Introduction
The progressive disease pulmonary arterial
hypertension (PAH) is characterized by increased pulmonary arterial
pressure and pulmonary vascular resistance, leading to right
ventricular failure and eventually death (1). The pathophysiology of PAH is
characterized by vascular remodeling and a vasoconstrictive and
proliferative thrombotic phenotype (2). Patients suffering from PAH always
experience symptoms such as exertional dyspnea, fatigue, chest pain
and dizziness (3). It has been
found that proliferation of pulmonary artery endothelial cells
(PAECs) and pulmonary artery smooth muscle cells (PASMCs)
contributes to the obstruction of the vascular lumen in the late
stage (4). Although progress has
been made in PAH treatment in recent years, PAH remains a disease
with limited treatment modalities (5). Most patients still have low overall
survival rates and their quality of life remains severely affected
(6). Therefore, a deeper
understanding of the molecular mechanisms of PAH is needed to
identify effective therapeutic targets for this complex disease
(7).
MicroRNAs (miRNAs/miR) are engaged in the regulation
of various biological processes, such as cell differentiation,
proliferation and apoptosis, and dysregulation of miRNAs is
involved in various human diseases (8). Recently, evidence has emerged
showing that abnormal expression of miRNAs participates in the
biological development of PAH (9,10).
Let-7 family members have been demonstrated to be key regulators of
cell development and differentiation and their tumor-suppressive
roles have been found in various human cancers (11). For example, miR-let-7d is a member
of the let-7 family and its upregulation can inhibit cell
proliferation and migration and promote apoptosis of trophoblast
cells in preeclampsia (12). Most
importantly, the let-7 family has also been demonstrated to be
abnormally expressed in cardiovascular diseases, such as heart
hypertrophy, dilated cardiomyopathy and hypertension (13). Autophagy plays an important role
in cardiovascular cells (14) and
miRNAs have been shown to function vitally in the regulation of
autophagy-related pathways (15).
In addition, autophagy-related 16-like 1 (ATG16L1) has been
identified as an autophagy-related gene that can regulate
autophagosomes (16). ATG16L1
belongs to a class of protein complexes that are considered vital
for autophagy (17). Emerging
data has illustrated the functionality of ATG16L1 in the
development of atherogenesis (18). Based on the aforementioned
findings, it has been demonstrated that both let-7d and ATG16L1 may
participate in PAH by affecting autophagy. The present study
further hypothesized that let-7d may interact with ATG16L1 to
participate in PAH and the present study was performed to elucidate
this interaction.
Materials and methods
Ethics statement
The current study was approved by the Ethics
Committee and the Experimental Animal Ethics Committee of Qingdao
Municipal Hospital. Written informed consent was obtained from all
participants prior to the study. The animal experiment strictly
adhered to the principles of using the least number of animals to
complete the experiment and minimizing the pain of the experimental
animals.
Patient enrollment
A total of 83 PAH patients hospitalized at the
Qingdao Municipal Hospital between June 2016 and June 2018 were
selected for the study. The patients were included if their
pulmonary artery systolic pressure was >30 mmHg as detected by
echocardiography. The patients were excluded if they suffered from
cardiomyopathy, myocardial infarction, heart failure, valvular
disease, pericardial disease, chronic thromboembolic disease, or
chronic obstructive pulmonary disease. All included patients (51
males and 32 females), aged 19-70 years with an average age of
45.70±13.89 years, had complete clinical data and did not receive
any surgeries or drug treatment prior to the operation (19,20). A total of 40 healthy individuals
were enrolled as the normal group. Patient characteristics are
presented in Table I. The levels
of let-7d in 2 ml plasma collected (stored at -80°C) from each PAH
patients and healthy individuals were measured.
 | Table IRelationship between let-7d
expression and clinicopathological features of PAH patients. |
Table I
Relationship between let-7d
expression and clinicopathological features of PAH patients.
| Clinicopathological
features | Case (n=83) | Let-7d expression
| P-value |
|---|
| Low expression
(n=39) | High expression
(n=44) |
|---|
| Age | | | | 0.805 |
| <50 years | 35 | 17 | 18 | |
| ≥50 years | 48 | 22 | 26 | |
| Sex | | | | 0.987 |
| Male | 51 | 24 | 27 | |
| Female | 32 | 15 | 17 | |
| Cardiac function
grade | | | | 0.003 |
| I | 28 | 9 | 19 | |
| II | 32 | 12 | 20 | |
| III | 13 | 9 | 4 | |
| IV | 10 | 9 | 1 | |
| Pulmonary
hypertension degree | | | | 0.021 |
| Mild | 34 | 10 | 24 | |
| Middle | 26 | 14 | 12 | |
| Severe | 23 | 15 | 8 | |
| Six-min walk test
performance classification | | | | 0.019 |
| Class 1 (<150
m) | 15 | 3 | 12 | |
| Class 2 (150-425
m) | 49 | 23 | 26 | |
| Class 3 (426-550
m) | 19 | 13 | 6 | |
Rat models simulating PAH
In total, 85 male healthy specific-pathogen-free
grade Sprague-Dawley rats (age, 7 weeks; weighing 220-250 g) were
purchased from the Qingdao Municipal Hospital Experimental Animal
Center. All animals were maintained at 22±2°C with a humidity of
55±5%, a 12:12 h light/dark cycle and free access to regular mouse
chow and water for 1 week. Monocrotaline (MCT; Sigma-Aldrich; Merck
KGaA) was dissolved in 0.1 mol/l HCl and titrated with 0.1 mol/l
NaOH to a pH of 7.4 and a final concentration of 30 mg/ml; the
final solution (60 mg/kg) was then injected intraperitoneally into
the rats of the PAH group. The rats in the control group were
injected with equal volumes of normal saline. After 28 days of MCT
induction, the rats in the PAH group presented with significant PAH
symptoms compared with those in the control group. A lentiviral
vector was constructed as previously reported (21); the ATG16L1 gene sequence was
amplified by PCR and then the amplified product was cloned into the
lentiviral gene overexpression vector pLV-EGFP-N at the
EcoRI and NotI sites using a Cold Fusion kit (System
Biosciences LLC). Cells were infected with empty lentiviral
particles or lentiviral overexpression particles and selected with
puromycin for 3 days to obtain stable cell lines. Both the
overexpressed (oe)-negative control (NC) and oe-ATG16L1 vectors
were constructed by Shanghai GenePharma Co., Ltd. 293T cells
(American Type Culture Collection) were used for lentiviral
packaging and the 293T cells were cultured in RPMI-1640 complete
medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), followed
by sub-culture every other day. The viruses (1.0×108
PFU/ml) were collected and the cells were infected with oe-ATG16L1
plasmid or NC plasmid. Rats were anesthetized with 3% sodium
pentobarbital (P3761; Sigma-Aldrich; Merck KGaA) 28 days after MCT
induction. The rats were then fixed on a sterilized test bench and
the tail of each rat was repeatedly rubbed with a cotton ball
soaked in alcohol. After the veins on both sides of the tail were
dilated, the residual air bubbles in the syringe were removed and
adenoviral vector (1×109 PFU/100 µl), let-7d
agomir (20 nM), or let-7d antagomir (20 nM) was injected into the
dilated tail vein at the proximal end at a 30° angle. After
infection for 48 h, green fluorescent protein expression efficiency
was observed under a fluorescence microscope and relevant
experiments were performed.
The modeling success was assessed using right
ventricular systolic pressure (RVSP) by right heart
catheterization, right ventricular hypertrophy with right
ventricular hypertrophy index (RVHI), and the morphology of the
pulmonary vessels by hematoxylin and eosin (H&E) staining. A
total of 10 normal rats were taken as the normal group. Among the
remaining 75 rats, the model was successfully induced in 70, with a
modeling success rate of 93.33%. The PAH rat models were assigned
into 7 groups with 10 rats in each group: The PAH group (PAH rat
models), the agomir-NC PAH group (PAH rat models injected with
agomir-NC), the let-7d agomir PAH group (PAH rat models injected
with let-7d agomir), the antagomir-NC PAH group (PAH rat models
injected with antagomir-NC), the let-7d antagomir PAH group (PAH
rat models injected with let-7d antagomir), the let-7d agomir +
oe-NC PAH group (PAH rat models injected with let-7d agomir and
oe-NC lentivirus) and the let-7d agomir + oe-ATG16L1 PAH group (PAH
rat models injected with let-7d agomir and oe-ATG16L1 lentivirus).
The rats were euthanized by deep anesthesia with pentobarbital
sodium (100 mg/kg) on the 28th day after MCT induction 48 h after
injection of agomir/antagomir or lentivirus.
Determination of RVSP and RVHI
RVSP and the RVHI were measured on the 28th day of
MCT induction 48 h after injection of the agomir/antagomir or
lentiviruses. Each rat was anesthetized with 3% pentobarbital
sodium. The right external jugular vein was cannulated and the
superior vena cava, right atrium, right ventricle, and pulmonary
artery were connected to a physiological polygraph (PowerLab;
ADInstruments). The location of the catheter was judged from the
pressure waveform changes displayed on the polygraph. After the
abdomen was opened, the left ventricle and right ventricle were
visualized through the complete diaphragm. Next, a 23 G needle was
inserted into the right ventricle, and RVSP was recorded.
Subsequently, hemodynamic data were collected and the heart was
isolated with the atria and major blood vessels removed. Dissection
of the right ventricle (RV) from the left ventricle (LV) and the
septum (S) was performed. The weights of the RV and the LV + S were
determined on an electronic balance and the RL gravity was
expressed as LV + S.
H&E staining
The lung tissues were fixed with 4% paraformaldehyde
at room temperature for 12-24 h, paraffin-embedded and cut into 5
µm sections. The sections were deparaffinized using xylene I
for 10 min and xylene II for 5 min. Then, the tissues were washed
with anhydrous alcohol to remove the xylene for 1 min, with 95%
alcohol for 1 min and with 85% alcohol for 1 min. After being
washed with tap water, the sections were stained with hematoxylin
(Beyotime Institute of Biotechnology) for 5 min at room
temperature, followed by an additional wash with tap water. Upon
removal of the water, the sections were differentiated with
hydrochloric acid ethanol, soaked in water for 15 min and stained
with eosin solution (Beyotime Institute of Biotechnology) for 2 min
at room temperature before being dehydrated with 85% alcohol for 20
sec and 95% alcohol for 1 min. After that, the sections were
incubated with anhydrous alcohol I and anhydrous alcohol II for 2
min each and soaked in xylene I and xylene II for 2 min each. The
sections were then mounted with neutral balsam or Canadian balsam
and observed under a light microscope (DMI3000, Leica GmbH) after
H&E staining. To assess vascular remodeling, the percentage of
medial wall thickness (% MT; diameter 50-250 µm) was
calculated using the following formula: MT = (2× medial wall
thickness) ×100/outer diameter.
Human PAEC culture and treatment
PAECs were purchased from Lonza Group, Ltd. In
strict accordance with the protocol, the cells were cultured with
10% FBS and high-glucose Dulbecco's modified eagle medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc.) and observed under a
light microscope. The cells at passage 3 were detached with
trypsin, inoculated into a 24-well plate at a density of
2×106 cells/well and cultured until they grew into
monolayers. When the cell density reached 75%, the cells were
considered to be in logarithmic phase and were inoculated into a
6-well cell culture plate. After 12 h, according to the protocol of
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
the cells were transfected with NC mimic, let-7d mimic, NC
inhibitor or let-7d inhibitor plasmids (final concentration, 50 nM)
for 48 h (22). The cells were
then assigned to the following groups: The blank group (without
transfection of any plasmid), the NC mimic group (transfected with
NC mimic plasmid), the let-7d mimic group (transfected with let-7d
mimic plasmid), the NC-inhibitor group (transfected with NC
inhibitor plasmid) and the let-7d inhibitor group (transfected with
let-7d inhibitor plasmid). The transfection plasmid was constructed
by Invitrogen; Thermo Fisher Scientific, Inc.
Dual luciferase reporter gene assay
The biological prediction website microRNA.org was employed to analyze the target genes
of let-7d and to verify whether ATG16L1 was a direct target gene of
let-7d. A dual luciferase reporter gene assay was employed to
confirm that ATG16L1 was a direct target gene of let-7d. A
synthesized ATG16L1 3' untranslated region (3'UTR) gene fragment
was introduced into the pMIR reporter (Beijing Huayueyang
Biotechnology Co., Ltd.) using the endonuclease sites SpeI
and HindIII. A complementary sequence with a mutation at the
site of the seed sequence was designed on the ATG16L1-wild type
(Wt) sequence and the target fragment was inserted into the pMIR
reporter plasmid by restriction endonuclease digestion using T4 DNA
ligase. The Wt and mutant (Mut) luciferase reporter plasmids with
the correct sequences were cotransfected into 293T cells with
let-7d mimic or let-7d NC (Shanghai Beinuo Biotechnology Co., Ltd.)
with Attractene Transfection reagent (cat. no. 301005; Qiagen
GmbH). After 48 h of transfection, the cells were collected and
lysed, and luciferase activity was measured using a GloMax 20/20
luminometer (Promega Corporation) and a luciferase assay kit
(K801-200; BioVision). Firefly luciferase activity was normalized
to Renilla luciferase activity.
RNA isolation and quantitation
Reverse transcription-quantitative PCR (RT-qPCR) was
employed to detect let-7d expression and ATG16L1 mRNA expression in
tissues or cells. Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) from the lung tissues of rats 48 h
after the different injections were administered and PAECs were
collected 24 h after transfection. The sequences of the primers are
shown in Table II. The primers
of let-7d and ATG16L1 were designed and synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). Total RNA was reverse transcribed
into complementary DNA (cDNA) using different reverse transcription
kits, including a TaqMan™ MicroRNA Reverse Transcription kit
(4366596; Thermo Fisher Scientific Inc.) and a High-Capacity cDNA
Reverse Transcription kit (4368813; Thermo Fisher Scientific, Inc.)
The temperature protocol for RT was as follows: 42°C for 15 min,
85°C for 5 sec and storage at 4°C. With U6 and β-actin used as
internal controls (Invitrogen; Thermo Fisher Scientific, Inc.), the
PCR system was set to a 25 µl volume using a qPCR kit
(Takara Bio, Inc.) on a real-time fluorescence quantitative PCR
instrument (Thermo Fisher Scientific, Inc.) for PCR. The qPCR
program consisted of initial denaturing at 95°C for 2 min, followed
by 45 cycles of 15 sec at 95°C and 45 sec at 60°C. The final data
were analyzed by the 2−ΔΔCq method (23).
| Table IIPrimer sequences for reverse
transcription-quantitative PCR. |
Table II
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Primer
sequence |
|---|
| Let-7d (Rattus
norvegicus) | F:
5′-GCGAGCACAGAATTAATACGAC-3′ |
| R:
5′-AGAGGTAGTAGGTTGCATAGTT-3′ |
| ATG16L1 (Homo
sapiens) | F: 5′-CAGTTACGTG
GCGGCAGGCT-3′ |
| R: 5′-ACAACGTGCG
AGCCAGAGGG-3′ |
| U6 (Rattus
norvegicus) | F:
5′-CTCGCTTCGGCAGCA-3′ |
| R:
5′-AACGCTTCACGAATTTGCGT-3′ |
| β-actin (Homo
sapiens) | F:
5′-TGGCACCCAGCACAATGAA-3′ |
| R:
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ |
Western blot analysis
Rat lung tissues were washed with PBS and incubated
with RIPA protein lysis buffer (Beyotime Institute of
Biotechnology) containing protease and alkaline phosphatase
inhibitors for 30 min at 4°C. The lysate was collected in a 1.5 ml
eppendorf tube and centrifuged at 6,700 × g for 15 min, after which
the supernatant was collected. Then, the supernatant was mixed with
loading dye and boiled for 5 min. Protein concentration was
measured using a bicinchoninic acid assay kit (Beyotime Institute
of Biotechnology). Next, the protein (50 µg) was separated
using 10% SDS-PAGE and transferred onto a polyvinylidene fluoride
membrane at 0.3 A and 20 V, which was then blocked with 5% skim
milk powder for 1 h at room temperature. After that, the membrane
was incubated overnight at 4°C with the following primary
antibodies diluted in Tris-buffered saline with 0.1% Tween 20
(TBST): Anti-p62 (cat. no. ab56416; Abcam; 1:1,000, anti-mouse),
anti-LC3B (L7543; Sigma-Aldrich; Merck KGaA; 1:10,000;
anti-rabbit), anti-ATG16L1 (cat. no. ab188642, Abcam; 1:1,000;
anti-rabbit) and anti-β-actin (cat. no. ab8226; Abcam; 1:1,000;
anti-mouse). After being washed with TBST three times, the membrane
was incubated with horseradish peroxidase (HRP)-labeled goat
anti-mouse immunoglobulin G (IgG; 1:5,000; cat. no. ab6789; Abcam)
or goat anti-rabbit IgG (1:5,000; cat. no. ab6721; Abcam) secondary
antibodies at room temperature for 1 h. Following this step, the
membrane was washed six times with TBST and visualized using
enhanced chemiluminescence reagent (Thermo Fisher Scientific,
Inc.). The gel image analysis software ImageJ (version 1.46;
National Institute of Health) was employed to analyze the gray
value of each band and the ratio of the gray value of the target
protein band to that of the internal control protein band was
calculated.
Immunofluorescence staining
The slides of human PAECs were prepared and then
fixed with 40 g/l polyformaldehyde at room temperature for 15 min,
washed three times with PBS, and sealed with 10% goat serum. Next,
the slides were incubated overnight at 4°C with anti-LC3B (L7543;
1:100; Sigma-Aldrich, Merck KGaA; anti-rabbit) and anti-PECAM-1
(cat. no. sc-18916; 1:100; Santa Cruz Biotechnology, Inc.;
anti-rat). After being washed with PBS three times, the tissues
were subjected to further incubation with fluorescein
isothiocyanate-labeled secondary antibodies (cat. no. Ab6717;
1:500; Abcam; anti-rabbit) and Cy3-labeled IgG secondary antibodies
(cat. no. Ab6939; 1:500; Abcam; anti-rat) and incubated with
protection from light for 60 min at room temperature. Confocal
imaging was performed using an LSM510 Meta inverted microscope with
a confocal laser scanning head (Carl Zeiss AG). Digital images were
collected for analysis (SPOT; Diagnostic Instruments, Inc.). ImageJ
(version 1.46; National Institute of Health) was employed to
analyze the fluorescence intensity and calculate the fluorescence
intensity value.
ELISA
For analysis of rat plasma, 2 ml blood samples were
collected using blood collection tubes containing anticoagulants
and then centrifuged at 600 × g for 20 min at 4°C, after which the
supernatant was collected for detection. Rat lung tissues were
extracted, washed with PBS, homogenized (with protease inhibitor)
and centrifuged at 6,700 × g for 5-10 min at 4°C, followed by
collection of the supernatant. A rat endo-thelin-1 (ET-1) ELISA kit
(CE-EL-R0167c) was purchased from Wuhan Elabscience Biotechnology
Co., Ltd. Standard or sample (100 µl) was added to each well
and the plate was then incubated at 37°C for 90 min before the
liquid was removed and the plate was dried. Then, 100 µl
biotinylated detection Ab was added to each well and the plate was
incubated at 37°C for 60 min, dried and washed three times. After
that, 100 µl HRP was added to each well and the plate was
incubated at 37°C for 30 min, dried and washed five times.
Substrate reagent (90 µl) was then added to each well and
the plate was incubated for 15 min at 37°C before 50 µl stop
solution was added to each well. The optical density (OD) value was
then measured at 450 nm. A standard curve was drawn with the
standard protein concentrations on the X-axis and the OD (A) values
on the Y-axis. The concentrations of ET-1 were obtained from the
standard curve according to the OD values of the sample wells.
Statistical analysis
The experimental data were analyzed using SPSS 21.0
software (IBM Corp.). Enumeration data were analyzed using the
Chi-square test. Measurement data were expressed as the mean ±
standard deviation. All data were subjected to normal distribution
and homogeneity of variance tests. The measurement data conforming
to normal distribution were expressed as the mean ± standard
deviation. The data with skewed distribution or heterogeneity of
variance were expressed as the median ± inter-quartile range. Data
between two groups were compared using an unpaired t-test while
data among multiple groups were analyzed using one-way analysis of
variance, followed by a Tukey's post hoc test. The nonparametric
Wilcoxon rank sum test was employed for data between two groups
with a skewed distribution while Kruskal-Wallis H test was used for
data among multiple groups with a skewed distribution. P<0.05
was considered to indicate a statistically significant
difference.
Results
Let-7d is poorly expressed in the plasma
of PAH patients
The expression of let-7d in the plasma of the PAH
group and the normal group was measured by RT-qPCR, and the results
showed that compared with the plasma in the normal group, the
plasma in the PAH group exhibited significantly reduced expression
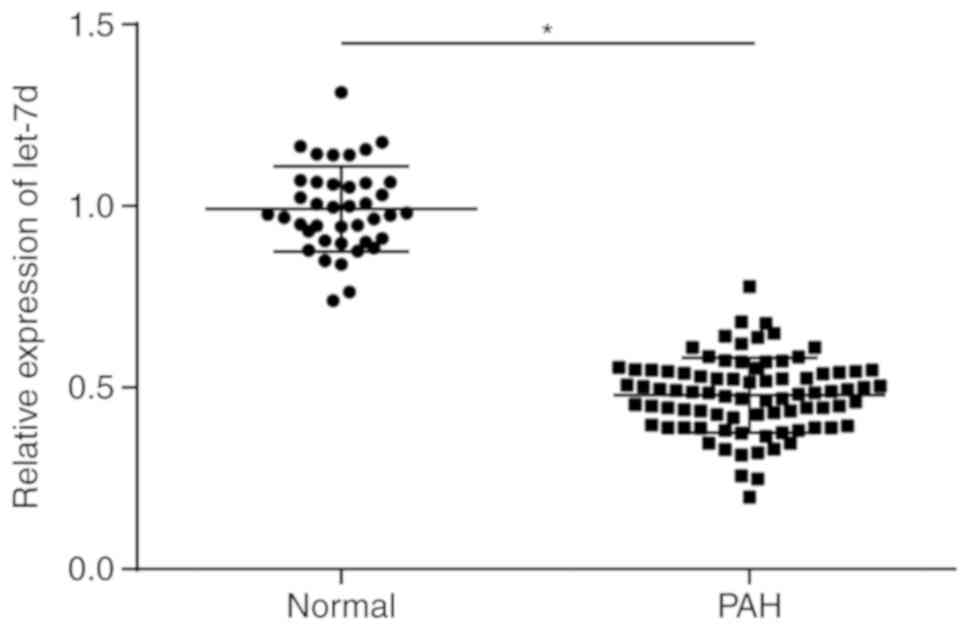
of let-7d (P<0.05; Fig. 1).
With the median let-7d expression as the dividing point, the PAH
patients were classified as PAH patients with highly expressed
let-7d or PAH patients with poorly expressed let-7d to analyze the
relationship between let-7d expression and the clinicopathological
features of PAH patients. The results (Table I) revealed that the expression of
let-7d was not correlated with the age or gender of the PAH
patients (P>0.05) but was significantly correlated with the
cardiac function grade, degree of pulmonary hypertension and 6-min
walk test performance (P<0.05). Collectively, the results
indicate that the plasma of PAH patients displays reduced
expression of let-7d compared with healthy subjects.
Let-7d is poorly expressed in the rat
model of PAH
RVSP was measured by right cardiac catheterization.
The RVHI was calculated to analyze right heart hypertrophy.
Morphological changes in pulmonary vessels were observed by H&E
staining. RT-qPCR was performed to measure the expression of
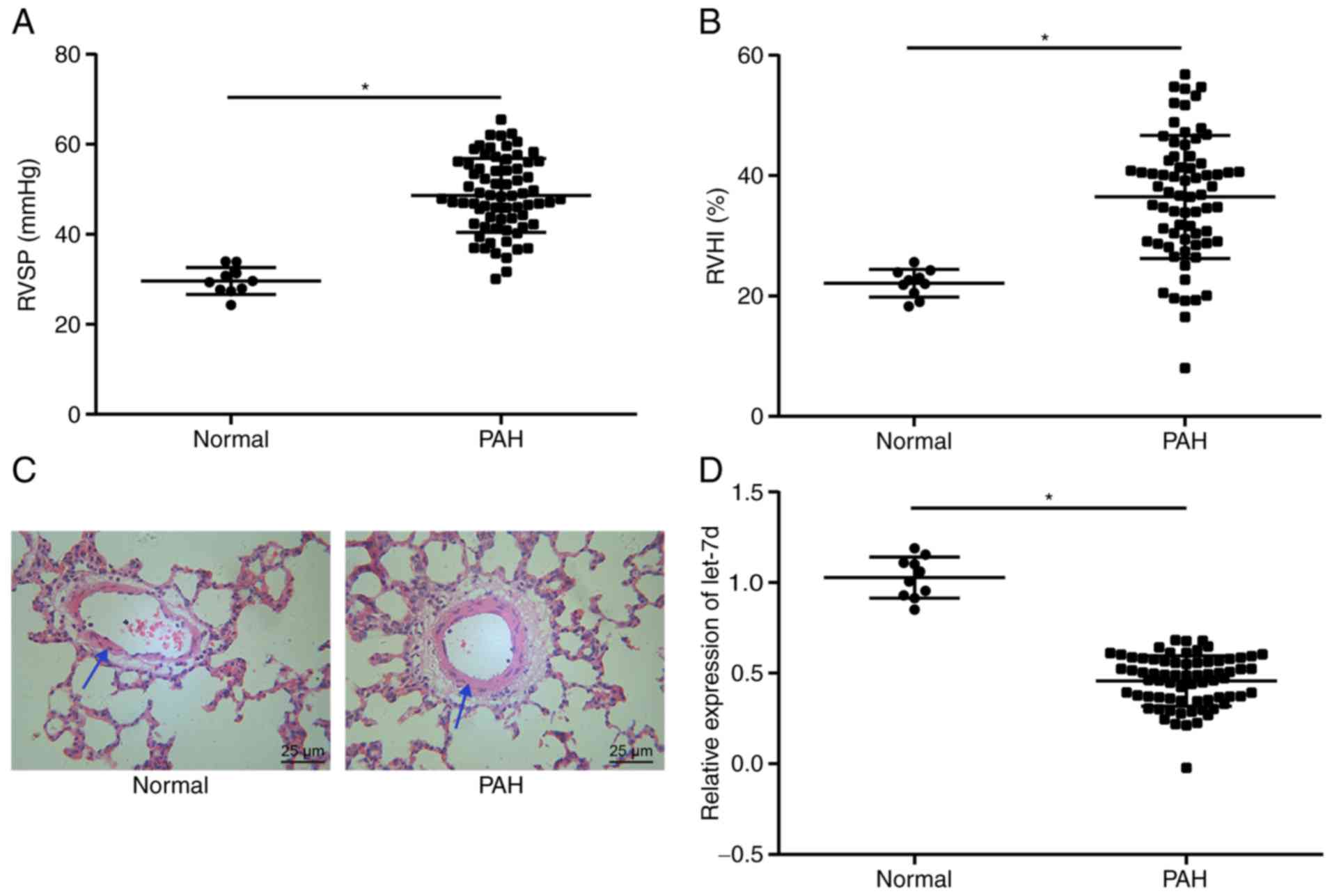
let-7d. The results demonstrated that the PAH group showed
significantly elevated RVSP and RVHI values (P<0.05; Fig. 2A and B), thicker pulmonary
vascular walls (34.65±3.58%) (Fig.
2C) and significantly reduced let-7d expression (Fig. 2D) compared with the normal group
(22.12±2.31%; P<0.05). The results above led to the conclusion
that let-7d expression was low in rat models of PAH.
Let-7d alleviates PAH
To further investigate the role of let-7d in PAH,
RT-qPCR was employed to detect the expression of let-7d, right
heart catheterization was performed to detect RVSP, RVHI values to
assess cardiac hypertrophy and H&E staining was performed to
observe morphological changes in the pulmonary vessels. The results
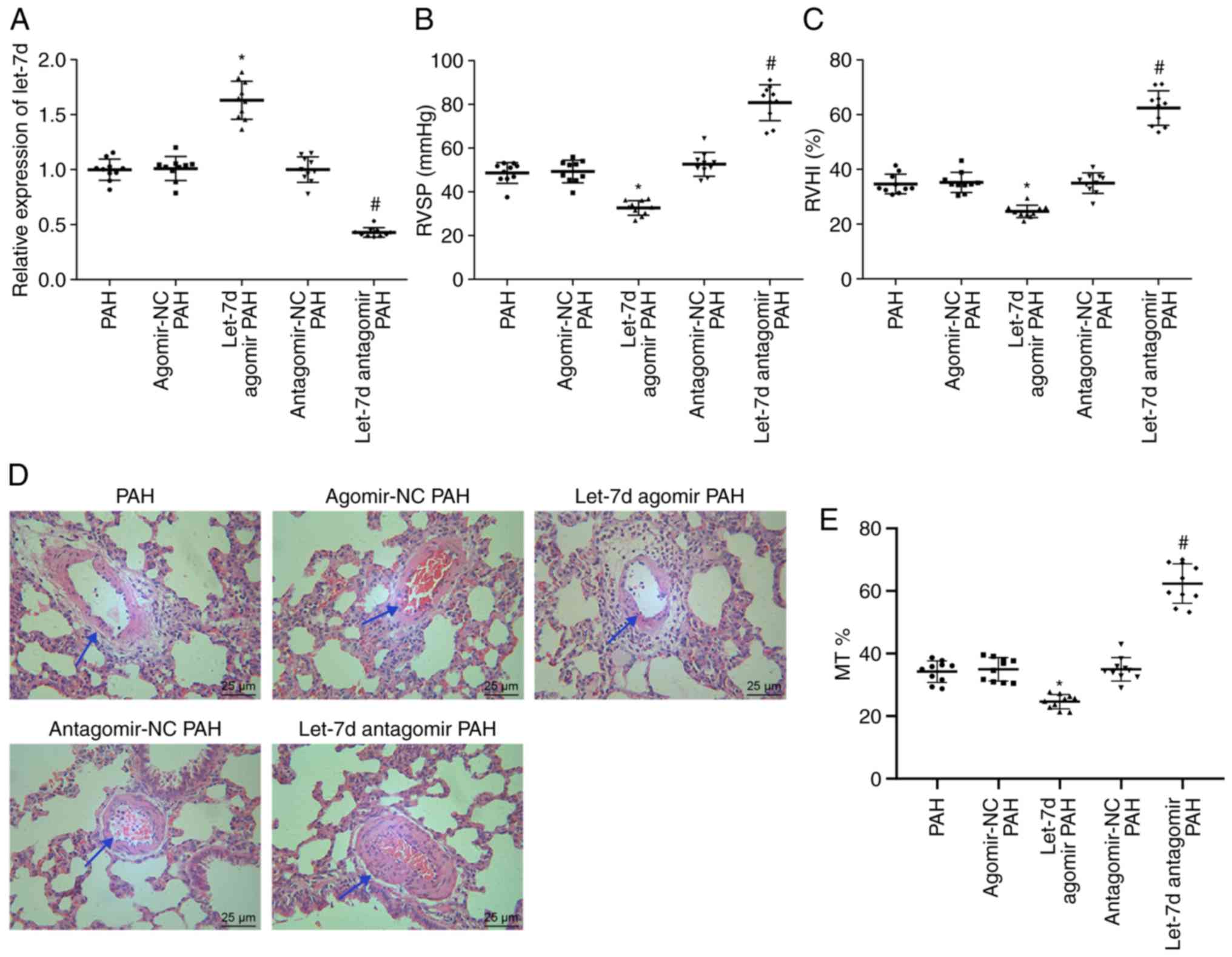
of RT-qPCR suggested that let-7d expression was significantly
increased in PAH rats injected with let-7d agomir compared with PAH
rats injected with agomir NC (P<0.05), while the expression of
let-7d was decreased in PAH rats injected with the let-7d antagomir
compared with rats injected with the antagomir NC (P<0.05;
Fig. 3A). Injection of let-7d
agomir resulted in reduced RVSP and RVHI, while injection of the
let-7d antagomir led to significantly elevated RVSP and RVHI values
(P<0.05; Fig. 3B and C). The
results of H&E staining proved that PAH rats injected with
let-7d agomir showed significantly thinner pulmonary vessel walls
(24.63±2.29%) than PAH rats injected with agomir NC (34.97±3.72%;
P<0.05), while PAH rats injected with let-7d antagomir presented
with significantly thicker pulmonary vessel walls (62.40±6.33%)
than PAH rats injected with antagomir NC (34.97±3.72%; P<0.05)
(Fig. 3D and E). The findings
above provided evidence that let-7d could relieve PAH.
Let-7d represses PAEC autophagy and
endothelin synthesis
Previous studies have shown the presence of
excessive autophagy (21) and
ET-1 accumulation in PAH models (24). To further investigate the
mechanism by which let-7d affects PAH, western blot analysis was
employed to assess the expression of autophagy-related proteins and
immunofluorescence staining was employed to analyze the expression
of LC3B, an autophagic marker, in PAECs. During autophagy, a small
segment of the cytosolic polypeptide LC3 (LC3-I) was enzymatically
cleaved, transforming the peptide into an (autophagosome) membrane
type (LC3-II). The ratio of LC3-II to LC3-I can be used to estimate
the level of autophagy and LC3B can be used as a marker for
autophagosomes. Western blot analysis showed that the expression of
autophagy-related protein p62 was reduced and that the ratio of
LC3-II to LC3-I was significantly increased in the PAH group
compared with the normal group (P<0.05). Injection of let-7d
agomir significantly elevated the levels of p62 and reduced the
ratio of LC3-II to LC3-I compared with injection of agomir NC
(P<0.05), while injection of the let-7d antagomir significantly
decreased the levels of p62 and increased the ratio of LC3-II to
LC3-I (P<0.05) compared with injection of the antagomir NC
(Fig. 4A and B).
Immunofluorescence staining showed that the expression of LC3B was
significantly increased in the PAH group compared with in the
normal group (P<0.05). The levels of LC3B were significantly
decreased in rats injected with the let-7d agomir compared with in
rats injected with the agomir NC (P<0.05). Injection of the
let-7d antagomir significantly increased the levels of LC3B in
comparison with injection of antagomir NC (P<0.05; Fig. 4C). The results of the ELISA, which
was performed to detect the ET-1 concentrations in plasma and lung
tissues, showed that the concentrations of ET-1 in plasma and lung
tissues were significantly increased in the PAH group compared with
in the normal group (P<0.05). Rats injected with the let-7d
agomir presented with significantly decreased concentrations of
ET-1 compared with those injected with agomir NC (P<0.05). Rats
injected with the let-7d antagomir showed significantly elevated
concentrations of ET-1 compared with those injected with the
antagomir NC (P<0.05; Fig. 4D and
E). In summary, let-7d inhibits autophagy and endothelin
synthesis in PAECs.
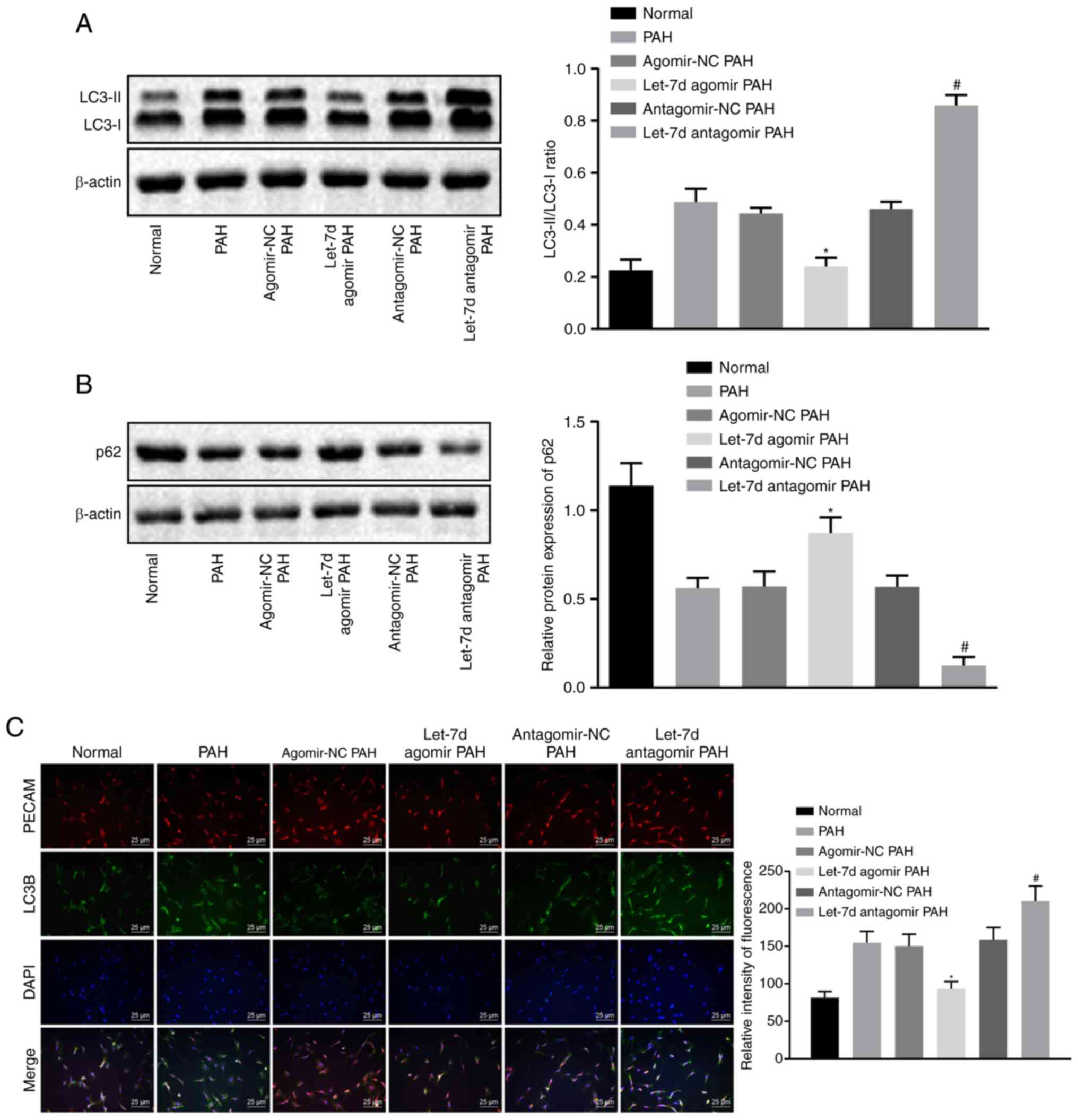 | Figure 4Let-7d inhibits autophagy and
endothelin synthesis in PAECs. (A) The protein bands and levels of
the autophagy-related protein LC3, as determined by western blot
analysis. (B) The protein bands and levels of p62 in rat lung
tissues, as measured by western blot analysis. Unprocessed blots
are shown in Figure S1. (C)
Quantification of the expression and relative fluorescence
intensity of LC3B in PAECs, as assessed by immunofluorescence
staining. (D) Levels of ET-1 in plasma, as determined by ELISA. (E)
The levels of ET-1 in lung tissue, as measured by ELISA.
*P<0.05 vs. the agomir NC PAH group.
#P<0.05 vs. the antagomir NC PAH group. The above
data were all measurement data and expressed as the mean ± standard
deviation. One-way analysis of variance was used for comparisons
among multiple groups, followed by a Tukey's post hoc test. N=10.
PAECs, pulmonary artery endothelial cells; ET-1, endothelin-1; LC3,
light chain 3; LC3B, light chain 3B; PAH, pulmonary arterial
hypertension. |
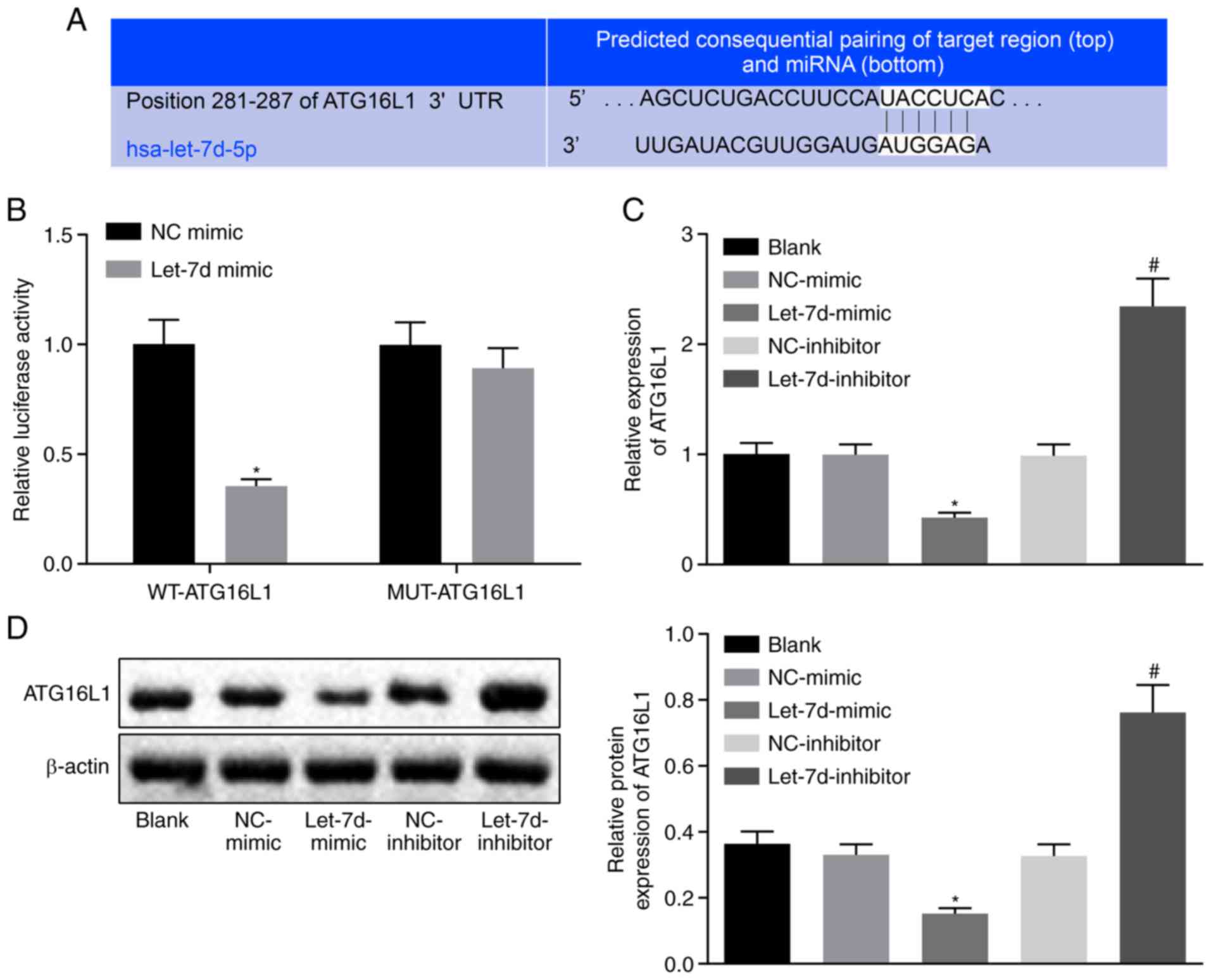
Let-7d could directly target ATG16L1
The binding site of let-7d and ATG16L1 was predicted
with a biological prediction website (Fig. 5A) and verified by luciferase
reporter assay (Fig. 5B). The
results suggested that the luciferase signal of the
Wt-let-7d/ATG16L1 cotransfection group was significantly decreased
in cells treated with the let-7d mimic compared with in cells
treated with the NC mimic (P<0.05), while there was no
significant difference in the luciferase activity of the
Mut-let-7d/ATG16L1 3'UTR (P>0.05). In comparison with the cells
treated with the NC plasmid, the cells treated with the let-7d
mimic showed significantly reduced mRNA and protein expression of
ATG16L1 (P<0.05), while the cells treated with the let-7d
inhibitor presented significantly increased mRNA and protein
expression of ATG16L1 (P<0.05; Fig. 5C and D), suggesting that let-7d
inhibited the expression of ATG16L1. The above results suggested
that ATG16L1 is a target gene of let-7d and that let-7d could
specifically bind to ATG16L1.
Let-7d inhibits autophagy in PAECs by
targeting ATG16L1
To further investigate the effect of let-7d on
autophagy in PAECs by targeting ATG16L1, western blot analysis was
performed to measure the expression of autophagy-related proteins
and immunofluorescence staining was employed to analyze the
expression of LC3B in PAECs. As determined by western blot
analysis, PAECs treated with the let-7d agomir and ATG16L1
overexpression vectors displayed significantly elevated levels of
ATG16L1 and significantly elevated ratios of LC3-II to LC3-I but
significantly reduced expression of p62 compared with PAECs treated
with the let-7d agomir and oe-NC vectors (P<0.05; Fig. 6A-C). The results of
immunofluorescence staining showed injection of both let-7d agomir
and the ATG16L1 over-expression vector significantly increased the
levels of LC3B compared with injection of both let-7d agomir and
the oe-NC vector (P<0.05; Fig.
6D). Based on these findings, let-7d could suppress autophagy
in PAECs by targeting ATG16L1.
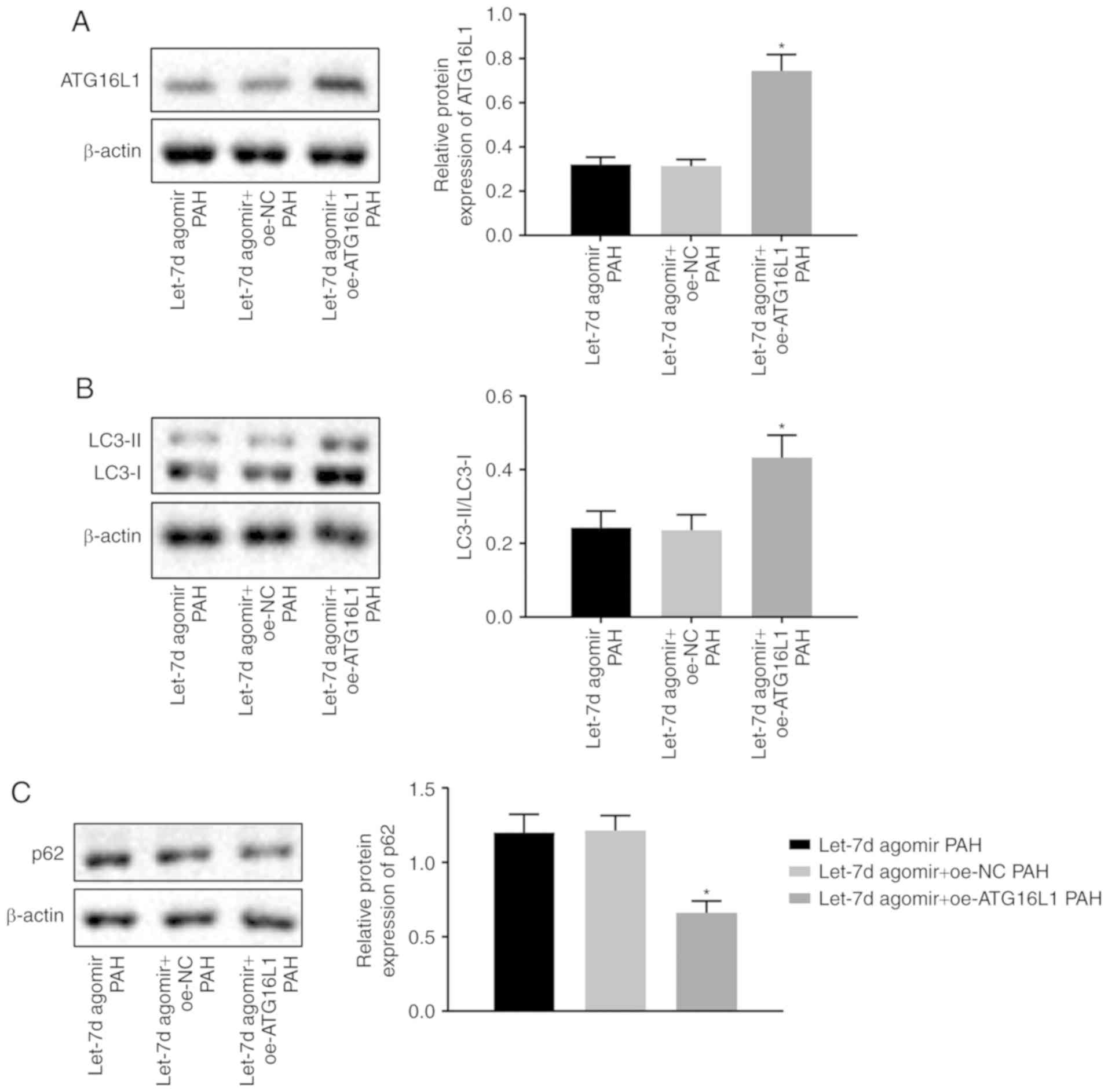 | Figure 6Let-7d suppresses autophagy in PAECs
by targeting ATG16L1. (A) The protein bands and levels of ATG16L1,
as determined by western blot analysis. (B) The protein bands and
levels of LC3 in rat lung tissues, as measured by western blot
analysis. (C) The protein bands and levels of p62 in rat lung
tissues, as assessed by western blot analysis. Unprocessed blots
are shown in Figure S1. Let-7d
suppresses autophagy in PAECs by targeting ATG16L1. (D) The
relative fluorescence intensity and levels of LC3B in rat PAECs, as
examined by immunofluorescence staining. *P<0.05 vs.
the let-7d agomir PAH + oe-NC group. The above data were all
measurement data and expressed as the mean ± standard deviation.
One-way analysis of variance was used for comparisons among
multiple groups. n=10. NC, negative control; PAECs, pulmonary
artery endothelial cells; PAH, pulmonary arterial hypertension;
ATG16L1, autophagy-related 16-like 1; LC3, light chain 3; LC3B,
light chain 3B; oe, overexpression. |
Let-7d alleviates PAH by inhibiting PAEC
autophagy and endothelin synthesis through targeting of
ATG16L1
To investigate the mechanism by which let-7d affects
PAH by targeting ATG16L1, an ELISA was employed to detect the
levels of ET-1 in plasma and lung tissues. Right heart
catheterization was conducted to detect RVSP. The RVHI was analyzed
to assess right heart hypertrophy. H&E staining was performed
to observe morphological changes in pulmonary vessels. The results
of ELISA suggested that injection of both the let-7d agomir and the
oe-ATG16L1 vector significantly elevated the ET-1 levels in the
plasma and lungs versus injection of the let-7d agomir and oe-NC
vector (P<0.05; Fig. 7A and
B). Rats treated with the let-7d agomir and oe-ATG16L1 vectors
showed significantly increased RVSP and RVHI values compared with
rats treated with both let-7d agomir and oe-NC vectors (P<0.05;
Fig. 7C and D). The results of
H&E staining demonstrated that PAH rats injected with both
let-7d agomir and oe-ATG16L1 vectors showed thicker medial walls of
pulmonary blood vessels (36.54±3.77%) than PAH rats injected with
let-7d agomir and oe-NC vectors (24.78±2.50%; Fig. 7E). In conclusion, let-7d relieves
PAH by reducing PAEC autophagy and endothelin synthesis through
targeting of ATG16L1.
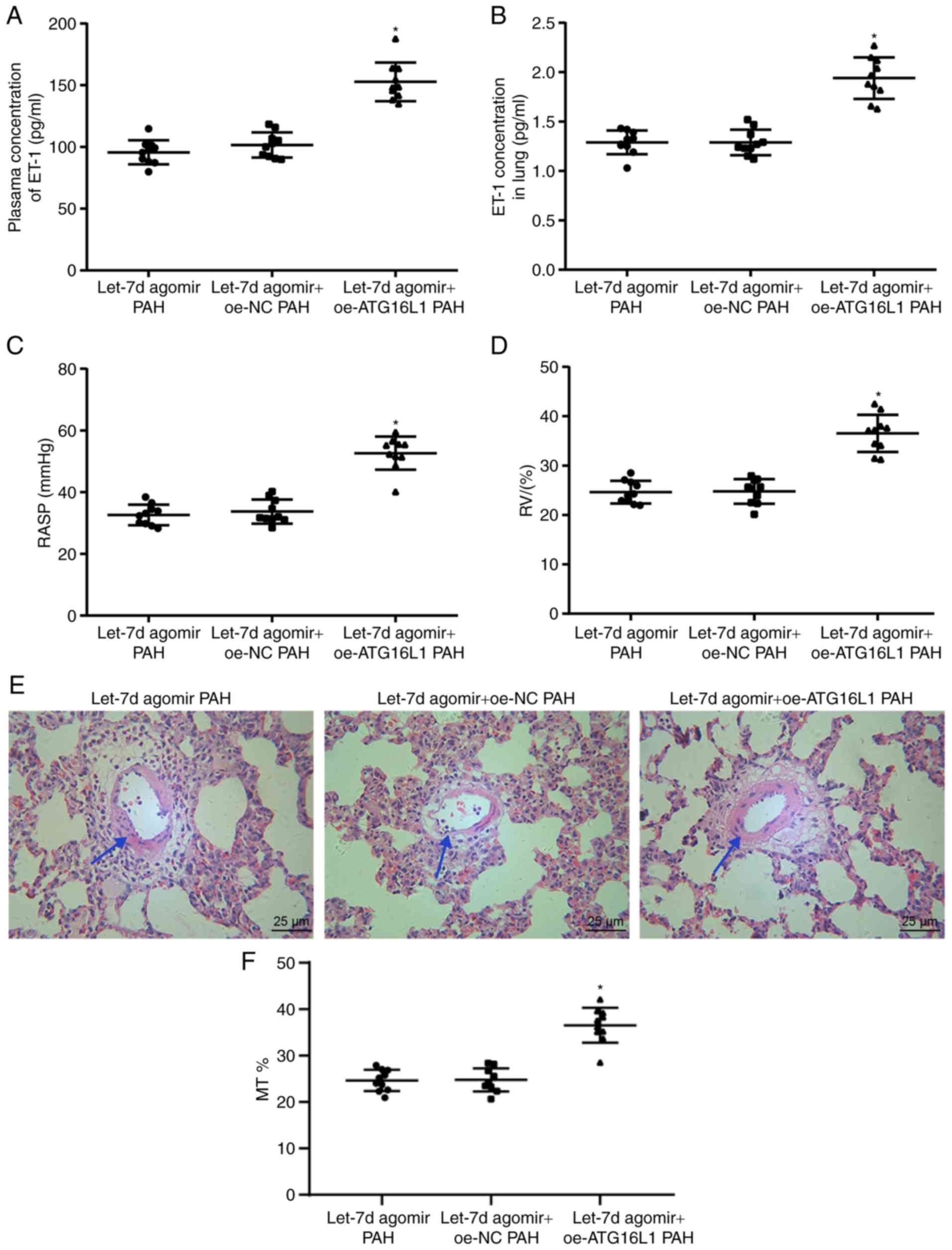 | Figure 7Let-7d ameliorates PAH via
suppression of PAEC autophagy and endothelin synthesis through
downregulation of ATG16L1. (A) The levels of ET-1 in rat plasma, as
determined by ELISA. (B) The levels of ET-1 in rat lungs, as
measured by ELISA. (C) RVSP changes in the rats in each group. (D)
RVHI changes in the rats in each group. (E) Hematoxylin and eosin
staining images of pulmonary arteries in the rats in each group;
the arrow indicates the part with obvious changes. (F) Percentage
thickness of pulmonary vascular wall of rats. *P<0.05
vs. the let-7d agomir PAH + oe-NC group. The above data were all
measurement data and expressed as mean ± standard deviation.
One-way analysis of variance was used for comparisons among
multiple groups, followed by a Tukey's post hoc test. n=10. NC,
negative control; ET-1, endothelin-1; PAH, pulmonary arterial
hypertension; ATG16L1, autophagy-related 16-like 1; RVSP, right
ventricular systolic pressure; RVHI, right ventricular hypertrophy
index; PAECs, pulmonary artery endothelial cells; oe,
overexpression |
Discussion
PAH is a serious disease with features including
vascular proliferation and remodeling of the pulmonary arteries
that leads to a gradual elevation in pulmonary vascular resistance,
right ventricular failure and ultimately death (25). Despite advances in treatment, PAH
remains incurable with high morbidity and mortality rates (26). There is evidence showing that
miRNAs function critically in the regulation of vascular remodeling
in PAH (27). Thus, with the
expectation to provide better treatment modalities for PAH
patients, this study investigated the effects of let-7d on
autophagy in PAECs and on endothelin synthesis. The results
revealed that upregulation of let-7d could downregulate ATG16L1 to
inhibit autophagy in PAECs and suppress endothelin synthesis, thus
ameliorating PAH.
The present study found that let-7d was poorly
expressed in PAH and that upregulation of let-7d could inhibit PAEC
autophagy and endothelin synthesis in PAH. Let-7d has been
demonstrated to regulate the mesenchymal phenotypic properties of
lung fibroblasts (28). The low
expression of let-7d in idiopathic pulmonary fibrosis and the
profibrotic effects of its downregulation indicate a critical role
for let-7d in attenuating lung fibrosis (29). Notably, the let-7 family has been
proven to be abnormally expressed in cardiovascular diseases,
including cardiac fibrosis and hypertension; this family is
expressed in human PAECs and is involved in the regulation of
cardiovascular functions (13).
For example, let-7d was found to be downregulated in cardiac
fibroblasts in mice and its upregulation could mitigate
fibrogenesis in cardiac fibrosis through regulation of
platelet-activating factor receptors; cardiac fibrosis is an
important feature of cardiovascular diseases (30). Wang et al (31) also demonstrated that let-7i was
expressed at low levels in angiotensin II-infused hearts and that
upregulation of let-7i could alleviate cardiac inflammation and
fibrosis. Furthermore, downregulation of let-7c has been reported
in lung cancer and overexpression of let-7c has been shown to exert
inhibitory effects on cell invasion, proliferation and migration
(32). More importantly, let-7d
has been found to be poorly expressed in patients with chronic
thromboembolic pulmonary hypertension and this low expression can
suppress the proliferation of PASMCs through upregulation of p21
(33).
Additionally, the present study demonstrated that
let-7d could suppress PAEC autophagy and endothelin synthesis by
negatively regulating ATG16L1. According to the current prediction
analysis and luciferase activity determination, ATG16L1 is a target
gene of let-7d, and let-7d can negatively regulate ATG16L1. It has
previously been reported that various biological factors and
chemical compounds are engaged in autophagy in vascular endothelial
cells (VECs), and autophagy has potential effects on endothelial
cells (34). There is evidence
showing the involvement of miRNAs in autophagy through the
regulation of ATGs or their regulators in human diseases (35). For example, miR-30d has been found
to suppress autophagy in human cancer cells by inhibiting autophagy
pathway-related genes, such as ATG12, ATG5 and ATG2; autophagosome
formation; and the conversion of LC3B-I to LC3B-II (36). Furthermore, ATG16L1 plays an
important role in autophagy and a previous study verified that
suppression of autophagy by miR-410 overexpression in osteo-sarcoma
cells was achieved partly through downregulation of ATG16L1
(17). Importantly, Xu et
al (37) found that ATG4B is
an underlying target gene of let-7i and that overexpression of
let-7i could inhibit autophagic activity in preeclampsia through
downregulation of ATG4B. Furthermore, endothelin is primarily
released from VECs and forced expression of ET-1 has been found to
be associated with elevations in right atrial pressure, pulmonary
vascular resistance and mortality in patients with PAH (38). In addition, the expression of the
ET-1 gene, which regulates the biological activities of ET-1, can
be mediated by miRNA regulation (39). For instance, miR-125a, miR-125b
and the let-7 family have been found to be upregulated in VECs, and
both miR-125a and miR-125b can inhibit ET-1 expression in VECs by
directly targeting preproET-1, which is essential for endothelin
synthesis (40).
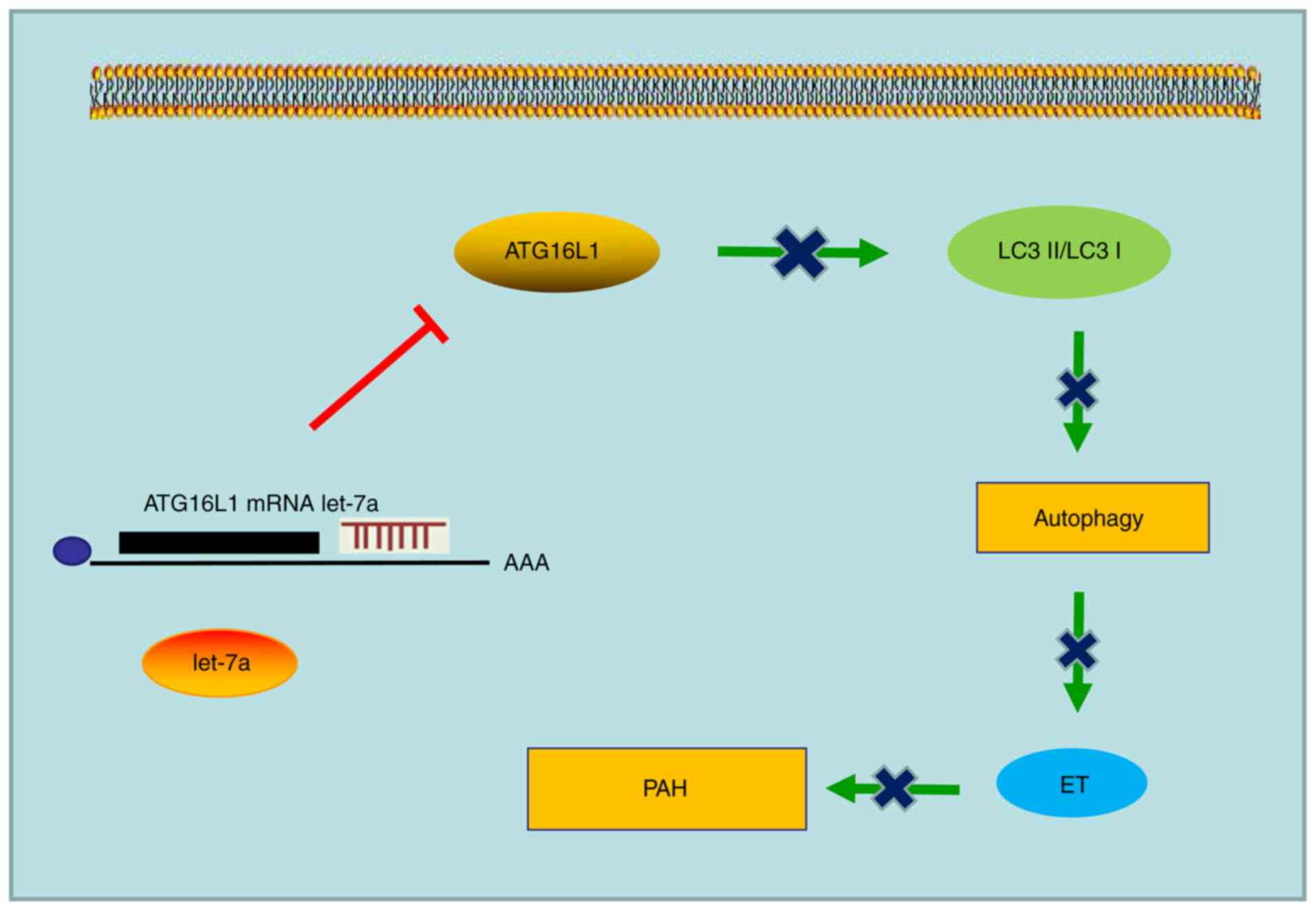
Taken together, the present results demonstrated
that over-expression of let-7d could relieve PAH by inhibiting PAEC
autophagy and endothelin synthesis through downregulation of
ATG16L1 (Fig. 8). Thus, let-7d
overexpression can serve as a potential therapeutic target for PAH
and this study provides new insight for the treatment of PAH.
Nevertheless, more studies are needed to analyze the specificity
and sensitivity of this molecular tool as a biomarker of PAH.
Supplementary Data
Funding
The present study was supported by the General
Program of National Natural Science Foundation (grant no.
81871187), the Regional Projects of National Natural Science
Foundation (grant no. 81460239) and the Natural Science Foundation
Project of Ningxia Hui Autonomous Region (grant no. NZ17195).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
MO designed the study. XL collated the data. SC and
SZ carried out data analyses and produced the initial draft of the
manuscript. JT participated in the design, interpretation of the
results and contributed to drafting the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Ethics
Committee and the Experimental Animal Ethics Committee of Qingdao
Municipal Hospital. Written informed consent was obtained from all
participants prior to the study. The animal experiment strictly
adhered to the principles of using the least number of animals to
complete the experiment and minimizing the pain of the experimental
animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Steele P, Strange G, Wlodarczyk J, Dalton
B, Stewart S, Gabbay E and Keogh A: Hemodynamics in pulmonary
arterial hypertension (PAH): Do they explain long-term clinical
outcomes with PAH-specific therapy? BMC Cardiovasc Disord.
10:92010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Preston IR, Roberts KE, Miller DP, Sen GP,
Selej M, Benton WW, Hill NS and Farber HW: Effect of warfarin
treatment on survival of patients with pulmonary arterial
hypertension (PAH) in the registry to evaluate early and long-term
PAH disease management (REVEAL). Circulation. 132:2403–2411. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sikirica M, Iorga SR, Bancroft T and
Potash J: The economic burden of pulmonary arterial hypertension
(PAH) in the US on payers and patients. BMC Health Serv Res.
14:6762014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mehta J, Parthasarathy PT, Lockey R and
Kolliputi N: New hope for a microRNA therapy for pulmonary arterial
hypertension. Front Genet. 4:1372013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubin LJ, Galie N, Grimminger F, Grunig E,
Humbert M, Jing ZC, Keogh A, Langleben D, Fritsch A, Menezes F, et
al: Riociguat for the treatment of pulmonary arterial hypertension:
A long-term extension study (PATENT-2). Eur Respir J. 45:1303–1313.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meloche J, Le Guen M, Potus F, Vinck J,
Ranchoux B, Johnson I, Antigny F, Tremblay E, Breuils-Bonnet S,
Perros F, et al: MiR-223 reverses experimental pulmonary arterial
hypertension. Am J Physiol Cell Physiol. 309:C363–C372. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Potus F, Graydon C, Provencher S and
Bonnet S: Vascular remodeling process in pulmonary arterial
hypertension, with focus on miR-204 and miR-126 (2013 grover
conference series). Pulm Circ. 4:175–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang L, Wang Y, Rong Y, Xu L, Chu Y,
Zhang Y and Yao Y: MiR-1179 promotes cell invasion through
SLIT2/ROBO1 axis in esophageal squamous cell carcinoma. Int J Clin
Exp Pathol. 8:319–327. 2015.PubMed/NCBI
|
|
9
|
Courboulin A, Paulin R, Giguere NJ,
Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher
S, Cote J, et al: Role for miR-204 in human pulmonary arterial
hypertension. J Exp Med. 208:535–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stevens HC, Deng L, Grant JS, Pinel K,
Thomas M, Morrell NW, MacLean MR, Baker AH and Denby L: Regulation
and function of miR-214 in pulmonary arterial hypertension. Pulm
Circ. 6:109–117. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramberg H, Alshbib A, Berge V, Svindland A
and Tasken KA: Regulation of PBX3 expression by androgen and Let-7d
in prostate cancer. Mol Cancer. 10:502011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dai X and Cai Y: Down-regulation of
microRNA let-7d inhibits the proliferation and invasion of
trophoblast cells in preeclampsia. J Cell Biochem. 119:1141–1151.
2018. View Article : Google Scholar
|
|
13
|
Bao MH, Feng X, Zhang YW, Lou XY, Cheng Y
and Zhou HH: Let-7 in cardiovascular diseases, heart development
and cardiovascular differentiation from stem cells. Int J Mol Sci.
14:23086–23102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L,
Pan Y and Li XJ: Curcumin induces autophagy to protect vascular
endothelial cell survival from oxidative stress damage. Autophagy.
8:812–825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JK, Yuk JM, Kim SY, Kim TS, Jin HS,
Yang CS and Jo EK: MicroRNA-125a inhibits autophagy activation and
antimicrobial responses during mycobacterial infection. J Immunol.
194:5355–5365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishimura T, Kaizuka T, Cadwell K, Sahani
MH, Saitoh T, Akira S, Virgin HW and Mizushima N: FIP200 regulates
targeting of Atg16L1 to the isolation membrane. EMBO Rep.
14:284–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen R, Li X, He B and Hu W: MicroRNA-410
regulates autophagy-related gene ATG16L1 expression and enhances
chemosensitivity via autophagy inhibition in osteosarcoma. Mol Med
Rep. 15:1326–1334. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Magne J, Gustafsson P, Jin H, Maegdefessel
L, Hultenby K, Wernerson A, Eriksson P, Franco-Cereceda A, Kovanen
PT, Goncalves I and Ehrenborg E: ATG16L1 expression in carotid
atherosclerotic plaques is associated with plaque vulnerability.
Arterioscler Thromb Vasc Biol. 35:1226–1235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chin KM, Rubin LJ, Channick R, Di Scala L,
Gaine S, Galie N, Ghofrani HA, Hoeper MM, Lang IM, McLaughlin VV,
et al: Association of N-terminal pro brain natriuretic peptide and
long-term outcome in patients with pulmonary arterial hypertension.
Circulation. 139:2440–2450. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruiz-Irastorza G, Garmendia M, Villar I,
Egurbide MV and Aguirre C: Pulmonary hypertension in systemic lupus
erythematosus: Prevalence, predictors and diagnostic strategy.
Autoimmun Rev. 12:410–415. 2013. View Article : Google Scholar
|
|
21
|
Zhai C, Shi W, Feng W, Zhu Y, Wang J, Li
S, Yan X, Wang Q, Zhang Q, Chai L, et al: Activation of AMPK
prevents monocrotaline-induced pulmonary arterial hypertension by
suppression of NF-kappaB-mediated autophagy activation. Life Sci.
208:87–95. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee SJ, Smith A, Guo L, Alastalo TP, Li M,
Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, et al: Autophagic
protein LC3B confers resistance against hypoxia-induced pulmonary
hypertension. Am J. Respir Crit Care Med. 183:649–658. 2011.
View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Montani D, Souza R, Binkert C, Fischli W,
Simonneau G, Clozel M and Humbert M: Endothelin-1/endothelin-3
ratio: A potential prognostic factor of pulmonary arterial
hypertension. Chest. 131:101–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Humbert M, Sitbon O, Chaouat A, Bertocchi
M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot
F, et al: Survival in patients with idiopathic, familial, and
anorex-igen-associated pulmonary arterial hypertension in the
modern management era. Circulation. 122:156–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee WT, Ling Y, Sheares KK, Pepke-Zaba J,
Peacock AJ and Johnson MK: Predicting survival in pulmonary
arterial hypertension in the UK. Eur Respir J. 40:604–611. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caruso P, Dempsie Y, Stevens HC, McDonald
RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, et al:
A role for miR-145 in pulmonary arterial hypertension: Evidence
from mouse models and patient samples. Circ Res. 111:290–300. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huleihel L, Ben-Yehudah A, Milosevic J, Yu
G, Pandit K, Sakamoto K, Yousef H, LeJeune M, Coon TA, Redinger CJ,
et al: Let-7d microRNA affects mesenchymal phenotypic properties of
lung fibroblasts. Am J Physiol Lung Cell Mol Physiol.
306:L534–L542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pandit KV, Corcoran D, Yousef H,
Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh
M, Handley D, et al: Inhibition and role of let-7d in idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med. 182:220–229. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang H, Pan Z, Zhao X, Liu L, Sun J, Su
X, Xu C, Zhou Y, Zhao D, Xu B, et al: LncRNA PFL contributes to
cardiac fibrosis by acting as a competing endogenous RNA of let-7d.
Theranostics. 8:1180–1194. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X, Wang HX, Li YL, Zhang CC, Zhou CY,
Wang L, Xia YL, Du J and Li HH: MicroRNA Let-7i negatively
regulates cardiac inflammation and fibrosis. Hypertension.
66:776–785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao B, Han H, Chen J, Zhang Z, Li S, Fang
F, Zheng Q, Ma Y, Zhang J, Wu N and Yang Y: MicroRNA let-7c
inhibits migration and invasion of human non-small cell lung cancer
by targeting ITGB3 and MAP4K3. Cancer Lett. 342:43–51. 2014.
View Article : Google Scholar
|
|
33
|
Wang L, Guo LJ, Liu J, Wang W, Yuan JX,
Zhao L, Wang J and Wang C: MicroRNA expression profile of pulmonary
artery smooth muscle cells and the effect of let-7d in chronic
thrombo-embolic pulmonary hypertension. Pulm Circ. 3:654–664. 2013.
View Article : Google Scholar
|
|
34
|
Jiang F: Autophagy in vascular endothelial
cells. Clin Exp Pharmacol Physiol. 43:1021–1028. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo L, Zhao J, Qu Y, Yin R, Gao Q, Ding S,
Zhang Y, Wei J and Xu G: MicroRNA-20a inhibits autophagic process
by targeting ATG7 and ATG16L1 and favors mycobacterial survival in
macrophage cells. Front Cell Infect Microbiol. 6:1342016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Zhong X, Tanyi JL, Shen J, Xu C,
Gao P, Zheng TM, DeMichele A and Zhang L: Mir-30d regulates
multiple genes in the autophagy pathway and impairs autophagy
process in human cancer cells. Biochem Biophys Res Commun.
431:617–622. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu Y, Huang X, Xie J, Chen Y, Fu J and
Wang L: Let-7i-induced Atg4B suppression is essential for autophagy
of placental trophoblast in preeclampsia. J Cell Physiol.
232:2581–2589. 2017. View Article : Google Scholar
|
|
38
|
Shao D, Park JE and Wort SJ: The role of
endothelin-1 in the pathogenesis of pulmonary arterial
hypertension. Pharmacol Res. 63:504–511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jacobs ME, Wingo CS and Cain BD: An
emerging role for microRNA in the regulation of endothelin-1. Front
Physiol. 4:222013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li D, Yang P, Xiong Q, Song X, Yang X, Liu
L, Yuan W and Rui YC: MicroRNA-125a/b-5p inhibits endothelin-1
expression in vascular endothelial cells. J Hypertens.
28:1646–1654. 2010. View Article : Google Scholar : PubMed/NCBI
|