Introduction
Hot climates are becoming more apparent in recent
years because of climate change and surges in temperature during
the spring and summer months (1).
The increase in body temperature caused by environmental heat
exposure can result in heat stress (HS) and even sudden death
(2,3), which has been reported frequently in
studies in the United States and warm countries, such as Saudi
Arabia (4,5). Previous studies have suggested that
heat stress-induced sudden death involves heart failure (2) and arterial hypotension (6), which are caused by disruption of the
function and structure of cardiomyocytes related to heat
stress-induced damage to the myocardial contractile force and the
blood circulation throughout the body (7). The results of our previous studies
demonstrated that a rat heart cell line exposed to heat for 5 h
presented disintegrated mitochondrial cristae, and even showed
apoptosis and necrosis (8,9).
Heat damage to myocardial cells is associated with increases in
certain serum enzymes, such as lactate dehydrogenase (LDH), and an
imbalance of the oxidation and antioxidant system (9).
Within cells exposed to heat stress, heat shock
proteins (HSPs) have a protective role in maintaining protein
homeostasis through their roles as molecular chaperones (10,11). On exposure to HS, heat shock
factors (HSFs), particularly HSF-1, are released from their protein
complexes and bind to the heat shock elements (HSEs) of target
genes following undergoing modification and nuclear import, which
regulates the transcription of various HSP genes (12,13). In our previous study, when
myocardial cells were stressed for 5 h, levels of Hsp90α protein
increased, implying its role in maintaining the homeostasis of
heat-stressed cells (8). Hsp90
participates in numerous survival signaling pathways. For instance,
its downstream target, protein kinase B (Akt) is activated by
various receptors for growth factors and cytokines, and pyruvate
kinase M2 (PKM2) interacts with Hsp90 to phosphorylate B-cell
lymphoma 2 (Bcl-2) after H2O2 treatment
(14,15). These two pathways are well
characterized in the regulation of cellular anti-apoptosis by
exerting antioxidant effects and inhibiting caspase-dependent
pathways, thereby reducing cellular loss and myocardial injury.
However, little is known regarding the specific influence of Hsp90
on these signaling pathways during heat stress.
Aspirin (ASA), a non-steroidal anti-inflammatory
drug (NSAID) used to treat fever, is now well established in the
prevention of cardiovascular disease (16,17). ASA lowers the temperature
threshold needed to induce Hsp gene transcription and promotes
thermotolerance (18). Our
previous studies in chickens confirmed that pre-treatment with ASA
(1 mg/kg) for 2 h could induce Hsp90 expression in heat-stressed
chicken myocardial cells in vivo and in vitro, and
that ASA's protective role may be weakened using the specific Hsp90
function inhibitor, geldanamycin (GA) (19,20). In the present study, ASA and GA
were used to regulate the expression or function of Hsp90.
The present study aimed to confirm the protective
effect of Hsp90 on mouse hearts, particularly in the first 5 h of
HS, via its expression induction and functional inhibition.
Furthermore, protection of the survival pathways associated with
Hsp90's chaperone function were also identified in myocardial
mitochondria.
Materials and methods
Animal treatment and sampling
C57BL/6JNju male mice (6 weeks old; 20–25 g) were
purchased from Shanghai GenePharma Co., Ltd. First, 40 mice were
divided randomly into four groups: The control (Con) group, ASA
group, HS group, and ASA + HS group. The mice were raised
conventionally for 1 week, and then used to assess the effect of
ASA on myocardial tissues of heat-stressed mice: The ASA and ASA +
HS groups were administered with ASA (Sigma Aldrich; Merck KGaA;
dissolved in distilled water) by intragastric administration (1
mg/kg body weight) (19), while
the Con and HS groups were treated with saline only. After 2 h of
ASA treatment, the HS and ASA + HS groups were exposed to HS by
rapidly increasing the air temperature from (25±1)°C to (42±1)°C in
a phytotron, while maintaining the humidity at 75%, for 5 h; the
Con and ASA groups were kept under normal conditions (25±1°C,
humidity 75%) for the same period.
Subsequently, another 40 mice were randomly divided
into Con, GA, HS, and GA + HS groups. The GA and GA + HS groups
were treated with GA (Beyotime Institute of Biotechnology);
dissolved in dimethyl sulfoxide (DMSO) and diluted with distilled
water; the DMSO concentration was <0.1% by intraperitoneal
injection (50 mg/kg body weight), according to the manufacturer's
protocol, while the Con and HS groups were treated with saline
only. After 14 h of GA or saline treatment, the HS and GA + HS
groups were exposed to heat stress for 5 h, while the Con and GA
groups were kept under normal conditions.
Finally, 40 mice were randomly divided into groups
of Con, TR, HS and TR + HS. The TR and groups were treated with
Triciribine (Beyotime Institute of Biotechnology; dissolved in DMSO
and diluted with distilled water; the DMSO concentration was
<0.1%), according to the manufacturer's protocol (1 mg/kg body
weight), while the Con and HS groups were treated with saline only.
After 14 h of TR treatment, the HS and TR + HS groups were exposed
to heat stress for 5 h, while the Con and TR groups were kept under
normal conditions.
All mice were allowed ad libitum access to
food and water during the experiment. After measuring physiological
indices and anesthesia with diethyl ether (after the mice had
fallen down and exhibited slow breathing, adequate anesthesia was
further confirmed by the skin pain reflex), orbital blood was
collected from the mice (~0.5 ml per mouse), prior to the mice
being sacrificed via cervical dislocation, and samples of their
heart tissues being taken. For each group, five ventricles were
used to extract mitochondria, and another five were cut into two
halves, of which one was used for morphological detection, and the
other was used for molecular biological analysis. All animal
experiments were performed according to the guidelines of the
regional Animal Ethics Committee and were approved by the
Institutional Animal Care and Use Committee of Nanjing Agricultural
University.
Measurement of physiological indices
Non-invasive Pulse Oxygen Respiratory Monitors
(MouseOx, Starr Life Sciences Corp.) were used to measure the
physiological parameters of the mice, including heart rate, blood
oxygenation content and respiratory rate, according to the
manufacturer's protocol.
Serum biochemical analyses
Mouse blood samples were centrifuged at 4,000 × g
for 5 min to obtain the serum, which was sent to Super Biotech Co.,
Ltd. to detect the activities of creatine kinase (CK), lactate
dehydrogenase (LDH), and aspartate aminotransferase (AST). Serum
Hsp70 levels were analyzed using the mouse Hsp70 ELISA kit (cat.
no. ANG-E21510M, Angle Gene), according to the manufacturer's
protocol.
Detection of the oxidation and
antioxidant capacity
The antioxidant status of the tested mouse hearts
was assessed by measuring the levels of glutathione peroxidase
(GSH-PX) and catalase (CAT). Peroxidation was estimated by
measuring the malondialdehyde (MDA) content. For these biochemical
analyses, hearts from five randomly selected animals per group were
used. All the aforementioned indices were determined using the
GSH-PX assay kit (cat. no. A005-1-2), the CAT assay kit (cat. no.
A007-1-1) and the MDA assay kit (cat. no. A003-1-2), all from
Jiancheng, according to the manufacturer's protocol.
Histopathological evaluations
For histopathological and immunohistofluorescence
analyses, ventricular tissues of five animals per group were
selected and fixed in 10% neutral formalin solution at room
temperature (RT) for at least 48 h. Serial 3-4 µm sections
were cut, followed by embedding in paraffin. The tissue sections
were then deparaffinized and stained with hematoxylin and eosin
(H&E) at RT for 3 and 5 min, respectively. Tissue damage was
evaluated under a light microscope at x400 magnification by two
pathologists who were blinded to the treatments. Damage was scored
as follows: No obvious pathological change (0 points), swelling of
myocytes or mild degeneration (1 point), middle myocardial
degeneration (2 points), serious myocardial degeneration (3
points), and necrosis or fracture of myocardial fiber and
microvascular hyperemia (4 points).
Deparaffinized sections were rehydrated in 100, 100,
95, 95, 85 and 75% alcohol, followed by antigen retrieval by
boiling at 98°C and washing with phosphate buffer solution. Tissue
sections were successively blocked with 0.1% Triton X-100 at RT for
20 min, and then with goat serum (AR0009, Boster Bio) at 37°C for
30 min. The slides were then incubated with primary antibodies
against Hsp70 and Hsp90 (cat. nos. ab79852 and ab2928, Abcam), Akt
and PKM2 (cat. nos. 9272 and 3198, Cell Signaling Technology, Inc.)
at a dilution of 1:50 overnight at 4°C. After washing in
phosphate-buffered saline with 0.05% Tween-20, the slides were
incubated with the FITC and TRITC-conjugated secondary antibody
(dilution, 1:50 and 1:3; cat. no., BA1105 and BA1090, Boster Bio)
at 37°C for 1 h, and then stained with
2-(4-amidinophenyl)-1H-indole-6-carbox-amidine (DAPI) at RT for 1
min. Images were acquired under a fluorescence microscope at a
magnification of ×400.
TdT-mediated dUTP nick-end labeling
(TUNEL) to detect cell apoptosis
TUNEL staining was conducted using a kit (KGA7021;
KeyGen Biotech. Co. Ltd.) on serial sections depa-raffinized as
mentioned above, according to the manufacturer's protocol. Nuclei
were re-stained with hematoxylin at RT for 35 sec. The sections
were mounted with neutral balsam and observed under a light
microscope at a magnification of ×400. Myocardial apoptotic nuclei
appeared brown and non-apoptotic nuclei appeared blue. The
percentage of apoptotic cells among the total cardiomyocytes was
used as the apoptotic rate.
Measuring the mitochondrial membrane
potential (MMP) and openness of permeability transition pores
Purified myocardial mitochondria were diluted to
0.25 g/l protein concentration. The mitochondrial absorbance (A520)
was observed at 520 nm before and after adding 200 µmol/l
CaCl2. The difference of A520 max-A520 min represents
the open level of the mitochondrial permeability transition pore
(mPTP) (21).
The tissue mitochondria isolation kit (cat. no.
C3606; Beyotime Institute of Biotechnology) was used to isolate
mitochondria of heart tissue according to the manufacturer's
protocol. A mitochondrial membrane potential detection kit with
JC-1 (cat. no. C2006; Beyotime Institute of Biotechnology) was used
to measure the MMP, according to the manufacturer's protocol.
Briefly, 0.1 ml purified mitochondria (20 µg) was incubated
with 0.9 ml of 5-times-diluted JC-1 staining solution for 20 min at
37°C. The fluorescence intensity of mitochondrial JC-1 monomers
(λex 490 nm, λem 530 nm) and aggregates (λex 525 nm, λem 590 nm)
were detected using a monochromator microplate reader (Safire II;
Tecan Group Ltd.). The MMP in each sample was calculated as the
fluorescence ratio of red (i.e., aggregates) to green (i.e.,
monomers).
Protein extraction and western blotting
analysis
Following the designated treatments, the collected
mouse heart tissues and purified mitochondria from mouse heart
tissues were frozen at −80°C for protein extraction. Total proteins
were extracted using lysis buffer (cat. no., CW2333, CWBio)
containing 1% phenylmethylsulfonyl fluoride and quantified using a
bicinchoninic acid assay kit. Equal amounts of proteins (20
µg) were mixed with 5X SDS sample buffer, boiled for 5 min,
and then separated by 10% SDS-PAGE. The proteins were transferred
onto nitrocellulose membranes after electrophoresis. The membranes
were incubated with 5% skimmed milk at RT for 2 h, prior to being
incubated with primary antibodies against the following: Hsp90α
(dilution, 1:1,000; cat. no. ab2928), Hsp70 (dilution, 1:10,000;
cat. no. ab79852) and COX IV (dilution, 1:1,000; cat. no. ab16056;
all Abcam) Akt (dilution, 1:1,000; cat. no. 9272), PKM2 (dilution,
1:1,000; cat. no. 3198), HSF-1 (dilution, 1:1,000; cat. no. 4356),
caspase-3 (dilution, 1:1,000; cat. no. 9662), cytochrome c
(cyt c; dilution, 1:1,000; cat. no. 4272), Bcl-2 (dilution,
1:1,000; cat. no. 3498) and β-actin (dilution, 1:1,000; cat. no.
4967; all Cell Signaling Technology, Inc.), phosphorylated (p)-Akt
(dilution, 1:500; cat. no. sc-377556) and p-Bcl-2 (dilution, 1:500;
cat. no. sc-377576) from Santa Cruz Biotechnology, Inc.
Subsequently, the membranes were incubated for 2 h with
corresponding horseradish peroxidase-conjugated secondary
antibodies (cat. nos. BA1051 and BA1055; dilution, 1:5,000; Wuhan
Boster Biological Technology, Ltd.). Immunoreactive protein bands
were visualized using an electrochemiluminescence substrate and a
gel-imaging system (Tanon Science and Technology, Co., Ltd.) with
Image Analysis software (version 1.46; National Institutes of
Health).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Differences among groups were determined using one-way analysis of
variance, followed by Duncan's multiple range test using SPSS 25.0
version (IBM Corp.). P<0.05 was considered to indicate
statistically significant differences.
Results
Higher Hsp90 levels alleviate the
heat-stress damage of mouse heart tissues
Whether ASA exerts a protective effect on mouse
heat-stressed hearts was first investigated through the examination
of physiological indices, serum markers and histopathology
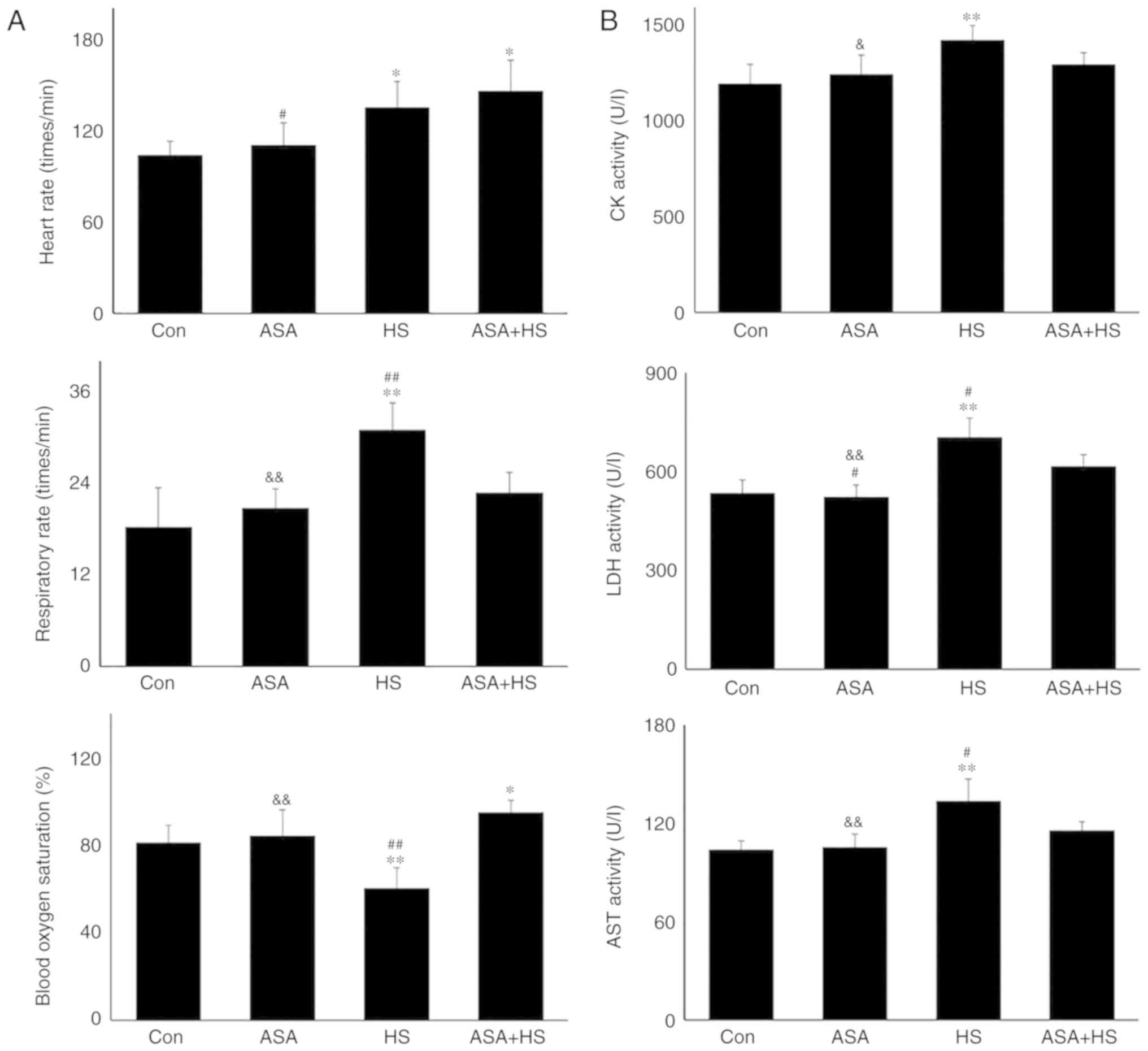
(Fig. 1). HS significantly
increased the heart rate and respiratory rate of the tested mice,
but significantly decreased their blood oxygen saturation
(P<0.01). Additionally, ASA administration enhanced the heart
rate to a higher degree during HS, accompanied by significantly
higher blood oxygen saturation, compared with that in the HS group
(P<0.01; Fig. 1A). Detection
of myocardial injury-related enzymes (Fig. 1B) revealed that, compared to the
Con group, HS raised the serum levels of CK, LDH and AST
(P<0.01), which were higher than those in the ASA + HS group,
especially for LDH and AST (P<0.05). Consistent with these
results, histopathological observation (Fig. 1C and D) revealed that HS caused
severe tissue damage, characterized by granular degeneration,
necrosis and rupture of myocardial fibers, and a higher
pathological score than that in the Con group. ASA administration
ameliorated the HS-induced myocardial necrosis and rupture, and
decreased the pathological score significantly. These results
revealed that ASA effectively alleviated HS-induced damage to heart
tissues.
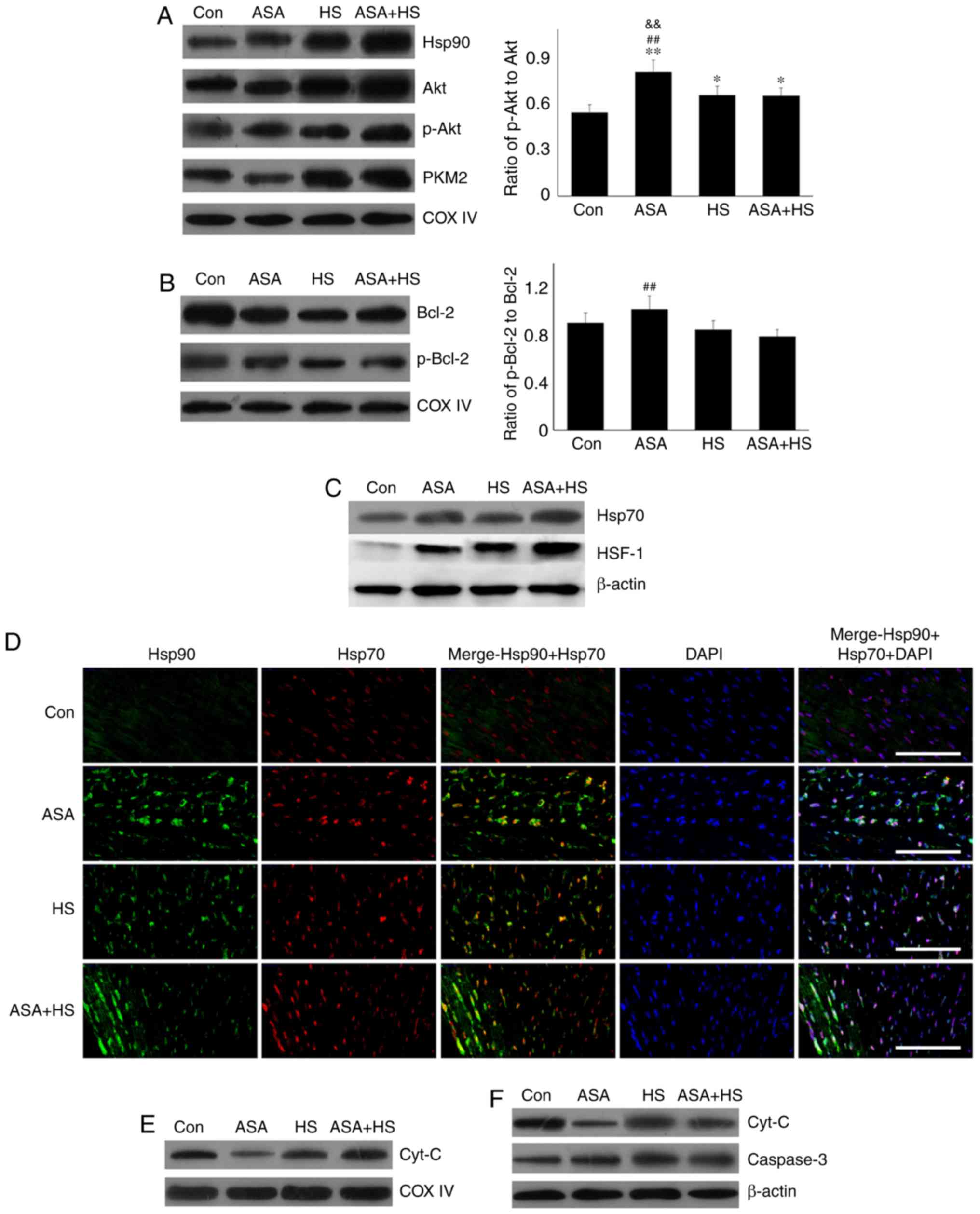
Western blotting revealed that the Hsp90 level in
heart tissues increased significantly (P<0.05) after 5 h of heat
exposure (Fig. 1E). ASA treatment
alone also increased the Hsp90 level. HS in addition to ASA (ASA +
HS) further increased the Hsp90 level significantly (P<0.01),
compared with that in the HS or ASA alone groups. These results
suggested that higher Hsp90 levels induced by ASA were associated
with the remission of heart damage during HS.
Hsp90 activates the Akt and PKM2
signaling pathways in myocardial tissues
Akt is a known client protein of Hsp90 that binds to
the middle domain of Hsp90 (22).
A previous study indicated that PKM2, a key apoptosis regulator
under oxidative stress, is a target of Hsp90. Hsp90 can induce a
conformational change to increase the function of PKM2 (15). Therefore, the levels of Akt,
p-Akt, and PKM2 in the tested heart tissues were analyzed using
western blotting (Fig. 2A).
Compared with the Con group, ASA alone increased the Akt level,
which was further increased by ASA + HS administration. However, HS
did not alter the Akt level. Additionally, compared with a slight
increase in the p-Akt level in the HS group, ASA and ASA + HS
administration further stimulated Akt phosphorylation, particularly
in the ASA + HS group. However, the ratios of p-Akt to Akt in the
ASA and ASA + HS groups were both decreased (P<0.01) but
increased in the HS group. ASA treatment significantly upregulated
the PKM2 level, regardless of the application of HS, while HS
caused only a slight stimulation. Western blotting also revealed
that, compared with the Con group, the Bcl-2 level was decreased
following ASA treatment, accompanied by an increase in the p-Bcl-2
level. The p-Bcl-2 level in the HS group was lower than that in the
ASA + HS group. Consistently, the ratios of p-Bcl-2 to Bcl-2 in the
ASA and ASA + HS groups were decreased (P<0.01), but unchanged
in the HS group.
 | Figure 2Effect of higher Hsp90 levels caused
by ASA on the tested proteins, and the co-localization, apoptosis
and mitochondria of myocardial cells. (A) Representative western
blotting results for Akt, p-Akt, PKM2, Bcl-2 and p-Bcl-2 are shown,
and the ratios of phosphorylated/total protein abundance are
evaluated. (B) Representative immunohistofluorescence staining
images showing the interaction between Hsp90 and Akt or PKM2
(Hsp90, green fluorescence; Akt/PKM2, red fluorescence; nucleus,
blue fluorescence; the merged image of Hsp90 and Akt/PKM2 in the
cytoplasm, yellow fluorescence; the merged image of Hsp90 and
Akt/PKM2 in the nucleus, white fluorescence) in myocardial tissues.
Effect of higher Hsp90 levels caused by ASA on the tested proteins,
and the co-localization, apoptosis and mitochondria of myocardial
cells. (C) Effect of higher Hsp90 abundance on the levels of
GSH-PX, CAT and MDA. (D) Effect of higher Hsp90 abundance on the
levels of cyt c and caspase-3. (E) The apoptotic rate of
myocardial cells in the different groups. (F) The detection of
myocardial mitochondrial function, mPTP and MMP, are shown. The
results are expressed as the mean ± SD; n=5. *P<0.05
and **P<0.01 vs. Con, #P<0.05 and
##P<0.01 vs. ASA + HS, and the comparison between ASA
and HS is indicated by &P<0.05 and
&&P<0.01. |
Hsp90 stabilizes the conformation of client proteins
or activates them, mainly depending on the binding of the
N-terminal ATP binding domain and intermediate domain with client
molecules (23). Therefore, the
co-localization (potential interactions) of Hsp90 with Akt and PKM2
were detected using immunofluorescence staining (Fig. 2B). Co-localization of Hsp90 and
Akt was not observed in the Con group. In the ASA and HS groups,
co-localization between Hsp90 and Akt was detected, but only in the
cytoplasm. In the ASA + HS group, the signal density of
co-localization between Hsp90 and Akt was markedly increased in the
cytoplasm. Notably, a certain amount of co-localization signal was
also observed in the nucleus. Meanwhile, PKM2 in the Con group was
mainly detected in the cytoplasm, with little in the nucleus, and
there was little intracellular co-localization between Hsp90 and
PKM2. In the ASA group, co-localization was markedly increased,
together with an upregulated PKM2 level; however, the
co-localization signal was still distributed only in the cytoplasm.
In the HS group, the co-localization signal was slightly increased,
and its nuclear translocation was observed. In the ASA + HS group,
the co-localization between Hsp90 and PKM2 was markedly increased
in the cytoplasm and nucleus, with an increased signal density to
separate them, compared with that in the ASA or HS groups.
These data suggested that higher levels of Hsp90
induced by ASA promoted Akt expression and activation, and
stimulated PKM2 signaling, possibly via their interaction, to exert
the myocardial protective effect against HS, through the Bcl-2
phosphorylation.
Higher Hsp90 levels enhance the
protection of mitochondria
Mitochondria are the most vulnerable organelles for
intracellular injury. In the myocardium, HS results in cell
peroxidation, inducing swelling of mitochondria, dilation of the
mitochondrial inner cavity, and fracture of the mitochondrial ridge
(24,25). The present study first evaluated
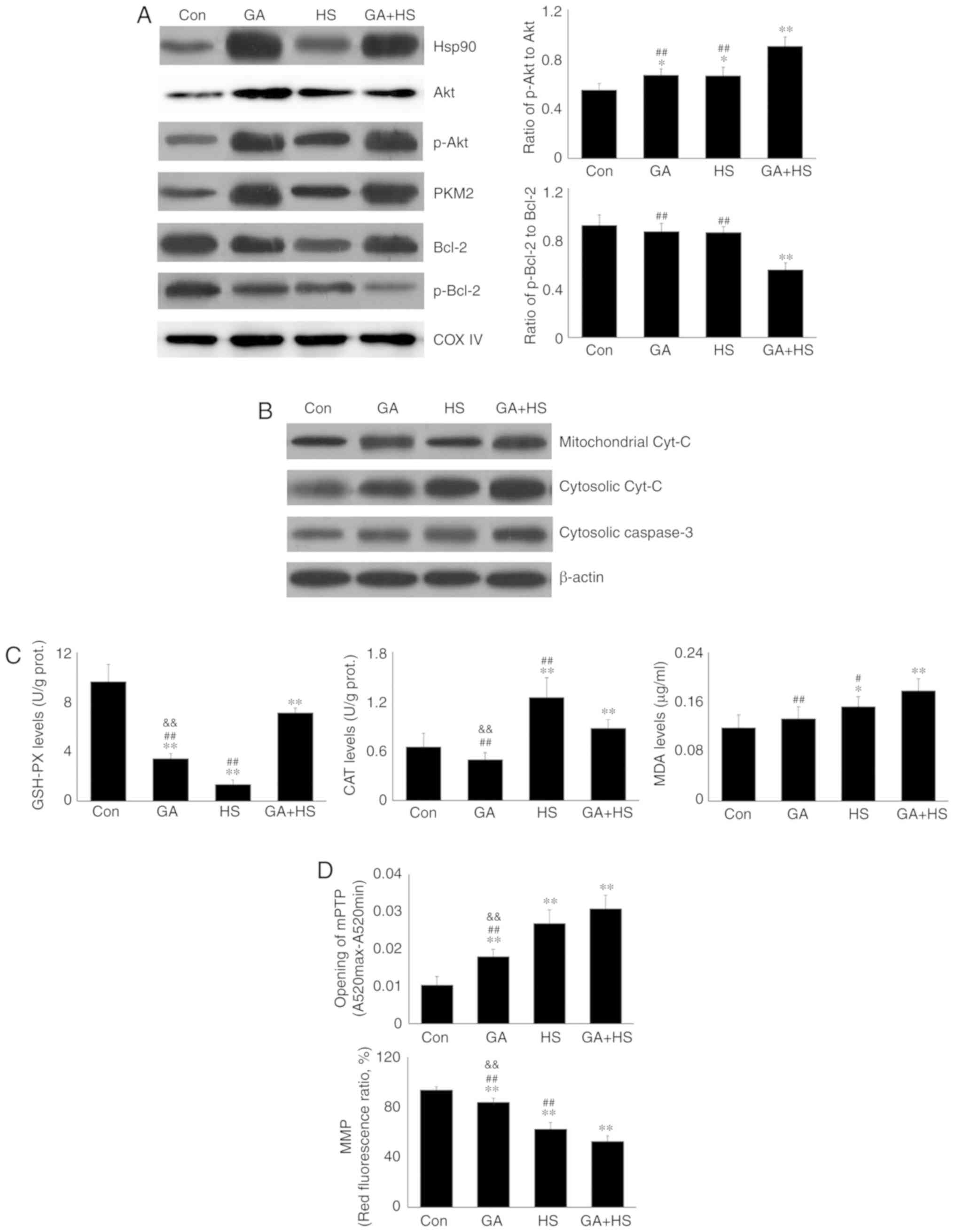
intramitochondrial oxidative stress parameters in the different
groups. HS significantly exacerbated the oxidative status,
characterized by a decrease in GSH-PX, and increased CAT and MDA
levels. Pre-induction of Hsp90 by ASA effectively strengthened the
antioxidant capacity during HS through retaining higher GSH-PX and
CAT activity, thereby decreasing the MDA level (Fig. 2C). An incomplete mitochondrial
structure often results in the release of internal cyt c,
which then activates caspase-3 and leads to the initiation of
apoptosis (26). Our results
revealed that higher levels of cyt c in the HS group
corresponded to increased caspase-3 activation and a higher
apoptosis rate of myocardial cells. Compared with those in the HS
group, ASA treatment reduced the cyt c and caspase-3 levels
during exposure to HS, and decreased the heat-stress-induced
apoptosis rate (Fig. 2D and E).
Consistently, HS induced further opening of the mPTP, and decreased
the MMP, implying the structural failure and functional degradation
of mitochondria. Higher Hsp90 levels in the ASA + HS group reversed
these adverse trends, characterized by decreased opening of the
mPTP and increased MMP (Fig. 2F).
Taken together, these results suggested that the ASA-induced
increase in Hsp90 could effectively reduce HS-related damage to
mitochondria and secondary cellular apoptosis.
Hsp90 induced by ASA promotes p-Akt and
PKM2 translocation into mitochondria
A previous study found that Akt activation in the
cytoplasm alone was not sufficient to induce myocardial protection
in mice with ischemia-reperfusion injury, and the translocation of
phosphorylated Akt from the cytoplasm into the mitochondria was the
key to inducing myocardial protection (27). However, the molecular mechanism of
its anti-apoptotic effect remains unclear. A recent study showed
that p-Bcl-2, the active form of Bcl-2 stimulated by Akt and PKM2,
functions in the mitochondria (15). The results of the present study
showed that HS upregulated the Hsp90, Akt, p-Akt and PKM2 levels
significantly in the mitochondria of rat hearts. Compared with
those in the HS group, in myocardial tissues of the ASA + HS group,
higher Hsp90 levels led to markedly increased levels of these
mitochondrial proteins (Fig. 3A).
Meanwhile, the ratios of mitochondrial p-Akt to Akt in ASA, HS, and
ASA + HS were all increased significantly, particularly in the ASA
group (significantly higher than HS and ASA + HS). Western blotting
revealed that mitochondrial Bcl-2 and p-Bcl-2 levels in the HS
group were decreased compared with those in the Con group. Although
the mitochondrial Bcl-2 levels also decreased markedly, the p-Bcl-2
level and the ratio of mitochondrial p-Bcl-2 to Bcl-2 were
increased in the ASA group, and largely decreased in the ASA + HS,
which was beneficial for resisting HS (Fig. 3B).
 | Figure 3Effect of higher Hsp90 levels
following ASA treatment on the mitochondrial proteins and the
influence of intracellular Hsp70 on Hsp90. (A) Representative
western blotting results of mitochondrial HSP90, Akt, p-Akt and
PKM2 are shown, and the ratios of phosphorylated/total protein
abundance are presented. (B) Representative western blotting
results of mitochondrial Bcl-2 and p-Bcl-2, and the ratios of
phosphorylated/total protein abundance are shown. (C)
Representative western blotting results of cellular Hsp70 and HSF-1
are shown. (D) Representative immunohistofluorescence staining
images showing the co-localization between Hsp90 and Hsp70 (Hsp90,
green fluorescence; Hsp70, red fluorescence; nucleus, blue
fluorescence; the merged image of Hsp90 and Hsp70 in the cytoplasm,
yellow fluorescence; the merged image of Hsp90 and Hsp70 in the
nucleus, white fluorescence) in myocardial tissues. Scale bar=100
µm. (E) Representative western blotting results of
mitochondrial cyt c are shown. (F) Representative western
blotting results of cytosolic cyt c and caspase-3 are shown.
The results are expressed as the mean ± SD; n=5.
*P<0.05 and **P<0.01 vs. Con,
##P<0.01 vs. ASA + HS, and the comparison between ASA
and HS is indicated by &&P<0.01. |
A previous study showed that the
phosphatidylino-sitol-4,5-bisphosphate 3-kinase (PI3K)-Akt
signaling pathway regulates Hsp70 expression by promoting HSF-1
expression and its nuclear translocation, which then critically
contributes toward the chaperone function of Hsp90 through the
interaction between Hsp70 and Hsp90 in multiple myeloma (28). The present study revealed that ASA
and/or HS could effectively induce high levels of HSF-1 and Hsp70.
The Hsp70 levels in the ASA and ASA + HS groups were also higher
than those in the HS group (Fig.
3C). Immunohistofluorescence detection revealed no
co-localization of Hsp70 and Hsp90 in the Con group, and although
ASA treatment alone increased the Hsp70 and Hsp90 levels in the
cytoplasm and nucleus, it did not promote their co-localization. HS
induced a notable co-localization between Hsp70 and Hsp90 in the
nucleus, while the co-localization in the ASA + HS group was
notably higher than that in the HS group, based on of the higher
levels and nuclear translocation of Hsp70 and Hsp90 (Fig. 3D).
Consistently, western blotting revealed that HS
stimulated the cytosolic cyt c level in myocardial cells,
despite no change in the mitochondria, thereby inducing higher
levels of cytosolic caspase-3 and cellular apoptosis, compared with
that in the Con group. The cytosolic cyt c level in the ASA
+ HS group was downregulated despite an increase in mitochondria,
which only caused a lower caspase-3 activity and cellular apoptosis
when compared with that in the HS group (Fig. 3E and F). These results suggested
that increased Hsp90 levels could accelerate the mitochondrial
translocation of p-Akt and PKM2, leading to phosphorylation of
mitochondrial Bcl-2 to protect the integrity of mitochondria from
apoptosis initiation.
Inhibition of Hsp90 function aggravates
the heat-stress injury of mouse hearts
To confirm the protective role of Hsp90-mediated
signaling against HS, GA was used to inhibit the function of Hsp90.
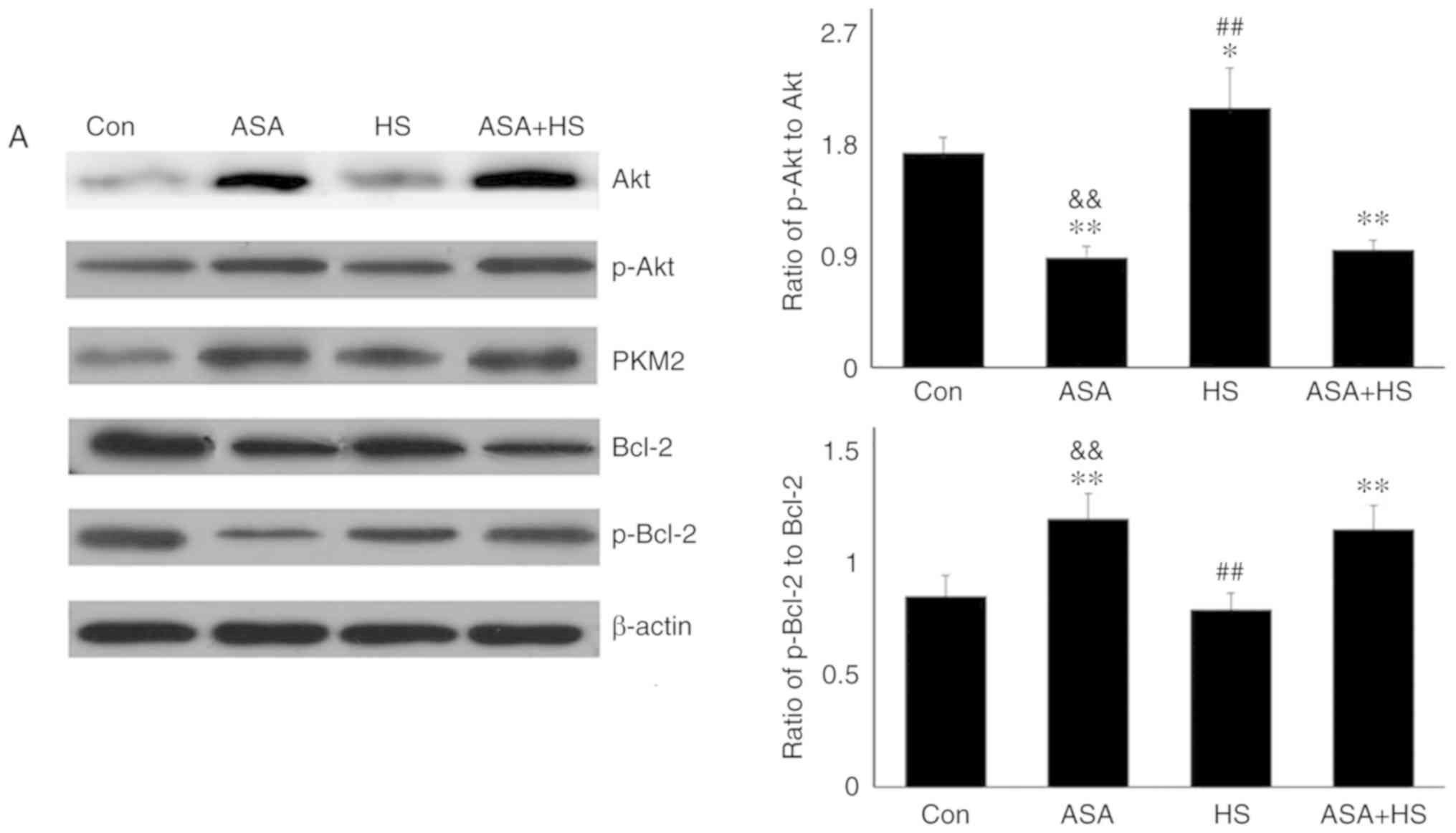
Western blotting revealed that GA efficiently inhibited the
function of Hsp90 in mouse hearts, characterized by marked
increases in Hsp70 and HSF-1 levels in the GA group, despite no
change in the Hsp90 level (Fig.
4A) (29,30). Compared with those in the HS
group, inhibition of Hsp90 in the GA + HS group caused a sharp
downregulation of Hsp90 and HSF-1 levels, and an increase in Hsp70
levels, during heat stress. GA treatment decreased the ratios of
p-Akt to Akt in GA and GA + HS groups, despite upregulation of the
Akt level and its activation. Inhibition of Hsp90 caused PKM2
levels to accumulate in the GA group, which was also observed in
the GA + HS group. GA decreased the levels of Bcl-2 and p-Bcl-2,
including the ratios of p-Bcl-2 to Bcl-2, and treatment with HS and
GA further exacerbated this effect. Immunohistofluorescence
analysis (Fig. 4B) suggested that
the merged signal of Hsp90 and Akt was slightly increased in the GA
group. In addition, in the GA + HS group, the merged signal of
Hsp90 and Akt was weakened and centralized around the nucleus.
Co-localization of Hsp90 and PKM2 was observed in the HS group, but
not in the GA group, while in the GA + HS group, co-localization of
Hsp90 and PKM2 could be detected in the cytoplasm and nucleus, but
was weaker compared with that in the HS group. A clear
co-localization signal of Hsp70 and Hsp90 was detected in the HS
group, but not in the GA and GA + HS groups.
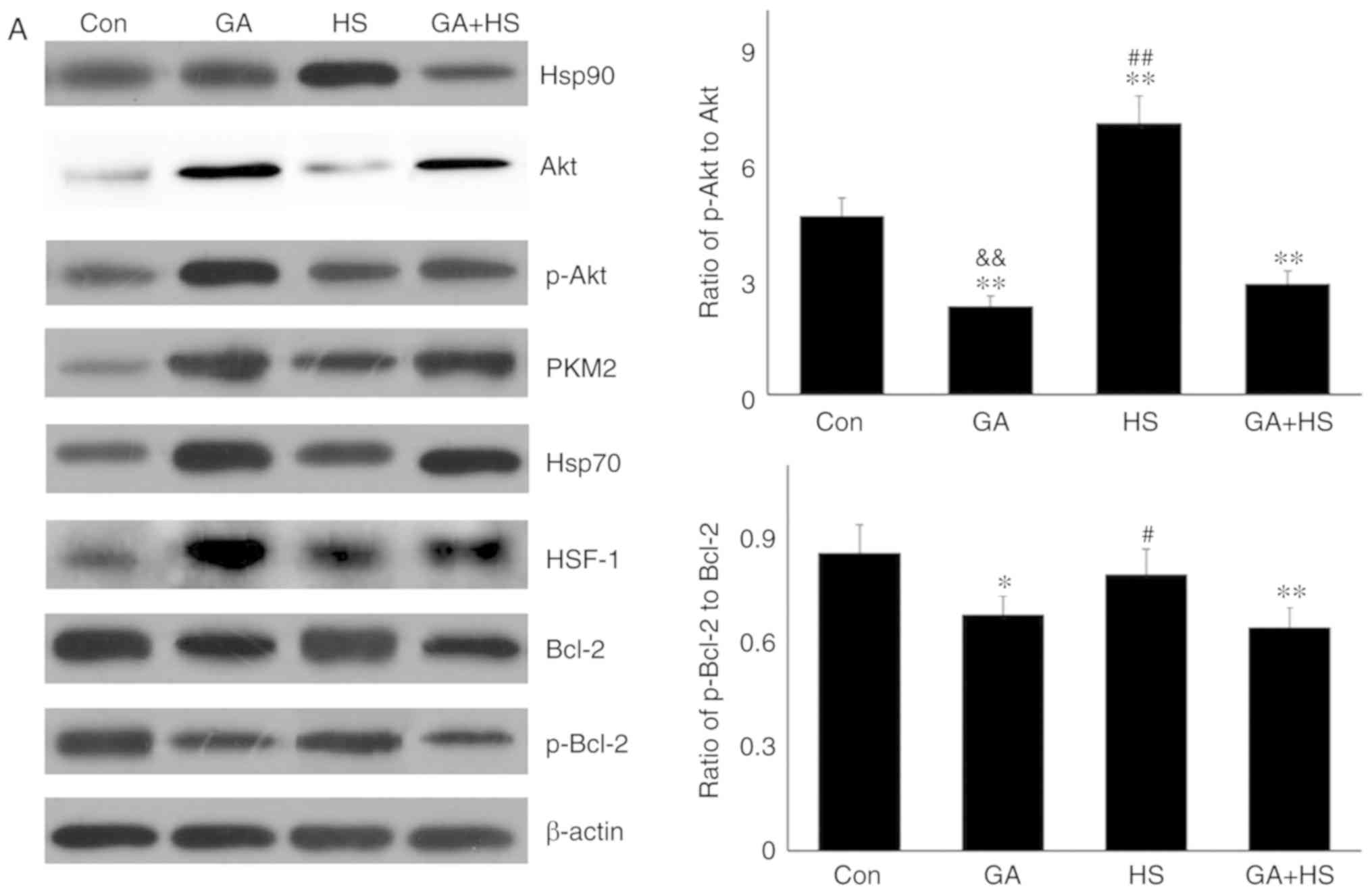 | Figure 4Effect of Hsp90 functional inhibition
on the levels of Hsp90, its associated proteins, and the
co-localization of Hsp90 with tested client proteins. (A)
Representative western blotting results for cellular Hsp90, Akt,
p-Akt, PKM2, Hsp70, HSF-1, Bcl-2 and p-Bcl-2 are shown, and the
ratios of phos-phorylated/total protein abundance are also
presented. (B) Representative immunohistofluorescence staining
images showing the co-localization of Hsp90 with Akt, PKM2 and
Hsp70 (Hsp90, green fluorescence; Akt/PKM2/Hsp70, red fluorescence;
nucleus, blue fluorescence; the merged image of Hsp90 and
Akt/PKM2/Hsp70 in the cytoplasm, yellow fluorescence; the merged
image of Hsp90 and Akt/PKM2/Hsp70 in the nucleus, white
fluorescence) in myocardial tissues. Scale bar=100 µm. The
results are expressed as the mean ± SD; n=5. *P<0.05
and **P<0.01 vs. Con, #P<0.05 and
##P<0.01 vs. ASA + HS, and the comparison between ASA
and HS is indicated by &&P<0.01. |
In consideration of the aforementioned results, the
physiological indices, damage level and apoptosis of mouse hearts
were examined. GA did not alter the heart rate, respiratory rate or
blood oxygen saturation significantly. Compared with that in the HS
group, the heart rate was markedly decreased in the GA + HS group
(P<0.01), while the respiratory rate and blood oxygen saturation
were also slightly inhibited (Fig.
5A). Biochemical analysis revealed that GA treatment increased
the serum LDH level, but CK and AST levels, and compared with those
in the HS group, whereas GA + HS further stimulated the release of
LDH and AST into the blood during HS exposure (P<0.01 and
P<0.05; Fig. 5A). H&E
staining also showed that GA pre-treatment aggravated the
HS-induced damage to myocardial tissues, indicated by increased
myocardial rupture and a higher injury score (Fig. 5B). TUNEL staining (Fig. 5C and D) revealed that the
inhibition of Hsp90 by GA increased HS-induced apoptosis
(P<0.05), compared with that in the HS group. Furthermore, GA
alone increased the levels of cyt c and caspase-3, and HS
further enhanced the induction of cyt c and caspase-3. These
results implied that Hsp90 was involved in the anti-damage response
in heat-stressed mouse hearts, and that this damage may be
associated with the reduced interaction of Hsp90 with Akt, PKM2 and
Hsp70, resulting in decreased Akt and Bcl-2 activation.
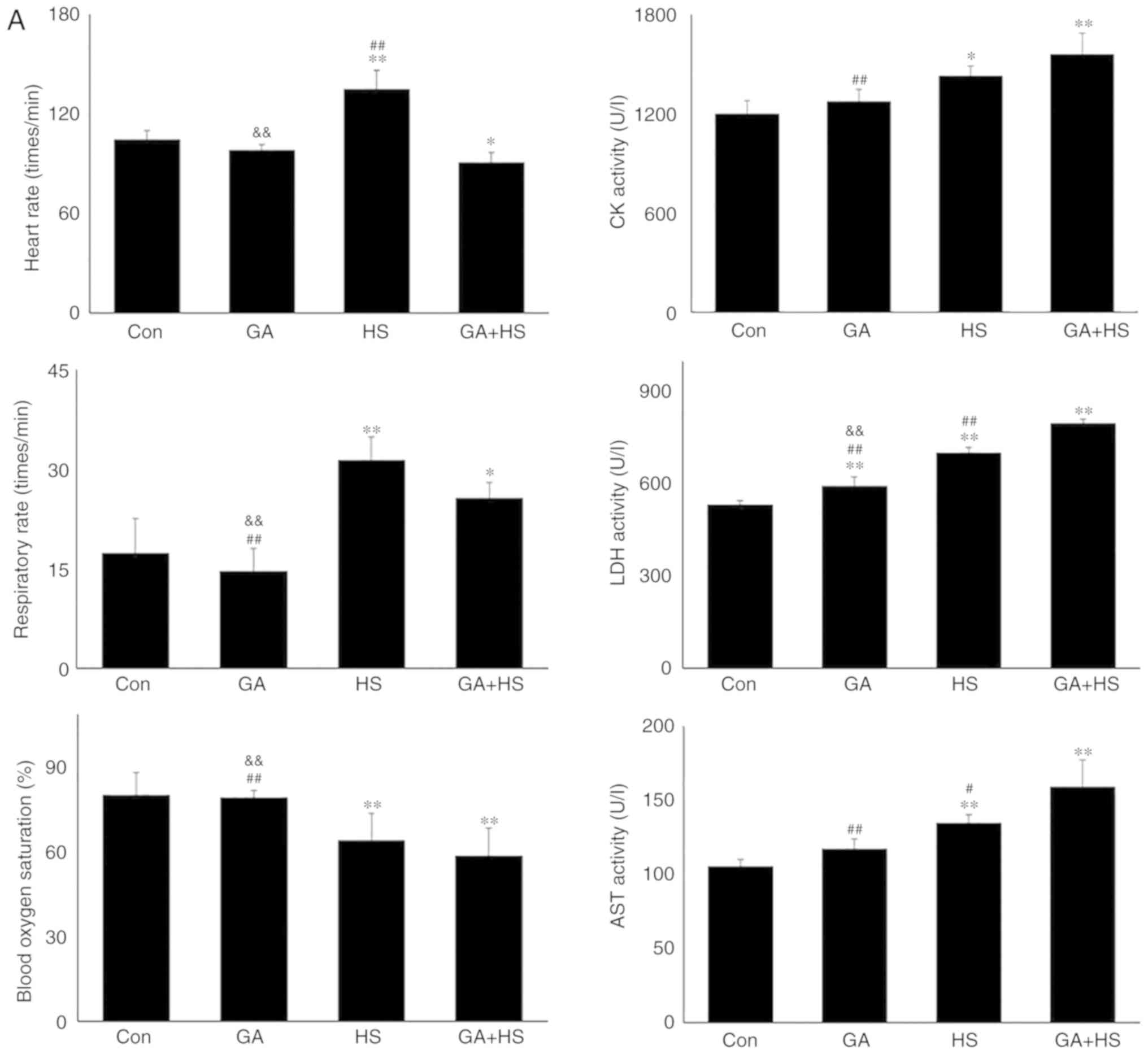 | Figure 5Effect of Hsp90 functional inhibition
on the levels of myocardial injury and apoptosis. (A) Effect of
Hsp90 functional inhibition on physiological indices, such as heart
rate, respiratory rate and blood oxygen saturation, and enzymes
activities associated with myocardial damage. (B) Representative
images showing H&E staining for histopathological examination
and its scores. Panels a-d represent the Con, GA, HS, and GA + HS
groups, respectively. Arrows indicate swelling cells and
degeneration, arrowheads point to hemorrhage, and asterisks mark
necrosis and myofiber rupture. Scale bar=100 µm. Effect of
Hsp90 functional inhibition on the levels of myocardial injury and
apoptosis. (C) Effect of Hsp90 functional inhibition on the cyt
c and caspase-3 levels in heart tissues. (D) Apoptosis rate
of myocardial cells under different treatments. The relative
abundance of all proteins was normalized to that of β-actin. All
the results are expressed as the mean ± SD; n=5.
*P<0.05 and **P<0.01 vs. Con,
#P<0.05 and ##P<0.01 vs. ASA + HS, and
the comparison between ASA and HS is indicated by
&&P<0.01. |
Inhibition of Hsp90 function restricts
the mitochondrial protection of Akt and PKM2
To confirm whether the functional deficiency of
Hsp90 influenced the mitochondrial effect of Akt and PKM2 against
HS in vivo, mitochondria from all related groups were
isolated and studied. As shown in Fig. 6A, the mitochondrial Hsp90 level
was increased by GA treatment to compensate for its functional
insufficiency. Meanwhile, in addition to Akt and p-Akt, the PKM2
levels in the myocardial mitochondria were also increased in
response to GA, regardless of heat stress. Due to the loss of
chaperone protection of Hsp90 on Akt, the ratio of mitochondrial
p-Akt to Akt increased in the GA group, which was further increased
in the GA + HS group. However, western blotting revealed that the
inhibition of Hsp90 by GA markedly antagonized the levels of
mitochondrial Bcl-2 and p-Bcl-2. Notably, compared with that in the
HS group, the mitochondrial p-Bcl-2 level in the GA + HS group
remained markedly restrained, consistent with the ratio of p-Bcl-2
to Bcl-2. These implied that Hsp90 did not affect the mitochondrial
translocation of Akt and PKM2 directly; however, it was responsible
for p-Akt and PKM2 assisting Bcl-2 in entering into mitochondria
and the initiation of its phosphorylation.
Western blotting (Fig.
6B) revealed that GA treatment markedly increased the
mitochondrial cyt c level, but not the cytosolic cyt
c level. HS following GA treatment increased the
mitochondrial and cytosolic cyt c levels simultaneously.
Furthermore, GA did not markedly influence the oxidation and
antioxidation indices. Compared with the HS group, GA followed by
HS markedly stimulated the mitochondrial GSH-PX and MDA activities
(Fig. 6C). GA promoted the
opening of the mPTP and decreased the MMP (Fig. 6D).
Compared with the HS group, GA followed by HS
further promoted mPTP opening and lowered the MMP (P<0.01).
These results indicated that inhibition of Hsp90 could aggravate
HS-induced mitochondrial damage by restricting Bcl-2 activity.
Inhibition of Akt activation exacerbates
the damage to mouse hearts
The aforementioned results demonstrated that Hsp70
and HSF-1 levels increased after the inhibition of Hsp90,
regardless of HS, suggesting that in the absence of Hsp90 function,
Akt was probably activated by an alternative pathway and increased
Hsp70 levels served to regulate the Hsp90 function. To confirm the
hypothesis and its association with PKM2 in vivo, the Akt
phosphorylation inhibitor TR was used to restrict the production of
p-Akt in mouse myocardial tissues. Western blotting (Fig. 7A) revealed that, compared with
that in the Con group, TR alone slightly decreased the Akt level,
but significantly decreased the p-Akt, Bcl-2 and p-Bcl-2 levels,
and decreased the ratio of p-Akt to Akt, accompanied by a decrease
in Hsp90. HS following TR treatment further decreased the levels of
Hsp90, p-Akt, Bcl-2 and p-Bcl-2, but increased the
non-phosphorylated Akt level (thereby causing further decrease in
the ratio of p-Akt to Akt), while HS alone could induce a high
level of Hsp90 (compared with the Con group). Compared with that in
the Con group, the PKM2 level increased in the TR group, while that
in the TR + HS group was higher than that in the HS group. This may
be responsible for the increase in the ratio of p-Bcl-2 to Bcl-2.
Compared with that in the Con group, the HSF-1 level increased
following TR treatment; however, it was not translocated into the
nucleus to exert its function, particularly considering that Hsp70
levels were not increased in the TR group. Compared with the high
level of HSF-1 in the HS group, in the TR + HS group, the HSF-1
level notably decreased, accompanied by the sharp downregulation of
Hsp70 synthesis.
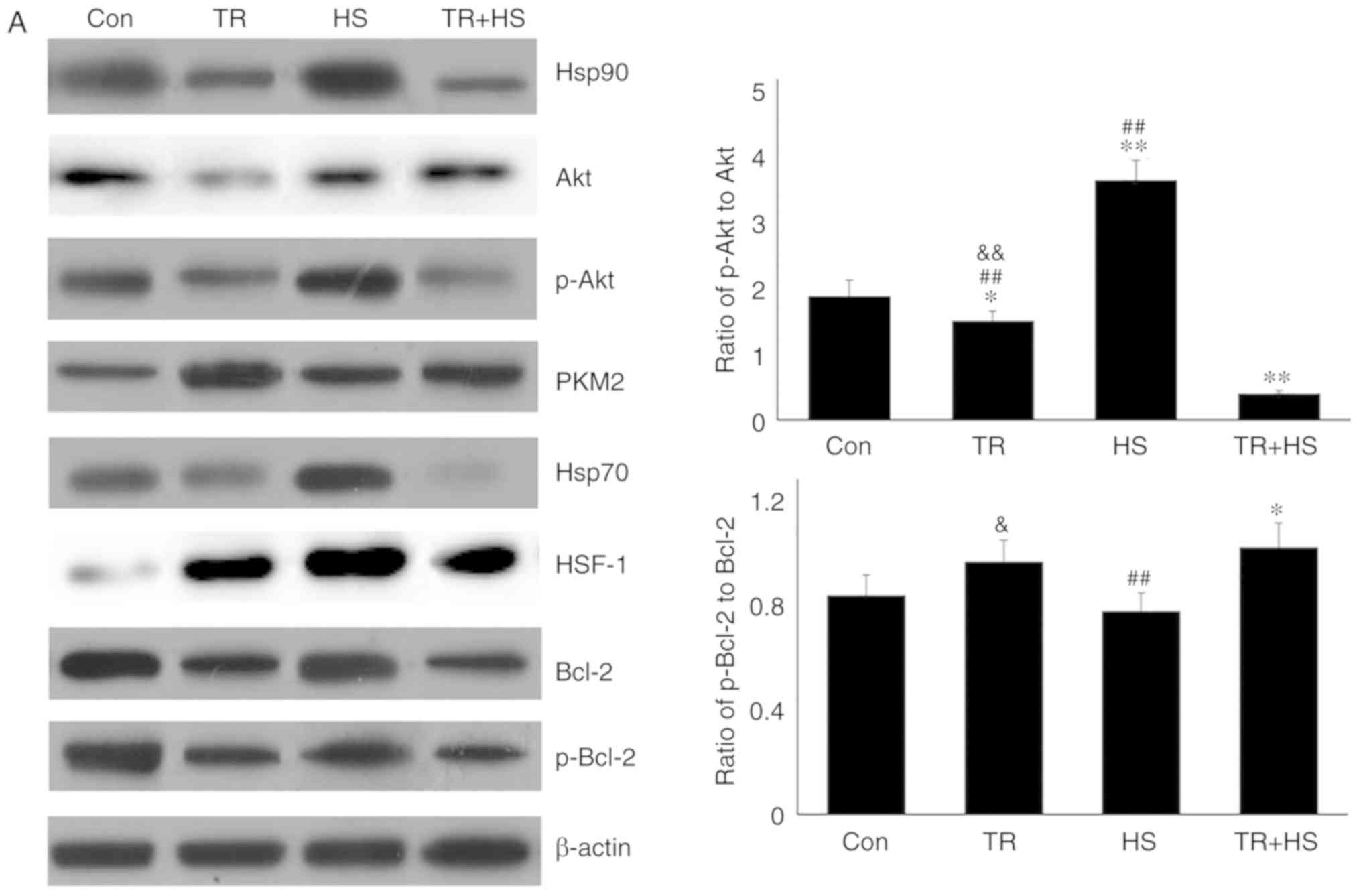 | Figure 7Effect of Akt activation inhibition
on the levels of Hsp90, its associated proteins, and the
co-localization of Hsp90 with tested client proteins. (A)
Representative western blotting results of Hsp90, Akt, p-Akt, PKM2,
Hsp70, HSF-1, Bcl-2 and p-Bcl-2 in the tested myocardial tissues,
and the ratios of phosphorylated/total protein abundance. (B)
Representative immunohistofluorescence staining images showing the
interaction of Hsp90 with Akt, PKM2 and Hsp70 (Hsp90, green
fluorescence; Akt/PKM2/Hsp70, red fluorescence; nucleus, blue
fluorescence; the merged image of Hsp90 and Akt/PKM2/Hsp70 in the
cytoplasm, yellow fluorescence; the merged of Hsp90 and
Akt/PKM2/Hsp70 in the nucleus, white fluorescence) in myocardial
tissues. Scale bar=100 µm. The relative abundance of all
proteins was normalized to that of β-actin. All the results are
expressed as the mean ± SD; n=5. *P<0.05 and
**P<0.01 vs. Con, ##P<0.01 vs. ASA +
HS, and the comparison between ASA and HS is indicated by
&P<0.05 and &&P<0.01. |
Immunohistofluorescence detection (Fig. 7B) revealed that the
co-localization of Hsp90 and Akt increased in TR-treated heart
tissues, and was mainly distributed in the cytoplasm of myocardial
cells. Compared with that in the HS group, the co-localization
between Hsp90 and Akt was reduced in the TR + HS group. The
co-localization of Hsp90 and PKM2 in the TR group was observed
primarily in the cytoplasm. The merged signal of Hsp90 and PKM2
could still be detected in the TR + HS group, but was not increased
compared with that in the TR group. The co-localization of Hsp70
and Hsp90 in the TR and TR + HS groups was not observed,
considering the decrease in the Hsp70 and Hsp90 signals.
As demonstrated in Fig. 8A, TR alone did not alter the heart
rate, respiratory rate and blood oxygen saturation. Furthermore,
compared with those induced by TR alone, HS following TR
administration further increased the heart rate (P<0.01) and
decreased blood oxygen saturation (P<0.01). Biochemical
detection revealed that TR significantly upregulated the serum LDH
level, but had no effect on CK and AST. HS exposure following TR
significantly increased the activities of CK, LDH and AST
(P<0.01), compared with those in the TR or HS groups.
Pathological examination revealed that the injury score following
TR treatment was slightly higher than that in the Con group, while
the score in the TR + HS group was slightly higher than that in the
HS group (Fig. 8B). In Fig. 8C, the cyt c level in the TR
+ HS group was significantly increased compared with that in the
Con group, but was lower than that in the HS group (P<0.05). In
addition, the caspase-3 levels in the TR + HS group were
significantly higher than those in the HS group (P<0.01).
Compared with that in the HS group, the apoptotic rate of
myocardial cells in the TR + HS group was further stimulated in the
tested hearts (Fig. 8D).
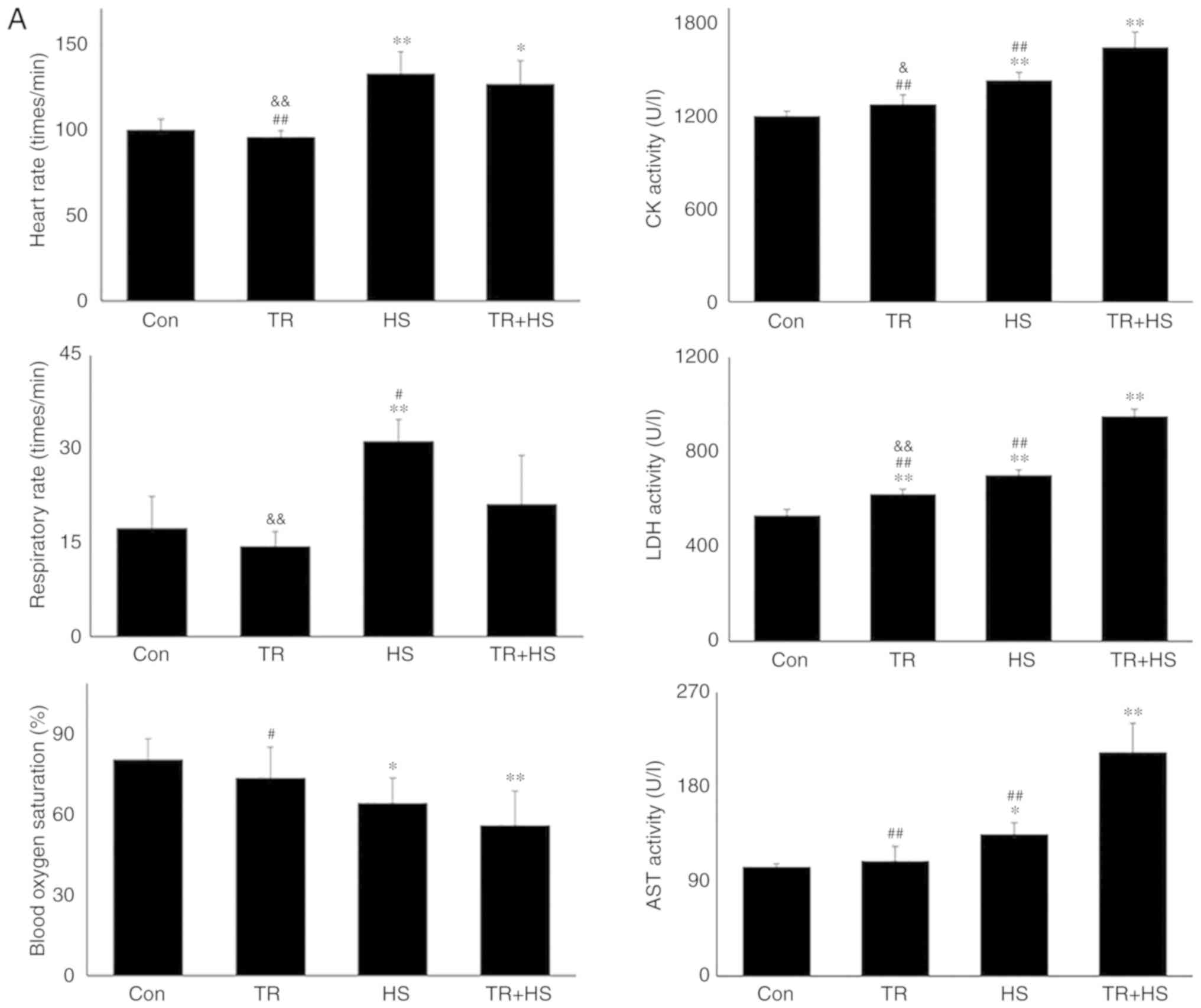 | Figure 8Effect of Akt activation inhibition
on the levels of myocardial injury and cell apoptosis. (A) The
changes in the heart rate, respiratory rate, blood oxygen
saturation and serum enzymes levels in response to TR. (B)
Representative H&E staining images of experimental mouse heart
tissues and pathological scores. Panels a-d represent the Con, TR,
HS, and TR + HS groups, respectively. Arrows indicate swelling
cells and degeneration, arrowheads point to hemorrhage, and
asterisks mark necrosis and myofiber rupture. Scale bar=100
µm. Effect of Akt activation inhibition on the levels of
myocardial injury and cell apoptosis. (C) Effect of Akt activation
inhibition on the cyt c and caspase-3 levels in heart
tissues. (D) The apoptotic rate of myocardial cells under different
treatments. The relative abundance of all proteins was normalized
to that of β-actin. All the results are expressed as the mean ± SD;
n=5. *P<0.05 and **P<0.01 vs. Con,
#P<0.05 and ##P<0.01 vs. ASA + HS, and
the comparison between ASA and HS is indicated by
&P<0.05 and &&P<0.01. |
These findings indicated that under conditions of
high Hsp90 levels induced by HS, inhibiting Akt activation markedly
decreased the Bcl-2 level and its phosphorylation, thereby inducing
more serious myocardial injury.
PKM2 cannot compensate the deletion of
mitochondrial protection caused by inhibition of Akt
activation
As shown in Fig.
9A, compared with that in the Con group, TR caused more Hsp90,
Akt, and p-Akt to move into mitochondria, but more decrease of the
ratio of p-Akt to Akt. It implied that when the levels of survival
molecules in myocardial cells were insufficient, mitochondria were
protected preferentially. However, the levels of mitochondrial
Hsp90, Akt, and p-Akt in the TR + HS group were significantly lower
(P<0.01) than those in the TR and HS groups, including the ratio
of p-Akt to Akt. Furthermore, compared with those in the Con group,
the Bcl-2 and p-Bcl-2 levels decreased following TR treatment, with
and without HS treatment, and their levels in the TR + HS group
were lower than those in HS group. The ratio of mitochondrial Bcl-2
and p-Bcl-2 was not significantly altered by TR treatment, as in
the HS alone group. As shown in Fig.
9B, TR exposure stimulated the synthesis of mitochondrial cyt
c, which leaked out of the mitochondria in the TR + HS
group. The level of cytosolic cyt c was not influenced by TR
alone, but was upregulated following subsequent HS exposure.
Mitochondrial biochemistry analysis (Fig. 9C) revealed that GSH-PX activities
in the TR, HS and TR + HS groups all decreased. TR increased the
CAT level, but not that of MDA. HS following TR further induced CAT
(P<0.01) and MDA levels, compared with those in the HS group.
Mitochondrial function detection (Fig. 9D) demonstrated that TR alone
increased mPTP opening and decreased the MMP. HS following TR
induced a further decrease in the MMP compared with that in the HS
group.
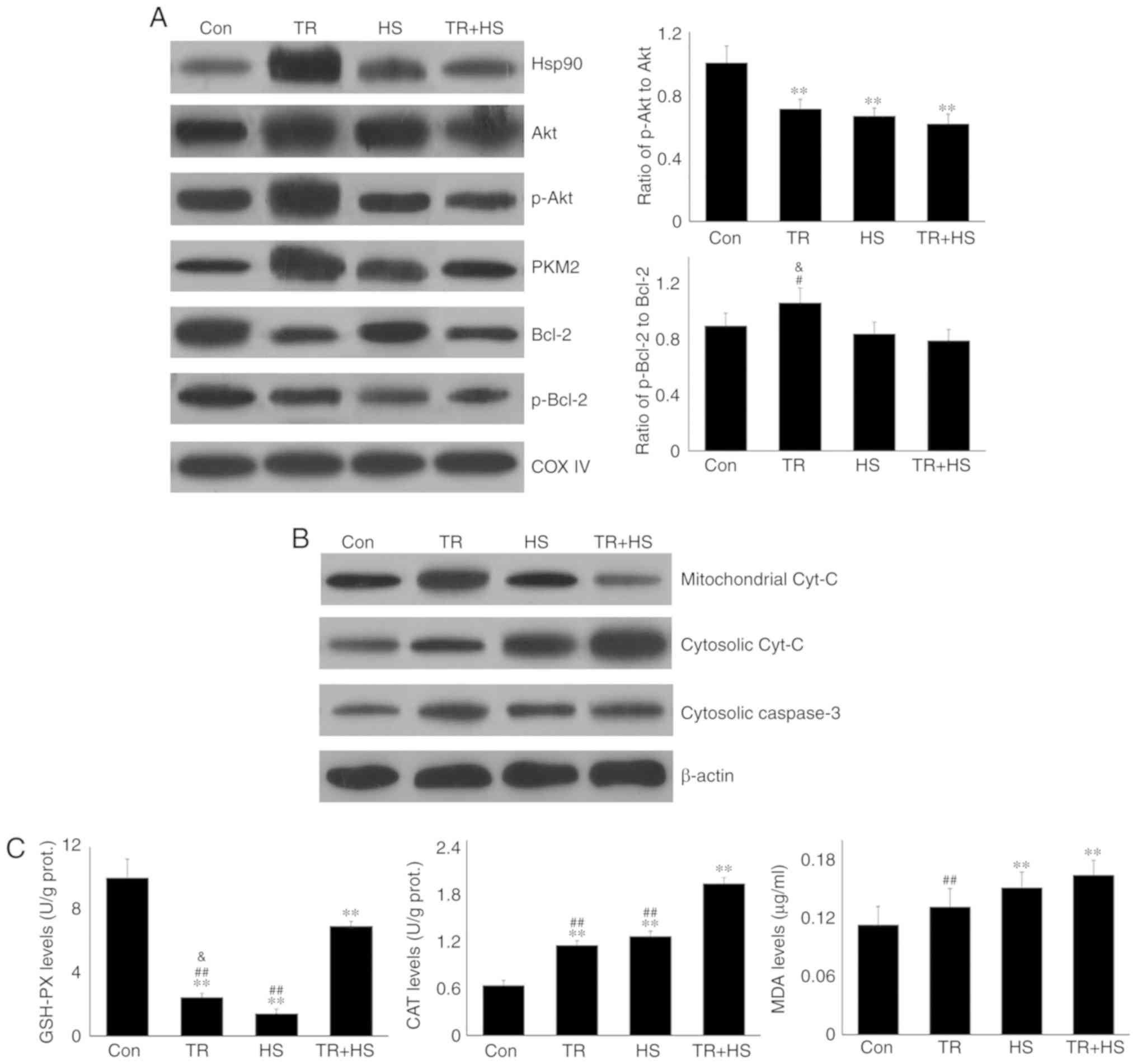 | Figure 9Effect of Akt activation inhibition
on the mitochondrial and cytosolic levels of Hsp90 and associated
proteins, the oxidation levels and function of myocardial
mitochondria, and different treatments on the serum levels of Hsp70
in the tested mice. (A) Representative western blotting results of
mitochondrial Hsp90, Akt, p-Akt, PKM2, Bcl-2 and p-Bcl-2. (B)
Representative western blotting results of mitochondrial cyt
c, and cytosolic cyt c and caspase-3. (C) Effects of
Akt activation inhibition on the oxidative levels of myocardial
mitochondria. (D) Effects of Akt activation inhibition on the
opening of mPTP, and MMP of myocardial mitochondria, respectively.
(E) Effects of different treatments on the serum levels of Hsp70 in
the tested mice. The relative abundance of all proteins was
normalized to that of β-actin. All the results are expressed as the
mean ± SD; n=5. *P<0.05 and **P<0.01
vs. Con, #P<0.05 and ##P<0.01 vs. ASA +
HS, and the comparison between ASA and HS is indicated by
&P<0.05 and &&P<0.01. |
These results suggested that inhibition of Akt
phosphorylation could further intensify mitochondrial injury of
heat-stressed myocardial cells by inhibiting mitochondrial Bcl-2
and its phosphorylation, even though PKM2 exerted its normal
function on Bcl-2.
Hsp90 expression promotes Hsp70 entry
into blood to improve cardiovascular function
Serum Hsp70 was also detected in the present study.
As shown in Fig. 9E, ASA, GA or
TR alone did not alter the level of serum Hsp70 significantly,
which contrasted with its significant downregulation by HS
(P<0.01 or P<0.05). Notably, ASA markedly increased the serum
Hsp70 level of heat-stressed mice in the ASA + HS group, while GA
and TR did not, implying that increasing the serum Hsp70 level may
be another method by which ASA protects the heat-stressed
heart.
Discussion
Cardiomyocytes are engaged continuously in
generating the powerful contractile force to pump blood to the
whole body, and heat stress can lead to a surge in metabolic
demand, followed by functional and structural disruption of these
cells, resulting in heart failure (31,32). Our previous in vivo and
in vitro studies confirmed that Hsp90 levels were high
during heat stress, and were closely associated with resisting heat
stress damage to the heart (8,19).
However, the underlying mechanism of how Hsp90 exerts these effects
remains elusive. In the present study, high levels of Hsp90 were
observed in heat-stressed heart tissues, and ASA, an effective
inducer of HSP90, was used to further enhance HSP90 levels to
investigate its signaling pathways. The higher Hsp90 levels induced
by ASA significantly reversed the HS-induced myocardial injury and
increased blood oxygen saturation effectively by regulating the
heart rate. Further investigation revealed that, during HS, higher
Hsp90 levels were accompanied by higher levels of Akt and its
phosphorylated version in cardiac tissue. The interaction of Akt
with Hsp90 is necessary to activate Akt, which works on downstream
survival signal proteins (33).
Consistently, in the present study, ASA treatment also stimulated
the interactive co-localization of Hsp90 and Akt, and promoted the
nuclear translocation of Hsp90 in myocardial cells. Activated Akt
functions to resist cellular oxidative stress and then inhibit
apoptosis, mainly through its regulation of Bcl-2 (34,35). We hypothesized that the decrease
in the ratio of p-Akt to Akt in myocardial tissue in the ASA
treatment groups was mainly due to effective chaperone protection
of higher Hsp90 on more Akt proteins. Meanwhile, PKM2 levels were
upregulated in response to the Hsp90 inducer, ASA. Evidence
suggests that the ATPase activity of Hsp90 facilitates the
interaction between PKM2 and Bcl-2, and then phosphorylates Bcl-2
(15). Co-localized interaction
is also necessary for Hsp90 to exert its auxiliary function on the
target complex (36). The
immunohistofluorescence analysis of the present study also showed
that higher Hsp90 levels after ASA treatment strengthened the
interaction between Hsp90 and PKM2.
The present study also revealed that, although Bcl-2
levels were decreased, the p-Bcl-2 levels and the ratio of p-Bcl-2
to Bcl-2 in the ASA and ASA + HS groups were increased compared
with those in the Con and HS groups, respectively. This indicated
that the activation of Bcl-2 was promoted by the Hsp90-Akt-p-Akt
and Hsp90-PKM2 axes, which contradicted a previous study
demonstrating that the Bcl-2 level should match the increase in Akt
activation (37). Bcl-2 acts as a
nodal point at the convergence of multiple pathways to inhibit
apoptosis in peroxidized cells, particularly in the protection of
mitochondrial structure and function (38,39). In mitochondria, Bcl-2
phosphorylation prevents the binding of Cul3-based E3 ligase to
Bcl-2, thereby enhancing cellular resistance to oxidative stress
(15).
In myocardial cells, mitochondria constitute ~45% of
the volume and manufacture ~90% of the ATP, and myocardial tissue
is particularly sensitive to free radicals due to its low levels of
antioxidant enzymes (40,41). Stress-induced ROS generation
causes mitochondrial dysfunction, resulting in irreversible
myocardial oxidative injury (40,42). Heat stress markedly increased the
degree of mitochondrial peroxidation and decreased the GSH-PX
activities, thereby disturbing the mitochondrial structure and
function, characterized by the increased opening of mPTP and the
decreased MMP. These adverse conditions were significantly reversed
by the higher Hsp90 levels resulting from ASA pre-treatment,
confirming a positive role of Hsp90 in HS-induced myocardial
mitochondria injury (43).
Mitochondrial-dependent intrinsic apoptosis of cardiomyocytes is
initiated by the leakage of mitochondrial cyt c and
initiation of caspase-3, and is the main cause of myocardial
damage, including myofiber loss and cardiac dysfunction (8,44).
The results of the present study indicated that higher Hsp90 levels
could inhibit HS-induced apoptosis by decreasing caspase-3 levels
in cardiac tissue.
Hsp90 also serves a key role in protein
mitochondrial translocation (45). The results of the present study
showed that increased mitochondrial Hsp90 protein further increased
the total amount of mitochondrial PKM2, Akt and p-Akt during heat
stress, including the ratio of mitochondrial p-Akt to Akt. The
stronger cytoplasmic co-localization of Akt/PKM2 with higher Hsp90
levels may suggest that mitochondrial translocation of Akt and PKM2
is completed with the assistance of Hsp90. This is in accordance
with the results of another study on hypothermic preservation of
rat hearts (21). The low levels
of mitochondrial Bcl-2 during exposure to HS were partially
reversed by the increased level of mitochondrial Hsp90 in the ASA +
HS group, making them beneficial for the production of p-Bcl-2.
Based on this increased protection from Bcl-2 and the consumption
of p-Bcl-2 on the mitochondrial structure, mitochondrial cyt
c could not leak into the cytoplasm to initiate caspase-3
dependent apoptosis. These all originated from the regulation of
Hsp90 on PKM2 and Akt, and enabled protection of the myocardium via
decreasing apoptosis.
In addition, the Akt pathway regulates the
expression and nuclear translocation of HSF-1, followed by
constitutive and inducible Hsp70 expression (28). Furthermore, Hsp70 serves an
essential role in the substrate-loading phase of the chaperone
function of Hsp90 by interacting functionally with the
Hsp90-complex (46). Consistent
with the aforementioned studies, the results of the present study
also demonstrated that a higher level and activation of Akt,
including its nuclear import in the ASA + HS group, induced HSF-1
and Hsp70 expression, followed by an increase in the
co-localization between Hsp70 and Hsp90, which critically
contributes toward the chaperone function of Hsp90 in myocardial
cells.
To confirm the aforementioned effect of Hsp90, GA
was used to inhibit its chaperone function in vivo. Previous
studies have indicated that GA did not alter Hsp90 levels, but
stimulated HSF-1 nuclear translocation and Hsp70 expression
(20,29,30). Activated HSF-1 in the nucleus can
stimulate the transcription of the HSP70 gene (47). Consistently, the present study
demonstrated that GA alone did not influence the Hsp90 level, but
increased the levels of Hsp70 and HSF-1. In addition, the ratio of
p-Akt to Akt and co-localization of Hsp90 with PKM2 and Hsp70 were
all decreased. These results proved that GA effectively inhibited
the function of Hsp90, which resulted in accumulation of Akt and
PKM2 in myocardial tissues, thereby decreasing the ratio of p-Akt
to Akt, and slightly strengthening the fake merge signal of Hsp90
and Akt. Finally, the levels of Bcl-2 and p-Bcl-2 (the executors
that protect myocardial cells) and the ratio of p-Bcl-2 and Bcl-2
were restricted effectively with and without HS exposure, which
indicated that the chap-erone function of Hsp90 was required to
assist Akt activation and p-Akt/PKM2-mediated phosphorylation of
Bcl-2 to resist heat-stress damage. It was also found that when
Hsp90 was functionally inhibited, mitochondrial p-Akt levels
remained increased, including Akt and PKM2, even resulting in an
increase in the ratio of p-Akt to Akt. This suggested that Hsp90
may serve an auxiliary role in their mitochondrial translocation,
instead of being strictly required. However, Hsp90 remained
necessary for p-Akt and PKM2 to regulate mitochondrial Bcl-2 and
its phosphorylation, evidenced by the significant decrease in the
ratio of mitochondrial p-Bcl-2 to Bcl-2, which was consistent with
previous studies showing that Hsp90 altered the conformation of
intermitochondrial client proteins (e.g., p-Akt and PKM2) and
facilitated the interaction between them and Bcl-2 to inhibit Bcl-2
degradation and secondary apoptosis (15,22). Inhibition of Hsp90 by GA
intensified the heat-stress damage to mitochondrial integrity,
followed by apoptosis of myocardial cells, which resulted in more
serious pathological injury, accompanied by lower blood oxygen
saturation.
Based on the aformentioned results, the preferred
target of Hsp90 should be p-Akt rather than Akt during heat stress.
To confirm this hypothesis and clarify the cross-talk between
Hsp90-Akt-Bcl-2 and Hsp90-PKM2-Bcl-2, Triciribine (TR) was used to
inhibit the phosphorylation of Akt at amino acids 308 and 473
(48). TR is highly selective for
Akt and does not inhibit the activation of other kinases, such as
phosphatidylinositol 3 kinase and phosphoinositol-dependent kinase
1 (48). In the present study,
although PKM2 exhibited higher expression and Hsp90 was extensively
degraded in the TR and TR + HS groups, the inhibition of Akt
activation by TR still significantly decreased the levels of Bcl-2
and p-Bcl-2. Additionally, HS aggravated this adverse impact,
thereby exacerbating heat-stress-induced tissue injury, even though
the ratio of p-Bcl-2 and Bcl-2 showed an increase. Previous studies
have suggested that PKM2 is capable of phosphorylating proteins as
a protein kinase, and PKM2-dependent Bcl-phosphorylation is
required to sustain the Bcl-2 protein level (15,49). This indicated that during heat
stress, p-Akt was necessary for Hsp90 to influence Bcl-2 and its
phosphorylation, and the Hsp90-PKM2 axis functions independently,
but was not sufficient enough to account for the deficiency in Akt
signaling. In mitochondria, TR also decreased the p-Akt level and
the ratio of mitochondrial p-Akt to Akt, and HS was further
downregulated it. Furthermore, despite the large consumption of
mitochondrial Hsp90 and PKM2, the reduction of the Bcl-2 and
p-Bcl-2 levels was not reversed during heat stress, and the ratio
of mitochondria p-Bcl-2 and Bcl-2 was also unaltered. The loss of
protective Bcl-2 by TR treatment induced more severe
intermitochondrial peroxidation and myocardial apoptosis and
damage.
An in vitro study on multiple myeloma cells
revealed substantial downregulation of HSF-1 upon PI3K inhibition,
which was accompanied by strong downregulation of Hsp70 (28). Similarly, the downregulation of
p-Akt by TR also abrogated the accumulation of Hsp70, particularly
under HS. However, the present study also noted that HSF-1 levels
increased following TR treatment, regardless of HS. Considering
that HSF-1 performs its function in the nucleus (20), and the fluorescence signal of
nuclear Akt was weakened in the TR and TR + HS groups, the results
suggested that Akt signaling may also be the regulator of the
nuclear translocation of HSF-1, and that its expression could be
affected by other factors. A previous study showed that
pharmacological inhibition of Hsp90 strongly induced Hsp70, and the
HSF-1-dependent induction of Hsp70 is controlled by PI3K (28). Consistently, Hsp70 was upregulated
by GA in the present study. However, the activation of the Akt
signaling pathway, as represented by the p-Akt level, remained at a
high level. Therefore, an alternative way to activate Akt should
exist in vivo, and remains to be further studied.
In addition, previous studies have confirmed that
extracellular Hsp70 exists in patients with arthritis,
atherosclerosis or other diseases, and is associated with the
disease severity (50,51). However, the source of
extracellular and serum Hsp70 remains unclear. The present study
analyzed serum Hsp70 levels in the experimental mice and found that
HS followed by ASA could significantly stimulate the serum Hsp70
level. This suggested that HS damages the myocardial cell membrane
and causes intracellular Hsp70 to flow into the blood (51,52). At this point, Hsp70 acts as a
cytokine, rather than a molecular chaperone, and combines with
Toll-like receptor (TLR)-4 on the myocardial cell membrane, which
activates downstream effectors to initiate the transcription of
cytokine genes (e.g., those encoding tumor necrosis factor-α,
interleukin-1, -6, -8 and -12) and accessory adhesion molecules
(CD80 and CD86), which are important self-protective cellular
factors (52). The results of the
present study also showed that inhibition of Akt activation
restricted serum Hsp70 levels by downregulating intracellular
Hsp70. However, the increase in intracellular Hsp70 induced by GA
did not cause upregulation of serum Hsp70, but the mechanism
underlying this requires further investigation. Furthermore,
PI3K-Akt is also a known classic autophagy pathway; therefore, the
association between Hsp90 and mitochondrial autophagy should be
further investigated in future experiments.
Taken together, as shown in Fig. 10, the results of the present
study provided in vivo mechanistic evidence that Hsp90 acts
as a myocardial protective molecule against heat-stress-induced
apoptosis and damage via the Akt-Bcl-2 and PKM2-Bcl-2 survival
signaling pathways at the cellular and mitochondrial levels, which
contributes toward preserving cardiac function and mitochondrial
homeostasis, thereby alleviating oxidative stress. The two
signaling pathways act independently, and the former is
predominant. Furthermore, the serum Hsp70 that leaks from
myocardial cells may also participate in resisting heat stress.
However, the present result would preferably be verified using a
specific inducer of Hsp90, as there are multi-target effects of
ASA. Therefore, identifying a specific Hsp90 agonist or developing
a gene transfection method would be beneficial in a future in
vivo study on heat stress. In conclusion, the present study may
serve as a foundation for further investigation of the protection
offered by Hsp90 induction against heat-stress damage, and
identifies several novel potential targets to develop drugs that
protect against heat stress.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation Youth Funding Project of China
(grant no. 31802157), the Postdoctoral Science Foundation of
Jiangsu Province (grant no. 2018K206C), the National Natural
Science Foundation of China (grant no. 31672520), and the Initial
Scientific Research Fund of Young Teachers in Nanjing Agricultural
University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
XHZ and EDB were involved in developing the concept
and design of the study. XHZ, JXW, JZS, BY and JRS performed the
experiments. XHZ analyzed the data and prepared the manuscript. EDB
revised the manuscript. All authors have read and approved the
final version of the manuscript submitted for publication.
Ethics approval and consent to
participate
All animal experiments were performed according to
the guidelines of the regional Animal Ethics Committee and were
approved by the Institutional Animal Care and Use Committee of
Nanjing Agricultural University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sandercock DA, Hunter RR, Nute GR,
Mitchell MA and Hocking PM: Acute heat stress-induced alterations
in blood acid-base status and skeletal muscle membrane integrity in
broiler chickens at two ages: Implications for meat quality. Poult
Sci. 80:418–425. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bettaieb A and Averill-Bates DA:
Thermotolerance induced at a fever temperature of 40 degrees C
protects cells against hyperthermia-induced apoptosis mediated by
death receptor signalling. Biochem Cell Biol. 86:521–538. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herbst J, Gilbert JD and Byard RW: Urinary
incontinence, hyperthermia, and sudden death. J Forensic Sci.
56:1062–1063. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jones TS, Liang AP, Kilbourne EM, Griffin
MR, Patriarca PA, Wassilak SG, Mullan RJ, Herrick RF, Donnell HD
Jr, Choi K and Thacker SB: Morbidity and mortality associated with
the July 1980 heat wave in St Louis and Kansas City, Mo. JAMA.
247:3327–3331. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ghaznawi HI and Ibrahim MA: Heat stroke
and heat exhaustion in pilgrims performing the Hajj (annual
pilgrimage) in Saudi Arabia. Ann Saudi Med. 7:323–326. 1987.
View Article : Google Scholar
|
|
6
|
Chang CP, Hsu YC and Lin MT: Magnolol
protects against cerebral ischaemic injury of rat heatstroke. Clin
Exp Pharmacol Physiol. 30:387–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zak R: Development and proliferative
capacity of cardiac muscle cells. Circ Res. 35(Suppl II): S17–S26.
1974.
|
|
8
|
Islam A, Lv YJ, Abdelnasir A, Rehana B,
Liu ZJ, Zhang M, Tang S, Cheng YF, Chen HB, Hartung J and Bao ED:
The role of Hsp90α in heat-induced apoptosis and cell damage in
primary myocardial cell cultures of neonatal rats. Genet Mol Res.
12:6080–6091. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin B, Tang S, Sun J, Zhang X, Xu J, Di L,
Li Z, Hu Y and Bao E: Vitamin C and sodium bicarbonate enhance the
antioxidant ability of H9C2 cells and induce HSPs to relieve heat
stress. Cell Stress Chaperones. 23:735–748. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kregel KC: Heat shock proteins: Modifying
factors in physiological stress responses and acquired
thermotolerance. J Appl Physiol (1985). 92:2177–2186. 2002.
View Article : Google Scholar
|
|
11
|
Powers ET, Morimoto RI, Dillin A, Kelly JW
and Balch WE: Biological and chemical approaches to diseases of
proteostasis deficiency. Annu Rev Biochem. 78:959–991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sonna LA, Wenger CB, Flinn S, Sheldon HK,
Sawka MN and Lilly CM: Exertional heat injury and gene expression
changes: A DNA microarray analysis study. J Appl Physiol (1985).
96:1943–1953. 2004. View Article : Google Scholar
|
|
13
|
Pérez-Salamó I, Papdi C, Rigó G, Zsigmond
L, Vilela B, Lumbreras V, Nagy I, Horváth B, Domoki M, Darula Z, et
al: The heat shock factor A4A confers salt tolerance and is
regulated by oxidative stress and the mitogen-activated protein
kinases MPK3 and MPK6. Plant Physiol. 165:319–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang J, Cao R, Wang X, Zhang Y, Wang P,
Gao H, Li C, Yang F, Zeng R, Wei P, et al: Mitochondrial PKM2
regulates oxidative stress-induced apoptosis by stabilizing Bcl2.
Cell Res. 27:329–351. 2017. View Article : Google Scholar :
|
|
16
|
Evans M, Fored CM, Bellocco R, Fitzmaurice
G, Fryzek JP, McLaughlin JK, Nyrén O and Elinder CG: Acetaminophen,
aspirin and progression of advanced chronic kidney disease. Nephrol
Dial Transplant. 24:1908–1918. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wali RK: Aspirin and the prevention of
cardiovascular disease in chronic kidney disease: Time to move
forward? J Am Coll Cardiol. 56:966–968. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amici C, Rossi A and Santoro MG: Aspirin
enhances thermo-tolerance in human erythroleukemic cells: An effect
associated with the modulation of the heat shock response. Cancer
Res. 55:4452–4457. 1995.PubMed/NCBI
|
|
19
|
Zhang XH, Zhu HS, Qian Z, Tang S, Wu D,
Kemper N, Hartung J and Bao ED: The association of Hsp90 expression
induced by aspirin with anti-stress damage in chicken myocardial
cells. J Vet Sci. 17:35–44. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang XH, Wu H, Tang S, Li QN, Xu J, Zhang
M, Su YN, Yin B, Zhao QL, Kemper N, et al: Apoptosis in response to
heat stress is positively associated with heat-shock protein 90
expression in chicken myocardial cells in vitro. J Vet Sci.
18:129–140. 2017. View Article : Google Scholar :
|
|
21
|
Yu GW, Chen J, Chen YY, Zheng MZ and Shen
YL: Heat-shock protein 90-dependent translocation of Akt to
mitochondria mediates insulin-like growth factor 1-induced
protection of rat hearts under hypothermic preservation. Chin J
Pathophysiol. 28:1773–1778. 2012.
|
|
22
|
Sato S, Fujita N and Tsuruo T: Modulation
of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA.
97:10832–10837. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chrousus GP and Gold PW: The concepts of
stress and stress system disorders. Overview of physical and
behavioral homeostasis. JAMA. 267:1244–1252. 1992. View Article : Google Scholar
|
|
24
|
Laszlo A: The effects of hyperthermia on
mammalian cell structure and function. Cell Prolif. 25:59–87. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yatvin MB and Cramp WA: Role of cellular
membranes in hyperthermia: Some observations and theories reviewed.
Int J Hyperthermia. 9:165–185. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ahmad N, Wang Y, Haider KH, Wang B, Pasha
Z, Uzun O and Ashraf M: Cardiac protection by mitoKATP channels is
dependent on Akt translocation from cytosol to mitochondria during
late preconditioning. Am J Physiol Heart Circ Physiol.
290:H2402–H2408. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chatterjee M, Andrulis M, Stühmer T,
Müller E, Hofmann C, Steinbrunn T, Heimberger T, Schraud H,
Kressmann S, Einsele H and Bargou RC: The PI3K/Akt signaling
pathway regulates the expression of Hsp70, which critically
contributes to Hsp90-chaperone function and tumor cell survival in
multiple myeloma. Haematologica. 98:1132–1141. 2013. View Article : Google Scholar :
|
|
29
|
Shen HY, He JC, Wang Y, Huang QY and Chen
JF: Geldanamycin induces heat shock protein 70 and protects against
MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem.
280:39962–39969. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Chung D, Yang YC, Neely L,
Tsurumoto S, Fan J, Zhang L, Biamonte M, Brekken J, Lundgren K and
Burrows F: Identification of new biomarkers for clinical trials of
Hsp90 inhibitors. Mol Cancer Ther. 5:1256–1264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu D, Xu J, Song E, Tang S, Zhang X,
Kemper N, Hartung J and Bao E: Acetyl salicylic acid protected
against heat stress damage in chicken myocardial cells and may
associate with induced Hsp27 expression. Cell Stress Chaperones.
20:687–696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao B, Sun G, Feng G, Duan W, Zhu X, Chen
S, Hou L, Jin Z and Yi D: Carboxy terminus of heat shock protein
(HSP) 70-interacting protein (CHIP) inhibits HSP70 in the heart. J
Physiol Biochem. 68:485–491. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fontana J, Fulton D, Chen Y, Fairchild TA,
McCabe TJ, Fujita N, Tsuruo T and Sessa WC: Domain mapping studies
reveal that the M domain of hsp90 serves as a molecular scaffold to
regulate Akt-dependent phosphorylation of endothelial nitric oxide
synthase and NO release. Circ Res. 90:866–873. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ikeyama S, Kokkonen G, Shack S, Wang XT
and Holbrook NJ: Loss in oxidative stress tolerance with aging
linked to reduced extracellular signal-regulated kinase and Akt
kinase activities. FASEB J. 16:114–116. 2002. View Article : Google Scholar
|
|
35
|
Kaufmann T, Schlipf S, Sanz J, Neubert K,
Stein R and Borner C: Characterization of the signal that directs
Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J
Cell Biol. 160:53–64. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jackson S: Molecular chaperones. Berlin
Heidelberg, Springer-verlag; pp. 1–272. 2013
|
|
37
|
Pugazhenthi S, Nesterova A, Sable C,
Heidenreich KA, Boxer LM, Heasley LE and Reusch JE: Akt/protein
kinase B up-regulates Bcl-2 expression through cAMP-response
element-binding protein. J Biol Chem. 275:10761–10766. 2000.
View Article : Google Scholar
|
|
38
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ling YH, Liebes L, Zou Y and Perez-Soler
R: Reactive oxygen species generation and mitochondrial dysfunction
in the apoptotic response to Bortezomib, a novel proteasome
inhibitor, in human H460 non-small cell lung cancer cells. J Biol
Chem. 278:33714–33723. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Govender J, Loos B, Marais E and
Engelbrecht AM: Mitochondrial catastrophe during
doxorubicin-induced cardio-toxicity: A review of the protective
role of melatonin. J Pineal Res. 57:367–380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang S, Wang Y, Zhang Z, Liu Q and Gu J:
Cardioprotective effects of fibroblast growth factor 21 against
doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell
Death Dis. 8:e30182017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Govender J, Loos B and Engelbrecht AM:
Melatonin: A protective role against doxorubicin-induced
cardiotoxicity. Future Oncol. 11:2003–2006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L,
Reiter RJ, Qiao S and Yuan J: AMPK/PGC1α activation by melatonin
attenuates acute doxorubicin cardiotoxicity via alleviating
mitochondrial oxidative damage and apoptosis. Free Radic Biol Med.
129:59–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Priya LB, Baskaran R, Huang CY and Padma
VV: Neferine ameliorates cardiomyoblast apoptosis induced by
doxorubicin: Possible role in modulating NADPH oxidase/ROS-mediated
NFκB redox signaling cascade. Sci Rep. 7:122832017. View Article : Google Scholar
|
|
45
|
Fan AC, Bhangoo MK and Young JC: Hsp90
functions in the targeting and outer membrane translocation steps
of Tom70-mediated mitochondrial import. J Biol Chem.
281:33313–33324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Daugaard M, Rohde M and Jaattela M: The
heat shock protein 70 family: Highly homologous proteins with
overlapping and distinct functions. FEBS Lett. 581:3702–3710. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chaudhury S, Welch TR and Blagg BSJ: Hsp90
as a target for drug development. ChemMedChem. 1:1331–1340. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dieterle A, Orth R, Daubrawa M, Grotemeier
A, Alers S, Ullrich S, Lammers R, Wesselborg S and Stork B: The Akt
inhibitor triciribine sensitizes prostate carcinoma cells to
TRAIL-induced apoptosis. Int J Cancer. 125:932–941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Johnson JD, Campisi J, Sharkey CM, Kennedy
SL, Nickerson M and Fleshner M: Adrenergic receptors mediate
stress-induced elevations in extracellular Hsp72. J Appl Physiol
(1985). 99:1789–1795. 2005. View Article : Google Scholar
|
|
51
|
Kumaraguru U, Pack CD and Rouse BT:
Toll-like receptor ligand links innate and adaptive immune
responses by the production of heat-shock proteins. J Leukoc Biol.
73:574–583. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Asea A, Kraeft SK, Kurt-Jones EA,
Stevenson MA, Chen LB, Finberg RW, Koo GC and Calderwood SK: HSP70
stimulates cytokine production through a CD14-dependant pathway,
demonstrating its dual role as a chaperone and cytokine. Nat Med.
6:435–442. 2000. View
Article : Google Scholar : PubMed/NCBI
|