1. Introduction
Pulmonary arterial hypertension (PAH), a chronic
lung disease with poor prognosis, is characterized by progressively
increasing blood pressure in the pulmonary vasculature. The normal
resting mean pulmonary arterial pressure in healthy adults is ~14
mmHg at rest, whereas it is >25 mmHg in adults with PAH
(1). PAH is usually secondary to
conditions such as collagen vascular diseases, cardiac
malformations and viral infections, and is currently believed to be
associated with endothelial dysfunction, vasoconstriction and
pulmonary vascular remodeling. Endothelial dysfunction, which is
associated with an imbalance between vasodilators [e.g., nitric
oxide (NO) and prostacyclin] and vasoconstrictors [e.g., endothelin
(ET), thromboxane A2 and serotonin], is considered to be an early
event during the process of PAH (2). A series of events leads to excessive
proliferation of lung smooth muscle cells, activation of lung
fibroblasts, induction of thrombotic mediators and release of
inflammatory cytokines, all of which increase pulmonary vascular
resistance and stress. One cause of vascular endothelial
dysfunction and vascular injury is activation of the
renin-angiotensin system (RAS). This results in overactivation of
the angiotensin-converting enzyme (ACE)-angiotensin II (Ang
II)-angiotensin II receptor type 1 (AT1R) axis, which involves ACE,
vasoactive peptides and blood vessels. Ang II and its receptor,
AT1R, exert adverse effects on pulmonary hemodynamics and may cause
PAH (3). Myocardial infarction
(MI) may lead to post-capillary PAH, increased left ventricular
filling pressure, ventricular remodeling, even heart failure
(4,5). Left ventricular failure leads to an
increase in PAH and right ventricular afterload, which, in turn,
leads to right ventricular remodeling and dysfunction. The PAH
caused by left heart disease is mainly associated with left
ventricular systolic or diastolic dysfunction or valvular heart
disease, and has a poor prognosis (1,6).
Persistent high pulmonary pressure may worsen endothelial
dysfunction, reduce NO utilization and increase ET expression
(4). Early-stage PAH associated
with left heart disease may be reversible. However, the cardiac
remodeling associated with long-term PAH may prevent reversal of
PAH. PAH is the most common complication of congestive heart
failure (7). Both hemodynamic
factors and various molecular mechanisms contribute to the
development of PAH after MI. In particular, in individuals with
chronic PAH, a combination of mechanical pressure and
histopathological reactions promotes progression of PAH, which
ultimately becomes irreversible. The aim of the present review was
to provide a comprehensive overview of the pathophysiological
effects of multiple systems in MI and PAH, particularly regarding
interactions between cardiomyocytes (CMs) and pulmonary vascular
cells in post-MI PAH.
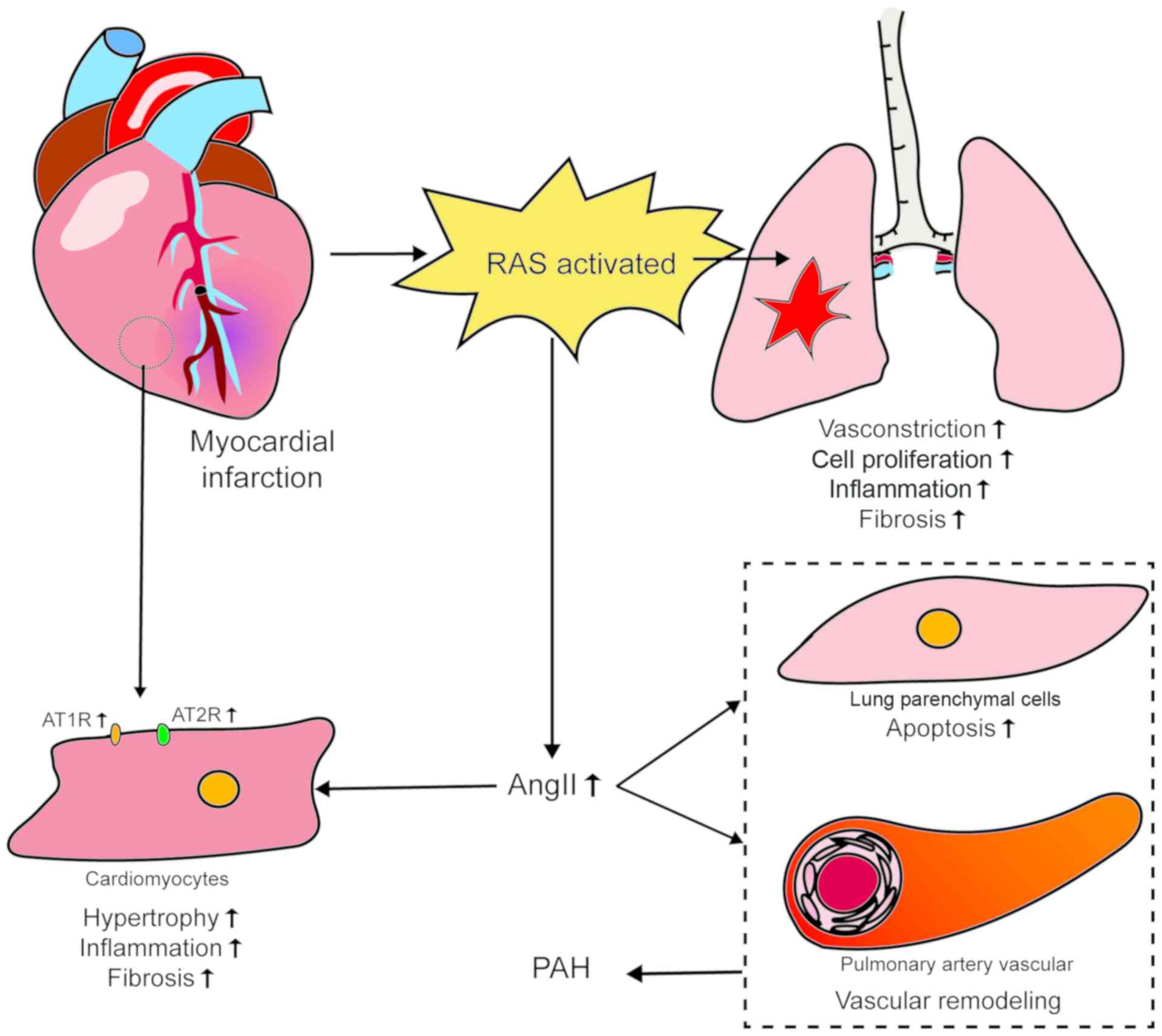
2. Renin-angiotensin system
There is considerable evidence that the RAS
contributes to the pathogenesis of PAH. High concentrations of
renin, ACE, Ang II and AT1R have been documented in experimental
models, as well as in patients with PAH (8-11).
Increased activity of the RAS, systemically and in the pulmonary
circulatory system, may adversely affect heart and lung function
and contribute to disease progression. However, drugs that block
the classical RAS, such as ACE inhibitors and AT1R blockers,
reportedly have adverse effects when used to treat PAH, including
drug-induced systemic hypotension, cough and angioedema (12). The advisability of blocking the
RAS is controversial, as it plays a vasoprotective role in certain
cardiovascular diseases.
As the pulmonary vasculature is more sensitive
compared with the systemic vasculature to the contractile effects
of AngII (8,9), AngII plays an important role in the
pulmonary vascular circulation. Myocardial tissue can express RAS
locally via a paracrine pathway (13). MI can trigger activation of the
RAS, the long-term activation of which may cause myocardial damage;
in this setting, AngII exerts a negative effect (14). When the circulating blood volume
or renal blood flow is reduced, the paracellular cells of the
juxtaglomerular apparatus secrete renin into the blood. This
hydrolyzes the angiotensinogen produced by the liver into the
decapeptide angiotensin I (AngI). In the pulmonary circulation,
AngI is hydrolyzed to the octapeptide AngII by a converting enzyme
present in the endothelial cells (ECs) of the pulmonary
vasculature. AngII exerts different biological effects by binding
to two subtype receptors, namely AT1R and AT2R. AT1R causes
vasoconstriction, cell proliferation, inflammation and fibrosis
through the ACE/AngII/AT1R signaling pathway, whereas AT2R protects
the lungs through the ACE2/Ang-(1-7)/Mas receptor signaling pathway
(15,16). In patients with PAH, as well as in
experimental models, renin, AngII, ACE and AT1R are all increased
to varying degrees in the blood. Additionally, the ratio of
AT1R/AT2R and the expression of AT1R are increased (11,17). However, it remains unclear whether
the increase in the levels of these hormones is a direct result of
PAH, or whether it is mainly caused by the decrease in cardiac
function caused by PAH.
Increased expression of AT1R and AT2R has been
demonstrated in CMs in non-infarcted areas following MI. In
vitro, acidosis promotes death of CMs and AngII enhances this
effect (18). There is increasing
evidence that AngII induces apoptosis of lung parenchymal cells,
causing pulmonary vascular remodeling that ultimately leads to PAH,
and that it also induces hypertrophy of CMs, inflammation and
fibrosis during cardiac ischemic injury. This leads to ventricular
remodeling and further impairment of cardiac function (12,19). It has been established that RAS is
activated by a decline in cardiac function after MI, and that PAH
is caused by both mechanical pressure and contraction mediated by
AngII and vascular remodeling. The resultant further impairment of
cardiac function in turn promotes the development of PAH (18,20). The initiation and development of
chronic PAH originate from pulmonary vascular EC dysfunction.
Endothelial damage increases the production of AngII in lung tissue
(20), which both exacerbates
contraction and remodeling of pulmonary vessels and adversely
affects CMs. The evidence mentioned above indicates that AngII
plays an important role in both MI and PAH. Of note, there are
different degrees of AT1R/AT2R imbalance in MI and PAH. These
findings indicate that there is a vicious circle between myocardial
damage and pulmonary vascular response; they also provide a new
perspective on the association between myocardial and pulmonary
blood vessels. Further study of these mechanisms may indicate new
approaches to the effective treatment of PAH (Fig. 1).
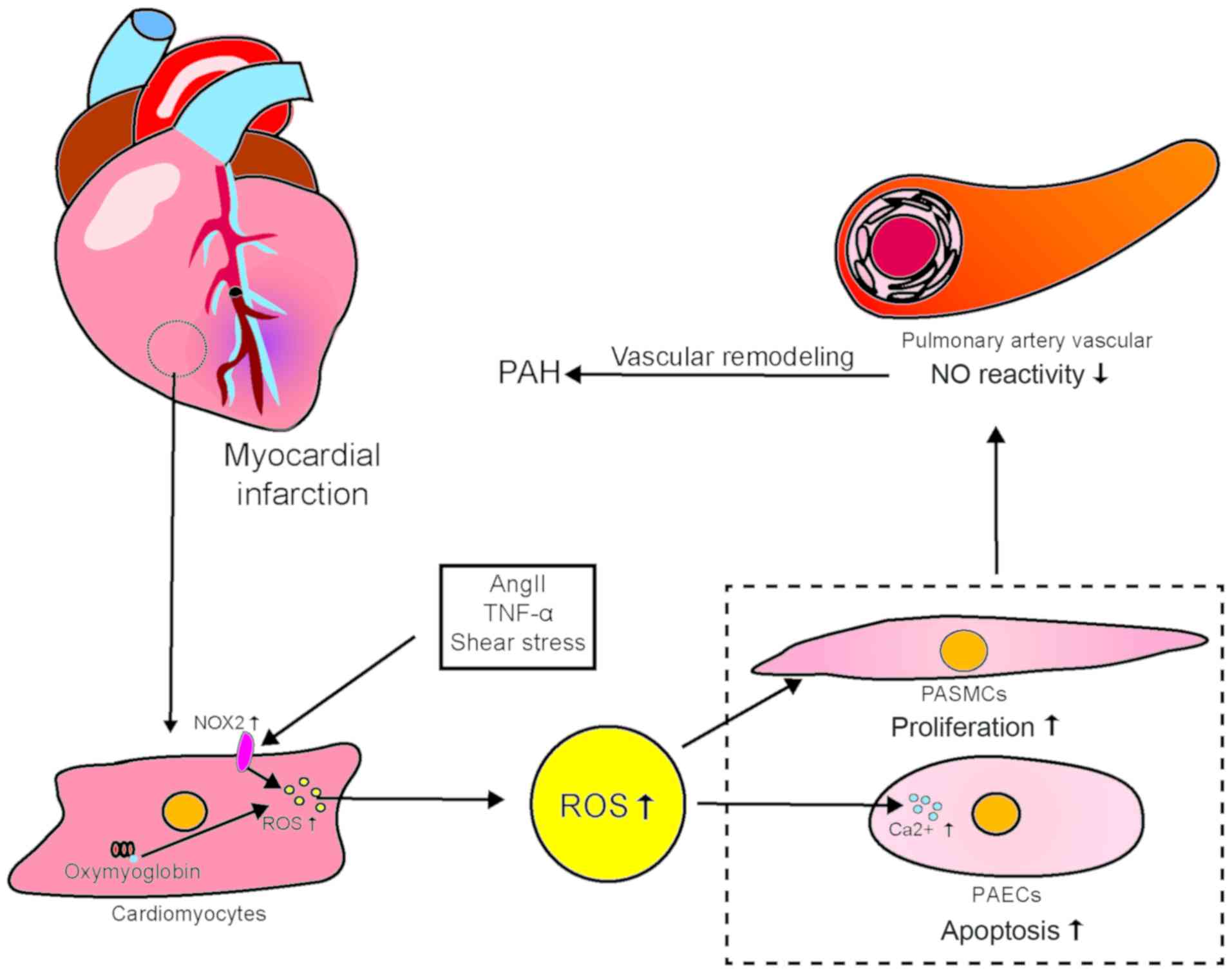
3. Reactive oxygen species
Reactive oxygen species (ROS) play a key role in
regulating contraction/expansion, cell growth, apoptosis,
migration, inflammation and fibrosis. Superoxide anions, hydrogen
peroxide, hydroxyl radicals, reactive nitrogen, NO and
peroxynitrite all serve important biological roles in the
cardiovascular system. NADPH oxidase 2 (NOX2), which is a major
source of ROS in the heart, is activated by MI, resulting in the
generation of large amounts of ROS. NOX may be activated by several
other noxious stimuli, including AngII, tumor necrosis factor
(TNF)-α, vascular endothelial growth factor (VEGF) and shear stress
(21). In addition, under hypoxic
conditions, oxygenated myoglobin in CMs can produce an abundance of
ROS (22). After an MI, ROS
increase significantly in non-infarcted myocardium. Pulmonary blood
vessels may be stimulated by the resultant changes in blood flow
(23). Increased ROS production
is involved in ventricular remodeling and heart failure after MI. A
growing body of evidence supports the role of large amounts of ROS
in the development and progression of PAH. Increased amounts of
oxidative stress markers have been detected in the urine and plasma
of patients with PAH. Additionally, histological examination of
lung sections from such patients has revealed large amounts of
by-products of oxidative stimuli (24,25). Experimental reduction of ROS
levels combined with the use of antioxidants may enhance the
response to the vasodilator NO in the pulmonary arteries.
Inhibition of NOX or treatment with ROS scavengers may inhibit the
development of chronic hypoxic PAH (26). ROS can reportedly promote pyruvate
kinase M2 (PKM2) phosphorylation and inhibit the glycolytic
activity of PKM2. This leads to proliferation of pulmonary artery
smooth muscle cells (PASMCs) and maintenance of their antioxidant
responses (27). Excessive ROS
generation not only promotes AngII-mediated vascular remodeling,
but also reduces vascular responses to NO. ROS have been
demonstrated to play a key role in promoting the proliferation of
pulmonary artery smooth muscle and EC damage associated with PAH
(28). Pulmonary vascular ECs can
also produce ROS. Additionally, ROS may act as signaling molecules
that induce an increase in the amount of intracellular
Ca2+, including in the cytoplasm of pulmonary ECs, which
causes cell damage (26). Of
note, AngII is also involved in the pathogenesis of ROS-mediated
myocardial and pulmonary vascular cell damage and plays a key role
in the cardiovascular system by activating NOX on cell membranes
(29). AngII not only exacerbates
CM injury to through direct action, but also promotes apoptosis of
pulmonary parenchymal and vascular cells and pulmonary vascular
remodeling, which stimulates almost all types of vascular cells to
produce large amounts of ROS (30). There is some evidence that ROS
produced post-MI can affect lung ECs and that AngII is also
involved in ROS-mediated EC injury. However, the pathological
mechanism of post-MI PAH has not been studied and warrants
investigation. In PAH, large amounts of ROS are harmful to the
already damaged heart, which further impairs cardiac function.
Deterioration in cardiac function increases blood flow shear force
in the pulmonary arteries, which impairs pulmonary vascular
function (Fig. 2).
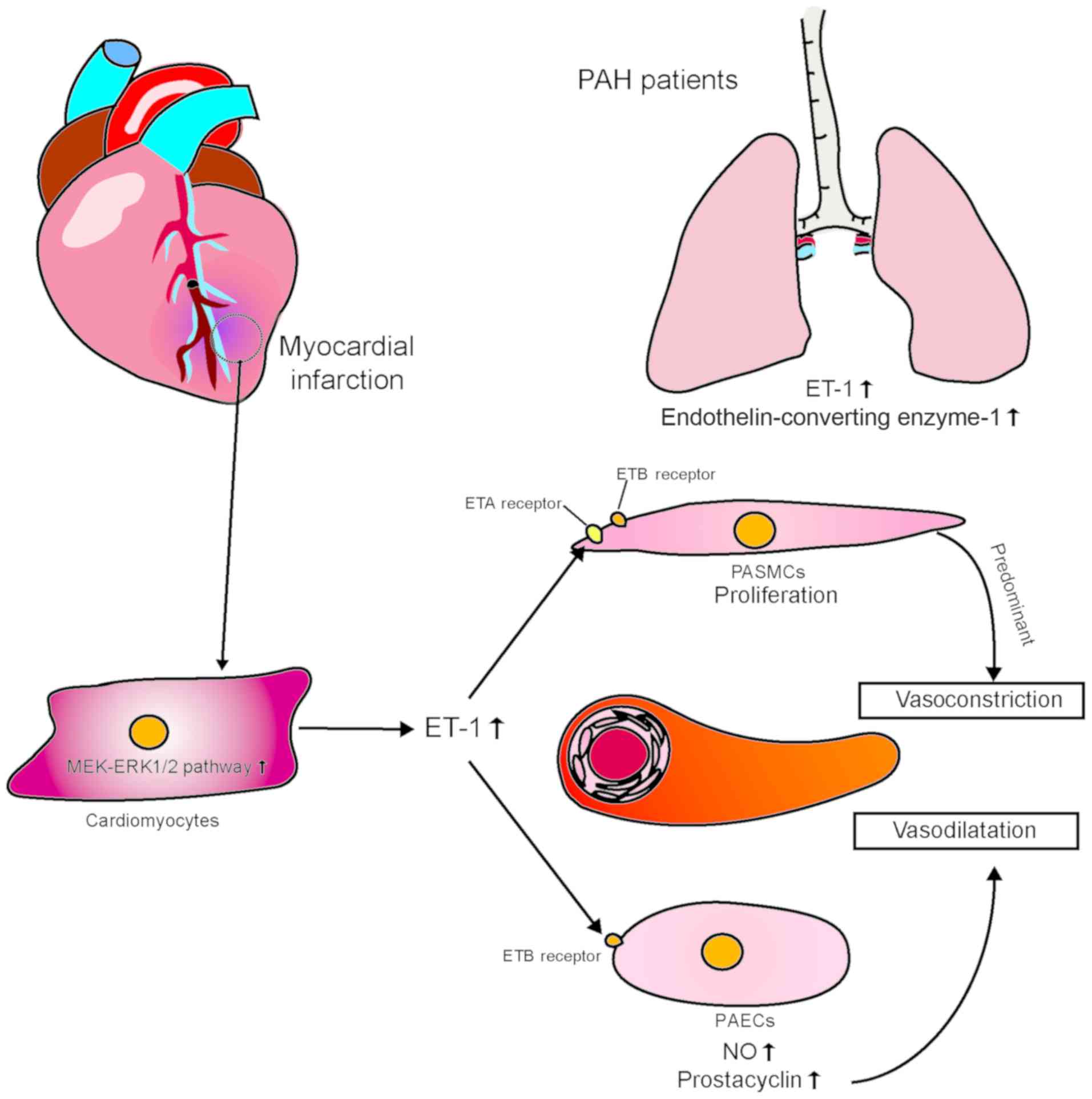
4. Endothelin-1
Endothelin (ET) has three isoforms, namely ET-1, -2
and -3. ET receptors are differentially expressed among different
tissues and organs, and they are coupled with at least four known G
proteins, the resultant complexes being ETA,
ETB1, ETB2 and ETC.
ETB1, ETB2 and ETC have different
binding affinities (31). The ET
pathway in the pulmonary circulation is composed of ET-1,
ETA and ETB (i.e., ETB1 and
ETB2), with ET-1 exhibiting high-affinity binding
(32). ETA and
ETB receptors are present in smooth muscle cells and
promote vasoconstriction and cell proliferation. ETB
receptors are also present in ECs; however, they promote
vasodilation by releasing NO, prostacyclin and other
endothelium-dependent vasodilators. Endogenous ET-1 mainly exerts
its vasoconstrictive effect through ETB on smooth muscle
cells. However, the role of ETA in the pulmonary
vascular circulation has not yet been clearly determined (33-36).
Enhancement of ET system activity is associated with
the severity of PAH. ET receptor antagonists have therefore been
used clinically to treat PAH. They have been shown to have
beneficial effects on PAH morbidity and mortality, emphasizing the
important role of ET-1 in the development of PAH. The drug
treatment of PAH is currently in its early stages, and there are
currently no known drugs that can completely reverse PAH. However,
two drugs appear to be somewhat effective, namely the dual
(ETA and ETB) ET receptor antagonist bosentan
and the selective ET receptor antagonist ambristan. However, their
efficacy is restricted by their limited ability to penetrate tissue
(37-39). Expression of ET-1 is mainly
detected in the ECs of the pulmonary artery and may lead to intimal
fibrosis and thickening of the media. Increased immunoreactivity of
ET-1 in the pulmonary microcirculation and high plasma
concentrations of ET-1 are directly associated with high right
atrial pressure, low pulmonary oxygen saturation, and high
pulmonary vascular resistance. Accumulating evidence suggests that
ET-1 is pathophysiologically involved in the development of
myocardial ischemia and infarction. It has been demonstrated that
plasma ET-1 concentrations are significantly higher in patients
with MI compared with those in healthy individuals (40).
Current research indicates that ET-1 plays an
important role in vasoconstriction and pulmonary microcirculation
remodeling. van Duin et al performed pulmonary vein banding
on pigs and found that, over time, pulmonary artery pressure and
resistance increased significantly. Pre-ET-1 and ET-converting
enzyme-1 also increased in the lungs, as did ET mRNA expression,
with resultant pulmonary vasoconstriction. However, sensitivity to
ET decreased with increased pulmonary vascular contractility, which
indicates that ET-1 plays an important role in pulmonary vascular
remodeling in post-MI PAH (41).
Similarly, in a study using a swine model of MI, Merkus et
al observed stronger ET-mediated vasoconstriction in the
pulmonary circulation, decreased bioavailability of NO, and
impairment of the vasodilation mediated by prostaglandin compared
with swine without MI. All these changes could be normalized by
ETA/ETB receptor blockade (42). Satwiko et al used ET
transgenic mice to study the correlation between PAH and ET and
found that targeted activation of ET-1 exacerbated hypoxia-induced
PAH. Their findings indicate that the pulmonary arteries are more
susceptible to ET-1-mediated vasoconstriction compared with the
systemic arteries, which further emphasizes the importance of ET-1
in the development of PAH (43).
Others have also reported evidence that enhanced ET system activity
may be a causative factor in the development of PAH (44,45), which provides a theoretical basis
for treating PAH by inhibiting the ET system.
ET-1 is synthesized and released from blood vessels
and endocardial ECs, as well as muscle cells. Of note, in patients
with coronary artery spasm, MI and congestive heart failure, the
amounts of ET-1 increase to varying degrees. In a porcine
myocardial ischemia model, it was found that even transient
blockage of coronary blood flow resulted in increased ET-1
production (10). It has also
been reported that the mitogen-activated protein
kinase/extracellular signal-regulated kinase (MEK-ERK1/2) signaling
pathway was a key regulator of ET-1 and ETB receptors in
the myocardium and coronary arteries after ischemia-reperfusion
(I/R) in a rat model (40).
Myocardial cells are subject to ischemia. Reperfusion stimulation
enhances transcriptional expression of ET-1 and vasoconstrictive
ETB receptors via the MEK-ERK1/2 signal transduction
pathway. The vasoconstrictor response to ET-1 in the heart is
mainly mediated by ETA receptors in vascular smooth
muscle cells (VSMCs), whereas vasodilation is mediated by
ETB receptors located in the endothelium. It has been
demonstrated that there is a phenotypic transformation of
ETB receptors in coronary VSMCs, the phenotype changing
from diastolic to contractile (46); this means that, in the case of
myocardial injury, ET-1 may cause coronary artery contraction and
exacerbate myocardial ischemia through systolic ETB
receptors. It remains unclear whether the ET-1 pathway is directly
involved in the development of PAH following myocardial ischemic
injury. Additionally, additional incompletely characterized
intercellular interactions may be responsible for the failure to
develop effective treatments for PAH (Fig. 3).
5. Vascular endothelial growth factor
VEGFs, a family comprising VEGF-A, -B, -C, -D and
-E, and placental growth factor, are named in accordance with the
number of constituent amino acids, e.g., VEGF121 and VEGF145.
VEGF165 is the predominant VEGF-A isoform, the others being
VEGF165, VEGF189 and VEGF206 (47). VEGF-A promotes vascular EC growth
and migration and induces angiogenesis in a variety of in
vivo animal models. VEGF-A165 is the most abundant and
biologically active VEGF-A isoform. VEGF-A165 is primarily
expressed in a variety of tumors, and its expression level is
associated with tumor activity (e.g., development, invasion and
metastasis) (48). The VEGF
receptor (VEGFR) family consists of three subtypes: VEGFR-1, -2 and
-3. All three receptor subtypes contain seven immunoglobulin-like
domains in the extracellular region and a tyrosine kinase domain in
the intracellular region. VEGFR-1 and VEGFR-2 are expressed in ECs
and hematopoietic stem cells (47,49). VEGFR-1 is also expressed in
monocytes and macrophages. By contrast, VEGFR-3 is only expressed
in lymphatic ECs. The VEGF protein has a different affinity for
each of the three VEGFR subtypes. VEGF-A is capable of activating
VEGFR-1 and VEGFR-2; however, VEGFR-1 binds VEGF-A with an affinity
10-fold that for VEGFR-2. VEGF-VEGFR-2 signaling is crucial for
vascular development and maintenance, whereas VEGFR-1 is an
anti-angiogenic decoy receptor for VEGF and is required for normal
vascular development (49,50).
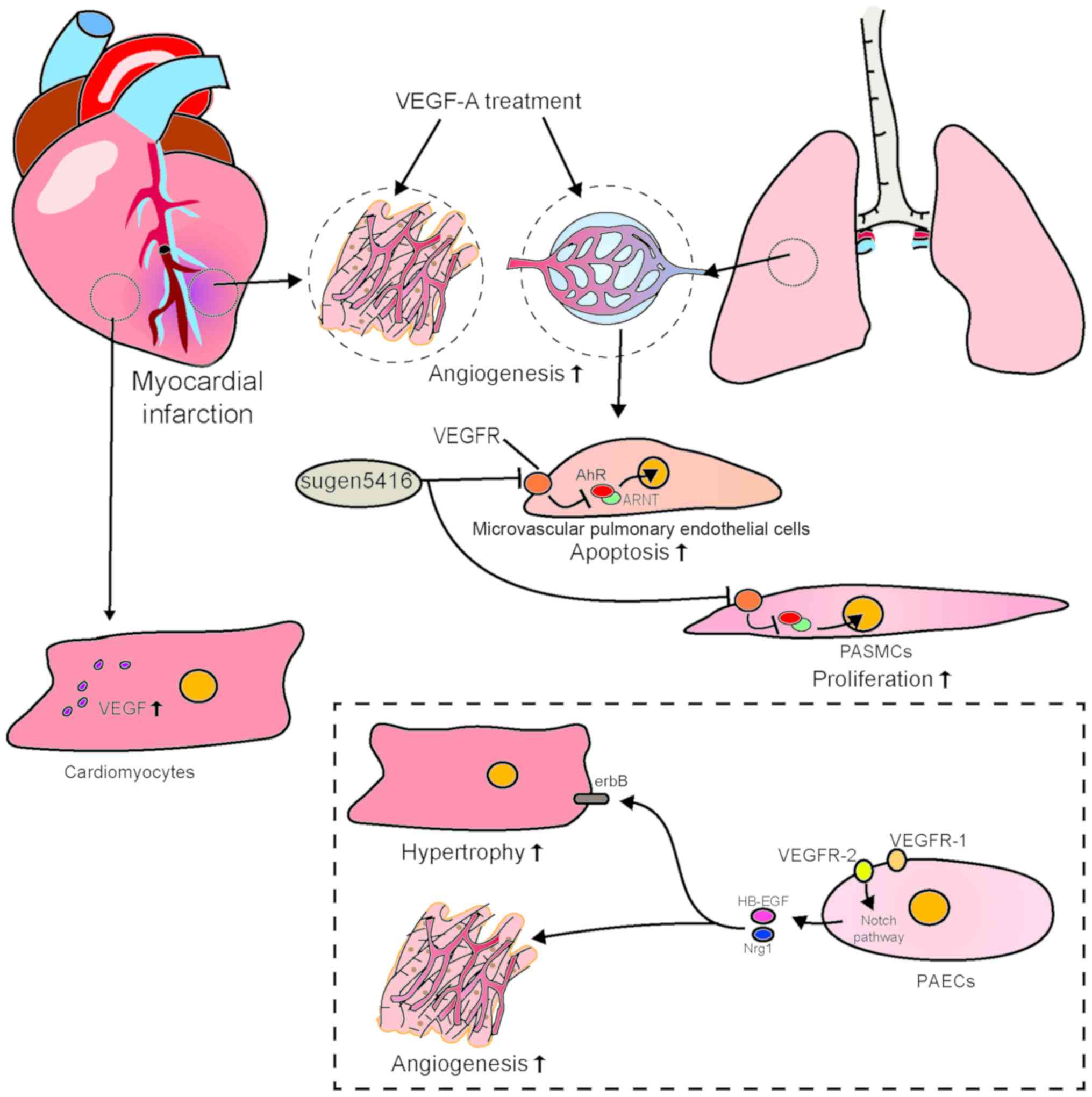
After an MI, the expression of VEGF is upregulated
in the myocardium and the peripheral blood, promoting angiogenesis
and improving cardiac function (51). Therefore, several studies have
used experimental animal models to investigate whether increasing
the amount of VEGF in myocardial tissue protects CMs and improves
cardiac function. One example is the use of collagen-binding domain
specific for myocardial extracellular matrix in animals with
chronic infarction. In one of these studies, modified VEGF was
injected into the myocardium adjacent to the infarct in pig hearts,
which resulted in angiogenesis and subsequent formation of CMs in
the infarcted area for up to 3 months (52). Similarly, other researchers have
used nanoparticle technology to increase the amounts of VEGF in
myocardial tissue, achieving similar results (53). As regard the treatment of
preclinical MI, there have been many reports of successful
promotion of angiogenesis in the infarcted myocardium after
treatment with VEGF gene or protein. However, the responses to VEGF
were found to be dose-dependent in clinical trials. Moreover,
administration of large doses of recombinant VEGF protein has been
found to result in various adverse effects (54,55).
VEGF-A, a mitogen and survival factor characteristic
of vascular ECs, is expressed in the epithelial cells of the
terminal respiratory region in the fetal and postnatal lung
(56) and it exerts a strong
effect on pulmonary angiogenesis. Some clinical and basic studies
have found that plasma VEGF concentrations and the expression of
VEGF and VEGFR in lung tissue increase with hypoxia (55-58). In animal models, blocking VEGF-A
receptors with the VEGF inhibitor Sugen 5416 induces PAH (59). Some studies have demonstrated that
Sugen 5416 induces proliferation of blood secretory ECs and
apoptosis of human microvascular pulmonary ECs by activating
aromatic hydrocarbon receptors (AhRs) (60-62). Sugen 5416 has also been
demonstrated to induce proliferation of human PASMCs by inducing
nuclear translocation of AhR under hypoxic conditions (62). The findings mentioned above
suggest that apoptosis of pulmonary microvascular ECs and
translocation of AhR into the nuclei of human PASMCs lead to
proliferation of pulmonary artery smooth muscle, which may be the
pathological mechanism underlying the development of PAH.
VEGF165b is a specific subtype of VEGF-A with
anti-angiogenic effects (63).
VEGF165b reportedly contributes to the pathophysiology of PAH,
particularly idiopathic PAH (64). VEGFR-2, a transmembrane tyrosine
kinase receptor that is primarily expressed in the pulmonary
endothelium, is the major receptor involved in VEGF-A angiogenesis
(65,66). Inhibition of VEGFR-2 reduces the
density of blood vessels, inhibits formation of alveoli, and
reduces the weight of the lungs of newborn rats compared with those
of the control group. These pathological changes eventually lead to
PAH (67,68). In addition, recombinant human
VEGF-A treatment and VEGFA gene therapy can restore pulmonary
vascular growth and lung structure, and help protect lung function
in rats (69,70). It has been reported that the
expression of VEGFR-2 is weak in patients with pulmonary bronchial
dysplasia (71). There is some
evidence indicating an important role of the VEGF-VEGFR-2 signaling
axis in maintaining endothelial homeostasis. Recent studies have
found that the activity of VEGFR-1 and VEGFR-2 is relatively
balanced in ECs. Kivelä et al found that VEGFR-1-knockout
ECs strongly express neuregulin-1 and heparin-binding epidermal
growth factor via VEGFR-2-Notch signaling. Binding to erbB
receptors on CMs results in induction of hypertrophy of CMs and
cardiac angiogenesis (72).
Neuroregulatory proteins (members of the epidermal growth factor
family) are produced by endothelial and myocardial cells containing
the receptor (erbB) of the ligand. Of note, neuroregulatory
proteins from cardiac ECs can slow hypoxia-related death of
CMS and reoxygenation through the paracrine effects of
CMs (73). Taken together, these
findings indicate that VEGF plays an important and unique role in
the heart and lungs. On the one hand, VEGF affects angiogenesis and
myocardial survival in infarcted myocardium (52,53); on the other hand, VEGF acts on
various complex pathophysiological mechanisms in ECs, particularly
the balance of VEGFR-1 and VEGFR-2 in ECs (72). This determines its signal
transduction direction, thereby affecting the function of ECs. Both
VEGF and its splice variants and several complex signaling pathways
play important roles in PAH, similarly to bone morphogenetic
protein receptor type II (BMPR2). The interaction between BMPR2 and
VEGF also requires attention. It has been reported that absence of
BMPR2 can inhibit VEGF signaling (74). Additionally, as mentioned earlier,
VEGF is also implicated in pulmonary hypertension and myocardial
injury by activating NOX to generate ROS, which indicates the
existence of a complex regulatory network between organs.
Differences in the pathophysiological effects of VEGF between CMs
and pulmonary vascular ECs require further investigations in
diseases such as post-MI PAH (Fig.
4).
6. Bone morphogenetic protein
The pathogenesis of PAH usually involves
abnormalities in the bone morphogenetic protein (BMP) pathway. BMP
is a secreted protein of the transforming growth factor (TGF)-β
superfamily. A series of studies have documented that BMP signaling
plays a key role in the cellular processes of proliferation,
differentiation, and apoptosis in various tissues (75). In particular, it plays an
important role in regulating cell pattern, differentiation,
proliferation and apoptosis in the process of embryogenesis. BMP
signaling is involved in promoting cell survival and proliferation
in distal lung epithelial cells (76). Typical BMP signaling cascades can
be divided into four subgroups as follows: BMP ligands, receptors,
extracellular secretory antagonists and cell-reactive kinases. Over
20 BMP ligands have been identified to date. BMP-binding receptors
are a group of transmembrane serine/threonine kinase receptors
involved in type I (ALK2, ALK3 and ALK6) and type II (BMPR-II,
ActR-IIa, and ActR-IIb) receptors (75). ALK1 is expressed on the surfaces
of pulmonary vascular ECs, but not in PASMCs (77); ALK3 and ALK2 are expressed in both
ECs and PASMCs (78); and BMPR-II
is mainly expressed on the surface of pulmonary vascular ECs, but
less strongly in smooth muscle and interstitial cells (79,80). BMP ligands initiate signal
transduction by binding to type II receptors. They then recruit
type I receptors and activate a range of intracellular kinases,
including classic Smad signals and various mitogen-activated
protein kinases. These signaling pathways are known to be involved
in regulating cell proliferation, differentiation, mitosis, cell
survival and apoptosis (75). BMP
antagonists are secreted proteins that compete with specific BMP
ligands for binding and, thus, inhibit their signal transduction.
The main members of this protein family are noggin, follistatin,
gremlin 1, matrix gla protein, chordin, twisted gastrin 1, and
Bmper (81). BMP antagonists can
inhibit BMP signaling by chelation of BMP ligands in a range of
different cell types.
The BMP signaling pathway is involved in the
pathophysiology of various diseases in adults. For example, an
imbalance of BMP activity is associated with osteoarthritis and
rheumatoid arthritis (82).
Mutations in components of the BMP pathway are also associated with
human gastrointestinal and cardiovascular diseases, such as PAH
(83). There is evidence that
ligands that are closely associated with BMP2 and BMP4 can cause
upregulation of oxidative stress and inflammatory pathways in lung
and coronary ECs. In addition, upregulation of BMP4 mRNA reportedly
occurs in the lung after exposure to a hypoxic environment
(84).
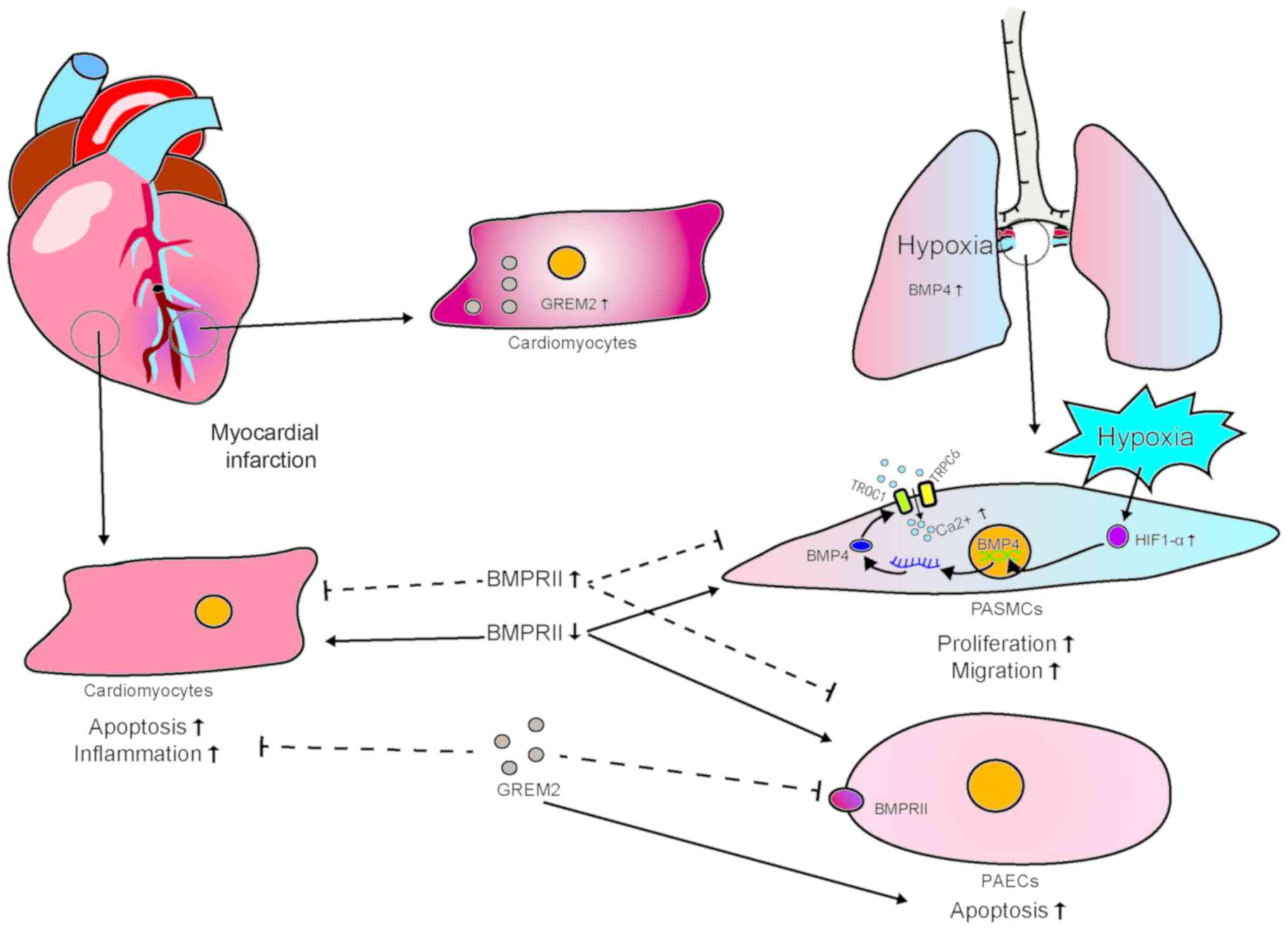
Evidence from patients with PAH and multiple
well-defined experimental PAH models suggests that dysfunction of
pulmonary artery ECs (PAECs) causes increased vascular permeability
and excessive proliferation of PASMCs, inducing perivascular
inflammation (85). Reduction in
the barrier function of PAECs and pulmonary vascular remodeling are
characteristic pathological characteristics of PAH. Abnormal
morphological changes in BMP ligands are also closely associated
with the pathogenesis of PAH. Hypoxia-responsive transcription
factor hypoxia-inducible factor (HIF)-1α reportedly regulates BMP4
transcription by directly binding to the promoter region of the
BMP4 gene and triggering BMP4 signaling (86). Han et al found that SMAD1
deficiency in endothelial or smooth muscle cells may render mice
susceptible to PAH. This deficiency results in impairment of the
balance between BMP4 and TGF-β1-mediated signaling pathways,
indicating BMP downstream mediators. SMAD1 is also crucial for
PAH-related BMPR-II signaling (87). In hypoxic PAH, high intracellular
calcium concentrations play a key role in promoting contraction and
proliferation of PASMCs. In PASMCs, hypoxia-triggered
storage-operated calcium entry greatly promotes high intracellular
calcium concentrations, which increase under hypoxic conditions.
Hypoxia induces stabilization of HIF-1α (86). By transcriptionally activating
BMP4 expression, BMP4 induces stronger expression of TORC1 and
TRPC6. This triggers storage-operated calcium entry, which results
in increased proliferation and migration of PASMCs (88). As regards BMPR-II-related EC
function, Yang et al further demonstrated that adenovirus
overexpression of BMPR-II mutant (kinase-deficient mutation, D485G)
in PAECs not only promotes their apoptosis, but also induces
secretion of growth factors (89). These findings suggest that
abnormalities in BMP signaling lead to increased EC dysfunction and
vascular permeability, and promote vascular smooth muscle cell
proliferation, further contributing to the development and
exacerbation of PAH. Recent research on the treatment of PAH has
shown that inhibiting BMPs by increasing the expression of BMPR-II
and inhibiting BMP ligand antagonists can ameliorate this
condition.
Recent studies have demonstrated that expression of
BMP-2 increases, whereas that of BMP-4 decreases when CMs are
mechanically stimulated, as occurs when BMP2 and BMP2
autocrine/paracrine factors regulate the mechanical conduction and
mechanical stretch of CMs (90).
There is evidence that BMP-mediated transient suppression of signal
transduction can promote differentiation of CMs, and that BMP2
increases the contractility of CMs. Therefore, previous studies
have investigated the potential of BMP for treating heart disease.
For example, administration of recombinant BMP2 has been shown to
reduce infarct size after MI in mice, even for mature CMs (91). Another study demonstrated that
BMP2 treatment can induce c-kit cardiac stem cells to differentiate
into functional CMs; thus, BMP2 can promote repair of infarcted
myocardium, thereby improving cardiac function (92). Activation of BMP4 signaling
promotes apoptosis after MI induced by I/R injury. Additionally,
in vivo treatment with noggin reduces infarct size and
inhibits pre-apoptotic signals while inhibiting Smad1
phosphorylation and JNK activation (91). The BMP antagonist gremlin 2
(Grem2) is required for early cardiac development and CM
differentiation. It has been found that the adult heart strongly
but transiently induces formation of Grem2 in the inflammatory
phase of myocardial tissue repair following experimental induction
of MI. In wild-type mice, intraperitoneal injection of Grem2
protein can reduce post-MI inflammation. It has been found that
BMP2 interacts with TNF-α to induce expression of proinflammatory
proteins and promote leukocyte adhesion in ECs. However, Grem2
specifically inhibits BMP2 and has been shown to control the extent
of inflammatory cell infiltration by inhibiting classical BMP
signaling (93).
The findings mentioned above indicate that BMP
signaling is important for the normal development of the heart and
pulmonary vessels, which suggests that there is BMP signal
crosstalk between the heart and the pulmonary vessels, for which,
however, there is currently no direct evidence. For example, the
endogenous BMP antagonist Grem2, which is induced by MI, can
alleviate inflammatory responses caused by MI. However, whether it
can affect the BMP signaling of pulmonary vascular cells deserves
further investigation. The role of BMP signaling in the early
stages of post-MI PAH also warrants further investigation.
Additionally, BMP2 exerts a positive effect on both CMs and
pulmonary vascular cells, whereas BMP4 has a negative impact. Thus,
when developing BMP-related treatments, signaling pathway crosstalk
between multiple organs must be taken into consideration to ensure
safety. However, the relevance of such crosstalk has not yet been
determined (Fig. 5).
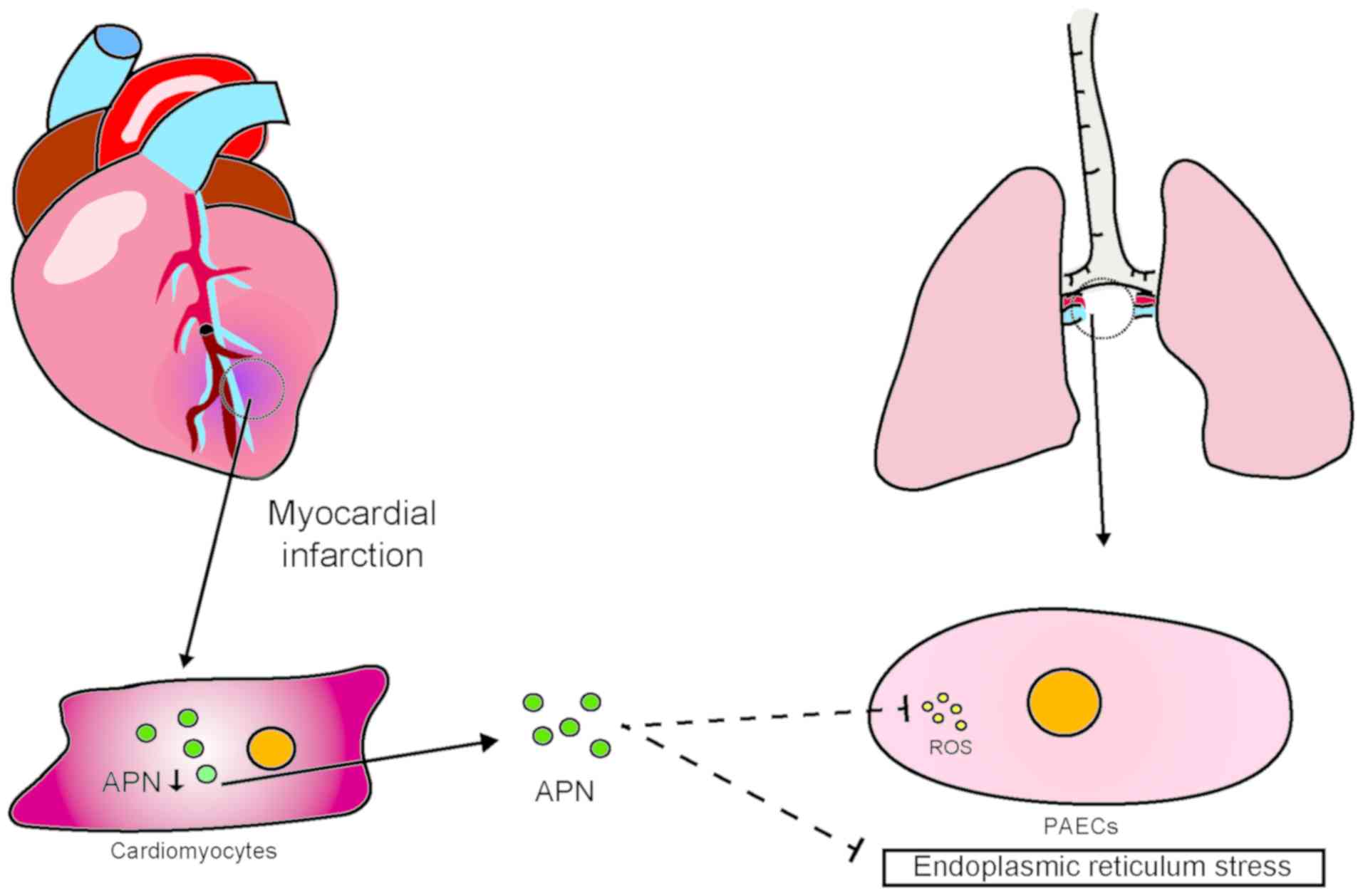
7. Adiponectin
Adiponectin, an insulin-sensitizing hormone secreted
by adipocytes that reduces endoplasmic reticulum stress and ROS in
ECs (94), plays an important
role in the resistance of these cells to oxidative stress. It has
been reported that plasma adiponectin concentrations are
significantly lower in patients with cardiovascular disease
compared with those in healthy individuals. Of note, cardiac cells
can also secrete adiponectin (95,96). Upregulation of the expression of
adiponectin in microvascular ECs and CMs of diabetic mice slows
ischemic injury of the myocardium and improves cardiac function.
Additionally, the adiponectin gene is regulated by HIF-1 (97). Experimental studies have
demonstrated that HIF-1 protects the heart from acute I/R injury
through transcriptional activation of cardiac protective genes
(such as erythropoietin, heme oxygenase-1 and inducible NO
synthase) (95). Interestingly,
HIF-1α is currently considered to play a negative role in the BMP
signaling pathway described earlier in this article. Thus, the
research on the role of adiponectin in pulmonary vessels has been
insufficient thus far. However, the abovementioned evidence
provides new clues regarding the role of adiponectin in the heart
and lungs and the pathological mechanism underlying post-MI PAH
(Fig. 6).
8. Conclusions
There is currently sufficient evidence indicating
that signaling pathways and action molecules are shared between the
heart and lungs, and that these molecules are expressed to
different degrees in the cardiac tissue and pulmonary vessels and
play different roles in the circulation of the heart and lungs. The
origins and development of diseases are not limited to the affected
organ; rather, internal imbalance and dysregulation between organs
usually underlies disease development. Research on the interaction
between the heart and lungs is currently focused mainly on the
effect of hypoxic PAH on the heart. The main therapeutic targets of
PAH are ET-1, NO and prostacyclin; however, there are also RAS,
VEGF, BMP, as well as other signaling pathways. The specific
regulatory mechanisms of these signaling pathways remain unclear.
Existing treatment protocols also have their limitations. Further
studies are required to elucidate pathogenetic and
pathophysiological factors, in order to enable diagnosis of these
diseases by measuring the levels of identified pathology-related
molecules, and reduction of their incidence by implementing
effective preventive drug interventions at an early stage of the
disease. This may both improve the effectiveness of treatment and
greatly reduce the incidence of PAH caused by MI. The current
challenge is to improve the understanding of the pathogenesis of
post-MI PAH, including the mechanisms through which signal
regulators affect PAH and the complexity of cardiopulmonary
physiological characteristics. This would improve the efficacy of
prevention and treatment of this condition. We believe that
elucidating the mechanisms underlying cardiopulmonary interaction
would not only improve our understanding of the pathological
process of PAH post-MI, but may also provide a new basis and
identify new therapeutic targets for the prevention and treatment
of PAH.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81970056,
81700269 and 81741129), the Southern Marine Science and Engineering
Guangdong Laboratory Zhanjiang (no. ZJW-2019-07), the Natural
Science Foundation of Guangdong Province (no. 2019A1515011925), and
the Science and Technology Plan Project of Zhanjiang (no.
2019A01002).
Availability of data and materials
Not applicable.
Authors' contributions
WL, SH and HL contributed to the conception of the
study, WY, HG, JX, SC, XS and YH performed the literature search
and wrote the manuscript. WY prepared the figures. All the authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Breitling S, Ravindran K, Goldenberg NM
and Kuebler WM: The pathophysiology of pulmonary hypertension in
left heart disease. Am J Physiol Lung Cell Mol Physiol.
309:L924–L941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lundgren J and Rådegran G: Pathophysiology
and potential treatments of pulmonary hypertension due to systolic
left heart failure. Acta Physiol (Oxf). 211:314–333. 2014.
View Article : Google Scholar
|
|
3
|
Raiesdana A and Loscalzo J: Pulmonary
arterial hypertension. Ann Med. 38:95–110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Duin RWB, Houweling B, Uitterdijk A,
Duncker DJ and Merkus D: Pulmonary vasodilation by
phosphodiesterase 5 inhibition is enhanced and nitric oxide
independent in early pulmonary hypertension after myocardial
infarction. Am J Physiol Heart Circ Physiol. 314:H170–H179. 2018.
View Article : Google Scholar
|
|
5
|
Hunt JM, Bethea B, Liu X, Gandjeva A,
Mammen PP, Stacher E, Gandjeva MR, Parish E, Perez M, Smith L, et
al: Pulmonary veins in the normal lung and pulmonary hypertension
due to left heart disease. Am J Physiol Lung Cell Mol Physiol.
305:L725–L736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fujimoto Y, Urashima T, Kawachi F, Akaike
T, Kusakari Y, Ida H and Minamisawa S: Pulmonary hypertension due
to left heart disease causes intrapulmonary venous arterialization
in rats. J Thorac Cardiovasc Surg. 154:1742–1753.e8. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghio S, Gavazzi A, Campana C, Inserra C,
Klersy C, Sebastiani R, Arbustini E, Recusani F and Tavazzi L:
Independent and additive prognostic value of right ventricular
systolic function and pulmonary artery pressure in patients with
chronic heart failure. J Am Coll Cardiol. 37:183–188. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lipworth BJ and Dagg KD: Vasoconstrictor
effects of angiotensin II on the pulmonary vascular bed. Chest.
105:1360–1364. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morrell NW, Upton PD, Higham MA, Yacoub
MH, Polak JM and Wharton J: Angiotensin II stimulates proliferation
of human pulmonary artery smooth muscle cells via the AT1 receptor.
Chest. 114(1 Suppl): 90S–91S. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Houweling B, Merkus D, Sorop O, Boomsma F
and Duncker DJ: Role of endothelin receptor activation in secondary
pulmonary hypertension in awake swine after myocardial infarction.
J Physiol. 574:615–626. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Man FS, Tu L, Handoko ML, Rain S,
Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel
E, et al: Dysregulated renin-angiotensin-aldosterone system
contributes to pulmonary arterial hypertension. Am J Respir Crit
Care Med. 186:780–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bruce E, Shenoy V, Rathinasabapathy A,
Espejo A, Horowitz A, Oswalt A, Francis J, Nair A, Unger T, Raizada
MK, et al: Selective activation of angiotensin AT2 receptors
attenuates progression of pulmonary hypertension and inhibits
cardiopulmonary fibrosis. Br J Pharmacol. 172:2219–2231. 2015.
View Article : Google Scholar :
|
|
13
|
Strawn WB, Richmond RS, Ann Tallant E,
Gallagher PE and Ferrario CM: Renin-angiotensin system expression
in rat bone marrow haematopoietic and stromal cells. Br J Haematol.
126:120–126. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Fan Y, Zhou L, Zhu HY, Song YC, Hu
L, Wang Y and Li QP: Pretreatment of mesenchymal stem cells with
angiotensin II enhances paracrine effects, angiogenesis, gap
junction formation and therapeutic efficacy for myocardial
infarction. Int J Cardiol. 188:22–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mendoza-Torres E, Oyarzún A, Mondaca-Ruff
D, Azocar A, Castro PF, Jalil JE, Chiong M, Lavandero S and
Ocaranza MP: ACE2 and vasoactive peptides: Novel players in
cardiovascular/renal remodeling and hypertension. Ther Adv
Cardiovasc Dis. 9:217–237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1-7) and
Mas: New players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar
|
|
17
|
Morrell NW, Atochina EN, Morris KG,
Danilov SM and Stenmark KR: Angiotensin converting enzyme
expression is increased in small pulmonary arteries of rats with
hypoxia-induced pulmonary hypertension. J Clin Invest.
96:1823–1833. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mann S, Bajulaiye A, Sturgeon K, Sabri A,
Muthukumaran G and Libonati JR: Effects of acute angiotensin II on
ischemia reperfusion injury following myocardial infarction. J
Renin Angiotensin Aldosterone Syst. 16:13–22. 2015. View Article : Google Scholar
|
|
19
|
Xu J, Carretero OA, Lin CX, Cavasin MA,
Shesely EG, Yang JJ, Reudelhuber TL and Yang XP: Role of cardiac
overexpression of ANG II in the regulation of cardiac function and
remodeling postmyocardial infarction. Am J Physiol Heart Circ
Physiol. 293:H1900–H1907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shenoy V, Qi Y, Katovich MJ and Raizada
MK: ACE2, a promising therapeutic target for pulmonary
hypertension. Curr Opin Pharmacol. 11:150–155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lassègue B, San Martín A and Griendling
KK: Biochemistry, physiology, and pathophysiology of NADPH oxidases
in the cardiovascular system. Circ Res. 110:1364–1390. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu X and Zuo L: Characterization of
oxygen radical formation mechanism at early cardiac ischemia. Cell
Death Dis. 4:e7872013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shiomi T, Tsutsui H, Matsusaka H, Murakami
K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H and
Takeshita A: Overexpression of glutathione peroxidase prevents left
ventricular remodeling and failure after myocardial infarction in
mice. Circulation. 109:544–549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bowers R, Cool C, Murphy RC, Tuder RM,
Hopken MW, Flores SC and Voelkel NF: Oxidative stress in severe
pulmonary hypertension. Am J Respir Crit Care Med. 169:764–769.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Shults NV and Suzuki YJ: Oxidative
profiling of the failing right heart in rats with pulmonary
hypertension. PLoS One. 12:e01768872017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suresh K and Shimoda LA: Endothelial cell
reactive oxygen species and Ca2+ signaling in pulmonary
hypertension. Adv Exp Med Biol. 967:299–314. 2017. View Article : Google Scholar
|
|
27
|
Guo D, Gu J, Jiang H, Ahmed A, Zhang Z and
Gu Y: Inhibition of pyruvate kinase M2 by reactive oxygen species
contributes to the development of pulmonary arterial hypertension.
J Mol Cell Cardiol. 91:179–187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jaitovich A and Jourd'heuil D: A brief
overview of nitric oxide and reactive oxygen species signaling in
hypoxia-induced pulmonary hypertension. Adv Exp Med Biol.
967:71–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shahzad S, Hasan A, Faizy AF, Mateen S,
Fatima N and Moin S: Elevated DNA damage, oxidative stress, and
impaired response defense system inflicted in patients with
myocardial infarction. Clin Appl Thromb Hemost. 24:780–789. 2018.
View Article : Google Scholar
|
|
30
|
Freund-Michel V, Guibert C, Dubois M,
Courtois A, Marthan R, Savineau JP and Muller B: Reactive oxygen
species as therapeutic targets in pulmonary hypertension. Ther Adv
Respir Dis. 7:175–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kedzierski RM and Yanagisawa M: Endothelin
system: The double-edged sword in health and disease. Annu Rev
Pharmacol Toxicol. 41:851–876. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Madonna R, Cocco N and De Caterina R:
Pathways and drugs in pulmonary arterial hypertension-focus on the
role of endothelin receptor antagonists. Cardiovasc Drugs Ther.
29:469–479. 2015. View Article : Google Scholar
|
|
33
|
Sato K, Oka M, Hasunuma K, Ohnishi M, Sato
K and Kira S: Effects of separate and combined ETA and ETB blockade
on ET-1-induced constriction in perfused rat lungs. Am J Physiol.
269:L668–L672. 1995.PubMed/NCBI
|
|
34
|
Taguchi K and Hattori Y: Unlooked-for
significance of cardiac versus vascular effects of endothelin-1 in
the pathophysiology of pulmonary arterial hypertension. Circ Res.
112:227–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Van Hung T, Emoto N, Vignon-Zellweger N,
Nakayama K, Yagi K, Suzuki Y and Hirata K: Inhibition of vascular
endothelial growth factor receptor under hypoxia causes severe,
human-like pulmonary arterial hypertension in mice: Potential roles
of interleukin-6 and endothelin. Life Sci. 118:313–328. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Merkus D, Houweling B, Mirza A, Boomsma F,
van den Meiracker AH and Duncker DJ: Contribution of endothelin and
its receptors to the regulation of vascular tone during exercise is
different in the systemic, coronary and pulmonary circulation.
Cardiovasc Res. 59:745–754. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Galiè N, Olschewski H, Oudiz RJ, Torres F,
Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker
EB, et al: Ambrisentan for the treatment of pulmonary arterial
hypertension: Results of the ambrisentan in pulmonary arterial
hypertension, randomized, double-blind, placebo-controlled,
multicenter, efficacy (ARIES) study 1-2. Circulation.
117:3010–3019. 2008. View Article : Google Scholar
|
|
38
|
Oudiz RJ, Galiè N, Olschewski H, Torres F,
Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker
EB, et al: Long-term ambrisentan therapy for the treatment of
pulmonary arterial hypertension. J Am Coll Cardiol. 54:1971–1981.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tanaka Y, Hino M and Gemma A: Potential
benefit of bosentan therapy in borderline or less severe pulmonary
hypertension secondary to idiopathic pulmonary fibrosis-an interim
analysis of results from a prospective, single-center, randomized,
parallel-group study. BMC Pulm Med. 17:2002017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Skovsted GF, Kruse LS, Berchtold LA, Grell
AS, Warfvinge K and Edvinsson L: Myocardial ischemia-reperfusion
enhances transcriptional expression of endothelin-1 and
vasoconstrictor ETB receptors via the protein kinase MEK-ERK1/2
signaling pathway in rat. PLoS One. 12:e01741192017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van Duin RWB, Stam K, Cai Z, Uitterdijk A,
Garcia-Alvarez A, Ibanez B, Danser AHJ, Reiss IKM, Duncker DJ and
Merkus D: Transition from post-capillary pulmonary hypertension to
combined pre- and post-capillary pulmonary hypertension in swine: A
key role for endothelin. J Physiol. 597:1157–1173. 2019. View Article : Google Scholar
|
|
42
|
Merkus D, Houweling B, de Beer VJ, Everon
Z and Duncker DJ: Alterations in endothelial control of the
pulmonary circulation in exercising swine with secondary pulmonary
hypertension after myocardial infarction. J Physiol. 580:907–923.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Satwiko MG, Ikeda K, Nakayama K, Yagi K,
Hocher B, Hirata K and Emoto N: Targeted activation of endothelin-1
exacerbates hypoxia-induced pulmonary hypertension. Biochem Biophys
Res Commun. 465:356–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hocher B, Thöne-Reineke C, Rohmeiss P,
Schmager F, Slowinski T, Burst V, Siegmund F, Quertermous T, Bauer
C, Neumayer HH, et al: Endothelin-1 transgenic mice develop
glomerulosclerosis, interstitial fibrosis, and renal cysts but not
hypertension. J Clin Invest. 99:1380–1389. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Giaid A, Yanagisawa M, Langleben D, Michel
RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP and Stewart
DJ: Expression of endothelin-1 in the lungs of patients with
pulmonary hypertension. N Engl J Med. 328:1732–1739. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wackenfors A, Emilson M, Ingemansson R,
Hortobagyi T, Szok D, Tajti J, Vecsei L, Edvinsson L and Malmsjö M:
Ischemic heart disease induces upregulation of endothelin receptor
mRNA in human coronary arteries. Eur J Pharmacol. 484:103–109.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tammela T, Enholm B, Alitalo K and
Paavonen K: The biology of vascular endothelial growth factors.
Cardiovasc Res. 65:550–563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kikuchi R, Stevens M, Harada K, Oltean S
and Murohara T: Anti-angiogenic isoform of vascular endothelial
growth factor-A in cardiovascular and renal disease. Adv Clin Chem.
88:1–33. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ruhrberg C, Gerhardt H, Golding M, Watson
R, Ioannidou S, Fujisawa H, Betsholtz C and Shima DT: Spatially
restricted patterning cues provided by heparin-binding VEGF-A
control blood vessel branching morphogenesis. Genes Dev.
16:2684–2698. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang Y, Shi C, Hou X, Zhao Y, Chen B, Tan
B, Deng Z, Li Q, Liu J, Xiao Z, et al: Modified VEGF targets the
ischemic myocardium and promotes functional recovery after
myocardial infarction. J Control Release. 213:27–35. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shi C, Zhao Y, Yang Y, Chen C, Hou X, Shao
J, Yao H, Li Q, Xia Y and Dai J: Collagen-binding VEGF targeting
the cardiac extracellular matrix promotes recovery in porcine
chronic myocardial infarction. Biomater Sci. 6:356–363. 2018.
View Article : Google Scholar
|
|
53
|
Oduk Y, Zhu W, Kannappan R, Zhao M,
Borovjagin AV, Oparil S and Zhang JJ: VEGF nanoparticles repair the
heart after myocardial infarction. Am J Physiol Heart Circ Physiol.
314:H278–H284. 2018. View Article : Google Scholar :
|
|
54
|
Henry TD, Annex BH, McKendall GR, Azrin
MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman
DS, et al: The VIVA trial: Vascular endothelial growth factor in
ischemia for vascular angiogenesis. Circulation. 107:1359–1365.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sato K, Wu T, Laham RJ, Johnson RB,
Douglas P, Li J, Sellke FW, Bunting S, Simons M and Post MJ:
Efficacy of intracoronary or intravenous VEGF165 in a pig model of
chronic myocardial ischemia. J Am Coll Cardiol. 37:616–623. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bhatt AJ, Amin SB, Chess PR, Watkins RH
and Maniscalco WM: Expression of vascular endothelial growth factor
and Flk-1 in developing and glucocorticoid-treated mouse lung.
Pediatr Res. 47:606–613. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Eddahibi S, Humbert M, Sediame S, Chouaid
C, Partovian C, Maître B, Teiger E, Rideau D, Simonneau G, Sitbon O
and Adnot S: Imbalance between platelet vascular endothelial growth
factor and platelet-derived growth factor in pulmonary
hypertension. Effect of prostacyclin therapy. Am J Respir Crit Care
Med. 162:1493–1499. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tuder RM, Flook BE and Voelkel NF:
Increased gene expression for VEGF and the VEGF receptors KDR/Flk
and Flt in lungs exposed to acute or to chronic hypoxia. Modulation
of gene expression by nitric oxide. J Clin Invest. 95:1798–1807.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Al-Husseini A, Kraskauskas D, Mezzaroma E,
Nordio A, Farkas D, Drake JI, Abbate A, Felty Q and Voelkel NF:
Vascular endothelial growth factor receptor 3 signaling contributes
to angioobliterative pulmonary hypertension. Pulm Circ. 5:101–116.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
60
|
Taraseviciene-Stewart L, Kasahara Y, Alger
L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF and Tuder RM:
Inhibition of the VEGF receptor 2 combined with chronic hypoxia
causes cell death-dependent pulmonary endothelial cell
proliferation and severe pulmonary hypertension. FASEB J.
15:427–438. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nicolls MR, Mizuno S,
Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A,
Gomez-Arroyo JG, Voelkel NF and Bogaard HJ: New models of pulmonary
hypertension based on VEGF receptor blockade-induced endothelial
cell apoptosis. Pulm Circ. 2:434–442. 2012. View Article : Google Scholar
|
|
62
|
Dean A, Gregorc T, Docherty CK, Harvey KY,
Nilsen M, Morrell NW and MacLean MR: Role of the Aryl hydrocarbon
receptor in sugen 5416-induced experimental pulmonary hypertension.
Am J Respir Cell Mol Biol. 58:320–330. 2018. View Article : Google Scholar :
|
|
63
|
Bates DO, Cui TG, Doughty JM, Winkler M,
Sugiono M, Shields JD, Peat D, Gillatt D and Harper SJ: VEGF165b,
an inhibitory splice variant of vascular endothelial growth factor,
is down-regulated in renal cell carcinoma. Cancer Res.
62:4123–4131. 2002.PubMed/NCBI
|
|
64
|
Suzuki S, Yoshihisa A, Yokokawa T, Misaka
T, Sakamoto N, Sugimoto K, Yamaki T, Kunii H, Nakazato K, Saitoh SI
and Takeishi Y: Association between levels of anti-angiogenic
isoform of vascular endothelial growth factor A and pulmonary
hypertension. Int J Cardiol. 222:416–420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signaling-in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yancopoulos GD, Davis S, Gale NW, Rudge
JS, Wiegand SJ and Holash J: Vascular-specific growth factors and
blood vessel formation. Nature. 407:242–248. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jakkula M, Le Cras TD, Gebb S, Hirth KP,
Tuder RM, Voelkel NF and Abman SH: Inhibition of angiogenesis
decreases alveolarization in the developing rat lung. Am J Physiol
Lung Cell Mol Physiol. 279:L600–L607. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Le Cras TD, Markham NE, Tuder RM, Voelkel
NF and Abman SH: Treatment of newborn rats with a VEGF receptor
inhibitor causes pulmonary hypertension and abnormal lung
structure. Am J Physiol Lung Cell Mol Physiol. 283:L555–L562. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kunig AM, Balasubramaniam V, Markham NE,
Morgan D, Montgomery G, Grover TR and Abman SH: Recombinant human
VEGF treatment enhances alveolarization after hyperoxic lung injury
in neonatal rats. Am J Physiol Lung Cell Mol Physiol.
289:L529–L535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Thébaud B, Ladha F, Michelakis ED, Sawicka
M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G
and Archer SL: Vascular endothelial growth factor gene therapy
increases survival, promotes lung angiogenesis, and prevents
alveolar damage in hyperoxia-induced lung injury: Evidence that
angiogenesis participates in alveolarization. Circulation.
112:2477–2486. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mahlman M, Huusko JM, Karjalainen MK,
Kaukola T, Marttila R, Ojaniemi M, Haataja R, Lavoie PM, Rämet M
and Hallman M; Gen-BPD Study Group: Genes encoding vascular
endothelial growth factor A (VEGF-A) and VEGF receptor 2 (VEGFR-2)
and risk for bronchopulmonary dysplasia. Neonatology. 108:53–59.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kivelä R, Hemanthakumar KA, Vaparanta K,
Robciuc M, Izumiya Y, Kidoya H, Takakura N, Peng X, Sawyer DB,
Elenius K, et al: Endothelial cells regulate physiological
cardiomyocyte growth via VEGFR2-mediated paracrine signaling.
Circulation. 139:2570–2584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hedhli N, Huang Q, Kalinowski A, Palmeri
M, Hu X, Russell RR and Russell KS: Endothelium-derived neuregulin
protects the heart against ischemic injury. Circulation.
123:2254–2262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Nagashima T, Li Q, Clementi C, Lydon JP,
DeMayo FJ and Matzuk MM: BMPR2 is required for postimplantation
uterine function and pregnancy maintenance. J Clin Invest.
123:2539–2550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Morrell NW, Bloch DB, ten Dijke P, Goumans
MJ, Hata A, Smith J, Yu PB and Bloch KD: Targeting BMP signalling
in cardiovascular disease and anaemia. Nat Rev Cardiol. 13:106–120.
2016. View Article : Google Scholar :
|
|
76
|
Eblaghie MC, Reedy M, Oliver T, Mishina Y
and Hogan BL: Evidence that autocrine signaling through Bmpr1a
regulates the proliferation, survival and morphogenetic behavior of
distal lung epithelial cells. Dev Biol. 291:67–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Trembath RC, Thomson JR, Machado RD,
Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE,
Humbert M, et al: Clinical and molecular genetic features of
pulmonary hypertension in patients with hereditary hemorrhagic
telangiectasia. N Engl J Med. 345:325–334. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Upton PD, Long L, Trembath RC and Morrell
NW: Functional characterization of bone morphogenetic protein
binding sites and Smad1/5 activation in human vascular cells. Mol
Pharmacol. 73:539–552. 2008. View Article : Google Scholar
|
|
79
|
Atkinson C, Stewart S, Upton PD, Machado
R, Thomson JR, Trembath RC and Morrell NW: Primary pulmonary
hypertension is associated with reduced pulmonary vascular
expression of type II bone morphogenetic protein receptor.
Circulation. 105:1672–1678. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Southwood M, Jeffery TK, Yang X, Upton PD,
Hall SM, Atkinson C, Haworth SG, Stewart S, Reynolds PN, Long L, et
al: Regulation of bone morphogenetic protein signalling in human
pulmonary vascular development. J Pathol. 214:85–95. 2008.
View Article : Google Scholar
|
|
81
|
Brazil DP, Church RH, Surae S, Godson C
and Martin F: BMP signalling: Agony and antagony in the family.
Trends Cell Biol. 25:249–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lories RJ and Luyten FP: Bone
morphogenetic protein signaling in joint homeostasis and disease.
Cytokine Growth Factor Rev. 16:287–298. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Deng Z, Morse JH, Slager SL, Cuervo N,
Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ,
et al: Familial primary pulmonary hypertension (gene PPH1) is
caused by mutations in the bone morphogenetic protein receptor-II
gene. Am J Hum Genet. 67:737–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Frank DB, Abtahi A, Yamaguchi DJ, Manning
S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE and de Caestecker MP:
Bone morphogenetic protein 4 promotes pulmonary vascular remodeling
in hypoxic pulmonary hypertension. Circ Res. 97:496–504. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sylvester JT, Shimoda LA, Aaronson PI and
Ward JP: Hypoxic pulmonary vasoconstriction. Physiol Rev.
92:367–520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang J, Fu X, Yang K, Jiang Q, Chen Y, Jia
J, Duan X, Wang EW, He J, Ran P, et al: Hypoxia inducible
factor-1-dependent up-regulation of BMP4 mediates hypoxia-induced
increase of TRPC expression in PASMCs. Cardiovasc Res. 107:108–118.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Han C, Hong KH, Kim YH, Kim MJ, Song C,
Kim MJ, Kim SJ, Raizada MK and Oh SP: SMAD1 deficiency in either
endothelial or smooth muscle cells can predispose mice to pulmonary
hypertension. Hypertension. 61:1044–1052. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Li X, Lu W, Fu X, Zhang Y, Yang K, Zhong
N, Ran P and Wang J: BMP4 increases canonical transient receptor
potential protein expression by activating p38 MAPK and ERK1/2
signaling pathways in pulmonary arterial smooth muscle cells. Am J
Respir Cell Mol Biol. 49:212–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yang X, Long L, Reynolds PN and Morrell
NW: Expression of mutant BMPR-II in pulmonary endothelial cells
promotes apoptosis and a release of factors that stimulate
proliferation of pulmonary arterial smooth muscle cells. Pulm Circ.
1:103–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tokola H, Rysä J, Pikkarainen S, Hautala
N, Leskinen H, Kerkelä R, Ilves M, Aro J, Vuolteenaho O, Ritvos O
and Ruskoaho H: Bone morphogenetic protein-2-a potential
auto-crine/paracrine factor in mediating the stretch activated
B-type and atrial natriuretic peptide expression in cardiac
myocytes. Mol Cell Endocrinol. 399:9–21. 2015. View Article : Google Scholar
|
|
91
|
Pachori AS, Custer L, Hansen D, Clapp S,
Kemppa E and Klingensmith J: Bone morphogenetic protein 4 mediates
myocardial ischemic injury through JNK-dependent signaling pathway.
J Mol Cell Cardiol. 48:1255–1265. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang YL, Zhang G, Wang HJ, Tan YZ and Wang
XY: Preinduction with bone morphogenetic protein-2 enhances
cardiomyogenic differentiation of c-kit+ mesenchymal
stem cells and repair of infarcted myocardium. Int J Cardiol.
265:173–180. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sanders LN, Schoenhard JA, Saleh MA,
Mukherjee A, Ryzhov S, McMaster WG Jr, Nolan K, Gumina RJ, Thompson
TB, Magnuson MA, et al: BMP antagonist gremlin 2 limits
inflammation after myocardial infarction. Circ Res. 119:434–449.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chen H, Montagnani M, Funahashi T,
Shimomura I and Quon MJ: Adiponectin stimulates production of
nitric oxide in vascular endothelial cells. J Biol Chem.
278:45021–45026. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chow WS, Cheung BM, Tso AW, Xu A, Wat NM,
Fong CH, Ong LH, Tam S, Tan KC, Janus ED, et al:
Hypoadiponectinemia as a predictor for the development of
hypertension: A 5-year prospective study. Hypertension.
49:1455–1461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Amin RH, Mathews ST, Alli A and Leff T:
Endogenously produced adiponectin protects cardiomyocytes from
hypertrophy by a PPARgamma-dependent autocrine mechanism. Am J
Physiol Heart Circ Physiol. 299:H690–H698. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Natarajan R, Salloum FN, Fisher BJ,
Kukreja RC and Fowler AA III: Hypoxia inducible factor-1
upregulates adipo-nectin in diabetic mouse hearts and attenuates
post-ischemic injury. J Cardiovasc Pharmacol. 51:178–187. 2008.
View Article : Google Scholar : PubMed/NCBI
|