Introduction
Unexplained cardiac death is mainly caused by
malignant ventricular tachycardia, ventricular fibrillation, or
both (1). On electrocardiography,
an early repolarization pattern (ERP) appears to be common among
cardiac death cases (2). ERP is
characterized by J point (QRS-ST junction) elevation of >0.1 mV
in at least two leads on the electrocardiogram, accompanied by
J-wave slurring and notching changes (3). Patients with an ERP and unexplained
cardiac arrest, but without structural heart disease, have early
repolarization syndrome (ERS) (4).
ERPs are common among the relatives of patients with
ERS. Therefore, heredity of a genetic mutation may play a key role
in the occurrence of ERPs (5).
The main mechanism of ERP is disordered ion currents during the
depolarization stage and during the early stages of repolarization
of cardiomyocyte action potentials (6). These abnormal ion currents mainly
include transient outward potassium currents (7), fast sodium currents (8), inward-rectifier ATP-dependent
K+ channel currents (9), and L-type calcium currents (10). Study results suggest that the
'loss of function' caused by mutations of CACNA1C (11), CACNB2b (12), CACNA2D1 (12), SCN5A (8) and SCN10A (13), and 'gain of function' caused by
mutations of KCNJ8 (14) and
ABCC9 (15), are associated with
ERS.
The glycerol-3-phosphate dehydrogenase 1-like
(GPD1-L) gene is located on chromosome 3. GPD1-L is mainly
expressed near, but not on the cell membrane (16), and it may affect the action
potentials of cardiomyocytes via regulation of sodium channel
function (16). The objective of
the present study was to examine the effects of changes in GPD1-L
using co-expression of GPD1-L with Nav1.5 in 293 cells in order to
determine whether there is an association between ERS and of a loss
of function in INa secondary to mutations in GPD1-L.
Materials and methods
Ethics approval
The present study was performed in accordance with
the principles for ethical human genetic disease research
formulated by the World Medical Association and the Helsinki
Declaration. The investigation was approved by the Ethics Committee
of the First Affiliated hospital of Sun Yat-Sen University. All
family members agreed to participate in the study and signed the
informed consent forms.
Clinical study
Patients with ERS admitted to the First Affiliated
Hospital of Sun Yat-Sen University in 2015 were selected for the
study. All clinical data, including those obtained from records of
routine and specialized cardiovascular disease examinations
(electrocardiogram, Holter electrocardiogram, X-ray,
echocardiography, coronary angiography, and electro-physiological
examination) were collected. ERP was defined as J point (QRS-ST
junction) elevation of >0.1 mV on the electrocardiogram, in at
least two leads, accompanied by J-wave slurring or notching
changes. Detailed family histories of the probands were
investigated, and the family pedigrees were drawn. Full and
reliable clinical phenotype data were collected as well as
possible. Peripheral venous blood was extracted from each proband
for genetic testing.
Genetic sequencing and analysis
QIAamp DNAKits (Qiagen GmbH) were used to extract
blood DNA according to the standard operating manual method
recommended by the manufacturer. The samples used to build the
libraries met the requirements for new-generation high-throughput
gene sequencing. Whole-genome sequencing was performed using Next
Generation Sequencing and the HiSeq X Ten (Illumina, Inc.).
The reference sequences were compared with the
sequencing results. Bioinformatics methods were used to detect and
annotate single-nucleotide variants (SNVs) and InDel results. The
gene frequency of each SNV was determined using comparison with
standard databases (e.g., the 1000 Genomes Project and dbSNP).
Candidate genes for malignant arrhythmias have been described
previously (17). SNVs with
allele frequencies <1% were selected during screening to
facilitate further investigation and identification of sudden
death-related genes. Protein function prediction software [PROVEAN
Protein, version 1.1 (http://provean.jcvi.org/seq_submit.php) and SIFT,
version 6.2.1 (http://sift.bii.a-star.edu.sg/)] and conservation of
gene sequences analysis software [PhyloP (https://ccg.epfl.ch//mga/hg19/phylop/phylop.html)
and Grantham (https://gist.github.com/danielecook/501f03650bca6a3db31ff3af2d413d2a)]
were used to predict the protein functions encoded by the mutant
genes. The candidate genetic mutation present in each proband was
verified using Sanger sequencing.
Mutagenesis, cell culture and
transfection
Nav1.5 cDNA (SCN5A, NM_198056) was subcloned into
the mammalian expression vector, pENTER. For western blotting and
electrophysiological study, GPD1-L cDNA (NM_015141.3) was amplified
using PCR and subcloned into pIRES2-EGFP. When the plasmid was
transfected into 293 cells, the 293 cells expressed enhanced green
fluorescent protein (eGFP) and GPD1-L protein. For the confocal
microscopy examination, GPD1-L cDNA (NM_015141.3) was amplified
using PCR and subcloned into pEGFP-N1. When the plasmid was
transfected into 293 cells, the 293 cells expressed eGFT-GPD1-L
fusion protein to trace the trafficking of GPD1-L. The p. P112L
mutation of GPD1-L was introduced by site-directed mutagenesis
using the QuikChange II kit (Stratagene; Agilent Technologies,
Inc.). 293 cells were cultured in DMEM containing 10% fetal bovine
serum, and under the conditions of 5% CO2 and 37̊C. 293
cells were transfected with SCN5A plasmid and WT/MT GPD1-L plasmid
(1:1 molar ratio) using Lipofectamine 3000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). The
electrophysiological recordings, western blotting, and confocal
microscopy observations were performed according to the
manufacturers' instructions.
Western blotting
After transfection for 24 h, the medium was
discarded and the cells were washed twice using pre-cooled PBS; 120
µl ice-cold RIPA buffer (Beyotime Institute of
Biotechnology) was added to each well of 6-well plates, and the
plates were shaken on ice for 15 min. Intermittent ultrasound (200
W, 25% power) was used to lyse the cells (working for 3 sec and
stopping for 5 sec), until the lysates were clear and bright. After
centrifugation at 12,580 × g for 15 min at 4°C, the supernatant was
collected as the protein sample.
The protein samples were quantified using the BCA
Protein Quantitation kit (Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Each protein sample was diluted
using 5X loading buffer and was denatured in 95̊C water for 10 min.
Protein samples (10 µg/lane) were separated by 10% SDS-PAGE
gel and transferred to PDVF membranes. The PVDF membranes were
blocked using 5% BSA solution (Thermo Fisher Scientific, Inc.) and
incubated with anti-GPD1L antibody (1:500, cat. no. NPB1-32279,
Novus Biologicals, Ltd.) and anti-tubulin antibody (1:5,000, cat.
no. 66031-1-Ig, Proteintech Group, Inc.) overnight on a shaker at
4̊C. The PVDF membranes were washed four times (10 min each time)
using TBS/0.1% Tween-20 (TBST) solution and were incubated with
HRP-conjugated goat-anti-mouse secondary antibody (1:10,000, cat.
no. 15134-1-AP, Proteintech Group, Inc.) for 1 h at room
temperature. After the secondary antibody incubation, the PBST
solution was used to wash the membranes four times (10 min each
time). The proteins were visualized using enhanced
chemiluminescence on an X-ray film in a dark room. The results were
quantified using Image J software, version 1.52a (National
Institutes of Health). For the analysis, the GPD1-L blots were
standardized to the corresponding tubulin blots in each sample.
Electrophysiological recordings
Electrophysiological recordings were performed using
a microelectrode manipulator (MP285, Sutter Instruments) under an
inverted microscope (IX71, Olympus Corporation). The recording
electrodes were prepared from capillary glass tubes (BF150-86-10,
Sutter Instruments), which were modified using three steps and a
microelectrode puller (P97, Sutter Instruments). The patch pipette
resistance was 2-3 MΩ when filled with pipette solution. The bath
solution contained 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2
mM CaCl2, 5 mM D-glucose monohydrate, and 10 mM HEPES
(pH 7.4, adjusted using NaOH). The pipette solution contained 145
mM CsCl, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM
NaCl, 0.5 mM Na2-GTP, 2 mM Mg-ATP, 1.1 mM EGTA, and 10
mM HEPES (pH 7.2, adjusted using CsOH). After the patch pipette
contacted the cell membrane, negative pressure was used to form a
GΩ seal and break the cell membrane to create the whole-cell
recording mode. All electrophysiological experiments were performed
at room temperature. Currents were filtered and digitized using
default settings. Series resistance (80%) was compensated by
computer. The data were collected using an EPC-10 amplifier (HEKA
Elektronik) and stored in PatchMaster software, version 2×90.5
(HEKA Elektronik). The results were analyzed using PatchMaster and
IGOR Pro 6.0 software.
The activation INa values were measured
by providing cell pulsing voltages ranging from −120 to 30 mV
(10-mV step pulse), with a holding potential at −120 mV; the peak
INa value was also recorded. The peak current was fitted
using the Boltzmann function: GNa/Gmax
=[1+exp −1 (V1/2−Vc)/k] , where k is the
slope factor and V1/2 is the half-maximal voltage of
activation. GNa=INa/(V−Vrev),
where V is the membrane potential and Vrev is the
reversal potential. The inactivation INa values were
measured using a two-step protocol, with a holding potential at
−120 mV. The first step was a 30-msec conditional pulse; the
voltage range was from −120 to −20 mV (10-mV step pulse). The
second step was the test pulse (−20 mV), and the peak sodium
current was recorded. The current was fitted using the Boltzmann
function: I/I =[1+exp −1 max (Vc-V1/2)/k] ,
where Vc is the membrane potential. The recovery
INa values were measured using a three-step protocol,
with a holding potential at −120 mV. The duration (30 msec) and
voltage (−20 mV) of the condition pulse and test pulse amplitudes
were consistent, and the two pulses were spaced from 1 to 60 msec,
in 1-msec steps. The values for the peak currents induced by the
test pulses were recorded. The current was fitted using an
exponential function: I/Imax=A+A1*exp(−t/t), where t
stands for time and t is the recovery time constant. All data were
plotted using Origin 8 software (OriginLab CorporationA).
Immunofluorescence and confocal
microscopy
All treated 293 cells expressing eGFP-GPD1-L fusion
protein were imaged at room temperature using a Zeiss LSM 780
confocal system with ZEN software (black edition, version
11.0.0190). The images were acquired using a ×63 oil immersion
objective and ×10 ocular lens.
After the 293 cells were transfected with the
plasmid for 48 h, they were digested using 0.25% trypsin (2520056,
Gibco; Thermo Fisher Scientific, Inc.), resuspended, and attached
to tissue culture-treated confocal dishes (BDD012035, Jet Biofil).
Cell membranes were stained using
1,1′-diocta-decyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
(Dil; 3 µM, D4010, US Everbright, Inc.) for 5 min at 37̊C.
The cells were then fixed using 4% paraformaldehyde for 15 min at
room temperature. DAPI (1 µg/ml, D4054, US Everbright, Inc.)
was then added. The cells were then exposed to a Dil excitation
wavelength of 549 nm, and emission sections were collected between
560 and 580 nm. The EGFP-GPD1-L fusion protein excitation
wavelength was 488 nm, and emission sections were collected between
505 and 530 nm. The DAPI excitation wavelength was 360 nm, and
emission sections were collected at 460 nm.
Statistical analysis
The data were analyzed using an SPSS 20.0 software
package (IBM Corp.) and the results were expressed as mean ±
standard deviation values. Student's t-tests were used for
between-group comparisons. One-way ANOVA and Student-Newman-Keuls
tests were used for comparisons of multiple groups. P<0.01 was
considered to indicate a statistically significant difference.
Results
Clinical data
A family whose electrocardiograms showed ERP from
January 2013 to June 2015 was investigated. The mean age of the
patients was 50 years (standard deviation, 22.62 years). The
details of the family members are presented in Table I. Blood samples were collected
from the proband (black arrow) for whole-genome sequencing
analysis. The whole-genome sequencing and bioinformatics analysis
of the proband genomic DNA revealed a total of 22,205 missense SNV
sites in the exon regions. After screening for candidate genes for
malignant arrhythmias, allele frequencies <1% and protein
function prediction, GPD1-L was identified as the suspected
pathogenic mutation (NCBI Reference Sequence: rs1201810677,
https://www.ncbi.nlm.nih.gov/snp/rs1201810677). The
results for the mutation sites of the GPD1-L gene are presented in
Fig. 1B. The mutation in the
proband was verified using Sanger sequencing (Fig. 1C). This mutation resulted in the
conversion of a hydrophilic proline to a hydrophobic leucine at
position 112 of the GPD1-L protein.
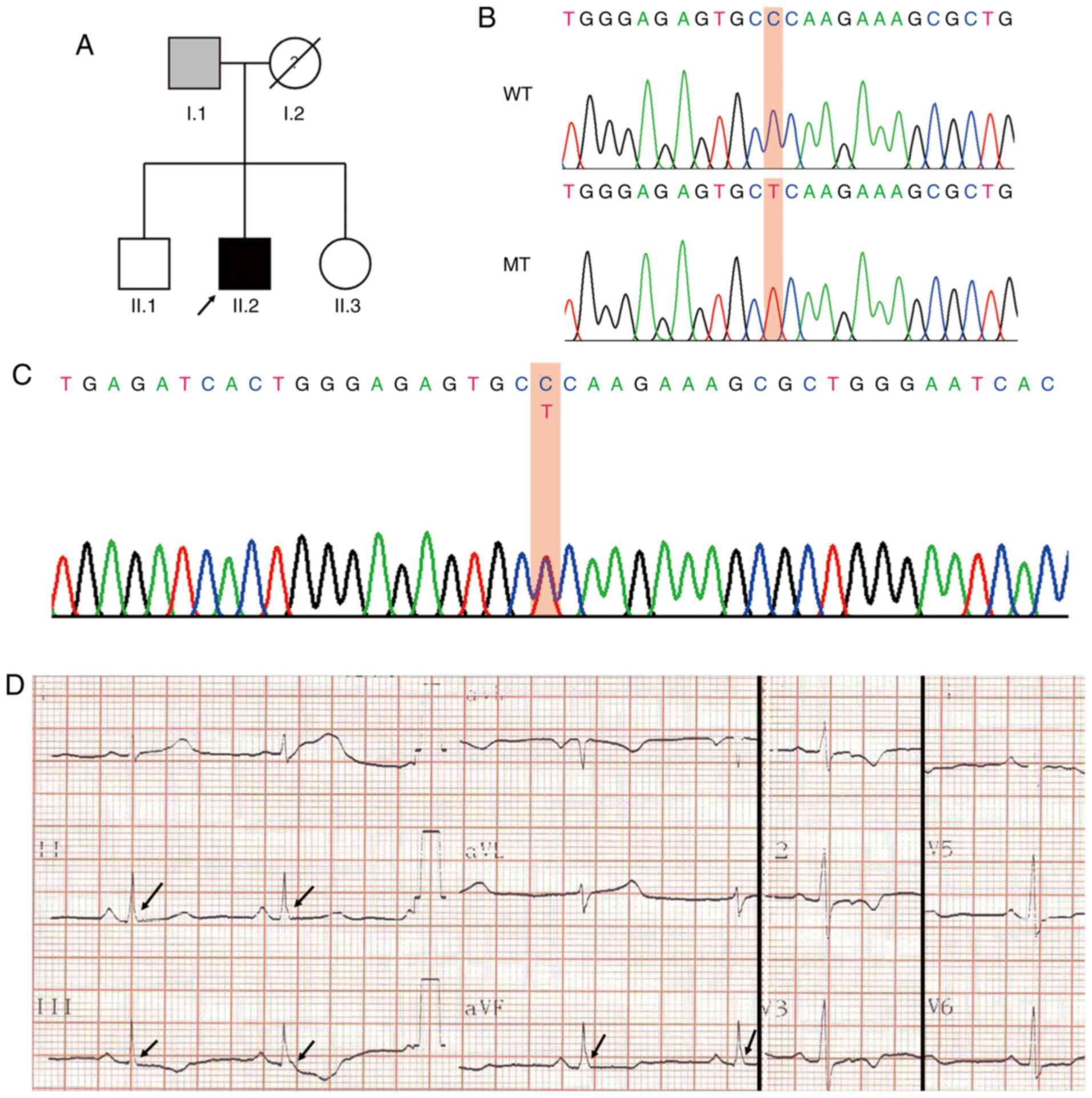 | Figure 1Clinical data of family members with
ERS. (A) Family pedigree of ERS syndrome (arrow, proband). Gray
square, presence of an ERP without sudden cardiac death; black
square, presence of an ERP with concurrent malignant ventricular
arrhythmia. (B) Proband DNA sequence of GPD1-L exon showing C>T
substitution generating a proline (P) to lysine (L) at residue 112
of GPD1-L. (C) Partial sequencing sequences containing P112L
mutations. (D) Electrocardiogram of the proband exhibits J-point
elevation (>0.1 mV) in leads II, III and aVF, and a slurring J
wave (arrows). ERS, early repolarization syndrome; ERP, early
repolarization pattern; GPD1-L, glycerol-3-phosphate dehydrogenase
1-like; WT, wild-type; MT, mutant type. |
 | Table IElectrocardiographic characteristics
of the patient's family members. |
Table I
Electrocardiographic characteristics
of the patient's family members.
| ID | Sex | Age (years) | HR (bpm) | J-wave
location | J-wave amplitude
(mV) | ST elevation
(mV) | R (mV) | QRS (msec) | QTc (msec) |
|---|
| I.1 | M | 66 | 88 | II, III, aVF
V4-V6 | 0.30 | 0.05 | 1.40 | 100 | 388 |
| II.1 | M | 36 | 68 | - | - | 0 | 2.10 | 80 | 426 |
| II.2 | M | 34 | 65 | II, III, aVF | 0.20 | 0 | 0.95 | 80 | 459 |
| II.3 | F | 29 | 71 | - | - | 0 | 1.20 | 70 | 392 |
The proband was a 34-year-old man who suffered from
recurrent palpitations with dizziness, fatigue and amaurosis fugax.
The patient's electrocardiogram results are presented in Fig. 1D. The electrocardiogram revealed
J-point elevations (>0.1 mV) in leads II, III and aVF, and a
slurring J wave. The chest X-ray, echocardiography, serum
electrolyte levels, coronary angiography, multiple myocardial
enzyme tests and treadmill test results were within the reference
ranges. The results of the patient's electrocardiogram monitoring
indicated the presence of ventricular premature contractions and
short polymorphic ventricular tachycardia. Based on these results,
ERS was diagnosed in the proband. A clinical investigation of all
members of the family (brother, sister and both parents of the
proband) revealed that the father's electrocardiography results
indicated the presence of ERPs, but the siblings'
electrocardiograms did not.
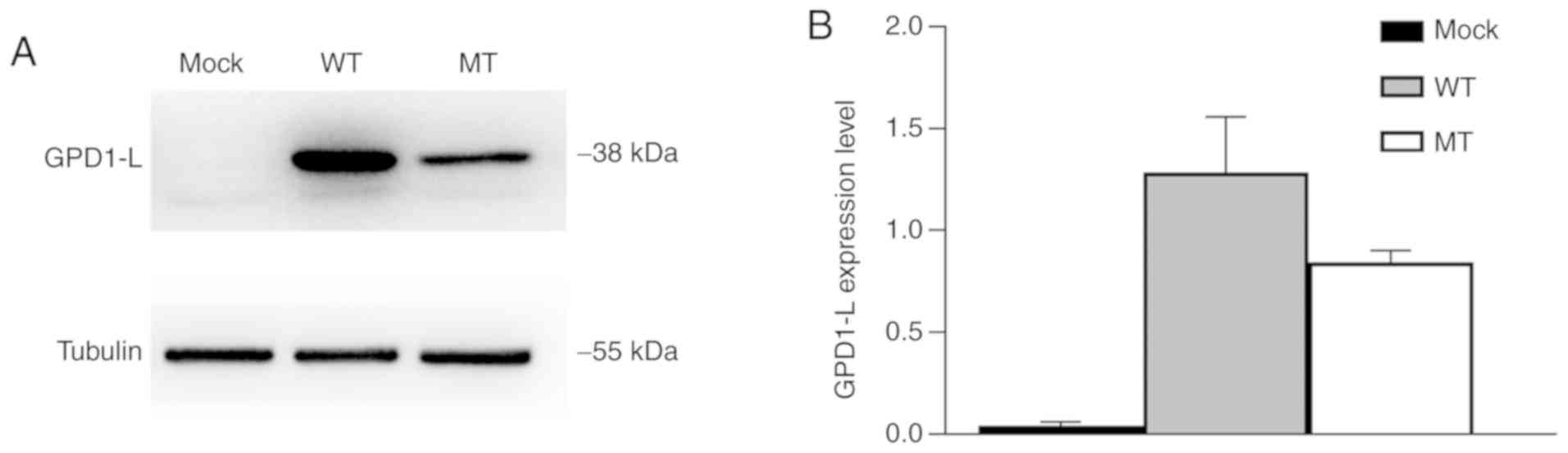
GPD1-L was successfully transfected in
293 cells and P112L mutant decreased the expression of GPD1-L
Western blotting of whole-cell lysates was performed
to verify the successful transfection of GPD1-L and study the
effects of gene mutation on protein expression. Tubulin, with a
molecular weight of 55 kDa, was used as an internal reference
protein. The results indicated that both the WT and MT groups
expressed the GPD1-L protein, but the mock group did not (Fig. 2A), which suggested that GPD1-L was
successfully transfected in 293 cells. The expression level of
MT-GPD-1L was significantly reduced in the MT group compared with
the WT group (P<0.01; Fig.
2B).
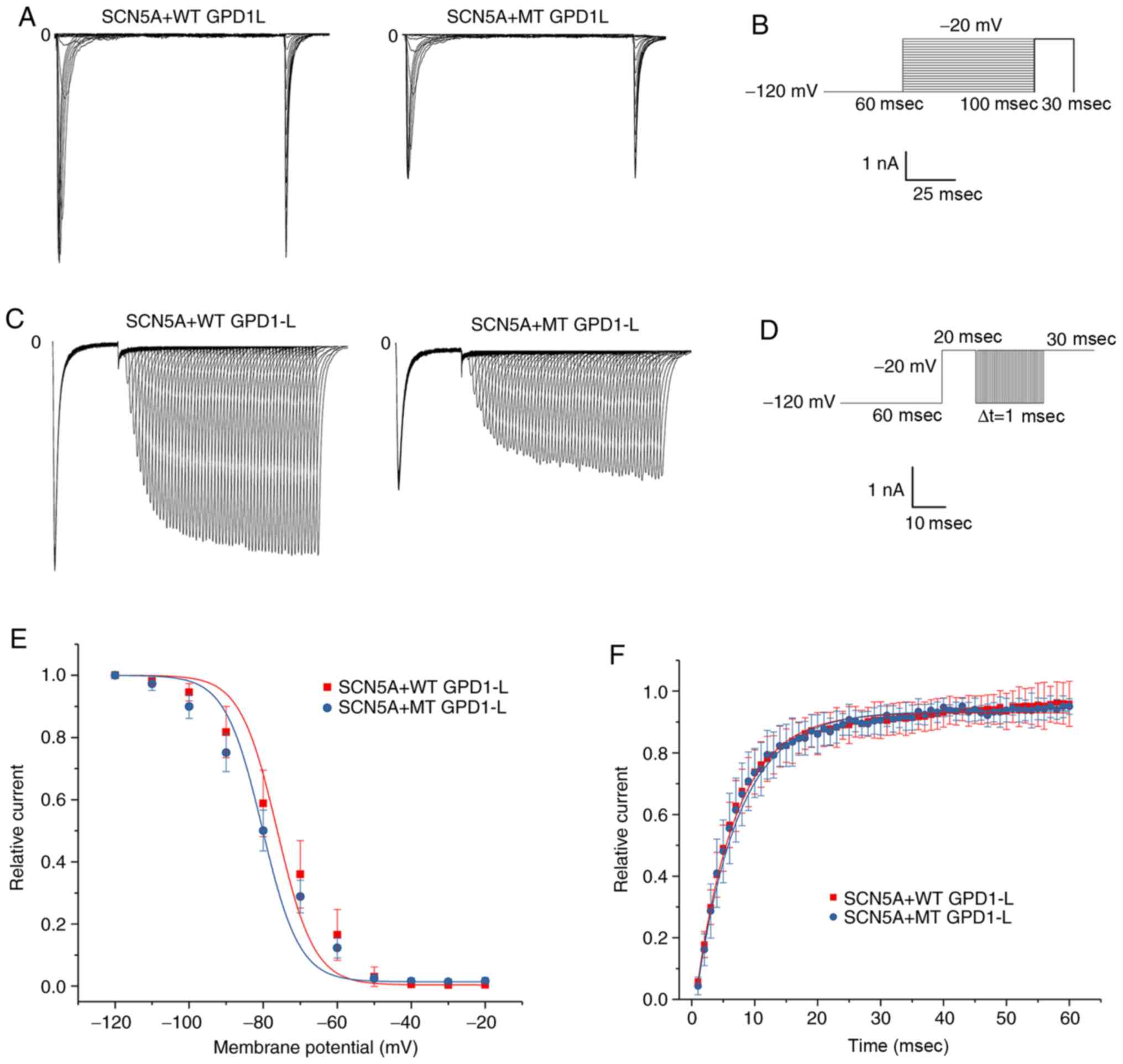
P112L mutation decreased INa
activation
293 cells were transfected with both SCN5A plasmid
and WT/MT GPD1-L plasmid. The results for representative activation
of INa in the 293 cells of the WT and MT groups are
presented in Fig. 3A. The voltage
stimulation protocol for activation of INa is presented
in Fig. 3B. The whole-cell
patch-clamp recording experiment revealed that the maximum current
densities for the WT and MT groups were −599.50±103.80 pA/pF (n=9)
and −197.25±78.53 pA/pF (n=9), respectively (Fig. 3C). The P112L mutation reduced the
current by ~60% compared with the WT group (P<0.01). The IV
curve morphology was basically the same between the two groups. The
Boltzmann function was used to obtain the V1/2 and slope
factor k (Fig. 3D). The results
indicated that the steady-activation curves for both groups began
to activate at ~ −60 mV. There were no significant between-group
differences for V1/2 (P=0.43) or slope factor k values
(P=0.17). These results indicated that the mutation exerted little
effect on activation of INa (Table II). Representative results for
inactivation and recovery of INa and stimulation
protocols are presented in Fig.
4A-D. The Boltzmann inactivation curve function was used to
obtain the V1/2 and slope factor k (Fig. 4E). Compared with the WT group, a
negative voltage change was detected in the MT group. There were no
significant between-group differences in V1/2 (P=0.54)
or slope factor k values (P=0.38). These results indicated that the
mutation exerted little effect on the inactivation of
INa (Table II). An
exponential reactivation curve function was fitted to obtain the τ
(Fig. 3F). The results indicated
that the mutation exerted little effect on recovery of
INa (P=0.77; Table
II).
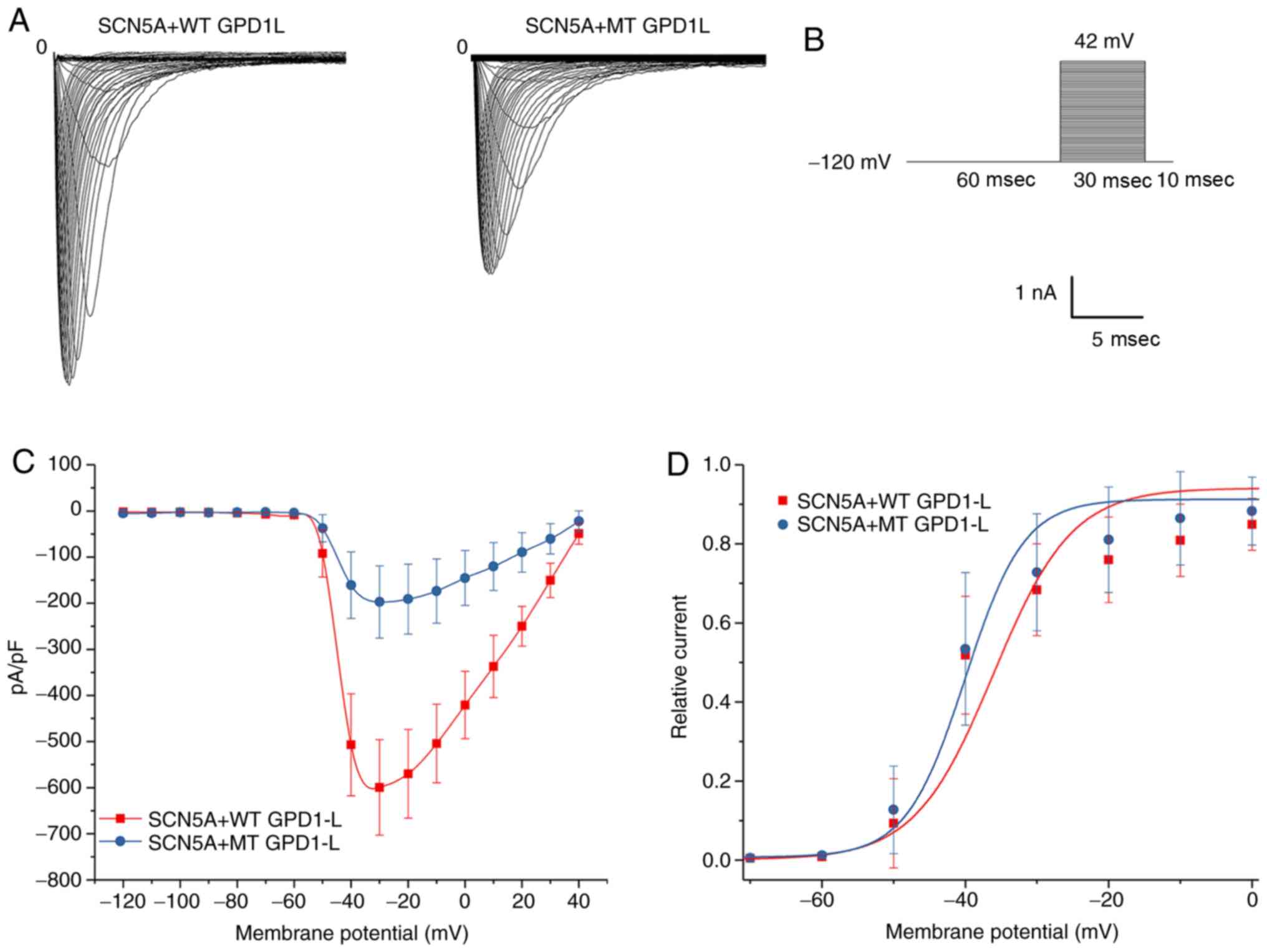 | Figure 3Activation of INa in 293
cells co-transfected with both SCN5A and WT/MT GPD1-L plasmids. (A)
Representative activation INa traces of two groups
induced using the voltage protocol. (B) Voltage protocol for
activation. (C) INa-V activation relationship in the WT
and MT groups, with INa normalized to the cell membrane
capacitance. The INa density was significantly greater
in the WT group compared with the MT group in the test potential
range of -60 to 20 mV (P<0.05). The mean ± standard deviation
membrane capacitance values were 11.96±2.84 and 11.47±3.70 pF for
the WT and MT groups, respectively (P>0.05). (D) Voltage
dependence of activation: The V1/2 and slope factor k
values were −39.15±4.80 and 7.28±2.44 mV, respectively, for the WT
group (n=9), and -39.37±5.34 and 7.05±2.55 mV, respectively, for
the MT group (n=9). There were no significant differences in
V1/2 or k between the WT and MT groups (P>0.05).
GPD1-L, glycerol-3-phosphate dehydrogenase 1-like; WT, wild-type;
MT, mutant type. |
| Table IIGating kinetics parameters of
INa in 293 cells co-expressed of SCN5A and WT/MT
GPD1-L. |
Table II
Gating kinetics parameters of
INa in 293 cells co-expressed of SCN5A and WT/MT
GPD1-L.
| Groups | Activation
| Inactivation
| Recovery
|
|---|
| V1/2
(mV) | k | n | V1/2
(mV) | k | n | τ (msec) | n |
|---|
| SCN5A + WT
GPD1-L | −39.15±4.80 | 7.28±2.44 | 7 | −75.97±4.58 | 9.34±0.60 | 9 | 6.26±0.86 | 5 |
| SCN5A + MT
GPD1-L | −39.37±5.34a | 7.05±2.55a | 7 | −79.93±2.71a | 9.71±0.65a | 9 | 6.24±1.49a | 5 |
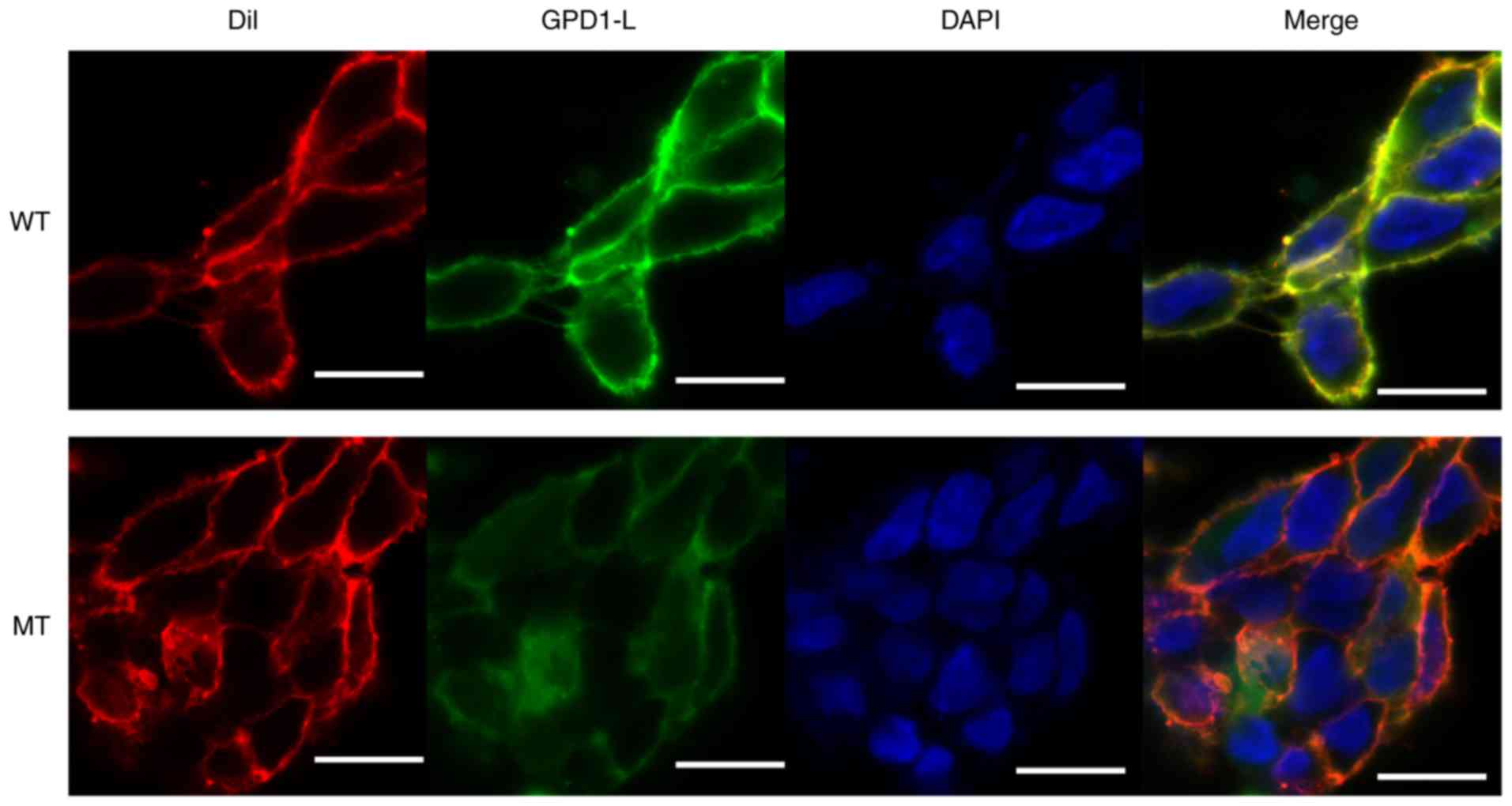
The P112L mutant reduces localization of
GPD1-L adjacent to the cell surface
To investigate the effect of the P112L mutation on
GPD1-L localization in cells, 293 cells were transfected with SCN5A
plasmid and eGFP-WT/MT GPD1-L fusion plasmid (1:1 ratio). After a
48-h transfection period, the cells were treated for confocal
microscopy observation. Dil was used to stain the cell membranes.
The eGFP-GPD1-L fusion protein was used to track the localization
of GPD1-L in the 293 cells. The results indicated that the
expression levels of GPD1-L near the membrane were higher in the WT
group compared with those in the MT group (Fig. 5). Both the WT and MT GPD1-L were
mainly distributed in the cytoplasm, whereas the distribution in
the nuclei was lower in both groups (Fig. 5).
Discussion
In the present study, the whole-cell patch-clamp
recording experiment results indicated that, compared with the WT
group, the activation of INa was 60% lower in the MT
group. The western blotting results indicated that GPD1-L was
successfully transfected in 293 cells and the expression of the
mutant GPD1-L protein was lower in the MT group. The evaluation
using confocal microscopy revealed that the MT GPD1-L was less
expressed near the cell membrane and more expressed in the
cytoplasm.
Research on the mechanism of ERS and severe
ventricular arrhythmia has reached the ion level (18). The change in the action potential
notch of the epicardial membrane is related to the J wave, and the
increase in repolarization current (secondary to the decrease in
inward current or the increase in outward current) leads to an
increase in the J waves or elevation of ST segments (19). The gene mutation leading to the
morphological change in the action potential notch may result in
the formation of ERPs. Currently, the genetic variations associated
with both ERP and ERS include seven types: KCNJ8 (9) and ABCC9 (15), which regulate pore formation of
the IK-ATP channel and the ATP-sensitive subunit;
CACNA1C (11), CACNB2b (12), CACNA2D1 (12), and related subunits of the L-type
calcium channel; and SCN5A (20)
and SCN10A (13) of the
INa channel. The enhanced repolarization dispersion is
prone to form 2-phase reentry, which may result in premature
ventricular contractions and ventricular tachycardia (19).
GPD1-L, a glycerol-3-phosphate dehydrogenase 1-like
gene, plays an important role in the function regulation of
myocardial sodium channels; it is an important susceptibility gene
for sudden unexpected death syndrome (21), sudden infant death syndrome
(21), and Brugada syndrome
(22). The human GPD1-L gene is
located on chromosome 3 and encodes a protein composed of 351 amino
acids. GPD1-L may interact with proteins associated with ion
channels, such as ankyrin-G, syntrophin, caveolin-3 and Nedd4-2
(23). GPD1-L regulates
endoplasmic reticulum signaling by affecting the protein kinase
A-dependent phosphorylation (24). GPD1-L also affects the oxidative
state to regulate the function of cardiac sodium channels (16).
The first reported association for GPD1-L was in
type II Brugada syndrome (25),
where it is co-expressed with Nav1.5 in the cardiomyocytes. Van
Norstrand (21) detected three
mutation sites for GPD1-L (E83K, I124V and R273C) in 83 cases of
sudden unexpected death syndrome and in 221 cases of subsequent
sudden infant death syndrome. All sites were highly conserved, and
none were detected in the 300 control group patients. Experiments
with 293 cells and neonatal rat myocardial cells transfected with
the aforementioned mutants demonstrated that the mutants may cause
an instantaneous decrease in sodium current density (21).
We found that the activation current density of the
MT group decreased by 60% compared with the WT group. A recent
study found that GPD1-L is expressed near the cell membrane
(26) and can regulate the redox
state of sodium channels via PKC-dependent phosphorylation, thus
affecting the function of sodium channels (16). It was inferred that the decrease
in the activation of INa can increase the relative
inward current of the action potential during repolarization
through the mechanisms mentioned above and, thus, alter the notch
morphology of action potentials in myocardial cells and form J
waves and J-point elevations in the electrocardiogram. Further
study is required to confirm the effects of GPD1-L P112L on action
potentials and elucidate the exact mechanisms underlying the
association between GPD1-L and sodium channels. The western
blotting results indicated that the mutation decreased the total
expression of GPD1-L. Although confocal microscopic observation
found that GPD1-L somewhat co-located with cell membrane markers, a
previous study demonstrated that GPD1-L is localized near rather
than on the cell membrane (26).
Therefore, the results may be interpreted as follows: MT GPD1-L was
less expressed near the cell membrane and more expressed in the
cytoplasm compared with WT GPD1-L. Our results indicate that the
difference in total expression level and distribution of GPD1-L
caused by mutations may also be implicated in the occurrence of
ERS. Further research is required to investigate the mechanisms of
expression and abnormal intracellular transport of GPD1-L affected
by mutations.
In summary, it was herein demonstrated that GPD1-L
P112L decreased the activation of INa. The decreased
total cell GPD1-L expression and the decreased expression of GPD1-L
near the cell membrane may also contribute to the development of
abnormal sodium currents. Our study results suggested that GPD1-L
P112L may be a novel pathogenic genetic mutation of ERS.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81370285 and 81970206), and
the Guangzhou City Science and Technology Program (grant no.
201508020057).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
WSH and JF made substantial contributions to the
design of the present study. JF, JCC, YH, CYJ, CXM and ZZH
performed the experiments; JF, JCC, YH, CYJ, CXM and ZZH analyzed
the data; JF, WSH, JCC, CYJ wrote the manuscript. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The investigation was approved by the Ethics
Committee of the First Affiliated Hospital of Sun Yat-Sen
University. All family members agreed to participate in the study
and signed the informed consent forms.
Patient consent for publication
All patients provided written informed consent
regarding the publication of their data, including clinical and
genetic information.
Competing interests
All the authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Tikkanen JT, Anttonen O, Junttila MJ, Aro
AL, Kerola T, Rissanen HA, Reunanen A and Huikuri HV: Long-term
outcome associated with early repolarization on
electrocardiography. N Engl J Med. 361:2529–2537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Junttila MJ, Tikkanen JT, Kenttä T,
Anttonen O, Aro AL, Porthan K, Kerola T, Rissanen HA, Knekt P and
Huikuri HV: Early repolarization as a predictor of arrhythmic and
nonarrhythmic cardiac events in middle-aged subjects. Heart Rhythm.
11:1701–1706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang Y, Stahl-Herz J and Sampson BA:
Molecular diagnostics of cardiovascular diseases in sudden
unexplained death. Cardiovasc Pathol. 23:1–4. 2014. View Article : Google Scholar
|
|
4
|
Gourraud JB, Le Scouarnec S, Sacher F,
Chatel S, Derval N, Portero V, Chavernac P, Sandoval JE, Mabo P,
Redon R, et al: Identification of large families in early
repolarization syndrome. J Am Coll Cardiol. 61:164–172. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCorquodale A, Poulton R, Hendry J,
Norrish G, Field E, Mead-Regan S, Lowe M and Kaski JP: High
prevalence of early repolarization in the paediatric relatives of
sudden arrhythmic death syndrome victims and in normal controls.
Europace. 19:1385–1391. 2017. View Article : Google Scholar
|
|
6
|
Koncz I, Gurabi Z, Patocskai B, Panama BK,
Szél T, Hu D, Barajas-Martinez H and Antzelevitch C: Mechanisms
underlying the development of the electrocardiographic and
arrhythmic manifestations of early repolarization syndrome. J Mol
Cell Cardiol. 68:20–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chauveau S, Janin A, Till M, Morel E,
Chevalier P and Millat G: Early repolarization syndrome caused by
de novo duplication of KCND3 detected by next-generation
sequencing. Heart Rhythm Case Rep. 3:574–578. 2017.
|
|
8
|
Watanabe H, Ohkubo K, Watanabe I,
Matsuyama TA, Ishibashi-Ueda H, Yagihara N, Shimizu W, Horie M,
Minamino T and Makita N: SCN5A mutation associated with ventricular
fibrillation, early repolarization, and concealed myocardial
abnormalities. Int J Cardiol. 165:e21–e23. 2013. View Article : Google Scholar
|
|
9
|
Medeiros-Domingo A, Tan BH, Crotti L,
Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp
D, et al: Gain-of-function mutation S422L in the KCNJ8-encoded
cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave
syndromes. Heart Rhythm. 7:1466–1471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X, Shen Y, Xie J, Bao H, Cao Q, Wan R,
Xu X, Zhou H, Huang L, Xu Z, et al: A mutation in the CACNA1C gene
leads to early repolarization syndrome with incomplete penetrance:
A Chinese family study. PLoS One. 12:e1775322017.
|
|
11
|
Chen Y, Barajas-Martinez H, Zhu D, Wang X,
Chen C, Zhuang R, Shi J, Wu X, Tao Y, Jin W, et al: Novel trigenic
CACNA1C/DES/MYPN mutations in a family of hypertro-phic
cardiomyopathy with early repolarization and short QT syndrome. J
Transl Med. 15:782017. View Article : Google Scholar
|
|
12
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Di Stolfo G, Palumbo P, Castellana S,
Mastroianno S, Biagini T, Palumbo O, Leone MP, De Luca G, Potenza
DR, Mazza T, et al: Sudden cardiac death in J wave syndrome with
short QT associated to a novel mutation in Nav 1.8 coding gene
SCN10A: First case report for a possible pharmacogenomic role. J
Electrocardiol. 51:809–813. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delaney JT, Muhammad R, Blair MA, Kor K,
Fish FA, Roden DM and Darbar D: A KCNJ8 mutation associated with
early repolarization and atrial fibrillation. Europace.
14:1428–1432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu D, Barajas-Martinez H, Terzic A, Park
S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C,
Schimpf R, et al: ABCC9 is a novel Brugada and early repolarization
syndrome susceptibility gene. Int J Cardiol. 171:431–442. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Valdivia CR, Ueda K, Ackerman MJ and
Makielski JC: GPD1L links redox state to cardiac excitability by
PKC-dependent phos-phorylation of the sodium channel SCN5A. Am J
Physiol Heart Circ Physiol. 297:H1446–H1452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang H, Chen YQ, Fan LL, Guo S, Li JJ,
Jin JY and Xiang R: Whole-exome sequencing identifies a novel
mutation of GPD1L (R189X) associated with familial conduction
disease and sudden death. J Cell Mol Med. 22:1350–1354. 2018.
|
|
18
|
Barbosa EC, Bomfim Ade S,
Benchimol-Barbosa PR and Ginefra P: Ionic mechanisms and vectorial
model of early repo-larization pattern in the surface
electrocardiogram of the athlete. Ann Noninvasive Electrocardiol.
13:301–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Antzelevitch C: Genetic, molecular and
cellular mechanisms underlying the J wave syndromes. Circ J.
76:1054–1065. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo Q, Ren L, Chen X, Hou C, Chu J, Pu J
and Zhang S: A novel mutation in the SCN5A gene contributes to
arrhythmo-genic characteristics of early repolarization syndrome.
Int J Mol Med. 37:727–733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Norstrand DW, Valdivia CR, Tester DJ,
Ueda K, London B, Makielski JC and Ackerman MJ: Molecular and
functional characterization of novel glycerol-3-phosphate
dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death
syndrome. Circulation. 116:2253–2259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Makiyama T, Akao M, Haruna Y, Tsuji K, Doi
T, Ohno S, Nishio Y, Kita T and Horie M: Mutation analysis of the
glycerol-3 phosphate dehydrogenase-1 like (GPD1L) gene in Japanese
patients with brugada syndrome. Circ J. 72:1705–1706. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abriel H and Kass RS: Regulation of the
voltage-gated cardiac sodium channel Nav1.5 by interacting
proteins. Trends Cardiovasc Med. 15:35–40. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hallaq H, Yang Z, Viswanathan PC, Fukuda
K, Shen W, Wang DW, Wells KS, Zhou J, Yi J and Murray KT:
Quantitation of protein kinase A-mediated trafficking of cardiac
sodium channels in living cells. Cardiovasc Res. 72:250–261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weiss R, Barmada MM, Nguyen T, Seibel JS,
Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM
and London B: Clinical and molecular heterogeneity in the brugada
syndrome: A novel gene locus on chromosome 3. Circulation.
105:707–713. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
London B, Michalec M, Mehdi H, Zhu X,
Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL,
Madhusudanan M, et al: Mutation in glycerol-3-phosphate
dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current
and causes inherited arrhythmias. Circulation. 116:2260–2268. 2007.
View Article : Google Scholar : PubMed/NCBI
|