Introduction
Myocardial fibrosis is a common pathological feature
of multiple end-stage cardiovascular diseases, including
hypertension, advanced coronary heart disease and cardio-myopathy
(1). Fibrosis is defined by
overproliferation and activation of cardiac fibroblasts (CFs), and
accumulation of extracellular matrix (ECM) components secreted by
activated fibroblasts (2,3). Therefore, prevention of CF
proliferation and abrogation of CF trans-differentiation into
myofibroblasts may become an effective strategy for treating
cardiac fibrosis.
Transforming growth factor-β1 (TGF-β1) is a major
pro-fibrotic factor (2,4). It can stimulate the proliferation of
CFs and the differentiation of CFs into myofibroblasts, which are
characterized by the upregulation of α-smooth muscle actin (α-SMA)
and the secretion of ECM proteins, such as collagen I and collagen
III (5,6). TGF-β1 ligand initiates a signaling
cascade through cell-surface receptors and intracellular Smad
signal proteins, such as the canonical signal transducer-Smad2/3
protein, whose activation is associated with the transcription of
numerous pro-fibrotic genes (7,8).
Furthermore, TGF-β1 can induce other non-canonical signaling
cascades independently of Smads, such as mitogen-activated protein
kinase (MAPK) signaling pathways, including extracellular
signal-regulated kinase 1/2 (ERK1/2), p38 MAPK and c-Jun N-terminal
kinase (JNK) (9). Accumulating
evidence has demonstrated that MAPK signaling plays a critical role
in ECM synthesis and in the proliferation of CFs (10,11). Thus, pharmacological interventions
in these signaling pathways could be considered as a promising
therapeutic strategy against cardiac fibrosis.
Statins, which are inhibitors of the
3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, have been
extensively applied to treat patients with hypercholesterolemia in
the past decade due to their lipid-reducing effects (12). Furthermore, statins have also been
reported to possess pleiotropic cardiovascular effects, including
reduced oxidative stress and inflammation, decreased platelet
adhesion, improved endothelial function and enhanced
atherosclerotic plaque stability (13-15). In addition, previous studies have
shown that atorvastatin (ATV), a member of the statin family,
exerts potential anti-myocardial fibrotic properties (16-20). Nevertheless, to the best of our
knowledge, the molecular mechanism involved in the effect of ATV on
cardiac fibrosis remains to be clarified, and limited information
is available concerning the possible impact of ATV on
TGF-β1-induced human ventricular fibroblast (hVF) proliferation and
myofibroblast differentiation. Thus, the aim of the present study
was to investigate the impact of ATV on TGF-β1-stimulated
fibrogenesis and its underlying molecular mechanism in hVFs. The
findings indicated that ATV prevents TGF-β1-induced fibrogenesis in
hVFs, at least in part, by inhibiting the Smad3 and MAPK signaling
pathways.
Materials and methods
Cells, chemicals and reagents
Adult hVFs (cat. no. 6310) and complete medium for
their culture (fibroblast medium-2; FM-2; cat. no. 2331) were
obtained from ScienCell Research Laboratories, Inc. Antibodies
against α-SMA, matrix metal-loproteinase-2 (MMP-2), collagen I and
collagen III were purchased from ProteinTech Group, Inc. Antibodies
against phosphorylated (p)-Smad3, total (t)-Smad3, p-ERK1/2,
t-ERK1/2, p-JNK, t-JNK, p-p38 and t-p38 were purchased from Cell
Signaling Technology, Inc. Anti-GAPDH antibody and Cell Counting
Kit-8 (CCK-8) were purchased from Beyotime Institute of
Biotechnology. ATV was obtained from Sigma-Aldrich (Merck KGaA).
Recombinant human TGF-β1 cytokine was purchased from PeproTech,
Inc.
Cell culture
hVFs were maintained in FM-2 supplemented with 5%
fetal bovine serum (cat. no. 0025; ScienCell Research Laboratories,
Inc.) and 1% penicillin-streptomycin in a humidified atmosphere
with 5% CO2 at 37°C. The cells were passaged ≤8 times
and were cultured for 16 h in serum-free medium before treatment.
In the present study, an in vitro cardiac fibrosis model was
established by treatment of hVFs with TGF-β1 (5 ng/ml) for 24 h at
37°C in a humidified atmosphere with 5% CO2, according
to the previous literature (3,21).
Cell viability assays
The survival rate of the cells was deter-mined by
CCK-8 assay according to the manufacturer's protocol. In brief,
fibroblasts (1×104 cells/well) were seeded into 96-well
plates until ~90% confluence. Subsequently, cells were subjected to
serum starvation for 16 h, followed by treatment with ATV (0, 2, 5,
10, 20 and 30 µM) for 24 h at 37°C. Following treatment, the
cells were washed with PBS, and then 10 µl CCK-8 was added
to each well and incubated for an additional 4 h at 37°C. Next, the
absorbance of each well at 450 nm was examined using an automatic
microplate reader (TECAN M200; Tecan Group, Ltd.). The results were
expressed as percentages of the control group (cells were treated
with an equal amount of DMSO).
Cell proliferation assay
hVFs were seeded into 96-well plates at
2×103 cells/well and cultured overnight. Cells were
deprived of serum for 16 h before being pretreated with ATV (2, 5
and 10 µM) or DMSO for 3 h, followed by stimulation with
TGF-β1 (5 ng/ml) for 24 h in a humidified atmosphere with 5%
CO2 at 37°C. Subsequently, the medium was removed from
each well, and CCK-8 solution (10 µl/well) was added into
the wells, followed by incubation for an additional 4 h at 37°C.
Finally, the optical density of each well was detected at an
absorbance wavelength of 450 nm using the aforementioned microplate
reader.
Western blotting
hVFs (5×105 cells/well) were plated into
6-well plates, cultured to ~80% confluence and starved of serum for
16 h. Then, the cells were pretreated for 3 h with ATV (10
µM) or DMSO before treatment with 5 ng/ml TGF-β1 for 24 h at
37°C. Subsequently, the cells were washed twice with cold PBS and
lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology) at
4°C. Lysates were centrifuged at 13,000 × g for 15 min at 4°C, and
protein concentrations were determined by BCA assay (cat. no.
23227; Pierce; Thermo Fisher Scientific, Inc.). Cell lysates (30
µg) were separated by 8-10% SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane (Merck KGaA). The membranes were
blocked with blocking buffer (5% nonfat dry milk in TBS plus 0.1%
Tween-20) for 1 h at room temperature and incubated with primary
antibodies overnight at 4°C, including antibodies against α-SMA
(1:1,000; cat. no. 14395-1-AP), MMP-2 (1:1,000; cat. no.
10373-2-AP), collagen I (1:2,000; cat. no. 14695-1-AP), collagen
III (1:1,000; cat. no. 22734-1-AP), p-Smad3 (1:1,000; cat. no.
9520), t-Smad3 (1:1,000; cat. no. 9523), p-ERK1/2 (1:1,000; cat.
no. 9101), t-ERK1/2 (1:1,000; cat. no. 9102), p-JNK (1:1,000; cat.
no. 4671), t-JNK (1:1,000; cat. no. 9252), p-p38 (1:1,000; cat. no.
4511), t-p38 (1:1,000; cat. no. 9212) and GAPDH (1:5,000; cat. no.
AG019). Following washing three times with washing buffer (TBS
containing 0.1% Tween-20), the membranes were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibodies
[HRP-conjugated goat anti-rabbit IgG, 1:2,000, cat. no. D110058;
HRP-conjugated goat anti-mouse IgG, 1:2,000, cat. no. D110087; all
from Sangon Biotech (Shanghai) Co., Ltd.] for 1 h at 25°C. The
protein signals were detected using Immobilon ECL HRP Substrate
(Merck KGaA), and images were acquired using Chemidoc XRS (Bio-Rad
Laboratories, Inc.). Image Pro-Plus 6.0 software (Media
Cybernetics, Inc.) was used to quantify the density of the
bands.
Immunofluorescence staining
In order to detect the effect of ATV on the
trans-differentiation of hVFs into activated myofibroblasts, cells
were stained with the fibroblast activation marker α-SMA. The
fibroblasts (1x104 cells/well) were cultured on
coverslips in the plate and were subjected to different treatments
as follows: i) Control group (DMSO treatment); ii) TGF-β1 group;
iii) ATV group; and iv) TGF-β1 combined with ATV group. Following
treatment, cells were washed three times with cold PBS, treated
with 4% paraformaldehyde for 20 min at room temperature and
permeabilized with 0.2% Triton-X 100 for 15 min at room
temperature. Following blocking with 5% bovine serum albumin [cat.
no. A602449; Sangon Biotech (Shanghai) Co., Ltd.] in PBS for 1 h at
room temperature, the slides were stained with the aforementioned
primary antibody against α-SMA (1:100) overnight at 4°C.
Subsequently, an Alexa Fluor 488-conjugated secondary antibody
(1:400; cat. no. A11008; Invitrogen; Thermo Fisher Scientific,
Inc.) was used to probe the primary antibody, and the nuclei were
stained with DAPI at room temperature for 5 min. Lastly, the cells
were covered with antifade mounting medium (cat. no. MM1401;
Shanghai Maokang Biotechnology Co., Ltd.), and images were captured
using an Olympus fluorescence microscope (Olympus Corporation;
magnification, x200).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using NucleoZOL
reagent (cat. no. 740404.200; Macherey-Nagel GmbH) and was reverse
transcribed into cDNA with PrimeScript™ RT Master mix (cat. no.
RR036A; Takara Biotechnology Co., Ltd.) according to the
manufacturer's protocol. Firstly, ordinary PCR was performed using
Rapid Taq Master mix (cat. no. P222; Vazyme Biotech Co., Ltd),
according to the manufacturer's protocol, to ensure that the PCR
product of each primer pair was specific. The temper-ature
conditions were 95°C for 3 min, followed by 30 cycles of 95°C for
15 sec, 60°C for 15 sec and 72°C for 15 sec, and a final extension
at 72°C for 5 min. Then, qPCR was performed on a Bio-Rad CFX
Connect Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.)
using SYBR Green Supermix (cat. no. RR820A; Takara Biotechnology
Co., Ltd.). The thermocy-cling conditions for qPCR were as follows:
Initial denaturation at 95°C for 30 sec; followed by 40 cycles of
95°C for 5 sec, 60°C for 30 sec and 72°C for 1 min. GAPDH was used
as a reference gene for normalization. The relative gene
expressions were calculated using the 2-ΔΔCq method
(22). The primer sequences are
presented in Table I.
 | Table ISequences of primers used for reverse
transcription-quantitative PCR. |
Table I
Sequences of primers used for reverse
transcription-quantitative PCR.
| Gene | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| α-SMA C |
TATGAGGGCTATGCCTTGCC |
GCTCAGCAGTAGTAACGAAGGA |
| MMP-2 |
GATACCCCTTTGACGGTAAGGA |
CCTTCTCCCAAGGTCCATAGC |
| COL-I |
GAGGGCCAAGACGAAGACATC C |
AGATCACGTCATCGCACAAC |
| COL-III |
GCCAAATATGTGTCTGTGACTCA |
GGGCGAGTAGGAGCAGTTG |
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCATGG |
Statistical analysis
All experiments were performed indepen-dently a
minimum of three times. The data are presented as the mean ±
standard error of mean. Statistical analysis was performed using
GraphPad Prism 7.0 software (GraphPad Software, Inc.). Statistical
differences were assessed using one-way analysis of variance
followed by Tukey's post hoc test for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ATV suppresses TGF-β1-induced
proliferation of hVFs
Firstly, to investigate the cytotoxic effect of ATV
on hVFs, cells were incubated with ATV for 24 h at different
concentrations (0, 2, 5, 10, 20 and 30 µM), and then cell
viability was detected using a CCK-8 assay. As presented in
Fig. 1A, ATV did not induce
cytotoxicity at concentrations ≤10 µM. Subsequently, to
assess the effect of ATV on the TGF-β1-stimulated proliferation of
hVFs, cells were exposed to 5 ng/ml TGF-β1 for 24 h after
pre-incubation with ATV (0, 2, 5 and 10 µM) for 3 h. The
rate of proliferation was then detected using the CCK-8 assay and
compared with that of the control group. The results demon-strated
that TGF-β1 significantly increased the proliferation of hVFs.
However, pre-incubation with ATV (10 µM) signifi-cantly
attenuated the increase in cell proliferation induced by TGF-β1
(Fig. 1B). Therefore, 10
µM was selected as the concentration of ATV for the
following experiments.
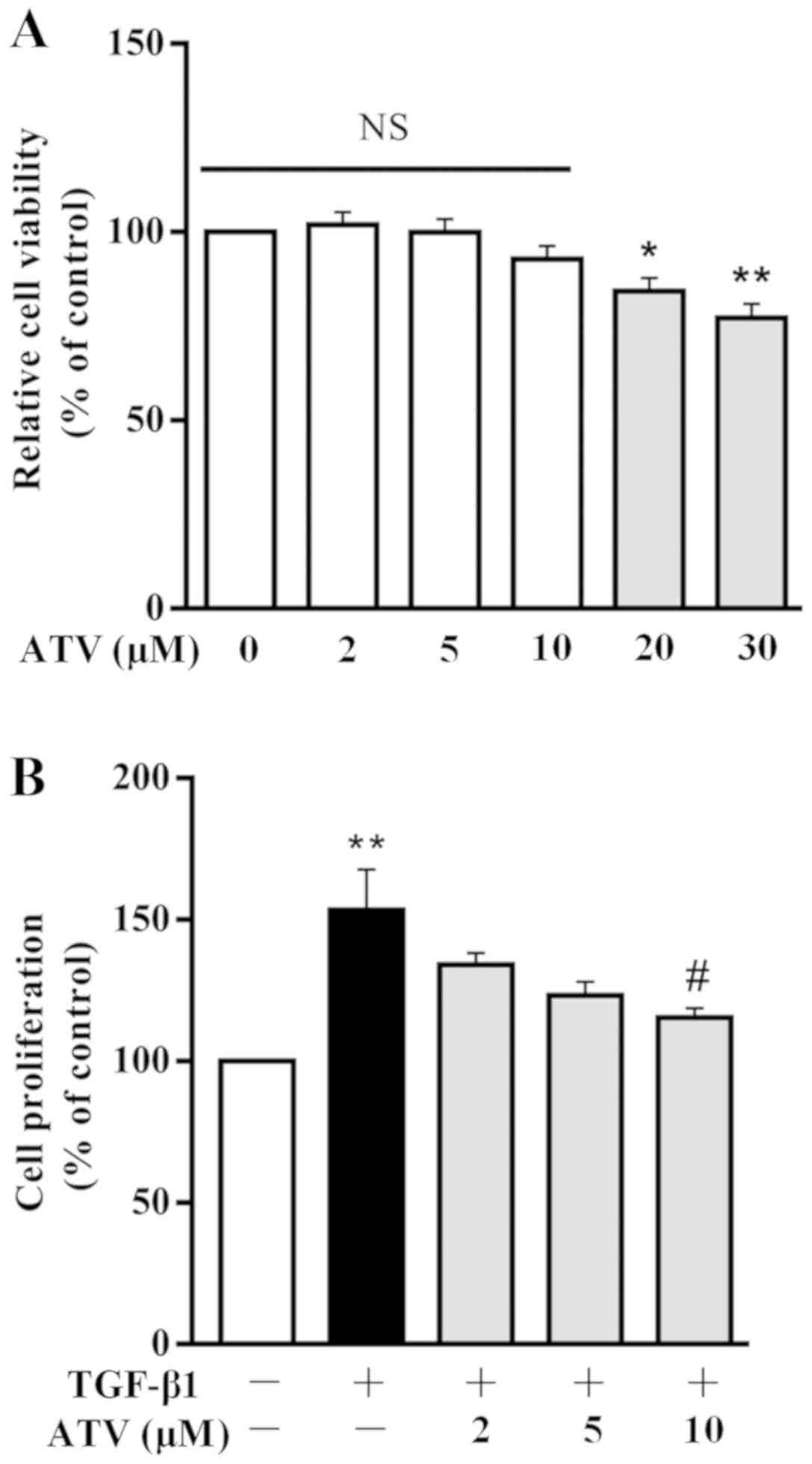 | Figure 1Effect of ATV on TGF-β1-induced
proliferation of hVFs. (A) Cells were exposed to different
concentrations of ATV, and then the viability of the cells was
assessed using a CCK-8 assay. (B) hVFs were pretreated with various
concentrations of ATV (0, 2, 5 and 10 µM) for 3 h, and then
exposed to 5 ng/ml TGF-β1 for 24 h. The proliferation of hVFs was
then assessed by CCK-8 assay. All data are presented as the mean ±
standard error of mean of three independent experiments and
described as a percentage of the control group.
*P<0.05, **P<0.01 vs. untreated control
group. #P<0.05 vs. TGF-β1 only-treated group. hVFs,
human ventricular fibroblasts; ATV, atorvastatin; TGF-β1,
transforming growth factor-β1; NS, not significant; CCK-8, Cell
Counting Kit-8. |
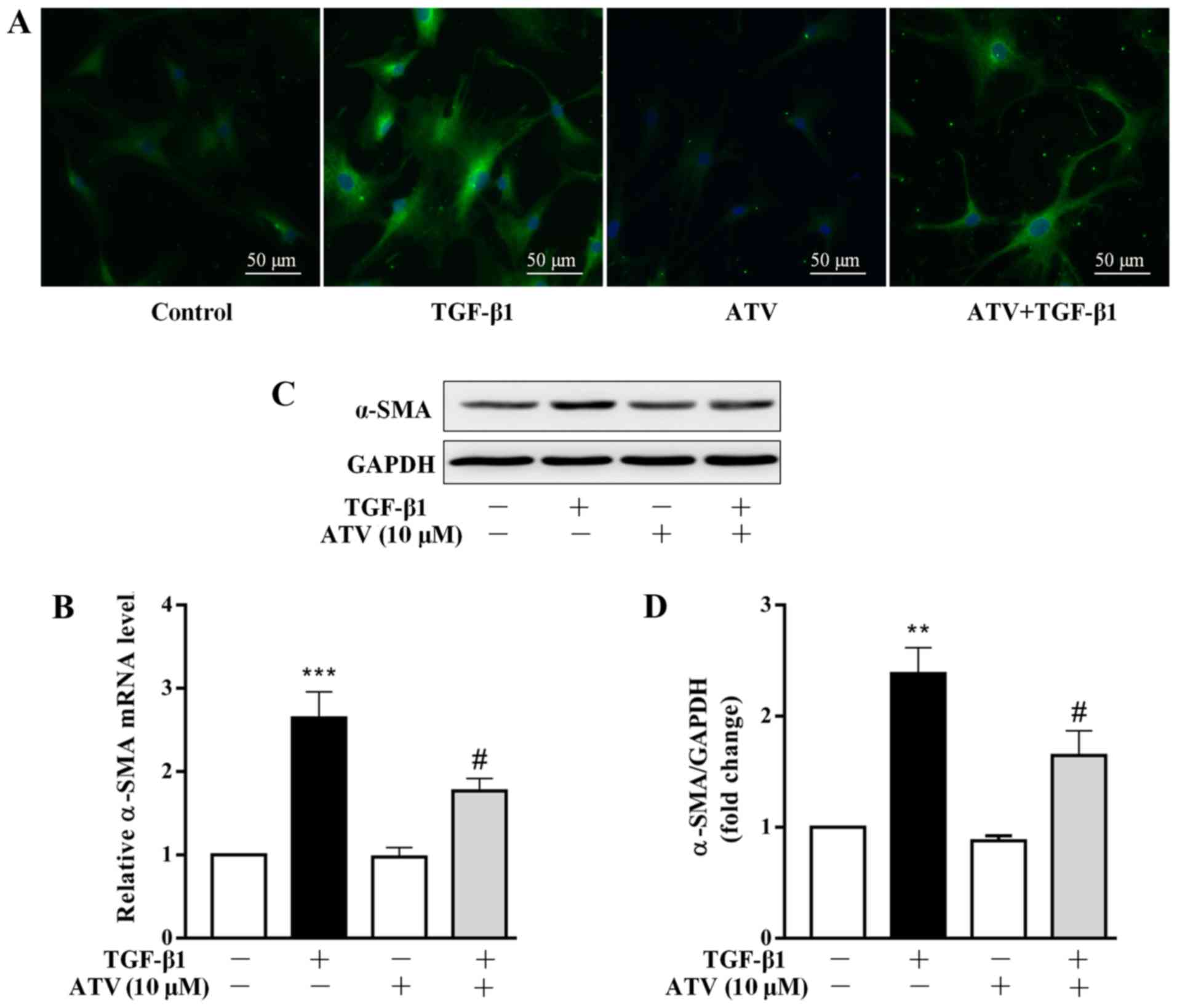
ATV treatment prevents the
trans-differentiation of hVFs into myofibroblasts
Several studies have demonstrated that the
differentiation of CFs into myofibroblasts plays a central role in
the development of cardiac fibrosis (23,24). The marker of fibroblast activation
is the overexpression of α-SMA (25). Therefore, the effect of ATV on
TGF-β1-induced α-SMA expression in hVFs was assessed. Firstly,
α-SMA expression was detected by immunofluorescence staining. As
presented in Fig. 2A, in
comparison with that of the control group, treatment with TGF-β1
alone for 24 h increased the expression level of α-SMA in hVFs.
However, pre-incubation with ATV markedly inhibited the
TGF-β1-induced α-SMA expression in hVFs. To confirm the
immunostaining results, α-SMA expression levels were further
detected using RT-qPCR and western blotting. Consistently, the
significant upregulation of α-SMA in hVFs exposed to TGF-β1 was
significantly attenuated by pre-incubation with ATV at both the
mRNA and protein levels (Fig.
2B-D).
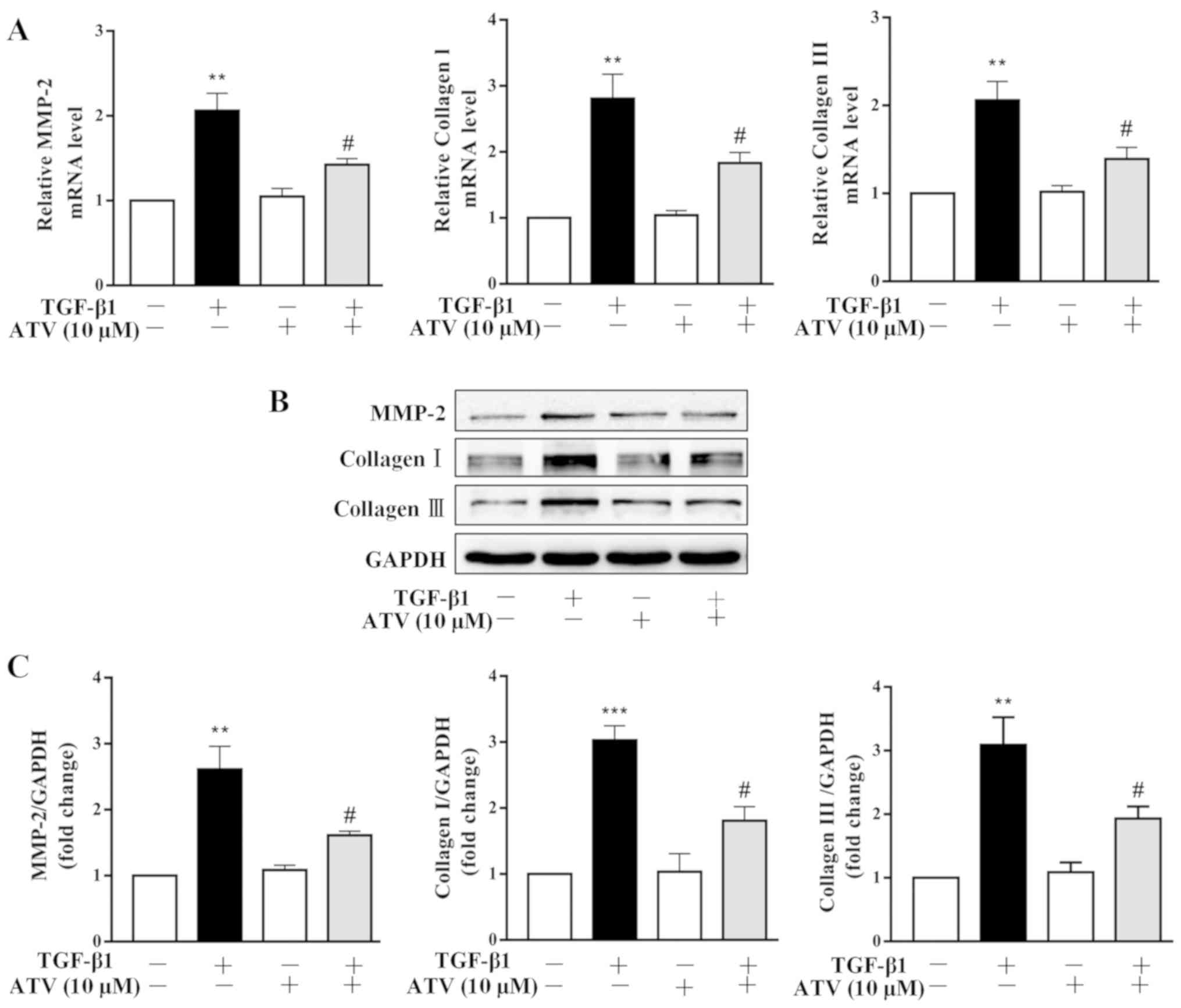
ATV inhibits TGF-β1-induced ECM
production in hVFs
It has been reported that ECM deposition plays an
important role in the pathogenesis of myocardial fibrosis (26). Thus, the effects of ATV on
TGF-β1-stimulated ECM expression in hVFs were further investigated.
Both RT-qPCR and immunoblot-ting assays revealed that TGF-β1
significantly increased the expression levels of MMP-2, collagen I
and collagen III in hVFs. Whereas, pretreatment of hVFs with ATV
significantly attenuated the TGF-β1-induced increases in the
expression levels of these ECM components (Fig. 3A–C).
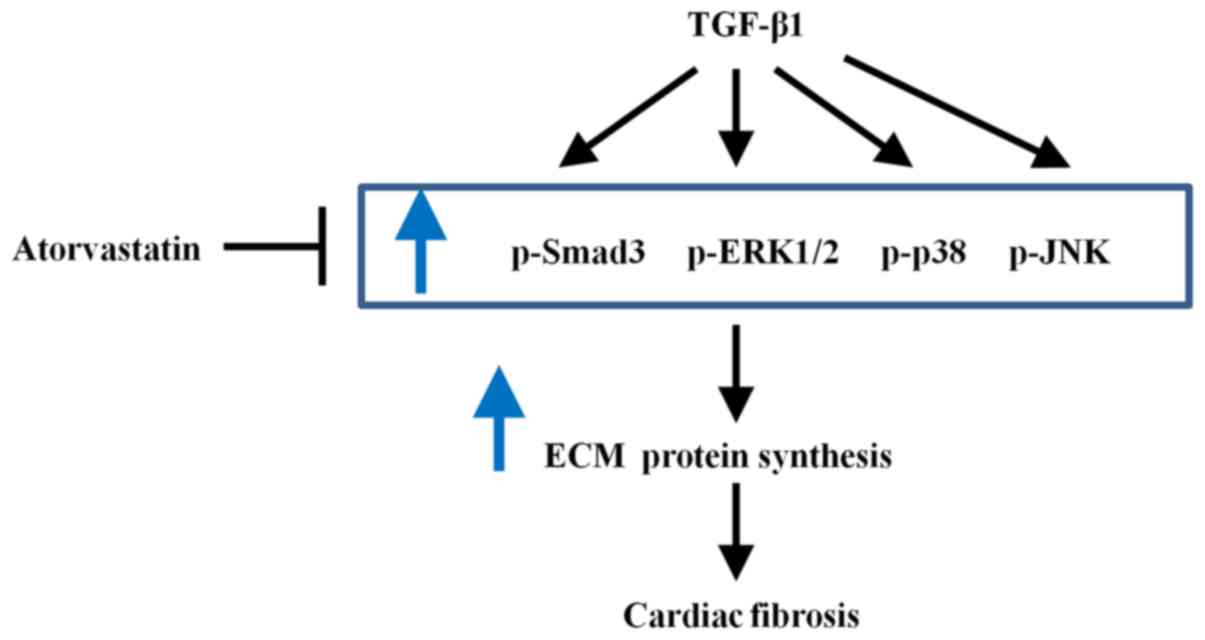
ATV inhibits TGF-β1-induced activation of
the Smad3 and MAPK signaling pathways in hVFs
To investigate the signaling pathways that are
involved in the ATV-suppressed fibrotic response in TGF-β1-treated
hVFs, the effect of ATV on the activation of Smad3, ERK1/2, p38 and
JNK signaling was detected. As presented in Fig. 4A-D, the western blot results
indicated that the phosphorylation of Smad3, ERK1/2, JNK and p38
was significantly increased in TGF-β1-induced hVFs. However, the
TGF-β1-induced activation of these signaling molecules was
significantly suppressed by ATV. These data demonstrated that the
ATV-decreased fibrotic response in hVFs treated by TGF-β1 was
likely associated with suppression of Smad3 and MAPK signaling
(Fig. 5).
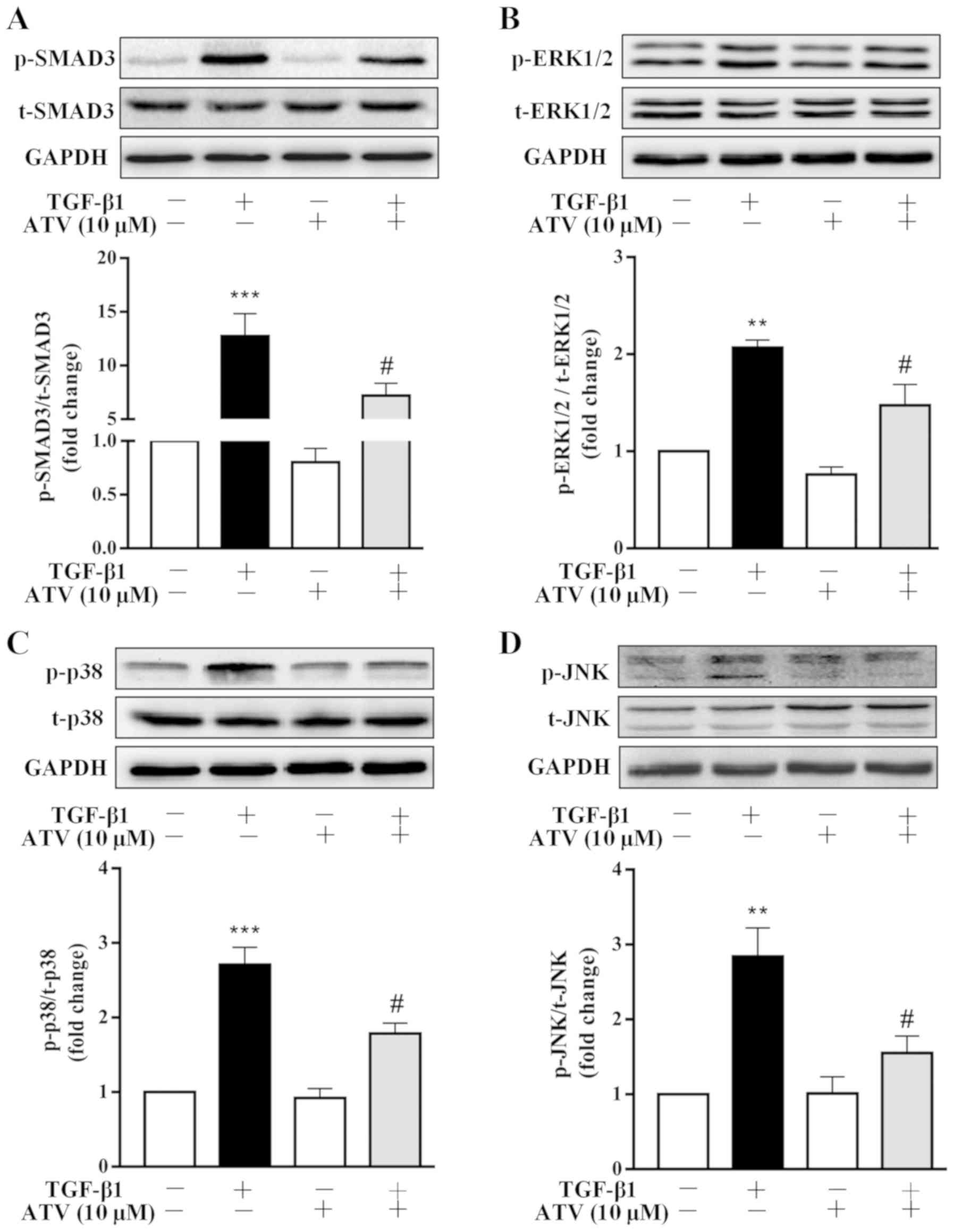 | Figure 4ATV inhibits TGF-β1-induced Smad3 and
MAPK signaling activation in hVFs. Fibroblasts were pre-incubated
with ATV (10 µM) for 3 h and then co-treated with 5 ng/ml
TGF-β1 for 30 min. The protein levels of (A) p-Smad3 and t-Smad3,
(B) p-ERK1/2 and t-ERK1/2, (C) p-p38 and t-p38, and (D) p-JNK and
t-JNK were detected using western blotting. The relative density
was expressed as the ratio of p-protein to the corresponding
t-protein. Values represent the mean ± standard error of mean of
three independent experiments. **P<0.01,
***P<0.001 vs. untreated control group.
#P<0.05 vs. TGF-β1 only-treated group. ATV,
atorvastatin; TGF-β1, transforming growth factor-β1; MAPK,
mitogen-activated protein kinase; hVFs, human ventricular
fibroblasts; p-, phosphorylated; t-, total; ERK1/2, extracellular
signal-regulated kinase 1/2; JNK, c-Jun N-terminal kinase. |
Discussion
To date, several specific anti-fibrotic therapies
have been evaluated for use, with unsatisfactory results (27). Therefore, identifying a novel and
effective strategy for the treatment of cardiac fibrosis is
urgently required. To the best of our knowledge, the present data
indicated for the first time that ATV prevents TGF-β1-stimulated
myofibroblast differentiation and ECM protein production in hVFs,
and the underlying mechanism may be, at least in part, due to the
suppression of Smad3 and MAPK signaling activation.
CFs account for ~2/3 of the total cell number in the
adult human heart, and contribute to the maintenance of cardiac
structure, function and ECM homeostasis in the normal state, and
the regulation of structural integrity and scar formation following
myocardial injury (28,29). Abnormal proliferation of CFs is
the major cellular pathological basis of cardiac fibrosis.
Increasing evidence suggests that TGF-β1, a key pro-fibrotic
cytokine, can promote CF proliferation (30,31), which was confirmed in the present
study. Furthermore, TGF-β1 has been demonstrated to be markedly and
consistently activated in various cardiovascular diseases,
including myocardial hyper-trophy and myocardial infarction
(32,33). The findings of the present study
indicated that ATV effectively decreased cell proliferation in
TGF-β1-stimulated hVFs. The results revealed that ATV can exert
protective effects against myocardial fibrosis through suppressing
hVF proliferation.
The trans-differentiation of CFs into myofibroblasts
has been shown to play a vital role in the progress of cardiac
fibrosis (34). Myofibroblasts
are defined by excessive α-SMA expression and ECM protein secretion
(5,25). The present study demonstrated that
TGF-β1 significantly promoted fibroblast differentiation and ECM
protein expression, which is in accordance with the findings
reported by previous studies (30,31,35). However, pre-incubation with ATV
inhibited the TGF-β1-stimulated fibrotic response in hVFs. The
results were in agreement with earlier studies, which reported that
ATV prevents advanced glycation end products-, angiotensin II-,
hypertension-, aldosterone- and doxorubicin-induced cardiac
fibrosis (16-20).
TGF-β1 canonically activates downstream Smad
pathways, and TGF-β1/Smad signaling plays a crucial role in the
development of myocardial fibrosis (36-38). During fibrogenesis, Smad3, a major
downstream mediator of TGF-β1, is phos-phorylated by activated
TGF-β type I receptor kinase (39-41). p-Smad3 protein then interacts with
Smad4 to form a complex and translocates to the cell nucleus, where
it functions as a transcriptional factor and induces the
transcriptional activation of numerous fibrotic genes, including
α-SMA, MMP-2 and collagens (7,8).
In addition to the Smad-mediated pathways, TGF-β1 can also act
through non-canonical signaling pathways, such as the MAPK
signaling pathway (9).
Accumulating evidence has reported that ERK/JNK/p38 MAPK signaling
is involved in regulating multiple cellular processes, including
cell proliferation, differentiation and survival (42,43). In addition, MAPK signaling
pathways have also been shown to play an essential role in the
progression of cardiac fibrosis. For example, ERK1/2 has been
reported to play an important signaling role in driving CF
proliferation (44), and it is
required for TGF-β1-induced pro-fibrotic phenotypes (3). Molkentin et al (11) reported that transgenic mice with
fibroblast-specific activation of p38 MAPK developed interstitial
and perivascular fibrosis in the heart. Thus, inhibiting these
signaling pathways may imply a promising therapeutic strategy
against cardiac fibrosis. The present study evaluated the
importance of Smad3 and MAPK signaling in TGF-β1-induced cardiac
fibrosis in hVFs. The results indicated that TGF-β1 significantly
activated both Smad3 and MAPK signaling in hVFs. However, ATV
pretreatment significantly decreased the TGF-β1-induced
phosphorylation levels of Smad3, ERK1/2, JNK and p38 MAPK in hVFs.
This suggested that ATV inhibited TGF-β1-induced fibrogenesis in
hVFs at least in part through inhibition of the Smad3 and MAPK
signaling pathways.
There are also some limitations in the present
study. Firstly, the results were obtained only from a series of
in vitro experiments, and were not validated in vivo.
Secondly, the exact targets of ATV in cardiac fibrosis remain
unclear. Further investigations need to be performed to resolve
these limitations.
Taken together, the results of the present study
report a protective role of ATV on TGF-β1-induced fibrogenesis in
hVFs and the potential mechanism involved. The results demonstrated
that ATV prevented TGF-β1-induced fibrogenesis in hVFs at least in
part by inhibiting Smad3 and MAPK signaling activation. These novel
findings suggest a potential therapeutic effect of ATV against
fibrotic disease in clinical practice.
Funding
This study was supported by The Natural Science
Foundation of Southwest Medical University and The Foundation of
The Affiliated Hospital of Southwest Medical University (grant no.
2017-PT-43).
Availability of data and materials
All data used or analyzed during the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
YD, HX, JW, XW, TL, JF, SZ, QY and JL performed the
experiments. ZF and GL conceived and designed the research. YD, HX,
GL and ZF analyzed the data and drafted the manuscript. QY and JL
reviewed and edited the manuscript. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
References
|
1
|
Rockey DC, Bell PD and Hill JA: Fibrosis-a
common pathway to organ injury and failure. N Engl J Med.
373:962015.
|
|
2
|
Davis J and Molkentin JD: Myofibroblasts:
Trust your heart and let fate decide. J Mol Cell Cardiol. 70:9–18.
2014. View Article : Google Scholar
|
|
3
|
Schafer S, Viswanathan S, Widjaja AA, Lim
WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E,
et al: IL-11 is a crucial determinant of cardiovascular fibrosis.
Nature. 552:110–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar
|
|
6
|
Eghbali M, Tomek R, Woods C and Bhambi B:
Cardiac fibro-blasts are predisposed to convert into myocyte
phenotype: Specific effect of transforming growth factor beta. Proc
Natl Acad Sci USA. 88:795–799. 1991. View Article : Google Scholar
|
|
7
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiller M, Javelaud D and Mauviel A:
TGF-beta-induced SMAD signaling and gene regulation: Consequences
for extracellular matrix remodeling and wound healing. J Dermatol
Sci. 35:83–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Evangelia P and Peter TD: TGFβ signaling
and cardiovascular diseases. Int J Bio Sci. 8:195–213. 2012.
View Article : Google Scholar
|
|
10
|
Chung CC, Kao YH, Yao CJ, Lin YK and Chen
YJ: A comparison of left and right atrial fibroblasts reveals
different collagen production activity and stress-induced
mitogen-activated protein kinase signalling in rats. Acta Physiol
(Oxf). 220:432–445. 2017. View Article : Google Scholar
|
|
11
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McFarlane SI, Muniyappa R, Francisco R and
Sowers JR: Clinical review 145: Pleiotropic effects of statins:
Lipid reduction and beyond. J Clin Endocrinol Metab. 87:1451–1458.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang CY, Liu PY and Liao JK: Pleiotropic
effects of statin therapy: Molecular mechanisms and clinical
results. Trends Mol Med. 14:37–44. 2008. View Article : Google Scholar
|
|
14
|
Ludman A, Venugopal V, Yellon DM and
Hausenloy DJ: Statins and cardioprotection-more than just lipid
lowering? Pharmacol Ther. 122:30–43. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao JK: Effects of statins on
3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond
low-density lipoprotein cholesterol. Am J Cardiol. 96:24F–33F.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen M, Li H, Wang G, Shen X, Zhao S and
Su W: Atorvastatin prevents advanced glycation end products
(AGEs)-induced cardiac fibrosis via activating peroxisome
proliferator-activated receptor gamma (PPAR-γ). Metabolism.
65:441–453. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choi SY, Park JS, Roh MS, Kim CR, Kim MH
and Serebruany V: Inhibition of angiotensin II-induced cardiac
fibrosis by atorvastatin in adiponectin knockout mice. Lipids.
52:415–422. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fang T, Guo B, Xue L and Wang L:
Atorvastatin prevents myocardial fibrosis in spontaneous
hypertension via inter-leukin-6 (IL-6)/signal transducer and
activator of transcription 3 (STAT3)/endothelin-1 (ET-1) pathway.
Med Sci Monit. 25:318–323. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Q, Cui W, Zhang HL, Hu HJ, Zhang YN,
Liu DM and Liu J: Atorvastatin suppresses aldosterone-induced
neonatal rat cardiac fibroblast proliferation by inhibiting ERK1/2
in the genomic pathway. J Cardiovasc Pharmacol. 61:520–527. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao G, Jiang S, Ge L, Zhang S, Zhai C,
Chen W and Sui S: Atorvastatin improves doxorubicin-induced cardiac
dysfunction by modulating Hsp70, Akt, and MAPK signaling pathways.
J Cardiovasc Pharmacol. 73:223–231. 2019. View Article : Google Scholar
|
|
21
|
Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M,
Sheng Q, Zhu Y and Zhang Y: Metformin attenuates cardiac fibrosis
by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc
Res. 87:504–513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Yi X, Li X, Zhou Y, Ren S, Wan W, Feng G
and Jiang X: Hepatocyte growth factor regulates the TGF-β1-induced
proliferation, differentiation and secretory function of cardiac
fibroblasts. Int J Mol Med. 34:381–390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Van Nieuwenhoven FA and Turner NA: The
role of cardiac fibro-blasts in the transition from inflammation to
fibrosis following myocardial infarction. Vascul Pharmacol.
58:182–188. 2013. View Article : Google Scholar
|
|
25
|
Abdalla M, Goc A, Segar L and Somanath PR:
Akt1 mediates α-smooth muscle actin expression and myofibroblast
differentiation via myocardin and serum response factor. J Biol
Chem. 288:33483–33493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lan TH, Huang XQ and Tan HM: Vascular
fibrosis in atherosclerosis. Cardiovasc Pathol. 22:401–407. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park S, Nguyen NB, Pezhouman A and
Ardehali R: Cardiac fibrosis: Potential therapeutic targets. Transl
Res. 209:121–137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jugdutt BI: Remodeling of the myocardium
and potential targets in the collagen degradation and synthesis
pathways. Curr Drug Targets Cardiovasc Haematol Disord. 3:1–30.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Souders CA, Bowers SL and Baudino TA:
Cardiac fibroblast: The renaissance cell. Circ Res. 105:1164–1176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li P, Wang D, Lucas J, Oparil S, Xing D,
Cao X, Novak L, Renfrow MB and Chen YF: Atrial natriuretic peptide
inhibits transforming growth factor beta-induced Smad signaling and
myofibroblast transformation in mouse cardiac fibroblasts. Circ
Res. 102:185–192. 2008. View Article : Google Scholar
|
|
31
|
Rizvi F, Siddiqui R, DeFranco A, Homar P,
Emelyanova L, Holmuhamedov E, Ross G, Tajik AJ and Jahangir A:
Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human
ventricular fibroblasts differentiation: Role of protein
phosphatase activation. Int J Cardiol. 270:228–236. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar
|
|
33
|
Li RK, Li G, Mickle DA, Weisel RD, Merante
F, Luss H, Rao V, Christakis GT and Williams WG: Overexpression of
transforming growth factor-beta1 and insulin-like growth factor-I
in patients with idiopathic hypertrophic cardiomyopathy.
Circulation. 96:874–881. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Petrov VV, Fagard RH and Lijnen PJ:
Stimulation of collagen production by transforming growth
factor-beta1 during differentiation of cardiac fibroblasts to
myofibroblasts. Hypertension. 39:258–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li B, Chen H, Yang X, Wang Y, Qin L and
Chu Y: Knockdown of eIF3a ameliorates cardiac fibrosis by
inhibiting the TGF-β1/Smad3 signaling pathway. Cell Mol Biol
(Noisy-le-grand). 62:97–101. 2016.
|
|
37
|
Zhang M, Pan X, Zou Q, Xia Y, Chen J, Hao
Q, Wang H and Sun D: Notch3 Ameliorates cardiac fibrosis after
myocardial infarction by inhibiting the TGF-β1/Smad3 pathway.
Cardiovasc Toxicol. 16:316–324. 2016. View Article : Google Scholar
|
|
38
|
Zhao M, Zheng S, Yang J, Wu Y, Ren Y, Kong
X, Li W and Xuan J: Suppression of TGF-β1/Smad signaling pathway by
sesamin contributes to the attenuation of myocardial fibrosis in
spontaneously hypertensive rats. PLoS One. 10:e01213122015.
View Article : Google Scholar
|
|
39
|
Khalil H, Kanisicak O, Prasad V, Correll
RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al:
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac
fibrosis. J Clin Invest. 127:3770–3783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Massagué J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar
|
|
41
|
Schmierer B and Hill CS: TGFbeta-SMAD
signal transduction: Molecular specificity and functional
flexibility. Nat Rev Mol Cell Biol. 8:970–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee SJ, Park K, Ha SD, Kim WJ and Moon SK:
Gleditsia sinensis thorn extract inhibits human colon cancer cells:
The role of ERK1/2, G2/M-phase cell cycle arrest and p53
expression. Phytother Res. 24:1870–1876. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan
W, Guo C, Zhou D, Wang Y, Xi M and Wen A: Cardioprotective effect
of Danshensu against myocardial ischemia/reperfusion injury and
inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2
phosphorylation. Eur J Pharmacol. 699:219–226. 2013. View Article : Google Scholar
|
|
44
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|