Introduction
Cyclin-dependent kinases (CDKs) are a group of
serine/threonine protein kinases family members whose main
biological functions are to regulate the cell cycle and
transcription (1,2). CDK5, as an atypical member of the
CDKs family, is orchestrated by p35/p39 (3) and hence does not directly regulate
the cell cycle, but plays an essential role in neurodevelopment and
neurotransmission. In addition to its role in the nervous system,
CDK5 also participates in numerous tumorigenic or non-tumorigenic
pathophysiological processes, including mediating cell migration,
apoptosis, cell cycle processes, as well as regulating the immune
system, angiogenesis, insulin secretion and tumorigenesis (4).
Previous studies showed that aberrant expression of
CDK5 had been observed in multiple human cancers, but not in normal
tissues. CDK5 were positively correlated with lymph node metastasis
and the stage of disease in head and neck squamous cell carcinoma,
cervical lesions, lung cancer, hepatocellular carcinoma and
nasopharyngeal carcinoma (5-10),
and its overexpression was also associated with the poor prognosis
in colorectal cancer and breast cancer (7,8).
Tumor tissues with high expression of CDK5 are usually insensitive
to chemoradiotherapy and targeted therapy (11,12). Therefore, targeting CDK5 may
circumvent the drawbacks of the current therapies (13,14).
Multiple myeloma (MM) is an incurable plasma cell
malignancy in bone marrow. The emergence of proteasome inhibitor
bortezomib is a milestone in the history of MM therapy. However,
most patients will eventually relapse, which can be partly put down
to resistance to bortezomib. Therefore, the exploration of new
drugs which can overcome the barrier of bortezomib resistance is
one of the main focuses of MM treatment. At present, the studies on
the relationship between CDK5 and cancer are mostly limited to
solid tumors. A small quantity of studies have shown that high
expression of CDK5 is associated with poor prognosis and bortezomib
resistance in MM patients, so further exploration of the specific
mechanism is still needed (15-17).
Dinaciclib is a specific inhibitor of CDK1/2/5/9.
Pre-clinical and early clinical studies have confirmed that
dinaciclib is a promising anti-myeloma drug (18-20). The purpose of this study is to
further explore the influence of CDK5 expression on prognosis,
bortezomib response in MM patients and to uncover the deep
synergistic anti-myeloma mechanism of dinaciclib and bortezomib.
These studies will lay a solid theoretical foundation for
dinaciclib in clinical treatment of MM.
Materials and methods
Cell culture and reagents
The isolation of primary MM cells from patients and
bone marrow mononuclear cells (BMMCs) from healthy donors was
carried out according to the authors' previous study (21). Human MM cell lines (RPMI8226,
H929, MM.1S and U226), primary MM cells from patients and BMMCs
from healthy donors were cultured in RPMI-1640 medium (Hyclone; GE
Healthcare) containing 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 units/ml penicillin, 100 mg/ml streptomycin,
2 mM L-glutamine and 5% CO2 at 37°C. Informed consent
was obtained from all patients, in accordance with the Helsinki
Protocol. These studies have been approved by the Ethics Committee
of Xijing Hospital. Dinaciclib was provided by Selleck Chemicals.
Bortezomib was obtained from Selleck Chemicals.
Western blotting
Cells were lysed with RIPA buffer (Beyotime
Institute of Biotechnology) and the concentration of protein was
assessed using a bicinchoninic acid assay (Beyotime Institute of
Biotechnology). A total of 20 µg supernatants were subjected
to 10 or 12% sodium dodecyl sulfate polyacrylamide gel
electrophoresis and transferred to PVDF followed by blocking in TBS
containing 5% non-fat milk for 1 h at room temperature.
Immunoblotting was performed overnight at 4°C using antibodies
against CDK5 (1:10,000; cat. no. ab40773; Abcam), CDK1 (1:10,000;
cat. no. ab133327; Abcam), CDK2 (1:5,000; cat. no. ab32147: Abcam),
CDK9 (1:5,000; ab76320, Abcam), CDC25c (1:2,000; cat. no. ab32444;
Abcam), cyclin B1 (1:50,000; cat. no. ab32053; Abcam), cyclin D1
(1:50,000; cat. no. ab134175; Abcam), cyclin E1 (1:2,000; cat. no.
ab133266; Abcam), p21 (1:5,000; cat. no. ab109520; Abcam), p16
(1:2,000; cat. no. ab108349; Abcam), p65 (1:5,000; cat. no.
ab32536; Abcam), phosphorylated- IκB kinase (p-) IKKα (1:500; cat.
no. ab38515; Abcam), IKKα (1:10,000; cat. no. ab32041; Abcam),
p-IκBα (1:5,000; cat. no. ab133462; Abcam), IκBα (1:5,000; cat. no.
ab32518; Abcam), Bcl-2 (1:500; cat. no. ab32124; Abcam), Bcl-xL
(1:500; cat. no. ab32370: Abcam), Lamin B (1:5,000; cat. no.
ab133741; Abcam), β-actin (1:2,000; cat. no. ab8226; Abcam), GAPDH
(1:5,000; cat. no. ab9484; Abcam) and cleaved poly (ADP-ribose)
polymerase (PARP; 1:2,000; cat. no. 5625; Cell Signaling
Technology, Inc.), cleaved caspase 8 (1:1,000; cat. no. 8592; Cell
Signaling Technology, Inc.), cleaved caspase 9 (1:500; cat. no.
20750; Cell Signaling Technology, Inc.), cleaved caspase 3 (1:500;
cat. no. 9661; Cell Signaling Technology, Inc.) and CDC2 (1:1,000;
cat. no. 9116; Cell Signaling Technology, Inc.). Then blots were
incubated with goat anti-rabbit horse-radish peroxidase-conjugated
secondary antibodies (1:4,000; cat. no. ab7090; Abcam) at room
temperature for 1 h. were developed by chemiluminescence using
SuperSignal reagent (EMD Millipore). To prepare nuclear and
cytosolic protein extracts, the harvested cells were resuspended in
cytosolic protein lysis buffer (Beyotime Institute of
Biotechnology) on ice for 15 min. After being centrifuged for 5 min
at 16,000 × g at 4°C, the supernatant was collected as cytosolic
protein extracts. Then the cell nuclear pellets were resuspended in
nuclear protein extraction buffer on ice for 40 min and centrifuged
for 5 min at 16,000 × g at 4°C. The supernatant was collected as
nuclear protein extracts.
Immunohistochemistry (IHC)
Bone marrow biopsies of 65 MM patients treated with
bortezomib and 10 healthy donors were obtained in Xijing Hospital
(Fourth Military Medical University) from January 2012 to January
2019 All patients and donors signed informed consent. The clinical
data, cytogenetic abnormalities and immunophenotypes of all
patients were collected from the medical records system in our
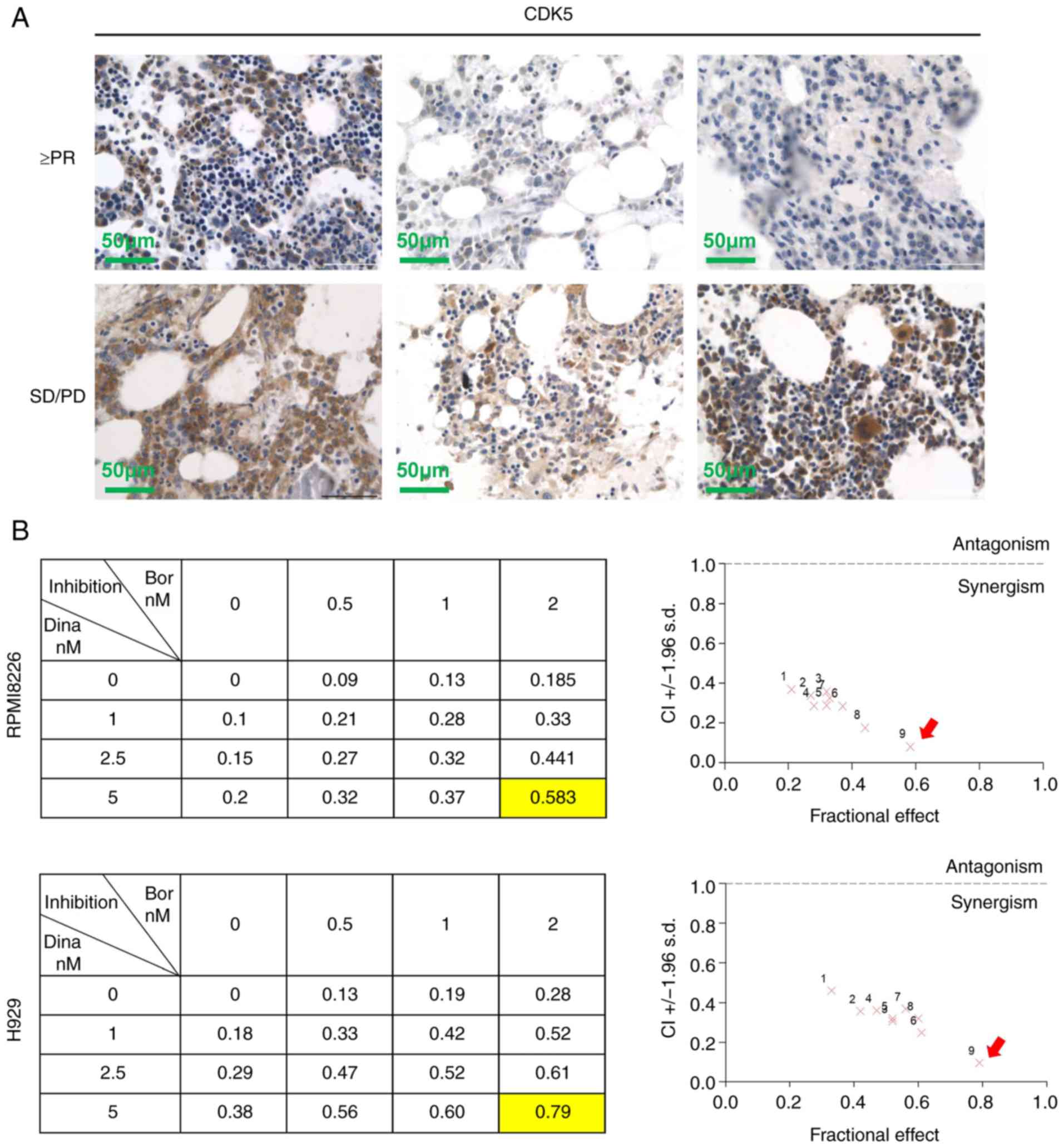
hospital. Patient characteristics are shown in Table I. The IHC staining of CDK5 and
scoring were carried out as previously described (6). The cutoff value of CDK5 was selected
according to the analysis of overall survival (OS). The score of
>4 was defined as high expression of CDK5 and <4 as low
expression of CDK5.
 | Table ICDK5 expression associated with the
multiple clinicopathological factors in MM |
Table I
CDK5 expression associated with the
multiple clinicopathological factors in MM
| MM | n CD | K5+
(n) | CDK5−
(n) | P-value |
|---|
| Bor response | | | | 0.020 |
| ≥PR | 51 | 20 | 31 | |
| SD/PD | 14 | 11 | 3 | |
| Sex | | | | 0.896 |
| Male | 33 | 16 | 17 | |
| Female | 32 | 15 | 17 | |
| Age (years) | | | | 0.870 |
| ≥65 | 11 | 5 | 6 | |
| <65 | 54 | 26 | 28 | |
| D-S | | | | 0.026 |
| I/II | 19 | 5 | 14 | |
| III | 46 | 26 | 20 | |
| ISS | | | | 0.291 |
| I | 13 | 4 | 9 | |
| II/III | 52 | 27 | 25 | |
| M protein | | | | 0.628 |
| IgG | 31 | 15 | 16 | |
| non-IgG | 18 | 10 | 8 | |
| M protein | | | | 0.063 |
| κ | 32 | 19 | 13 | |
| λ | 33 | 12 | 21 | |
| Plasma cells | | | | 0.188 |
| ≥50% | 24 | 14 | 10 | |
| <50% | 41 | 17 | 24 | |
| HGB (g/l) | | | | 0.135 |
| ≥100 | 25 | 9 | 16 | |
| <100 | 40 | 22 | 18 | |
| Ca2+
(mmol/l) | | | | 0.927 |
| ≥2.65 | 15 | 7 | 8 | |
| <2.65 | 50 | 24 | 26 | |
| Cr
(µmol/l) | | | | 0.614 |
| ≥177 | 17 | 9 | 8 | |
| <177 | 48 | 22 | 26 | |
Transient transfection
RPMI 8226 cells were transiently transfected with
genome scramble-siRNA or siRNA against CDK5 (Smart pool siRNA; GE
Healthcare Dharmacon, Inc.) using Nucleofector kit V in accordance
with the manufacturer's protocol (Amaxa Biosystems). CDK5-siRNA was
designed with Block-iT RNAi Designer (Invitrogen; Thermo Fisher
Scientific, Inc,) and synthesized (Wuhan GeneCreate Biological
Engineering Co., Ltd.). The sequences of CDK5-siRNA were 5′-CCT TAT
AGT CTG GCA GCT T-3′ and the scramble shRNA sequences were 5′-CCT
ATT GTC GGA CGT ACT T-3′. After 48 h of transfection with 100 nM
siRNA (shRNA 10 pmol/sample), the cells were collected for CDK5
expression and cell viability detection.
Cell viability assay
MM cell lines, primary MM cells and normal BMMCs
were inoculated into 96-well plates (1×104 cells/well)
and treated with different concentrations of dinaciclib according
to indicated experimental design. After incubation with 10
µl 5 mg/ml MTT for 4 h, 100 µl DMSO was added after
the supernatant was removed by centrifugation at a speed of 114 × g
for 15 min at room temperature and the absorbance value was
measured at 570 nM using a spectrophotometer.
Cell cycle analysis
MM cells were seeded in a 6-well plate
(10×104 cells/well) and incubated with various doses of
dinaciclib for 24 h, washed twice with PBS and fixed with 70% ice
ethanol at −20°C for 24 h. Cells were washed twice with PBS and
incubated with 1 mg/ml RNase A at 37°C for 20 min. Finally, PI (50
µg/ml; Sigma-Aldrich; Merck KGaA) was added to incubate at
room temperature for 20 min and then Epics XL flow cytometry
(Beckman Coulter, Inc.) was used to detect the cell cycle
distribution. At least 10,000 events per group were measured within
an acquisition rate of 100-300 events/sec. Data were analyzed by
FlowJo. 7.6.1 software (Flowjo, LLC).
Apoptosis assay
MM cells were seeded in a 6-well plate
(10×104 cells/well) and incubated with various doses of
dinaciclib for 24 h. Cell apoptosis was detected with Annexin V-PI
Staining kit (BD Biosciences; Becton, Dickinson and Company) in
accordance with the manufacturer's protocol.
Myeloma xenograft mouse model
A total of 20 male Balb/C athymic nude (weight, 15
g; age, 4-6 weeks) were obtained from the Core Animal Facility of
Fourth Military Medical University and were maintained under
pathogen-free conditions (25°C; 50% humidity; 12 h light/dark
cycle) and access to food and water. RPMI8226 cells
(1×107) were suspended in 80 µl serum-free
RPMI1640 medium and mixed with 70 µl matrix glue (BD
Biosciences; Becton, Dickinson and Company). A total of 150
µl suspension was subcutaneously injected into the right
lower limb of mice and the tumor formation rate was 30% (6/20).
When the tumors were measurable (100-150 mm3) after ~two
weeks, mice were randomly divided into two groups (3 for each
group): Control group (equal volume of vehicle) and dinaciclib
group (intraperitoneal injection, 45 mg/kg, 4 days a week)
(19). The size of the tumors and
the weight of mice were measured every three days with vernier
calipers. All the six mice were euthanized by cervical dislocation
~6 weeks after the cell injection and tumor xeno-grafts were
carefully excised for further use after the heartbeat and
respiratory arrest of the mice. The volume was calculated by the
following formula: V = 0.5xaxb2, a and b were the length
and width of the tumors respectively. Apoptosis of tumors excised
from mice was detected by TUNEL staining and hematoxylin and eosin
staining (3 min in the two dyes respectively at room temperature).
The expression of CDK5 was detected by immunohistochemistry. All
the animal studies performed were approved by the Animal Ethics
Committee of Fourth Military Medical University (Xi'an, China).
TUNEL staining
The in situ cell death detection kit, POD
(cat. no. 11684817910; Roche Diagnostics GmbH), was used for TUNEL
staining in accordance with the manufacturer's protocol.
paraffin-embedded specimens were de-paraffinized using
dimethylbenzene twice, rehydrated with a gradient elution of
alcohol (100, 95, 90, 80 and 70%) and permeabilized using
proteinase K at 37°C for 10 min. Sections were subsequently treated
with 50 µl TUNEL reaction mixture for 60 min in a humidified
container at 37°C in the dark. Sections were then incubated with 50
µl converter-POD at 37°C in the dark for 30 min. A total of
50 µl DAB was added to the sections for 10 min at room
temperature followed by hematoxylin staining for 3 sec at room
temperature. Apoptotic cells were viewed using a light microscope
(Olympus Corporation) at ×400 magnification. Five random fields of
view were captured for each section and the total nuclei and
TUNEL-positive nuclei were counted.
Synergetic effect analysis
Three concentrations <IC50 (50% of inhibiting
concentration) of dinaciclib (MM cell lines: 0, 1, 2.5 and 5 nM and
primary MM cells: 0, 10, 25, 50 nM) and bortezomib (MM cell lines:
0, 0.5, 1 and 2 nM and primary MM cells: 0, 5, 10 and 20 nM) were
chosen. The inhibition rates of MM cell lines and primary MM cells
treated with individual drugs or the drug combination were
calculated by MTT method. Calcusyn software 2.0 (Premier Biosoft
International) was used to analyze the reduction indices and
isobologram plots. CI<1 represents synergism, CI>1 represents
antagonism.
Statistical analysis
All data was determined by GraphPad Prism5 software
(GraphPad Software, Inc.). Survival curves were plotted by the
Kaplan-Meier method and compared using log-rank test. A two-tailed
independent Student's t-test was used for analyzing two groups. A
Chi square test was used to analyze the relationship between CDK5
expression and the clinicopathological features, cytogenetic
abnormalities or immunophenotypes of MM. All data were presented as
the mean ± standard deviation from at least three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
CDK5 expression and prognostic relevance
in MM
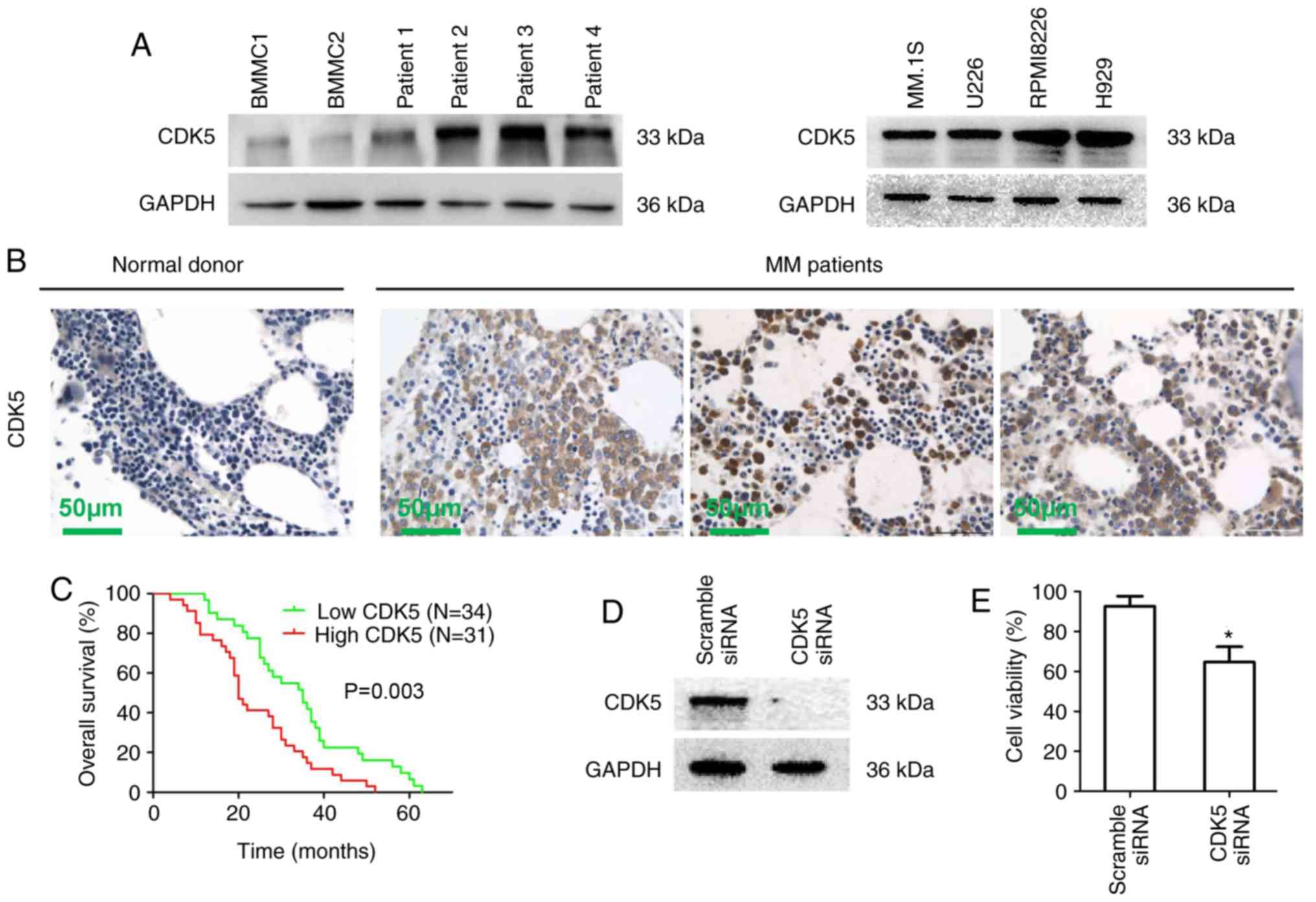
Western blot results showed that the expression of
CDK5 in MM cells from patients (patient 1-4) and multiple MM cell
lines was significantly increased compared with normal BMMCs
(Figs. 1A and S1A). Similar results were observed
using immunohistochemical staining of bone marrow biopsies from 65
newly diagnosed MM patients treated with bortezomib induction
therapy and healthy donors (Fig.
1B). To understand the potential roles of CDK5 in the
development of MM, the correlation of CDK5 expression with the
clinical indicators, cytogenetic abnormalities and immunophenotype
was analyzed and the results showed that high expression of CDK5
was correlated with the high DS stage (P=0.026), but not with other
indicators (including gender, age, clinicopathological
characteristics, cytogenetic abnormalities and immunophenotype;
Tables I and II). Kaplan-Meier survival analysis
showed a significant correlation between high CDK5 expression and
poor overall survival statistically in MM patients (P=0.003;
Fig. 1C). Next, an siRNA strategy
was used to evaluate the importance of CDK5 for MM cell viability.
A marked down expression of CDK5 was observed by CDK5 smart pool
siRNAs compared with scrambled siRNA in RPMI8226 cells (Figs. 1D and S1B). MTT results showed that the
viability of MM cells transfected with CDK5 siRNA was inhibited
compared with scramble siRNA (Fig.
1E). These results revealed that CDK5 might be involved in the
pathogenesis of MM and had a prognostic relevance in MM, providing
a rationale for targeting CDK5 in MM.
| Table IICorrelation of CDK5 with cytogenetic
abnormalities and immunophenotypes in MM. |
Table II
Correlation of CDK5 with cytogenetic
abnormalities and immunophenotypes in MM.
| MM | n CD | K5+
(n) | CDK5−
(n) | P-value |
|---|
| 17p13 deletion | | | | 0.914 |
| Positive | 5 | 3 | 2 | |
| Negative | 60 | 28 | 32 | |
|
1q21amplification | | | | 0.730 |
| Positive | 35 | 16 | 19 | |
| Negative | 30 | 15 | 15 | |
| IgH
rearrangement | | | | 0.204 |
| Positive | 15 | 5 | 10 | |
| Negative | 50 | 26 | 24 | |
| 13q14 deletion | | | | 0.179 |
| Positive | 35 | 14 | 21 | |
| Negative | 30 | 17 | 13 | |
| CD117 | | | | 0.172 |
| Positive | 16 | 10 | 6 | |
| Negative | 49 | 21 | 28 | |
| CD20 | | | | 0.385 |
| Positive | 9 | 6 | 3 | |
| Negative | 56 | 25 | 31 | |
| CD56 | | | | 0.428 |
| Positive | 43 | 19 | 24 | |
| Negative | 22 | 12 | 10 | |
| CD81 | | | | 0.789 |
| Positive | 43 | 20 | 23 | |
| Negative | 22 | 11 | 11 | |
| CD45 | | | | 0.818 |
| Positive | 24 | 11 | 13 | |
| Negative | 41 | 20 | 21 | |
| CD27 | | | | 0.180 |
| Positive | 18 | 11 | 7 | |
| Negative | 47 | 20 | 27 | |
| CD28 | | | | 0.881 |
| Positive | 9 | 5 | 4 | |
| Negative | 56 | 26 | 30 | |
| CD33 | | | | 0.456 |
| Positive | 13 | 5 | 8 | |
| Negative | 52 | 26 | 26 | |
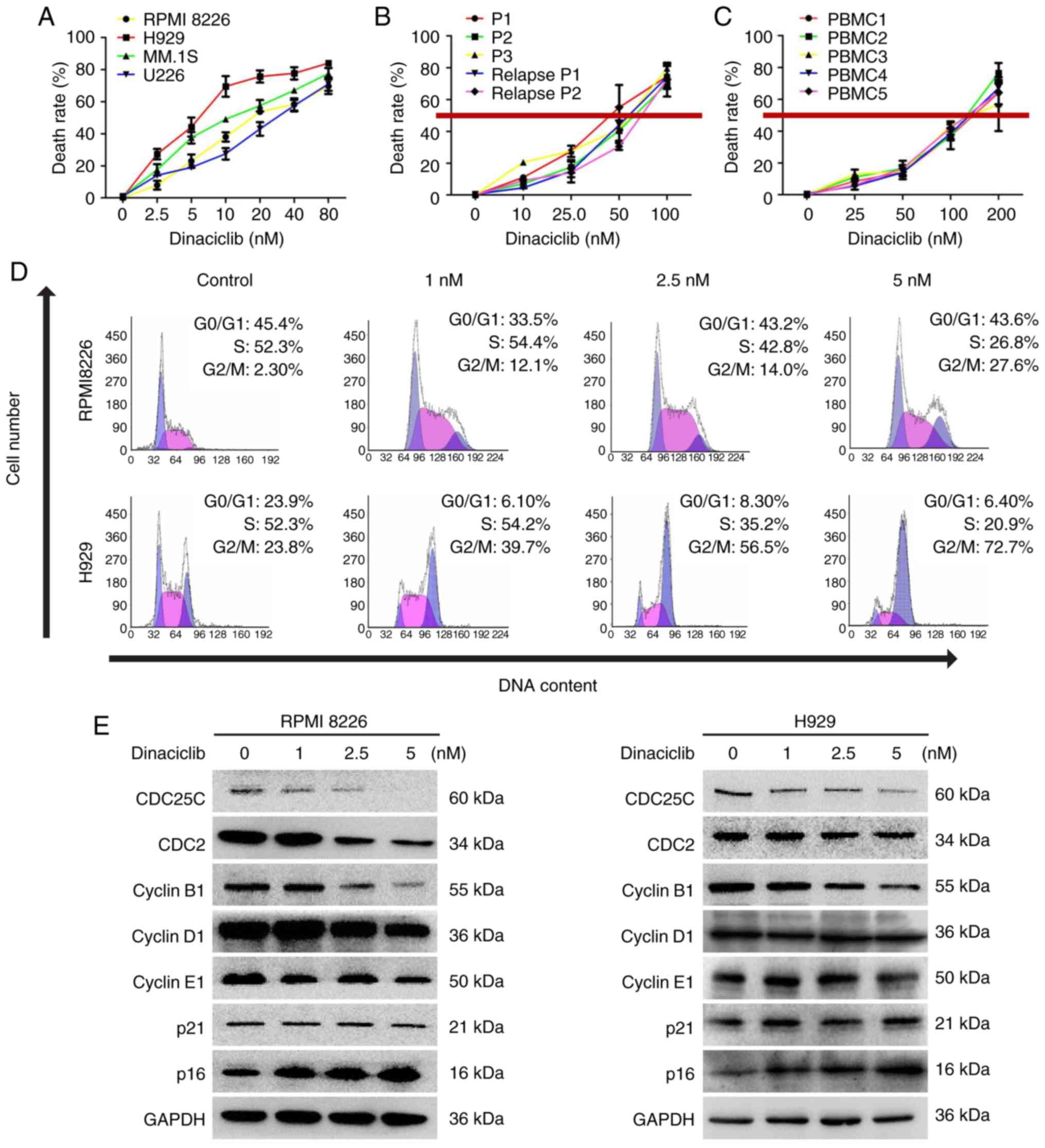
Effect of dinaciclib on the viability of
MM cells in vitro
Dinaciclib is a small molecule CDK1/2/5/9 inhibitor,
but the IC50 is different for distinct target molecules. The
present study evaluated the expression of CDK1/2/5/9 in RPMI8226
and H929 cell lines treated with different concentrations (0, 1, 2
and 5 nM) of dinaciclib. A total of 5 nM dinaciclib significantly
downregulated the expression of CDK5 without affecting that of
other target molecules (Fig.
S2). Therefore, dinaciclib could be used as a selective
inhibitor of CDK5 to explore its effect on the biological function
in MM cells.
Firstly, various MM cell lines (RPMI 8226, H929, MM.
1S and U226), primary MM cells and BMMCs from healthy people were
treated with different concentrations of dinaciclib for 24 h,
followed by cell viability assessment. A concentration-dependent
decrease of cell viability was observed in all MM cell lines in
response to dinaciclib (Fig. 2A).
Dinaciclib also significantly inhibited the viability of primary MM
cells, including those who relapsed after bortezomib treatment with
an IC50 of 40-80 nM (Fig. 2B),
which was much lower than the that (~150 nM) in BMMCs from healthy
donors (Fig. 2C).
In order to further disclose the mechanism of action
under-lying dinaciclib-triggered MM cell viability inhibition, cell
cycle distribution assessment was conducted in RPMI8226 and H929
cells. The results showed that dinaciclib induced G2/M phase arrest
in both cell lines (Fig. 2D). In
accordance with these findings, dinaciclib also inhibited the
expression of cyclin regulatory protein CDC25C and its downstream
proteins such as CDC2 and cyclin B1 in G2/M phase (Figs. 2E and S1C). The marker proteins in G1/S phase
were also detected and the expression of cyclin D1, cyclin E1 or
p21 did not change significantly (Fig. 2E), and the expression of p16
increased significantly (Figs. 2E
and S1C).
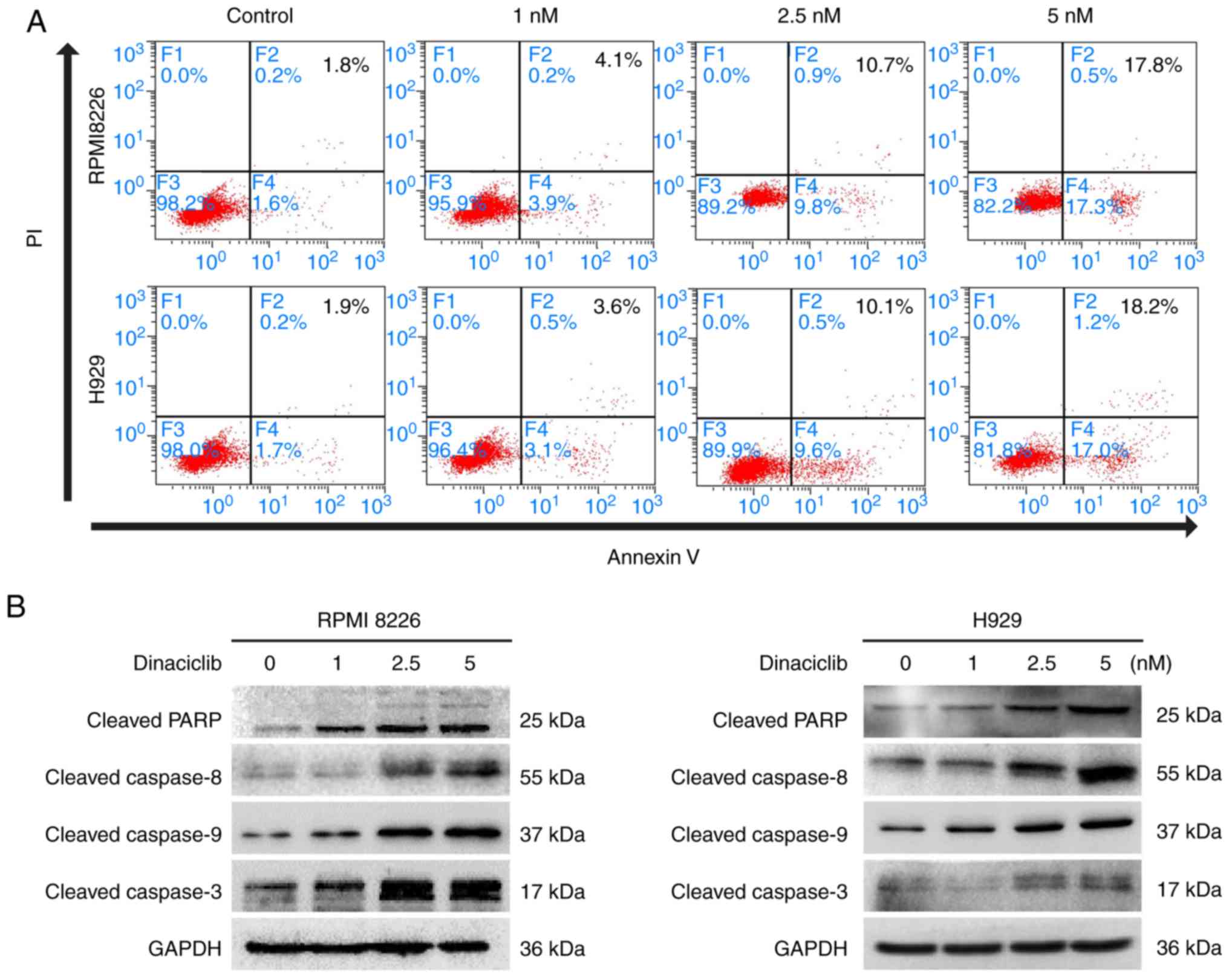
Furthermore, dinaciclib induced apoptosis in
RPMI8226 and H929 cells demonstrated by Annexin V/PI staining
(Fig. 3A). Moreover, dinaciclib
also increased the cleavage of PARP, a hallmark of apoptosis. The
present study further examined the expression of representative
endogenous (caspase 9) and exogenous (caspase 8) apoptotic
proteins, and found that dinaciclib activated caspase 8 and caspase
9 followed by activation of caspase 3 (Figs. 3B and S1D).
These results suggested that restraining the
expression of CDK5 by dinaciclib mediated the arrest of cell cycle
and programmed cell death in MM cells. Dinaciclib induced evident
inhibition of viability in both primary MM cells and MM cell lines
with little killing effect on normal blood cells. This might be the
theoretical basis for the slight hematological toxicity and good
tolerance in human study (20).
Anti-myeloma effect of dinaciclib in
vivo
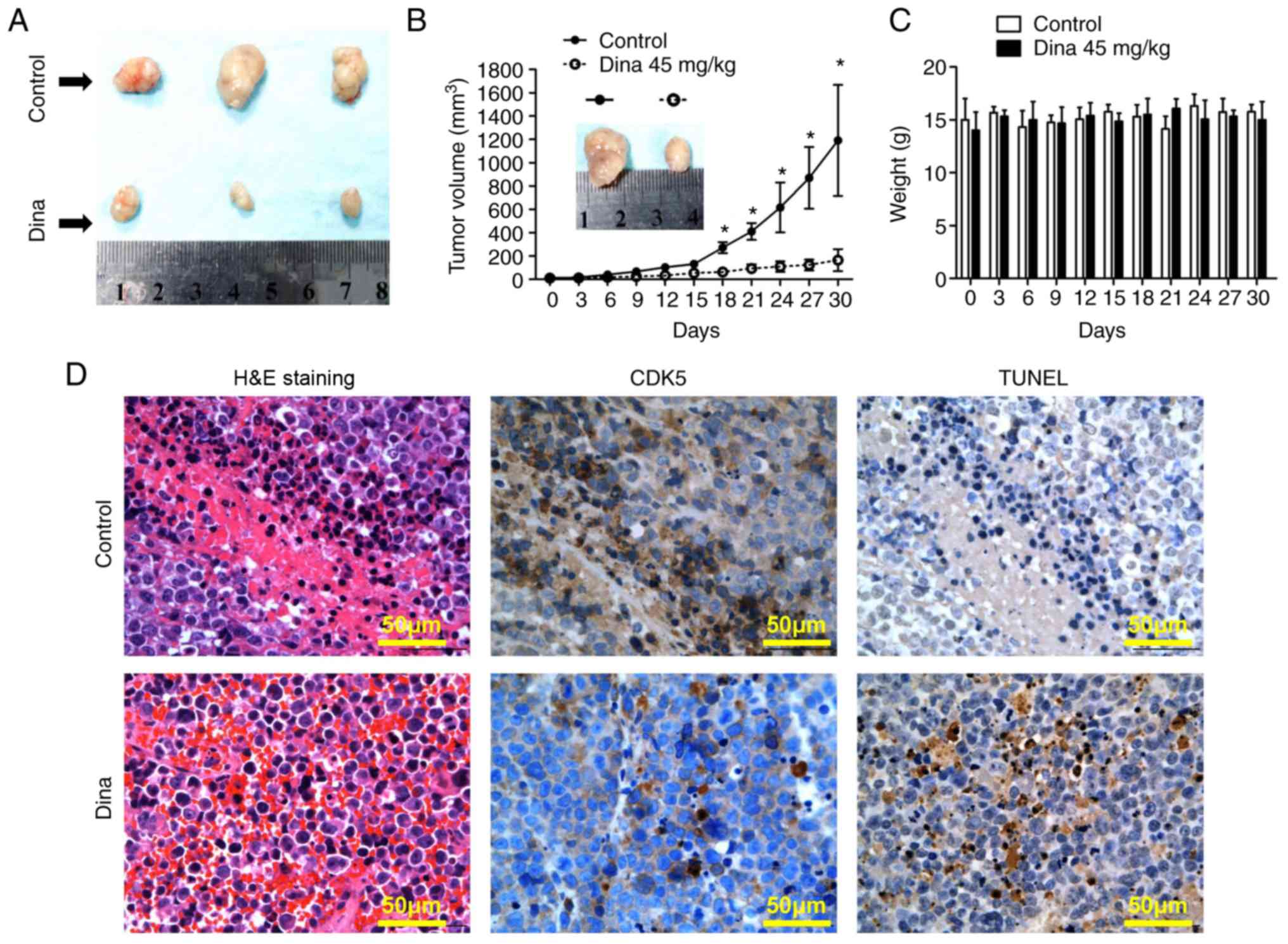
Next, the anti-myeloma efficacy in vivo was
detected using a MM xenograft mouse model. Fig. 4A showed MM tumors excised from
mice with/without dinaciclib treatment on day 30. Treatment of
RPMI8226 tumor-bearing mice with dinaciclib injection started to
inhibit MM tumor growth from the day 18 compared with that of
control mice (the maximum tumor diameters between the two groups
were 7.8×5.1 mm vs. 19.1×13.5 mm on day 30, Fig. 4A and B). During the whole in
vivo experiment, no significant change in body weight was noted
in the dinaciclib group, suggesting that dinaciclib was well
tolerated (Fig. 4C). Mechanistic
study by IHC showed that the expression of CDK5 was significantly
decreased and the TUNEL staining was significantly enhanced in
tumors excised from mice receiving dinaciclib (Fig. 4D). Taken together, dinaciclib
showed potent anti-myeloma effect in vivo by inducing
apoptosis of MM cells and was well tolerated.
Combining dinaciclib with bortezomib has
synergistic anti-myeloma activity
Dinaciclib exerted encouraging single-agent activity
in patients with relapsed MM in an early clinical trial (20). However, at present, the treatment
of MM is still based on multi-drug 'cocktail therapy'. If
dinaciclib could play a synergistic role with existing drugs or
overcome drug resistance, it would have a broader application
prospect. First, it was observed that patients with high expression
of CDK5 in bone marrow biopsies were less responsive to induction
therapy of bortezomib (Table I,
Fig. 5A). The results further
supported the view that pharmacological inhibition of CDK5 might
overcome bortezomib resistance. Next synergistic experiments in
RPMI8226, H929 cells and primary MM cells exposed to dinaciclib
with bortezomib across a range of concentrations for 24 h revealed
that dinaciclib and bortezomib triggered obvious synergistic
anti-myeloma effect with a CI <1.0 (Figs. 5B and S3). A total of 5 nM dinaciclib and 2 nM
bortezomib, which had the most obvious synergistic effect, were
selected to explore the follow-up mechanism.
Combining dinaciclib with bortezomib
cooperatively suppresses the nuclear factor (NF)-κB signaling
pathway in MM cells
It is well established that the activation of NF-κB
pathway is found in numerous tumors, including MM (22). One of the main anti-myeloma
mechanisms of bortezomib is to inhibit the activation of the NF-κB
pathway by disturbing the ubiquitin-proteasome degradation of the
inhibitor IκBα of the NF-κB pathway (23). Therefore, the present study
speculated whether the synergistic anti-myeloma effect of
dinaciclib and bortezomib involved the inhibition of the NF-κB
pathway at the same time. The activation of NF-κB pathway is
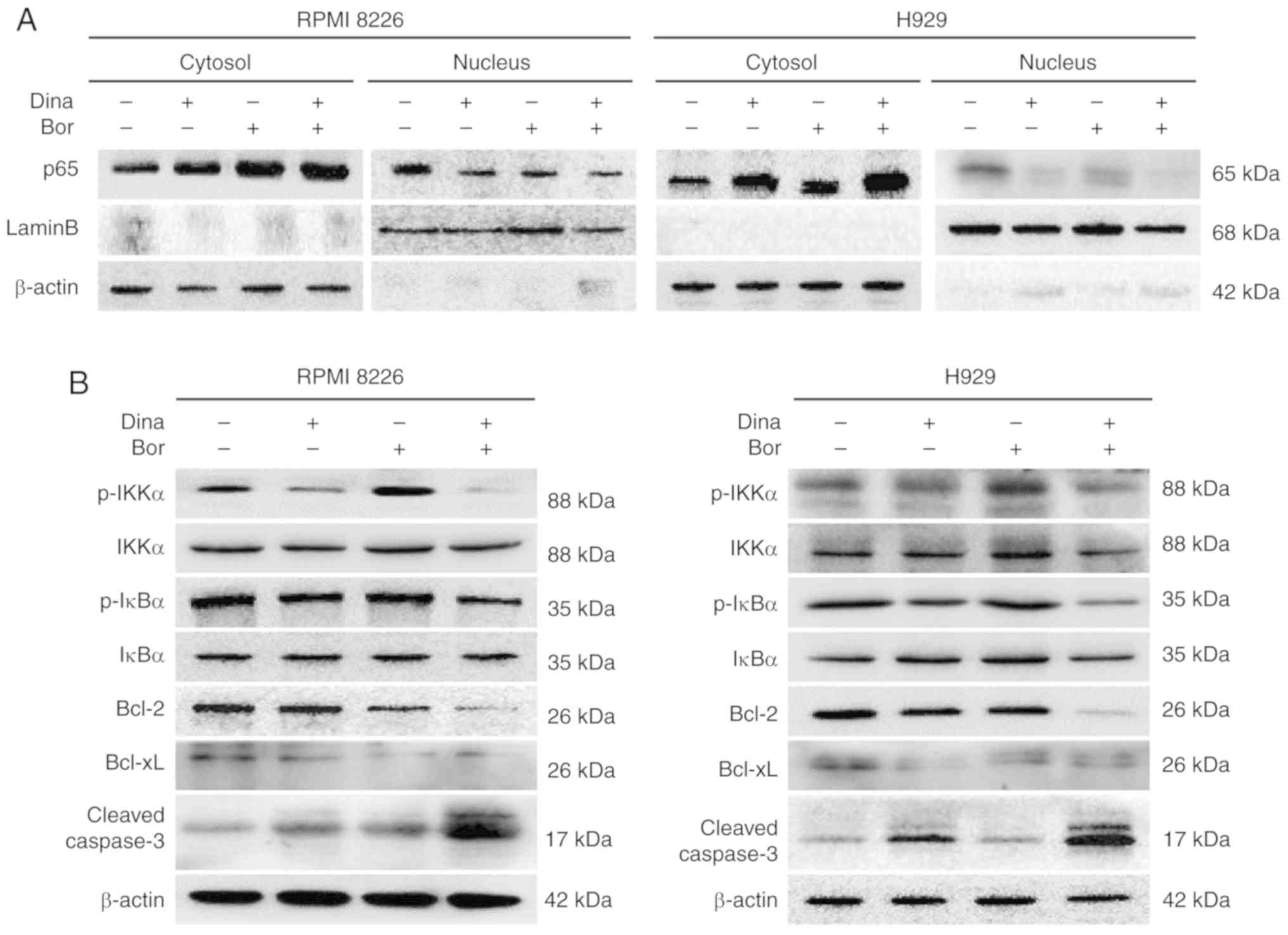
characterized by the nuclear import of the NF-κB protein (24). The present study measured the
subcellular distribution of NF-κB p65. The results showed that the
intranuclear transfer of NF-κB p65 in RPMI8226 and H929 cells
treated with dinaciclib or bortezomib was lower than that in the
control group; and the intranuclear transfer of p65 in the combined
group was much lower than that in the single drug group (Figs. 6A and S4A), suggesting the combination of the
two drugs could further inhibit the activation of the NF-κB
pathway. At the same time, the present study detected the
expression levels of p-IKKα and p-IκBα. Inhibited phosphorylation
levels of IKKα and IκBα were found in MM cells treated by
dinaciclib alone or in combination with bortezomib (Figs. 6B and S4B), further indicating that the
synergistic anti-myeloma effect of dinaciclib and bortezomib
involved the simultaneous inhibition of classical NF-κB pathway.
The downstream anti-apoptotic proteins Bcl-2 and Bcl-xL were
decreased and the cleaved caspase 3 was increased in MM cells
treated with the combination of dinaciclib and bortezomib (Figs. 6B and S4B). These results suggested that the
synergistic anti-myeloma mechanism of dinaciclib and bortezomib is
involved the synchronous inhibition of the classical NF-κB pathway,
which resulted in the decrease of downstream anti-apoptotic protein
expression and the increase of apoptosis in MM cells.
Discussion
As an unorthodox member of CDK family, CDK5 has
attracted much attention for its unconventional functions. CDK5
involves in a variety of signaling pathways and plays an important
role in tumorigenesis (4). At
present the role of CDK5 in the MM development, prognosis and drug
resistance is still not clear. The purpose of this study is to
explore the role of CDK5 in MM.
Consistent with previous study (16), the present study also found that
the expression of CDK5 increased in both primary MM cells, MM cell
lines and MM bone marrow biopsies. For the first time, to the best
of our knowledge the current study confirmed that the
overexpression of CDK5 suggested the adverse prognosis of MM.
Knockout of CDK5 by siRNA suppressed the viability of MM cells, all
of which suggested that CDK5 plays a key role of the pathogenesis
in MM and favors a solid clinical application significance for the
current research.
Dinaciclib, a small molecule inhibitor of
CDK1/2/5/9, has been shown to interfere with DNA damage repair in
MM cells (18) and suppress the
proliferation of MM cells by inducing endoplasmic reticulum stress
in a CDK1/5 dependent manner (19). However, the mechanism of its
inhibition on MM cell viability remains to be further studied.
RPMI8226 and H929 cells were treated with different concentrations
of dinaciclib and detected the expression of target molecules. It
was unexpectedly found that the inhibitory effect of 5 nM
dinaciclib on CDK5 was better than that on other molecules.
Therefore, dinaciclib could be used as a specific inhibitor of CDK5
to carry out follow-up studies on the function of CDK5 in MM.
First, the effect of pharmacological inhibition of
CDK5 by dinaciclib on MM cells was studied. It was shown that
dinaciclib caused an obvious decrease in cell viability in various
MM cell lines and MM cells from patients including those relapsed
after bortezomib treatment, suggesting that it might overcome
proteasome inhibitors resistance. At the same time, the present
study found that dinaciclib did not affect the normal viability of
BMMCs, exhibiting its high selectivity to cancer cells and safety
to normal cells, which was also consistent with good tolerance as
suggested by an early clinical study of dinaciclib (20).
Unlike other CDKs, CDK5 was not considered to have
the function of regulating the cell cycle previously. However, it
has been found that retinoblastoma protein (Rb), as a downstream
target molecule of CDK5, can be directly phosphorylated by CDK5,
thus affecting the process of the cell cycle (25). The current results showed that
pharmacological inhibition of CDK5 resulted in the stagnation of MM
cells in the G2/M phase, while the expression of CDC25c, CDC2 and
cyclinB1 in G2/M phase decreased; and cyclinD1 and cyclinE1 in G1/S
phase remained unchanged. Unlike the results of Abbas and Dutta
(26), the expression of p21 did
not change significantly, but the expression of p16 increased.
Previous studies have shown that CDK5 and p16 are both upstream
regulators of Rb. Inhibition of CDK5 or upregulation of p16 both
result in a decrease in phosphorylated Rb expression (25,27). Therefore, the present study
supposed that after pharmacological inhibition of CDK5, the
expression of phosphorylated Rb decreased, resulting in a feedback
enhancement of p16 expression (28). Of course, the complete mechanisms
still need to be further studied.
The results from flow cytometry and western blotting
suggested that pharmacological inhibition of CDK5 significantly
induced apoptosis of MM cells via activation of caspases. The
expression of endogenous (caspase 9) and exogenous (caspase 8)
apoptotic proteins was detected and it was found that dinaciclib
activated both endogenous and exogenous apoptotic pathways followed
by activation of caspase 3.
Having conducted in vitro studies, the in
vivo effects of dinaciclib were also verified. A MM-bearing
mice model was established, which was administered by intermittent
injection of dinaciclib. A marked reduction in tumor volume was
noted in mice experiencing dinaciclib vs. mice receiving vehicle
alone. With the prolongation of administration time, there was no
significant change in body weight, indicating that dinaciclib was
well tolerated in vivo. The outstanding anti-myeloma
activity of dinaciclib in vivo was also confirmed by IHC
analysis for apoptosis (TUNEL stain) and CDK5 expression of tumor
sections from control or dinaciclib-treated mice.
In conclusion, it was confirmed that dinaciclib had
an excellent anti-myeloma effect, including in vivo/vitro
experiments and primary MM cells, which provided a further
theoretical basis for dinaciclib as a new drug for the treatment of
MM. In terms of the current treatment strategy of MM, combination
therapy is a better choice. The resistance of first-line bortezomib
has always been an urgent clinical problem. So, the combined
effects of dinaciclib and bortezomib on MM cells were subsequently
tested in order to find new strategies to enhance the sensitivity
of bortezomib.
It has been reported that CDK5 may touch upon the
resistance of MM to proteasome inhibitors, but only the preliminary
mechanism has been discussed, which is thought be related to
miR-27a-5p or PSMB5 (15,17,29). The correlation between the
expression of CDK5 in bone marrow biopsies and the response of MM
patients to proteasome inhibitors bortezomib was analyzed. The
results indicated that MM patients with high expression of CDK5 at
diagnosis had a poor response to bortezomib, suggesting that CDK5
might play an important role in bortezomib resistance. It was also
demonstrated that MM patients with high CDK5 expression tended to
have higher DS stage, indicating that target organ damage was more
serious in patients with high CDK5 expression. Further in
vitro experiments were carried out and it was found that CDK5
inhibitors and bortezomib showed synergistic anti-myeloma effect,
further suggesting that targeting CDK5 is probably one of the ways
to solve the problem of bortezomib resistance.
The mutation of NF-κB pathway and the interaction
with the MM microenvironment lead to the over-activation of the
NF-κB pathway in MM, which promotes the transcriptional activation
of downstream oncogenes, the enhanced expression of gene encoding
anti-apoptotic proteins, and the suppressed expression of gene
encoding proapoptotic proteins (22). The present study wondered whether
CDK5 was also involved in the activation of the NF-κB pathway. The
current results confirmed that dinaciclib combined with bortezomib
further inhibited the activation of the NF-κB pathway, resulting in
the decrease of downstream anti-apoptotic protein expression,
thereby inducing apoptosis and exerting a synergistic anti-myeloma
effect. The regulation of the classical pathway of NF-κB involves
numerous kinds of phosphorylation events. IKKα phosphorylation
participates in the activation of the trimeric IκB kinase (IKK)
complex (IKKα/IKKβ/NEMO) and the activation of IKK tripolymer
phosphorylates IκB, which leads to the degradation of
ubiquitin-proteasome pathway of IκB and the activation of the
classical pathway of NF-κB (30).
After dinaciclib exposure, the expression of p-IKKα and p-IKBα in
MM cells decreased, indicating that the phosphorylation levels of
some key proteins in the classical pathway of NF-κB changed after
targeted inhibition of CDK5. Whether CDK5 could directly
phosphorylate IKKα, IKBα or other molecules needs further
study.
Collectively, the present study established the
potent role of CDK5 in the pathogenesis and prognosis of MM, and
confirmed the promising anti-myeloma effect of CDK5 inhibitors
through functional experiments in vitro and in vivo.
Dinaciclib combined with bortezomib exert synergistic anti-myeloma
effect by collectively inhibiting the activation of the NF-κB
pathway in MM. This study provided the proof of concept for
targeting CDK5 as a new method to improve outcome of MM patients
and overcome the obstacle of bortezomib resistance.
Supplementary Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Social Development Science and Technology Fund of Shaanxi Province
2016SF071 (to HT).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HT, LX, XC, LY, JF, GL, HZ, SG, YY, YZ, ZT, LH and
SY performed experiments; HT, XC and GG wrote the paper. HT and GG
conceived the strategy, oversaw the experiments and provided
overall guidance and interpretation of the results. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All studies have been approved by the Ethics
Committee of Xijing Hospital and the Animal Ethics Committee of
Fourth Military Medical University, Xi'an, Shaanxi, China. Informed
consent was obtained from all patients, in accordance with the
Helsinki Protocol. These studies have been approved by the Ethics
Committee of Xijing Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no potential conflicts of
interest.
References
|
1
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Casimiro MC, Crosariol M, Loro E, Li Z and
Pestell RG: Cyclins and cell cycle control in cancer and disease.
Genes Cancer. 3:649–657. 2012. View Article : Google Scholar
|
|
3
|
Dhavan R and Tsai LH: A decade of CDK5.
Nat Rev Mol Cell Biol. 2:749–759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shupp A, Casimiro MC and Pestell RG:
Biological functions of CDK5 and potential CDK5 targeted clinical
treatments. Oncotarget. 8:17373–17382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Zhong T, Dang Y, Li Z, Li P and
Chen G: Aberrant expression of CDK5 infers poor outcomes for
nasopharyngeal carcinoma patients. Int J Clin Exp Pathol.
8:8066–8074. 2015.PubMed/NCBI
|
|
6
|
Zhuang K, Zhang J, Xiong M, Wang X, Luo X,
Han L, Meng Y, Zhang Y, Liao W and Liu S: CDK5 functions as a tumor
promoter in human colorectal cancer via modulating the ERK5-AP-1
axis. Cell Death Dis. 7:e24152016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang R, Lin P, Yang H, He Y, Dang YW,
Feng ZB and Chen G: Clinical role and biological function of CDK5
in hepatocellular carcinoma: A study based on immunohistochemistry,
RNA-seq and in vitro investigation. Oncotarget. 8:108333–108354.
2017.
|
|
8
|
Pan DH, Zhu ML, Lin XM, Lin XG, He RQ,
Ling YX, Su ST, Wickramaarachchi MM, Dang YW, Wei KL and Chen G:
Evaluation and clinical significance of cyclin-dependent kinase5
expression in cervical lesions: A clinical research study in
Guangxi, China. Eur J Med Res. 21:282016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei K, Ye Z, Li Z, Dang Y, Chen X, Huang
N, Bao C, Gan T, Yang L and Chen G: An immunohistochemical study of
cyclin-dependent kinase 5 (CDK5) expression in non-small cell lung
cancer (NSCLC) and small cell lung cancer (SCLC): A possible
prognostic biomarker. World J Surg Oncol. 14:342016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun SS, Zhou X, Huang YY, Kong LP, Mei M,
Guo WY, Zhao MH, Ren Y, Shen Q and Zhang L: Targeting STAT3/miR-21
axis inhibits epithelial-mesenchymal transition via regulating CDK5
in head and neck squamous cell carcinoma. Mol Cancer. 14:2132015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang S, Lu Z, Mao W, Ahmed AA, Yang H,
Zhou J, Jennings N, Rodriguez-Aguayo C, Lopez-Berestein G, Miranda
R, et al: CDK5 regulates paclitaxel sensitivity in ovarian cancer
cells by modulating AKT activation, p21Cip1- and p27Kip1-Mediated
G1 cell cycle arrest and apoptosis. PLoS One. 10:e01318332015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li R, Liu GZ, Luo SY, Chen R and Zhang JX:
Cyclin I promotes cisplatin resistance via Cdk5 activation in
cervical cancer. Eur Rev Med Pharmacol Sci. 19:4533–4541.
2015.PubMed/NCBI
|
|
13
|
Ehrlich SM, Liebl J, Ardelt MA, Lehr T, De
Toni EN, Mayr D, Brandl L, Kirchner T, Zahler S, Gerbes AL and
Vollmar AM: Targeting cyclin dependent kinase 5 in hepatocellular
carcinoma-A novel therapeutic approach. J Hepatol. 6:102–113. 2015.
View Article : Google Scholar
|
|
14
|
Merk H, Zhang S, Lehr T, Müller C, Ulrich
M, Bibb JA, Adams RH, Bracher F, Zahler S, Vollmar AM and Liebl J:
Inhibition of endothelial Cdk5 reduces tumor growth by promoting
non-productive angiogenesis. Oncotarget. 7:6088–6104. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ri M: Endoplasmic-reticulum stress
pathway-associated mechanisms of action of proteasome inhibitors in
multiple myeloma. Int J Hematol. 104:273–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levacque Z, Rosales JL and Lee KY: Level
of cdk5 expression predicts the survival of relapsed multiple
myeloma patients. Cell Cycle. 11:4093–4095. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu YX, Tiedemann R, Shi CX, Yin H,
Schmidt JE, Bruins LA, Keats JJ, Braggio E, Sereduk C, Mousses S
and Stewart AK: RNAi screen of the druggable genome identifies
modulators of proteasome inhibitor sensitivity in myeloma including
CDK5. Blood. 117:3847–3857. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alagpulinsa DA, Ayyadevara S, Yaccoby S
and Shmookler Reis RJ: A cyclin-dependent kinase inhibitor,
dinaciclib, impairs homologous recombination and sensitizes
multiple myeloma cells to PARP inhibition. Mol Cancer Ther.
15:241–250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nguyen TK and Grant S: Dinaciclib
(SCH727965) inhibits the unfolded protein response through a CDK1-
and 5-dependent mechanism. Mol Cancer Ther. 13:662–674. 2014.
View Article : Google Scholar
|
|
20
|
Kumar SK, LaPlant B, Chng WJ, Zonder J,
Callander N, Fonseca R, Fruth B, Roy V, Erlichman C, Stewart AK, et
al: Dinaciclib, a novel CDK inhibitor, demonstrates encouraging
single-agent activity in patients with relapsed multiple myeloma.
Blood. 125:443–448. 2015. View Article : Google Scholar :
|
|
21
|
Tang H, Shu M, Dai B, Xu L, Dong B, Gao G
and Chen X: DNA damage response-initiated cytokine secretion in
bone marrow stromal cells promotes chemoresistance of myeloma
cells. Leuk Lymphoma. 59:2220–2226. 2018. View Article : Google Scholar
|
|
22
|
Roy P, Sarkar UA and Basak S: The NF-κB
activating pathways in multiple myeloma. Biomedicines. 6:pii: E59.
2018. View Article : Google Scholar
|
|
23
|
Hideshima T, Chauhan D, Richardson P,
Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A,
Palombella V, et al: NF-kappa B as a therapeutic target in multiple
myeloma. J Biol Chem. 277:16639–16647. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoffmann A, Natoli G and Ghosh G:
Transcriptional regulation via the NF-kappaB signaling module.
Oncogene. 25:6706–6716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pozo K, Castro-Rivera E, Tan C, Plattner
F, Schwach G, Siegl V, Meyer D, Guo A, Gundara J, Mettlach G, et
al: The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell.
24:499–511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burkhart DL and Sage J: Cellular
mechanisms of tumour suppression by the retinoblastoma gene. Nat
Rev Cancer. 8:671–682. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang H, Xu L, Liang X and Gao G: Low dose
dinaciclib enhances doxorubicin-induced senescence in myeloma
RPMI8226 cells by transformation of the p21 and p16 pathways. Oncol
Lett. 16:6608–6614. 2018.PubMed/NCBI
|
|
29
|
Ballabio E, Armesto M, Breeze CE,
Manterola L, Arestin M, Tramonti D, Hatton CS and Lawrie CH:
Bortezomib action in multiple myeloma: MicroRNA-mediated synergy
(and miR-27a/CDK5 driven sensitivity)? Blood Cancer J. 2:e832012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|