Introduction
Cardiac fibrosis is the major pathological process
in ventricular remodelling occurring after myocardial infarction
(MI), and is characterised by deposition of extracellular matrix
proteins and ventricular dysfunction (1). Multiple pathophysiological factors
contribute to the process, including the inflammatory response,
activation of the renin-angiotensin-aldosterone system, oxidative
stress and apoptosis (2,3). Chronic infiltration of inflammatory
cells, particularly macrophages, following MI can lead to the
formation of scar tissue, and macrophage-secreted transforming
growth factor β (TGF-β) is a major molecule involved in fibrosis
following MI (4). In addition,
experimental evidence has indicated that the increased production
of reactive oxygen species (ROS) contributes to myocardial fibrosis
occurring after MI (5). ROS from
multiple sources participate in this process, including xanthine
oxidase, inflammatory cells, mitochondria and NADPH oxidases (Noxs)
(6,7). Nox2 and Nox4 were identified as the
major sources of ROS in the heart, while Nox2 deficiency has been
reported to mitigate angiotensin II-induced cardiac hypertrophy in
mice (8). Studies have also
demonstrated that Nox2 knockout mice exhibited reduced interstitial
fibrosis and improved survival rates in a myocardial-infarction
model, when compared with those in the control group (9). An in vitro study verified
that Nox4 knockdown led to decreased fibronectin and collagen
synthesis in cardiac fibroblasts treated with angiotensin II
(10). In addition, a large
amount of ROS exacerbated apoptosis and inflammatory cell
infiltration following MI, thus worsening tissue injury and cardiac
fibrosis (11).
Imbalances in the production of ROS and the
antioxidant capability of the biological system are also important
causes of cardiac dysfunction. AMP-activated protein kinase (AMPK)
is a critical regulator of cardiomyocyte energy homeostasis and
survival. Activation of AMPK has been reported to display a
protective effect in myocardial ischaemia-induced damage. AMPK also
suppresses oxidative stress through the activation of nuclear
factor erythroid 2-related factor 2 (Nrf2) and haem oxygenase
(HO)-1, and ameliorates tissue damage (12,13).
Corosolic acid (CRA) is a triterpenoid compound
discovered in numerous medicinal herbs, particularly in
Lagerstroemia speciosa L. (also known as Banaba). CRA
initially attracted much attention for its anti-diabetic function
(14), while further studies
demonstrated that CRA has more functions, including antitumour
(15) and anti-atherosclerotic
properties (16). Previous
studies have confirmed that CRA inhibits acute inflammation by
regulating IRAK-1 phosphorylation via an NF-κB-independent pathway
in macrophages (17). In
addition, in endothelial dysfunction, CRA protects mitochondrial
function by regulating Drp1 phosphorylation (Ser637) in an
AMPK-dependent manner, which contributes to inhibiting Nox2 oxidase
signalling and suppressing NLRP3 inflammasome activation (18). However, to the best of our
knowledge, the effects of CRA on post-MI remodelling have not been
reported to date.
Therefore, in the present study, the aim was to
evaluate the effects of CRA on MI induced by coronary artery
ligation in mice and to explore the underlying mechanism.
Materials and methods
Animals
All animal experimental protocols were approved by
the Animal Care and Use Committee of Renmin Hospital of Wuhan
University (Wuhan, China) and were conducted in accordance with the
National Institutes of Health (NIH) Guide for the Care and Use of
Laboratory Animals. Male C57BL/6J mice (n=120; weight, 23.5–27.5 g;
age, 8 weeks) were purchased from the Institute of Laboratory
Animal Science, CAMS & PUMC (Beijing, China). The animals were
housed at a controlled temperature and humidity under a 12-h
light-dark cycle with free access to food and water at the
Cardiovascular Research Institute of Wuhan University (Wuhan,
China). The animals were allowed to acclimatize to the laboratory
environment for at least one week, and were then randomly assigned
to the control [phosphate-buffered saline (PBS)-treated] and
CRA-treated groups (10 and 20 mg/kg; purity, >98%; Baoji Herbest
Bio-Tech Co., Ltd.) (19,20). After 14 days of pre-treatment, the
mice were subjected to either sham surgery (sham group) or MI by
left anterior descending coronary artery ligation. A total of four
groups (n=30) were formed, including: Sham group (PBS-treated), MI
group (PBS-treated), MI+CRA 10 group (treated with 10 mg/kg CRA),
and MI+CRA 20 group (treated with 20 mg/kg CRA). Following surgery,
all animals were treated with PBS or CRA for 4 weeks. In the sham,
MI, MI+CRA 10 and MI+CRA 20 groups, the number of surviving mice
were 30, 15, 22 and 24, respectively at 4 weeks after surgery.
Induction of MI
Briefly, the mice were intraperitoneally
anaesthetised with sodium pentobarbital (60 mg/kg), intubated and
ventilated with a ventilator. Following a left thoracotomy, the
heart was rapidly exposed, and the left anterior descending branch
of the coronary artery was quickly identified approximately 2–3 mm
away from the inferior margin of the left auricle and ligated with
a 7-0 silk suture. In sham-operated mice, the left coronary artery
was encircled without ligation. Subsequent to the surgery, all
animals were treated with PBS or CRA for 4 weeks.
Echocardiography and haemodynamic
analysis
At 4 weeks after surgery, the mice were
anaesthetised by inhalation of 1.5–2% isoflurane. Echocardiography
was performed to evaluate the function of the left ventricle using
a MyLab 30CV system (Biosound Esaote, Inc.) equipped with a 15-MHz
probe. M-mode tracings derived from the short axis of the left
ventricle at the level of the papillary muscles were recorded. For
haemodynamic analysis, insertion of a 1.4-French catheter-tip
micromanometer catheter (Millar Instruments) into the left
ventricle via the right carotid artery was performed. The heart
rates, pressure and volume signals were continuously recorded using
an Aria pressure-volume conductance system (Millar Instruments)
coupled with a PowerLab/4SP A/D converter. According to the
guidelines of the Chinese Animal Welfare Committee, subsequent to
pressure-volume measurement, the mice were anaesthetised with 1.5%
pentobarbital sodium (60 mg/kg) and then sacrificed by cervical
dislocation under anaesthesia.
Injury regions
According to a previous study (21), the heart was described as infarct
zone (left ventricle free wall), border zone (left ventricle
anterior and posterior walls) and distal zone (interventricular
septum). Western blotting and reverse transcription-quantitative
PCR (RT-qPCR) were studied using border zone tissue.
Histology
The hearts were removed from the mice, arrested in
diastole with 10% KCl, weighed and fixed with 4% formaldehyde,
followed by embedding in paraffin. The mouse hearts were cut
transversely close to the apex to visualize the left and right
ventricles. Tissue sections (4–5 μm) were stained with haematoxylin
and eosin (H&E) to assess the infarct size, or stained with
picrosirius red (PSR) and Masson’s trichrome to assess the collagen
accumulation as an indication of fibrosis. The ratio of
interstitial fibrosis to the total left ventricular area was
calculated based on the examination of 10 microscopic fields that
were randomly selected in three individual sections per heart, and
the images were further analysed by Image Pro Plus software
(version 6.0; Media Cybernetics, Inc.).
After dewaxing and sequentially deparaffinised,
tissue sections were stained with haematoxylin for 5 min at room
temperature and then moved into differentiation fluid (1%
hydrochloric acid alcohol) for 3 sec. After washing with flowing
water for 15 min, sections were placed in eosin liquid for 2 min,
and then washed with water for 1 min. Finally, sections were washed
in graded alcohol (75, 90 and 100%) and dehydrated with xylene 3
times for 5 min each time at room temperature, and covered by the
coverslips. For PSR straining, previous steps up to stained into
haematoxylin were the same as H&E staining, and the slides were
then immersed in 2% phosphomolybdic acid for 2 min, before covering
sections in picro-sirius red solution and incubating for 90 min at
room temperature. Finally, sections were washed with acetic acid
solution for 2 sec, dehydrated, cleared and covered by the
coverslips. For the Masson’s trichrome staining, after dewaxing,
the paraffin sections were transferred from the PBS directly in
iron hematoxylin for 8 min at room temperature, rinsed for 1–3 sec
in 1% hydrochloric acid alcohol, followed by washing with running
tap water for 5 min. Following incubation with 1% Acid Ponceau
Fuchsin for 20 min, slides were rinsed in distilled water 5 times.
Sections were then placed in 1% Phosphormolybdenic acid for 4 min,
and then 2% Anilinblue for 5 min, distilled water for 5 min and
then 0.2% Acetic Acid for 2 min. Finally, sections were washed with
acetic acid solution, dehydrated, cleared and covered by the
coverslips.
Immunohistochemistry
Paraffin-embedded heart sections were sequentially
deparaffinised and blocked with 10% normal goat serum in
Tris-buffered saline with 1% bovine serum albumin at 37°C for 2 h.
The sections were incubated overnight at 4°C with primary
antibodies against Nox4 (1:200; Abcam; ab154244), HO-1 (1:200;
Abcam; ab13243) and CD68+ (1:200; Abcam; ab125212),
followed by incubation with EnVision™+/HRP reagent at 37°C for 1 h,
and staining with a DAB detection kit (GK600710; Gene Tech). Images
of stained cells were captured with a light optical microscope at
×400 magnification.
Western blot analysis
The ventricular tissues were homogenised by a
lapping machine and lysed in RIPA lysis buffer. The protein lysates
were collected, and the protein concentration was measured with a
BCA kit (Synergy HT; BioTek Instruments, Inc.). Next, protein
lysates were separated by SDS-PAGE (10% gel) and transferred onto
Immobilon-PL transfer membranes (Millipore). The membranes were
blocked with 5% skim milk and then incubated overnight at 4°C with
the following primary antibodies: Nox2 (1:1,000; ab129068), Nox4
(1:1,000; ab154244), HO-1 (1:1,000; ab13243), Nrf2 (1:1,000;
ab31163), TGFβ1 (1:1,000; ab64715), monocyte chemotactic protein 1
(1:1,000; Mcp-1; ab151538), C-C chemokine receptor type 2 (1:1,000;
CCR2; ab203128), P-inhibitor of NF-κB kinase β (1:1,000; P-Ikkβ;
ab59195) and T-Ikkβ (1:1,000; ab178870), which were purchased from
Abcam; P-AMPKα (1:1,000; 2535), T-AMPKα (1:1,000; 2603P), B-cell
lymphoma 2 (1:1,000; Bcl2; 2870), Bcl2-associated X protein
(1:1,000; Bax; 2772), T-p65 (1:1,000; 8242), P-Smad2 (1:1,000;
3108S), T-Smad2 (1:1,000; 3103s), P-Smad3 (1:1,000; 8769), T-Smad3
(1:1,000; 9513s) and GAPDH (1:1,000; 2118), which were obtained
from Cell Signaling Technology, Inc.; and P-p65 (1:1,000; s276;
cat. no. BS4135), obtained from Bioworld Technology, Inc. The
samples were subsequently incubated with the goat anti-mouse IgG
(P/N 925-32210; 1:1,250; LI-COR Biosciences) and goat anti-rabbit
IgG (P/N 925-32211; 1:1,250; LI-COR Biosciences) for 1 h at room
temperature. (Thermo Fisher Scientific, Inc.). Next, the membranes
were incubated with enhanced chemiluminescence reagent (HP193406;
Wuhan Servicebio Technology). Finally, the blots were scanned using
a ChemiDoc Imaging System (cat. no. 733BR2234; Bio-Rad
Laboratories, Inc.) and analysed using ImageJ software (NIH).
RT-qPCR
The relative mRNA expression levels of atrial
natriuretic peptide (ANP), B-type natriuretic peptide (BNP),
α-myosin heavy chain (α-MHC), β-MHC, connective tissue growth
factor (CTGF), Collagen I α1, Collagen III α1, fibronectin, Nox4,
Nox2, HO-1, interleukin-1β (IL-1β), tumour necrosis factor α
(TNF-α) and IL-6 were determined by RT-qPCR. Briefly, total RNA was
isolated from the snap-frozen tissues and cardiomyocytes using an
RNA isolation kit (15596-026; Invitrogen; Thermo Fisher Scientific,
Inc.). The yield and purity of RNA samples were calculated
spectrophotometrically according to the A260/A280 and A230/260
ratios using a SmartSpec Plus spectrophotometer (Bio-Rad
Laboratories, Inc.). Next, RNA (2 μg of each sample) was reverse
transcribed into cDNA using oligo(dT) primers and the Transcriptor
First Strand cDNA Synthesis kit (4897030001; Roche). PCR
amplifications were performed using a LightCycler 480 SYBR-Green I
Master Mix (04887352001; Roche). All PCR primers are listed in
Table SI. The thermal profile
consisted of 10 min of pre-incubation step at 95°C for FastStart
Taq DNA polymerase activation, followed by 45 cycles of PCR at 95°C
for 10 sec (denaturation), 60°C for 20 sec (annealing), and 72°C
for 30 sec (elongation). Amplified cDNA products were detected by
melting curve analysis which consisted of 95°C for 5 sec and 65°C
for 1 min, and heated to 97°C to detect continuous changes in
fluorescence of SYBR-Green I. After 45 cycles, the housekeeping
gene GAPDH was used to normalize the gene expression (22).
Cell culture
H9C2 cells were cultured in Dulbecco’s modified
Eagle medium containing 10% foetal bovine serum (FBS) with
streptomycin (100 mg/ml) and penicillin (100 U/ml) under standard
conditions at 37°C with 5% CO2. Cells in exponential
growth were dissociated with 0.25% trypsin (Gibco; Thermo Fisher
Scientific, Inc.) and seeded in 6-well or 24-well culture plates at
a density of 1×105 cells/ml prior to incubation for 24
h. Next, different concentrations of CRA were added to the medium 1
h before hypoxia injury. Among the five CRA concentrations tested
(0.1, 1, 5, 10 and 20 μM), the H9C2 cells in the 0.1 and 20
μM-treated groups are in poor condition under hypoxia, thus only
three CRA concentrations were selected for this analysis, including
1, 5 and 10 μM (data not shown). For the induction of hypoxia
injury, the cells were cultured in D-Hank’s solution in an MCO-18M
O2/CO2 incubator (Sanyo Electric Co., Ltd.)
with 1% O2, 5% CO2 and 94% N2 for
24 h. Oxidative stress was detected by western blotting, PCR and
ROS detection, and apoptosis was detected by TUNEL assay.
Transfection experiment
H9c2 cells were transfected with 50 μg AMPK α2 siRNA
(GCCCAGATGAACGCTAAGATA) or 50 μg control siRNA; (Guangzhou RiboBio
Co. Ltd.) and Lipo6000™ (Beyotime Institute of Biotechnology)
according to the manufacturer’s protocol. Briefly, DMEM (Gibco;
Thermo Fisher Scientific, Inc.) medium without serum and Lipo6000
were mixed in a PE tube for 5 min, AMPK α2 siRNA (50 μM) and DMEM
medium without serum were mixed in another PE tube for 5 min, and
the two PE tubes were evenly mixed into another PE tube. After 5
min, they were added to a 6-well plate or a 24-well plate. After 6
h, the medium was replaced and the culture continued for 48 h under
standard conditions at 37°C with 5% CO2.
ROS detection
2,7-Dichlorodihydrofluorescein diacetate (DCFH-DA;
Invitrogen; Thermo Fisher Scientific, Inc.) was used to detect the
ROS levels in H9C2 cells following treatment and hypoxia. Briefly,
the cells were cultured in 24-well plates with or without CRA, and
then exposed to hypoxia for 24 h. Next, the cells were incubated
with DCFH-DA for 30 min at 37°C. The production of ROS by the cells
was observed under an Olympus IX53 fluorescence microscope (Olympus
Corporation).
TUNEL assay
Apoptotic cells and heart tissue sections were
detected by TUNEL straining according to the manufacturer’s
instructions (S7111; EMD Millipore Crop). For cells, they were
fixed with 4% paraformaldehyde in 4°C overnight, rinsed with PBS
and then incubated in 0.1% Triton X-100 for 5 min at room
temperature. For tissue sections, after routinely dewaxing and
hydration, TUNEL straining was performed according to the protocols
provided with each kit. Briefly, equilibration of buffer (EMD
Millipore Corp.) was added to cells or tissue sections and
incubated for 10 sec at room temperature, the TDT Enzyme (EMD
Millipore Corp.) was added to the cells or tissue sections and
incubated for 1 h at 37°C in a dark humidified chamber. Stop/Wash
buffer (EMD Millipore Corp.) was added and incubated for 10 min at
room temperature, rinsed with PBS before Anti-Digoxigenin
Fluorescein (EMD Millipore Corp.) was added, and then incubated for
30 min at room temperature in a dark humidified chamber. Finally,
stained with 4′, 6-diamidino-2′-phenylindole dihydrochloride (DAPI)
(Invitrogen; Thermo Fisher Scientific, Inc.) before washed by PBS.
Cells were detected using a fluorescence microscope at ×200
magnification, and the ratio of TUNEL-positive cells to total cells
was calculated after at least 20 viewed fields.
Statistical analysis
The data are presented as the mean ± standard error
of the mean. Data were analysed with a one-way analysis of variance
followed by a post-hoc Tukey’s test using SPSS software (version
22.0; SPSS, Inc.). A value of P<0.05 was considered to denote a
statistically significant difference.
Results
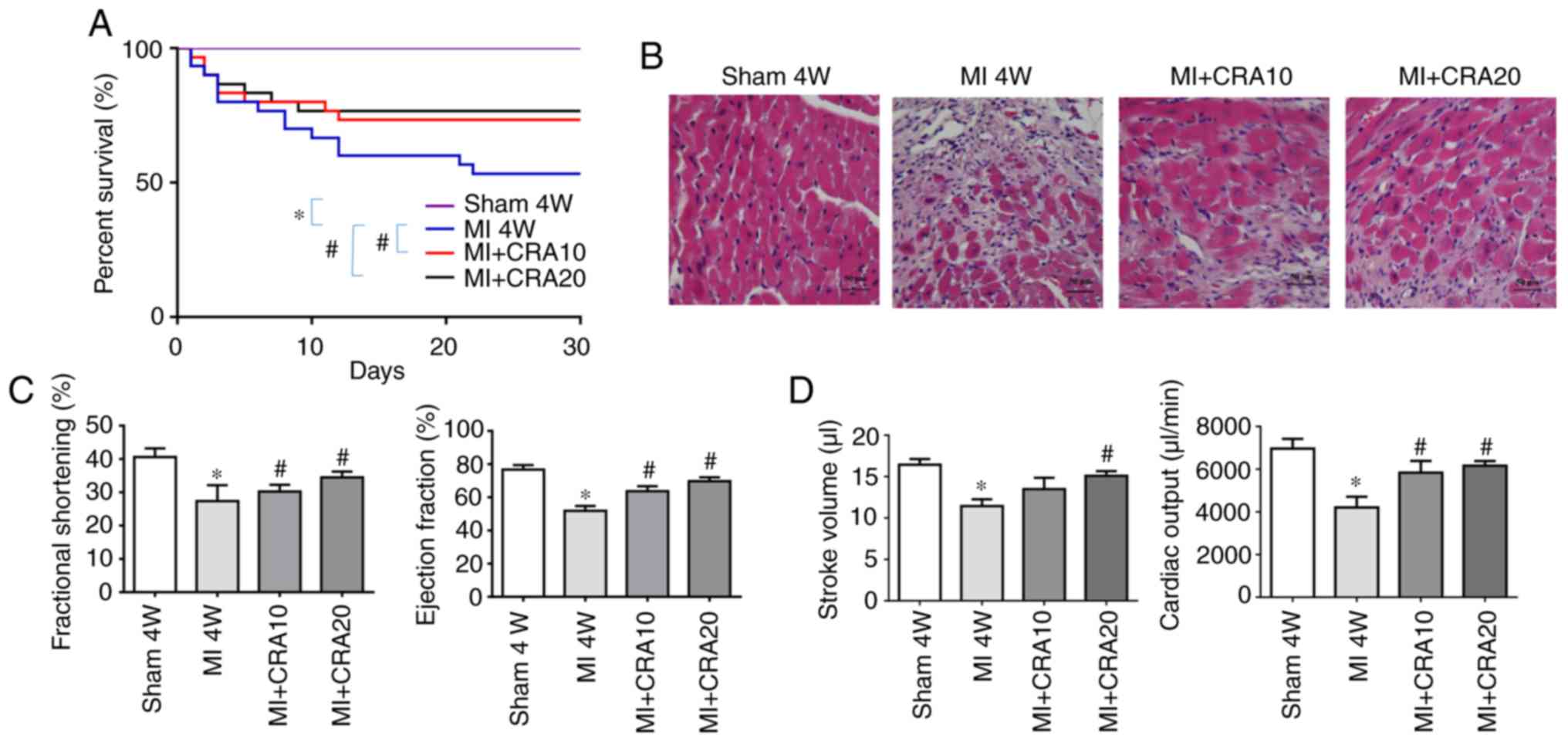
CRA improves the survival rates and
post-infarction cardiac function of mice
The survival rates of mice in the MI+CRA 10 and
MI+CRA 20 groups were significantly higher in comparison with those
in the MI group (Fig. 1A).
H&E staining revealed that the CRA-treated groups displayed a
higher number of cardiomyocytes and reduced infarct size in the
border zone at 4 weeks after MI, as shown in Fig. 1B. Echocardiography analysis
demonstrated that CRA treatment improved the left ventricular
function of mice after MI, as evidenced by increased fractional
shortening, ejection fraction, stroke work and cardiac output
compared with the untreated MI group (Fig. 1C and D).
CRA attenuates cardiac fibrosis
To explore whether CRA affects cardiac fibrosis
following MI, PSR staining (Fig.
2A) and Masson’s trichrome staining (Fig. S1) were performed. It was observed
that CRA decreased the interstitial fibrosis caused by MI.
Additionally, CRA increased the transcription of α-MHC, and
inhibited the transcription of hypertrophic markers (ANP, BNP, and
β-MHC) and fibrotic markers (CTGF, Collagen Iα, Collagen IIIα and
FN) at 4 weeks after MI, as compared with the untreated MI group
(Fig. 2B). These data indicated
that CRA treatment alleviated the cardiac remodelling following
MI.
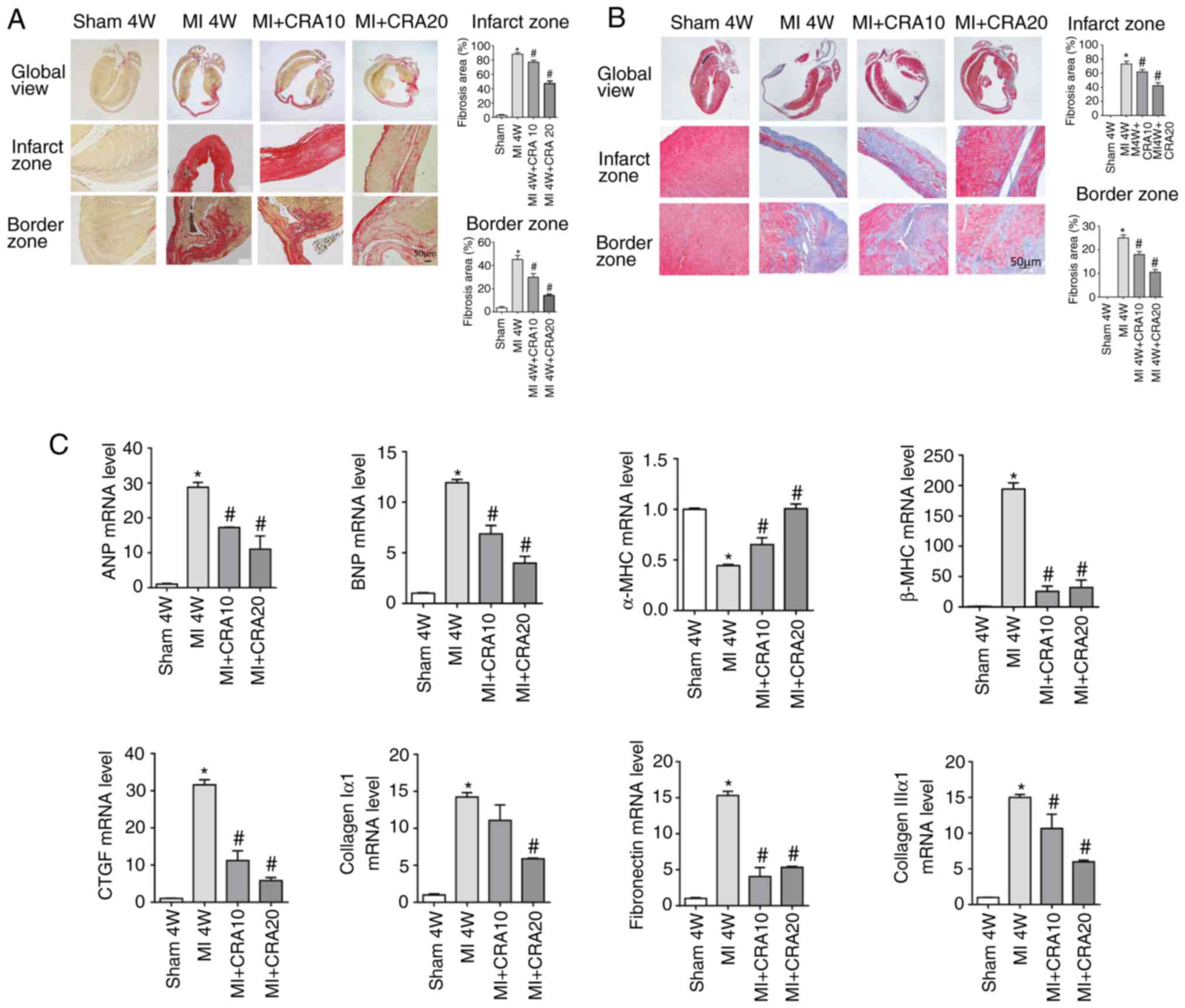 | Figure 2Effects of CRA on cardiac fibrosis
following MI. (A) Picrosirius red staining of histological sections
at 4 weeks after MI (magnification, ×100; n=5). (B) Masson’s’
trichrome staining of histological sections at 4 weeks after MI
(magnification: ×100) (n=5). (C) Reverse transcription-quantitative
polymerase chain reaction analyses of ANP, BNP, α-SMA, β-MHC, CTGF,
Collagen I α1, Collagen III α1 and fibronectin (n=6).
*P<0.05 vs. sham group; #P<0.05 vs. MI
group. CRA, corosolic acid; MI, myocardial infarction; ANP, atrial
natriuretic peptide; BNP, B-type natriuretic peptide; MHC, myosin
heavy chain; CTGF, connective tissue growth factor. |
CRA reverses MI-induced inactivation of
the AMPKα/Nrf2/HO-1 signalling pathway and attenuates oxidative
stress
Nox-mediated oxidative stress serves a significant
role in the progression of myocardial fibrosis (9,11).
In the present study, H9C2 cells were exposed to hypoxia (1%
O2) for 24 h with or without CRA treatment. Western blot
analysis revealed that Nox2, HO-1 and Nox4 were upregulated in
hypoxia-induced cardiomyocytes. However, CRA treatment inhibited
the expression levels of Nox2 and Nox4 proteins, while it increased
HO-1 expression in the hypoxia-induced cardiomyocytes (Fig. 3A). RT-qPCR data confirmed that CRA
pre-treatment led to reduced mRNA expression levels of Nox2 and
Nox4, and higher mRNA expression levels of HO-1 (Fig. 3B). Furthermore, as shown in
Fig. 3C, immunohistochemical
analysis indicated that CRA treatment evidently increased HO-1 and
decreased Nox4 expression following MI. Consistently, in
vivo studies indicated that CRA decreased the expression levels
of Nox2 and Nox4, and increased HO-1 expression in myocardial
tissue after 4 weeks of MI (Fig.
3D). In order to identify the underlying mechanism of the
effect of CRA, proteins associated with the signalling pathways
involved in oxidative stress were also detected in the myocardial
tissues. It was observed that MI-induced inactivation of
AMPKα/Nrf2/HO-1 signalling was reversed by CRA treatment (Fig. 3D).
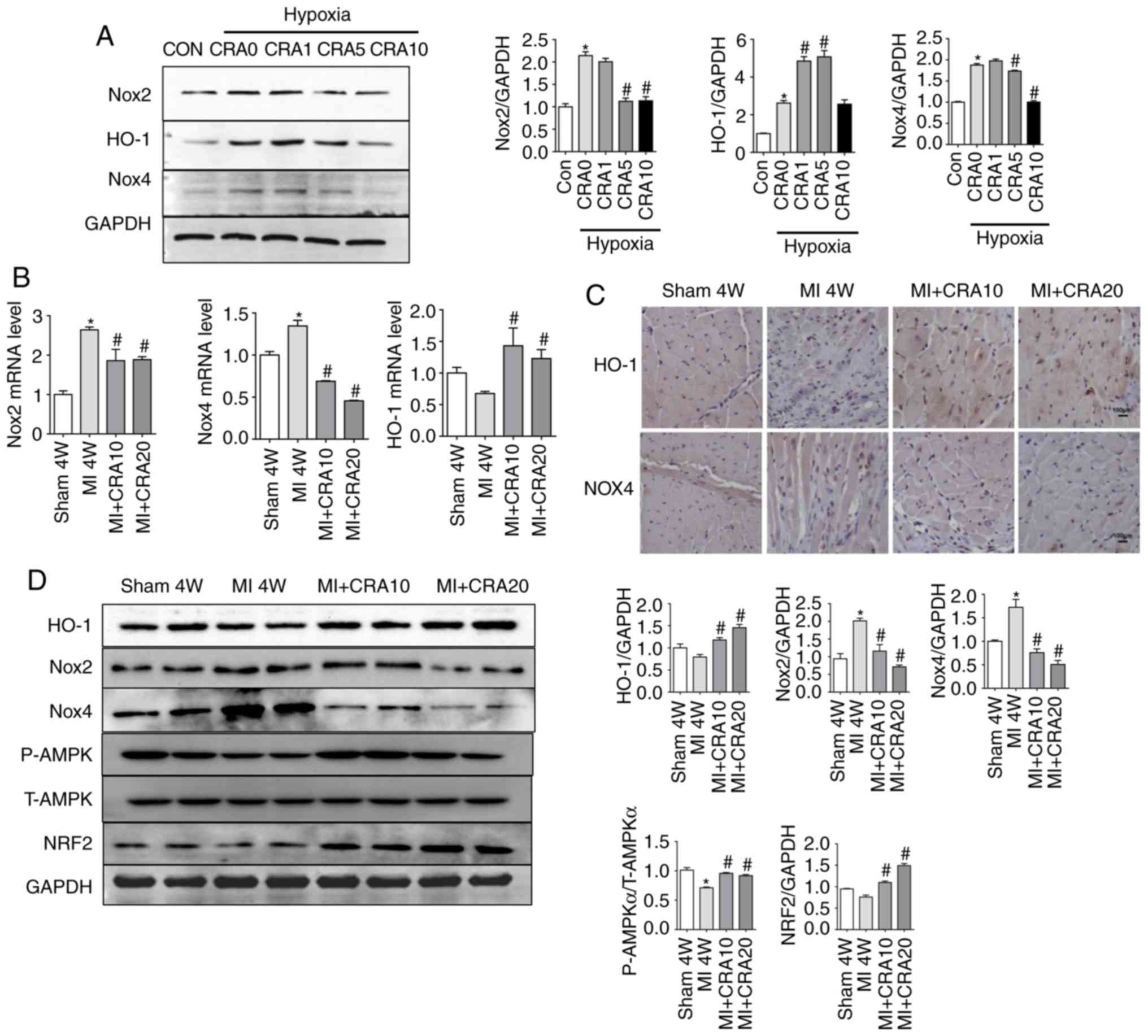 | Figure 3Effects of CRA on oxidative stress
following MI. (A) Western blot analysis showing the protein levels
of Nox2, HO-1, Nox4 and GAPDH in H9C2 cells treated with hypoxia in
the presence or absence of CRA (n=5). *P<0.05 vs. Con
group; #P<0.05 vs. hypoxia group. (B) Reverse
transcription-quantitative polymerase chain reaction analyses of
Nox2, Nox4 and HO-1 mRNA levels in the border zone of cardiac
tissues obtained from mice subjected to sham surgery or MI, with or
without CRA treatment (n=6). (C) HO-1 and Nox4 expression in the
border zone was also examined by immunohistochemistry. (D) Western
blot analysis indicating the protein levels of HO-1, Nox2, Nox4,
AMPKα, Nrf2 and GAPDH in cardiac tissue (n=5).
*P<0.05 vs. sham group; #P<0.05 vs. MI
group. CRA, corosolic acid; MI, myocardial infarction; Nox, NADPH
oxidase; HO-1, haem oxygenase 1; AMPKα, AMP-activated protein
kinase α; Nrf2, nuclear factor erythroid 2-related factor 2. |
CRA regulates the TGF-β1/Smad signalling
pathway and reduces the infiltration of macrophages in vivo
The TGF-β1/Smad signalling pathway is known to serve
a crucial role in the pathogenesis of numerous fibrotic diseases,
and thus the TGF-β1/Smad cascade activation was tested in the
current study. As shown in Fig.
4A, the MI-induced increase in the protein levels of TGF-β1,
P-Smad2 and P-Smad3 was attenuated in CRA-treated mice (Fig. 4A). Since macrophage-secreted TGF-β
is a major molecule involved in fibrosis after MI, the infiltration
of macrophages in the myocardial tissues of each group was then
examined. The Mcp-1 and CCR2 expression levels decreased in the
MI+CRA 10 and MI+CRA 20 groups, as compared with those in the
untreated MI group (Fig. 4B). In
addition, immunohistochemical staining for CD68 in the infarct and
border zones revealed decreased macrophage infiltration in
CRA-treated hearts post-MI as compared with that observed in the MI
alone group (Fig. 4C; IgG
negative control staining is shown in Fig. S2).
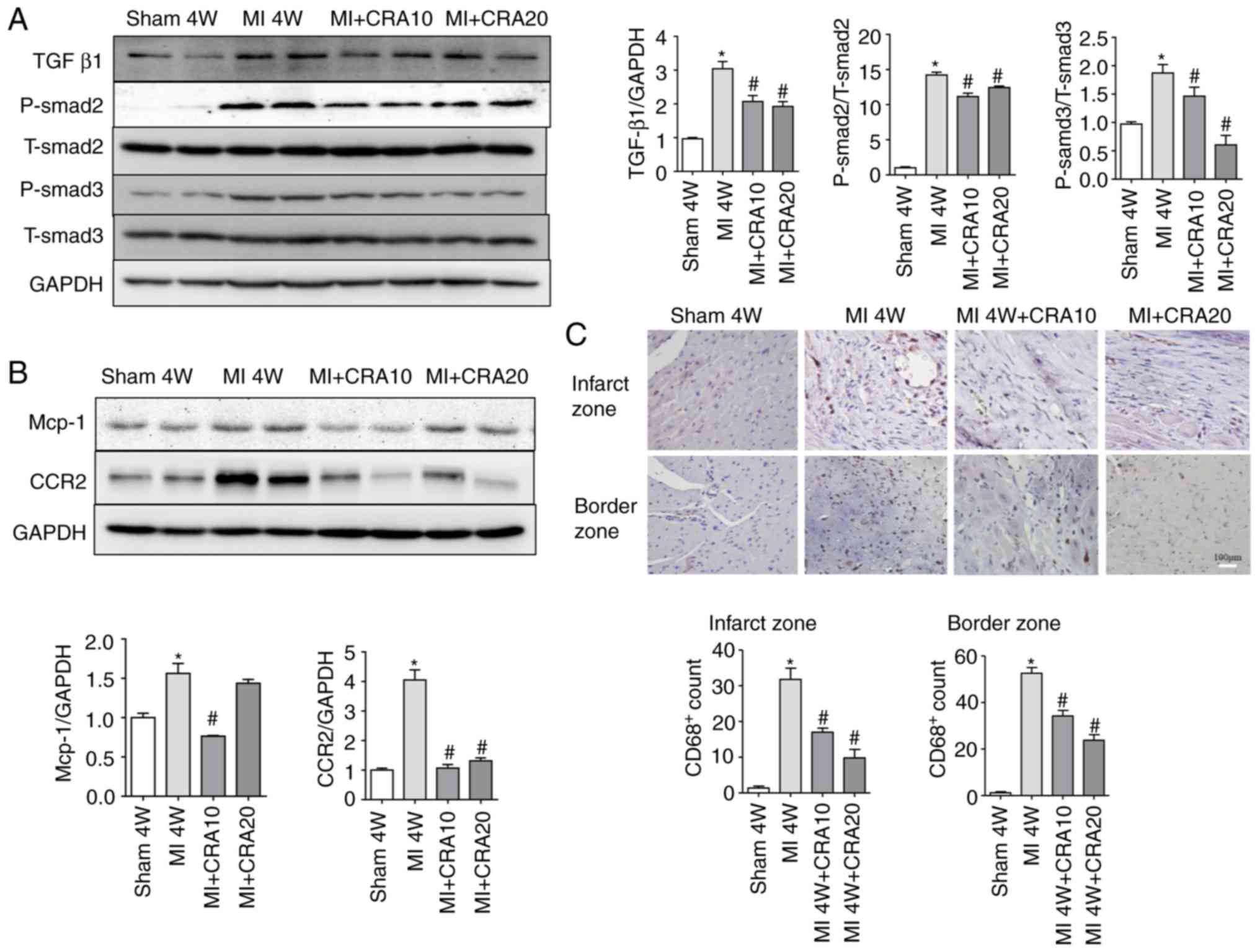 | Figure 4CRA regulates TGF-β1/Smads and
macrophage infiltration. (A) Western blot analysis of the protein
levels of TGF-β1/Smad signals in cardiac tissue, including TGF-β1,
P-Smad2, T-Smad2, P-Smad3, T-Smad3 and GAPDH (n=5). (B) Western
blot analysis of the protein levels of Mcp-1, CCR2 and GAPDH in
cardiac tissue (n=5). (C) CD68 protein in the infarct and border
zones of cardiac tissue was determined by immunohistochemistry
(magnification, ×200; n=6). *P<0.05 vs. sham group;
#P<0.05 vs. MI group. CRA, corosolic acid; MI,
myocardial infarction; TGF-β1, transforming growth factor β1;
Mcp-1, monocyte chemotactic protein 1; CCR2, C-C chemokine receptor
type 2. |
CRA inhibits inflammation and apoptosis
in myocardial tissues
The expression levels of several inflammatory
markers, including IL-1β, TNF-α and IL-6, in the myocardial tissues
were also detected. As indicated by the RT-qPCR results, CRA
decreased the mRNA expression levels of IL-1β, TNF-α and IL-6 in
the myocardium post-MI (Fig. 5A),
and repressed the expression levels of nuclear transcription factor
NF-κB p65 and p-Ikkβ (Fig. 5B).
TUNEL assay revealed that MI induced apoptosis, as indicated by the
large number of TUNEL-positive cells, while CRA reduced the number
of apoptotic cells (Fig. 5C).
Western blot analysis further demonstrated increased expression of
the pro-apoptotic protein Bax and decreased expression of the
anti-apoptotic protein Bcl-2 in the MI group, while CRA inhibited
these MI-induced changes (Fig.
5D).
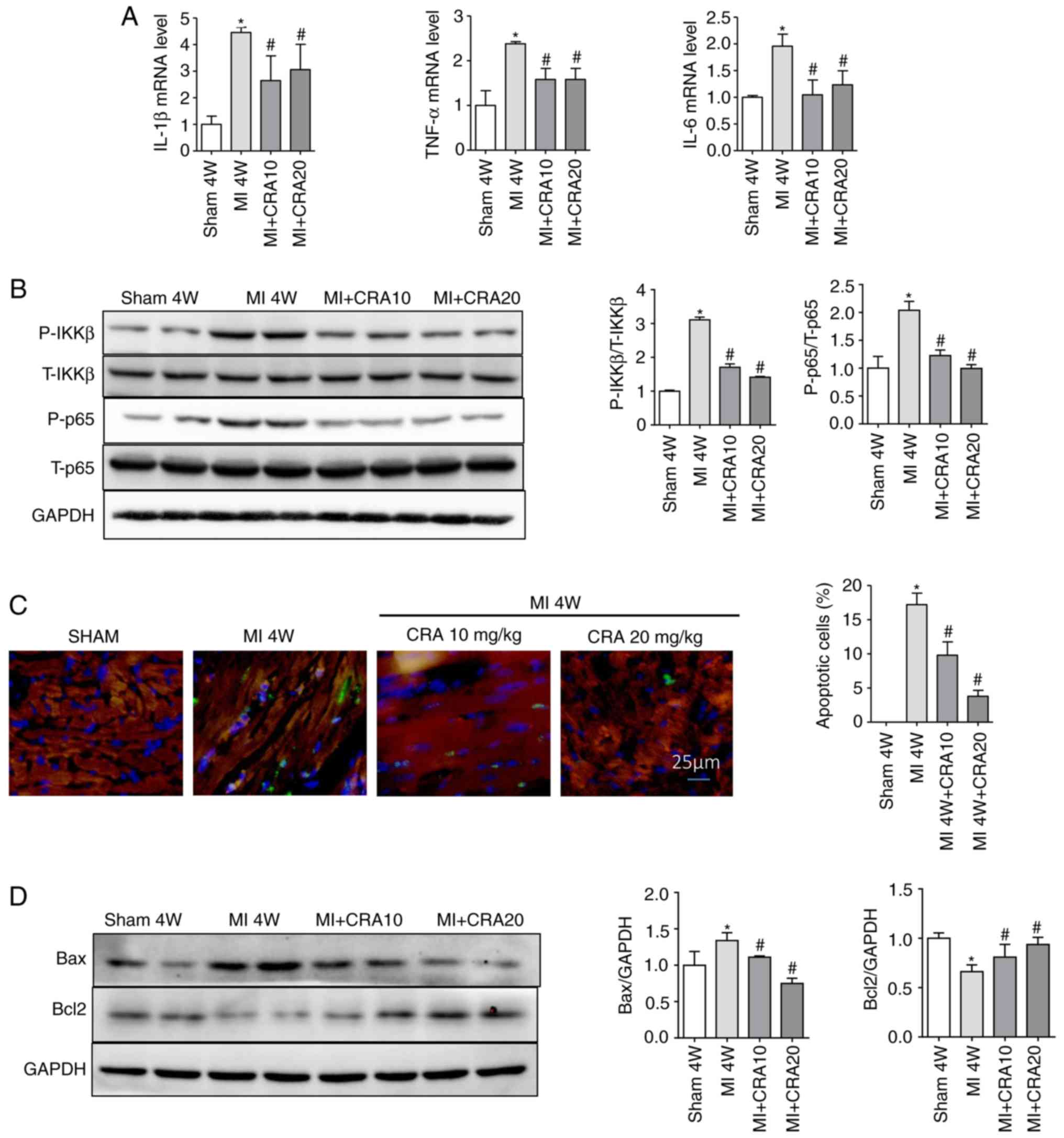 | Figure 5CRA regulates inflammation and
apoptosis in cardiac tissues. (A) Reverse
transcription-quantitative polymerase chain reaction analyses of
IL-1β, TNF-α and IL-6 in border zone of cardiac tissue (n=6). (B)
Western blot analysis of the protein levels of p-Ikkβ, T-Ikkβ,
p-P65 and T-P65 in border zone tissues (n=5). (C) Representative
TUNEL staining of border zone tissues (magnification, ×400; n=5).
(D) Western blot analysis of the protein levels of Bcl2, Bax and
GAPDH in cardiac tissue (n=5). *P<0.05 vs. sham
group; #P<0.05 vs. MI group. CRA, corosolic acid; MI,
myocardial infarction; IL, interleukin; TNF-α, tumour necrosis
factor α; Ikkβ, inhibitor of nuclear factor-κB kinase β; Bcl2,
B-cell lymphoma 2; Bax, Bcl2-associated X protein. |
Inhibition of AMPKα reverses the
protective effect of CRA in H9C2 cells
As detected by the DCFH-DA method, CRA treatment
partly blocked the hypoxia-induced ROS upregulation in H9C2 cells;
however, the antioxidant capacity of CRA was reversed by AMPKα
siRNA (Fig. 6A). In addition, the
protective effects of CRA against hypoxia-induced changes were
reversed by AMPKα siRNA, as indicated by the results of TUNEL
staining (Fig. 6B), and Nox2,
P-p65, Nrf2 and HO-1 protein expression levels (Fig. 6C). These results suggested that
AMPKα may mediate the protective effects of CRA following MI.
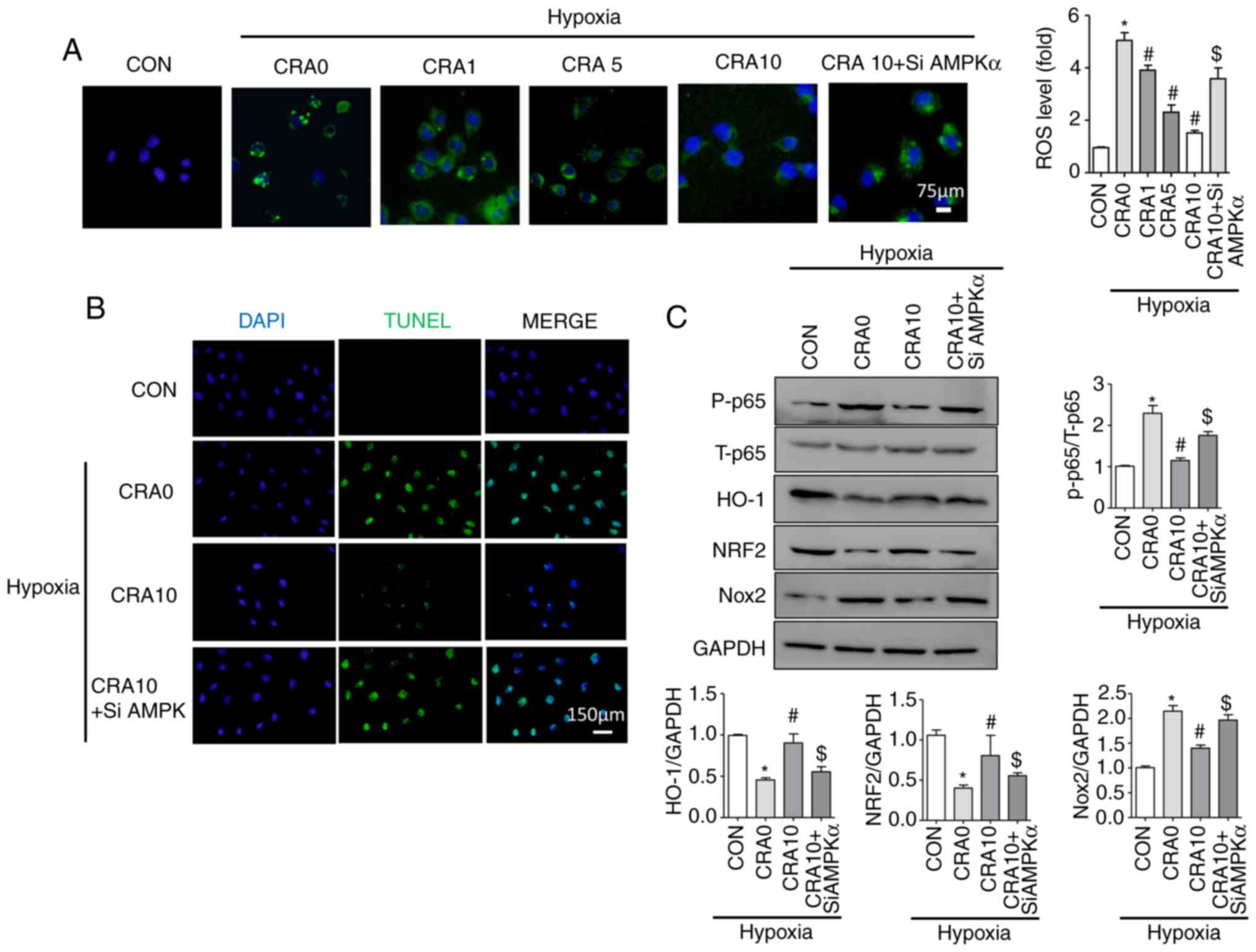 | Figure 6Inhibition of AMPKα reversed the
protective effect of CRA against hypoxia in H9C2 cells. (A) ROS
production was examined by DCFH-DA staining of H9C2 cells
(magnification, ×400; n=5). (B) Representative TUNEL staining in
H9C2 cells (magnification, ×200). (C) Western blot analysis of the
protein levels of P-p65, T-p65, HO-1, Nrf2, Nox2 and GAPDH in H9C2
cells (n=5). *P<0.05 vs. CON group;
#P<0.05 vs. CRA 0 group; $P<0.05 vs.
CRA 10 group. AMPKα, AMP-activated protein kinase α; CRA, corosolic
acid; ROS, reactive oxygen species; HO-1, haem oxygenase 1; Nox,
NADPH oxidase; Nrf2, nuclear factor erythroid 2-related factor 2;
CON, control; siAMPKα, AMPKα siRNA. |
Discussion
Fibrosis is the major cause of the deterioration of
cardiac function in patients who survive acute MI. Targeting
cardiac fibrosis may significantly delay the progression of heart
failure and improve the quality of life of patients. In the present
study, the following results were observed: i) CRA inhibited
cardiac fibrosis and improved left ventricular dysfunction
following MI; ii) CRA reduced the production of ROS, which was
associated with regulating the activity of the AMPKα/Nrf2/HO-1
signalling pathway and Noxs, particularly Nox2 and Nox4; iii) CRA
inhibited inflammation and apoptosis caused by MI; and iv) AMPKα
inhibition reversed the protective effect of CRA. Collectively,
these findings suggest that CRA attenuates MI-induced cardiac
fibrosis and dysfunction through modulation of inflammation and
oxidative stress associated with AMPKα.
The abnormal deposition of extracellular matrix
between non-ischaemic myocardial cells is the main mechanism of
fibrosis following MI and is associated with increased mortality
(23). It was confirmed that ROS
are involved in the synthesis and degradation of collagen (24,25), indicating that oxidative stress
serves an important role in synthesis of collagen. It has been
reported that Nox2 deficiency attenuates fibrosis and improves left
ventricular dysfunction following MI, while inhibition of Nox2 can
reduce oxidative stress and apoptosis in hypoxia-induced cells
(26). Nox4 is a major source of
superoxide (27), and mediates
mitochondrial dysfunction and fibronectin synthesis (28). HO-1 is a rate-limiting enzyme in
haem degradation and displays a strong protective effect on
oxidative damage induced by ROS. The increased expression of HO-1
and the production of bilirubin may regulate the production of
endogenous ROS in cells (29).
Previous data have also indicated that HO-1 improves mitochondrial
damage induced by hypoxia and inhibits the production of Noxs
(30). In addition, AMPKα, which
is closely associated with cell survival during myocardial
ischaemia (31), stimulates Nrf2
and its downstream antioxidant enzyme HO-1 to resist oxidative
stress (12). CRA may attenuate
cardiac fibrosis by regulating the AMPKα/Nrf2/HO-1/Nox signalling
pathway.
In addition to oxidative stress, it has been
demonstrated that macrophages are also involved in the process of
cardiac fibrosis during MI (32).
During the later inflammatory phase of infarct healing, the
activity of macrophages is an important cause of fibrosis, while
these cells are also an important source of TGF-β following MI
(33). In the current study, a
decrease in macrophage infiltration was observed in the CRA-treated
group, while western blot analysis revealed the downregulation of
CCR2 and Mcp-1 following CRA treatment, indicating that CRA
attenuated macrophage infiltration through the Mcp-1/CCR2 axis.
Previous studies have reported that ROS are associated with the
activation of macrophages by participating in the activation of the
Mcp-1/CCR2 signalling pathway (34–36). In addition, the current study
observed that a high dose of CRA was less effective in blocking
Mcp-1 expression, which does not appear to be consistent with the
immunohistochemistry results. In fact, there are numerous factors
affecting macrophage infiltration in addition to Mcp-1, which may
account for the discrepancy in the results. Although the blocking
effect of high-dose CRA on Mcp-1 was poor, a downward trend was
observed. Compared with western blot assay results, the findings of
immunohistochemistry may reflect the infiltration of macrophages
more intuitively and have more credibility. Therefore, it can be
deduced that, although high dose of CRA is less effective in
blocking Mcp-1 expression, it still has a strong inhibitory effect
on macrophages. Furthermore, previous studies have demonstrated
that the anti-inflammatory effect of AMPKα/Nrf2/HO-1 is due to the
anti-peroxidation effect and reduced ROS (20,37). Thus, it was hypothesise that the
anti-inflammatory ability of CRA may be due to decreased ROS,
although further research is needed to verify this hypothesis.
Increased expression of inflammatory cytokines is
also an important factor causing apoptosis (38). Apoptosis caused by myocardial
ischaemia is one of the reasons for fibrosis and affected cardiac
function following MI. Bax and Bcl2 are important regulatory
molecules of apoptosis that belong to the Bcl2 family. Bax is
located in the cytoplasm and moves to the mitochondria under the
stimulation of apoptosis signals, damaging the permeability of the
mitochondrial membrane and promoting apoptosis. Furthermore, Bax
inhibits the activity of the anti-apoptotic protein Bcl2. In heart
failure, the expression of the pro-apoptotic protein Bax in
patients is increased (39),
while the activity of Bcl2 is decreased; therefore, enhancing the
expression of Bcl2 can effectively reduce the occurrence of
apoptosis. In the present study, CRA decreased the expression
levels of pro-apoptotic protein Bax and increased the activity of
anti-apoptotic protein Bcl2 both in vivo and in
vitro, confirming the function of CRA in inhibiting apoptosis
of cardiomyocytes.
A previous study revealed that, after 4 weeks of MI,
abnormal remodelling occurred in the infarction border zone,
accompanied by oxidative stress, inflammation and apoptosis,
suggesting that anti-oxidation, anti-apoptotic and
anti-inflammatory therapy is an important measure to reduce
abnormal remodelling of the border zone (1). The current study attempted to
investigate the effect of CRA on ventricular remodelling and heart
failure following infarction, and thus the time point of 4 weeks
after infarction was selected for investigation. Studies have
confirmed that men and women have different risk of cardiovascular
disease, which may be associated with metabolism and inflammation
(40). To eliminate gender
interference in the results, mice of the same sex were selected.
However, certain limitations exist in the present study. Firstly,
CRA was administered 2 weeks before MI. A previous review on CRA
safety reported that using a gel product containing 10 mg CRA can
improve the symptoms of patients without causing any adverse
effects (41). The current study
explored whether CRA can be used as an adjunct drug to improve
heart failure in patients with MI or high-risk groups; therefore,
prophylactic administration of CRA was provided to the mice. Future
studies should verify the effects of CRA on MI when it is only
administered to animals following the induction of MI. In addition,
the distribution of CRA in the blood following intragastric
administration and the proper dosage should be determined in
further studies. Finally, whether CRA influences other cell types
and signalling pathways in the process of MI requires further
investigation.
In conclusion, the data of the present study
revealed that CRA attenuated MI-induced cardiac fibrosis and
dysfunction through modulation of inflammation, apoptosis and
oxidative stress associated with AMPKα. It is, thus, proposed that
CRA may be a suitable adjuvant therapy for the treatment of MI and
heart failure in clinical practice.
Supplementary Information
Acknowledgements
The authors would like to thank Professor Tang
Qizhu, Renmin Hospital of Wuhan University, for the help and
support provided.
Funding
This research was supported by the National Natural
Science Foundation of China (grant nos. 81530012 and 81700218),
National Key R&D Programme of China (grant no. 2018YFC1311300)
and the National Natural Science Foundation of Hubei Province
(grant no. 2017CFB320).
Availability of data and materials
The datasets used during the current study are
available from the corresponding author upon reasonable
request.
Authors’ contributions
ZPW and YY designed this study. HZ and YYM performed
the data collection. QQW performed the data analysis. YC and YGJ
performed the animal experiments. YY and HMW supervised the project
and controlled the administration. SSW and HMW performed cell
culture; YC wrote the original draft of the manuscript. ZPW and YY
reviewed the article. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experimental protocols were approved by
the Animal Care and Use Committee of Renmin Hospital of Wuhan
University and were conducted in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang L, Gregorich ZR, Cai W, Zhang P,
Young B, Gu Y, Zhang J and Ge Y: Quantitative proteomics and
immunohistochemistry reveal insights into cellular and molecular
processes in the infarct border zone one month after myocardial
infarction. J Proteome Res. 16:2101–2112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Saxena A, Li N, Sun J, Gupta A,
Lee DW, Tian Q, Dobaczewski M and Frangogiannis NG: Endogenous
IRAK-M attenuates postinfarction remodeling through effects on
macrophages and fibroblasts. Arterioscler Thromb Vasc Biol.
32:2598–2608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu X, Wan N, Zhang XJ, Zhao Y, Zhang Y,
Hu G, Wan F, Zhang R, Zhu X, Xia H and Li H: Vinexin-β exacerbates
cardiac dysfunction post-myocardial infarction via mediating
apoptotic and inflammatory responses. Clin Sci (Lond). 128:923–936.
2015. View Article : Google Scholar
|
|
4
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar
|
|
5
|
Neri M, Fineschi V, Di Paolo M, Pomara C,
Riezzo I, Turillazzi E and Cerretani D: Cardiac oxidative stress
and inflammatory cytokines response after myocardial infarction.
Curr Vasc Pharmacol. 13:26–36. 2015. View Article : Google Scholar
|
|
6
|
Lambeth JD: NOX enzymes and the biology of
reactive oxygen. Nat Rev Immunol. 4:181–9. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson AJ, Gill EK, Abudalo RA, Edgar KS,
Watson CJ and Grieve DJ: Reactive oxygen species signalling in the
diabetic heart: Emerging prospect for therapeutic targeting. Heart.
104:293–299. 2018. View Article : Google Scholar
|
|
8
|
Byrne JA, Grieve DJ, Bendall JK, Li JM,
Gove C, Lambeth JD, Cave AC and Shah AM: Contrasting roles of NADPH
oxidase isoforms in pressure-overload versus angiotensin II-induced
cardiac hypertrophy. Circ Res. 93:802–805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Doerries C, Grote K, Hilfiker-Kleiner D,
Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C,
Schieffer B, Drexler H and Landmesser U: Critical role of the
NAD(P)H oxidase subunit p47phox for left ventricular
remodeling/dysfunction and survival after myocardial infarction.
Circ Res. 100:894–903. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Somanna NK, Valente AJ, Krenz M, Fay WP,
Delafontaine P and Chandrasekar B: The Nox1/4 dual inhibitor
GKT137831 or Nox4 knockdown inhibits angiotensin-II-induced adult
mouse cardiac fibroblast proliferation and migration. AT1
physically associates with Nox4. J Cell Physiol. 231:1130–1141.
2016. View Article : Google Scholar
|
|
11
|
Qi Z, Yin F, Lu L, Shen L, Qi S, Lan L,
Luo L and Yin Z: Baicalein reduces lipopolysaccharide-induced
inflammation via suppressing JAK/STATs activation and ROS
production. Inflamm Res. 62:845–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao C, Zhang Y, Liu H, Li P, Zhang H and
Cheng G: Fortunellin protects against high fructose-induced
diabetic heart injury in mice by suppressing inflammation and
oxidative stress via AMPK/Nrf-2 pathway regulation. Biochem Biophys
Res Commun. 490:552–559. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chi PL, Liu CJ, Lee IT, Chen YW, Hsiao LD
and Yang CM: HO-1 induction by CO-RM2 attenuates TNF-α-induced
cytosolic phospholipase A2 expression via inhibition of
PKCα-dependent NADPH oxidase/ROS and NF-kappaB. Mediators Inflamm.
2014:279171. 2014. View Article : Google Scholar
|
|
14
|
Miura T, Takagi S and Ishida T: Management
of diabetes and its complications with banaba (Lagerstroemia
speciosa L.) and corosolic acid. Evid Based Complement Alternat
Med. 2012:871495. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JH, Kim YH, Song GY, Kim DE, Jeong YJ,
Liu KH, Chung YH and Oh S: Ursolic acid and its natural derivative
corosolic acid suppress the proliferation of APC-mutated colon
cancer cells through promotion of beta-catenin degradation. Food
Chem Toxicol. 67:87–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen H, Yang J, Zhang Q, Chen LH and Wang
Q: Corosolic acid ameliorates atherosclerosis in apolipoprotein
E-deficient mice by regulating the nuclear factor-κB signaling
pathway and inhibiting monocyte chemoattractant protein-1
expression. Circ J. 76:995–1003. 2012. View Article : Google Scholar
|
|
17
|
Kim SJ, Cha JY, Kang HS, Lee JH, Lee JY,
Park JH, Bae JH, Song DK and Im SS: Corosolic acid ameliorates
acute inflammation through inhibition of IRAK-1 phosphorylation in
macrophages. BMB Rep. 49:276–281. 2016. View Article : Google Scholar :
|
|
18
|
Yamaguchi Y, Yamada K, Yoshikawa N,
Nakamura K, Haginaka J and Kunitomo M: Corosolic acid prevents
oxidative stress, inflammation and hypertension in SHR/NDmcr-cp
rats, a model of metabolic syndrome. Life Sci. 79:2474–2479. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takagi S, Miura T, Ishibashi C, Kawata T,
Ishihara E, Gu Y and Ishida T: Effect of corosolic acid on the
hydrolysis of disaccharides. J Nutr Sci Vitaminol (Tokyo).
54:266–8. 2008. View Article : Google Scholar
|
|
20
|
Yang J, Leng J, Li JJ, Tang JF, Li Y, Liu
BL and Wen XD: Corosolic acid inhibits adipose tissue inflammation
and ameliorates insulin resistance via AMPK activation in high-fat
fed mice. Phytomedicine. 23:181–190. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leach JP, Heallen T, Zhang M, Rahmani M,
Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF: Hippo
pathway deficiency reverses systolic heart failure after
infarction. Nature. 550:260–264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fellahi S, El Harrak M, Kuhn JH, Sebbar G,
Bouaiti el A, Khataby K, Fihri OF, El Houadfi M and Ennaji MM:
Comparison of SYBR green I real-time RT-PCR with conventional
agarose gel-based RT-PCR for the diagnosis of infectious bronchitis
virus infection in chickens in Morocco. BMC Res Notes. 9:2312016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dobaczewski M, Gonzalez-Quesada C and
Frangogiannis NG: The extracellular matrix as a modulator of the
inflammatory and reparative response following myocardial
infarction. J Mol Cell Cardiol. 48:504–511. 2010. View Article : Google Scholar :
|
|
24
|
Siwik DA, Pagano PJ and Colucci WS:
Oxidative stress regulates collagen synthesis and matrix
metalloproteinase activity in cardiac fibroblasts. Am J Physiol
Cell Physiol. 280:C53–C60. 2001. View Article : Google Scholar
|
|
25
|
Kunkemoeller B and Kyriakides TR: Redox
signaling in diabetic wound healing regulates extracellular matrix
deposition. Antioxid Redox Signal. 27:823–838. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sirker A, Murdoch CE, Protti A, Sawyer GJ,
Santos CX, Martin D, Zhang X, Brewer AC, Zhang M and Shah AM:
Cell-specific effects of Nox2 on the acute and chronic response to
myocardial infarction. J Mol Cell Cardiol. 98:11–17. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuroda J, Ago T, Nishimura A, Nakamura K,
Matsuo R, Wakisaka Y, Kamouchi M and Kitazono T: Nox4 is a major
source of superoxide production in human brain pericytes. J Vasc
Res. 51:429–438. 2014. View Article : Google Scholar
|
|
28
|
Ago T, Kuroda J, Pain J, Fu C, Li H and
Sadoshima J: Upregulation of Nox4 by hypertrophic stimuli promotes
apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ
Res. 106:1253–1264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cao J, Tsenovoy PL, Thompson EA, Falck JR,
Touchon R, Sodhi K, Rezzani R, Shapiro JI and Abraham NG: Agonists
of epoxyeicosatrienoic acids reduce infarct size and ameliorate
cardiac dysfunction via activation of HO-1 and Wnt1 canonical
pathway. Prostaglandins Other Lipid Mediat. 116–117:76–86. 2015.
View Article : Google Scholar
|
|
30
|
Datla SR, Dusting GJ, Mori TA, Taylor CJ,
Croft KD and Jiang F: Induction of heme oxygenase-1 in vivo
suppresses NADPH oxidase derived oxidative stress. Hypertension.
50:636–642. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qi D and Young LH: AMPK: Energy sensor and
survival mechanism in the ischemic heart. Trends Endocrinol Metab.
26:422–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yan X, Zhang H, Fan Q, Hu J, Tao R, Chen
Q, Iwakura Y, Shen W, Lu L, Zhang Q and Zhang R: Dectin-2
deficiency modulates Th1 differentiation and improves wound healing
after myocardial infarction. Circ Res. 120:1116–1129. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Prabhu SD and Frangogiannis NG: The
biological basis for cardiac repair after myocardial infarction:
From inflammation to fibrosis. Circ Res. 119:91–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ullevig S, Zhao Q, Lee CF, Seok Kim H,
Zamora D and Asmis R: NADPH oxidase 4 mediates monocyte priming and
accelerated chemotaxis induced by metabolic stress. Arterioscler
Thromb Vasc Biol. 32:415–426. 2012. View Article : Google Scholar :
|
|
35
|
Kim MJ, Kadayat T, Um YJ, Jeong TC, Lee ES
and Park PH: Inhibitory effect of
3-(4-Hydroxyphenyl)-1-(thiophen-2-yl) prop-2-en-1-one, a chalcone
derivative on MCP-1 expression in macrophages via inhibition of ROS
and Akt signaling. Biomol Ther (Seoul). 23:119–127. 2015.
View Article : Google Scholar
|
|
36
|
Park SY, Jin ML, Yi EH, Kim Y and Park G:
Neochlorogenic acid inhibits against LPS-activated inflammatory
responses through up-regulation of Nrf2/HO-1 and involving AMPK
pathway. Environ Toxicol Pharmacol. 62:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao HH, Zhu JX, Feng H, Ni J, Zhang N,
Chen S, Liu HJ, Yang Z, Deng W and Tang QZ: Myricetin possesses
potential protective effects on diabetic cardiomyopathy through
inhibiting IκBα/NFκB and enhancing Nrf2/HO-1. Oxid Med Cell Longev.
2017:8370593. 2017. View Article : Google Scholar
|
|
38
|
Li M, Ye J, Zhao G, Hong G, Hu X, Cao K,
Wu Y and Lu Z: Gas6 attenuates lipopolysaccharideinduced TNFα
expression and apoptosis in H9C2 cells through NFkappaB and MAPK
inhibition via the Axl/PI3K/Akt pathway. Int J Mol Med. 44:982–994.
2019.PubMed/NCBI
|
|
39
|
Liu W, Ru L, Su C, Qi S and Qi X: Serum
levels of inflammatory cytokines and expression of BCL2 and BAX
mRNA in peripheral blood mononuclear cells and in patients with
chronic heart failure. Med Sci Monit. 25:2633–2639. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Henstridge DC, Abildgaard J, Lindegaard B
and Febbraio MA: Metabolic control and sex: A focus on
inflammatory-linked mediators. Br J Pharmacol. 176:4193–4207. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stohs SJ, Miller H and Kaats GR: A review
of the efficacy and safety of banaba (Lagerstroemia speciosa L.)
and corosolic acid. Phytother Res. 26:317–324. 2012.
|