Introduction
Lung cancer is one of the most common types of
cancer and a leading cause of death worldwide (1). After the diagnosis of lung cancer at
stage 3 or 4, the 5-year survival rate is ~15% (2). Cancer research and clinical trials
are in progress for the development of a suitable treatment
(3,4). At present, the primary treatment
regimens employed include surgery, radiotherapy, chemotherapy, or
various combinations of the three. Combination therapy may prevent
the development of advanced progressive tumors that are resistant
to monotherapy, and has played a significant role in cancer
management for several years. In addition, combination strategies
with effective chemotherapeutic drugs may offer advantages against
cancers such as non-small cell lung adenocarcinoma (5-7).
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is a commonly known transmembrane cytokine that
selectively kills cancer cells by binding to death receptors, while
also being non-toxic to normal cells (8,9).
Cancer cells are often resistant to TRAIL due to their insufficient
expression of death receptors (DR4/DR5), excessive expression of
decoy receptors, or genetic and epigenetic modification of TRAIL
receptors (10). The binding of
TRAIL to death receptors results in the induction of the apoptotic
pathway to activate apoptotic signals (11). TRAIL binds to its receptors, DR4
and DR5, and forms death-inducing signaling complexes (DISC), which
in association with adaptor molecules, such as Fas-associated
protein with death domain and caspase-8, activate caspase-9, and
consequently activate caspase-3 to ensure apoptotic cell death
(12,13). Several types of cancer cells,
including lung A549 cells, are resistant to the apoptotic effects
of TRAIL (14). However, TRAIL
resistance can be prevented through the effective use of
TRAIL-sensitizing pharmacological agents (15,16).
Autophagy is an intracellular catabolic mechanism
associated with a well-maintained self-degrading lysosomal pathway.
It is vital to maintain programmed cell death and cellular
homeostasis. In this process, cytosolic substances are sequestered
into autophagosomes that fuse with lysosomes to form autolysosomes,
wherein their substances are degraded (17). The process of autophagosome
formation is associated with the autophagy-related gene
(Atg)12-Atg5-Atg16 complex and the conversion of
microtubule-associated protein 1 light chain 3 (LC3)-I isoform to
an autophagosome-associated LC3-II, which commonly serves as an
autophagy marker (18). A
well-known autophagy marker, sequestosome 1 (p62), can be
integrated into autophagosomes upon direct interaction with LC3.
Autophagic flux inhibition may result in increased levels of
cellular p62 due to the inhibition of lysosomal degradation
(19). Inhibiting autophagy may
have several effects, including sensitizing cancer cells to
chemotherapy and conventional radiotherapy treatment (20), and serving as an effective
strategy for cancer management. Chloroquine and 3-methyladenine
(3-MA) are commonly used autophagy inhibitors in the study of
autophagy. Chloroquine inhibits lysosome acidification and prevents
the fusion of autophagosomes with lysosomes (21). The compound 3-MA is a specific
inhibitor of phosphoinositide 3-kinase (PI3K) and autophagy
(22). AMP-activated protein
kinase (AMPK), a key conserved sensor, maintains the energy balance
to regulate cellular energy homeostasis. Autophagic flux formation
involves the activation of AMPK via the inhibition of mammalian
target of rapamycin (mTOR) under cellular stress. A number of
studies have reported that the downregulation of AMPK
phosphorylation mediates anticancer effects and autophagy plays a
protective function as an anticancer mechanism (23,24). Inhibition of AMPK phosphorylation
can result in cellular stress and apoptosis induction by
downregulating autophagy (25).
Selective serotonin reuptake inhibitors (SSRIs) are
commonly used to treat depression, anxiety and some social
behavioral disorders, and are often suggested for the treatment of
depression in patients with cancer (26,27). Sertraline is an SSRI that is
broadly used as an antidepressant drug and exerts antitumor
activities against various types of cancers, including colorectal
cancer, liver cancer and lymphoma (28). In the present study, the use of
sertraline as a sensitizing agent to TRAIL-mediated apoptosis in
lung cancer cells was investigated and the molecular mechanisms
underlying the anticancer effects of sertraline in combination with
TRAIL were explored. It was found that this effect was mediated
through the inhibition of autophagy via downregulation of AMPK
phosphorylation and activation of DR5, indicative of the effective
sensitization of TRAIL-resistant lung cancer cells.
Materials and methods
Cell culture
A549 and HCC-15 cells originating from lung tumors
were obtained from the American Type Culture Collection. Calu-3
cancer cells were purchased from the Korean Cell Line Bank (Korean
Cell Line Research Foundation). Roswell Park Memorial Institute
(RPMI)-1640 cell culture media (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% (v/v) fetal bovine serum (Atlas
Biologicals, Inc.) and 100 µg/ml penicillin-streptomycin
were used for cell culture at 37°C in 5% CO2.
Reagents
Sertraline was procured from Cayman Chemical
Company. Chloroquine (10 µM) and 3-MA (5 mM) were obtained
from Sigma-Aldrich (Merck KGaA), and TRAIL (100 ng/ml) was
purchased from AFrontier Co., Ltd.
Cell viability assay
A549, HCC-15 and Calu-3 cells were seeded into
12-well plates at a density of 1×104 cells and incubated
at 37°C for 24 h. Cultured cells were pretreated with sertraline at
different concentrations (0, 2.5, 5 and 10 µM) for 18 h, and
then treated with recombinant TRAIL (100 ng/ml) for 2 h and 30 min.
Some cells were also preincubated with chloroquine (10 µM)
or 3-MA (5 mM) for 18 h, and then treated with or without
recombinant TRAIL (100 ng/ml) for 2 h 30 min. Cell morphology was
observed under an inverted microscope (Nikon Corporation). Crystal
violet staining was used for the assessment of cell viability. In
this method, viable cells were stained with a crystal violet
staining solution (0.5% crystal violet in 30% ethanol and 3%
formaldehyde) for 10-15 min at room temperature, washed 3-4 times
with phosphate-buffered saline (PBS), and dried. Viability of cells
was also assessed by an MTT assay. In brief, 350 µl of 5
mg/ml MTT solution was added to each well and the plate was
incubated at 37°C for 2 h and 30 min. The medium was removed and
500 µl DMSO was added to each well. The absorbance was
recorded at 570 nm wavelength using a spectrophotometer (Bio-Rad
Laboratories, Inc.). All experiments were performed at least three
times. Viability was expressed relative to the percentage of the
control group, which was set to 100%.
Lactate dehydrogenase (LDH) assay
Cell culture supernatants were collected and
cytotoxicity was analyzed using an LDH detection kit (cat. no.
MK401; Takara Bio, Inc.) according to the manufacturer's protocol.
LDH was assessed by measuring absorbance at 490 nm wavelength using
a microplate reader (Spectra Max M2; Molecular Devices, LLC).
Western blot analysis
Cultured A549 cells were washed with 1X cold PBS,
harvested with a lysis buffer [25 mM HEPES (pH 7.4), 100 mM
ethylenediaminetetraacetic acid (EDTA), 5 mM magnesium chloride
(MgCl2), 0.1 mM dithiothreitol (DTT) and protease
inhibitor cocktail], sonicated to obtain cell lysates (4 sec/20
kHz) and centrifuged at 11,200 × g for 10 min at 4°C. The protein
concentration was assessed using a BCA protein assay kit (Thermo
Fisher Scientific, Inc.). The cell supernatant was collected and
the proteins (30 µg) were separated by SDS-PAGE on 10-15%
gels. The separated protein bands were transferred to
nitrocellulose or PVDF membranes, which were blocked with 5%
non-fat dried milk at 25°C for 1 to 2 h. Next, specific primary
antibodies in a dilution buffer [1% milk and 1% PBS with 1%
Tween-20 (PBST)] for 1 h at 25°C. The following primary antibodies
were used for immunoblotting: LC3 (1:1,000; cat. no. 4108), p62
(1:1,000; cat. no. 5114s), cleaved caspase-3 (1:1,000; cat. no.
39665), AMPK (1:1,000; cat. no. 2532), phosphorylated (p)-AMPKα
(1:1,000; cat. no. 2535), mTOR (1:1,000; cat. no. 2983) and p-mTOR
(1:1,000; cat. no. 5536; all from Cell Signaling Technology, Inc.);
cleaved caspase-8 (1:1,000; cat. no. 551242; BD Pharmingen); DR5
(1:10,000; cat. no. ab181846) and DR4 (1:1,000; cat. no. ab8414;
both from Abcam); and β-actin (1:1,000; cat. no. A5441;
Sigma-Aldrich; Merck KGaA). Then, membranes were probed with
horseradish peroxidase-conjugated secondary antibodies (1:5,000;
cat. nos. ADI-SAB-100 and ADI-SAB-300; Enzo Life Sciences, Inc.) at
25°C for 1 h. The targeted protein bands were detected with
enhanced chemiluminescence reagents (Cytvia). Bands were visualized
using a Fusion-FX7 image capturing system (Vilber Lourmat).
Immunocytochemistry (ICC)
A549 cells (1×105 cells/well) were
cultured on glass coverslips at 37°C for 24 h and treated with
sertraline for 18 h, followed by washing with 1% PBS and fixing
with 4% paraformaldehyde in PBS at room temperature for 15 min.
Cells were washed twice with ice-cold PBS and incubated with PBS
containing 0.25% Triton X-100 at room temperature for 10 min. After
incubation, cells were washed three times with PBS and blocked with
1% bovine serum albumin (BSA; GenDEPOT) in PBST for 30 min at 4̊C.
Cells were probed with primary antibodies [anti-p62 (1:100; cat.
no. 5114s; Cell Signaling Technology, Inc.) and anti-DR5 (1:100;
cat. no. ab181846; Abcam) diluted in PBST containing 1% BSA] in a
5% CO2 incubator at 37°C for 3 h, followed by washing
three times with PBS. These cells were then incubated with the
Alexa Fluor® 488-conjugated donkey polyclonal
anti-rabbit secondary antibody (1:1,000; cat. no. A-21206; Thermo
Fisher Scientific, Inc.) for 2 h at 25°C in a dark condition. The
solution was washed off and cells were further washed 3-4 times
with PBS and stained with DAPI for 10 min at room temperature
(25°C). Cells were washed three times and mounted with a
fluorescent mounting medium. Images were captured using a
fluorescence microscope (Nikon ECLIPSE 80i; Nikon Corporation) at
×400 magnification.
Transmission electron microscopy
(TEM)
Adherent A549 cells (1×106 cells/well)
were detached using trypsin and then fixed with 2% glutaraldehyde
(Electron Microscopy Sciences) and 2% paraformaldehyde in 0.05 M
sodium cacodylate buffer (pH 7.2; Electron Microscopy Sciences) for
2 h at 4°C. Next, cells were treated with 2% osmium tetroxide
(Electron Microscopy Sciences) for 1 h at 4°C and dehydrated with
graded ethanol (25, 50, 70, 90 and 100%) for 5 min each. After
dehydration, samples were embedded in epoxy resin (Embed 812;
Electron Microscopy Sciences) for 48 h at 60°C, according to the
manufacturer's instructions. Ultra-thin sections (60 nm) were
prepared using an LKB-III ultramicrotome (Leica Microsystems GmbH)
and stained with 0.5% uranyl acetate (Electron Microscopy Sciences)
for 20 min and 0.1% lead citrate (Electron Microscopy Sciences) for
7 min at room temperature. Images were captured at ×10,000
magnification on a Hitachi H7650 electron microscope (Hitachi,
Ltd.) installed at the Center for University-Wide Research
Facilities at Jeonbuk National University (Republic of Korea).
RNA interference
Small interfering RNA (siRNA) targeting DR5 and
scramble control siRNA were purchased from Ambion (Thermo Fisher
Scientific, Inc.). The transfection reagent
Lipofectamine® 2000 was obtained from Invitrogen (Thermo
Fisher Scientific, Inc.). A549 lung cancer cells (1×105
cells/well) were transfected with 40 nM DR5 siRNA (siRNA ID 104279;
Sequence 5′-UUU AGC CAC CUU UAU CUC AUU GUC C-3′; Ambion; Thermo
Fisher Scientific, Inc.) using Lipofectamine 2000, according to the
manufacturer's protocol. The cells were incubated with siRNA for 6
h and the medium was then changed to RPMI-1640 with 10% FBS for 24
h. Cells were then treated with sertraline, or sertraline in
combination with TRAIL. At 24-h post transfection, knockdown
efficiency was assessed by the cell viability assay and
immunoblotting at the protein level.
Data analysis
Data are expressed as the mean ± SD. The
significance of the differences between treatments were analyzed
using one-way ANOVA followed by Tukey's test. Statistical analyses
were performed using GraphPad Prism 5 software (GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Sertraline induces TRAIL-mediated
apoptosis in lung cancer cell lines
Effects of sertraline on TRAIL-mediated apoptosis
were evaluated following the treatment of lung adenocarcinoma
cells. Human lung cancer cell lines (A549, HCC-15 and Calu-3) were
preincubated with the indicated doses of sertraline for 18 h and
then treated with TRAIL for 2 h and 30 min. Cells were imaged and
variations in morphology related to apoptosis, such as cell
shrinkage, were investigated under a light microscope. Cells
treated with sertraline or TRAIL alone had slight changes in
viability and showed no obvious morphological changes compared with
control cells (Fig. 1). Thus,
indicating that A549, HCC-15 and Calu-3 cells were highly resistant
to TRAIL-mediated apoptosis. However, treatment with different
concentrations of sertraline along with TRAIL significantly
increased the number of apoptotic cells (Fig. 1A, B, E, F, I and J). The result of
the MTT assay showed that combination treatment with sertraline and
TRAIL reduced cell viability, which indicated that the number of
apoptotic cells increased, for all cell lines tested (Fig. 1C, G and K). Sertraline or TRAIL
alone failed to cause any significant increase in levels of LDH.
Whereas, sertraline in combination with TRAIL significantly
increased the level of LDH in all cell lines, indicative of
apoptosis induction in a sertraline dose-dependent manner (Fig. 1D, H and L). These results
demonstrated that sertraline could sensitize TRAIL-resistant human
lung adenocarcinoma A549, HCC-15 and Calu-3 cells to TRAIL-mediated
apoptosis.
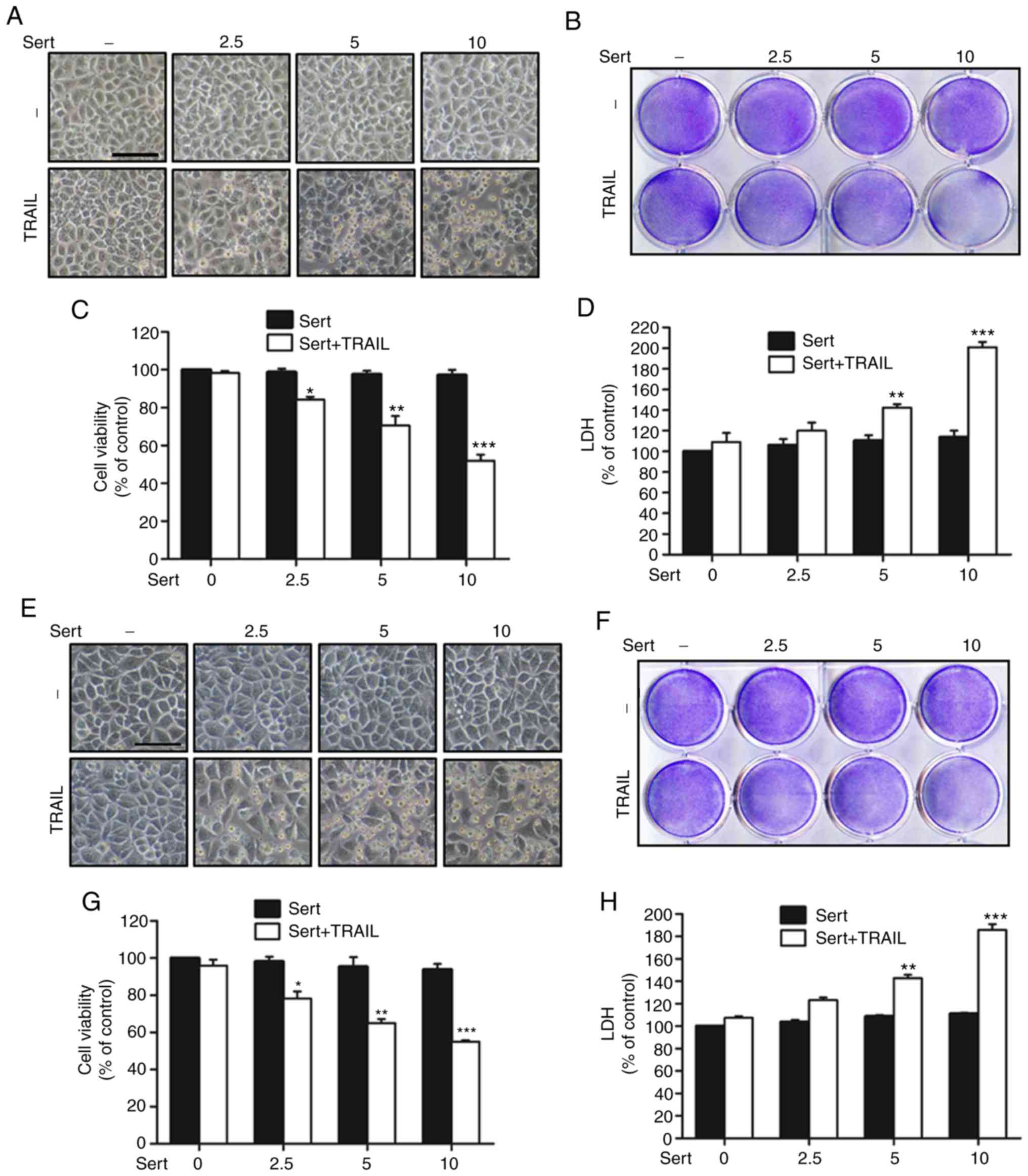 | Figure 1Sert induces TRAIL-mediated apoptosis
in lung cancer cell lines. (A-D) A549, (E-H) HCC-15 and (I-L)
Calu-3 cells were preincubated with the indicated concentrations of
sert for 18 h and 100 ng/ml TRAIL for 2 h and 30 min. (A, E and I)
Cells were imaged and variations in morphology were examined under
a light microscope (magnification, ×100; scale bar, 50 µm).
(B, F and J) Crystal violet staining was performed to determine
cell viability. (C, G and K) MTT assays were conducted to reveal
cell viability, which are expressed as bar graphs. (D, H and L)
Secretion of LDH during co-treatment was assessed in the collected
supernatants. *P<0.05, **P<0.01,
***P<0.001 vs. untreated (control) group Sert,
sertraline; TRAIL, tumor necrosis factor-related apoptosis-inducing
ligand; LDH, lactate dehydrogenase. |
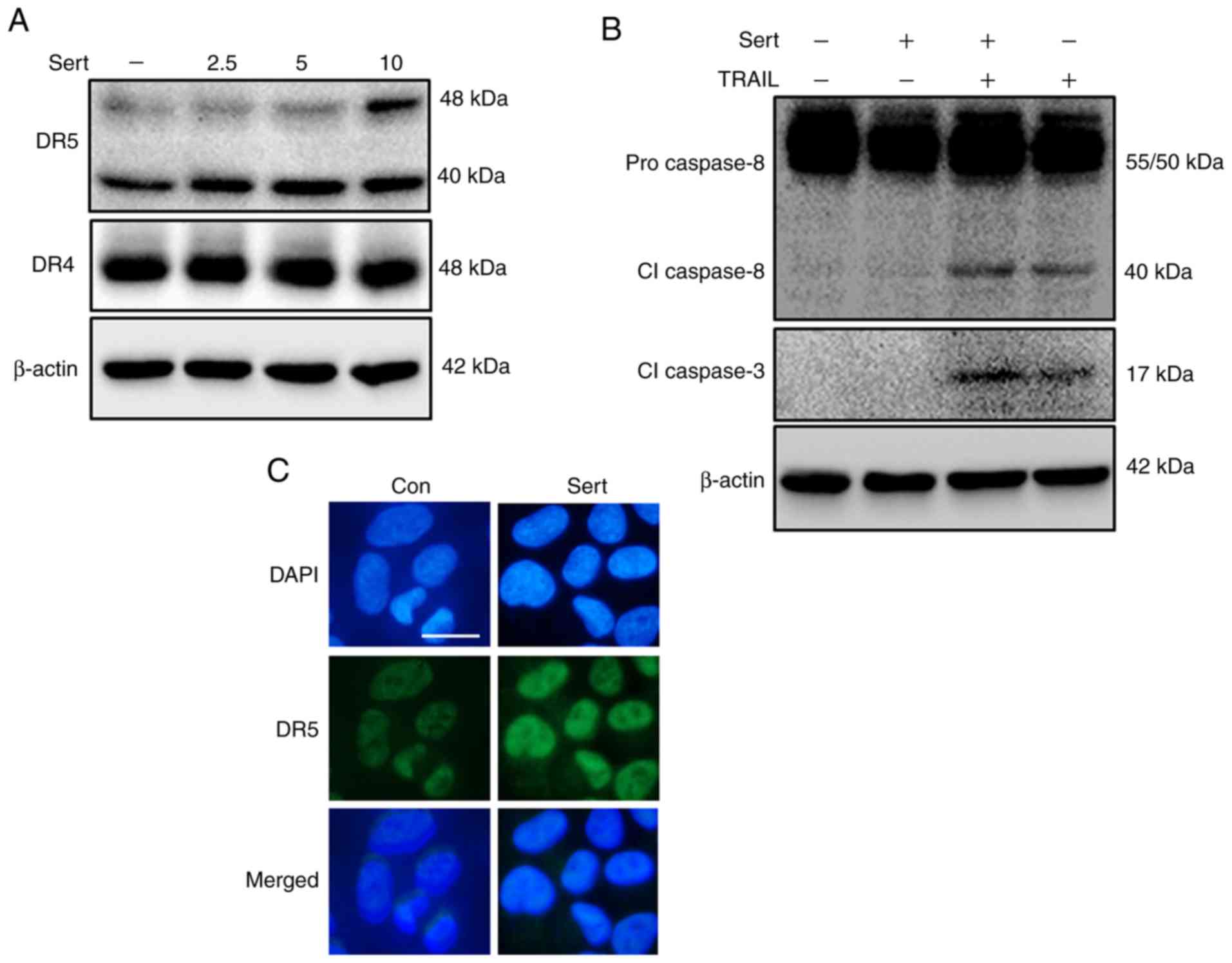
Sertraline triggers the upregulation of
DR5 expression to induce TRAIL-mediated apoptosis
Next, the molecular function of sertraline in
TRAIL-mediated apoptosis of A549 lung cancer cells was investigated
(Fig. 2). A549 cells were treated
with indicated doses of sertraline for 18 h, and the harvested cell
lysates were analyzed by western blotting to determine DR4 and DR5
expression. Sertraline upregulated DR5 expression in a
dose-dependent manner in A549 cells; however, the expression of DR4
was unaltered (Fig. 2A). A549
cells were preincubated with the indicated doses of sertraline for
18 h and then exposed to TRAIL for 2 h. Intracellular
apoptosis-regulatory proteins, cleaved caspase-8 and cleaved
caspase-3, were activated in cells subjected to a combined
treatment of sertraline and TRAIL compared with cells treated with
sertraline alone (Fig. 2B).
Furthermore, ICC results revealed the increased expression of DR5
in sertraline-treated cells compared with that in non-treated cells
(Fig. 2C). These findings
indicated that sertraline could induce TRAIL-mediated apoptosis in
lung adenocarcinoma cells by upregulating DR5 expression.
Sertraline inhibits autophagic flux via
the downregulation of AMPK phosphorylation
To explore the outcomes of sertraline exposure on
autophagic flux, LC3-II and p62 expression levels were assessed by
western blotting (Fig. 3). The
expression levels of LC3-II and p62 increased after sertraline
treatment, indicating the inhibition of autophagic flux (Fig. 3A). Treatment with sertraline alone
or in combination with TRAIL upregulated LC3-II and p62 expression
compared with TRAIL treatment alone (Fig. 3B). ICC results also showed that
sertraline increased the expression of p62 (Fig. 3C). The AMPK pathway is involved in
stabilizing cellular energy through the control of autophagic flux
via downregulation of mTOR expression (29). Sertraline inhibited
phosphorylation of AMPK, resulting in the inhibition of autophagic
flux (Fig. 3D). TEM revealed the
inhibition of autophagic flux, as confirmed by the accumulation of
autophagic vacuoles containing intracellular material in the
treatment group (Fig. 3E). These
findings indicated that the inhibition of AMPK phosphorylation by
sertraline may result in the suppression of autophagic flux in lung
cancer cells.
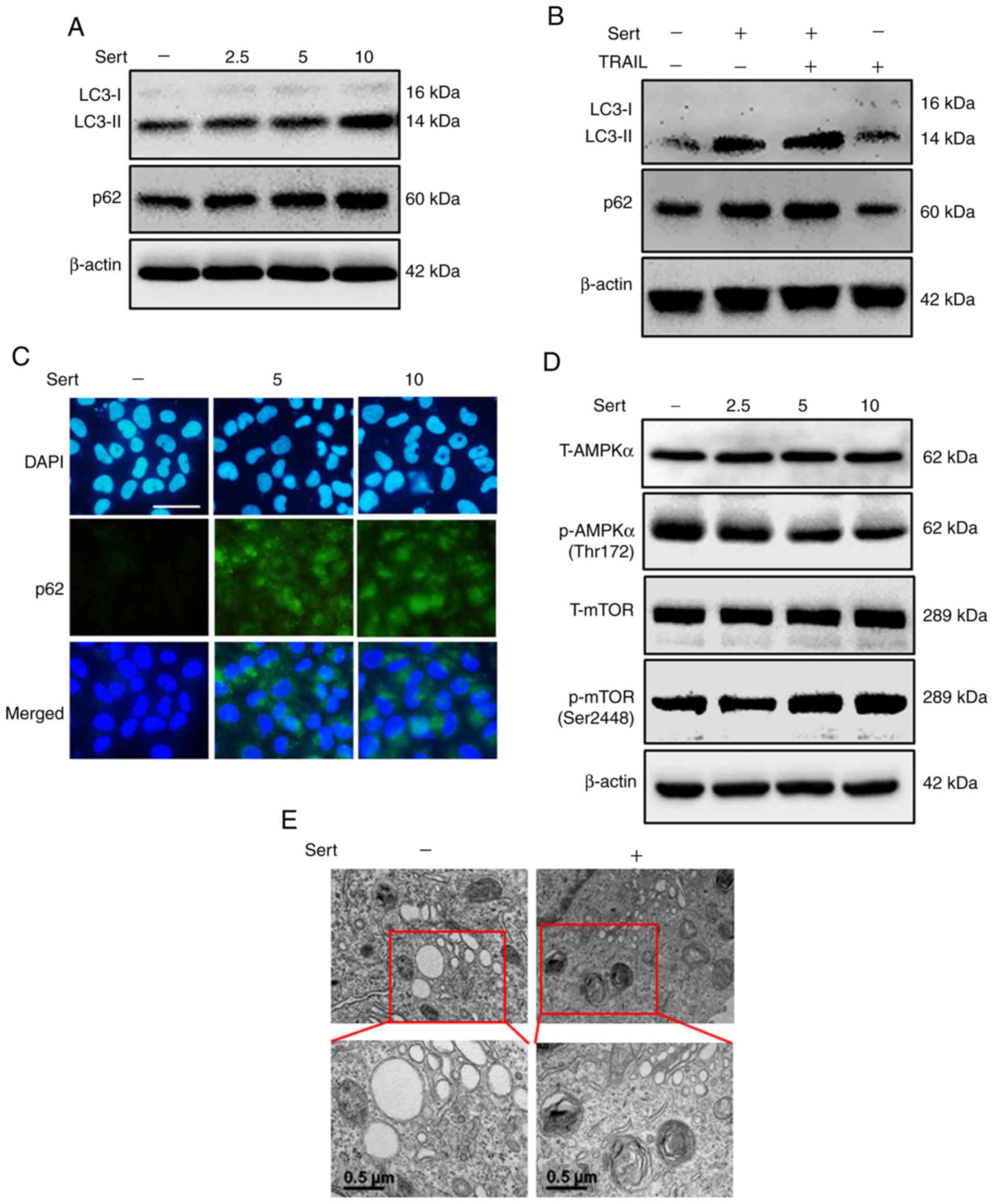 | Figure 3Sert inhibits autophagic flux via the
downregulation of AMPK phosphorylation. A549 cells were incubated
with the indicated doses of sert for 18 h. (A and D) LC3 conversion
and p62, p-AMPKα and p-mTOR levels were evaluated by western blot
analysis. (B) Sert (10 µM)-treated cells were incubated for
18 h and exposed to TRAIL protein (100 ng/ml) for 2 h. The
expression levels of p62, LC3-I and -II were analyzed by western
blotting. (C) The increase in the expression of p62 mediated by
sert was analyzed by immunocytochemistry (scale bar, 50 µm).
(E) Transmission electron microscopy results showed the
accumulation of autophagosomes (Scale bar, 0.5 µm). Sert,
sertraline; TRAIL, tumor necrosis factor-related apoptosis-inducing
ligand; AMPK, AMP-activated protein kinase; LC3,
microtubule-associated protein 1 light chain 3; T-, total protein;
p-, phosphorylated; mTOR, mammalian target of rapamycin. |
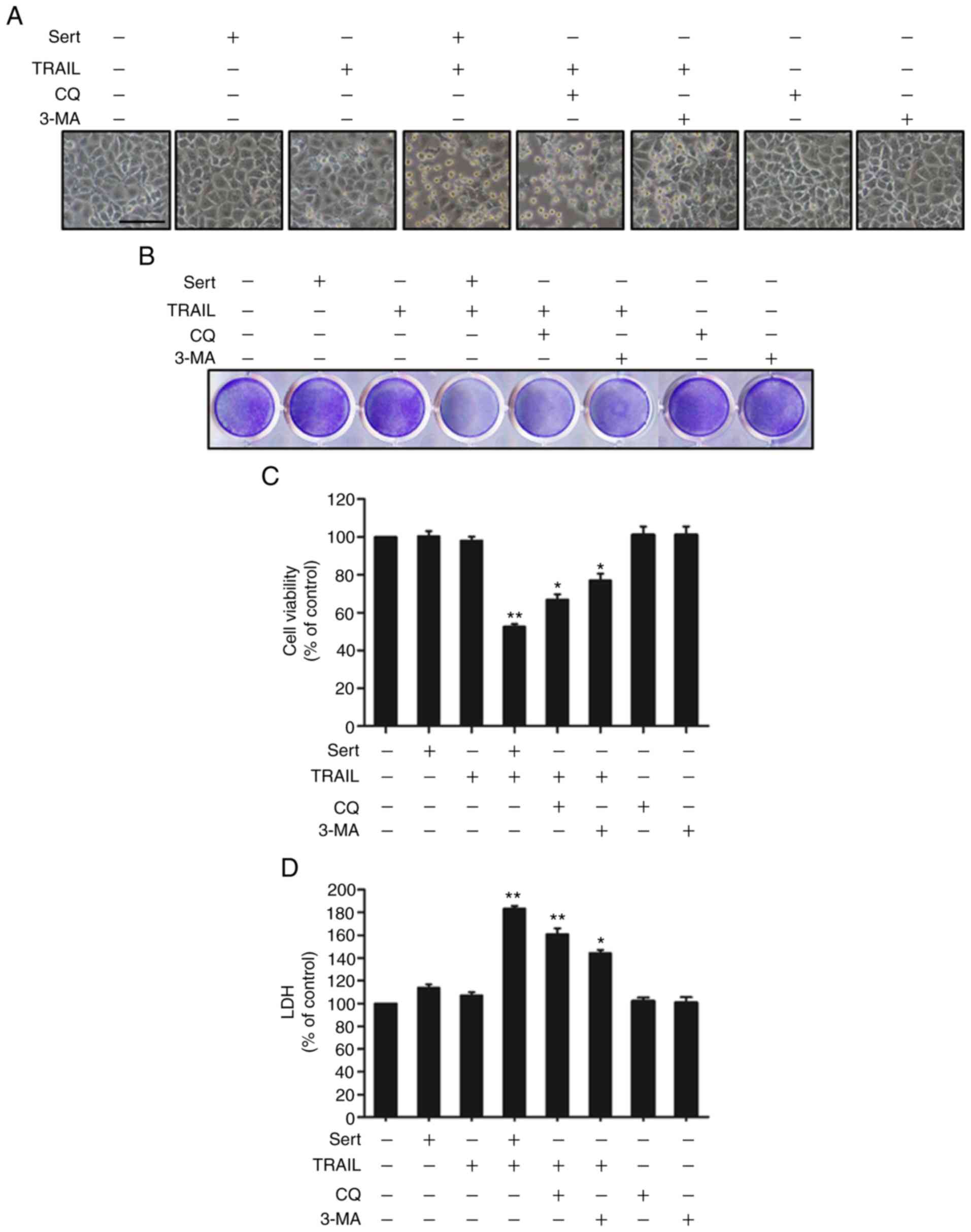
Sertraline induces TRAIL-mediated
apoptosis of cancer cells by inhibiting autophagic flux
The well-known autophagy inhibitors chloroquine and
3-MA were used to examine the effects of sertraline-induced
TRAIL-mediated apoptosis in A549 cells (Fig. 4). A549 cells were incubated with
chloroquine, 3-MA and sertraline at the indicated concentrations
for 18 h. Then, cells were exposed to TRAIL for an additional 2 h
and 30 min. Cells were photographed to investigate any
morphological changes under light microscope. Cell viability was
analyzed by crystal violet staining and an MTT assay. The number of
dead A549 cells increased slightly after treatment with either
TRAIL or sertraline alone. However, the combination of TRAIL and
chloroquine or 3-MA notably increased cell death. Based on cellular
morphology, it was observed that TRAIL with sertraline, chloroquine
or 3-MA enhanced cell death compared with sertraline or TRAIL alone
(Fig. 4A and B). The MTT assay
showed that both autophagy inhibitors, chloroquine and 3-MA, in
combination with TRAIL significantly decreased the viability of
A549 cells compared with control cells (Fig. 4C). Additionally, the combination
of TRAIL and chloroquine or 3-MA also enhanced LDH release compared
with control cells (Fig. 4D).
These results indicated that sertraline enhanced the TRAIL-mediated
apoptosis of lung cancer cells by inhibiting autophagic flux.
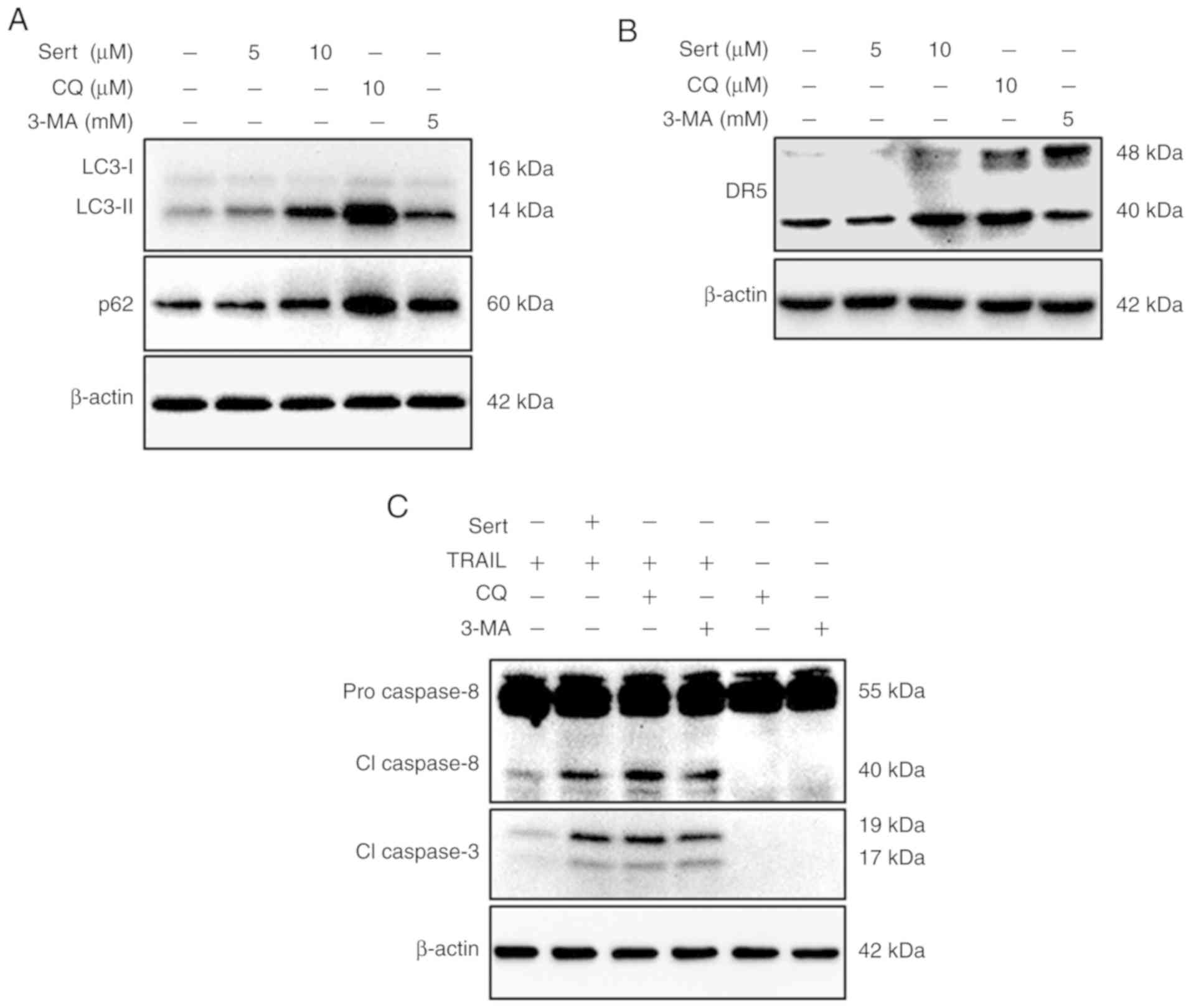
Autophagic flux inhibition leads to the
upregulation of DR5 expression and enhances sertraline-induced
TRAIL-mediated apoptosis
To further explore the mechanism underlying
sertraline-induced TRAIL-mediated apoptosis, autophagic flux was
inhibited using autophagy inhibitors, chloroquine and 3-MA.
Inhibition of autophagic flux with chloroquine and 3-MA resulted in
the upregulation of DR5 expression, thereby increasing apoptosis
(Fig. 5B). Cells were treated
with the indicated concentration of sertraline and two autophagy
inhibitors [chloroquine (10 µM) and 3-MA (5 mM)] for 18 h.
Cell lysates were collected for western blotting and it was found
that sertraline and chloroquine treatment notably increased the
expression levels of p62 and LC3-II; 3-MA could also slightly
increase p62 and LC3-II expression (Fig. 5A). Moreover, DR5 expression in the
chloroquine or 3-MA groups was similar to that observed with
sertraline treatment alone (Fig.
5B). The expression of key apoptosis indicators, cleaved
caspase-8 and cleaved caspase-3, were also evaluated in the lysates
of the cells treated with chloroquine, 3-MA and sertraline with the
indicated doses for 18 h, and TRAIL for 2 h. Autophagy inhibitors
chloroquine and 3-MA in the presence of TRAIL could trigger the
expression of cleaved caspase-8 and cleaved caspase-3 (Fig. 5C). Taken together, these findings
suggested that the inhibition of autophagy may result in the
upregulation of DR5 expression and sertraline-induced
TRAIL-mediated apoptosis.
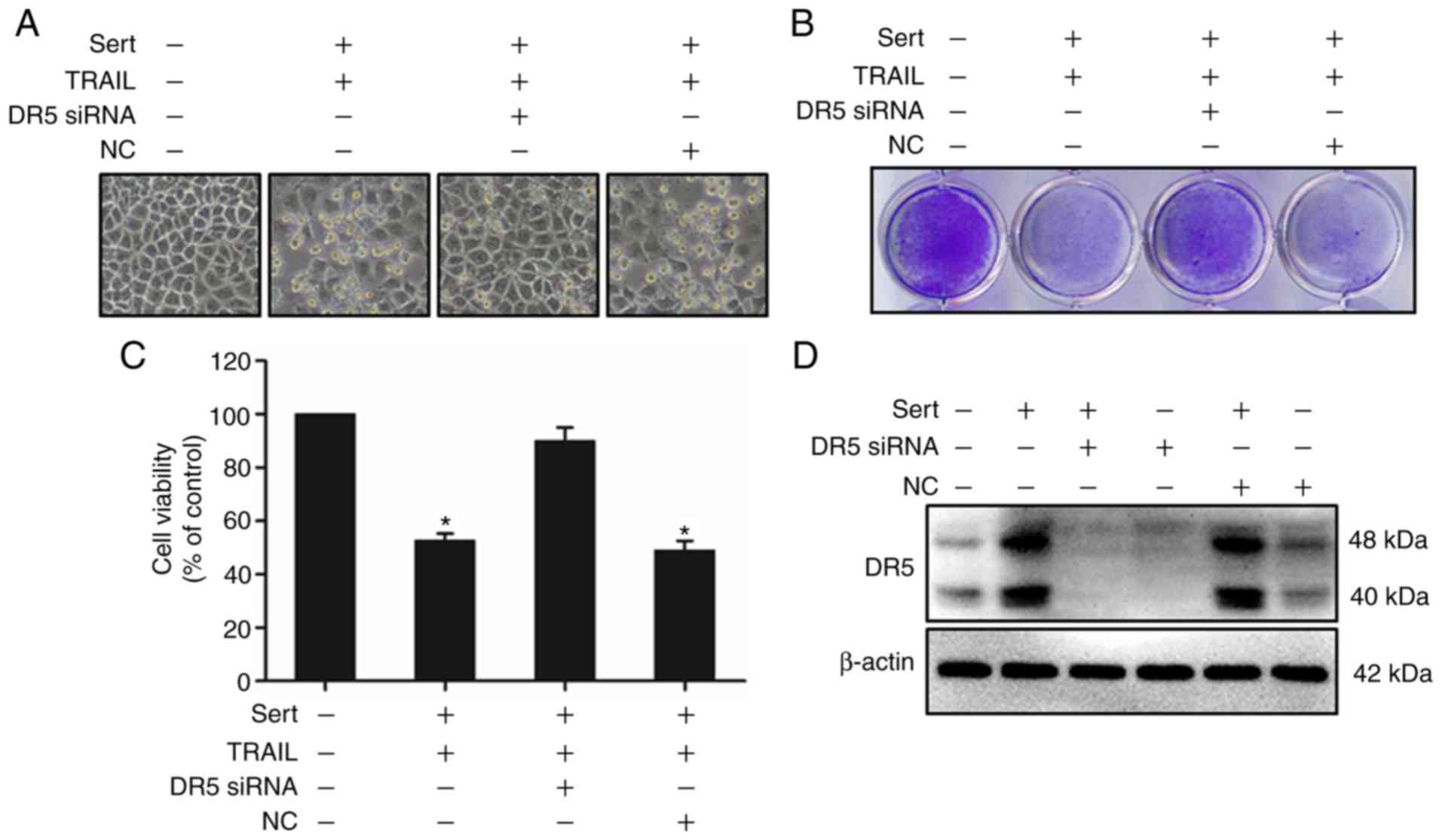
Silencing of DR5 expression alters
sertraline-induced TRAIL-mediated apoptosis
Silencing DR5 expression using DR5 siRNA
significantly altered the effect on cell viability (Fig. 6). This experiment showed that DR5
plays a key role in sertraline-induced TRAIL-mediated apoptosis of
cancer cells. After 24 h of transfection with DR5 siRNA or scramble
control siRNA, cells were treated with sertraline for 18 h and then
with TRAIL (100 ng/ml) for 2 h and 30 min. Cells were then
subjected to viability and western blot analyses. As a result, it
was found that DR5 siRNA-transfected cells blocked cell death
despite co-treatment with sertraline and TRAIL. The viability of
cells treated with scramble siRNA was similar to that of the cells
exposed to sertraline and TRAIL co-treatment (Fig. 6A-C). Western blot analysis showed
that DR5 expression was inhibited in DR5 siRNA-transfected cells
compared with that in non-transfected cells. These experimental
findings indicated that sertraline-mediated upregulation of DR5
plays an important role in weakening TRAIL resistance. Overall,
these findings demonstrated that the induction of TRAIL-mediated
apoptosis with sertraline via AMPK-mediated autophagic flux
inhibition is a possible therapeutic strategy to target the
TRAIL-DR5 apoptotic pathway.
Discussion
In the present study, the effect of sertraline alone
and in combination with TRAIL on A549 lung cancer cells was
investigated. The findings highlighted that sertraline increased
the expression of DR5 via the inhibition of autophagic flux,
thereby resulting in an increase in the TRAIL-induced apoptosis of
A549 cells.
The treatment of patients with lung cancer using
antidepressants was found to increase their survival rate, but the
underlying mechanism of action is not completely understood
(2). Antidepressants are commonly
used in patients with cancer to release emotional stress, such as
depression and dysthymia. Antidepressants, such as fluoxetine and
sertraline, can help to prevent stress-induced tumor progression
(30). Chronic stress decreases
the antitumor immune response, thereby increasing tumor growth
(31). A number of studies in
animal models have shown that behavioral stress induces the rapid
progression of ovarian cancer (32), pancreatic cancer (33), prostate cancer (34), breast carcinomas (35) and malignant melanomas (36). It has already been demonstrated
that SSRI treatment may reduce the risk of lung cancer (37). The antitumor effects of several
SSRIs in various types of cancer cells have been described.
Paroxetine, an SSRI, was reported to induce the apoptotic death of
human osteosarcoma cells via the activation of p38 MAPK and
caspase-3 pathways (38).
Fluoxetine, another SSRI, could prevent the propagation of prostate
cancer cells both in vitro and in vivo, and induce
apoptosis of glioma cells (39,40). In addition, SSRIs sertraline and
paroxetine were demonstrated to increase the activity of caspase-3,
decrease the expression of Bcl-2, and significantly reduce the
viability of malignant T cells (41).
TRAIL is considered as one of the most favorable
anticancer agents, given its specific action involving the
induction of apoptosis in cells and stimulation of cancer cell
death without affecting the functions of normal cells (42,43). Previous experiments have reported
that the repetitive application of TRAIL can markedly prevent tumor
growth without damaging normal cells (44,45). However, several cancer cells,
including lung cancer cells, have developed resistance to the
apoptotic effects of TRAIL (46).
TRAIL resistance can be overcome through the use of combination
therapy with efficient TRAIL-sensitizing pharmacological agents
(47).
The present study demonstrated that small doses of
sertraline in combination with TRAIL could notably increase the
apoptosis of cancer cells. These experiments showed that lung
cancer cells (A549, HCC-15 and Calu-3) are TRAIL resistant.
Furthermore, it was confirmed that sertraline in combination with
TRAIL upregulated the expression of DR5, thereby promoting cancer
cell death. Although TRAIL can bind to the decoy receptors DcR1 and
DcR2 and soluble osteoprotegerin, only DR4 and DR5 can trigger
apoptotic signals through their intracellular death domains
(43). These results clarified
that sertraline could attenuate TRAIL resistance and activate the
apoptotic caspase cascade (Figs.
1 and 2).
Autophagy is a self-regulated mechanism in cells and
is related to cell death and survival. It plays a vital role in
cell survival by eliminating damaged cellular components and
facilitating the degradation of misfolded or aggregated proteins
(48). Autophagy supports the
recycling of essential cell components to fuel bioenergetics
machinery. A number of studies have suggested that the prevention
of lysosomal degradation in starved cells may enhance the rate of
apoptosis via the activation of death receptors (20,49). AMPK plays an important role in
cellular energy homeostasis by inducing autophagy via mTOR
inhibition. Downregulation of AMPK phosphorylation induces
apoptotic cell death via autophagic flux inhibition (50). LC3-II is a well-known marker
indicating the formation of a complete autophagosome, while p62 is
involved in the lysosome- and proteasome-dependent degradation of
proteins. Inhibition of autophagy results in the accumulation of
cellular p62 (51). The findings
of the present study suggested that sertraline increases the number
of autophagosomes, as evident from the enlarged volume of LC3-II,
and triggers the degradation of lysosomes, consistent with the
accumulation of p62. The consequences of these two events is the
inhibition of autophagic flux (Fig.
3). This study further revealed that combined treatment with
TRAIL and sertraline, TRAIL and chloroquine, or TRAIL and 3-MA
could increase cell viability compared with a single treatment
regimen. 3-MA inhibits autophagy by preventing autophagosome
formation via the suppression of PI3K, while chloroquine inhibits
the autophagic flux by blocking the acidification of lysosomes
(52,21). The present study also demonstrated
that autophagic flux inhibition by sertraline facilitates
TRAIL-induced apoptosis, as confirmed by the use of autophagy
inhibitors chloroquine and 3-MA (Fig.
4) (53,54). The inhibition of autophagic flux
using sertraline and autophagy inhibitors, 3-MA and chloroquine,
mediated the upregulation of DR5 expression and increased
TRAIL-mediated apoptotic cell death of lung cancer cells (Fig. 5). The silencing of DR5 expression
using DR5 siRNA reduced TRAIL-mediated apoptosis of cancer cells
(Fig. 6).
Taken together, the present results suggested that
sertraline may serve as a prospective candidate to prevent TRAIL
resistance, and in combination with TRAIL may be an active
treatment regimen to treat lung cancer. The present findings are
based on cell culture experiments. Thus, further investigations are
required with an animal model. However, this study lays the
foundation of future studies to determine patient-specific
treatment strategies in those affected by both depression and
cancer.
Funding
This study was supported by a grant from National
Research Foundation of the Korea (NRF) funded by the Ministry of
Education (grant no. 2019R1A6A1A03033084).
Availability of data and materials
All datasets generated or analyzed during the
present study are available from the corresponding author upon
reasonable request.
Authors' contributions
KZ and SP designed and performed the study, and KZ,
JS and SP analyzed data and wrote the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Ethical approval for the project was granted by the
Institutional Review Board of The Jeonbuk National University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zingone A, Brown D, Bowman ED, Vidal O,
Sage J, Neal J and Ryan BM: Relationship between anti-depressant
use and lung cancer survival. Cancer Treat Res Commun. 10:33–39.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schelhaas S, Held A, Wachsmuth L, Hermann
S, Honess DJ, Heinzmann K, Smith DM, Griffiths JR, Faber C and
Jacobs AH: Gemcitabine mechanism of action confounds early
assessment of treatment response by
3′-Deoxy-3′-[18F]Fluorothymidine in preclinical models of lung
cancer. Cancer Res. 76:7096–7105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garcia G and Odaimi M: Systemic
combination chemotherapy in elderly pancreatic cancer: A review. J
Gastrointest Cancer. 48:121–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun W, Sanderson PE and Zheng W: Drug
combination therapy increases successful drug repositioning. Drug
Discov Today. 21:1189–1195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uchibori K, Inase N, Araki M, Kamada M,
Sato S, Okuno Y, Fujita N and Katayama R: Brigatinib combined with
anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated
non-small-cell lung cancer. Nat Commun. 8:147682017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aggarwal BB: Signalling pathways of the
TNF superfamily: A double-edged sword. Nat Rev Immunol. 3:745–756.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mellier G, Huang S, Shenoy K and Pervaiz
S: TRAILing death in cancer. Mol Aspects Med. 31:93–112. 2010.
View Article : Google Scholar
|
|
9
|
Nazim UM, Rasheduzzaman M, Lee YJ, Seol DW
and Park SY: Enhancement of TRAIL-induced apoptosis by
5-fluorouracil requires activating Bax and p53 pathways in
TRAIL-resistant lung cancers. Oncotarget. 8:18095–18105. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Leary L, van der Sloot AM, Reis CR,
Deegan S, Ryan AE, Dhami SPS, Murillo LS, Cool RH, Sampaio PC,
Thompson K, et al: Decoy receptors block TRAIL sensitivity at a
supracellular level: The role of stromal cells in controlling
tumour TRAIL sensitivity. Oncogene. 35:1261–1270. 2016. View Article : Google Scholar
|
|
11
|
Wang S and El-Deiry WS: TRAIL and
apoptosis induction by TNF-family death receptors. Oncogene.
22:8628–8633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu GS: TRAIL as a target in anti-cancer
therapy. Cancer Lett. 285:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin CY, Park C, Hwang HJ, Kim GY, Choi BT,
Kim WJ and Choi YH: Naringenin up-regulates the expression of death
receptor 5 and enhances TRAIL-induced apoptosis in human lung
cancer A549 cells. Mol Nutr Food Res. 55:300–309. 2011. View Article : Google Scholar
|
|
15
|
Dai X, Zhang J, Arfuso F, Chinnathambi A,
Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G: Targeting
TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural
products as a potential therapeutic approach for cancer therapy.
Exp Biol Med (Maywood). 240:760–773. 2015. View Article : Google Scholar
|
|
16
|
Ding J, Polier G, Köhler R, Giaisi M,
Krammer PH and Li-Weber M: Wogonin and related natural flavones
overcome tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) protein resistance of tumors by down-regulation of c-FLIP
protein and up-regulation of TRAIL receptor 2 expression. J Biol
Chem. 287:641–649. 2012. View Article : Google Scholar :
|
|
17
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar
|
|
19
|
Gómez-Sánchez R, Yakhine-Diop SMS,
Rodríguez-Arribas M, Pedro JMB, Martínez-Chacón G, Uribe-Carretero
E, de Castro DCJ, Pizarro-Estrella E, Fuentes JM and González-Polo
RA: mRNA and protein dataset of autophagy markers (LC3 and p62) in
several cell lines. Data Brief. 7:641–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boya P, González-Polo R, Casares N,
Perfettini J, Dessen P, Larochette N, Métivier D, Meley D, Souquere
S, Yoshimori T, et al: Inhibition of macroautophagy triggers
apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema K, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heckmann BL, Yang X, Zhang X and Liu J:
The autophagic inhibitor 3-methyladenine potently stimulates
PKA-dependent lipolysis in adipocytes. Br J Pharmacol. 168:163–171.
2013. View Article : Google Scholar :
|
|
23
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou C, Gu J, Zhang G, Dong D, Yang Q,
Chen M and Xu D: AMPK-autophagy inhibition sensitizes
icaritin-induced anti-colorectal cancer cell activity. Oncotarget.
8:14736–14747. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cho SW, Na W, Choi M, Kang SJ, Lee S and
Choi CY: Autophagy inhibits cell death induced by the anti-cancer
drug morusin. Am J Cancer Res. 7:518–530. 2017.PubMed/NCBI
|
|
26
|
Laoutidis ZG and Mathiak K:
Antidepressants in the treatment of depression/depressive symptoms
in cancer patients: A systematic review and meta-analysis. BMC
Psychiatry. 13:1402013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Serafin MB, Bottega A, da Rosa TF, Machado
CS, Foletto VS, Coelho SS, da Mota AD and Hörner R: Drug
repositioning in oncology. Am J Ther. 2019. View Article : Google Scholar
|
|
28
|
Xia D, Zhang Y, Xu G, Yan W, Pan X and
Tong J: Sertraline exerts its antitumor functions through both
apoptosis and autophagy pathways in acute myeloid leukemia cells.
Leuk Lymphoma. 58:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar :
|
|
30
|
Di Rosso ME, Sterle HA, Cremaschi GA and
Genaro AM: Beneficial effect of fluoxetine and sertraline on
chronic stress-induced tumor growth and cell dissemination in a
mouse model of lymphoma: Crucial role of antitumor immunity. Front
Immunol. 9:13412018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dhabhar FS, Saul AN, Holmes TH, Daugherty
C, Neri E, Tillie JM, Kusewitt D and Oberyszyn TM: High-anxious
individuals show increased chronic stress burden, decreased
protective immunity, and increased cancer progression in a mouse
model of squamous cell carcinoma. PLoS One. 7:e330692012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim-Fuchs C, Le CP, Pimentel MA,
Shackleford D, Ferrari D, Angst E, Hollande F and Sloan EK: Chronic
stress accelerates pancreatic cancer growth and invasion: A
critical role for beta-adrenergic signaling in the pancreatic
microenvironment. Brain Behav Immun. 40:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hassan S, Karpova Y, Baiz D, Yancey D,
Pullikuth A, Flores A, Register T, Cline JM, D'Agostino Jr, Danial
N, et al: Behavioral stress accelerates prostate cancer development
in mice. J Clin Invest. 123:874–886. 2013.PubMed/NCBI
|
|
35
|
Sloan EK, Priceman SJ, Cox BF, Yu S,
Pimentel MA, Tangkanangnukul V, Arevalo JMG, Morizono K,
Karanikolas BDW, Wu L, et al: The sympathetic nervous system
induces a metastatic switch in primary breast cancer. Cancer Res.
70:7042–7052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hasegawa H and Saiki I: Psychosocial
stress augments tumor development through beta-adrenergic
activation in mice. Jpn J Cancer Res. 93:729–735. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Toh S, Rodríguez LAG and Hernández-Díaz S:
Use of antidepressants and risk of lung cancer. Cancer Causes
Control. 18:1055–1064. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chou CT, He S and Jan CR:
Paroxetine-induced apoptosis in human osteosarcoma cells:
Activation of p38 MAP kinase and caspase-3 pathways without
involvement of [Ca2+]i elevation. Toxicol Appl Pharmacol.
218:265–273. 2007. View Article : Google Scholar
|
|
39
|
Abdul M, Logothetis CJ and Hoosein NM:
Growth-inhibitory effects of serotonin uptake inhibitors on human
prostate carcinoma cell lines. J Urol. 154:247–250. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spanová A, Kovárů H, Lisá V, Lukásová E
and Rittich B: Estimation of apoptosis in C6 glioma cells treated
with antidepressants. Physiol Res. 46:161–164. 1997.PubMed/NCBI
|
|
41
|
Amit BH, Gil-Ad I, Taler M, Bar M, Zolokov
A and Weizman A: Proapoptotic and chemosensitizing effects of
selective serotonin reuptake inhibitors on T cell lymphoma/leukemia
(Jurkat) in vitro. Eur Neuropsychopharmacol. 19:726–734. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
LeBlanc H, Lawrence D, Varfolomeev E,
Totpal K, Morlan J, Schow P, Fong S, Schwall R, Sinicropi D and
Ashkenazi A: Tumor-cell resistance to death receptor-induced
apoptosis through mutational inactivation of the proapoptotic Bcl-2
homolog Bax. Nat Med. 8:274–281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Srivastava RK: TRAIL/Apo-2L: Mechanisms
and clinical applications in cancer. Neoplasia. 3:535–546. 2001.
View Article : Google Scholar
|
|
44
|
Bellail AC, Qi L, Mulligan P, Chhabra V
and Hao C: TRAIL agonists on clinical trials for cancer therapy:
The promises and the challenges. Rev Recent Clin Trials. 4:34–41.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Walczak H, Miller RE, Ariail K, Gliniak B,
Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Klionsky DJ: Autophagy: From phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Park EJ, Min K, Choi KS, Kubatka P,
Kruzliak P, Kim DE and Kwon TK: Chloroquine enhances TRAIL-mediated
apoptosis through up-regulation of DR5 by stabilization of mRNA and
protein in cancer cells. Sci Rep. 6:229212016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu X, Chhipa RR, Nakano I and Dasgupta B:
The AMPK inhibitor compound C is a potent AMPK-independent
antiglioma agent. Mol Cancer Ther. 13:596–605. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zinnah KMA and Park SY: Duloxetine
enhances TRAIL-mediated apoptosis via AMPK-mediated Inhibition of
autophagy flux in lung cancer cells. Anticancer Res. 39:6621–6633.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang Y, Hu L, Zheng H, Mao C, Hu W, Xiong
K, Wang F and Liu C: Application and interpretation of current
autophagy inhibitors and activators. Acta Pharmacol Sin.
34:625–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu X, Shi S, Wang H, Yu X, Wang Q, Jiang
S, Ju D, Ye L and Feng M: Blocking autophagy improves the
anti-tumor activity of afatinib in lung adenocarcinoma with
activating EGFR mutations in vitro and in vivo. Sci Rep.
7:45592017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin YC, Lin JF, Wen SI, Yang SC, Tsai TF,
Chen HE, Chou KY and Hwang TI: Chloroquine and hydroxychloroquine
inhibit bladder cancer cell growth by targeting basal autophagy and
enhancing apoptosis. Kaohsiung J Med Sci. 33:215–223. 2017.
View Article : Google Scholar : PubMed/NCBI
|