1. Introduction
The mature large intestine is composed of the cecum,
colon and rectum. Embryologically, the large intestine is a part of
the developing gastrointestinal (GI) tract and shares the same
progenitor tissues with the other organs of the GI tract, as it
arises during the development of the endoderm. Later during
embryogenesis, it incorporates tissue from all three germ cell
layers of the trilaminar embryo (1). The epithelium and associated glands
derive from the endoderm, while mesenteries, connective tissues,
smooth muscle and blood vessels come from mesoderm, and the
intrinsic and extrinsic innervation originate from ectoderm. In
order to achieve functional and structural harmony, excellent
molecular tissue crosstalk is required during the development of
the large intestine (2).
During embryological development, several factors
can derail the normal sequence of events which normally lead to the
formation of an intact and functioning GI tract. Usually, the
disruption of gut tube morphogenesis occurs due to problems in
specification and maintenance (3). Some of the developmental colonic
abnormalities, which embryos may experience, are atresia, stenosis,
duplication, situs inversus, intestinal malrotation and intestinal
aganglionosis, malformations of cecum, persistent colonic mesentery
and other rare conditions. There is also evidence to suggest a
possible connection between colon embryogenesis and carcinogenesis
(4).
Hence, in the present review article summarizes the
current knowledge regarding the embryology of the large intestine,
focusing on the responsible molecular mechanisms along with their
disturbances.
2. Overview of gastrointestinal
embryogenesis
Initially, the GI tract, which emerges from the
endoderm during gastrulation (week 3), extends from the
buccopharyngeal membrane to the cloacal membrane. During and
immediately following gastrulation, the development of the gut tube
occurs in simultaneity with the turning and folding movements of
the embryo (1). Thus, three
regions begin to form in the sagittal plane, the foregut
cephalically in the head fold, hindgut caudally with its allantoic
outgrowth and midgut in the middle. Initially, definitive endoderm
invaginates at a cranial and a caudal region forming the anterior
intestinal portal (AIP) and the caudal intestinal portal (CIP),
respectively. As a consequence, the two ends of the GI system are
formed. AIP and CIP extend toward one another and the lateral
endoderm of midgut folds ventrally to form a sealed gut tube. At
the same time, the subjacent splanchnic mesenchyme grows around the
endoderm and differentiates to smooth muscle (2). After week 4, the foregut, midgut and
hindgut are craniocaudally discernible and they evolve into the
different compartments of the GI tract. These three divisions are
later distinguished by their different arterial supply. The GI
epithelial cells proliferate and obliterate the gut lumen by week
6. By week 8, the central cells degenerate, and the tube is thus
again patent.
The parts of the large intestine derive from the
midgut and hindgut. The midgut is formed during week 4, as the
embryo laterally folds and the pocket of the yolk sac protrudes
ventrally. The yolk sac continues to communicate with the primitive
gut through the omphalomesenteric duct (vitelline duct). The
vitelline duct normally obliterates later, usually before day 36
(5). The arterial supply of the
midgut comes from the superior mesenteric artery and it is drained
by the corresponding venous and lymphatic vessels. The main GI
tract organs which form between weeks 6 and 10 from the midgut are
the distal duodenum, jejunum, ileum, appendix, cecum, ascending
colon and the proximal two thirds of the transverse colon. The
mesenteries of the GI tract are generated from the common dorsal
mesentery, while the ventral mesentery participates in the lesser
omentum and falciform ligament. During embryological development,
much of the midgut herniates at the umbilicus externally to the
abdomen, providing a potential position of rotation, which must
occur in order to place the GI tract in the correct abdominal
position with its associated mesentery (6). Between weeks 5 and 8, the midgut
elongates inside its mesentery and forms loops. As a result, a
normal umbilical herniation of the midgut occurs, as the midgut
loop gradually protrudes through the umbilical ring. At week 8, the
total intraumbilical loop rotates counterclockwise 90° and
positions the midgut along the horizontal plane. By 10 weeks, the
abdomen has developmentally enlarged sufficiently so that the
entire midgut can be accommodated inside it. Following a further
180° counterclockwise rotation around the superior mesenteric
artery, the small intestine returns to the abdominal cavity.
Concurrently, the large intestine follows its rotation and also
moves 180° counterclockwise. Following the return of midgut in the
abdomen, the mesenteries of cecum and ascending colon fix to the
dorsal wall, making these parts immobile (6).
The hindgut initially consists of the cloaca, which
later gets segregated by a septum to a dorsal GI compartment and a
ventral urogenital compartment (7). Its blood supply mainly comes from
inferior mesenteric artery, with corresponding venous and lymphatic
drainage. The hindgut later differentiates to urinary epithelium,
distal one third of transverse colon, descending colon, sigmoid
colon, rectum and superior anus. The transverse colon and sigmoid
colon maintain a mobile mesentery, whereas the descending colon
becomes immobile as its mesentery fixes to the dorsal wall. As
regards the rectum, the upper third is intraperitoneal, the middle
third retroperitoneal, whereas the lower third is infraperitoneal
along with the superior anus (8).
In the large intestine, the nervous system is
represented by intrinsic and extrinsic innervation. The enteric
nervous system (ENS), which is the intrinsic innervation,
originates from cells of the vagal neural crest migrating into and
across the wall of the GI tract (9). The ENS is considered as part of the
autonomous nervous system (10,11), and it is composed of two plexuses;
the external myenteric plexus of Auerbach between the circular and
the longitudinal muscular layers across the entire length of the GI
tract, and the internal submucosal plexus, which does not exist in
the esophagus and principally includes Meissner's and secondly
Schabadasch's plexus (12).
Although Schabadasch's plexus is described as a part of the ENS,
placed between Meissner's and Auerbach's plexuses, there is a lack
of information regarding its embryological formation. The extrinsic
innervation of the large intestine is mediated through sympathetic
nerves, originating from T5 to L2 levels, and parasympathetic
nerves from the dorsal vagus complex and pelvic nerves S2-S4. The
major events during gut embryogenesis are summarized in Table I.
 | Table IMajor embryological events during gut
development. |
Table I
Major embryological events during gut
development.
| Embryonic week | Developmental
stage |
|---|
| Week 3 | - Gastrulation |
| - Primitive gut
tube formation |
| - Elongation and
invagination of endoderm anteriorly, caudally, and ventrally |
| - Development of
splanchnic mesenchyme around endoderm |
| Week 4 | - Discernibility of
foregut, midgut and hindgut |
| - Buccopharyngeal
membrane resorption |
| - Invasion of
foregut by enteric neural crest cells |
| Week 6 | - Obliteration of
gut lumen |
| - Initiation of
midgut herniation through the umbilical ring |
| Week 7 | - Obliteration of
vitelline duct |
| - Completion of
enteric neural crest cell migration |
| Week 8 | - Recanalization of
gut tube |
| - Rotation of
intraumbilical loop 90° counterclockwise |
| Week 9 | - Initiation of
villus formation |
| Week 10 | - Rotation of
midgut 180° clockwise back to the peritoneal cavity |
| Week 11 | - Development of
circular and longitudinal smooth muscle layers |
| Week 12 | - Initiation of
crypt formation |
| Week 14 | - Formation of
mucosal muscle layer |
| Week 24 | - Development of
intestinal absorption function |
| Week 32 | - Fetal intestinal
absorption equal to adult levels |
3. Early development of the gut tube
The gut consists of two types of tissue in a tubular
arrangement. The outer layer(s) of the tube is mainly smooth muscle
derived from lateral plate splanchnic mesoderm, while the inner
lining is derived from endoderm in the majority of the gut
epithelium. On the contrary, the epithelium of the most cephalic
(mouth) and caudal (anus) regions of the gut is derived from
ectoderm (2).
Gastrulation begins at the 7th day of embryonal
development, when the primitive streak is formed in the pluripotent
epiblast layer. The endoderm and mesoderm are derived from the
former mesendoderm, through the transforming growth factor-β
(TGF-β) family member Nodal (13)
and possibly wingless-related integration site (Wnt) signaling
(14,15). Depending on the intensity of Nodal
signaling, the mesendodermal cells differentiate to the endoderm,
whereas lower Nodal signaling induces differentiation towards
mesoderm (13,16-19). Transcriptional factors which are
members of the T-box, SRY-related HMG-box (Sox), Mix, GATA and
forkhead-box (Fox)-A families have been found to regulate the
promotion of Nodal-induced endoderm formation in all vertebrate
species (20-24). Activin is another TGF-β family
member, which binds to the same receptors with Nodal and can mimic
the role of Nodal in the induction of mesendodermal differentiation
towards the endoderm (25,26).
The anterior definitive endoderm is created by the
first cells which emerge through the primitive streak and migrate
anteriorly (27,28). On the contrary, the endoderm of
the midgut and hindgut are derived from the mid-streak stage of
gastrulation and the entire definitive endoderm is completely
formed before somite formation (29,30). It is noteworthy that the
definitive endoderm intercalates with the existing extraembryonic
visceral endoderm (31), mostly
in hindgut final endoderm (35%), possibly masking genetic disorders
of its development.
The initial biological events leading to AIP and CIP
formation following gastrulation have not yet been determined. The
AIP is created by cells of cephalic endoderm, while the CIP forms
by caudal endodermal cells (29,32,33). In the beginning, endodermal cells
from the cephalic and caudal pits invaginate and form pockets,
further elongating until they meet each other and merge.
Simultaneously, the lateral endoderm of the midgut ventrally folds
and completes the closing of the gut tube (34). A study using Xenopus frogs
proposed that both the elongation and gut tube formation, require
Rho, Rho kinase (ROCK) and myosin II in order to regulate cellular
and tissue arrangement (35).
Several factors have been investigated to evaluate
their participation in the mechanisms which promote the
invaginations. The GATA family of transcriptional factors seems to
be implicated in the events of invagination. GATA-4 has been found
to be expressed in the very early definitive endoderm of the AIP
and is necessary to close the body wall. GATA-4−/−
embryos develop a malformed AIP and therefore, no foregut. They
also evolve without the normal placement of the yolk sac due to the
defective lateral-ventral body folding and the abnormal AIP.
However, GATA-4−/− embryos maintain the anterior
endoderm, indicating that other factors are crucial for its
development (36).
There is evidence to indicate that in mice, the
formation and elongation of the gut tube is guided by the
regulation of the ultimate extension movements of the embryo. A
Kruppel associated box (KRAB) zinc-finger protein, Chato, directs
body elongation of all three germ layers. Chato has been shown to
affect endodermal elongation, and embryos lacking Chato experience
failure of endodermal elongation and unsuccessful gut tube closure
(37). Contrary to Chato, dapper
(Dact)-1 is implicated in gut tube morphogenesis via modulating the
non-canonical Wnt/planar cell polarity (PCP) signaling pathway.
Dact-1−/− mice experience impaired posterior development
with the failure of the hindgut endoderm to form CIP, failure of
the ventral endodermal folding and failure to form cloaca or
hindgut (38).
The molecular mechanisms controlling the development
of the mesoderm are better understood in comparison with those
regarding the endoderm, since their interaction is crucial for the
formation of the gut tube (2).
It is a fact that cultures of endodermal cells from
the foregut cannot differentiate without co-culturing with
meso-dermal tissues (39). There
is a developmental window before which the differentiation of
primitive endoderm relies on the anteroposterior (AP) position of
its subjacent mesoderm (39).
After this window, the morphologically undifferentiated primitive
GI endoderm is committed to develop into regionally specific
epithelium (40). Furthermore,
studies have confirmed that the endoderm can also alter the
differentiation of mesoderm. When somatic, non-gut mesoderm is
co-cultured with gut endoderm, it is directed to differentiate
towards smooth muscle rather than skeletal muscle and this is
confirmed by histology and by tracing the expression of mesodermal
proteins, e.g., tenascin (41)
and of smooth muscle actin (42).
The majority of endodermal gut regions exhibit
morphological and cellular plasticity to the influence of mesoderm
except for the midgut, where the endodermal cells are more
autonomous (43,44). Some molecular events which
coordinate the endodermal-mesodermal co-development have been
described. Sonic hedgehog (Shh), a vertebrate homologue of
Drosophila hedgehog (hh), encodes a signaling molecule which
participates in the development of limbs (45), somites (46) and neural tube (47). Shh is expressed in the endoderm of
the GI tract and its derivatives (48) it has been hypothesized to initiate
the early endodermally-derived inductive signal for the gut
morphogenesis. Initially, it is expressed only in AIP and CIP even
before the beginning of invaginations (49). However, it has been proven that
this is not the molecule which signals for the beginning of
invaginations (50), as
Shh−/− mice develop a GI tract even with major
abnormalities in the foregut, such as malformed esophagus with
wider lumen and without a surrounding mesoderm (51). Therefore, Shh endodermal signal
towards mesoderm probably mediates development, recruitment and
other aspects of mesoderm in foregut. It has been confirmed that
Shh receptors exist only in the mesodermal part of the GI tract
(52), whereas the overexpression
of Shh in the primitive gut leads to the overdevelopment of the
mesoderm rather than endoderm (44).
Wherever the endoderm expresses Shh, the subjacent
mesenchymal mesoderm expresses a homologue of Drosophila's
decapentaplegic (dpp) (48). Bone
morphogenetic protein (BMP)-2 and BMP-4 are the two dpp homologs
which are expressed in the vertebrate gut. Even in the primitive
gut, before invagination becomes apparent, by the time Shh
expression is traceable in the CIP region, BMP-4 is expressed in
the closely associated mesenchymal mesoderm (49). Shh has been proven to induce BMP-4
in the splanchnic mesoderm even ectopically (44,49,52).
BMP-4 may negatively regulate the growth and
hypertrophy of smooth muscle or it may facilitate differentiation
to smooth muscle. At a later developmental stage, when the smooth
muscle layers of the gut have already formed, BMP-4 is expressed in
the submucosa, which is the mesodermal tissue subjacent to the Shh
expressing endoderm. On the contrary, the expression of BMP-4
cannot be traced in the differentiated smooth muscle region of
mesoderm, which does not have direct contact with endoderm. It is
noteworthy that both Shh expression and BMP-4 do not induce smooth
muscle differentiation of mesoderm. Shh has even been found to
reduce the development of smooth muscle proteins in explant
cultures (52).
The role of BMP-2 in the development of a functional
gut has been evaluated and seems to be mainly implicated in its
enervation. BMP-2 is expressed in gut mesoderm and has been found
to promote the maturation of enteric neurons (53). Nevertheless, its overexpression
deregulates the contribution of other crucial factors on the
incorporation of neural cells in gut, including colon, impeding the
survival of neural cells in the gut (54).
4. Molecular control of the antero-posterior
(AP) pattern
The GI tract is patterned into distinct AP
compartments, a fact that can be validated by the expression of
hematopoietically-expressed homeobox (Hhex), Fox-A2 and Sox-2
anteriorly and caudal-related homeobox (Cdx)-1,2,4 posteriorly
(30,55-57). Even following gastrulation, the
endoderm continues to receive signals mainly from mesoderm,
according to which it gets further patterned (57). The signaling continues in each
developmental step, thus being vulnerable to possible alterations
and/or disorders. Some of the most important molecular pathways
which mediate the patterning of the AP axis of the endoderm include
fibroblast growth factor (FGF), Wnt, BMP and retinoic acid (RA)
signaling (10,58-61).
Role of FGF
The exposure of human embryonal stem cells (hESCs)
to a particular concentration of 1.1 ng/ml of FGF-4 augments their
pancreatic and duodenal homeobox (Pdx)-1 expression, which is
primarily induced by RA signaling. Moreover, FGF-4 and RA possibly
cooperate to increase Cdx-2 expression of the posterior gut
endoderm (62). It has also been
demonstrated that as far as the concentration of added FGF-2
increases, endodermal hESCs are patterned towards either an
anterior phenotype, or a posterior phenotype, respectively to the
increase of FGF-2 concentration (63). Finally, FGF and Wnt collaborate to
direct endodermal hESCs towards a hindgut Cdx-2-positive epithelial
phenotype (64). Cdx-2 is
recognized as one of the most conserved markers of hindgut, while
the majority of the colon is derived from it.
While the endodermally-derived epithelium develops,
FGF-9 is required to be expressed by the epithelium so as to signal
towards the surrounding mesenchyme and harmonically direct it to
proliferate and elongate according to the respective growth of its
underlying epithelium (65). In
response, the mesenchyme simultaneously expresses FGF-10, which
signals back to the epithelium through FGF receptor (FGFR)2b and
directs it to proliferate and form the cecal budding. However,
FGF-10 does not induce the specific differentiation of the cecal
epithelium (66-68).
Role of Cdx
The endoderm, indeed, possess intrinsic molecular
pathways, which wait for the time to respond and guide endoderm to
its proper differentiation and functional specification in
accordance with extrinsic inductive signals. Pertaining to the
colon, the hindgut endoderm maintains a fixed epithelial identity
following a certain stage of development. As a consequence,
mesoderm can only limitedly alter the AP developmental pattern of
endoderm prior to cytodifferentiation (69).
One of the most significant intrinsic molecular
factors for hindgut intestinal differentiation is Cdx-2, which is a
member of the ParaHox gene cluster (70,71). It has been demonstrated that the
lack of Cdx-1 or Cdx-4 results in a non-intestinal phenotype
(72). Furthermore, if Cdx-2
loss-of-function is imposed, the endodermal differentiation is
disoriented, the intestinal phenotype is not rescued and the gut
epithelium is altered to esophageal epithelium. It is interesting
that, even though Cdx-2 loss-of-function leads to a posterior to
anterior alteration, the expression of other important factors of
the AP differentiation, such as homeobox (Hox) genes, Pdx-1 and
Barx-1 were unaffected (70).
Therefore, Cdx-2 indeed is a key molecular factor and it is
required for both the establishment and maintenance of the
posterior endodermal phenotype. The combined action of the most
important extrinsic factors, Wnt, FGF, BMP and RA at each
developmental stage may be necessary to induce the proper Cdx-2
activity (73-75). T-cell factor (Tcf)-1 and
Tcf-4−/− mice develop the loss of the most caudal
hindgut region and alteration in the differentiation of the
duodenum, which, instead of Cdx-2, express Sox-2 which is a stomach
marker (76). This evidence
appears to be very similar to the results of the Cdx-2
loss-of-function (71).
Role of Hox genes
The molecular pathways which control the overall
body plan are probably implicated in the AP pattern of the gut as
well. There are indications that Hox genes, which participate in
the patterning of the total body plan, initially were meant to
pattern the gut. The abdominal (Abd)-B-like Hox genes are expressed
in the mesoderm of the posterior midgut and hindgut, whereas
clusters A and D of these genes are regionally expressed, defining
morphological landmarks of the mid- and hindgut (49,77). It has been assumed that Hox genes
are important in setting up limits between major AP regions, such
us sphincters. Nevertheless, Hox genes mutations seem to provoke
only minor defects in the developmental of endoderm (78-80).
The most posterior region of the gut, the cloaca is
the tissue that later evolves to both the anorectum and urogenital
opening. In humans, a missense mutation in Hox-A13 results in
protein truncation and finally in a syndrome that includes a
genital phenotype (81).
Elaborating on the role of Hox genes, it seems that they also
participate in the patterning of the gut epithelium. The
misexpression of Hox-D13 in the chick midgut mesoderm induces a
hindgut differentiation of the midgut mesoderm epithelium. Shh
expressed in the endoderm has been identified as an activator of
Abd-b like Hox genes (49), while
Shh also induces Hox genes in the limb (45). Thus, Shh may be a general inducer
of Hox genes.
Role of Wnt pathways
Another very important system of signaling for the
AP pattern is the Wnt. Wnt genes encode a variety of molecules
which signal through the Frizzled (Fz) family of cell surface
receptors, activating several intracellular pathways, such as the
Wnt/β-catenin canonical pathway, the Wnt/Ca2+ pathway
and the planar polarity pathway (58,82). Of the Wnt ligands, Fz7, Fz8, Fz9,
Wnt5b, Wnt 6 and Wnt 14 are traced during the early or late stages
of gut development (83,84). Of the Wnt antagonists,
Frizzled-related protein (FRZ)-b1 and secreted frizzled-related
protein (sFRP)-2 are expressed in the developing gut. Of the
downstream targets of the canonical pathway, Tcf-4 and Tcf-7L2 are
expressed in the colon (83,85). While the embryonal development
evolves, various Wnt ligands, antagonists and downstream molecules
are detected. A combination of different proteins is found in very
distinct regions along the AP axis of the gut tube, in
correspondence with the boundaries of the future organs of the GI
tract (83). For instance,
lymphoid enhancer-binding factor (Lef)-1, Fz1 and Fz8 are expressed
in the duodenum and large intestine.
The canonical Wnt pathway
Along the canonical pathway, which is the most
extensively studied of the Wnt pathways, β-catenin translocates to
the nucleus and it associates with members of the Tcf/Lef family of
high mobility group (HMG) transcriptional factors to transduce the
Wnt signal (86). If Wnt is
absent or insufficient, β-catenin becomes phosphorylated and is
degraded by β-transducin repeats-containing protein (β-TrCP), an E3
ubiquitin ligase (87). This
phosphorylation requires glycogen synthase kinase (GSK)-3β, axin,
and adenomatous polyposis coli (APC) (86,88). The active, functioning canonical
Wnt pathway allows β-catenin to escape phosphorylation and
degradation (89).
The expression profiles of molecules of the
canonical Wnt pathway define domains that give rise to the
duodenum, ceca and large intestine, whereas non-canonical pathway
members define regions which will become the posterior small
intestine and cecum. To elaborate, there is evidence that the Wnt
canonical signaling pathway guides the AP patterning of the gut by
directly augmenting the expression of Cdx-2. However, another study
demonstrated that Cdx-2 expression was also decreased after the
withdrawal of a Wnt antagonist and the subsequent reactivation of
Wnt signaling, which seems a bit confusing (90). Therefore, the canonical and
non-canonical Wnt pathways may define distinct domains in the AP
axis and they may have different functions in regions of the gut
where they overlap, such as the cecum (83).
A number of disorders can deregulate the dynamic
equilibrium between the activation and degradation of β-catenin and
of the activity of the overall canonical pathway. Axin mutations
prevent axin from binding to β-catenin, blocking the deactivation
of β-catenin by APC. β-catenin mutations can also rescue β-catenin
from phosphorylation for degradation by β-TrCP (87). As a consequence of the above
mutations, β-catenin levels remain stable and can continue the
activation of the canonical Wnt pathway endlessly (91).
Lef-1 is a transcriptional mediator in canonical Wnt
pathway, but is not normally expressed in the colon (92). Lef-1−/− chicks exhibit
stenosis of the cecal lumen due to the overproliferation of
epithelial cells. Tcf-1 is a downstream target of Tcf-4/β-catenin
(93) Tcf-1 and
Tcf-4−/− mice exhibit a lack of crucial caudal
structures, while the main defect in these mice is localized in the
endoderm, contrary to the majority of canonical Wnt pathway
disorders which primarily disrupt the mesoderm. These mice do not
form a CIP, their hindgut endoderm does not express Fox-A1 and
Sox-17 and they end up with an open midgut and a total lack of
hindgut structures related to the complete absence of Shh
expression (76). The maintenance
of the stem cell compartment is potentially mediated by Cdx-1,
which is a downstream target of Tcf-4 and is expressed in the
crypts of the intestinal epithelium (94). Tcf-4 and Lef-1 are evidently
important for the adjustment of the balance between proliferation
and differentiation in the developing gut.
The lack of Wnt3a, Wnt5a, low-density lipoprotein
receptor-related protein (LRP)-6 or Tcf-1/Lef-1 all lead to
mesodermally mediated posterior developmental disorders (95,96).
Non-canonical pathways
At the non-canonical planar cell polarity or Wnt/PCP
pathway, ROCK and the Jun-N-terminal kinase (JNK) are activated
(97,98) and participate in the direction of
the cytoskeletal organization and of the epithelial cell
polarization (99). This pathway
is also implicated in the extension movements of the mesoderm
during gastrulation, in which Wnt11 and Fz7 ligands are required
(100).
A second non-canonical Wnt pathway, the
Wnt/Ca2+ pathway, is implicated in the extension
movements of the mesoderm and in the ventral development of the
embryo (101,102). It acts by the release of
intracellular Ca2+ following the activation of
phospholipase C, protein kinase C and calmodulin-dependent kinase
II. Wnt5a, which participates in the canonical pathway as well, and
Fz2 are necessary for the intracellular release of Ca2+
(103).
Not only the activation, but also the deactivation
of Wnt signaling is crucial for the patterning of the total body
plan (104,105). Antagonists of Wnt, such as the
sFRP, block the activity of Wnt ligands with relative specificity.
For instance, FRZ-b1 and sFRP-2 inhibit Wnt1, but only FRZ-b1 can
block Wnt8 and only sFRP-2 can block Wnt4 (105), while all Wnt1, 4 and 8 are
ligands in the canonical pathway (106). It has also been observed that in
regions with the absence of sFRP-1, the Tcf-4 transcript is found,
proposing a positive link between the antagonist and the downstream
target of Wnt.
After the gut tube is fully formed, the intestine
commences to lengthen. Wnt5a seems to be implicated in this
elongation via mediating in the non-canonical Wnt signaling
pathway. Wnt5a−/− mice develop a 63% shorter intestine
than Wnt efficient mice (107).
It is noteworthy that mice that have ineffective proteins of the
sFRP family, which normally antagonize Wnt5a, also exhibit a
shorter intestinal length (108). This evidence suggests that the
development of proper intestinal length requires the balanced
regulation of Wnt signaling, and neither the its hyperactivation
nor hypoactivation.
5. Molecular control of the left-right (LR)
pattern
The molecular events which are implicated in the
configuration of the LR pattern are among the most conserved events
among all species (109).
Two crucial molecules which are linked to the
creation of LR asymmetry are Shh and Activin (110). The left side of the embryo
restrictedly expresses Shh, whereas Activin is expressed only in
the left side. Shh expression leads to a cascade of unilateral
expression of factors, such as Nodal, paired-like homeodomain
transcription factor (Pitx)-2, bagpipe homeobox homolog (Bapx)-1
and FGF-8 (111-113). Bilateral Shh expression would
lead to the randomization of the situs of each organ independently
(112). Pertaining to the gut,
the levositus is defined by the same left-sided factors, such as
Shh, Pitx-2 and Bapx-1, whereas the bilateral expression of these
molecules would lead to heterotaxy syndromes (112).
Herniation and rotation
The primary midgut loop, which is formed during
midgut herniation into the umbilical cord at week 5, as mentioned
before, contains only one part of the colon, the cecum, which is
part of the 4th secondary ileocecal loop of the distal limb
(114). The colon develops
2-fold at a slower rate than the small intestine, in accordance
with its dorsal mesentery and does not require to loop (115). Before the resolution of the
hernia, the intra-abdominal part of colon and its dorsal mesentery
is sagittally positioned in the midline of abdomen. Between weeks
8.5 and 9.5 of gestation, the development rate of both small and
large intestine significantly decreases (115), whereas the body axis and wall
growth create enough space to drive midgut back inside abdomen
(116). As the midgut returns
inside the abdomen, the duodenum is placed dorsocaudally to the
superior mesenteric artery (SMA) at the left side and jejunum
occupies most of the left side. The ileocecal loop is the last to
return inside the abdomen at week 9.5, and becomes positioned
initially ventrally at the midline and half a week later it can be
found in the right side (115).
The ascending and proximal transverse colon, which
are also suspended by the midgut mesentery, initially have a
ventrocranial position relative to SMA, and move inwards following
the movements of the intra-abdominally returning small intestine
and maintain their continuation with cecum, thus occupying a right
sided position. In the meantime, at week 6 the last part of the
midgut, the future distal transverse colon, is sagittally
continuous with hindgut and they are hardly discernible. At week 8,
the transition of the midgut to the hindgut, as it can be
recognized by the overlap of perfusion by branches of both SMA and
inferior mesenteric artery (IMA), begins to lift and move leftward
to form the splenic bend. As gestation proceeds, the distal part of
this transition extends downward and forms the descending colon. At
week 10, while the cecum is positioned cranioventrally relative to
small intestine and subhepatically due to the relatively large size
of liver, proximal colon runs its course obliquely towards the
splenic bend (114,117). Between weeks 10 and 20, while
the relative size of the liver decreases (118), the hepatic bend develops, the
ascending colon extends downward at the right abdominal side and
the cecum follows and occupies its final position in the right
iliac fossa (117).
The molecular and structural interactions that
promote the phenomenon of rotation, which are crucial for the
proper positioning of the colon, have not been investigated in
detail; however, there is evidence to indicate that the dorsal
mesentery of the midgut loop presents molecular and architectural
left-right asymmetry. The mesenchymal cells of the left side of the
mesentery are more condense than those of the right side, whereas
the midgut epithelium of the left side is columnar, while the
right-side epithelium is cuboidal. Therefore, there seems to be a
greater proliferation of the mesenchyme and epithelium on the left
side, which condenses the cellular and non-cellular structures
creating mechanical forces which tilts the mesentery and the midgut
loop counterclockwise (119).
To elaborate on the molecular factors, the
transcriptional factor Pitx-2, which can be activated by Nodal, is
restrictedly expressed in the left side of the dorsal mesentery in
its whole dorsal-ventral extent (111). This may be one of the results of
the leftward flow, which is generated by the asymmetric beating of
nodal cilia (63,120,121). Concurrently, islet (Isl)-1, a
LIM homeodomain-containing transcriptional factor, is exclusively
expressed in the left side of the mesentery at the time of
rotation, whereas T-box (Tbx)-18 factor is expressed at higher
levels in the right side than the left side of the midgut loop
mesentery (119,122).
Shroom3 and N-cadherin have been proposed as
downstream targets of Pitx-2, which mediate the cellular shape
changes that characterize the left-right asymmetry (123). In silico has analysis
established that N-cadherin is exclusively expressed in the left
side of the mesentery and it fosters the asymmetry of the
extracellular matrix (ECM), being partly responsible for the
mesenchymal left-right side asymmetry. The mesenchymal asymmetry
also exists owing to different cell to cell adhesion (124). Another possible Pitx-2
downstream target is dishevelled associated activator of
morphogenesis (Daam)-2 and it is activated in the dorsal mesentery
both directly and indirectly by Pitx-2. The indirect activation is
mediated by Wnt signaling (125).
Disorders: Misexpression of key
factors
Either the ectopic bilateral expression of Nodal,
which is known to activate Pitx-2, or the ectopic expression of
Pitx-2 itself bilaterally in the splanchnic mesoderm forces a
symmetrical bilateral expression of Isl-1 and a loss of right sided
Tbx-18 expression. Isl-1 probably augments Pitx-2 expression, as
the ectopic expression of Isl-1 in the right side of the mesentery
also leads to bilateral Pitx-2 expression and the loss of
right-sided Tbx-18 expression. The ectopic expression of either of
the above-mentioned factors, Nodal, Pitx-2 or Isl-1, lead to the
disruption of the left-right asymmetry of the dorsal mesentery,
creating a bilateral symmetry with the replacement of the cuboidal
cells of the right side by columnar cells and an increase in
mesenchymal density of the right side. Pitx-2−/− embryos
do not express Isl-1; they express Tbx-18 bilaterally and present
bilateral symmetry as the left side acquires the structural
characteristics of the right side (119). As a conclusion, Pitx-2 and Isl-1
expression seem to be significant in creating the dynamic force,
which directs the initial events of midgut rotation.
Disorders: Situs inversus
In situs inversus, either abdominus or totalis,
beyond the other clinical manifestations, the organs of abdomen
swap sides. Therefore, pertaining to the colon, the cecum, appendix
and ascending part are located in the left side, whereas the
descending and sigmoid colon are on the right side. Relatively rare
complications of situs inversus are cecal volvulus and intestinal
atresia (126). This syndrome
occurs due to problems of the nodal function after the stage of 3
somites if there are mutations or defect in genes, such as
inversin, kinesin family member (KIF)3B, Dishevelled (Dvl),
polycystin (Pkd)-2 and KIF3A. The improper function of inversin
reduces the forwarding effectiveness of the cilial movement
(127,128), and the lack of KIF3B impairs
ciliogenesis, resulting in prenatal death and LR asymmetry
(129). Furthermore, Dvl-1, 2
and 3 intervene in both the canonical and non-canonical Wnt
pathways. The non-canonical Wnt/PCP pathway is crucial for the
proper polarization and function of the node (130) and the malfunction of Dvls leads
to the PCP deregulation-mediated randomization of LR asymmetry
(131,132). Finally, Pkd-2 or KIF3A loss
diminishes the mechanosensory ability of the node, which is
necessary to maintain the leftward nodal fluid flow. As a
consequence, LR asymmetry distribution randomly occurs (133).
Disorders: Heterotaxy syndromes
Intestinal obstruction, due to cecal volvulus or
intestinal atresia, is far more common in heterotaxy syndrome
(134). Heterotaxy syndrome
differs from situs inversus in that the internal organs are
abnormally positioned in the chest and abdomen without some order
whereas in situs inversus the order is maintained. Abdominal
abnormalities can be present in both of its subgroups, although
they are more frequent in the subgroup of isomerism of the left
atrial appendage. Intestinal malrotation can derive from failure of
the 270° counterclockwise rotation of the midgut, whereas
intestinal obstruction with the danger of necrosis can occur due to
flaws of mesenteric fixation to the dorsal wall (135). Moreover, the cecum does not
reach its normal position in the right lower quadrant because the
cecal mesentery improperly fuses with posterior parietal peritoneum
increasing the hazard of cecal volvulus (136-138). Possible molecular causes of
heterotaxy syndromes, affecting normal intestinal embryogenesis,
may include insufficiency of Fox-A2, which is known to be a Nodal
regulator, and disturbances of the function of FGF-12, RNA binding
Fox homolog (RBFox)-1, microRNA (miRNA/miR)-302F and polypeptide
N-acetylgalactosaminyltransferase (GALNT)11 (139-141). GALNT11 dysfunction deregulates
the mechanisms that ensure harmonic maintenance of left side flow
in the node, creating insurmountable obstacles to the proper
designing of left-right asymmetry.
6. Mesenteric fixation
The fixation of the colon at the dorsal wall is the
least structurally and molecularly understood procedure pertaining
to the embryology of the colon. Fixation is a process which occurs
only in primates, making it difficult to perform experiments
because the available animal models usually are not primates
(142). As far as the colon is
concerned, at 10 weeks of gestation, directly following the
resolution of the umbilical hernia, the ascending and descending
colon initially adhere to the dorsal body wall and their dorsal
mesentery gradually fuses irreversibly with the parietal
peritoneum, which line the internal surface of the abdominal
cavity. In such a manner, the cranial part of the ascending colon
attaches to the ventral surface of the duodenum and the mesentery
extends downwards and leftward, while the greater omentum attaches
to the ventral side of the transverse colon and the descending
colon mesenteric fusion begins cranially and continues caudally
(115,143). The product of fusion, Toldt's
fascia or membrane mesenterii propria, contains nerves, blood and
lymphatic vessels, lymph nodes and fat tissue (144). Toldt has been the principal
investigator of mesenteric fixation and the majority of our
knowledge is derived from his observations (144).
The absence of fixation of the colon, termed
'persistent colonic mesentery' or 'mobile colon', normally exists
in 20% of infantile autopsies without intestinal malrotation, in
14% of adult autopsies, and in 10% of patient with intestinal
volvulus (145).
At 9-10 weeks of gestation, the similar fusion of
peritoneal layers seems to exist in the most caudal region of the
peritoneal cavity, the prerectal peritoneal pouch, at the
transverse level of S3 sacral vertebra. The adhesion and fusion of
the peritoneal walls lead to the creation of symphysis that
constitutes the rectoprostatic fascia (146,147).
7. Cecum and appendix embryology
The cecum and the appendix derive from the 'bud of
cecum' which forms in the midgut just next to the apex of the
umbilical herniation at week 6. This bud can be used to recognize
the transition of ileum to colon. The cecum changes positions
following the rotation of the midgut and the elongation of the
ascending colon. The increasing accumulation of meconium inside the
cecum is possibly responsible for its increased diameter (148).
The appendix becomes traceable at week 8 of
gestation, whereas it may remain thin because it cannot be filled
with content due to the existence of mucosal folds in the distal
cecum, which confine the flow towards the appendix (148). Lymphatic cells begin to colonize
the epithelium of appendix during weeks 14 and 15, positioned
directly under the epithelial compartment, which contains
relatively fewer goblet cells than the rest of colon. It is
noteworthy that the appendix does not possess any lymphatic
vessels. Postpartum, while the cecum dislocates laterally, the
appendix remains in a more medial position (148).
Cecal and appendical malformations
Possible embryological malformations, which partly
or individually implicate the cecum, include non-rotation,
malrotation, hyper-rotation, as well as subhepatic, mobile,
inverse, retroperitoneal cecum and internal hernias (148). In non-rotation, there is failure
of the last 180° rotation of the midgut, resulting in the left-side
positioning of the whole colon, including the cecum. In
malrotation, only the last 90° of the midgut rotation do not
succeed, placing the ileocecal loop below the pylorus. As an
unfortunate consequence, the cecum may become attached to the
dorsal body wall with ligaments, which may compress the cecal
opening towards the duodenum, leading to ileus (149). In hyper-rotation, the cecum is
positioned at the splenic bend of colon either directly or
indirectly, due to hyperdescent and travelling through the pelvis
and all the way cranially until the left colic flexure (150). In the subhepatic cecum, the
elongation of the ascending colon either does not occur or is
insufficient, impeding the normal descent of cecum. Therefore, the
cecum-appendix complex may be found anywhere from the subhepatic
region until the right iliac fossa. The malformation of the mobile
cecum occurs after failure of the fixation of the mesentery of
ascending colon to the dorsal parietal peritoneum. This situation
predisposes to cecal volvulus or malposition of the cecum and may
sometimes require urgent surgery for a condition that may resemble
acute appendicitis (151,152).
The inverse cecum untimely fixes in the subhepatic region and the
forthcoming elongation of the ascending colon forces it to bend
cranially. In the case of retroperitoneal cecum, a membrane is
created, Jackson's membrane, which encloses the cecum and ascending
colon and differs from peritoneal adhesions in that it contains
blood vessels. The most frequent internal hernia in the cecum,
which is the second most usual type of intestinal hernias, is
created towards the left paracecal region (148).
The most frequent embryological malformations of the
appendix are agenesis and duplication, even though they are
relatively rare (148,153).
8. Molecular control of the radial (RAD)
pattern
Early obliteration of the lumen
Patterning along the RAD axis is the last to begin
chronologically and it continues throughout the life of an organism
(154). There are significant
indications that, during week 6 of gestation, before epithelial
maturation, the GI lumen normally obliterates as a result of
epithelial hyperproliferation. In 1900, Julius Tandler observed
that, at day 42 of human embryologic development, the epithelium of
the duodenal endoderm markedly thickens to such a degree, that the
former lumen converts to a solid string without lumen. Between days
44 and 46, duodenum begins to recanalize, while numerous tiny
canals are created, which eventually merge into one common lumen
(155). However, over the years,
it has been difficult to reproduce and investigate the same
phenomenon in animal models as neither rats nor mice undergo
luminal obliteration of their gut lumen (156).
Recanalization disorders: Atresia
In atresia of the colon, which usually coexist with
atresia of other GI compartments, most frequently the duodenum, the
mesodermal surrounding and blood supply are absent. Colonic atresia
represents approximately 10% of total congenital intestinal atresia
cases (157). The categorization
system of Louw, is often being utilized even today, even for
different sections of intestine rather than duodenum (158). In some theoretical basis, a
number of factors have been implicated for this malformation, such
as maternal psychiatric conditions, bile secretion problems,
intestinal malrotation, mechanical compression and obliterative
embryonic conditions.
As far as mechanical compression is concerned, two
pathologic entities have been implicated in the development of
intestinal atresia, gastroschisis and volvulus. The main hypothesis
in both of these conditions is that the limitation of blood flow
leads to atresia. Gastroschisis leads to the consequent herniation
of intestinal loops and strangulation of their vasculature
(159,160). On the other side, intestinal
volvulus, either if it is a result of impaired rotation and
fixation or after twisting of the intestine around some adhesive
band, impedes intestinal blood flow (161-163). Cystic fibrosis has also been
epidemiologically related with intestinal atresia without any
recognized pathophysiological mechanism (164). However, the vast majority of
these atresia cases does not pertain to the colon (161).
Based on the initial hypothesis that the
obstruction of blood flow can elicit intestinal atresia,
thromboembolic events have also been investigated as possible
causes (165). A study
established statistical correlation between the presence of factor
V Leiden or R353R mutation of factor VII and the development of
intestinal atresia (166).
Nevertheless, this scenario is not very convincing, as factor V
Leiden is not usually associated with arterial thrombosis (167) and the levels of factor VII are
low during gestation due to the insufficiency of vitamin K
(168).
Atresias are usually accompanied by developmental
disturbances of other organs, such as esophagus, pancreatic duct,
bile duct, heart and rectum. All of these structures emerge in the
midline and either develop from the endoderm or, in the example of
the heart, are significantly affected by the neighboring endoderm.
Omphalocele has also been reported as a pathological result of
intestinal atresia in association with abdominal wall defects;
however, the exact common defective mechanisms remain unknown
(169). Nevertheless, some
defect early during endodermal development can justify intestinal
atresia and the effect on the other organs (170). Disorders of three different
molecular complexes have been proven to be able to provoke
intestinal obliteration. Firstly, mutations of either FGFR2IIIb or
its ligand, FGF-10, which function only in the endoderm, can lead
to colonic and duodenal atresia by altering the equilibrium between
epithelial apoptosis and proliferation even before any vascular
alterations (171,172). In FGFR2IIIb−/− mice,
atresia does not occur owing to some epithelial plug inside the
lumen. Moreover, in FGFR2IIIb−/− mice with atresia, in
which Fox-F1 expression is disrupted, early epithelial apoptosis
and total epithelial loss seem to be the core causes of the
pathophysiologic event (173).
The established Fox-F1 disruption is not the most crucial problem
as, even though the presence of exogenous Shh restores Fox-F1
expression that was previously disrupted, it does not alter the
phenotype (174,175). Furthermore, in
FGFR2IIIb−/− mice, the downregulation of the expression
of retinaldehyde dehydrogenase (Raldh)-2 is observed, indicating
some alteration of RA signaling (176). However, haploin sufficiency of
Raldh-2 in FGFR2IIIb−/− mice diminishes the risk for
intestinal atresia (177).
Secondly, Hedgehog signaling and its disruption through Shh
mutations can also evoke phenotypes of the spectrum of intestinal
atresia (178). The role of
other molecules which are implicated in Hedgehog signaling, such as
glioma-associated oncogene homolog (Gli)-1, Gli-2, Gli-3, Indian
hedgehog homolog (Ihh) and Fox-F1 in the development of intestinal
atresias has been excluded by recent studies (178,179). Last but not least, mutations of
Cdx-2, which is known to be expressed only in the hindgut, elicit
colonic atresia as well (70). It
is a fact that atresias begin to emerge during the stage of rapid
intestinal elongation when the developmental rate of intestine
greatly outpaces the developmental rate of the whole embryo.
Recanalization disorders:
Duplication
Another rare, yet possible malformation during
colonic embryonal development is colonic duplication. The extra
lumen may be spherical or tubular and it may or may not communicate
with the major lumen (180).
However, to date, no clear etiology for this condition has been
recognized. Among various hypotheses, one refers to persistent
intestinal outpouchings after their creation during week 6 to 8 of
gestation. Moreover, another possible cause is attachment between
gut endoderm and neural tube ectoderm, which pulls gut towards the
vertebra and does not allow it to separate from the ectoderm.
Developmental traction later leads to the creation of tubular
outpouchings, which produce the duplication. Another theory
suggests that insufficient recanalization following the
obliteration of the GI tract at week 6 of gestation eventually
creates two lumens instead of one (181). Finally, some other theory
involves vascular events in the pathogenesis of duplication
(182).
Morphogenesis of the epithelium and
adjacent mesenchyme
Following the recanalization of the GI lumen, the
endoderm begins to mature in a site-specific manner. The 9th week
of gestation is the time when the initial cuboidal stratified
epithelium of midgut and hindgut begins to alter into the future
simple columnar epithelium. In the meantime, the mesenchyme and
outer mesothelium surrounds the epithelium (183). During week 10, villus-like
structures emerge and longitudinal shafts form secondary lumens
(184,185). Simultaneously, mesenchyme
invaginates into the epithelial shafts and forms longitudinal
folds, which convert to villi with stratified epithelium. The
epithelial cells differentiate and goblet cells are visible for the
first time during 11-12th week of gestation (186). Subsequently, epithelial and
mesenchymal rearrangements lead to dissolution of the primary
villi. Crypts emerge as crypt-shaped formations of the secondary
lumens in the basal layers of the epithelium. In the meantime,
mesenchymal cells invade between adjacent epithelial layers and
divide primary villi into smaller villi, consisting of mesenchymal
lamina propria and an overlying simple columnar epithelium
(185). Notably, the initial
embryonal structure of large intestine not only includes crypts,
but also villi (87,184).
The presence of villi polarizes the epithelium and
the underlying mesoderm creating a basal side, with crypts inside
the submucosa, and a luminal villus, which contains epithelium that
changes pertaining to the differentiation stage and the
proliferation rate along the crypt-villus axis. Generally, crypts
contain less well-differentiated cells with higher proliferation
rate, whereas in the villus tip cells are terminally
differentiated, specialized, with minimum proliferation rate.
Epithelial reorganization, which constantly occurs during
development, requires this apicobasal polarity, which is controlled
by various signaling pathways (108). There is sufficient evidence to
indicate that Wnt5a is one of the regulators of the intestinal
epithelial architecture, as it mediates part of non-canonical Wnt
signaling. Wnt5a null mice exhibit the deregulation of the
apical-basal polarity of the intestinal epithelium, as post-mitotic
cells remain in apical layers and do not attach to the basal
membrane, resulting in the hindering of gut elongation (107). Concurrently, the insufficient
functioning of the sFRP inhibition over Wnt5a also lead to
deregulation of the intestinal epithelial apical-basal polarity. To
elaborate, the inactivation of sFRP provokes the formation of
epithelial clumps due to defective intracellular organization of
the epithelial cells rather than epithelial overproliferation
(108). Therefore, Wnt5a
balanced activity is important for the proper development over both
the AP and RAD axis. Ezrin also seems to be a key factor in Wnt/PCP
signaling and its absence leads to improper polarization with
villus fusion and mucosal disorganization, even though no
disturbance is apparent early during gut development (187).
Epithelial-mesenchymal interaction is of major
significance during the patterning of the epithelium along the RAD
axis. Various experiments have been conducted to evaluate the
effects of the endoderm over the mesoderm and vice versa when they
originate from different tissues. When the proximal jejunum
endoderm is co-cultured with proximal colon mesenchyme, it forms
villi, which contain jejunum specific sucrase-isomaltase expressing
enterocytes and endocrine cells that express the jejunum specific
cholecystokinin (CCK) hormone. However, small intestine-specific
Paneth cells are not apparent in this epithelium. If proximal colon
endoderm is mixed with proximal jejunum mesenchyme, villi form
again and they contain sucrase-isomaltase enterocytes as well.
However, Paneth cells are apparent, whereas the endocrine cells
express distal type hormones peptide-YY (PYY) and glucagon-like
peptide (GLP)-1 instead of CCK (188).
The mesenchyme expresses and secretes various
signaling molecules which significantly affect endodermal
differentiation. If the secretion of vesicles through the the
plasma membrane of mesenchymal cells is deregulated, for example by
a mutation in epimorphin, epithelial morphogenesis becomes is and
epithelial proliferation is accelerated (189,190). Epimorphin is a protein which is
implicated in the vesicle secretory system of mesenchymal cells
(191) and its inhibition
probably disrupts endodermal-mesenchymal intercellular signaling
pathways, such as BMP and Hedgehog. Plenty of other signaling
molecules are implicated in the intercellular coordination, such as
Wnt, FGF, epidermal growth factor (EGF), platelet-derived growth
factor (PDGF) and TGF-β.
The maturation of the mesenchyme is largely
regulated by signals of the overlying endoderm. Hedgehog molecules
are among the most important mediators of these signals. During the
early development of the pseudostratified epithelium, Shh and Ihh
are expressed by the intestinal endoderm. As long as development
proceeds, Ihh continues to be traced throughout the whole endoderm,
whereas Shh expression gradually becomes limited to the villus
base, being restricted to the less differentiated progenitor cells
and eventually stops (192). In
Shh mutations, villi overgrow, whereas in Ihh mutations, epithelial
proliferation decreases and the villi become fewer and smaller.
Therefore, Shh and Ihh may provoke opposite results (178). Total Hedgehog inhibition via
various mechanisms leads to the defective development of the
intestinal mesenchyme with immature epithelium and the disruption
of the organization of villi. Moderate inhibition of Hedgehog
disorganizes (193,194) the differentiation of the
epithelium along the RAD axis, as crypts ectopically form and
branch inside villi (193,195).
The transcription factors, Gli-2 and Gli-3, have
been found to act as subepithelial mesenchymal mediators of
Hedgehog signaling by activating transcription factors of the
Forkhead box winged-helix superfamily (196). Mutations to Fox-L1 or forkhead
homologue (Fkh)-6, which are genetic targets of Gli, lead to the
developmental reorganization of the epithelium and villi, with
concurrent hyperproliferation and deregulation of crypts and their
branching (197). Fox-F1 and
Fox-F2 are also targets of Hedgehog signaling and participate in
mesenchymal differentiation, elongation and maintenance. The
silencing of these factors provokes the disintegration of the
mesenchyme shortly after the formation of villi (198). Gli and Fox molecules cooperate
with other signaling pathways, such as Wnt and BMP, which
hormonally adapt endodermal development in coordination with
mesenchyme.
BMPs, particularly BMP-2, 4 and 7 are expressed in
the subepithelial mesenchyme, mostly under nascent villi and have
been reported as downstream targets of Hedgehog (49). In the chick hindgut, the
derangement of BMP signaling results in impaired development and
the differentiation of all three layers of gastrulation (199). Mutation of the BMP receptor
(BMPR)1 facilitates proliferation, crypt development in villi and
polyp formation, leading to juvenile polyposis (200,201). If BMPR1 is inhibited only in the
epithelium, proliferation still increases, although no polyps form
(202). The epithelial
inhibition of BMP by the antagonist, Noggin, leads to the less
compact development of subepithelial mesenchyme with an unaffected
epithelium. As a consequence, larger, but fewer villi form,
whereas, in later developmental stages, crypts ectopically
proliferate in the villi and polyps form in the intestines.
Probably, Wnt, PDGF, Hedgehog and other signaling pathways
contribute to this pathology as well (193,203). It is evident that BMP is crucial
for the configuration of the RAD intestinal axis.
PDGF signals from the epithelium towards the
mesenchyme, such as Hedgehog. PDGF-A is expressed in the endoderm
even before the formation of villi and gradually becomes restricted
in the lower parts of the villi and the crypts. Its receptor,
PDGFR-α, is expressed in the mesenchyme simultaneously with PDGF-A,
while its expression is enhanced beneath nascent villi, at the
growing spot of the elongating villi. The genetic inhibition of
PDGF signaling disrupts the mucosal architecture in colon, possibly
due to the early differentiation of mesenchymal smooth muscle, in
spite the fact that proliferation continues to occur in crypts and
in the intervillus epithelium (194).
EGF and its receptor, EGFR, are also implicated in
intestinal development. The deletion of EGFR delays villus
emergence, with consequent diminished proliferation, villus
blunting and disintegration of tissue. However, these observations
vary among different species and different developmental stages of
intervention.
E74-like factor (Elf)-3 cooperates with
CR6-interacting factor (Crif)-1, which is a transcriptional
co-activator, to facilitate the emergence of villi. Mice with
either Elf-3 or Crif-1 deficiency exhibit a diminished expression
of TGF-β receptor (TGF-βR)II, with concurrent fewer, malformed
villi, disorganized lamina propria and malfunctioning epithelial
cells. The re-expression of TGF-βRII has been shown to rescue the
normal phenotype with proper epithelial differentiation, and it has
been suggested that Elf-3/Crif-1 co-mediate villus emergence via a
procedure that is mediated by TGF-β signaling (204-206).
Mutations of the mesenchymal factor, NK2 homeobox 3
(Nkx2.3), lead to mesenchymal cell reduction, lower epithelial
proliferation, the delayed emergence of villi and a high risk of
intrauterine death. If mice manage to survive, they exhibit a
rebound epithelial hyperproliferation and mucosal thickening with
branched villi and abnormal architecture (207).
Intestinal development also requires chromatin
remodeling. Mutation of the p300 histone acetyl-transferase (HAT)
delays villus emergence, with failure of subepithelial mesenchymal
condensation and decreased BMP-4 expression at the points of
perspective villi. However, a similar mutation of the HAT protein
CREB-binding protein (CBP) does not alter intestinal embryological
development (208).
The opposite procedure of HATs is conducted by
histone deacetylases (HDACs). HDAC-1 and 2 are highly expressed in
the early intestinal endoderm, whereas they become confined to the
villi following the emergence of the villus. The overexpression of
HDACs has been found to block epithelial differentiation, while
HDAC inhibition leads to the increased histone acetylation of
epithelial cells and the subsequent immaturity of villus
development and epithelial differentiation (209).
Epithelial cytodifferentiation
Cytodifferentiation along the RAD axis is based on
interaction with the underlying mesoderm, interaction with basal
membrane proteins and contact with luminal nutrients in some
species. There is no evidence that functional cytodifferentiation
begins before villus emergence, when proliferation is very rapid,
even though at that time there is some functional barrier to the
passive diffusion of macromolecules (210). During the initial stages of
endodermal differentiation in the colon, there is a simultaneous
conversion of the pseudostratified epithelium towards a simple
columnar epithelium, the emergence of villi and the augmentation of
the epithelial proliferation rate at the villus bases. The centers
of proliferation are later altered, traced initially in the
intervillus epithelium and finally inside the crypts of
Lieberkühn.
Four main epithelial cell types are distinct during
villus emergence in the large intestine using molecular and
functional markers: i) The main absorptive cells are columnar
enterocytes, while the class of secretory cells includes; ii)
mucous-producing goblet cells; iii) caveolated or tuft cells; and
iv) various hormone-producing enteroendocrine cells. It is not
clear whether there are specific stem cells which differentiate to
each one of these cellular categories. Two of the major molecular
pathways which are implicated in the modulation of proliferation
and cytodifferentiation are Wnt/β-catenin and Notch.
As it has already been stated, Wnt/β-catenin
mediates part of the stem cell maintenance, proliferation and
cytodifferentiation. Several factors of the Wnt/β-catenin pathway
are traceable in the developing endoderm and mesoderm. It seems
noteworthy that β-catenin is transcriptionally active after villus
emergence, being detectable restrictedly in the post-mitotic cells
before birth (90,211). After birth, β-catenin
redistributes and is expressed in the intervillus epithelium.
However, there is much controversy about this evidence.
Wnt/β-catenin signaling and its final purpose can
be disrupted via various mechanisms. In the adult intestine, if
Wnt/β-catenin is directly inhibited, the acute loss of
proliferation occurs, with the depletion of progenitor cells and
the concurrent arrest of the cytodifferentiation of secretory cells
(212,213). Therefore, the canonical Wnt
pathway is necessary at least for the maintenance of the
proliferative potential of the intestinal epithelium. In the
embryonic intestine, Tcf-4 is expressed in the intervillus
epithelium, whereas Tcf-3 is expressed in the villus epithelium.
Taking into consideration that β-catenin is traceable only in the
embryonic mature villus epithelium and Tcf-4 is restricted to the
intervillus epithelium, it is evident that β-catenin probably
utilizes different Tcf mediators, such as Tcf-3, or totally
different families of signaling factors, such as Sox, to achieve
its purpose (211,214).
However, if β-catenin is primarily or secondarily
prematurely activated in the intestinal endoderm, villi do not
emerge normally and cytodifferentiation is disrupted, even though
this may be a consequence of the fact that the endodermal
differentiation radically alters towards non-intestinal tissues as
endoderm no longer expresses Cdx-2 (90,215).
The Notch signaling pathway consists of
transmembrane receptors, which bind several adjacently secreted
ligands and respond by releasing a cytoplasmic transcriptional
activation domain, the Notch intracellular domain (NICD). This
domain travels intracellularly, enters the nucleus and binds to
CBF-1, suppressor of hairless, Lag-1 (CSL)/recombination
signal-binding protein-J (RBP-J) proteins facilitating the
transcription of target genes. Hairy and enhancer of split (Hes)-1
is one of these genes and its activation alters the equilibrium of
the cytodifferentiation, favoring the formation of enterocytes
against secretory cells (216).
The activity of Hes1 represses atonal homolog (Atoh)-1/Math-1,
which are significant for the manifestation of the secretory
phenotype (217-219). Thus, Notch signaling, orients
epithelial cytodifferentiation towards absorptive or secretory
cells just by adjusting the balance between Hes-1 and Atoh-1
expression.
Notch signaling does not act independently, but it
cross-talks with Wnt/β-catenin signaling (212). Usually, it seems that Notch
antagonize Wnt signaling trying to facilitate absorptive
cytodifferentiation. Notch signaling enhances the GSK-3β-mediated
degradation of β-catenin (220).
Concurrently, Notch blockade augments Wnt signaling with subsequent
differentiation towards the secretory phenotype. When Wnt
inhibition and Notch inhibition coexist, secretory
cytodifferentiation normalizes (221). Of note, the intestinal
inhibition of Wnt suppresses epithelial proliferation and its
manifestations resemble Atoh-1 mutation as secretory
cyto-differentiation is repressed (217). When the Wnt/β-catenin pathway
functions properly, it enhances the effects of Notch signaling by
upregulating its receptors and the expression of its downstream
ligands, and by activating the targets of Notch signaling by
itself. One of these targets is Hes-1 (222). Simultaneously, in the presence
of usual Wnt activity, the APC/axin complex degrades Atoh-1
altering the equilibrium towards an absorptive phenotype, whereas
β-catenin has been shown to have Atoh-1 as a transcriptional
target, reinforcing its activity (223). Various factors can affect both
Notch and Wnt signaling, such as the reactive oxygen species
(ROS)-producing NADPH oxidase (Nox)-1 (224).
Crypt development
Crypt formation begins with the transposition of
stem cells in the intervillus epithelium and synchronized tissue
remodeling, resulting in the final form of the crypts. Crypts
extend by the translocation of the crypt-villus junction upwards
(225). In utero, stem
cells in crypts are polyclonal, whereas the postnatal stem cell
population becomes monoclonal (226). Polyclonality possibly derives
from two major facts, the rapid epithelial proliferation of the
intestine and the fission-way of crypt expansion. Rapid
proliferation induces each stem cell to create its own perimeter of
daughter cells, resulting in patches with monoclonality in the
center and polyclonality in the periphery of each patch. In the
meantime, the fission of existing crypts forms new crypts
contributing to the increase in polyclonality. After birth, the
rapid proliferation of resident stem cells gradually restores the
initial monoclonality.
The main molecular pathways which are implicated in
the development of crypts are BMP and Hedgehog. BMP signals
restrict stem cells in the emerging crypts, partly via the
inhibition of the Wnt/β-catenin pathway through the phosphatase and
tensin homolog (PTEN)/phosphoinositide 3-kinase (PI3K)/AKT pathway
(227). Wnt/β-catenin mediates
the expression of ephrin type-B (EphB)2 and EphB3 in the
intervillus epithelium and the restriction of the EphrinB1 in the
villus epithelial cells. Eph/EphB function modulates the formation
of crypts by allowing the maintenance of proliferation initially in
the intervillus epithelium and later inside the crypts (228). Moreover, β-catenin-targeted
genes can be used as markers of the crypt-base stem cells. For
instance, the expression of achaetescute complex homolog (Ascl)2,
which is a β-catenin target, augments proliferation and boosts
crypt formation, whereas its deletion leads to the inhibition of
stem cell replication and subsequent vanish of crypts (229). Nevertheless, further research is
required in order to elucidate the mechanisms of the morphogenesis
of the colonic crypts. The developmental stages of the intestinal
epithelium are depicted in Fig.
1.
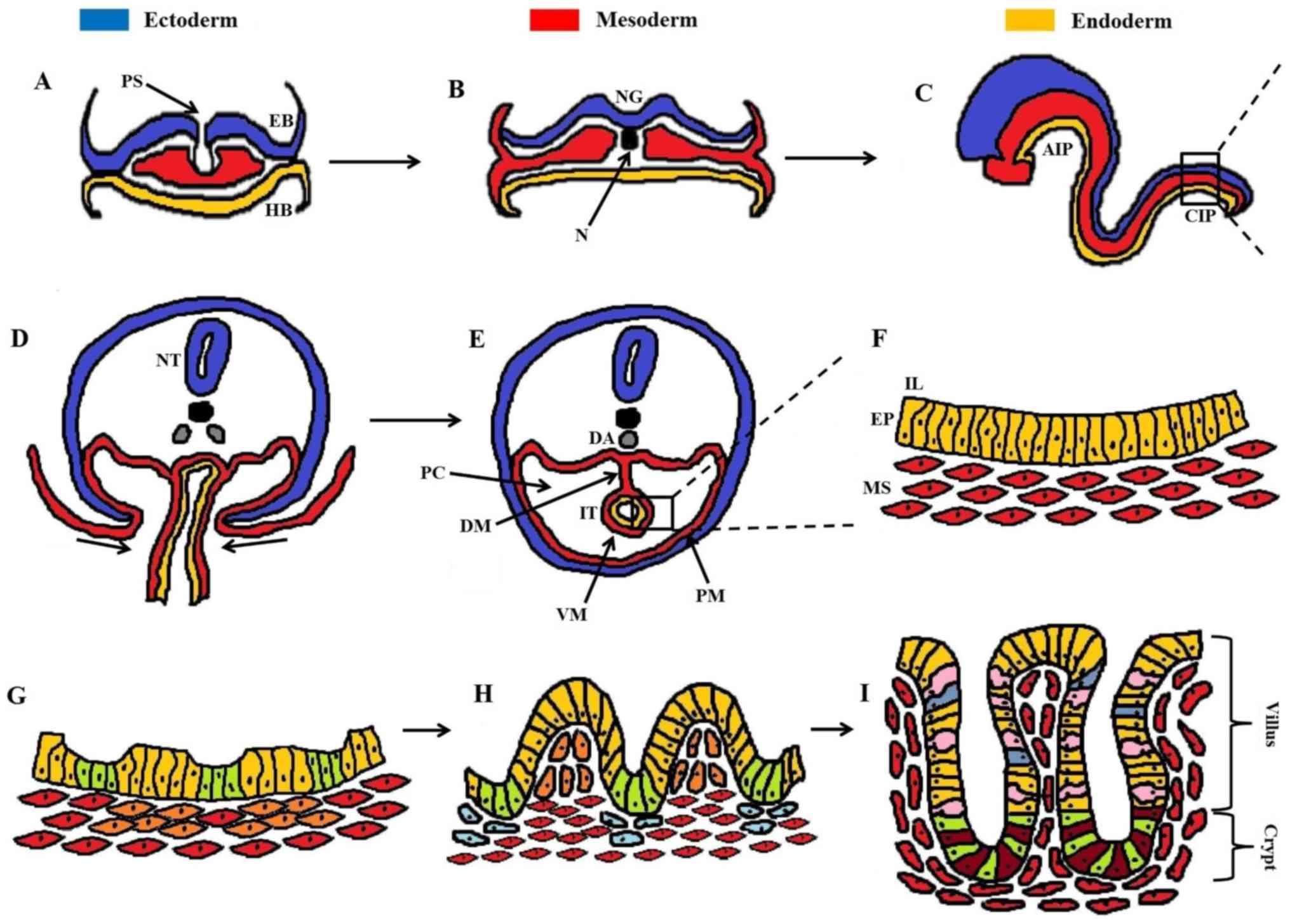 | Figure 1Developmental stages of the
intestinal epithelium. (A) Gastrulation with migration of cells
from the primitive streak, performing either EMT and/or MET. (B)
Formation of the three germinal layers (ectoderm, mesoderm,
endoderm). (C) The primitive gut tube is formed through
invaginations in the AIP and CIP, consisting of the endoderm
(internally) underlying the mesoderm (externally). (D)
Cross-section of the rectangular area in (C), showing the ventral
invagi-nations of mesoderm. (E) Formation of the peritoneal cavity
around the closed IT, with PM and VM. (F) Magnification of the
rectangular area in (E), showing the pseudostratified structure of
the intestinal epithelium (EP, yellow cells) and underlying
mesenchyme (MS, red cells). (G) Formation of mesenchymal clusters
(orange cells) as a response to epithelial signaling, marking the
onset of villus morphogenesis. (H) Progressive epithelial
remodeling via mesenchymal signaling polarizes the columnar
epithelial cells into shaping stereotypical villi (yellow cells)
and proliferative intervilli (green cells). Mesenchymal clusters at
the top of villi prevent further epithelial proliferation through
post-mitotic signaling, whereas clusters below the intervilli (blue
cells) regulate the division of intestinal stem cells. (I)
Maturation of intestinal epithelium with definite formation of
villi and crypts housing mostly enterocytes (yellow cells), goblet
cells (pink cells), and enteroendocrine cells (blue cells). At the
base of the crypt the cellular populations are mainly dominated by
secretory Paneth cells (dark red cells) and proliferative
intestinal stem cells (green cells). A dense network of
myofibroblasts (red cells) underlies the intestinal epithelium.
AIP, anterior intestinal portal; CIP, caudal intestinal portal; DA,
dorsal aorta; DM, dorsal mesentery; EB, epiblast; EMT,
epithelial-mesenchymal transition; EP, epithelium; HB, hypoblast;
IL, intestinal lumen; IT, intestinal tube; MET,
mesenchymal-epithelial transition; MS, mesenchyme; N, notochord;
NG, neural groove; NT, neural tube; PC, peritoneal cavity; PM,
parietal mesoderm; PS, primitive streak; VM, visceral mesoderm. |
9. Enteric nervous system (ENS)
The development of the ENS is a necessary condition
for the functionality of the mature colon. In the ENS, >100
million sensory, motor neural cells and interneurons exist, being
supported by glial cells, both of which are products of the same
progenitor cells (230,231).
The impairment of ENS embryonal development can
result in possible developmental disorders, such as colonic
dysmotility (232). The most
usual congenital clinical syndrome owing to abnormalities during
the development of the ENS is Hirschsprung disease (HSCR), in which
the ENS fails to establish its position along the GI tract,
resulting in functional intestinal obstruction of various lengths.
It usually involves only the distal colon, although there are cases
of the total absence of the ENS across the entire GI tract
(233,234). In this condition, the
pathological intestinal section is contracted, empty of content and
unable to participate in a peristaltic wave.
Developmental principles of the ENS
Firstly, the compartment of the progenitor cells of
the ENS derive from neural crest and these specific cells are
called enteric neural crest cells (ENCCs). These ectodermal cells,
delaminate from the gradually closing cranial neural folds, migrate
through the mesenchyme and endoderm, colonize the GI tract and
differentiate towards the different cell types of the ENS (235,236) If the dorsal neural tube of
chicks is removed, they fail to develop an ENS (237). The major initial source of ENCCs
is the vagal neural crest, from neural tube at the levels of
somites 1-7 (236,238). The sacral neural crest also
contributes to the ENS, by providing progenitor cells for the
development of enteric neurons and glial cells caudally to the
umbilicus (239). Different
sections of the GI tube receive ENCCs form different axial levels
of the neural tube (238). The
somite levels 1-2 neural crest contributes only in esophagus, ENCCs
from somite levels 3-5 colonize regions from stomach to hindgut and
somite levels 6-7 provide progenitor cells only to the hindgut
(238,240). Therefore, it is evident that the
ENS of the colon is created by cells from neural crest cells (NCCs)
below somite level 3.
As far as the ENS is concerned, at somite stage 13,
a population of NCCs originating from somites 1-3 migrate ventrally
and gradually form the sympathetic and dorsal root ganglia, while a
proportion of them colonizes either the heart or foregut to begin
the formation of ENS (238,241). NCCs originating from the rest of
the vagal neural crest, at somites levels 4-7, migrate only
ventrally and reinforce the colonization of GI tube. In contrast to
vagal-derived NCCs (242), the
more caudal truncal NCCs are not able to enter the foregut, as they
express Robo, which interacts with Slit-2 that is expressed in the
foregut, resulting in rejection from the foregut (243). After exiting the neural crest,
vagal NCCs cross the mesoderm between the sclerotome and
dermomyotome. Before they enter the intestinal mesenchyme, they
form ganglia next to it (244).
It seems that during this passage, paraxial mesoderm produces RA,
which binds to RA receptors of NCCs, activating the expression of
receptor tyrosine kinase rearranged during transfection (RET) by
NCCS. This step probably represents the future restriction of these
NCCs as progenitors only for ENS. The deficiency of the necessary
RA signaling mediator Raldh-2 leads to ENS agenesis, linked to the
decreased expression of RET.
After the entering of vagally-derived ENCCs into
the foregut, they migrate caudally as a waveform process with a
speed of 40 µm/h (245).
Their migration occurs randomly inside the outer mesenchyme, where
smooth muscle has not yet emerged (246,247). In the midgut, while the caudal
extension of ENS continues, smooth muscle begins to form and ENCCs
become limited between the circular and the longitudinal muscle
layers, forming the myenteric plexus. A secondary migration wave
guides ENCCs from the myenteric plexus towards the submucosal
region, establishing the submucosal plexus (247). It is noteworthy that the two
plexuses develop early and simultaneously in human and avian
organisms (248). In humans, the
total AP colonization of the GI tract by ENCCs is completed by week
7 of gestation (248).
Apart from the vagal neural crest, the sacral
neural crest, which is located caudally to sacral level 28, also
contributes to ENS of the GI tract, particularly in the colon and
rectum. To elaborate, sacral NCCs also travel ventrally, migrate
through somites and mesenchyme, and reach regions laterally of the
cloaca and caudal hindgut in order to give rise to the pelvic
plexus. Following the arrival of NCCs from the vagal neural crest
in the hindgut, cells from the pelvic plexus set out to colonize
the distal hindgut and provide additional neurons and glial cells
to the ENS (240,249).
Compared to sacral NCCs, vagal NCCs are far more
invasive, partly being justified by the fact that they express
four-fold higher levels of RET (250). If RET is upregulated in sacral
NCCs, their invasiveness and potential for migration increases,
whereas if the sacral neural tube gets replaced by vagal neural
tube, these NCCs also reach distal colon much earlier (251). Alternative paths for the neural
colonization of the gut and particularly colon have been presented.
It has recently been proven that Schwann cell progenitors in
extrinsic nerve fibers also contribute 20% of the hindgut ENS
(252). Moreover, the waveform
process may not be totally continuous along the AP axis of the
intestine. Specifically, some of the caudally migrating ENCCs skip
cecal mesenchyme, cross the midgut mesentery and reenter gut more
distally. This phenomenon is so profound that >50% of the ENCCs
of distal hindgut follow this method of migration (253).
The key features of the developing ENS, which have
to be carefully regulated, are cell proliferation, cell survival,
migration and its direction, the creation of concentric plexuses,
cytodifferentiation and the formation of ganglia, axis and
synapsis.
One the most important factors during ENS formation
is the quantity of ENCCs, since the reduction of the size of vagal
neural crest leads to distal aganglionosis (254). Moreover, the extension of ENCCs
occurs in a chain pattern and is facilitated by close cellular
contact, whereas individual ENCCs migrate at a much lower speed
(255,256). Proliferation increases the
cellular density and augments contact of cells. It seems that the
cellular contact is perceived by cell to cell adhesion and requires
adhesion molecules, such as L1 cell adhesion molecule (L1CAM)
(257). It is noteworthy that,
when ENCC density is relatively low, the decreased speed of their
migration is not sufficient, as the mesenchyme matures and alters
its conditions, which are no longer favorable for the arrival of
ENCCs (258).
Another principal is that, across the AP axis of
migration, cells in the wavefront have to maintain their
proliferative potential and migrate distally, whereas cells behind
the wavefront need to slower their proliferation rate and start to
maturate. The migration progresses from regions of high cellular
density and low availability of neurotrophic factors, such as
glial-derived neurotrophic factor (GDNF) due to depletion, towards
regions of higher ENCC carrying capacity, which happen to exist
distally (251,259). Another mechanism that may
regulate this migration stream and its termination could be the
existence of some negative signal among ENCCs when there is no
neighboring tissue, without ENCCs for them to head, and they
inevitably contact each other (260).
Once the wavefront reaches each compartment of the
GI tube, including the colon, ENCCs have to stop proliferating and
begin differentiating. If proliferation occurs at a relatively slow
rate, there is no sufficient number of ENCCs to colonize the whole
gut, with the colon being the most affected part as the last
compartment which becomes colonized. The premature differentiation
of otherwise proliferative ENCCs also decreases the migratory
potential of the wavefront, leading to a variable extent of
aganglionosis. On the contrary, differentiation rates which are too
slow result in abnormal neural and glial maturation as the
mesenchyme, which maturates more rapidly, no longer facilitates
cytodifferentiation at the late time when that begins (261).
The innervation of colon has been found to be
embryo-logically configured by several concurrent and complex
mechanisms and provide a broad area of research.
Molecular signaling in enteric
innervation
RET is a tyrosine kinase transmembrane receptor,
expressed by ENCCs and mediating migration (262,263). Both Sox-10 and paired-like
homeobox (PHOX)-2B are necessary for the expression of RET, or
total aganglionosis otherwise occurs (264,265). One of the RET activators is
GDNF, which is expressed in the mesenchyme. GDNF binds to the
complex RET-GDNF family receptor (GFR)a1, provoking the RET
phosphorylation and activation of multiple intracellular pathways,
including Ras/mitogen activated protein kinase (MAPK), JNK and PI3K
(266). PI3K activation is of
upmost significance as it mediates the GDNF-guided proliferation,
migration and survival (267,268). PI3K normally mediates the
conversion of phosphatidylinositol (3-5)-trisphosphate (PIP3) to
phosphatidylinositol 4,5-bisphosphate (PIP2), whereas its
inhibition leads to distal aganglionosis due to limited migration
(269,270). PTEN antagonizes PI3K, catalyzing
the opposite reaction, the conversion of PIP2 to PIP3. As is
evident, PTEN is not normally expressed in the tip of the wavefront
as its expression would compromise the migration potential. RA
signaling concurrently induces RET expression in NCCs, patenting
them as ENCCs, and accomplishes PTEN absence, maintaining the
effectiveness of PI3K in facilitating proliferation and migration
(270,271). On the other side, PTEN is
present and is very useful in regions proximal to the wavefront,
where it is needed to terminate proliferation and migration
(270). Additional molecular
factors, which try to balance the function of RET-GDNF interaction
are Sprouty homolog (SPRY)-2 and KIF26A. Enteric nerve hyperplasia
occurs if either of their genes is silenced (272,273).
Apart from proliferation, RET also maintains the
survival of ENCCs, which is necessary for migration (274,275). The intestinal length where
aganglionosis occurs is proportional to the degree of RET
insufficiency. RET+/− mice exhibit an almost normal
intestine with proper innervation, the reduction of RET expression
to one-third of its normal value, provoking moderate colorectal
aganglionosis, whereas RET−/− mice present with total
aganglionosis (276,277). GDNF possibly emits some
chemoattractive signals, which direct migration (278), whereas the absence of GFRa1
attenutes ENCC migration (279).
It is noteworthy that the RET-GDNF interaction contributes to the
migration of ENCCs towards the submucosa layer as well (279).
There are indications that RET-GDNF somehow
regulates cytodifferentiation, even though the research results are
controversial (275,279,280).
Endothelin receptor (EDNR)B is a G-protein receptor
expressed by ENCCs, whereas its ligand, endothelin (ET)-3 is
expressed in the mesenchyme. EDNRB signaling is required in regions
caudal to the cecum (255) and
it serves to facilitate proliferation and inhibit
cytodifferentiation (274,281). The deletion of the genes of
EDNRB or ET-3 or of the endothelin converting enzyme (ECE)-1 gives
rise to colorectal aganglionosis (282). The loss of EDNRB signaling
suppresses proliferation and promotes cytodifferentiation. There is
evidence of the EDNRB interaction with RET signaling, since either
RET heterozygous mutation or EDNRB homozygous mutation do not raise
any severe abnormality, whereas their combination provokes
aganglionosis (283,284). Furthermore, ET-3 reinforces
GDNF-mediated proliferation (274), even though it antagonizes
GDNF-mediated ENCCs cytodifferentiation towards neural cells
(285).
Sox-10, an HMG box-containing transcriptional
factor, expressed by NCC and later by ENCCs, is required for the
survival of ENCCs, along with the maintenance of the proliferative
potential and the inhibition of cytodifferentiation (286). Heterozygous Sox-10 mutation
decreases the ENCC population and promotes premature
differentiation to neurons and distal aganglionosis (287), whereas homozygous Sox-10
mutation provokes total aganglionosis due to the failure of NCCs to
survive until they reach the foregut (264). Sox-10 may succeed its purpose
via the direct activation of RET and EDNRB (288,289). Following the cytodifferentiation
of ENCCs, Sox-10 expression is terminated in neurons, whereas it
continues in glial cells (261).
PHOX-2B, a transcriptional factor expressed in
ENCCs following their invasion inside the mesenchyme, promotes
proliferation and survival and mediates the formation of the
enteric ganglia (290). Its
deletion results in total intestinal aganglionosis (265). There is antagonism between
Sox-10 and PHOX-2B inside cells, as they suppress each other. ENCCs
differentiating towards neurons express PHOX-2B, in contrast with
glial cells, which express Sox-10 (291). The msutation of PHOX-2B leads to
Sox-10 dominant expression and differentiation towards glial cells,
not neurons (292).
Heart- and neural crest derivative (HAND)-2, also a
base helix-loop-helix transcription factor expressed in ENCCs
following their arrival in the foregut, mediates differentiation to
mature neural cells and the specification of neuro-transmitters
(293). When HAND-2 expression
is expressed, intestinal motility becomes impaired (294), whereas HAND-2 deletion leads to
lower numbers of enteric neurons and disrupted ENCC differentiation
into neurons (293,294).
BMP-2 and BMP-4 affect migration and regulate the
balance between neural versus the glial differentiation of ENCCs
(295-297). They also facilitate the
formation of enteric ganglia and a lower neural density. If BMP
signaling is inhibited, for instance due to Noggin, the
neural-to-glial ratio increases (297). Moreover, BMPs promote the
domination of neural cells which express tropomyosin receptor
kinase (Trk)C, which is a tyrosine kinase receptor that binds ET-3
and reinforces survival and differentiation (296). The deletion of either TrkC or
ET-3 diminishes the neural cell pool (298).
Role of extracellular matrix in ENS
development
Apart from the receptors and transcription factors,
which affect the key features of the development of the ENS,
another necessary condition for the proper distribution and
differentiation of ENCCs is a suitable microenvironment. This is
constructed by components of the ECM. When ET-3 is ablated,
laminin, perlecan and collagen type VI are traced in increased
concentrations, supporting the statement for the crucial
contribution of the ECM in the patterning of ENS (299,300). ENCCs have receptors in their
cellular surface, while ECM can alter survival, proliferation,
migration, differentiation, gangliogenesis and the formation of the
two plexuses.
Migration has been proven to be facilitated by the
presence of laminin, fibronectin, vitronectin and collagen type I,
whereas it is inhibited by collagen type VI and possibly by
chondroitin sulfate proteoglycans, such as versican (301-305). The ECM significantly contributes
to the RAD patterning of ENS, creating mesenchymal boundaries which
are not permissive for the development and extension of ENCCs. As a
consequence, ENCCs are forced to position themselves along two
permissive regions where the two enteric plexuses form (305). Moreover, it is noteworthy that
while ENS formation proceeds, ECM in the midgut and hindgut
expresses greater concentrations of collagen type I and gradually
becomes stiffer, decreasing the potential speed of ENCC migration
(306).
However, it seems that ENCCs produce their own ECM
proteins, which they secrete in their surrounding ECM in order to
ameliorate their microenvironment and augment their migratory
potential. This is particularly observed in vagal NCCs, not in
sacral NCCs, partly justifying their different migratory capability
(301). ENCCs have also been
found to secrete metalloproteinases (MMPs) in the ECM to degrade
part of it and make it less dense and stiff in order to ease their
passage, according to their direction. When MMP-2 is inhibited, the
migratory potential of ENCCs is reduced (307). Furthermore, in the case of the
upregulation of collagen-6a4 gene, the secreted collagen VI in ECM
is increased and ENCC migration is confined (304).
ENCCs interact with the ECM via their surface
adhesion molecules, which bind to ECM molecules, such as L1CAM,
N-cadherin and numerous integrins (302,303,308). L1CAM inhibition disrupts the
close contact of ENCCs in the wavefront tip, which is required for
migration to proceed (309),
while the deficiency of either L1CAM or N-cadherin attenuates ENCC
migration (257,310). As far as laminin is concerned,
even though it initially is non-specifically expressed in
mesenchyme, it later becomes restricted at the basal lamina of
epithelia, endothelial cells and smooth muscle cells. The pertinent
endothelial cells form two concentric capillary plexuses in
mesenchyme, the endothelial cells of which maintain laminin
expression. The migratory ENCC wave colonizes the mesenchyme in
close contact with these endothelial cells, suggesting that the two
concentric capillary plexuses act as definers of the future
position of the two concentric ENS plexuses, in a process which
also requires integrin-β1 (302). The disruption of the formation
of capillary plexuses provokes distal aganglionosis which
implicates the colon (302).
Epithelial signals regulate ENS
formation
The epithelium, either with direct signals or with
indirect signals via the mesenchyme, regulates ENS formation vai
signaling that implicates Netrin and hedgehog (305). Epithelial cells produce netrins,
which are extracellularly secreted and direct axon formation,
acting in a chemoattractive manner on neural cells that express
deleted in colorectal cancer (DCC). Netrins have also been proven
to guide ENCC migration across the RAD axis, from the myenteric to
the submucosal plexus. It is noteworthy that DCC mutant mice do not
develop the submucosal neural plexus, even though they exhibit a
normal Netrin expression (311).
On the other side, endodermally-derived Hedgehog
signaling, which includes Shh and Ihh, can alter mesenchymal
proliferation and differentiation, which can alter the
microenvironment where the ENS develops. Shh also contributes to
the concentric pattern of the ENS (2). Hedgehog signals reach the
mesenchyme, which responds by changing the degree of BMP-4
expression. BMP-4 inhibits mesenchymal differentiation towards
smooth muscle, potentiating ENCC migration, particularly towards
the submucosal layer (49,52,295).
Moreover, Hedgehog induces the mesenchymal expression of Fox-F1 and
Fox-F2, the inactivation of which diminishes the presence of
collagen type-I and IV in ECM. As a result, the ECM is less
conducive for the development of ENS and colorectal aganglionosis
occurs (198).
Shh signaling over the mesenchyme is partly
mediated by the mesenchymal receptor patched-1 (PTC1) and the
downstream smoothened-Gli cascade. Both Shh inhibition and
deficiency provoke the emergence of ectopic ganglia in the
submucosal layer with increased neurons and malformed neural
projections (178). On the
contrary, Shh overexpression provokes intestinal aganglionosis,
supporting some inhibitory action of Shh over the development of
the ENS (305). Endorsing this
statement, Shh overexpression has also been found to increase the
mesenchymal expression of chondroitin sulfate proteoglycans even
ectopically, resulting in the hampering of ENCC migration and
consequent intestinal aganglionosis (305). The role of Ihh in ENS
development has not been adequately investigated.
It is noteworthy that, even though the significance
of Shh in ENS development seems undoubtable, its effects on ENCCs
probably occur indirectly by other mediating tissues. To elaborate,
no receptors for Shh have been traced in ENCCs, whereas PTC1 is
expressed only by the intestinal mesenchyme (305).
HSCR
As it has already been reported, HSCR is a
congenital condition which usually involves the distal GI tract,
most frequently sigmoid and rectum, and is also referred as
congenital megacolon or intestinal aganglionosis. It has an
incidence of 1:5,000 and a male predominance of 4:1 (312). In HSCR, the ENS does not succeed
to develop in the respective intestinal segment resulting in
inability of propelling the intestinal content out of the GI tract.
The aganglionic segment is collapsed, without content, whereas the
more proximal intestinal parts distend, contributing to the
phenotype of mega-colon. It is noteworthy that some intestinal
length proximal to the totally aganglionic compartment also
exhibits some developmental hypoganglionosis (233).
HSCR is one of the few congenital conditions which
has attracted extensive research into its underlying defects. RET
mutations are the most frequent of the known mutations that have
been traced in HSCR. However, in >50% of cases, none of the
previously noted mutations are apparent. A number of different
genes and signaling pathways have been implicated in its
pathophysiology (262,263).
A particular RET mutation has been found to delay
the transmesenteric passage of ENCCs on their way to reach the
hindgut, which later makes it difficult for ENCCs to cross the
already maturated mesenchyme and increases the risk of developing
agangliogenesis of the distal colon (253). Mutations of non-coding regions
in RET have also been proven to increase the risk of HSCR, which
probably still occurs owing to the existence of other mutations
(263,313,314).
Another possible reason for HSCR is reduction of
the progenitor pool. This can occur either due to GDNF
insufficiency, which confines ENCC proliferation and migration
(315), or owing to irregular
interaction among Sox-10, ET-3 and EDNRB, which normally maintains
ENCCs in an undifferentiated, proliferative state (274,316,317). In particular, ET-3-EDNRB
signaling has been proven to be more important for the development
of ENS in distal colon, during the latest phases of intestinal
colonization (318). If
heterozygous Sox-10 mutant mice also experience some loss of EDNRB,
they exhibit a phenotype similar to that of HSCR, without any
apparent increase in ENCC apoptosis (319). Moreover, EDNRB mutations can
also delay the arrival of ENCCs in distal intestinal compartments,
which, similar to some RET mutations, restrict the ability of ENCCs
to invade and colonize distal gut compartments (258). Furthermore, it has been
established that haploin sufficiency of treacle ribosome biogenesis
factor (Tcof)-1 alone in mice reduces the population vagal NCCs and
delays their migration along the length of the gut during early
development, although it is not sufficient to provoke
aganglionosis, such us in HSCR. Heterozygosity of paired box gene
(Pax)-3 does not result in ENS defects either. However, if
Tcof-1+/− is combined with a coexisting heterozygogosity
of Pax-3, mice present colonic aganglionosis with cumulative
apoptosis of neural crest cells leading to consequent reduction of
the population of migrating ENCCs into the foregut and diminishing
of the proliferating capacity of the remaining ENCCs (320).
A variety of other factors have also been proposed
as possible contributors to the defects in HSCR, based on recent
research. The lack of zinc finger protein X-linked (ZFX) h1B
(321) or PHOX-2B (322), the inhibition of BMPs (323), the mutation of Sox-10 (324) and abnormal signaling via RA
(325) are some of these
factors.
10. The embryological hypothesis of
colorectal carcinogenesis
Colorectal cancer (CRC) is the third most
frequently diagnosed neoplasm, being the second cause of high
mortality related to cancer, with the majority of cases (95%) being
sporadic (326). The principles
of developmental biology provide novel insight into CRC
carcinogenesis and progression, creating unconventional theories
regarding its origin, since human embryonic cells present major
phenotypic similarity with cancer cells (327).
As regards the genetic origins of cancer,
particularly CRC, the prevailing theory states that somatic
mutations alter the colonic epithelium, triggering carcinogenesis
(328). However, these mutations
are not evenly distributed in the genome, forming complex
'mutational landscapes', which are mostly determined by the
epigenetics (329). Furthermore,
during cell differentiation in embryogenesis, various patterns of
gene expression are defined by cardinal epigenetic alterations
(330). Notably, different
populations of stem cells are characterized by different epigenomic
patterns (331). The
above-mentioned observations suggest that a genomic mutational
landscape related to CRC includes information regarding the
epigenomic organization and identity of its origin cell, along with
the reliable process of its embryogenesis. Nevertheless, the
current knowledge regarding the oncogenic activation of specific
genes related to CRC pathogenesis and their embryonic origin is
very limited, relying mostly on theories.
In order to link cell differentiation and
embryology, Pierce proposed that the cause of carcinogenesis could
be related to development (332). According to his hypotheses, the
phenotype of cancerous cells should be encoded in normal cell
genome, indicating that the development of normal tissue is similar
to tumorigenesis (e.g., colonic epithelium and CRC) (333). Studies have revealed that
carcinogenesis can derive from the awakening of repressed genes
that are normally active during embryogenesis, particularly in
gastrulation, suggesting that the CRC transcriptome impersonates an
embryologically active gene network as a 'developmental signature'
(334). This phenomenon has been
identified in several human tumors, including CRC (334,335). Another aspect that should be
taken into consideration is the occurrence of keystone events
during gastrulation; epithelial-mesenchymal transition (EMT) and
mesenchymal-epithelial transition (MET) (336). These two processes are able to
regulate cell proliferation, differentiation, invasion and
migration, which are also natural traits of cancer cells during
metastasis. Migratory mesenchymal cells respond differently to
microenvironments adjacent to the primitive streak, resulting in
the alteration of their epigenome (337), altering the transcriptional
control (338), permitting the
differentiation of endodermal and mesodermal cell lineages.
Additionally, endodermal cells present the capacity of remodeling
the basal membrane, suggesting a higher invasiveness of tumors of
endodermal origin (339), and
therefore a poorer prognosis compared to cancers of mesodermal or
ectodermal origin. Thus, the future cancerogenic potential of
tumors, such as CRC, will be proportionate to the capacity of basal
membrane remodeling in response to various embryogenic signals
directed by epigenomic signatures produced during gastrulation.
Since cancer may be an impaired demonstration of
the physiological tissue renewal process, the role of cancer stem
cells (CSCs) in carcinogenesis and development should also be
considered. The current hypothesis regarding colonic CSCs states
that an initial somatic mutation occurs in a normal colonic stem
cell at the bottom of the colonic crypt. Following this mutational
transformation, these stem cells divide in a symmetrical or
asymmetrical manner producing other progenitors and CSCs,
colonizing the entire niche. Furthermore, the accumulation of
various alterations eventually concludes in CRC tumorigenesis and
progression (340). To further
investigate the morphological features of colonic stem cells,
Gostjeva et al, through colonic tissue sampling that
preserves cell architecture (341), demonstrated unique bell-shaped
nuclei, which were extremely uncommon in adult normal colon,
although they were abundant in fetal colon, colonic adenomas and
adenocarcinomas (342). In CRC,
such nuclei were mainly detected in the center of the tumoric mass
and at the bottom of cancerous crypts. These results support the
hypothesis of embryonic basis of CRC carcinogenesis, which is
depended on colonic stem cells via transformation to a more
juvenile proliferation state, turning them into CSCs and finally
leading to CRC. A very recent theoretical model tries to unify the
CSC and embryonic hypothesis in the context of carcinogenesis based
on the principles of gastrulation (343). According to this proposal, a
normal adult differentiated cell can transform into a CSC, after
undergoing deprogramming (dedifferentitation) and reprogramming in
the lack of genomic stability. This CSC can be considered as a
para-embryonic stem cell, which actually constitutes the initial
CSC (CSC0) in the process of tumorigenesis. The CSC0 can generate a
primary tumor, which resembles a pre-implantation blastocyst. In a
suitable niche, regarding signaling factors, this primary tumor
would be implanted, resembling a post-implantation blastocyst. From
these cells, pluripotent, slow self-renewing primary CSCs (CSC1s)
would be produced. CSC1s resemble the epiblast cells, and being
epigenetically blocked in this primed state would act as
tumor-initiating cells. CSC1s would then produce secondary CSCs
(CSC2s) resembling hypoblast cells, which would act as tumor-growth
cells. CSC1s or CSC2s would produce tertiary CSCs (CSC3s),
possessing a mesenchymal phenotype, which would act as
tumor-migrating cells, mimicking the mesodermal precursors at the
primitive streak. If the microenvironment of the primary tumor
creates preferable conditions such as normoxia, the CSC3s will then
proceed into asymmetrical division creating cancer progenitor cells
(CPCs), which then would differentiate into cancer differentiated
cells (CDCs), thus completing a defined cell hierarchy resembling
somito-histo-organogenesis. However, unfavorable conditions, such
as hypoxia, would trigger the migration/delamination of CSC3s,
behaving as migrating micrometastases, corresponding to the
mesenchymal cells during gastrulation. In distant metastatic
niches, CSC3s would be localized in a dormant state as an effect of
hypoxia and EMT signaling. Contrarily, when normoxia or MET
signaling prevail in the metastatic niche, a CSC3/CSC1 reversion
would then be induced, and the newly formed CSC1s, as
tumor-initiating cells would reproduce the same cell hierarchy of
the primary tumor as macro-metastases (343).
Various embryonic molecular signaling pathways are
emerging as major determinants of CRC carcinogenesis and
progression, being common between embryogenesis and cancer
(Fig. 2). The canonical
Wnt/β-catenin signaling pathway is one of the most commonly
involved in cancer. In the vast majority of sporadic CRC cases
(90%) a mutation in the tumor suppressive APC gene as the initial
molecular change, leading to the loss of expression of the
corresponding protein and to the hyperstimulation of the
Wnt/β-catenin pathway (344).
Although canonical Wnt components are of major importance for the
regulation of the integrity of normal stem cells and CSCs, the
deregulation of β-catenin levels is not solely sufficient for CRC
carcinogenesis. Specifically, APC mutation creates a basal
impairment of β-catenin activity and then further genetic
alterations induce the hyper-stimulation of Wnt/β-catenin signaling
pathway, leading to the production of colonic cells with CSC
characteristics and enhanced metastatic capacity (345). Moreover, the canonical Wnt
signaling pathway through positive or negative crosstalk with other
developmental pathways, including PI3K, Notch, BMP and Hedgehog can
regulate the CSC population (346). Notably, components of the
non-canonical Wnt signaling pathways seem to be involved in the
association between colon embryogenesis and CRC carcinogenesis. The
detection of methylated DNA by Kaiso, via methyl-CpG binding
location, is crucial for the epigenomic silencing of
tumor-suppressive genes, an essential role that has already been
described in CRC cancerogenesis (347). The nuclear translocation of
Kaiso/p120ctn stabilizes cells that have already undergone MET,
reduces MMP-7 gene expression along with cell migration, and
triggers E-cadherin expression, ultimately inducing the endodermal
formation including the primitive colon (348), thus functioning as an epigenetic
control system connecting CRC and embryogenesis. The TGF-β/BMP
signaling pathway is another essential pathway in both development
and cancer, promoting cell migration and invasion (349). Despite their compartmental-ized
activity in the normal colonic tissue, the TGF-β/BMP and
Wnt/β-catenin signaling pathways are altered in CRC, crosstalking
in inducing CRC carcinogenesis (350). BMP-2 and BMP-4 in particular
participate in the regulation of colonic CSCs by furthering CSC
differentiation and antagonizing the canonical Wnt signaling
(351). Finally, concerning the
molecular basis of EMT, during embryo-genesis, both Wnt and TGF-β
signaling pathways regulate the formation of the primitive streak
through induction of EMT by upregulating SNAIL and TWIST
transcription factors (352).
The same EMT regulators also promote EMT and consequent migration
and metastasis in CRC (353,354). Notch signaling pathway is
another link between embryo-genesis and CRC carcinogenesis, acting
as a controller of colonic CSCs, since it is crucial for the
homeostasis of the normal colon stem cell (355). Evidence demonstrates the control
of stem cell expansion and self-renewal by a signaling consortium
of SNAIL, miR-146a, and Numb, a Notch inhibitor, leading to
fine-tuning of Wnt signaling (356).
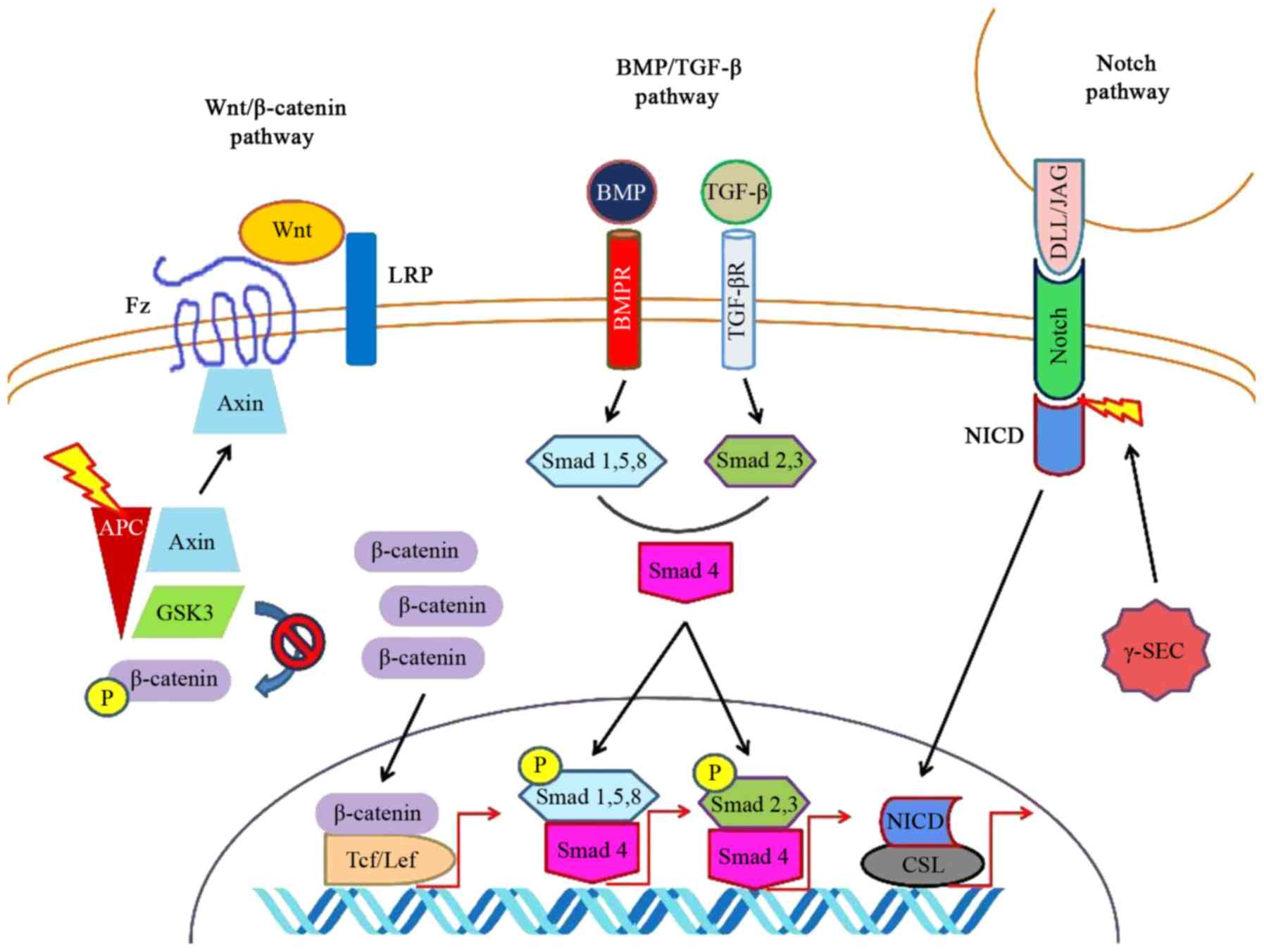 | Figure 2Common developmental pathways in gut
embryogenesis and colorectal cancer. Mutations in APC result in
instability of the APC-axin-GSK3 destruction complex, which
insufficiently targets β-catenin for degradation after
phosphorylation. Thus, increased levels of β-catenin accumulate in
the nucleus, forming complexes with Tcf/Lef proteins. BMP or TGF-β
bind to their receptors (BMPR and TGF-βR) and activate Smad 1,5,8
and Smad 2,3, respectively. Combined with Smad 4, they form
transcriptional complexes which translocate to the nucleus. Ligands
of Notch (DLL/JAG) enhance proteolysis through γ-SEC, releasing the
activated NCID which translocates into the nucleus and subsequently
binds with CSL. All the above signaling pathways upregulate the
expression of target genes, resulting in the dysregulation of
colonic stem cells and increasing their carcinogenic potential.
APC, adenomatous polyposis coli; BMP, bone morphogenetic protein;
BMPR, bone morphogenic protein receptor; CSL, CBF-1, Suppressor of
Hairless, Lag-1; DLL, delta-like; Fz, frizzled; GSK, glycogen
synthase kinase; JAG, jagged; Lef, lymphoid enhancer-binding
factor; LRP, low-density lipoprotein receptor-related protein;
NICD, notch intracellular domain; Tcf, T-cell factor; TGF,
transforming growth factor; TGF-βR, transforming growth factor-beta
receptor; Wnt, wingless-related integration site; γ-SEC,
γ-secretase. |
11. Conclusions
Large intestine embryogenesis constitutes an
extremely complex procedure, involving a vast plethora of cellular
populations cross-talking through various ways. These interactions
are mediated and controlled via numerous molecular pathways
(Table II). However,
deregulation may occur, resulting in developmental disorders which
could be fatal in embryonic life, or cause long-term issues in
adult life (Table III).
Moreover, novel insight into these pathways reveals a possible
connection between embryology and carcinogenesis. Future studies
are required to focus on further investigating and clarifying the
detailed role of the molecular components in the development of
large intestine and related pathological conditions.
| Table IISummary of the expression and
function of molecular factors that regulate embryogenesis in the
large intestine. |
Table II
Summary of the expression and
function of molecular factors that regulate embryogenesis in the
large intestine.
| Molecular
factors | Site of
expression | Functions |
|---|
| Activin | Former
mesendoderm | - Endoderm
formation |
| Splanchnic
mesoderm | - LR pattern
control |
| BMP | Mesenchymal
mesoderm | - Smooth muscle
formation (BMP-4) |
| - Maturation of
enteric neurons (BMP-2) |
| Subepithelial
mesenchyme | - RAD pattern
control |
| - Crypt
formation |
| - Epithelial and
mesenchymal development and differentiation (BMP-2,4,7) |
| Mesenchyme | - ENCC migration,
survival, differentiation |
| - Enteric ganglia
formation (BMP-2,4) |
| Cdx | Posterior
endoderm | - Hindgut formation
and differentiation (Cdx-2) |
| Chato | Primitive
endoderm | - Endoderm
elongation |
| Dact | Posterior
endoderm | - Hindgut and CIP
formation (Dact-1) |
| Elf | Epithelium | - Villi formation
(Elf-3/Crif-1 through TGF-β) |
| Epimorphin | Mesenchyme | - Secretion of
molecules for endodermal differentiation |
| ET | Mesenchyme | - ENCCs
proliferation, inhibition of cytodifferentiation (ET-3 through
EDNR-B) |
| FGF | Endoderm | - AP pattern of
hESCs (FGF-2) |
| Posterior
endoderm | - Hindgut formation
(FGF-4) |
| Epithelium | - Proliferation and
elongation of mesenchyme (FGF-9) |
| Mesenchyme | - Formation of
cecal budding (FGF-10) |
| Splanchnic
mesoderm | - LR pattern
control (FGF-8) |
| Fox | Former
mesendoderm | - Endoderm
formation of foregut and midgut (Fox-A) |
| Anterior
endoderm | - AP pattern
control (Fox-A2) |
| Subepithelial
mesenchyme | - Epithelial
organization and maturation, RAD pattern control |
| - Mesenchymal
maturation |
| - Villi
formation |
| - Crypt formation
(Fox-l1, Fox-F1, Fox-F2) |
| Mesenchyme | - ENCCs migration
through modulation of ECM (Fox-F1, Fox-F2) |
| GATA | Former
mesendoderm | - Endoderm
formation |
| Early definitive
endoderm of AIP | - Endoderm
invagination (GATA-4) |
| GDNF | Mesenchyme | - ENCC
proliferation, migration, and survival (through RET/PI3K
activation) |
| Gli | Subepithelial
mesenchyme | - Epithelial
organization and maturation, RAD pattern control |
| - Mesenchymal
maturation |
| - Villi
formation |
| - Crypt formation
(Gli-2, 3) |
| HAND | ENCCs | - ENCCs maturation,
neuro-transmitter specification (HAND-2) |
| HAT | Endoderm | - RAD pattern
control |
| - Villi
formation |
| HDAC | Endoderm | - RAD pattern
control |
| - Villi formation
(HDAC1,2) |
| Hox | Mesoderm of midgut
and hindgut | - AP patterning of
midgut and hindgut (Abd-B subfamily of Hox-A and D clusters) |
| Isl | Left side
splanchnic mesoderm | - LR pattern
control (Isl-1) |
| L1CAM | ENCCs | - ENCCs migration
through interaction with ECM |
| MMP | ENCCs | - Migration through
degradation of ECM (MMP-2) |
| Netrin | Epithelium | - Direction of axon
formation |
| - ENCCs migration
through RAD axis |
| Nodal | Former
mesendoderm | - Endoderm
differentiation (high signaling) |
| - Mesoderm
differentiation (low signaling) |
| Splanchnic
mesoderm | - LR pattern
control |
| Notch | Endoderm | - Epithelial
cytodifferentiation (through Hes-1, Atoh-1, and crosstalk with
Wnt/β-catenin) |
| PDGF | Endoderm | - RAD pattern
control |
| - Epithelial and
mesenchymal maturation |
| - Crypt and villi
formation (PDGF-A) |
| PHOX | ENCCs | - ENCCs
proliferation, survival |
| - Enteric ganglia
formation |
| - Neuronal
formation (PHOX-2B) |
| Pitx | Left side
splanchnic mesoderm | - LR pattern
control (Pitx-2, through Shroom3, N-cadherin) |
| RA | Paraxial
mesoderm | - Directing NCCs
into the formation of ENS (through RET) |
| Mesoderm | - AP pattern
control |
| Mesenchyme | - ENCCs
proliferation, migration (through inhibition of PTEN) |
| Shh/Ihh | Endoderm of AIP and
CIP | - Endoderm
invagination |
| - Mesodermal
formation |
| Splanchnic
mesoderm | - LR pattern
control |
| Endoderm | - Villi
formation |
| - Crypt
formation |
| - Maturation of
mesenchyme and epithelium |
| Epithelium | - ENCCs migration
through RAD axis (through PTC-1) |
| Sox | Former
mesendoderm | - Endoderm
formation of midgut and hindgut (Sox-17) |
| Anterior
endoderm | - AP pattern
control (Sox-2) |
| ENCCs | - ENCC survival,
proliferation, inhibition of cytodifferentiation |
| - Glial cell
formation (Sox-10) |
| T-box | Former
mesendoderm | - Endoderm
formation |
| Right side
splanchnic mesoderm | - LR pattern
control (Tbx-18) |
|
Wnt/Ca2+ | Mesoderm | - Elongation of
mesenchyme and ventral development (through Wnt5a and Fz2) |
| Wnt/PCP | Epithelium | - Cytoskeleteal
organization |
| - Apical-basal
epithelial polarization, RAD pattern control (through Wnt5a,
Ezrin) |
| - Crypt
development |
| Mesoderm | - Elongation of
mesenchyme (through Wnt11 and Fz7) |
| Wnt/β-catenin | Endoderm | - AP pattern
control (through Tcf-1,4 and Lef-1) |
| - Epithelial
proliferation and cytodifferentiation |
| - Villus formation
(through Tcf-3,4) |
| - Crypt formation
(through Ephrin-B) |
| Table IIIMolecular factors and related
disorders in intestinal embryogenesis. |
Table III
Molecular factors and related
disorders in intestinal embryogenesis.
| Molecular
factors | Disorders |
|---|
| BMP | - (General
inhibition) |
| polyp formation,
larger and fewer villi, less compact subepithelial mesenchymal
development |
| - (BMP-2,4,7
misexpression/mutation) |
| impairment of all
layer development and differentiation, disruption of RAD
pattern |
| - (BMP-2
overexpression) |
| decreased survival
of gut neural cells |
| - (BMP-2,4
inhibition) |
| disruption of ENCCs
migration, imbalanced neural-glial ENCC differentiation, HSCR |
| - (BMPR1
misexpression/mutation) |
| juvenile
polyposis |
| Canonical Wnt
pathway | - (General
inhibition) |
| loss of epithelial
proliferation, depletion of progenitor cells, arrest of secretory
cell cytodifferentiation |
| - (Tcf-1,4
knockout) |
| disruption of AP
pattern, loss of caudal hindgut region, altered differentiation of
duodenum, endodermal defects, CIP formation failure, loss of
Fox-A1, Sox-17 and Shh expression in hindgut, midgut
disclosure |
| - (Lef-1
knockout) |
| cecal stenosis,
posterior mesodermal disorders |
| - (Wnt3a,5a
lack) |
| posterior
mesodermal disorders |
| - (Ascl2
deletion) |
| inhibited stem cell
replication, crypt diminishment |
| Cdx | - (Cdx-1,4
lack) |
| disruption of AP
pattern, absence of intestinal phenotype |
| - (Cdx-2 loss) |
| disruption of AP
pattern, disoriented endodermal differentiation, absence of
intestinal phenotype, alteration of gut epithelium to esophageal
epithelium |
| - (Cdx-2
misexpression/mutation) |
| colonic atresia,
disruption of RAD pattern |
| Chato | - (Lack) |
| ineffective
endodermal elongation, unsuccessful gut tube closure |
| Dact-1 | - (Knockout) |
| CIP formation
failure, ventral endodermal folding failure, hindgut and cloaca
formation failure |
| DCC | -
(Misexpression/mutation) |
| failed development
of submucosal neural plexus |
| EGFR | - (Deletion) |
| delayed villus
emergence, diminished epithelial proliferation, villus blunting,
tissue disintegration |
| Elf-3/Crif-1 | - (Knockout) |
| diminished TGF-βRII
expression, fewer malformed villi, lamina propria disorganization,
epithelial malfunction |
| Epimorphin | -
(Misexpression/mutation) |
| disruption of RAD
axis, inhibition of epithelial morphogenesis, acceleration of
epithelial proliferation, disruption of BMP and Hedgehog
pathways |
| ET | - (ET-3, EDNRB,
ECE-1 deletion) |
| colorectal
agangliosis, HSCR |
| FGF | - (FGF-12 improper
function) |
| heterotaxy
syndrome, disruption of LR pattern |
| - (FGF-10
misexpression/mutation) |
| colonic and
duodenal atresia, disruption of RAD pattern |
| - (FGFR2IIIb
knockout) |
| intestinal atresia,
disruption of RAD pattern, altered RA signaling |
| Fox | - (Fox-A2 improper
function) |
| heterotaxy
syndrome, disruption of LR pattern |
| - (Fox-F1
misexpression/mutation) |
| intestinal atresia,
disruption of RAD pattern |
| - (Fox-L1
misexpression/mutation) |
| reorganization of
epithelium and villi, deregulated crypt development and
branching |
| - (Fox-F1,F2
silencing) |
| mesenchymal
disintegration after villi formation |
| GALNT11 | - (Improper
function) |
| heterotaxy
syndrome, disruption of LR pattern, disruption of left side nodal
flow |
| GATA-4 | - (Knockout) |
| AIP malformation,
foregut absence, yolk sac displacement, defective lateral-ventral
folding |
| HAND-2 | - (Deletion) |
| fewer enteric
neurons, disrupted ENCCs differentiation |
| HAT | -
(Misexpression/mutation) |
| delayed villus
emergence, failed subepithelial mesenchymal condensation, decreased
BMP-4 expression |
| HDAC | -
(Overexpression) |
| blocked epithelial
differentiation |
| - (Inhibition) |
| immature villus
development, abnormal epithelial differentiation |
| Hox | - (Hox-A13
misexpression/mutation) |
| disruption of AP
pattern, protein truncation, endodermal defects |
| - (Hox-D13
misexpression/mutation) |
| disruption of AP
pattern, hindgut differentiation of midgut epithelium |
| Ihh | -
(Misexpression/mutation) |
| decreased
epithelial proliferation, reduced size and number of villi |
| Inversin | - (Improper
function) |
| disruption of
forward cilial movement, situs inversus |
| Isl-1 | - (Ectopic
expression) |
| disruption of LR
pattern, bilateral symmetry, bilateral Pitx-2 expression, loss of
right-sided |
| Tbx-18
expression |
| KIF | - (KIF3B lack) |
| ciliogenesis
impairment, disruption of LR pattern, prenatal death, situs
inversus |
| - (KIF3A loss) |
| diminishment of
nodal mechanosensory ability, disruption of LR pattern, situs
inversus |
| L1CAM | - (Inhibition) |
| ENCCs close contact
abruption, slow ENCCs migration |
| MMP-2 | - (Inhibition) |
| reduced ENCCs
migration |
| Nkx2.3 | -
(Misexpression/mutation) |
| mesenchymal cell
reduction, decreased epithelial proliferation, delayed villus
emergence, intrauterine death, epithelial hyperproliferation,
mucosal thickening, branched villi, abnormal epithelial
architecture |
| Nodal | - (Ectopic
expression) disruption of LR pattern, bilateral symmetry |
| Non-canonical Wnt
pathway | - (Wnt5a
knockout) |
| shorter intestinal
length, deregulated apical-basal polarity of intestinal
epithelium |
| - (sFRP improper
function) |
| shorter intestinal
length, deregulated apical-basal polarity of intestinal
epithelium |
| - (Dvl-1,2,3
improper function) |
| PCP deregulation,
disruption of LR pattern, situs inversus |
| - (Ezrin
absence) |
| improper epithelial
polarization, villus fusion, mucosal disorganization |
| Notch | - (Atoh-1
misexpression/mutation) |
| inhibition of
secretory cytodifferentiation, abnormal epithelial
proliferation |
| PDGF | - (Inhibition) |
| disrupted colonic
mucosal architecture |
| PHOX-2B | -
(Misexpression/mutation) |
| total agangliosis,
enhanced glial differentiation, HSCR |
| Pitx-2 | - (Ectopic
expression) |
| disruption of LR
pattern, bilateral symmetry, symmetrical bilateral Isl-1
expression, loss of right-sided Tbx-18 expression |
| - (Knockout) |
| loss of Isl-1
expression, bilateral expression of Tbx-18, bilateral symmetry |
| Pkd-2 | - (Loss) |
| diminishment of
nodal mechanosensory ability, disruption of LR pattern, situs
inversus |
| RA | - (Raldh-2
deficiency) |
| ENS agenesis,
decreased RET expression |
| - (abnormal
signaling) |
| HSCR |
| RET-GDNF | - (RET
knockout) |
| total
agangliosis |
| - (RET
misexpression/mutation) |
| HSCR |
| - (GDNF
insufficiency) |
| HSCR |
| - (PI3K
inhibition) |
| distal agangliosis,
limited ENCCs migration |
| - (SPRY-2, KIF26A
silencing) |
| enteric nerve
hyperplasia |
| Shh | - (Knockout) |
| esophageal
malformation, foregut mesodermal absence, defective mesenchymal
development, defective epithelial maturation, disrupted villi
organization, disruption of RAD pattern, ectopic submucosal ganglia
formation, malformed neural projections |
| -
(Overexpression) |
| mesodermal
overdevelopment, intestinal agangliosis, inhibited ENCCs
migration |
| -
(misexpression/mutation) |
| intestinal atresia,
villi overgrowth |
| Sox-10 | -
(Misexpression/mutation) |
| decreased ENCCs
survival, premature ENCCs differentiation, distal agangliosis,
HSCR |
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JT conceived and designed the study. AK, IK and KN
researched the literature, processed the figures and drafted the
manuscript. KN, DAS, AT and JT edited the manuscript, assisted in
the literature search, critically revised the article for important
intellectual content and provided the final approval of the version
to be published. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors confirm that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
AIP
|
anterior intestinal portal
|
|
AP
|
anteroposterior
|
|
BMP
|
bone morphogenetic protein
|
|
CIP
|
caudal intestinal portal
|
|
CRC
|
colorectal cancer
|
|
CSCs
|
cancer stem cells
|
|
ENCCs
|
enteric neural crest cells
|
|
ENS
|
enteric nervous system
|
|
FGF
|
fibroblast growth factor
|
|
Fox
|
forkhead-box
|
|
Hox
|
homeobox
|
|
HSCR
|
Hirschsprung disease
|
|
LR
|
left-right
|
|
PHOX
|
paired-like homeobox
|
|
RAD
|
radial
|
|
Shh
|
sonic hedgehog
|
|
Sox
|
SRY-related HMG-box
|
|
Tbx
|
T-box
|
|
TGF-β
|
transforming growth factor-β
|
|
Wnt
|
wingless-related integration site
|
References
|
1
|
Spence JR, Lauf R and Shroyer NF:
Vertebrate intestinal endoderm development. Dev Dyn. 240:501–520.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roberts DJ: Molecular mechanisms of
development of the gastrointestinal tract. Dev Dyn. 219:109–120.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Santa Barbara P, Van den Brink GR and
Roberts DJ: Molecular etiology of gut malformations and diseases.
Am J Med Genet. 115:221–230. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kelleher FC, Fennelly D and Rafferty M:
Common critical pathways in embryogenesis and cancer. Acta Oncol.
45:375–388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park JJ, Wolff BG, Tollefson MK, Walsh EE
and Larson DR: Meckel diverticulum: The mayo clinic experience with
1476 patients (1950-2002). Ann Surg. 241:529–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kluth D, Jaeschke-Melli S and Fiegel H:
The embryology of gut rotation. Semin Pediatr Surg. 12:275–279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kluth D, Fiegel HC and Metzger R:
Embryology of the hindgut. Semin Pediatr Surg. 20:152–160. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee JM and Kim NK: Essential anatomy of
the anorectum for colorectal surgeons focused on the gross anatomy
and histologic findings. Ann Coloproctol. 34:59–71. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Uesaka T, Young HM, Pachnis V and Enomoto
H: Development of the intrinsic and extrinsic innervation of the
gut. Dev Biol. 417:158–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bayha E, Jørgensen MC, Serup P and
Grapin-Botton A: Retinoic acid signaling organizes endodermal organ
specification along the entire antero-posterior axis. PLoS One.
4:e58452009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bayliss WM and Starling EH: The movements
and innervation of the small intestine. J Physiol. 24:99–143. 1899.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Timmermans JP, Hens J and Adriaensen D:
Outer submucous plexus An intrinsic nerve network involved in both
secretory and motility processes in the intestine of large mammals
and humans. Anat Rec. 262:71–78. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen M: Nodal signaling: Developmental
roles and regulation. Development. 134:1023–1034. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu P, Wakamiya M, Shea MJ, Albrecht U,
Behringer RR and Bradley A: Requirement for Wnt3 in vertebrate axis
formation. Nat Genet. 22:361–365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haegel H, Larue L, Ohsugi M, Fedorov L,
Herrenknecht K and Kemler R: Lack of beta-catenin affects mouse
development at gastrulation. Development. 121:3529–3537.
1995.PubMed/NCBI
|
|
16
|
Lowe LA, Yamada S and Kuehn MR: Genetic
dissection of nodal function in patterning the mouse embryo.
Development. 128:1831–1843. 2001.PubMed/NCBI
|
|
17
|
Vincent SD, Dunn NR, Hayashi S, Norris DP
and Robertson EJ: Cell fate decisions within the mouse organizer
are governed by graded Nodal signals. Genes Dev. 17:1646–1662.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weng W and Stemple DL: Nodal signaling and
vertebrate germ layer formation. Birth Defects Res C Embryo Today.
69:325–332. 2003. View Article : Google Scholar
|
|
19
|
Tada S, Era T, Furusawa C, Sakurai H and
Nishikawa S, Kinoshita M, Nakao K, Chiba T and Nishikawa S:
Characterization of mesendoderm: A diverging point of the
definitive endoderm and mesoderm in embryonic stem cell
differentiation culture. Development. 132:4363–4374. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Horb ME and Thomsen GH: A vegetally
localized T-box transcription factor in Xenopus eggs specifies
mesoderm and endoderm and is essential for embryonic mesoderm
formation. Development. 124:1689–1698. 1997.PubMed/NCBI
|
|
21
|
Hart AH, Hartley L, Sourris K, Stadler ES,
Li R, Stanley EG, Tam PP, Elefanty AG and Robb L: Mixl1 is required
for axial mesendoderm morphogenesis and patterning in the murine
embryo. Development. 129:3597–3608. 2002.PubMed/NCBI
|
|
22
|
Maduro MF, Meneghini MD, Bowerman B,
Broitman-Maduro G and Rothman JH: Restriction of mesendoderm to a
single blastomere by the combined action of SKN-1 and a GSK-3beta
homolog is mediated by MED-1 and -2 in C. elegans. Mol Cell.
7:475–485. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanai-Azuma M, Kanai Y, Gad JM, Tajima Y,
Taya C, Kurohmaru M, Sanai Y, Yonekawa H, Yazaki K, Tam PP and
Hayashi Y: Depletion of definitive gut endoderm in Sox17-null
mutant mice. Development. 129:2367–2379. 2002.PubMed/NCBI
|
|
24
|
Zhu J, Fukushige T, McGhee JD and Rothman
JH: Reprogramming of early embryonic blastomeres into endodermal
progenitors by a Caenorhabditis elegans GATA factor. Genes Dev.
12:3809–3814. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith JC, Price BM, Van Nimmen K and
Huylebroeck D: Identification of a potent Xenopus mesoderm-inducing
factor as a homologue of activin A. Nature. 345:729–731. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Henry GL, Brivanlou IH, Kessler DS,
Hemmati-Brivanlou A and Melton DA: TGF-beta signals and a pattern
in Xenopus laevis endodermal development. Development.
122:1007–1015. 1996.PubMed/NCBI
|
|
27
|
Lawson A and Schoenwolf GC: Epiblast and
primitive-streak origins of the endoderm in the gastrulating chick
embryo. Development. 130:3491–3501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kimura W, Yasugi S, Stern CD and Fukuda K:
Fate and plasticity of the endoderm in the early chick embryo. Dev
Biol. 289:283–295. 2006. View Article : Google Scholar
|
|
29
|
Franklin V, Khoo PL, Bildsoe H, Wong N,
Lewis S and Tam PP: Regionalisation of the endoderm progenitors and
morphogenesis of the gut portals of the mouse embryo. Mech Dev.
125:587–600. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zorn AM and Wells JM: Vertebrate endoderm
development and organ formation. Annu Rev Cell Dev Biol.
25:221–251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kwon GS, Viotti M and Hadjantonakis AK:
The endoderm of the mouse embryo arises by dynamic widespread
intercalation of embryonic and extraembryonic lineages. Dev Cell.
15:509–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tam PP, Khoo PL, Wong N, Tsang TE and
Behringer RR: Regionalization of cell fates and cell movement in
the endoderm of the mouse gastrula and the impact of loss of
Lhx1(Lim1) function. Dev Biol. 274:171–187. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lawson KA and Pedersen RA: Cell fate,
morphogenetic movement and population kinetics of embryonic
endoderm at the time of germ layer formation in the mouse.
Development. 101:627–652. 1987.PubMed/NCBI
|
|
34
|
Lewis SL and Tam PP: Definitive endoderm
of the mouse embryo: Formation, cell fates, and morphogenetic
function. Dev Dyn. 235:2315–2329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reed RA, Womble MA, Dush MK, Tull RR,
Bloom SK, Morckel AR, Devlin EW and Nascone-Yoder NM: Morphogenesis
of the primitive gut tube is generated by Rho/ROCK/myosin
II-mediated endoderm rearrangements. Dev Dyn. 238:3111–3125. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Molkentin JD, Lin Q, Duncan SA and Olson
EN: Requirement of the transcription factor GATA4 for heart tube
formation and ventral morphogenesis. Genes Dev. 11:1061–1072. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garcia-Garcia MJ, Shibata M and Anderson
KV: Chato, a KRAB zinc-finger protein, regulates convergent
extension in the mouse embryo. Development. 135:3053–3062. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wen J, Chiang YJ, Gao C, Xue H, Xu J, Ning
Y, Hodes RJ, Gao X and Chen YG: Loss of Dact1 disrupts planar cell
polarity signaling by altering dishevelled activity and leads to
posterior malformation in mice. J Biol Chem. 285:11023–11030. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koike T and Yasugi S: In vitro analysis of
mesenchymal influences on the differentiation of stomach epithelial
cells of the chicken embryo. Differentiation. 65:13–25. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sumiya M: Differentiation of the digestive
tract epithelium of the chick embryo cultured in vitro enveloped in
a fragment of the vitelline membrane, in the absence of mesenchyme.
Roux Arch Dev Biol. 179:1–17. 1976. View Article : Google Scholar
|
|
41
|
Aufderheide E and Ekblom P: Tenascin
during gut development: Appearance in the mesenchyme, shift in
molecular forms, and dependence on epithelial-mesenchymal
interactions. J Cell Biol. 107:2341–2349. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kedinger M, Simon-Assmann P, Bouziges F,
Arnold C, Alexandre E and Haffen K: Smooth muscle actin expression
during rat gut development and induction in fetal skin
fibro-blastic cells associated with intestinal embryonic
epithelium. Differentiation. 43:87–97. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yasugi S, Takeda H and Fukuda K: Early
Determination of developmental fate in presumptive intestinal
endoderm of the chicken embryo. Dev Growth Differ. 33:235–241.
1991. View Article : Google Scholar
|
|
44
|
Roberts DJ, Smith DM, Goff DJ and Tabin
CJ: Epithelial-mesenchymal signaling during the regionalization of
the chick gut. Development. 125:2791–2801. 1998.PubMed/NCBI
|
|
45
|
Riddle RD, Johnson RL, Laufer E and Tabin
C: Sonic hedgehog mediates the polarizing activity of the ZPA.
Cell. 75:1401–1416. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fan CM and Tessier-Lavigne M: Patterning
of mammalian somites by surface ectoderm and notochord: Evidence
for sclerotome induction by a hedgehog homolog. Cell. 79:1175–1186.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roelink H, Augsburger A, Heemskerk J,
Korzh V, Norlin S, Ruiz i Altaba A, Tanabe Y, Placzek M, Edlund T,
Jessell TM, et al: Floor plate and motor neuron induction by vhh-1,
a vertebrate homolog of hedgehog expressed by the notochord. Cell.
76:761–775. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Narita T, Ishii Y, Nohno T, Noji S and
Yasugi S: Sonic hedgehog expression in developing chicken digestive
organs is regulated by epithelial-mesenchymal interactions. Dev
Growth Differ. 40:67–74. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roberts DJ, Johnson RL, Burke AC, Nelson
CE, Morgan BA and Tabin C: Sonic hedgehog is an endodermal signal
inducing Bmp-4 and Hox genes during induction and regionalization
of the chick hindgut. Development. 121:3163–3174. 1995.PubMed/NCBI
|
|
50
|
Chiang C, Litingtung Y, Lee E, Young KE,
Corden JL, Westphal H and Beachy PA: Cyclopia and defective axial
patterning in mice lacking Sonic hedgehog gene function. Nature.
383:407–413. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pepicelli CV, Lewis PM and McMahon AP:
Sonic hedgehog regulates branching morphogenesis in the mammalian
lung. Curr Biol. 8:1083–1086. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sukegawa A, Narita T, Kameda T, Saitoh K,
Nohno T, Iba H, Yasugi S and Fukuda K: The concentric structure of
the developing gut is regulated by Sonic hedgehog derived from
endodermal epithelium. Development. 127:1971–1980. 2000.PubMed/NCBI
|
|
53
|
Pisano JM, Colón-Hastings F and Birren SJ:
Postmigratory enteric and sympathetic neural precursors share
common, developmentally regulated, responses to BMP2. Dev Biol.
227:1–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chalazonitis A: Neurotrophin-3 in the
development of the enteric nervous system. Prog Brain Res.
146:243–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chawengsaksophak K, de Graaff W, Rossant
J, Deschamps J and Beck F: Cdx2 is essential for axial elongation
in mouse development. Proc Natl Acad Sci USA. 101:7641–7645. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kinkel MD, Eames SC, Alonzo MR and Prince
VE: Cdx4 is required in the endoderm to localize the pancreas and
limit beta-cell number. Development. 135:919–929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sherwood RI, Chen TY and Melton DA:
Transcriptional dynamics of endodermal organ formation. Dev Dyn.
238:29–42. 2009. View Article : Google Scholar
|
|
58
|
Wells JM and Melton DA: Vertebrate
endoderm development. Annu Rev Cell Dev Biol. 15:393–410. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tiso N, Filippi A, Pauls S, Bortolussi M
and Argenton F: BMP signalling regulates anteroposterior endoderm
patterning in zebrafish. Mech Dev. 118:29–37. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Grapin-Botton A: Antero-posterior
patterning of the vertebrate digestive tract: 40 years after Nicole
Le Douarin's PhD thesis. Int J Dev Biol. 49:335–347. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Marikawa Y: Wnt/beta-catenin signaling and
body plan formation in mouse embryos. Semin Cell Dev Biol.
17:175–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Johannesson M, Ståhlberg A, Ameri J, Sand
FW, Norrman K and Semb H: FGF4 and retinoic acid direct
differentiation of hESCs into PDX1-expressing foregut endoderm in a
time- and concentration-dependent manner. PLoS One. 4:e47942009.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ameri J, Ståhlberg A, Pedersen J,
Johansson JK, Johannesson MM, Artner I and Semb H: FGF2 specifies
hESC- derived definitive endoderm into foregut/midgut cell lineages
in a concentration-dependent manner. Stem Cells. 28:45–56.
2010.
|
|
64
|
Spence JR, Mayhew CN, Rankin SA, Kuhar MF,
Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn
AM, et al: Directed differentiation of human pluripotent stem cells
into intestinal tissue in vitro. Nature. 470:105–109. 2011.
View Article : Google Scholar :
|
|
65
|
Geske MJ, Zhang X, Patel KK, Ornitz DM and
Stappenbeck TS: Fgf9 signaling regulates small intestinal
elongation and mesen-chymal development. Development.
135:2959–2968. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang X, Stappenbeck TS, White AC, Lavine
KJ, Gordon JI and Ornitz DM: Reciprocal epithelial-mesenchymal FGF
signaling is required for cecal development. Development.
133:173–180. 2006. View Article : Google Scholar
|
|
67
|
Burns RC, Fairbanks TJ, Sala F, De Langhe
S, Mailleux A, Thiery JP, Dickson C, Itoh N, Warburton D, Anderson
KD and Bellusci S: Requirement for fibroblast growth factor 10 or
fibroblast growth factor receptor 2-IIIb signaling for cecal
development in mouse. Dev Biol. 265:61–74. 2004. View Article : Google Scholar
|
|
68
|
Sala FG, Curtis JL, Veltmaat JM, Del Moral
PM, Le LT, Fairbanks TJ, Warburton D, Ford H, Wang K, Burns RC and
Bellusci S: Fibroblast growth factor 10 is required for survival
and proliferation but not differentiation of intestinal epithelial
progenitor cells during murine colon development. Dev Biol.
299:373–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Grapin-Botton A and Melton DA: Endoderm
development: From patterning to organogenesis. Trends Genet.
16:124–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gao N, White P and Kaestner KH:
Establishment of intestinal identity and epithelial-mesenchymal
signaling by Cdx2. Dev Cell. 16:588–599. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Grainger S, Savory JG and Lohnes D: Cdx2
regulates patterning of the intestinal epithelium. Dev Biol.
339:155–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Beck F and Stringer EJ: The role of Cdx
genes in the gut and in axial development. Biochem Soc Trans.
38:353–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Allan D, Houle M, Bouchard N, Meyer BI,
Gruss P and Lohnes D: RARgamma and Cdx1 interactions in vertebral
patterning. Dev Biol. 240:46–60. 2001. View Article : Google Scholar
|
|
74
|
Ikeya M and Takada S: Wnt-3a is required
for somite specification along the anteroposterior axis of the
mouse embryo and for regulation of cdx-1 expression. Mech Dev.
103:27–33. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Keenan ID, Sharrard RM and Isaacs HV: FGF
signal transduction and the regulation of Cdx gene expression. Dev
Biol. 299:478–488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gregorieff A, Grosschedl R and Clevers H:
Hindgut defects and transformation of the gastro-intestinal tract
in Tcf4(-/-)/Tcf1(-/-) embryos. EMBO J. 23:1825–1833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yokouchi Y, Sakiyama J and Kuroiwa A:
Coordinated expression of Abd-B subfamily genes of the HoxA cluster
in the developing digestive tract of chick embryo. Dev Biol.
169:76–89. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Boulet AM and Capecchi MR: Targeted
disruption of hoxc-4 causes esophageal defects and vertebral
transformations. Dev Biol. 177:232–249. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Warot X, Fromental-Ramain C, Fraulob V,
Chambon P and Dollé P: Gene dosage-dependent effects of the Hoxa-13
and Hoxd-13 mutations on morphogenesis of the terminal parts of the
digestive and urogenital tracts. Development. 124:4781–4791.
1997.
|
|
80
|
Zacchetti G, Duboule D and Zakany J: Hox
gene function in vertebrate gut morphogenesis: The case of the
caecum. Development. 134:3967–3973. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mortlock DP and Innis JW: Mutation of
HOXA13 in hand-foot-genital syndrome. Nat Genet. 15:179–180. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Huelsken J and Birchmeier W: New aspects
of Wnt signaling pathways in higher vertebrates. Curr Opin Genet
Dev. 11:547–553. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Theodosiou NA and Tabin CJ: Wnt signaling
during development of the gastrointestinal tract. Dev Biol.
259:258–271. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
McBride HJ, Fatke B and Fraser SE: Wnt
signaling components in the chicken intestinal tract. Dev Biol.
256:18–33. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang X, Chen Y, Ye Y, Wang J, Wang H,
Yuan G, Lin Z, Wu Y, Zhang Y and Lin X: Wnt signaling promotes
hindgut fate commitment through regulating multi-lineage genes
during hESC differentiation. Cell Signal. 29:12–22. 2017.
View Article : Google Scholar
|
|
86
|
Behrens J, von Kries JP, Kühl M, Bruhn L,
Wedlich D, Grosschedl R and Birchmeier W: Functional interaction of
beta-catenin with the transcription factor LEF-1. Nature.
382:638–642. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hart M, Concordet JP, Lassot I, Albert I,
del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F,
Benarous R and Polakis P: The F-box protein beta-TrCP associates
with phosphorylated beta-catenin and regulates its activity in the
cell. Curr Biol. 9:207–210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hart MJ, de los Santos R, Albert IN,
Rubinfeld B and Polakis P: Downregulation of beta-catenin by human
Axin and its association with the APC tumor suppressor,
beta-catenin and GSK3 beta. Curr Biol. 8:573–581. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Noordermeer J, Klingensmith J, Perrimon N
and Nusse R: Dishevelled and armadillo act in the wingless
signalling pathway in Drosophila. Nature. 367:80–83. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Joo JH, Taxter TJ, Munguba GC, Kim YH,
Dhaduvai K, Dunn NW, Degan WJ, Oh SP and Sugrue SP: Pinin modulates
expression of an intestinal homeobox gene, Cdx2, and plays an
essential role for small intestinal morphogenesis. Dev Biol.
345:191–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
92
|
van Genderen C, Okamura RM, Farinas I, Quo
RG, Parslow TG, Bruhn L and Grosschedl R: Development of several
organs that require inductive epithelial-mesenchymal interactions
is impaired in LEF-1-deficient mice. Genes Dev. 8:2691–2703. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Roose J, Huls G, van Beest M, Moerer P,
van der Horn K, Goldschmeding R, Logtenberg T and Clevers H:
Synergy between tumor suppressor APC and the beta-catenin-Tcf4
target Tcf1. Science. 285:1923–1926. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lickert H, Domon C, Huls G, Wehrle C,
Duluc I, Clevers H, Meyer BI, Freund JN and Kemler R:
Wnt/(beta)-catenin signaling regulates the expression of the
homeobox gene Cdx1 in embryonic intestine. Development.
127:3805–3813. 2000.PubMed/NCBI
|
|
95
|
Takada S, Stark KL, Shea MJ, Vassileva G,
McMahon JA and McMahon AP: Wnt-3a regulates somite and tailbud
formation in the mouse embryo. Genes Dev. 8:174–189. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Yamaguchi TP, Bradley A, McMahon AP and
Jones S: A Wnt5a pathway underlies outgrowth of multiple structures
in the vertebrate embryo. Development. 126:1211–1223.
1999.PubMed/NCBI
|
|
97
|
Mlodzik M: Spiny legs and prickled bodies:
New insights and complexities in planar polarity establishment.
Bioessays. 22:311–315. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Li L, Yuan H, Xie W, Mao J, Caruso AM,
McMahon A, Sussman DJ and Wu D: Dishevelled proteins lead to two
signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase
in mammalian cells. J Biol Chem. 274:129–134. 1999. View Article : Google Scholar
|
|
99
|
Winter CG, Wang B, Ballew A, Royou A,
Karess R, Axelrod JD and Luo L: Drosophila Rho-associated kinase
(Drok) links Frizzled-mediated planar cell polarity signaling to
the actin cytoskeleton. Cell. 105:81–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Marlow F, Topczewski J, Sepich D and
Solnica-Krezel L: Zebrafish Rho kinase 2 acts downstream of Wnt11
to mediate cell polarity and effective convergence and extension
movements. Curr Biol. 12:876–884. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Moon RT, Campbell RM, Christian JL, McGrew
LL, Shih J and Fraser S: Xwnt-5A: A maternal Wnt that affects
morphogenetic movements after overexpression in embryos of Xenopus
laevis. Development. 119:97–111. 1993.PubMed/NCBI
|
|
102
|
Kuhl M, Sheldahl LC, Malbon CC and Moon
RT: Ca(2+)/calmodulin-dependent protein kinase II is stimulated by
Wnt and Frizzled homologs and promotes ventral cell fates in
Xenopus. J Biol Chem. 275:12701–12711. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Slusarski DC, Corces VG and Moon RT:
Interaction of Wnt and a Frizzled homologue triggers
G-protein-linked phosphati-dylinositol signalling. Nature.
390:410–413. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xu Q, D'Amore PA and Sokol SY: Functional
and biochemical interactions of Wnts with FrzA, a secreted Wnt
antagonist. Development. 125:4767–4776. 1998.PubMed/NCBI
|
|
105
|
Wang S, Krinks M and Moos M Jr: Frzb-1, an
antagonist of Wnt-1 and Wnt-8, does not block signaling by Wnts
-3A, -5A, or -11. Biochem Biophys Res Commun. 236:502–504. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lee CS, Buttitta LA, May NR, Kispert A and
Fan CM: SHH-N upregulates Sfrp2 to mediate its competitive
interaction with WNT1 and WNT4 in the somitic mesoderm.
Development. 127:109–118. 2000.PubMed/NCBI
|
|
107
|
Cervantes S, Yamaguchi TP and Hebrok M:
Wnt5a is essential for intestinal elongation in mice. Dev Biol.
326:285–294. 2009. View Article : Google Scholar :
|
|
108
|
Matsuyama M, Aizawa S and Shimono A: Sfrp
controls apicobasal polarity and oriented cell division in
developing gut epithelium. PLoS Genet. 5:e10004272009. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Capdevila I and Izpisúa Belmonte JC:
Knowing left from right: The molecular basis of laterality defects.
Mol Med Today. 6:112–118. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Levin M and Mercola M: The compulsion of
chirality: Toward an understanding of left-right asymmetry. Genes
Dev. 12:763–769. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Logan M, Pagan-Westphal SM, Smith DM,
Paganessi L and Tabin CJ: The transcription factor Pitx2 mediates
situs-specific morphogenesis in response to left-right asymmetric
signals. Cell. 94:307–317. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Izraeli S, Lowe LA, Bertness VL, Good DJ,
Dorward DW, Kirsch IR and Kuehn MR: The SIL gene is required for
mouse embryonic axial development and left-right specification.
Nature. 399:691–694. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
113
|
Boettger T, Wittler L and Kessel M: FGF8
functions in the specification of the right body side of the chick.
Curr Biol. 9:277–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Soffers JH, Hikspoors JP, Mekonen HK,
Koehler SE and Lamers WH: The growth pattern of the human intestine
and its mesentery. BMC Dev Biol. 15:312015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Hikspoors JPJM, Kruepunga N, Mommen GMC,
Peeters J, Hülsman CJM, Eleonore Köhler S and Lamers WH: The
development of the dorsal mesentery in human embryos and fetuses.
Semin Cell Dev Biol. 92:18–26. 2019. View Article : Google Scholar
|
|
116
|
Mekonen HK, Hikspoors JP, Mommen G, Köhler
SE and Lamers WH: Development of the ventral body wall in the human
embryo. J Anat. 227:673–685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Frazer JE and Robbins RH: On the factors
concerned in causing rotation of the intestine in man. J Anat
Physiol. 50:75–110. 1915.PubMed/NCBI
|
|
118
|
Szpinda M, Paruszewska-Achtel M, Woźniak
A, Badura M, Mila-Kierzenkowska C and Wiśniewski M:
Three-dimensional growth dynamics of the liver in the human fetus.
Surg Radiol Anat. 37:439–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Davis NM, Kurpios NA, Sun X, Gros J,
Martin JF and Tabin CJ: The chirality of gut rotation derives from
left-right asymmetric changes in the architecture of the dorsal
mesentery. Dev Cell. 15:134–145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Okada Y, Takeda S, Tanaka Y, Belmonte JI
and Hirokawa N: Mechanism of nodal flow: A conserved symmetry
breaking event in left-right axis determination. Cell. 121:633–644.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ermakov AS: Establishment of visceral
left-right asymmetry in mammals: The role of ciliary action and
leftward fluid flow in the region of Hensen's node. Ontogenez.
44:341–356. 2013.In Russian. PubMed/NCBI
|
|
122
|
Yuan S and Schoenwolf GC: Islet-1 marks
the early heart rudiments and is asymmetrically expressed during
early rotation of the foregut in the chick embryo. Anat Rec.
260:204–207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Plageman TF Jr, Zacharias AL, Gage PJ and
Lang RA: Shroom3 and a Pitx2-N-cadherin pathway function
cooperatively to generate asymmetric cell shape changes during gut
morphogenesis. Dev Biol. 357:227–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Kurpios NA, Ibanes M, Davis NM, Lui W,
Katz T, Martin JF, Izpisua Belmonte JC and Tabin CJ: The direction
of gut looping is established by changes in the extracellular
matrix and in cell: Cell adhesion. Proc Natl Acad Sci USA.
105:8499–8506. 2008. View Article : Google Scholar
|
|
125
|
Welsh IC, Thomsen M, Gludish DW,
Alfonso-Parra C, Bai Y, Martin JF and Kurpios NA: Integration of
left-right Pitx2 transcription and Wnt signaling drives asymmetric
gut morphogenesis via Daam2. Dev Cell. 26:629–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Gupta R, Soni V, Valse PD, Goyal RB, Gupta
AK and Mathur P: Neonatal intestinal obstruction associated with
situs inversus totalis: Two case reports and a review of the
literature. J Med Case Rep. 11:2642017. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Morgan D, Turnpenny L, Goodship J, Dai W,
Majumder K, Matthews L, Gardner A, Schuster G, Vien L, Harrison W,
et al: Inversin, a novel gene in the vertebrate left-right axis
pathway, is partially deleted in the inv mouse. Nat Genet.
20:149–156. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mochizuki T, Saijoh Y, Tsuchiya K,
Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H,
Nakatsuji N, et al: Cloning of inv, a gene that controls left/right
asymmetry and kidney development. Nature. 395:177–181. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Nonaka S, Tanaka Y, Okada Y, Takeda S,
Harada A, Kanai Y, Kido M and Hirokawa N: Randomization of
left-right asymmetry due to loss of nodal cilia generating leftward
flow of extraembryonic fluid in mice lacking KIF3B motor protein.
Cell. 95:829–837. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Hashimoto M, Shinohara K, Wang J, Ikeuchi
S, Yoshiba S, Meno C, Nonaka S, Takada S, Hatta K, Wynshaw-Boris A
and Hamada H: Planar polarization of node cells determines the
rotational axis of node cilia. Nat Cell Biol. 12:170–176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Song H, Hu J, Chen W, Elliott G, Andre P,
Gao B and Yang Y: Planar cell polarity breaks bilateral symmetry by
controlling ciliary positioning. Nature. 466:378–382. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Mahaffey JP, Grego-Bessa J, Liem KF Jr and
Anderson KV: Cofilin and Vangl2 cooperate in the initiation of
planar cell polarity in the mouse embryo. Development.
140:1262–1271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yoshiba S, Shiratori H, Kuo IY, Kawasumi
A, Shinohara K, Nonaka S, Asai Y, Sasaki G, Belo JA, Sasaki H, et
al: Cilia at the node of mouse embryos sense fluid flow for
left-right determination via Pkd2. Science. 338:226–231. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Gottschalk I, Stressig R, Ritgen J,
Herberg U, Breuer J, Vorndamme A, Strizek B, Willruth A, Geipel A,
Gembruch U and Berg C: Extracardiac anomalies in prenatally
diagnosed hetero-taxy syndrome. Ultrasound Obstet Gynecol.
47:443–449. 2016. View Article : Google Scholar
|
|
135
|
Applegate KE, Anderson JM and Klatte EC:
Intestinal malrotation in children: A problem-solving approach to
the upper gastrointestinal series. Radiographics. 26:1485–1500.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Nagel BH, Williams H, Stewart L, Paul J
and Stumper O: Splenic state in surviving patients with visceral
heterotaxy. Cardiol Young. 15:469–473. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Mahalik SK, Khanna S and Menon P:
Malrotation and volvulus associated with heterotaxy syndrome. J
Indian Assoc Pediatr Surg. 17:138–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Sedik A, Bar EA and Ismail M: Cecal
volvulus: Case report and review of literature. Saudi Surg J.
3:47–49. 2015. View Article : Google Scholar
|
|
139
|
Tsai EA, Grochowski CM, Falsey AM,
Rajagopalan R, Wendel D, Devoto M, Krantz ID, Loomes KM and Spinner
NB: Heterozygous deletion of FOXA2 segregates with disease in a
family with heterotaxy, panhypopituitarism, and biliary atresia.
Hum Mutat. 36:631–637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Hagen EM, Sicko RJ, Kay DM, Rigler SL,
Dimopoulos A, Ahmad S, Doleman MH, Fan R, Romitti PA, Browne ML, et
al: Copy-number variant analysis of classic heterotaxy highlights
the importance of body patterning pathways. Hum Genet.
135:1355–1364. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Martin V and Shaw-Smith C: Review of
genetic factors in intestinal malrotation. Pediatr Surg Int.
26:769–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Straus WL Jr: The thoracic and abdominal
viscera of primates with special reference to the orangutan. Proc
Am Philo Soc. 76:1–85. 1936.
|
|
143
|
Bresalier RS, Boland CR and Kim YS:
Regional differences in normal and cancer-associated
glycoconjugates of the human colon. J Natl Cancer Inst. 75:249–260.
1985.PubMed/NCBI
|
|
144
|
Congdon ED, Blumberg R and Henry W:
Fasciae of fusion and elements of the fused enteric mesenteries in
the human adult. Am J Anat. 70:251–279. 1942. View Article : Google Scholar
|
|
145
|
Smith GM: A statistical review of the
variations in the anatomic positions of the caecum and the
processus vermiformis in the infant. Anat Rec. 5:549–566. 1911.
View Article : Google Scholar
|
|
146
|
Uhlenhuth E, Wolfe WM, Smith EM and
Middleton EB: The rectogenital septum. Surg Gynecol Obstet.
76:148–163. 1948.
|
|
147
|
Tobin CE and Benjamin JA: Anatomical and
surgical restudy of Denonvilliers' fascia. Surg Gynec Obst.
80:373–388. 1945.
|
|
148
|
Schumpelick V, Dreuw B, Ophoff K and
Prescher A: Appendix and cecum. Embryology, anatomy, and surgical
applications. Surg Clin North Am. 80:295–318. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Torres AM and Ziegler MM: Malrotation of
the intestine. World J Surg. 17:326–331. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Roux M and Delavierre P: Mesocelial
abscess of probable sigmoid diverticular origin. Hyper-rotation of
the intestinal lopp with the cecum at the left. Concours Med.
87:47–48. 1965.In French. PubMed/NCBI
|
|
151
|
Garude K and Rao S: Mobile cecum: An
incidental finding. Indian J Surg. 75:265–267. 2013. View Article : Google Scholar :
|
|
152
|
Rogers RL and Harford FJ: Mobile cecum
syndrome. Dis Colon Rectum. 27:399–402. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Collins DC: Agenesis of the vermiform
appendix. Am J Surg. 82:689–696. 1951. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Karam SM: Lineage commitment and
maturation of epithelial cells in the gut. Front Biosci.
4:D286–D298. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
155
|
Tandler J: Zur entwicklungsgeschichte des
Menschlichen Duodenum in Fruhen Embryonalstadien. Morph Jahrb.
29:187–216. 1900.
|
|
156
|
Cheng W and Tam PK: Murine duodenum does
not go through a 'solid core' stage in its embryological
development. Eur J Pediatr Surg. 8:212–215. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Dalla Vecchia LK, Grosfeld JL, West KW,
Rescorla FJ, Scherer LR and Engum SA: Intestinal atresia and
stenosis: A 25-year experience with 277 cases. Arch Surg.
133:490–497. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Louw JH: Jejunoileal atresia and stenosis.
J Pediatr Surg. 1:8–23. 1966. View Article : Google Scholar
|
|
159
|
Abdullah F, Arnold MA, Nabaweesi R,
Fischer AC, Colombani PM, Anderson KD, Lau H and Chang DC:
Gastroschisis in the United States 1988-2003: Analysis and risk
categorization of 4344 patients. J Perinatol. 27:50–55. 2007.
View Article : Google Scholar
|
|
160
|
Baglaj M, Carachi R and MacCormack B:
Colonic atresia: A clinicopathological insight into its etiology.
Eur J Pediatr Surg. 20:102–105. 2010. View Article : Google Scholar
|
|
161
|
Barnard CN and Louw JH: The genesis of
intestinal atresia. Minn Med. 39:7451956.PubMed/NCBI
|
|
162
|
Louw JH and Barnard CN: Congenital
intestinal atresia; observations on its origin. Lancet.
269:1065–1067. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Black PR, Mueller D, Crow J, Morris RC and
Husain AN: Mesenteric defects as a cause of intestinal volvulus
without malrotation and as the possible primary etiology of
intestinal atresia. J Pediatr Surg. 29:1339–1343. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Stollman TH, Wijnen RM and Draaisma JM:
Investigation for cystic fibrosis in infants with jejunoileal
atresia in the Netherlands: A 35-year experience with 114 cases.
Eur J Pediatr. 166:989–990. 2007. View Article : Google Scholar
|
|
165
|
Erskine JM: Colonic stenosis in the
newborn: The possible thromboembolic etiology of intestinal
stenosis and atresia. J Pediatr Surg. 5:321–333. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Johnson SM and Meyers RL: Inherited
thrombophilia: A possible cause of in utero vascular thrombosis in
children with intestinal atresia. J Pediatr Surg. 36:1146–1149.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Cooley BC, Chen CY and Schmeling G:
Increased venous versus arterial thrombosis in the Factor V Leiden
mouse. Thromb Res. 119:747–751. 2007. View Article : Google Scholar
|
|
168
|
Greer FR: Vitamin K the basics-what's new?
Early Hum Dev. 86(Suppl 1): S43–S47. 2010. View Article : Google Scholar
|
|
169
|
Cortese MG, Morra I, Marchese C,
Costantino S, Forni M and Canavese F: Association between multiple
intestinal atresia and omphalocele: A case report. Pediatr Pathol
Mol Med. 20:203–207. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Pameijer CR, Hubbard AM, Coleman B and
Flake AW: Combined pure esophageal atresia, duodenal atresia,
biliary atresia, and pancreatic ductal atresia: Prenatal diagnostic
features and review of the literature. J Pediatr Surg. 35:745–747.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Fairbanks TJ, Sala FG, Kanard R, Curtis
JL, Del Moral PM, De Langhe S, Warburton D, Anderson KD, Bellusci S
and Burns RC: The fibroblast growth factor pathway serves a
regulatory role in proliferation and apoptosis in the pathogenesis
of intestinal atresia. J Pediatr Surg. 41:132–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Ornitz DM and Itoh N: Fibroblast growth
factors. Genome Biol. 2:REVIEWS30052001. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Botham RA, Franco M, Reeder AL, Lopukhin
A, Shiota K, Yamada S and Nichol PF: Formation of duodenal atresias
in fibroblast growth factor receptor 2IIIb-/- mouse embryos occurs
in the absence of an endodermal plug. J Pediatr Surg. 47:1369–1379.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Reeder AL, Botham RA, Franco M, Zaremba KM
and Nichol PF: Formation of intestinal atresias in the Fgfr2IIIb-/-
mice is not associated with defects in notochord development or
alterations in Shh expression. J Surg Res. 177:139–145. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Reeder AL, Zaremba KM, Liebl RM,
Kowalkowski A and Nichol PF: Exogenous Sonic hedgehog protein does
not rescue cultured intestine from atresia formation. J Surg Res.
187:14–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Nichol PF, Tyrrell JD and Saijoh Y:
Retinaldehyde dehydroge-nase 2 is down-regulated during duodenal
atresia formation in Fgfr2IIIb-/- mice. J Surg Res. 175:82–87.
2012. View Article : Google Scholar
|
|
177
|
Reeder AL, Botham RA, Zaremba KM and
Nichol PF: Haploinsufficiency of retinaldehyde dehydrogenase 2
decreases the severity and incidence of duodenal atresia in the
fibroblast growth factor receptor 2IIIb-/- mouse model. Surgery.
152:768–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Ramalho-Santos M, Melton DA and McMahon
AP: Hedgehog signals regulate multiple aspects of gastrointestinal
development. Development. 127:2763–2772. 2000.PubMed/NCBI
|
|
179
|
Mo R, Kim JH, Zhang J, Chiang C, Hui CC
and Kim PC: Anorectal malformations caused by defects in sonic
hedgehog signaling. Am J Pathol. 159:765–774. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Al-Jaroof AH, Al-Zayer F and Meshikhes AW:
A case of sigmoid colon duplication in an adult woman. BMJ Case
Rep. 2014:pii: bcr2014203874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Ramakrishna HK: Intestinal duplication.
Indian J Surg. 70:270–273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Favara BE, Franciosi RA and Akers DR:
Enteric duplications. Thirty-seven cases: A vascular theory of
pathogenesis. Am J Dis Child. 122:501–506. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Colony PC, Kois JM and Peiffer LP:
Structural and enzymatic changes during colonic maturation in the
fetal and suckling rat. Gastroenterology. 97:338–347. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Helander HF: Morphological studies on the
development of the rat colonic mucosa. Acta Anat (Basel). 85. pp.
155–176. 1973, View Article : Google Scholar
|
|
185
|
Bell L and Williams L: A scanning and
transmission electron microscopical study of the morphogenesis of
human colonic villi. Anat Embryol (Berl). 165:437–455. 1982.
View Article : Google Scholar
|
|
186
|
Lev R and Orlic D: Histochemical and
radioautographic studies of normal human fetal colon.
Histochemistry. 39:301–311. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Saotome I, Curto M and McClatchey AI:
Ezrin is essential for epithelial organization and villus
morphogenesis in the developing intestine. Dev Cell. 6:855–864.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Ratineau C, Duluc I, Pourreyron C,
Kedinger M, Freund JN and Roche C: Endoderm- and
mesenchyme-dependent commitment of the differentiated epithelial
cell types in the developing intestine of rat. Differentiation.
71:163–169. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Fritsch C, Swietlicki EA, Lefebvre O,
Kedinger M, Iordanov H, Levin MS and Rubin DC: Epimorphin
expression in intestinal myofibroblasts induces epithelial
morphogenesis. J Clin Invest. 110:1629–1641. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Shaker A, Swietlicki EA, Wang L, Jiang S,
Onal B, Bala S, DeSchryver K, Newberry R, Levin MS and Rubin DC:
Epimorphin deletion protects mice from inflammation-induced colon
carcinogenesis and alters stem cell niche myofibroblast secretion.
J Clin Invest. 120:2081–2093. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Hirai Y, Takebe K, Takashina M, Kobayashi
S and Takeichi M: Epimorphin: A mesenchymal protein essential for
epithelial morphogenesis. Cell. 69:471–481. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Kolterud A, Grosse AS, Zacharias WJ,
Walton KD, Kretovich KE, Madison BB, Waghray M, Ferris JE, Hu C,
Merchant JL, et al: Paracrine Hedgehog signaling in stomach and
intestine: New roles for hedgehog in gastrointestinal patterning.
Gastroenterology. 137:618–628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Madison BB, Braunstein K, Kuizon E,
Portman K, Qiao XT and Gumucio DL: Epithelial hedgehog signals
pattern the intestinal crypt-villus axis. Development. 132:279–289.
2005. View Article : Google Scholar
|
|
194
|
Karlsson L, Lindahl P, Heath JK and
Betsholtz C: Abnormal gastrointestinal development in PDGF-A and
PDGFR-(alpha) deficient mice implicates a novel mesenchymal
structure with putative instructive properties in villus
morphogenesis. Development. 127:3457–3466. 2000.PubMed/NCBI
|
|
195
|
Wang LC, Nassir F, Liu ZY, Ling L, Kuo F,
Crowell T, Olson D, Davidson NO and Burkly LC: Disruption of
hedgehog signaling reveals a novel role in intestinal morphogenesis
and intestinal-specific lipid metabolism in mice. Gastroenterology.
122:469–482. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Madison BB, McKenna LB, Dolson D, Epstein
DJ and Kaestner KH: FoxF1 and FoxL1 link hedgehog signaling and the
control of epithelial proliferation in the developing stomach and
intestine. J Biol Chem. 284:5936–5944. 2009. View Article : Google Scholar :
|
|
197
|
Kaestner KH, Silberg DG, Traber PG and
Schutz G: The mesenchymal winged helix transcription factor Fkh6 is
required for the control of gastrointestinal proliferation and
differentiation. Genes Dev. 11:1583–1595. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Ormestad M, Astorga J, Landgren H, Wang T,
Johansson BR, Miura N and Carlsson P: Foxf1 and Foxf2 control
murine gut development by limiting mesenchymal Wnt signaling and
promoting extracellular matrix production. Development.
133:833–843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
De Santa Barbara P, Williams J, Goldstein
AM, Doyle AM, Nielsen C, Winfield S, Faure S and Roberts DJ: Bone
morpho-genetic protein signaling pathway plays multiple roles
during gastrointestinal tract development. Dev Dyn. 234:312–322.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
He XC, Zhang J, Tong WG, Tawfik O, Ross J,
Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, et al: BMP
signaling inhibits intestinal stem cell self-renewal through
suppression of Wnt-beta-catenin signaling. Nat Genet. 36:1117–1121.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
201
|
Howe JR, Bair JL, Sayed MG, Anderson ME,
Mitros FA, Petersen GM, Velculescu VE, Traverso G and Vogelstein B:
Germline mutations of the gene encoding bone morphogenetic protein
receptor 1A in juvenile polyposis. Nat Genet. 28:184–187. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Shroyer NF and Wong MH: BMP signaling in
the intestine: Cross-talk is key. Gastroenterology. 133:1035–1038.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Batts LE, Polk DB, Dubois RN and Kulessa
H: Bmp signaling is required for intestinal growth and
morphogenesis. Dev Dyn. 235:1563–1570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Flentjar N, Chu PY, Ng AY, Johnstone CN,
Heath JK, Ernst M, Hertzog PJ and Pritchard MA: TGF-betaRII rescues
development of small intestinal epithelial cells in Elf3-deficient
mice. Gastroenterology. 132:1410–1419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Jedlicka P and Gutierrez-Hartmann A: Ets
transcription factors in intestinal morphogenesis, homeostasis and
disease. Histol Histopathol. 23:1417–1424. 2008.PubMed/NCBI
|
|
206
|
Kwon MC, Koo BK, Kim YY, Lee SH, Kim NS,
Kim JH and Kong YY: Essential role of CR6-interacting factor 1
(Crif1) in E74-like factor 3 (ELF3)-mediated intestinal
development. J Biol Chem. 284:33634–33641. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Pabst O, Zweigerdt R and Arnold HH:
Targeted disruption of the homeobox transcription factor Nkx2-3 in
mice results in postnatal lethality and abnormal development of
small intestine and spleen. Development. 126:2215–2225.
1999.PubMed/NCBI
|
|
208
|
Shikama N, Lutz W, Kretzschmar R, Sauter
N, Roth JF, Marino S, Wittwer J, Scheidweiler A and Eckner R:
Essential function of p300 acetyltransferase activity in heart,
lung and small intestine formation. EMBO J. 22:5175–5185. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Tou L, Liu Q and Shivdasani RA: Regulation
of mammalian epithelial differentiation and intestine development
by class I histone deacetylases. Mol Cell Biol. 24:3132–3139. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Grand RJ, Watkins JB and Torti FM:
Development of the human gastrointestinal tract. A review.
Gastroenterology. 70:790–810. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Kim BM, Mao J, Taketo MM and Shivdasani
RA: Phases of canonical Wnt signaling during the development of
mouse intestinal epithelium. Gastroenterology. 133:529–538. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Pinto D, Gregorieff A, Begthel H and
Clevers H: Canonical Wnt signals are essential for homeostasis of
the intestinal epithelium. Genes Dev. 17:1709–1713. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Kuhnert F, Davis CR, Wang HT, Chu P, Lee
M, Yuan J, Nusse R and Kuo CJ: Essential requirement for Wnt
signaling in proliferation of adult small intestine and colon
revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci
USA. 101:266–271. 2004. View Article : Google Scholar
|
|
214
|
Sinner D, Kordich JJ, Spence JR, Opoka R,
Rankin S, Lin SC, Jonatan D, Zorn AM and Wells JM: Sox17 and Sox4
differentially regulate beta-catenin/T-cell factor activity and
proliferation of colon carcinoma cells. Mol Cell Biol.
27:7802–7815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Wong MH, Huelsken J, Birchmeier W and
Gordon JI: Selection of multipotent stem cells during morphogenesis
of small intestinal crypts of Lieberkuhn is perturbed by
stimulation of Lef-1/beta-catenin signaling. J Biol Chem.
277:15843–15850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Jensen J, Pedersen EE, Galante P, Hald J,
Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P and Madsen
OD: Control of endodermal endocrine development by Hes-1. Nat
Genet. 24:36–44. 2000. View
Article : Google Scholar
|
|
217
|
Shroyer NF, Helmrath MA, Wang VY, Antalffy
B, Henning SJ and Zoghbi HY: Intestine-specific ablation of mouse
atonal homolog 1 (Math1) reveals a role in cellular homeostasis.
Gastroenterology. 132:2478–2488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
van Es JH, de Geest N, van de Born M,
Clevers H and Hassan BA: Intestinal stem cells lacking the Math1
tumour suppressor are refractory to Notch inhibitors. Nat Commun.
1:182010. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
VanDussen KL and Samuelson LC: Mouse
atonal homolog 1 directs intestinal progenitors to secretory cell
rather than absorptive cell fate. Dev Biol. 346:215–223. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Koo BK, Lim HS, Chang HJ, Yoon MJ, Choi Y,
Kong MP, Kim CH, Kim JM, Park JG and Kong YY: Notch signaling
promotes the generation of EphrinB1-positive intestinal epithelial
cells. Gastroenterology. 137:145–155. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Tian H, Biehs B, Chiu C, Siebel CW, Wu Y,
Costa M, de Sauvage FJ and Klein OD: Opposing activities of Notch
and Wnt signaling regulate intestinal stem cells and gut
homeostasis. Cell Rep. 11:33–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Peignon G, Durand A, Cacheux W, Ayrault O,
Terris B, Laurent-Puig P, Shroyer NF, Van Seuningen I, Honjo T,
Perret C and Romagnolo B: Complex interplay between β-catenin
signalling and Notch effectors in intestinal tumorigenesis. Gut.
60:166–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Tsuchiya K, Nakamura T, Okamoto R, Kanai T
and Watanabe M: Reciprocal targeting of Hath1 and beta-catenin by
Wnt glycogen synthase kinase 3beta in human colon cancer.
Gastroenterology. 132:208–220. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Coant N, Ben Mkaddem S, Pedruzzi E,
Guichard C, Treton X, Ducroc R, Freund JN, Cazals-Hatem D, Bouhnik
Y, Woerther PL, et al: NADPH oxidase 1 modulates WNT and NOTCH1
signaling to control the fate of proliferative progenitor cells in
the colon. Mol Cell Biol. 30:2636–2650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Calvert R, Beaulieu JF and Ménard D:
Epidermal growth factor (EGF) accelerates the maturation of fetal
mouse intestinal mucosa in utero. Experientia. 38:1096–1097. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Griffiths DF, Davies SJ, Williams D,
Williams GT and Williams ED: Demonstration of somatic mutation and
colonic crypt clonality by X-linked enzyme histochemistry. Nature.
333:461–463. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Tian Q, He XC, Hood L and Li L: Bridging
the BMP and Wnt pathways by PI3 kinase/Akt and 143-3zeta. Cell
Cycle. 4:215–216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Batlle E, Henderson JT, Beghtel H, van den
Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering
M, Pawson T and Clevers H: Beta-catenin and TCF mediate cell
positioning in the intestinal epithelium by controlling the
expression of EphB/ephrinB. Cell. 111:251–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
van der Flier LG and Clevers H: Stem
cells, self-renewal, and differentiation in the intestinal
epithelium. Annu Rev Physiol. 71:241–260. 2009. View Article : Google Scholar
|
|
230
|
Schemann M: Control of gastrointestinal
motility by the 'gut brain'-the enteric nervous system. J Pediatr
Gastroenterol Nutr. 41(Suppl 1): S4–S6. 2005. View Article : Google Scholar
|
|
231
|
Furness JB: Types of neurons in the
enteric nervous system. J Auton Nerv Syst. 81:87–96. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Goldstein AM, Thapar N, Karunaratne TB and
De Giorgio R: Clinical aspects of neurointestinal disease:
Pathophysiology, diagnosis, and treatment. Dev Biol. 417:217–228.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Kapur RP: Practical pathology and genetics
of Hirschsprung's disease. Semin Pediatr Surg. 18:212–223. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Amiel J and Lyonnet S: Hirschsprung
disease, associated syndromes, and genetics: A review. J Med Genet.
38:729–739. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Bronner ME and LeDouarin NM: Development
and evolution of the neural crest: An overview. Dev Biol. 366:2–9.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Le Douarin NM and Teillet MA: The
migration of neural crest cells to the wall of the digestive tract
in avian embryo. J Embryol Exp Morphol. 30:31–48. 1973.PubMed/NCBI
|
|
237
|
Yntema CL and Hammond WS: The origin of
intrinsic ganglia of trunk viscera from vagal neural crest in the
chick embryo. J Comp Neurol. 101:515–541. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Kuo BR and Erickson CA: Regional
differences in neural crest morphogenesis. Cell Adh Migr.
4:567–585. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Burns AJ and Le Douarin NM: Enteric
nervous system development: Analysis of the selective developmental
potentialities of vagal and sacral neural crest cells using
quail-chick chimeras. Anat Rec. 262:16–28. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Burns AJ, Champeval D and Le Douarin NM:
Sacral neural crest cells colonise aganglionic hindgut in vivo but
fail to compensate for lack of enteric ganglia. Dev Biol.
219:30–43. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Fu M, Lui VC, Sham MH, Cheung AN and Tam
PK: HOXB5 expression is spatially and temporarily regulated in
human embryonic gut during neural crest cell colonization and
differentiation of enteric neuroblasts. Dev Dyn. 228:1–10. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
242
|
De Bellard ME, Rao Y and Bronner-Fraser M:
Dual function of Slit2 in repulsion and enhanced migration of
trunk, but not vagal, neural crest cells. J Cell Biol. 162:269–279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Zuhdi N, Ortega B, Giovannone D, Ra H,
Reyes M, Asención V, McNicoll I, Ma L and de Bellard ME: Slit
molecules prevent entrance of trunk neural crest cells in
developing gut. Int J Dev Neurosci. 41:8–16. 2015. View Article : Google Scholar :
|
|
244
|
Nagy N, Brewer KC, Mwizerwa O and
Goldstein AM: Pelvic plexus contributes ganglion cells to the
hindgut enteric nervous system. Dev Dyn. 236:73–83. 2007.
View Article : Google Scholar
|
|
245
|
Young HM, Bergner AJ, Anderson RB, Enomoto
H, Milbrandt J, Newgreen DF and Whitington PM: Dynamics of neural
crest-derived cell migration in the embryonic mouse gut. Dev Biol.
270:455–473. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Allan IJ and Newgreen DF: The origin and
differentiation of enteric neurons of the intestine of the fowl
embryo. Am J Anat. 157:137–154. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Young HM and Newgreen D: Enteric neural
crest-derived cells: Origin, identification, migration, and
differentiation. Anat Rec. 262:1–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Wallace AS and Burns AJ: Development of
the enteric nervous system, smooth muscle and interstitial cells of
Cajal in the human gastrointestinal tract. Cell Tissue Res.
319:367–382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Burns AJ and Douarin NM: The sacral neural
crest contributes neurons and glia to the post-umbilical gut:
Spatiotemporal analysis of the development of the enteric nervous
system. Development. 125:4335–4347. 1998.PubMed/NCBI
|
|
250
|
Delalande JM, Barlow AJ, Thomas AJ,
Wallace AS, Thapar N, Erickson CA and Burns AJ: The receptor
tyrosine kinase RET regulates hindgut colonization by sacral neural
crest cells. Dev Biol. 313:279–292. 2008. View Article : Google Scholar
|
|
251
|
Burns AJ, Delalande JM and Le Douarin NM:
In ovo transplantation of enteric nervous system precursors from
vagal to sacral neural crest results in extensive hindgut
colonisation. Development. 129:2785–2796. 2002.PubMed/NCBI
|
|
252
|
Uesaka T, Nagashimada M and Enomoto H:
Neuronal differentiation in schwann cell lineage underlies
postnatal neurogenesis in the enteric nervous system. J Neurosci.
35:9879–9888. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Nishiyama C, Uesaka T, Manabe T, Yonekura
Y, Nagasawa T, Newgreen DF, Young HM and Enomoto H:
Trans-mesenteric neural crest cells are the principal source of the
colonic enteric nervous system. Nat Neurosci. 15:1211–1218. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Peters-van der Sanden MJ, Kirby ML,
Gittenberger-de Groot A, Tibboel D, Mulder MP and Meijers C:
Ablation of various regions within the avian vagal neural crest has
differential effects on ganglion formation in the fore-, mid- and
hindgut. Dev Dyn. 196:183–194. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Druckenbrod NR and Epstein ML: The pattern
of neural crest advance in the cecum and colon. Dev Biol.
287:125–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Young HM, Bergner AJ, Simpson MJ, McKeown
SJ, Hao MM, Anderson CR and Enomoto H: Colonizing while migrating:
How do individual enteric neural crest cells behave? BMC Biol.
12:232014. View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Anderson RB, Turner KN, Nikonenko AG,
Hemperly J, Schachner M and Young HM: The cell adhesion molecule l1
is required for chain migration of neural crest cells in the
developing mouse gut. Gastroenterology. 130:1221–1232. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Druckenbrod NR and Epstein ML:
Age-dependent changes in the gut environment restrict the invasion
of the hindgut by enteric neural progenitors. Development.
136:3195–3203. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Simpson MJ, Zhang DC, Mariani M, Landman
KA and Newgreen DF: Cell proliferation drives neural crest cell
invasion of the intestine. Dev Biol. 302:553–568. 2007. View Article : Google Scholar
|
|
260
|
Carmona-Fontaine C, Matthews HK, Kuriyama
S, Moreno M, Dunn GA, Parsons M, Stern CD and Mayor R: Contact
inhibition of locomotion in vivo controls neural crest directional
migration. Nature. 456:957–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Nagy N and Goldstein AM: Enteric nervous
system development: A crest cell's journey from neural tube to
colon. Semin Cell Dev Biol. 66:94–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Gabriel SB, Salomon R, Pelet A, Angrist M,
Amiel J, Fornage M, Attié-Bitach T, Olson JM, Hofstra R, Buys C, et
al: Segregation at three loci explains familial and population risk
in Hirschsprung disease. Nat Genet. 31:89–93. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Emison ES, McCallion AS, Kashuk CS, Bush
RT, Grice E, Lin S, Portnoy ME, Cutler DJ, Green ED and Chakravarti
A: A common sex-dependent mutation in a RET enhancer underlies
Hirschsprung disease risk. Nature. 434:857–863. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Southard-Smith EM, Kos L and Pavan WJ:
Sox10 mutation disrupts neural crest development in Dom
Hirschsprung mouse model. Nat Genet. 18:60–64. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
265
|
Pattyn A, Morin X, Cremer H, Goridis C and
Brunet JF: The homeobox gene Phox2b is essential for the
development of autonomic neural crest derivatives. Nature.
399:366–370. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
266
|
Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis
V, Takahashi M and Costantini F: Targeted mutation of serine 697 in
the Ret tyrosine kinase causes migration defect of enteric neural
crest cells. Development. 133:4507–4516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
267
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Mograbi B, Bocciardi R, Bourget I, Busca
R, Rochet N, Farahi-Far D, Juhel T and Rossi B: Glial cell
line-derived neurotrophic factor-stimulated phosphatidylinositol
3-kinase and Akt activities exert opposing effects on the ERK
pathway: Importance for the rescue of neuroectodermic cells. J Biol
Chem. 276:45307–45319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
269
|
Natarajan D, Marcos-Gutierrez C, Pachnis V
and de Graaff E: Requirement of signalling by receptor tyrosine
kinase RET for the directed migration of enteric nervous system
progenitor cells during mammalian embryogenesis. Development.
129:5151–5160. 2002.PubMed/NCBI
|
|
270
|
Fu M, Sato Y, Lyons-Warren A, Zhang B,
Kane MA, Napoli JL and Heuckeroth RO: Vitamin A facilitates enteric
nervous system precursor migration by reducing Pten accumulation.
Development. 137:631–640. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
271
|
Simkin JE, Zhang D, Rollo BN and Newgreen
DF: Retinoic acid upregulates ret and induces chain migration and
population expansion in vagal neural crest cells to colonise the
embryonic gut. PLoS One. 8:e640772013. View Article : Google Scholar : PubMed/NCBI
|
|
272
|
Taketomi T, Yoshiga D, Taniguchi K,
Kobayashi T, Nonami A, Kato R, Sasaki M, Sasaki A, Ishibashi H,
Moriyama M, et al: Loss of mammalian Sprouty2 leads to enteric
neuronal hyper-plasia and esophageal achalasia. Nat Neurosci.
8:855–857. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
273
|
Zhou R, Niwa S, Homma N, Takei Y and
Hirokawa N: KIF26A is an unconventional kinesin and regulates
GDNF-Ret signaling in enteric neuronal development. Cell.
139:802–813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Barlow A, de Graaff E and Pachnis V:
Enteric nervous system progenitors are coordinately controlled by
the G protein-coupled receptor EDNRB and the receptor tyrosine
kinase RET. Neuron. 40:905–916. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
275
|
Taraviras S, Marcos-Gutierrez CV, Durbec
P, Jani H, Grigoriou M, Sukumaran M, Wang LC, Hynes M, Raisman G
and Pachnis V: Signalling by the RET receptor tyrosine kinase and
its role in the development of the mammalian enteric nervous
system. Development. 126:2785–2797. 1999.PubMed/NCBI
|
|
276
|
Schuchardt A, D'Agati V, Larsson-Blomberg
L, Costantini F and Pachnis V: Defects in the kidney and enteric
nervous system of mice lacking the tyrosine kinase receptor Ret.
Nature. 367:380–383. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Uesaka T, Nagashimada M, Yonemura S and
Enomoto H: Diminished Ret expression compromises neuronal survival
in the colon and causes intestinal aganglionosis in mice. J Clin
Invest. 118:1890–1898. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
278
|
Mwizerwa O, Das P, Nagy N, Akbareian SE,
Mably JD and Goldstein AM: Gdnf is mitogenic, neurotrophic, and
chemoattractive to enteric neural crest cells in the embryonic
colon. Dev Dyn. 240:1402–1411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
279
|
Uesaka T, Nagashimada M and Enomoto H:
GDNF signaling levels control migration and neuronal
differentiation of enteric ganglion precursors. J Neurosci.
33:16372–16382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
280
|
Flynn B, Bergner AJ, Turner KN, Young HM
and Anderson RB: Effect of Gdnf haploinsufficiency on rate of
migration and number of enteric neural crest-derived cells. Dev
Dyn. 236:134–141. 2007. View Article : Google Scholar
|
|
281
|
Nagy N and Goldstein AM: Endothelin-3
regulates neural crest cell proliferation and differentiation in
the hindgut enteric nervous system. Dev Biol. 293:203–217. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Baynash AG, Hosoda K, Giaid A, Richardson
JA, Emoto N, Hammer RE and Yanagisawa M: Interaction of
endothelin-3 with endothelin-B receptor is essential for
development of epidermal melanocytes and enteric neurons. Cell.
79:1277–1285. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
283
|
Carrasquillo MM, McCallion AS,
Puffenberger EG, Kashuk CS, Nouri N and Chakravarti A: Genome-wide
association study and mouse model identify interaction between RET
and EDNRB pathways in Hirschsprung disease. Nat Genet. 32:237–244.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
284
|
McCallion AS, Stames E, Conlon RA and
Chakravarti A: Phenotype variation in two-locus mouse models of
Hirschsprung disease: Tissue-specific interaction between Ret and
Ednrb. Proc Natl Acad Sci USA. 100:1826–1831. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
285
|
Hearn CJ, Murphy M and Newgreen D: GDNF
and ET-3 differentially modulate the numbers of avian enteric
neural crest cells and enteric neurons in vitro. Dev Biol.
197:93–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
286
|
Paratore C, Eichenberger C, Suter U and
Sommer L: Sox10 haploinsufficiency affects maintenance of
progenitor cells in a mouse model of Hirschsprung disease. Hum Mol
Genet. 11:3075–3085. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
287
|
Kapur RP: Early death of neural crest
cells is responsible for total enteric aganglionosis in
Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatr Dev Pathol. 2:559–569.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
288
|
Lang D, Chen F, Milewski R, Li J, Lu MM
and Epstein JA: Pax3 is required for enteric ganglia formation and
functions with Sox10 to modulate expression of c-ret. J Clin
Invest. 106:963–971. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
289
|
Zhu L, Lee HO, Jordan CS, Cantrell VA,
Southard-Smith EM and Shin MK: Spatiotemporal regulation of
endothelin receptor-B by SOX10 in neural crest-derived enteric
neuron precursors. Nat Genet. 36:732–737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
290
|
Anderson RB, Stewart AL and Young HM:
Phenotypes of neural-crest-derived cells in vagal and sacral
pathways. Cell Tissue Res. 323:11–25. 2006. View Article : Google Scholar
|
|
291
|
Sasselli V, Pachnis V and Burns AJ: The
enteric nervous system. Dev Biol. 366:64–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
292
|
Nagashimada M, Ohta H, Li C, Nakao K,
Uesaka T, Brunet JF, Amiel J, Trochet D, Wakayama T and Enomoto H:
Autonomic neurocristopathy-associated mutations in PHOX2B
dysregulate Sox10 expression. J Clin Invest. 122:3145–3158. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
293
|
D'Autréaux F, Morikawa Y, Cserjesi P and
Gershon MD: Hand2 is necessary for terminal differentiation of
enteric neurons from crest-derived precursors but not for their
migration into the gut or for formation of glia. Development.
134:2237–2249. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
294
|
D'Autréaux F, Margolis KG, Roberts J,
Stevanovic K, Mawe G, Li Z, Karamooz N, Ahuja A, Morikawa Y,
Cserjesi P, et al: Expression level of Hand2 affects specification
of enteric neurons and gastrointestinal function in mice.
Gastroenterology. 141:576–587. 587.e1–e6. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
295
|
Fu M, Vohra BP, Wind D and Heuckeroth RO:
BMP signaling regulates murine enteric nervous system precursor
migration, neurite fasciculation, and patterning via altered Ncam1
polysialic acid addition. Dev Biol. 299:137–150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
296
|
Chalazonitis A, Pham TD, Li Z, Roman D,
Guha U, Gomes W, Kan L, Kessler JA and Gershon MD: Bone
morphogenetic protein regulation of enteric neuronal phenotypic
diversity: Relationship to timing of cell cycle exit. J Comp
Neurol. 509:474–492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
297
|
Chalazonitis A, D'Autréaux F, Pham TD,
Kessler JA and Gershon MD: Bone morphogenetic proteins regulate
enteric gliogenesis by modulating ErbB3 signaling. Dev Biol.
350:64–79. 2011. View Article : Google Scholar :
|
|
298
|
Chalazonitis A, Pham TD, Rothman TP,
DiStefano PS, Bothwell M, Blair-Flynn J, Tessarollo L and Gershon
MD: Neurotrophin-3 is required for the survival-differentiation of
subsets of developing enteric neurons. J Neurosci. 21:5620–5636.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
299
|
Payette RF, Tennyson VM, Pomeranz HD, Pham
TD, Rothman TP and Gershon MD: Accumulation of components of basal
laminae: Association with the failure of neural crest cells to
colonize the presumptive aganglionic bowel of ls/ls mutant mice.
Dev Biol. 125:341–360. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
300
|
Parikh DH, Tam PK, Van Velzen D and Edgar
D: Abnormalities in the distribution of laminin and collagen type
IV in Hirschsprung's disease. Gastroenterology. 102:1236–1241.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
301
|
Akbareian SE, Nagy N, Steiger CE, Mably
JD, Miller SA, Hotta R, Molnar D and Goldstein AM: Enteric neural
crest-derived cells promote their migration by modifying their
microenvironment through tenascin-C production. Dev Biol.
382:446–456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
302
|
Nagy N, Mwizerwa O, Yaniv K, Carmel L,
Pieretti-Vanmarcke R, Weinstein BM and Goldstein AM: Endothelial
cells promote migration and proliferation of enteric neural crest
cells via beta1 integrin signaling. Dev Biol. 330:263–272. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
303
|
Breau MA, Dahmani A, Broders-Bondon F,
Thiery JP and Dufour S: Beta1 integrins are required for the
invasion of the caecum and proximal hindgut by enteric neural crest
cells. Development. 136:2791–2801. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
304
|
Soret R, Mennetrey M, Bergeron KF, Dariel
A, Neunlist M, Grunder F, Faure C, Silversides DW and Pilon N: A
collagen VI-dependent pathogenic mechanism for Hirschsprung's
disease. J Clin Invest. 125:4483–4496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
305
|
Nagy N, Barad C, Graham HK, Hotta R, Cheng
LS, Fejszak N and Goldstein AM: Sonic hedgehog controls enteric
nervous system development by patterning the extracellular matrix.
Development. 143:264–275. 2016. View Article : Google Scholar :
|
|
306
|
Chevalier NR, Gazguez E, Bidault L,
Guilbert T, Vias C, Vian E, Watanabe Y, Muller L, Germain S,
Bondurand N, et al: How tissue mechanical properties affect enteric
neural crest cell migration. Sci Rep. 6:209272016. View Article : Google Scholar : PubMed/NCBI
|
|
307
|
Anderson RB: Matrix metalloproteinase-2 is
involved in the migration and network formation of enteric neural
crest-derived cells. Int J Dev Biol. 54:63–69. 2010. View Article : Google Scholar
|
|
308
|
Nagy N, Burns AJ and Goldstein AM:
Immunophenotypic char-acterization of enteric neural crest cells in
the developing avian colorectum. Dev Dyn. 241:842–851. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
309
|
Jackson SR, Guner YS, Woo R, Randolph LM,
Ford H and Shin CE: L1CAM mutation in association with X-linked
hydrocephalus and Hirschsprung's disease. Pediatr Surg Int.
25:823–825. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
310
|
Broders-Bondon F, Paul-Gilloteaux P,
Carlier C, Radice GL and Dufour S: N-cadherin and β1-integrins
cooperate during the development of the enteric nervous system. Dev
Biol. 364:178–191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
311
|
Jiang Y, Liu MT and Gershon MD: Netrins
and DCC in the guidance of migrating neural crest-derived cells in
the developing bowel and pancreas. Dev Biol. 258:364–384. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
312
|
Spouge D and Baird PA: Hirschsprung
disease in a large birth cohort. Teratology. 32:171–177. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
313
|
Brooks AS, Oostra BA and Hofstra RM:
Studying the genetics of Hirschsprung's disease: Unraveling an
oligogenic disorder. Clin Genet. 67:6–14. 2005. View Article : Google Scholar
|
|
314
|
Griseri P, Lantieri F, Puppo F, Bachetti
T, Di Duca M, Ravazzolo R and Ceccherini I: A common variant
located in the 3'UTR of the RET gene is associated with protection
from Hirschsprung disease. Hum Mutat. 28:168–176. 2007. View Article : Google Scholar
|
|
315
|
Gianino S, Grider JR, Cresswell J, Enomoto
H and Heuckeroth RO: GDNF availability determines enteric neuron
number by controlling precursor proliferation. Development.
130:2187–2198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
316
|
Bondurand N, Natarajan D, Barlow A, Thapar
N and Pachnis V: Maintenance of mammalian enteric nervous system
progenitors by SOX10 and endothelin 3 signalling. Development.
133:2075–2086. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
317
|
Hosoda K, Hammer RE, Richardson JA,
Baynash AG, Cheung JC, Giaid A and Yanagisawa M: Targeted and
natural (piebald-lethal) mutations of endothelin-B receptor gene
produce megacolon associated with spotted coat color in mice. Cell.
79:1267–1276. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
318
|
Leibl MA, Ota T, Woodward MN, Kenny SE,
Lloyd DA, Vaillant CR and Edgar DH: Expression of endothelin 3 by
mesenchymal cells of embryonic mouse caecum. Gut. 44:2461999.
View Article : Google Scholar : PubMed/NCBI
|
|
319
|
Stanchina L, Baral V, Robert F, Pingault
V, Lemort N, Pachnis V, Goossens M and Bondurand N: Interactions
between Sox10, Edn3 and Ednrb during enteric nervous system and
melanocyte development. Dev Biol. 295:232–249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
320
|
Barlow AJ, Dixon J, Dixon M and Trainor
PA: Tcof1 acts as a modifier of Pax3 during enteric nervous system
development and in the pathogenesis of colonic aganglionosis. Hum
Mol Genet. 22:1206–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
321
|
Yamada K, Yamada Y, Nomura N, Miura K,
Wakako R, Hayakawa C, Matsumoto A, Kumagai T, Yoshimura I, Miyazaki
S, et al: Nonsense and frameshift mutations in ZFHX1B, encoding
Smad-interacting protein 1, cause a complex developmental disorder
with a great variety of clinical features. Am J Hum Genet.
69:1178–1185. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
322
|
Elworthy S, Pinto JP, Pettifer A, Cancela
ML and Kelsh RN: Phox2b function in the enteric nervous system is
conserved in zebrafish and is sox10-dependent. Mech Dev.
122:659–669. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
323
|
Goldstein AM, Brewer KC, Doyle AM, Nagy N
and Roberts DJ: BMP signaling is necessary for neural crest cell
migration and ganglion formation in the enteric nervous system.
Mech Dev. 122:821–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
324
|
Pingault V, Bondurand N, Kuhlbrodt K,
Goerich DE, Préhu MO, Puliti A, Herbarth B, Hermans-Borgmeyer I,
Legius E, Matthijs G, et al: SOX10 mutations in patients with
Waardenburg-Hirschsprung disease. Nat Genet. 18:171–173. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
325
|
Niederreither K, Vermot J, Le Roux I,
Schuhbaur B, Chambon P and Dollé P: The regional pattern of
retinoic acid synthesis by RALDH2 is essential for the development
of posterior pharyngeal arches and the enteric nervous system.
Development. 130:2525–2534. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
326
|
Xiao JB, Leng AM, Zhang YQ, Wen Z, He J
and Ye GN: CUEDC2: Multifunctional roles in carcinogenesis. Front
Biosci (Landmark Ed). 24:935–946. 2019. View Article : Google Scholar
|
|
327
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
328
|
Sokic-Milutinovic A: Appropriate
management of attenuated familial adenomatous polyposis: Report of
a case and review of the literature. Dig Dis. 37:400–405. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
329
|
Schuster-Böckler B and Lehner B: Chromatin
organization is a major influence on regional mutation rates in
human cancer cells. Nature. 488:504–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
330
|
Khavari DA, Sen GL and Rinn JL: DNA
methylation and epigenetic control of cellular differentiation.
Cell Cycle. 9:3880–3883. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
331
|
Aranda P, Agirre X, Ballestar E, Andreu
EJ, Román-Gómez J, Prieto I, Martin-Subero JI, Cigudosa JC, Siebert
R, Esteller M and Prosper F: Epigenetic signatures associated with
different levels of differentiation potential in human stem cells.
PLoS One. 4:e78092009. View Article : Google Scholar : PubMed/NCBI
|
|
332
|
Pierce GB: Neoplasms, differentiations and
mutations. Am J Pathol. 77:103–118. 1974.PubMed/NCBI
|
|
333
|
Pierce GB: The cancer cell and its control
by the embryo. Rous-whipple award lecture. Am J Pathol.
113:117–124. 1983.PubMed/NCBI
|
|
334
|
Hu M and Shivdasani RA: Overlapping gene
expression in fetal mouse intestine development and human
colorectal cancer. Cancer Res. 65:8715–8722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
335
|
Kaiser S, Park YK, Franklin JL, Halberg
RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, et
al: Transcriptional recapitulation and subversion of embryonic
colon development by mouse colon tumor models and human colon
cancer. Genome Biol. 8:R1312007. View Article : Google Scholar : PubMed/NCBI
|
|
336
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: Parallels between
normal development and tumor progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
337
|
Arney KL and Fisher AG: Epigenetic aspects
of differentiation. J Cell Sci. 117:4355–4363. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
338
|
Lemaire P, Darras S, Caillol D and
Kodjabachian L: A role for the vegetally expressed Xenopus gene
Mix.1 in endoderm formation and in the restriction of mesoderm to
the marginal zone. Development. 125:2371–2380. 1998.PubMed/NCBI
|
|
339
|
Costello I, Biondi CA, Taylor JM, Bikoff
EK and Robertson EJ: Smad4-dependent pathways control basement
membrane deposition and endodermal cell migration at early stages
of mouse development. BMC Dev Biol. 9:542009. View Article : Google Scholar : PubMed/NCBI
|
|
340
|
Abdul Khalek FJ, Gallicano GI and Mishra
L: Colon cancer stem cells. Gastrointest Cancer Res. (Suppl 1):
S16–S23. 2010.
|
|
341
|
Leedham SJ, Brittan M, McDonald SA and
Wright NA: Intestinal stem cells. J Cell Mol Med. 9:11–24. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
342
|
Gostjeva EV, Zukerberg L, Chung D and
Thilly WG: Bell-shaped nuclei dividing by symmetrical and
asymmetrical nuclear fission have qualities of stem cells in human
colonic embryogenesis and carcinogenesis. Cancer Genet Cytogenet.
164:16–24. 2006. View Article : Google Scholar
|
|
343
|
Manzo G: Similarities between embryo
development and cancer process suggest new strategies for research
and therapy of tumors: A new point of view. Front Cell Dev Biol.
7:202019. View Article : Google Scholar : PubMed/NCBI
|
|
344
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
345
|
Moon BS, Jeong WJ, Park J, Kim TI, Min do
S and Choi KY: Role of oncogenic K-Ras in cancer stem cell
activation by aberrant Wnt/β-catenin signaling. J atl Cancer Inst.
106:djt3732014.
|
|
346
|
Todaro M, Gaggianesi M, Catalano V,
Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S,
Cocorullo G, et al: CD44v6 is a marker of constitutive and
reprogrammed cancer stem cells driving colon cancer metastasis.
Cell Stem Cell. 14:342–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
347
|
Lopes EC, Valls E, Figueroa ME, Mazur A,
Meng FG, Chiosis G, Laird PW, Schreiber-Agus N, Greally JM,
Prokhortchouk E and Melnick A: Kaiso contributes to DNA
methylation-dependent silencing of tumor suppressor genes in colon
cancer cell lines. Cancer Res. 68:7258–7263. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
348
|
Ouko L, Ziegler TR, Gu LH, Eisenberg LM
and Yang VW: Wnt11 signaling promotes proliferation,
transformation, and migration of IEC6 intestinal epithelial cells.
J Biol Chem. 279:26707–26715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
349
|
Pan D: The hippo signaling pathway in
development and cancer. Dev Cell. 19:491–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
350
|
Takaku K, Oshima M, Miyoshi H, Matsui M,
Seldin MF and Taketo MM: Intestinal tumorigenesis in compound
mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 92:645–656.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
351
|
Lombardo Y, Scopelliti A, Cammareri P,
Todaro M, Iovino F, Ricci-Vitiani L, Gulotta G, Dieli F, de Maria R
and Stassi G: Bone morphogenetic protein 4 induces differentiation
of colorectal cancer stem cells and increases their response to
chemotherapy in mice. Gastroenterology. 140:297–309. 2011.
View Article : Google Scholar
|
|
352
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
353
|
De Craene B, Gilbert B, Stove C, Bruyneel
E, van Roy F and Berx G: The transcription factor snail induces
tumor cell invasion through modulation of the epithelial cell
differentiation program. Cancer Res. 65:6237–6244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
354
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
355
|
Pellegrinet L, Rodilla V, Liu Z, Chen S,
Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J and Radtke F:
Dll1- and dll4-mediated notch signaling are required for
homeostasis of intestinal stem cells. Gastroenterology.
140:1230–1240. e1–e7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
356
|
Hwang WL, Jiang JK, Yang SH, Huang TS, Lan
HY, Teng HW, Yang CY, Tsai YP, Lin CH, Wang HW and Yang MH:
MicroRNA-146a directs the symmetric division of Snail-dominant
colorectal cancer stem cells. Nat Cell Biol. 16:268–280. 2014.
View Article : Google Scholar : PubMed/NCBI
|














