
|
Santoni M, Massari F, Del Re M, Ciccarese
C, Piva F, Principato G, Montironi R, Santini D, Danesi R, Tortora
G and Cascinu S: Investigational therapies targeting signal
transducer and activator of transcription 3 for the treatment of
cancer. Expert Opin Investig Drugs. 24:809–824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Subramaniam A, Shanmugam MK, Perumal E, Li
F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK, et al:
Potential role of signal transducer and activator of transcription
(STAT)3 signaling pathway in inflammation, survival, proliferation
and invasion of hepatocellular carcinoma. Biochim Biophys Acta.
1835:46–60. 2013.
|
|
42
|
Tai WT, Cheng AL, Shiau CW, Huang HP,
Huang JW, Chen PJ and Chen KF: Signal transducer and activator of
transcription 3 is a major kinase-independent target of sorafenib
in hepatocellular carcinoma. J Hepatol. 55:1041–1048. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Delibrias CC, Floettmann JE, Rowe M and
Fearon DT: Downregulated expression of SHP-1 in Burkitt lymphomas
and germinal center B lymphocytes. J Exp Med. 186:1575–1583. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu C, Sun M, Liu L and Zhou GW: The
function of the protein tyrosine phosphatase SHP-1 in cancer. Gene.
306:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakase K, Cheng J, Zhu Q and Marasco WA:
Mechanisms of SHP-1 P2 promoter regulation in hematopoietic cells
and its silencing in HTLV-1-transformed T cells. J Leukoc Biol.
85:165–174. 2009. View Article : Google Scholar :
|
|
46
|
Warner N, Burberry A, Franchi L, Kim YG,
McDonald C, Sartor MA and Núñez G: A genome-wide siRNA screen
reveals positive and negative regulators of the NOD2 and NF-κB
signaling pathways. Sci Signal. 6:rs32013. View Article : Google Scholar
|
|
47
|
Avalle L, Camporeale A, Morciano G,
Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P
and Poli V: STAT3 localizes to the ER, acting as a gatekeeper for
ER-mitochondrion Ca2+ fluxes and apoptotic responses.
Cell Death Differ. 26:932–942. 2019. View Article : Google Scholar
|