Introduction
Epilepsy is the term used to describe a common
neurological disorder characterized by long-term, recurrent
epileptic seizures that is accompanied by various neurobiological,
cognitive, psychological and social consequences (1). Temporal lobe epilepsy (TLE) is one
of the most common forms of partial epilepsy and its animal model
can be divided into acute, silent, and chronic phases (2). Abundant alterations can be observed
in the hippocampus during the acute and silent period after status
epilepticus (SE), such as neuronal loss and degeneration, gliosis,
mossy fiber sprouting, scattering of dentate granule cells, and
synaptic modifications in the dentate gyrus, all of which
contribute to the epileptogenic process (3,4).
The accumulated evidence shows that neuronal cell death in the
hippocampus is the most important factor in initiating and
precipitating the development of epileptic disorders (5-7).
In addition, previously discovered datum also provided novel
insight into two mechanisms in connection with cell survival and
death, autophagy and apoptosis, and their function in regulating
neuronal fate (8-10).
Autophagy plays a vital role in various aspects of
cell homeostasis, such as cell survival, cell death, cell
metabolism, development, neuroprotection and even neurodegeneration
(11). Mammalian target of
rapamycin (mTOR) is a central regulator of autophagy that
strategically influences synaptic plasticity at pre- and
postsynaptic sites by regulating the balance between protein
synthesis and degradation (12-15). Activation of mTOR can inactivate
assembly of Unc-51-like autophagy-activating kinase 1 (ULK-1) at
S757 and in turn prevent phosphorylation and activation of Beclin-1
at S14 to halt autophagy (16,17). Beclin-1 promotes autophagosome
nucleation, through the generation of PI3P in complex with a class
III phosphatidylinositol 3 kinase. Autophagosome maturation and the
lipidation of LC3-II to the autophagosome membrane is completed by
two ubiquitin-like conjugation systems [ATG5-ATG12-ATG16L and
(ATG8) LC3] (18,19). The limiting autophagosome membrane
elongates to recognize and encapsulate degradation substrates via
p62 protein (20). Several
findings have implied the participation of autophagy in progressive
central nervous system (CNS) diseases such as Parkinson's,
Huntington's and Alzheimer's disease (21,22), amyotrophic lateral sclerosis
(23,24) and global ischemia (25). Furthermore, in the chronic phase
of pilocarpine-induced seizures, cannabidiol post-treatment
alleviates rat epileptic-related behaviors and activates the
hippocampal cell autophagy pathway and antioxidant defense
mechanisms (26). However,
detailed understanding of the role played by autophagy in epilepsy
is lacking.
The endogenous endocannabinoids and their receptors
are important regulators of neuronal activity (27,28). Cannabinoid receptor type 1 (CB1R),
which is abundantly expressed throughout the CNS (29), has been a research focus for
numerous years. In contrast, cannabinoid receptor type 2 (CB2R) is
mainly distributed in immune organs and cells, including the
spleen, lymphocytes and even neutrophil granulocytes (30,31), and plays important roles in
regulating immune function. Previous studies have paid much less
attention to the role of CB2R in epilepsy, mainly because of its
limited CNS expression. However, research has suggested that CB2R
is expressed in the brain (32-34), raising the possibility that it
could also play a direct role in mediating CNS function. Growing
evidence shows that CB2R stimulation exerts anti-convulsant
properties in seizure models. Previous studies suggested that
activation of CB2Rs may regulate neuronal excitability in the
hippo-campus and increase excitatory synaptic transmission
(35), trigger cell type-specific
hippocampal pyramidal cell hyperpolarization (36), and suppress epileptic seizures
(37). The author's previous
study revealed the expression of CB2R in hippocampal neurons of
developing rats and its time-dependent changes after SE, which are
accompanied by a fall in the number of neurons (38). However, the activation of CB2R on
neuronal repair in response to the damage after SE is still
unclear. The present study hypothesized that the anti-convulsant
effects of CB2R during the neuronal damage-repair process may be
contributing to induction of autophagy. In this experiment, a
highly selective CB2R agonist JWH133 and antagonist AM630 was
administered to verify the present hypothesis.
Materials and methods
Animal models
A total of 42 rats healthy male Sprague-Dawley rats
(from the Medical Animal Center, Shengjing Hospital of China
Medical University), 3 weeks of age and weighing 45.0-56.5 g, were
used in this study. The rats were provided with ad libitum
access to food and purified water and were maintained under
controlled temperature, humidity, and lighting conditions (20-25°C,
50-60% humidity, 12-h light/ dark cycle). The China Medical
University Institutional Animal Care and Use Committee (no.
2016PS260K) approved all procedures.
A commonly used SE model was used, as previously
reported by the authors' lab (38,39). Male rats 3-weeks old weighing
45.0-56.5 g were used. A total of 42 rats were randomly divided
into 7 groups (n=6 per group): Control group and 2 h, 1, 3, 7, 14
and 21 d groups. Furthermore, 30 male rats were randomly divided
into 5 groups (n=6 per group) according to pre-treatment: SE group,
Vehicle group, JWH133 1 µg/side group, JWH133 3
µg/side group and JWH133+AM630 group. Rats were treated with
lithium chloride (3 mEq/kg, intraperitoneal; Sigma-Aldrich; Merck
KGaA) 16-18 h prior to the injection of pilocarpine and then
methylscopolamine (1 mg/kg, intraperitoneal; Sigma-Aldrich; Merck
KGaA) 30 min prior to injection of pilocarpine for reducing the
peripheral cholinergic effects. Experimental animals were then
injected intraperitoneally (i.p.) with a single dose of 30 mg/kg of
pilocarpine in order to induce SE. Control rats were administered a
comparable volume of vehicle after the initial methylscopolamine
treatment. Behavioral observation after pilocarpine injection was
performed and pilocarpine-induced seizures were evaluated according
to the Racine scale (40).
Seizure intensity was that stage 1 (facial clonus with scratching,
chewing and grooming), stage 2 (head nodding, tail wagging and
chirping), stage 3 (forelimb clonus, wet dog shakes, standing
tonus, and occasional loss of posture), stage 4 (forelimb clonus
with rearing without falling), and stage 5 (four limb clonus with
rearing and falling or jumping). SE was defined by continuous
seizure activity for at least 30 min without full recovery between
seizures when rats experienced continuous Stage 4 or 5 seizures.
Then the rats were injected (i.p.) with 3% sodium pentobarbital (30
mg/kg) for anesthetization. After decapitation, each brain specimen
was equally divided into two blocks. One block was used for
morphological research and the other was used for molecular
biological studies.
Pharmacological treatment
The CB2R agonist JWH133 (Ki=3.4 nM for CB2R; Tocris
Bioscience) and the CB2R antagonist AM630 (Ki=31.2 nM for CB2R;
Tocris Bioscience) were dissolved in 20% dimethyl sulfoxide (in
normal saline). Subjects randomly received one
intra-cerebroventricular injection dose of JWH133 (1 or 3 µg
in 1 µl/side), AM630 (3 µg in 1 µl/side), a
mixed solution of AM630 (3 µg in 1 µl/side) and
JWH133 (3 µg in 1 µl/side), or vehicle (1
µl/side) 60 min before pilocarpine hydrochloride injection.
In the present study, the SE group was used as the control group
and injected with comparable volume of vehicle. All rats were
administered lithium chloride-pilocarpine to induce SE. At 1 day
after SE, six animals of each group were sacrificed.
Epileptic symptom observations after
intracerebroventricular injection of CB2R agonist/antagonist in the
lithium-pilocarpine SE model
After pilocarpine injection, the latency period to
SE, Racine stage, success rate of the model and 24-h death rate
after SE were examined to evaluate the potential of CB2R to remit
seizure onset. The epileptic symptoms of each rat were observed and
rated from 0 to 5 according to the Racine scale.
Immunofluorescence
For double-labelled immunofluorescence, 4-µm
brain sections were deparaffinized in xylene, hydrated with a
graded alcohol series, blocked with 5% non-immune fetal bovine
serum (CAS: 9048-46-8; Sigma-Aldrich; Merck KGaA) in PBST (0.5%
Tween-20 in PBS) at 37°C for 2 h and incubated with primary
antibodies overnight at 4°C with rabbit anti-CB2R antibody (1:100;
cat. no. ab3561; Abcam), rabbit anti-mTOR antibody (1:200; cat. no.
20657-1-AP; Proteintech, Inc.), rabbit anti-LC3B antibody (1:400;
cat. no. 2775; Cell Signaling Technology, Inc.), and mouse
anti-NeuN antibody (1:800; cat. no. MAB377; EMD Millipore).
Afterward, an Alexa Fluor 594-conjugated donkey anti-rabbit IgG
(1:200; cat. no. ab150076; Abcam) and an Alexa Fluor 488-conjugated
donkey anti-mouse IgG (1:200; cat. no. ab150105; Abcam) was used as
secondary antibodies conjugated with CB2R and NeuN, mTOR and NeuN,
or LC3B and NeuN. After washing with PBS three times, the sections
were covered with the VECTASHIELD Mounting Medium glasses. For the
evaluation of CB2R+/NeuN+ cells,
mTOR+/ NeuN+ cells and
LC3+/NeuN+ cells, 10 microscope fields of the
CA1, CA3, and DG regions were randomly selected under ×200
magnification by fluorescence light microscopy (Nikon Eclipse 80i;
Nikon Corporation).
TdT-mediated dUTP nick end labeling
assay
As previously described (36), TdT-mediated dUTP nick end labeling
(TUNEL) assay was performed for identifying neuronal death numbers.
The assay was performed according to the manufacturer's protocol.
TUNEL-positive cells were those with blue-green nuclear staining.
For the evaluation of TUNEL-positive cells, 10 microscope fields of
the CA1, CA3 and DG regions were randomly captured under ×200
magnification by fluorescence light microscopy (Nikon Eclipse 80i)
and the results were calculated as the means.
Protein extraction and western blotting
assay
Protein preparation was performed as previously
described (36). Hippocampal
samples were homogenized with a sonicator in double-distilled water
containing protease inhibitors and centrifuged at 14,000 × g for 15
min at 4°C, and the supernatant was collected. Protein
concentrations were measured with the DC Protein Assay (Bio-Rad
Laboratories, Inc.). Sample proteins (40 µg) were denatured
by heating at 90°C for 5 min and were size-fractionated by means of
Bis-Tris 10% SDS-PAGE (Beyotime Institute of Biotechnology).
Samples were then transferred to polyvinylidene fluoride (PVDF)
membranes (EMD Millipore) and were blocked with 5% fat-free milk
for 2 h at room temperature. The membranes were incubated overnight
at 4°C with the following primary antibodies: Rabbit anti-mTOR
polyclonal antibody (1:1,000; cat. no. 20657-1-AP; Proteintech,
Inc.), rabbit anti-p-mTOR monoclonal antibody (1:1,000; cat. no.
5536; Cell Signaling Technology, Inc.), rabbit anti-ULK1 polyclonal
antibody (1:1,000; 20986-1-AP; Proteintech, Inc.), rabbit
anti-p-ULK1 monoclonal antibody (1:1,000; cat. no. 14202; Cell
Signaling Technology, Inc.), rabbit anti-Beclin-1 polyclonal
antibody (1:1,000; cat. no. 11306-1-AP; Proteintech, Inc.), rabbit
anti-p62 polyclonal antibody (1:1,000; cat. no. 18420-1-AP;
Proteintech, Inc.), rabbit anti-LC3B monoclonal antibody (1:1,000;
cat. no. 2775; Cell Signaling Technology, Inc.), and rabbit
anti-GAPDH polyclonal antibody (1:5,000; cat. no. 10494-1-AP;
Proteintech, Inc.). The following day, the membranes were washed
and then incubated at room temperature for 2 h with the horseradish
peroxidase conjugated goat anti-rabbit IgG antibody (1:5,000; cat.
no. SA00001-2; Proteintech, Inc.). The membrane images were
collected and analyzed with the Infrared Odyssey Imaging System.
GAPDH was used as a control. Densitometric analysis of the bands
was semiquantitatively performed using GEL-PRO 4.0 software (Media
Cybernetics, Inc.).
Statistical analysis
All experiments were performed at least in
triplicate. Data are reported as the mean ± standard deviation and
were analyzed using SPSS 18.0 for Windows (IBM, Corps). Multiple
group comparisons were performed by one-way analysis of variance,
followed by the LSD post hoc multiple comparison test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Spatiotemporal expression changes in
CB2R, mTOR and LC3 in hippocampal neurons after SE
To determine the localization of neuronal CB2R, LC3B
and mTOR and their expression changes in the hippocampus under
normal conditions and after SE, double-label immunofluorescence and
western blotting essays were performed. CB2R-, LC3B- and
mTOR-positive neurons could be detected under normal conditions and
after SE in the CA1, CA3, and DG regions of the hippocampus. CB2R
and mTOR were mainly localized to the cytomembrane in neurons, with
some occurring in the cytoplasm (Fig.
1A-C). In addition, the mTOR protein expression in all three
regions was decreased at 2 h and 1 and 3 days after SE compared
with the control group, whereas the expression levels of CB2R and
LC3B were increased (Fig. 1D).
Interestingly, the clearest differences in mTOR, CB2R and LC3B
expression emerged 1 day after SE (Fig. 1D). Therefore, subsequent
intervention experiments were all conducted 1 day after SE.
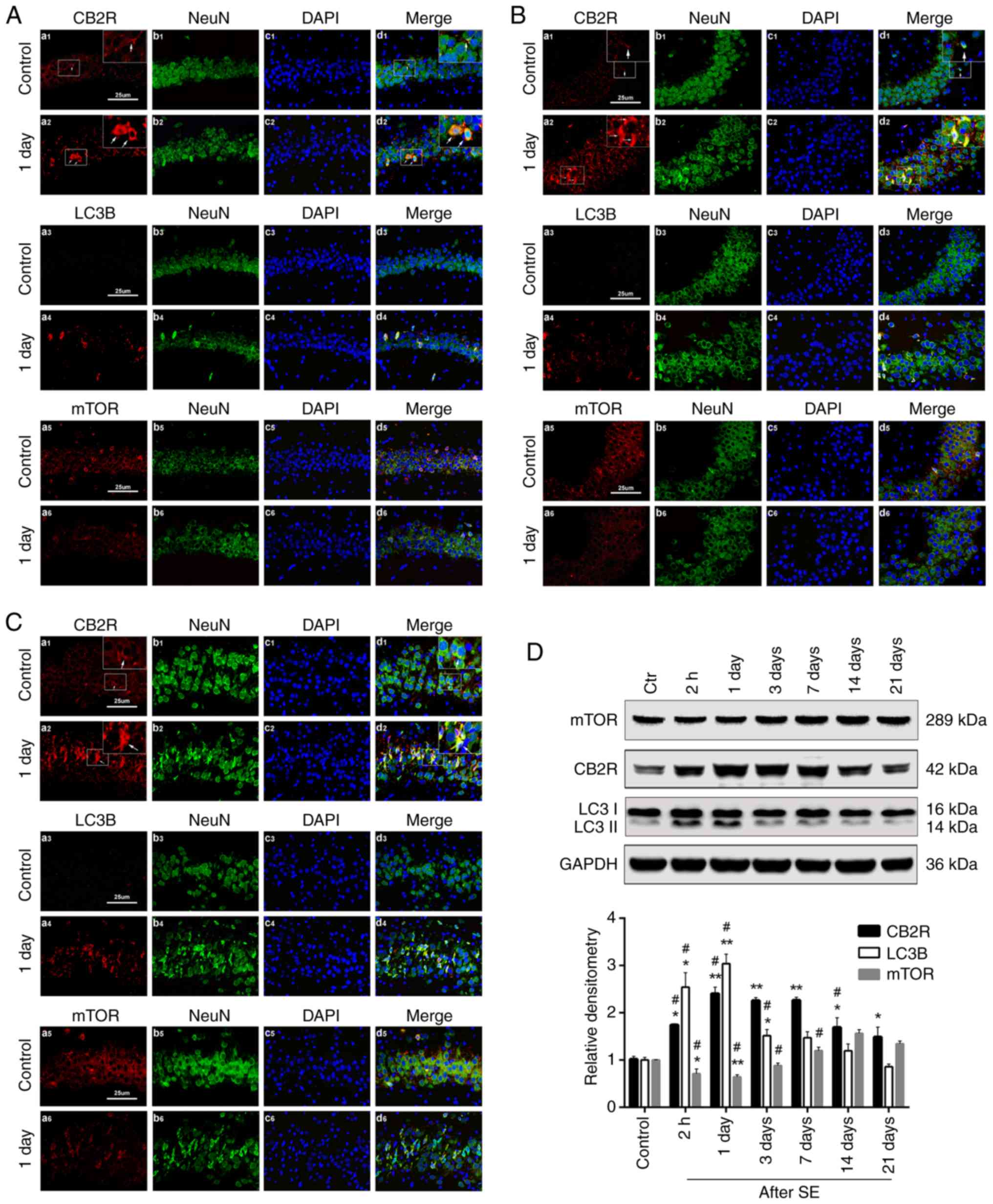 | Figure 1Spatiotemporal expression changes in
CB2R, mTOR and LC3 in hippocampal neurons after status epilepticus.
[(A) CA1, (B) CA3 and (C) DG regions)] Alexa Fluor 594-labeled
CB2R+, LC3B+ and mTOR+ cells in
the hippocampal CA1, CA3, and DG regions (red). Alexa Fluor
488-labeled neuronal nuclear antigen NeuN (green). DAPI-labeled
nucleus (blue). Images of NeuN marker with CB2R, LC3B and mTOR
colocalization (yellow) were digitally merged within the CA1, CA3,
and DG regions. (D) Western blot analysis of CB2R, LC3B, mTOR and
GAPDH protein from hippocampal specimens. Data are presented as the
mean ± standard deviation. *P<0.05 and
**P<0.01 vs. the control group, n=6 rats per group;
#P<0.05 vs. the previous group, n=6 rats per group).
mTOR, mammalian target of rapamycin; CB2R, cannabinoid receptor
type 2. |
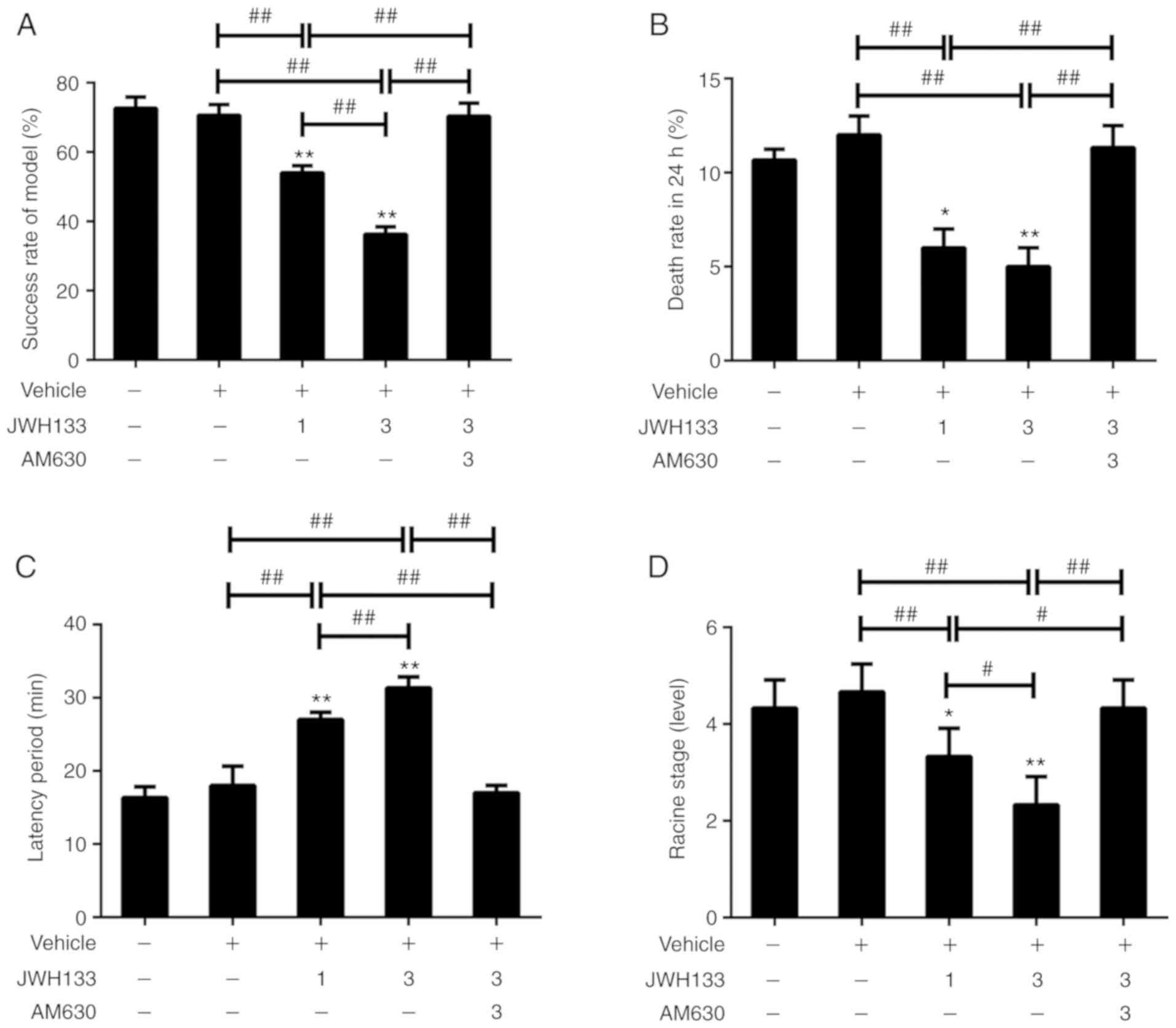
Effects of CB2R on epileptic symptoms
after SE
The behavior of seizure activity was recorded
following lithium-pilocarpine treatment. The rats began to exhibit
definite SE behavior within 15-33 min after pilocarpine
intraperitoneal injection. Significant differences could be
observed in the success rate of the SE model between the vehicle
group, JWH133 low-dose group and JWH133 high-dose group (Fig. 2A). Rats injected with low- or
high-dose JWH133 showed a lower mortality rate compared with the
vehicle group (Fig. 2B). The time
between pilocarpine injection and seizures reaching Racine stage 4
or 5 was defined as the latency period. The latency periods of the
JWH133 low-dose and high-dose groups were dose-dependently longer
than that of the vehicle group (Fig.
2C). Rats injected with low- or high-dose JWH133 showed a
significantly lower Racine stage compared with the vehicle group
(Fig. 2D). Rats injected with
low- or high-dose JWH133 showed alleviated epileptic symptoms
compared with rats injected with vehicle or JWH-133+AM630. In
addition, no significant difference could be observed in the
success rate of the SE model, 24-h death rate, latency period and
Racine stage between the SE and vehicle groups (Fig. 2). In each pilocarpine-treated
group, rats with a Racine stage higher than 3 were included in the
next two parts of the present study.
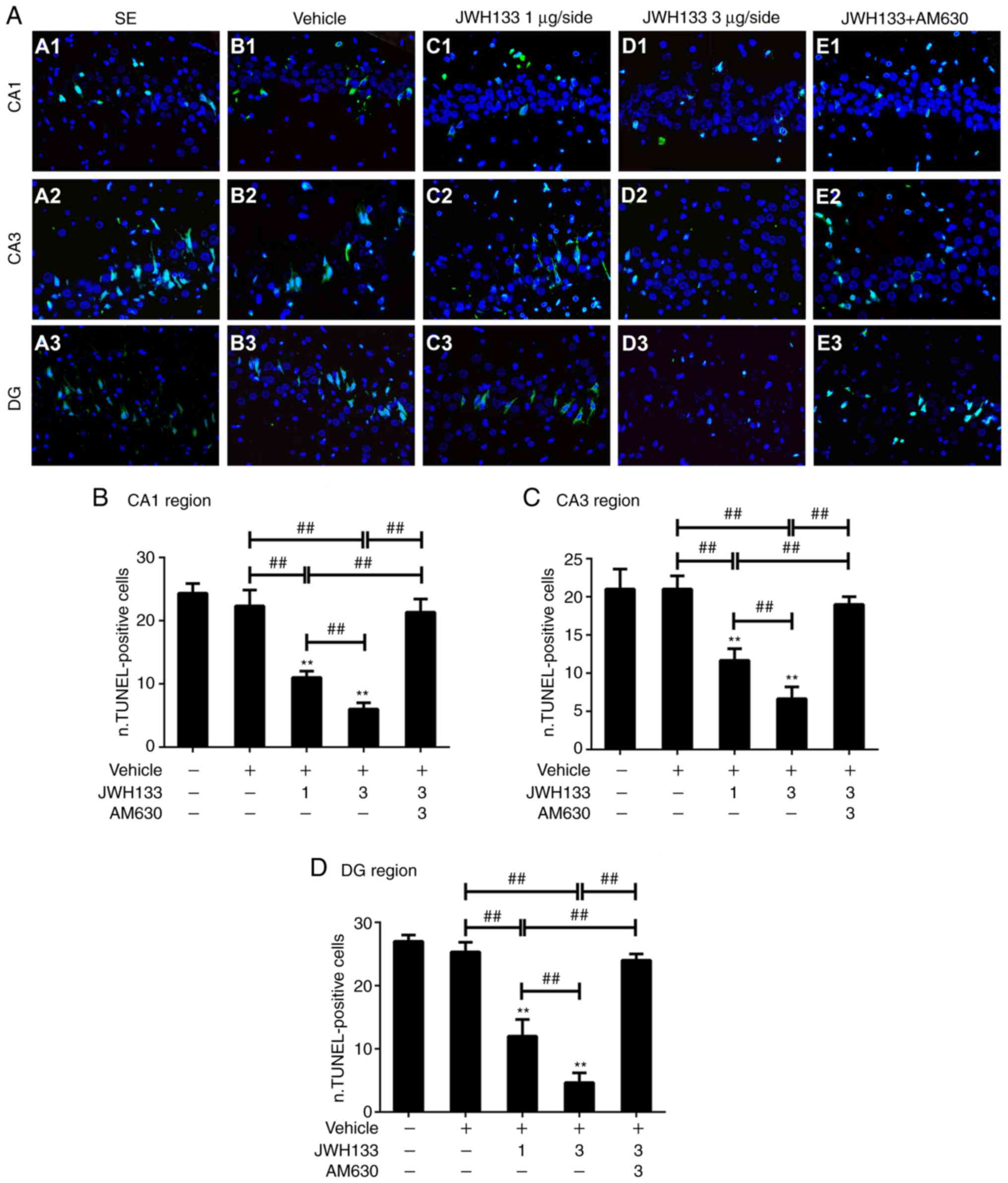
Hippocampal neuron apoptosis after
SE
A TUNEL assay was performed to observe neuronal
apoptosis in the hippo-campus in each treatment group (Fig. 3A). The present study counted
TUNEL-positive cell numbers in the CA1, CA3 and DG regions after
SE. There were TUNEL-positive cells in all groups 1 day after SE
and there was no significant difference in the average numbers of
TUNEL-positive cells between the SE group and the vehicle group
(P>0.05; Fig. 3B-D). However,
the number of apoptotic neurons was seriously decreased by
pretreatment with JWH133 at 1 day after SE. As shown in Fig. 3B-D, the numbers of apoptotic
neurons were significantly and dose-dependently decreased in all
three regions of the post-SE hippocampus in the JWH133-treated
groups compared with the vehicle group 1 day after SE, whereas the
numbers of apoptotic cells were increased again in the JWH133+AM630
group at 1 day after SE.
Effects of CB2R on the expression of
autophagy-related proteins in the hippocampus after SE
According to the authors' preliminary experiment
(Fig. S1), JWH133 1 µg in
1 µl/side and JWH133 3 µg in 1 µl/side were
selected as the JWH133 low-dose and high-dose groups, respectively.
The expression of autophagy-related proteins in the hippocampus
after SE could be tested in all specimens of each group by western
blotting. The levels of p-mTOR/mTOR, p-ULK1/ ULK1, Beclin-1,
LC3II/LC3I and p62 expression were compared among all groups 1 day
after SE (Fig. 4). Compared with
the vehicle group, western blotting showed significant and
dose-dependent decreases in p-ULK1/ULK1 and p62 expression in the
JWH133 low-dose and high-dose groups at 1 day after SE (P<0.05;
Fig. 4) and dose-dependent
increases in p-mTOR/mTOR, Beclin-1, and LC3II/LC3I expression in
the JWH133 low-dose and high-dose groups 1 day after SE (P<0.05;
Fig. 4). In the JWH133+AM630
group, the results clearly indicated no significant difference in
the expression of p-mTOR/mTOR, p-ULK1/ULK1, Beclin-1, LC3II/LC3I
and p62 compared with the vehicle group (P>0.05; Fig. 4). Compared with the JWH133
high-dose group, western blotting showed significant increases in
p-ULK1/ULK1 and p62 expression in the JWH133+AM630 group 1 day
after SE (P<0.05; Fig. 4) and
decreases in p-mTOR/mTOR, Beclin-1, and LC3II/LC3I expression in
the JWH133+AM630 group 1 day after SE (P<0.05; Fig. 4).
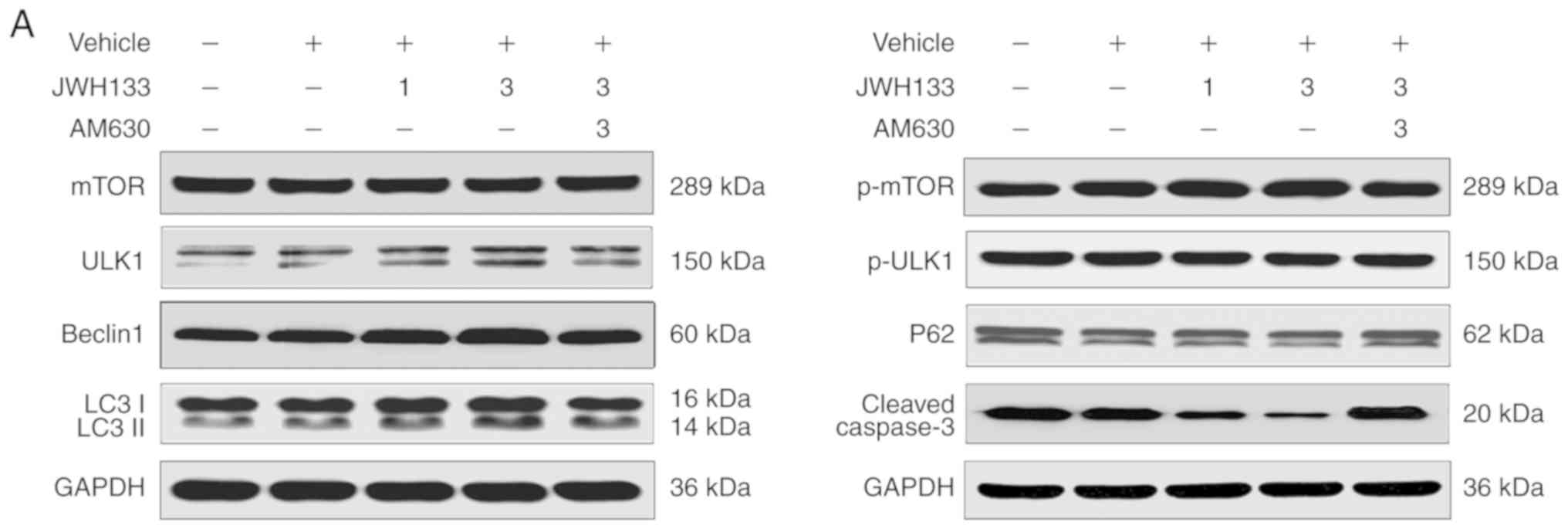 | Figure 4Western blotting was used to
simultaneously determine the expression of p-mTOR/mTOR,
p-ULK1/ULK1, Beclin-1, p62, LC3II/LC3I and caspase-3 at 1 day
post-SE. (A) Immunoblotting results of mTOR, p-mTOR, ULK1, p-ULK1,
Beclin-1, p62, LC3II/LC3I and caspase-3 expression at 1 day
post-SE. (B) Western blot analysis of p-mTOR/mTOR, p-ULK1/ULK1,
Beclin-1, p62, LC3II/LC3I and caspase-3 expression at 1 day
post-SE. Data are presented as the mean ± standard deviation.
*P<0.05 vs. the SE group, (n=6 rats per group),
#P<0.05 between comparison groups, (n=6 rats per
group). SE, status epilepticus; p-mTOR, phosphorylated-mammalian
target of rapamycin. |
Effects of CB2R on the expression of
apoptosis-related proteins in the hippocampus after SE
Paralleling the changes in neuronal cell fate
determined using a TUNEL assay, the apoptotic process was also
altered in the post-SE rat hippo-campus. Briefly, the expression of
the apoptosis-related protein cleaved caspase-3 could be detected
in all specimens of each group by western blotting. The levels of
cleaved caspase-3 were examined in all groups at 1 day after SE.
There was no significant difference in the expression of cleaved
caspase-3 between the SE and vehicle groups (P>0.05; Fig. 4). However, the upregulation of
cleaved caspase-3 protein was significantly and dose-dependently
inhibited in the JWH133-treated group; the reduction in its
expression was more marked in the JWH133 high-dose group at 1 day
after SE (P<0.05; Fig. 4). In
the JWH133+AM630 group, the results clearly indicated no
significant difference in the expression of cleaved caspase-3
compared with the vehicle group (P>0.05; Fig. 4). Compared with the JWH133
high-dose group, western blotting showed significant increases in
cleaved caspase-3 expression in the JWH133+AM630 group at 1 day
after SE (P<0.05; Fig. 4).
Discussion
Epilepsy as the most common neurological disorder is
characterized by the clinical manifestation of 'abnormal excessive
or synchronous neuronal activity in the brain' (1). Although the pathogenesis of epilepsy
remains unclear, it is generally acknowledged that persistent
seizures can directly kill neurons. In addition, a number of
studies in previous years has hinted that autophagy and apoptosis
contribute to the neuronal damage process through the modulation of
Beclin family members and their cascades (41,42). Consistently, the neuronal damage
induced by kainate can be worsened by autophagy blockers while
being prevented by autophagy inducers (43,44). In detail, mTOR is activated both
acutely and chronically after Kainic acid-induced SE, which is
prominent in the hippocampus and other cortical areas (45). An early observation from 7 years
ago showed an increase in the autophagy markers LC3-II, p-mTOR/mTOR
ratio and p-protein kinase B (Akt)/Akt ratio after kainate
administration to mice (46). A
total of 2 years later, a sudden increase in autophagy markers
(LC3II/LC3I and Beclin-1) was also found after pilocarpine
administration to rats (47). In
this study, it was found that the tendency for a decrease in mTOR
expression and an increase in CB2R and LC3 expression was clearest
in all three regions at 1 day (the early stage of neuronal repair
and damage) after SE. Interestingly, it was hinted that CB2R may
participate in the autophagy process and the expression level of
CB2R may be related to the extent of the damage in hippocampal
neurons.
Several studies revealed that CB2R exerts
neuroprotective effects on alcohol addiction (48), depression (49), cerebral ischemia (50) and Alzheimer's disease (51). The endocan-nabinoid system also
participates in the epileptiform activity in the hippocampus
(52-54). Cannabinoid and nitric oxide
signaling can interplay in the modulation of hippocampal
hyperexcitability in the temporal lobe epilepsy model of rats
(55). Carletti et al
(56) also found that neuronal
nitric oxide synthase is involved in CB/TRPV1 signaling for
controlling hippocampal hyperexcitability. Furthermore, a mixed
CB1/2 agonist WIN 55,212-2 exerts anti-convulsant effects in acute
hypoxia-induced seizures or tonic-clonic seizures evoked by
pentylenetetrazole, while a CB2R antagonist AM630 increases
pentylenetetrazol-induced seizure severity in a rat model (57). However, Rizzo et al
(58) argued that CB2R antagonist
AM630 seems to cooperate with WIN 55,212-2 and lowered neuronal
hyperexcitability by targeting nitrergic-dependent cGMP pathway.
Additionally, Tchekalarova et al (59) discovered that CB2R stimulation by
β-caryophyllene could restrain the spread of seizures. Single
administration of JWH133 before 30 min of pilocarpine treatment
postpones the initiation of SE and significantly reduces the
frequency of various epileptic behaviors according to the Racine
scale (40). The decrease in
epilepsy-related scores is in line with the dose-dependent increase
in the levels of autophagy-related proteins in the hippocampus of
epileptic rats after single administration of JWH133. Because
elevation of autophagy-related proteins is related to neural
plasticity (60), and epileptic
behavior also is related to neural plasticity, autophagy by this
mechanism may affect epilepsy-related behaviors. Thus, CB2R may
take part in the early stage of pilocarpine-induced SE.
The present results have shown an alteration in the
expression of p-mTOR/mTOR, p-ULK1/ULK1, Beclin-1, LC3II/LC3I and
p62 proteins for autophagy and caspase-3 proteins for apoptosis in
CA1, CA3, and DG regions of the rat hippocampus after SE. In short,
the expression levels of the autophagy-related proteins
p-mTOR/mTOR, Beclin-1 and LC3II/LC3I were dose-dependently
increased in the rat hippocampus 1 day after SE especially in the
JWH133 high-dose group. At the same time, the expression levels of
the autophagy proteins p-ULK1/ULK1 and p62 were down-regulated. The
results were reversed by pretreament with the CB2R antagonist,
AM630. A dose-dependent decrease in the number of apoptotic neurons
and the expression of the apoptosis protein cleaved caspase-3 was
evident at 1 day after SE in the JWH133 treated group, and was
further decreased in the group receiving the highest dose of
JWH133. In the present study, intracerebroventricular injections
(JWH133, 1 or 3 µg in 1 µl/side; AM630, 3 µg
in 1 µl/side) was used and an AM630 alone group was not
used. There are two reasons why the groups that were used were
chosen. One reason is that some conclusions are obtained according
to the references. Bilateral microinjections of JWH133 (1 or 3
µg in 1 µl/ side) into the ventral tegmental area
significantly reduced cocaine self-administration in WT mice and
this effect was blocked by coadministration of AM630 and JWH133,
whereas microinjections of AM630 alone had no effect on cocaine
self-administration (61).
Administration of AM630 alone exerts no effects on hippocampal
hyperexcitability in the maximal dentate activation model of
hippocampal epilepsy (58). The
other reason is that this paper mainly focuses on the effect of
CB2R agonist JWH133 on neuronal autophagy in the early stage of
neuronal damage and repair after SE, to find a new target for
epilepsy treatment. CB2R antagonist AM630 is only used to
antagonize the effect of the agonist JWH133. In summary, the
results from the present study clearly demonstrated that CB2R
significantly reduced neuronal damage in the rat hippocampus after
SE through the induction of autophagy and inhibition of apoptosis.
Furthermore, the results from this study also suggest that the CB2R
could be promising for use in the treatment of epilepsy.
Autophagy can exert both harmful and beneficial
effects in different situations (62-65). The present results suggests that
the increase in autophagy signaling induced by SE or seizure
attacks has a protective role in hippocampal neurons, while
apoptosis signaling seems to lead to neuronal death in the
hippocampus after SE. Furthermore, the current study hints that
apoptosis can be considered an outcome following autophagy failure,
because these two mechanisms cooperate to influence hippocampal
neuronal fate after SE. In other words, autophagy acts as a
self-protection mechanism to keep neuronal dynamic equilibrium at
early stages (about 24 h) post-SE, however, apoptosis is activated
to ameliorate neuron death when hippocampal neuronal autophagy is
insufficient. The autophagy and apoptosis mechanism participating
in hippocampal neuronal damage after SE is still unclear. The
present results only reveal that CB2R may be involved in neuronal
damage and CB2R stimulation may protect hippocampal neurons by
modulating autophagy and apoptosis.
Of course, the paper has a limit in terms of scope.
The mTOR plays multiple roles in neuronal and non-neuronal cells
development, neuronal plasticity, as well as in the expression of
different neuronal molecules involved in cell excitability. These
effects may not be directly dependent on autophagy (66). Thus, attribution of mTOR-related
epilepsy to autophagy impairment could remain a mere speculation,
if not supported by the analyses on the other molecular target
implicated in autophagy such as LC3 and ULK1 as was inserted in the
present study (67). Regarding
this point, the 'ATG7 KO' strain of mice which possess a normal
mTOR signaling but disrupted autophagy activity is being considered
for use in the present study (68) and the relevant experiments have
already been started.
In summary, the present study indicated that CB2R
orchestrates neuronal autophagy through regulation of the mTOR
signaling pathway in the hippocampus of developing rats with SE.
These novel findings might provide an important basis for further
investigation of the therapeutic role of CB2R in the treatment or
prevention of epilepsy-related neuronal damage.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
Department of Science and Technology of Liaoning Province, China
(grant nos. 2014225007 and 2019-BS-280), the Department of
Education and Technology of Liaoning Province, China (grant no.
L2014309) and the National Key Research and Development Program of
China (grant no. 2016YFC1306203).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QW, MZ and HW were involved in the conception and
design of the study and the drafting of the manuscript. QW and MZ
were involved in the analysis and interpretation of the data. JZ
and XL assisted in setting up the protocols as well as in their
administration, and XL offered financial support for the project
leading to this publication. QW and MZ performed all the
experiments and acquired all data. All authors read and approved
the manuscript.
Ethics approval and consent to
participate
The China Medical University Institutional Animal
Care and Use Committee (no. 2016PS260K) approved all
procedures.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fisher RS, Acevedo C, Arzimanoglou A,
Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA,
Glynn M, et al: ILAE official report: A practical clinical
definition of epilepsy. Epilepsia. 55:475–482. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Engel J Jr: Mesial temporal lobe epilepsy:
What have we learned? Neuroscientist. 7:340–352. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shibley H and Smith BN:
Pilocarpine-induced status epilepticus results in mossy fiber
sprouting and spontaneous seizures in C57BL/6 and CD-1 mice.
Epilepsy Res. 49:109–120. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morimoto K, Fahnestock M and Racine RJ:
Kindling and status epilepticus models of epilepsy: Rewiring the
brain. Prog Neurobiol. 73:1–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Friedman WJ: Proneurotrophins, seizures,
and neuronal apoptosis. Neuroscientist. 16:244–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baram TZ, Jensen FE and Brooks-Kayal A:
Does acquired epileptogenesis in the immature brain require
neuronal death. Epilepsy Curr. 11:21–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Varvel NH, Neher JJ, Bosch A, Wang W,
Ransohoff RM, Miller RJ and Dingledine R: Infiltrating monocytes
promote brain inflammation and exacerbate neuronal damage after
status epilepticus. Proc Natl Acad Sci USA. 113:E5665–E5674. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hung SY, Huang WP, Liou HC and Fu WM: LC3
overexpression reduces Aβ neurotoxicity through increasing α7nAchR
expression and autophagic activity in neurons and mice.
Neuropharmacology. 93:243–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vieira M, Fernandes J, Carreto L,
Anuncibay-Soto B, Santos M, Han J, Fernández-López A, Duarte CB,
Carvalho AL and Santos AE: Ischemic insults induce necroptotic cell
death in hippocampal neurons through the up-regulation of
endogenous RIP3. Neurobiol Dis. 68:26–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu M and Zhang HL: Death and survival of
neuronal and astrocytic cells in ischemic brain injury: A role of
autophagy. Acta Pharmacol Sin. 32:1089–1099. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hochfeld M, Lamecker H, Thomale UW, Schulz
M, Zachow S and Haberl H: Frame-based cranial reconstruction. J
Neurosurg Pediatr. 13:319–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ravikumar B, Vacher C, Berger Z, Davies
JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ and
Rubinsztein DC: Inhibition of mTOR induces autophagy and reduces
toxicity of polyglutamine expansions in fly and mouse models of
Huntington disease. Nat Genet. 36:585–595. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoeffer CA and Klann E: mTOR signaling: At
the crossroads of plasticity, memory and disease. Trends Neurosci.
33:67–75. 2010. View Article : Google Scholar
|
|
14
|
Costa-Mattioli M, Sossin WS, Klann E and
Sonenberg N: Translational control of long-lasting synaptic
plasticity and memory. Neuron. 61:10–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harris H and Rubinsztein DC: Control of
autophagy as a therapy for neurodegenerative disease. Nat Rev
Neurol. 8:108–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hara T, Nakamura K, Matsui M, Yamamoto A,
Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I,
Okano H and Mizushima N: Suppression of basal autophagy in neural
cells causes neurodegenerative disease in mice. Nature.
441:885–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Menzies FM, Fleming A, Caricasole A, Bento
CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez
Sanchez M, Karabiyik C, et al: Autophagy and neurodegeneration:
Pathogenic mechanisms and therapeutic opportunities. Neuron.
93:1015–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pasquali L, Longone P, Isidoro C, Ruggieri
S, Paparelli A and Fornai F: Autophagy, lithium, and amyotrophic
lateral sclerosis. Muscle Nerve. 40:173–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fornai F, Longone P, Ferrucci M, Lenzi P,
Isidoro C, Ruggieri S and Paparelli A: Autophagy and amyotrophic
lateral sclerosis: The multiple roles of lithium. Autophagy.
4:527–530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hwang JY, Gertner M, Pontarelli F,
Court-Vazquez B, Bennett MV, Ofengeim D and Zukin RS: Global
ischemia induces lysosomal-mediated degradation of mTOR and
activation of autophagy in hippocampal neurons destined to die.
Cell Death Differ. 24:317–329. 2017. View Article : Google Scholar :
|
|
26
|
Hosseinzadeh M, Nikseresht S, Khodagholi
F, Naderi N and Maghsoudi N: Cannabidiol post-treatment alleviates
rat epileptic-related behaviors and activates hippocampal cell
autophagy pathway along with antioxidant defense in chronic phase
of pilocarpine-induced seizure. J Mol Neurosci. 58:432–440. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soltesz I, Alger BE, Kano M, Lee SH,
Lovinger DM, Ohno-Shosaku T and Watanabe M: Weeding out bad waves:
Towards selective cannabinoid circuit control in epilepsy. Nat Rev
Neurosci. 16:264–277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Regehr WG, Carey MR and Best AR:
Activity-dependent regulation of synapses by retrograde messengers.
Neuron. 63:154–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jansen EM, Haycock DA, Ward SJ and Seybold
VS: Distribution of cannabinoid receptors in rat brain determined
with aminoalkylindoles. Brain Res. 575:93–102. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Muñoz-Luque J, Ros J, Fernández-Varo G,
Tugues S, Morales-Ruiz M, Alvarez CE, Friedman SL, Arroyo V and
Jiménez W: Regression of fibrosis after chronic stimulation of
cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther.
324:475–483. 2008. View Article : Google Scholar
|
|
31
|
Galiègue S, Mary S, Marchand J, Dussossoy
D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G and
Casellas P: Expression of central and peripheral cannabinoid
receptors in human immune tissues and leukocyte subpopulations. Eur
J Biochem. 232:54–61. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y and Kim J: Neuronal expression of CB2
cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience.
311:253–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lanciego JL, Barroso-Chinea P, Rico AJ,
Conte-Perales L, Callén L, Roda E, Gómez-Bautista V, López IP,
Lluis C, Labandeira-García JL and Franco R: Expression of the mRNA
coding the cannabinoid receptor 2 in the pallidal complex of Macaca
fascicularis. J Psychopharmacol. 25:97–104. 2011. View Article : Google Scholar
|
|
34
|
Viscomi MT, Oddi S, Latini L, Pasquariello
N, Florenzano F, Bernardi G, Molinari M and Maccarrone M: Selective
CB2 receptor agonism protects central neurons from remote
axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci.
29:4564–4570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim J and Li Y: Chronic activation of CB2
cannabinoid receptors in the hippocampus increases excitatory
synaptic transmission. J Physiol. 593:871–886. 2015. View Article : Google Scholar :
|
|
36
|
Stempel AV, Stumpf A, Zhang HY, Özdoğan T,
Pannasch U, Theis AK, Otte DM, Wojtalla A, Rácz I, Ponomarenko A,
et al: Cannabinoid type 2 receptors mediate a cell type-specific
plasticity in the hippocampus. Neuron. 90:795–809. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sugaya Y, Yamazaki M, Uchigashima M,
Kobayashi K, Watanabe M, Sakimura K and Kano M: Crucial roles of
the endocannabinoid 2-arachidonoylglycerol in the suppression of
epileptic seizures. Cell Rep. 16:1405–1415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu Q and Wang H: The spatiotemporal
expression changes of CB2R in the hippocampus of rats following
pilocarpine-induced status epilepticus. Epilepsy Res. 148:8–16.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Curia G, Longo D, Biagini G, Jones RS and
Avoli M: The pilocarpine model of temporal lobe epilepsy. J
Neurosci Methods. 172:143–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Racine RJ: Modification of seizure
activity by electrical stimulation. II. Motor seizure.
Electroencephalogr Clin Neurophysiol. 32:281–294. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li X, Lou X, Xu S, Wang Q, Shen M and Miao
J: Knockdown of miR-372 inhibits nerve cell apoptosis induced by
spinal cord ischemia/reperfusion injury via enhancing autophagy by
up-regulating Beclin-1. J Mol Neurosci. 66:437–444. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Salminen A, Kaarniranta K, Kauppinen A,
Ojala J, Haapasalo A, Soininen H and Hiltunen M: Impaired autophagy
and APP processing in Alzheimer's disease: The potential role of
Beclin 1 interactome. Prog Neurobiol. 106-107:33–54. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fornai F, Longone P, Cafaro L,
Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A,
Bellio N, Lenzi P, et al: Lithium delays progression of amyotrophic
lateral sclerosis. Proc Natl Acad Sci USA. 105:2052–2057. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Calderó J, Brunet N, Tarabal O, Piedrafita
L, Hereu M, Ayala V and Esquerda JE: Lithium prevents excitotoxic
cell death of motoneurons in organotypic slice cultures of spinal
cord. Neuroscience. 165:1353–1369. 2010. View Article : Google Scholar
|
|
45
|
Macias M, Blazejczyk M, Kazmierska P,
Caban B, Skalecka A, Tarkowski B, Rodo A, Konopacki J and Jaworski
J: Spatiotemporal characterization of mTOR kinase activity
following kainic acid induced status epilepticus and analysis of
rat brain response to chronic rapamycin treatment. PLoS One.
8:e644552013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth
KA and Zhang J: Kainic acid induces early and transient autophagic
stress in mouse hippocampus. Neurosci Lett. 414:57–60. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cao L, Xu J, Lin Y, Zhao X, Liu X and Chi
Z: Autophagy is upregulated in rats with status epilepticus and
partly inhibited by Vitamin E. Biochem Biophys Res Commun.
379:949–953. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Al Mansouri S, Ojha S, Al Maamari E, Al
Ameri M, Nurulain SM and Bahi A: The cannabinoid receptor 2
agonist, β-caryophyllene, reduced voluntary alcohol intake and
attenuated ethanol-induced place preference and sensitivity in
mice. Pharmacol Biochem Behav. 124:260–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Katsuyama S, Mizoguchi H, Kuwahata H,
Komatsu T, Nagaoka K, Nakamura H, Bagetta G, Sakurada T and
Sakurada S: Involvement of peripheral cannabinoid and opioid
receptors in β-caryophyllene-induced antinociception. Eur J Pain.
17:664–675. 2013. View Article : Google Scholar
|
|
50
|
Choi IY, Ju C, Anthony Jalin AM, Lee DI,
Prather PL and Kim WK: Activation of cannabinoid CB2
receptor-mediated AMPK/CREB pathway reduces cerebral ischemic
injury. Am J Pathol. 182:928–939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cheng Y, Dong Z and Liu S: β-Caryophyllene
ameliorates the Alzheimer-like phenotype in APP/PS1 Mice through
CB2 receptor activation and the PPARγ pathway. Pharmacology.
94:1–12. 2014. View Article : Google Scholar
|
|
52
|
Wallace MJ, Martin BR and DeLorenzo RJ:
Evidence for a physiological role of endocannabinoids in the
modulation of seizure threshold and severity. Eur J Pharmacol.
452:295–301. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Monory K, Massa F, Egertová M, Eder M,
Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, et
al: The endocannabinoid system controls key epileptogenic circuits
in the hippocampus. Neuron. 51:455–466. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ludányi A, Eross L, Czirják S, Vajda J,
Halász P, Watanabe M, Palkovits M, Maglóczky Z, Freund TF and
Katona I: Downregulation of the CB1 cannabinoid receptor and
related molecular elements of the endocannabinoid system in
epileptic human hippocampus. J Neurosci. 28:2976–2990. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carletti F, Gambino G, Rizzo V, Ferraro G
and Sardo P: Cannabinoid and nitric oxide signaling interplay in
the modulation of hippocampal hyperexcitability: Study on
electro-physiological and behavioral models of temporal lobe
epilepsy in the rat. Neuroscience. 303:149–159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Carletti F, Gambino G, Rizzo V, Ferraro G
and Sardo P: Neuronal nitric oxide synthase is involved in CB/TRPV1
signalling: Focus on control of hippocampal hyperexcitability.
Epilepsy Res. 138:18–25. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huizenga MN, Wicker E, Beck VC and
Forcelli PA: Anticonvulsant effect of cannabinoid receptor agonists
in models of seizures in developing rats. Epilepsia. 58:1593–1602.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Rizzo V, Carletti F, Gambino G, Schiera G,
Cannizzaro C, Ferraro G and Sardo P: Role of CB2 receptors and cGMP
pathway on the cannabinoid-dependent antiepileptic effects in an in
vivo model of partial epilepsy. Epilepsy Res. 108:1711–1718. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tchekalarova J, da Conceição Machado K,
Gomes Júnior AL, de Carvalho Melo Cavalcante AA, Momchilova A and
Tzoneva R: Pharmacological characterization of the cannabinoid
receptor 2 agonist, β-caryophyllene on seizure models in mice.
Seizure. 57:22–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Otabe H, Nibuya M, Shimazaki K, Toda H,
Suzuki G, Nomura S and Shimizu K: Electroconvulsive seizures
enhance autophagy signaling in rat hippocampus. Prog
Neuropsychopharmacol Biol Psychiatry. 50:37–43. 2014. View Article : Google Scholar
|
|
61
|
Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang
HJ, Gardner EL, Wu J and Xi ZX: Cannabinoid CB2 receptors modulate
midbrain dopamine neuronal activity and dopamine-related behavior
in mice. Proc Natl Acad Sci USA. 111:E5007–E5015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What's the connection? Trends Cell Biol. 21:387–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Leber B and Andrews DW: Closing in on the
link between apoptosis and autophagy. F1000. Biol Rep.
2:882010.
|
|
64
|
Linkermann A and Green DR: Necroptosis. N
Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lasarge CL and Danzer SC: Mechanisms
regulating neuronal excitability and seizure development following
mTOR pathway hyperactivation. Front Mol Neurosci. 7:182014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Giorgi FS, Biagioni F, Lenzi P, Frati A
and Fornai F: The role of autophagy in epileptogenesis and in
epilepsy-induced neuronal alterations. J Neural Transm (Vienna).
122:849–862. 2015. View Article : Google Scholar
|
|
68
|
McMahon J, Huang X, Yang J, Komatsu M, Yue
Z, Qian J, Zhu X and Huang Y: Impaired autophagy in neurons after
disinhibition of mammalian target of rapamycin and its contribution
to epileptogenesis. J Neurosci. 32:15704–15714. 2012. View Article : Google Scholar : PubMed/NCBI
|