Introduction
Connexin 43 (Cx43) is a major gap-junction protein
that mediates intercellular communication by promoting the passage
of small molecules and ions (1).
In cardiomyocytes, Cx43 regulates electrical coupling and
synchronous contraction (2).
Alterations to Cx43 expression and distribution have been reported
for several cardiopathological conditions, such as heart failure.
The decreased expression and/or heterogeneous distribution of Cx43
has been demonstrated in the myocardium of patients with
hypertrophic cardiomyopathies, dilated cardiomyopathies, ischemic
cardiomyopathies and clinical congestive heart failure (3).
In addition to the well-known role of Cx43 in the
cell membrane, in recent years, ample attention has been paid to
the mitochondrial isoform of Cx43 (mitoCx43). It is well known that
Cx43 translocates to the mitochondrial membrane through a mechanism
that involves Hsp90 as its chaperone and Tom20 as a mitochondrial
translocase (4). Previous studies
have reported that mitoCx43 plays an important role in the balance
of reactive oxygen species (ROS) for redox signalling (5,6).
MitoCx43 is also involved in cardioprotection, as it has been
demonstrated that it is overexpressed in ischemic pre-conditioning
(7), as well as in other forms of
cardioprotection, through the control of the initiation of
apoptosis (8). The authors have
previously demonstrated an increase in mitoCx43 expression in H9c2
cells (9) and in hearts of mice
treated with doxorubicin (Doxo) (10). This overexpression is an adaptive
response that can be activated to counteract the intracellular
Ca2+ overload, ROS production and the propagation of
apoptotic signals induced by Doxo. Indeed, Doxo is widely used as a
chemotherapeutic agent, although its clinical use is hampered by
its potential to elicit cardiotoxicity. The onset of Doxo-induced
cardiomyopathy can occur without warning, and this condition can be
rapidly abrogated; moreover, oxidative stress has been recognized
as the main cause in this process (11). A variety of mechanisms are
involved in Doxo-induced cardiotoxicity, such as oxidative stress,
the dysregulation of calcium homeostasis and inflammation (12), and these all converge on the
mitochondria (13). The induction
of free radical stress by Doxo results in mitochondrial membrane
depolarization and the release of cytochrome c into the
cytoplasm, which can then trigger apoptosis (9).
In addition to oxidative stress, there is evidence
to indicate that nitrosative stress is involved in Doxo-induced
cardio-toxicity, as increases in reactive nitrogen species (such as
peroxynitrite) have been reported (14). Peroxynitrite is a potent, reactive
and cytotoxic free radical that is formed through a reaction that
involves nitric oxide (NO) and the superoxide anion
(O2−). In Doxo-treated cardiomyocytes, large
amounts of NO are produced by the inducible isoform of NO synthase
(iNOS), a downstream effector of the nuclear transcription factor
NF-κB (15). Both of these
oxidative species (i.e., NO, O2−), are
mediators of inflammatory conditions.
ROS are oxygen-based chemical species that show high
reactivity. These include free radicals, such as OH− and
O2−, and non-radicals that can generate free
radicals, such as hydrogen peroxide (H2O2). O
−2 is a primary radical that can lead to the formation
of other types of ROS, such as H2O2 and
OH−. OH− is also generated by the reduction
of H2O2 in the presence of endogenous iron,
through the Fenton reaction. In addition, OH− can arise
from electron exchange between O2− and
H2O2, via the Harber-Weiss reaction.
Furthermore, when both O2− and NO are
synthesized within a few cell diameters, they can combine
spontaneously to form peroxynitrite (ONOO−), which is a
potent, reactive and cytotoxic free radical (16,17).
Three isoforms of NOS have been defined: Endothelial
NOS (eNOS or NOS3), neuronal NOS (nNOS or NOS1) and inducible NOS
(iNOS or NOS2). NOS1 and NOS3 are constitutive enzymes that are
controlled by intracellular Ca2+/calmodulin; NOS2 is
inducible at the level of gene transcription, and it is
Ca2+-independent and expressed by macrophages and other
tissues in response to (pro)inflammatory mediators, such as NF-κB,
which was observed to be increased in the present study.
There is ample evidence to indicate that oxidative
stress and inflammation are implicated in the pathogenesis of
congestive heart failure. Indeed, Doxo itself is a potent activator
of NF-κB (18), the main nuclear
transcription factor that is involved in inflammation. A sustained
inflammatory/oxidative environment leads to damage to cells, which
can then remain in a vicious circle of impaired pathways (17,19,20). Furthermore, an increase in NO
production in the myocardium in itself can result in the nitration
of actin and other cytoskeletal proteins, thus altering their
structures and resulting in harmful effects on the contractile
function of myofilaments (21).
NO is necessary for the regulation of cardiac
function during normal cardiac physiology, including for coronary
vasodilatation, the inhibition of platelet and neutrophil adhesion
and activation, and the modulation of cardiac contractile function.
NO also plays a protective role against the ischemic and/or failing
heart. This protective role is mediated through several mechanisms,
which include the stimulation of soluble guanylyl cyclase. This
leads to a decrease in the concentration of intracellular
Ca2+ and inhibition of oxidative stress. Therefore,
O2− can exert cytotoxic effects not only
directly due to O2− itself, but also mediated
through the inactivation of cytoprotective NO and the formation of
the highly reactive oxidant ONOO−, which is produced
following interactions between NO and
O2−.
It was hypothesized that as a regulator of
mitochondrial ROS production, mCx43 is also involved in the
regulation of antioxidant enzymes and in the production of
downstream effectors of oxidative/nitrosative signaling pathways.
For this purpose, a well-designed in-vitro model of
Doxo-induced cardiotoxicity with embryonic rat heart
cardiomyoblasts (H9c2 cells) was used, as these cells are generally
accepted as a good model to study the mechanisms of cellular and
cardiac protection (9). To
achieve this goal, the H9c2 cells were treated with Doxo and
radicicol, a well-established Hsp90 inhibitor (22) that, as previously reported, was
used herein to inhibit the translocation of Cx43 to the
mitochondria (9). The present
study provides evidence that Doxo-induced cardiotoxicity occurs not
only via caspase-mediated apoptosis, but also through the NF-κB
signaling pathway. Oxidative and nitrosative stress-induced damage
is amplified in H9c2 cells in which the translocation of Cx43 to
the mitochondrion is inhibited, which thus confirms the
cardioprotective effects of mitoCx43 (Fig. S1).
Materials and methods
Materials and cell culture
Doxo was obtained from Baxter Manufacturing Spa.
Radicicol and L-NAME were from Sigma-Aldrich; Merck KGaA. The H9c2
embryonic rat heart cardiomyocyte-derived cell line was purchased
from the American Tissue Culture Collection (ATCC), with mycoplasma
testing carried out. The H9c2 cells were grown to confluence in
Dulbecco's modified Eagle's medium (DMEM; Microgem) with 10% fetal
bovine serum (FBS; Microgem) and antibiotics (25 U/ml penicillin;
25 U/ml streptomycin) under an atmosphere of 95% air/ 5%
CO2 at 37°C.
Experimental protocol
The H9c2 cells were treated with 1 µM Doxo
for 3or 6 h in DMEM, 10% FBS. MitoCx43 translocation was inhibited
with 1 µM radicicol, an Hsp90 inhibitor. Radicicol was
administered 30 min prior to the Doxo treatments.
Cell morphology
The H9c2 cells were seeded on glass cover-slips, and
after 24 h they were treated with Doxo and radicicol according to
the protocol described above. At the indicated time points, the
H9c2 cells were fixed with 70% ethanol and stained with 1%
toluidine blue solution (TAAB Laboratories Equipment Ltd.) for 15
min at room temperature. Images of approximately 50 random fields
of view at a magnification of ×20 were obtained using a light
microscope (Axioskop 40; Carl Zeiss) equipped with a videocamera
(Coolsnap; Photometrics). The images were analyzed using Axio
Vision software, SE64.
Total RNA isolation and RT-qPCR
analysis
The H9c2 cells were seeded at a density of
7.0×105 cells/well into 100-mm culture dishes (Corning,
Inc.) and treated as described above. Total RNA was extracted using
EuroGold TriFast (EuroClone S.p.A.), according to the manufacturer
instructions. RNA concentrations and quality were determined by
absorbance measurements using a UV-Vis spectrophotometer (NanoDrop
2000; Thermo Fisher Scientific, Inc.). The ratios of the absorbance
at 260 to 280 nm and at 260 to 230 nm were used to determine the
purity of the total RNA samples, which were then stored at −70°C.
Reverse transcription reactions were carried out using reverse
transcription-quantitative PCR (RT-qPCR; GoTaq 2-Step system;
Promega Corp.). The cDNAs were synthesized starting from 1
µg purified total RNA, according to the manufacturer
instructions. The expression level of the catalase (CAT),
manganese superoxide dismutase (MnSOD) and iNOS genes
were evaluated using SYBR-Green RT-qPCR analysis (StepOne 2.0;
Applied Biosystems; Thermo Fisher Scientific, Inc.). The cycling
conditions were performed as follows: 10 min at 95°C and 40 cycles
of 15 sec at 95°C followed by 1 min at 60°C and final elongation of
15 sec at 95°C. The data were analyzed using the comparative Cq
method, and are illustrated graphically as 2−ΔΔCq ±
standard deviation (SD) (23).
Targets and reference genes were amplified separately in
triplicate, in 10 µl containing 1 µl template cDNA,
0.2 µl primers mixture, and 5 µl enzyme mix (GoTaq
2-Step RT-qPCR system; Promega Corp.). For the RT-qPCR assays, the
primers were designed using Allele ID (Premier Biosoft
International) and IDT SciTools, Inc. (TEMA Ricerca), and are
presented in Table I. The
products of real-time PCR were electrophoresed on a 1,5% agarose
gel with ethidium bromide and then captured using the Gel Doc EZ
Imaging System with Image Lab software 5.0 (Bio-Rad Laboratories,
Inc.).
 | Table INucleotide base sequences of primers
designed for the RT-qPCR assays. |
Table I
Nucleotide base sequences of primers
designed for the RT-qPCR assays.
| Genes | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| ACTB |
GGGAAATCGTGCGTGACATT |
TACCCAGGAAGGAAGGCTGG |
| CAT |
ACAACTCCCAGAAGCCTAAGAATG |
GGCTTGTGCCCTGCTTCATG |
| NOS2 |
CAAGGTCTACGTTCAAGACAT |
AAAGTGGTAGCCACATCCCG |
| MnSOD |
ATTAACGCGCAGATCATGCA |
GGCTGAAGAGCAACCTGAGTT |
| HPRT1 |
CAGTCCCAGCGTCGTGATTAGT | ATCCAGCAGGTCAG |
Measurement of mitochondrial superoxide
with MitoSOX red
Mitochondrial superoxide formation was evaluated
using MitoSOX red (Molecular Probes, Invitrogen; Thermo Fisher
Scientific, Inc.). Briefly, the H9c2 cells were plated at
4.0×105 cells/well in 6-well tissue culture plates, and
were treated as described above. Following the indicated
incubations, 2.5 µM MitoSOX red was added for 15 min at
37°C. MitoSOX red is a fluorogenic dye for the highly selective
detection of superoxide in the mitochondria of living cells and,
once targeted to the mitochondria, it is oxidized by superoxide and
emits red fluorescence. MitoSOX red is readily oxidized by
superoxide, but not by other ROS-generating systems. Cell
fluorescence was measured using fluorescence-activated cell sorting
(FACS, BD Biosciences) by counting 10,000 events/min, and analyzed
using CellQuest software, (version 5.2.1). These data are reported
as percentages of positive cells, as referred at the 'gate', which
was the sequential identification and refinement of the cellular
population using MitoSOX red as the marker that was visualized by
fluorescence as a unique emission spectrum.
Measurement of mitochondrial membrane
depolarization
Mitochondrial membrane depolarization was measured
using FACS scans and the fluorescent dye tetramethylrhodamine
methyl ester (TMRE). Due to its positive charge, TMRE readily
accumulates in active mitochondria in inverse proportion to Δψm
according to the Nernst equation. The H9c2 cells were seeded at
4.0×105 cells/well in 6-well tissue culture plates, and
treated as described above. The H9c2 cells were then collected,
washed twice with phosphate-buffered saline (PBS), and incubated in
PBS containing 5 nM TMRE at 37°C. After 30 min, cell fluorescence
was evaluated using FACS, and analyzed using CellQuest software,
version 5.2.1.
Mitochondrial protein extraction and
western blot analysis for mitochondrial Cx43
The H9c2 cells were seeded at 1.0×106
cells/well in 100-mm culture dishes (Corning, Inc.), and treated as
described above. Mitochondrial protein extraction was carried out
as previously described (9).
Briefly, the H9c2 cells were lysed in buffer A (250 mM sucrose, 20
mM K+ Hepes, pH 7.5, 10 mM KCl, 1.5 mM MgCl2,
1 mM EDTA, 1 mM EGTA, protease inhibitors, 50 mM NaF, 0.2 mM
Na3VO, 1 mM phenylmethylsulfonyl fluoride, 1 mM
dithiothreitol and 0.025% digitonin). The H9c2 cells were then
centrifuged at 16,000 × g for 2 min at 4°C. The supernatants were
discarded, and the pellets were resuspended in lysis buffer B [150
mM NaCl, 0.1% Triton-X, 0.5% sodium deuteroxide, 1% sodium dodecyl
sulfate (SDS), 50 mM Tris/HCl, pH 7.4; all from Sigma-Aldrich;
Merck KGaA], to obtain the mitochondrial proteins. Protein
concentrations were determined using protein assays (Bio-Rad
Laboratories, Inc.). Equal amounts of protein were loaded onto an
acrylamide gel (50 µg protein/lane), and separated by 10%
SDS-PAGE under denaturing conditions. The blots were incubated
overnight at 4°C with the primary antibodies as anti-Cx43 (1:8,000,
cat. no. 610062; BD Transduction Laboratories) or anti-TOM20, used
as a loading control, (1:250, cat. no. sc-17764; Santa Cruz
Biotechnology, Inc.). Following incubation with the primary
antibodies and washing in PBS/0.1% Tween, the appropriate secondary
antibodies were added for 1 h at room temperature, as anti-rabbit
or anti-mouse (each diluted 1:4,000, rabbit conjugate horseradish
peroxidase, cat. no. GtxRb-003-DHRPX and mouse conjugate
horseradish peroxidase, cat. no. GtxMu-003-DHRPX; Microtech).
Immunoreactive protein bands were detected by chemiluminescence
using enhanced chemiluminescence reagents and blot imaging (LAS
4000; GE Healthcare); densitometry was performed using ImageJ,
1.52t (Wayne Rasband) To determine the purity of mitochondrial
protein extraction, western blot analysis was performed for
proteins expressed only in the mitochondria (Ox-Phos Complex II;
Abcam) without including proteins expressed in other cellular
compartments (e.g., Na+/K+ ATPase; Abcam)
(24).
Total protein extraction and western blot
analysis
The H9c2 cells were seeded at 7.0×105
cells/well in 100-mm culture dishes (Corning, Inc.), and treated as
described above. Total protein was extracted from the cells by
freeze/thawing in lysis buffer (50 mM NaF, 150 mM NaCl, 50 mM
Tris/HCl, pH 7.4, 1% Nonidet P40, 1 mM EDTA, 0.2 mM
Na3VO4, 1 mM phenylmethylsulfonyl fluoride
and protease inhibitors; all from Sigma-Aldrich; Merck KGaA).
Protein concentrations were determined using protein assays
(Bio-Rad Laboratories, Inc.). Total cell extracts containing equal
amounts of protein (50 µg protein/lane) were separated on
10% SDS-polyacrylamide gels under denaturing conditions, and
transferred to nitrocellulose membranes (Amersham, GE Healthcare)
using a semi-dry transfer apparatus (Bio-Rad Laboratories, Inc.).
The blots were blocked with 5% non-fat dry milk powder in PBS for 1
h at room temperature. The membranes were then incubated for 1 h at
4°C with the primary antibodies, as anti-procaspase 3, anti-caspase
9, anti-iKKα (all diluted 1:200; cat. nos. sc-56052, sc-76548,
sc-7606, Santa Cruz Biotechnology, Inc.), and anti-iNOS (diluted
1:8,000; cat. no. sc-7271, Santa Cruz Biotechnology, Inc.). Gapdh
(diluted 1:1,000; cat. no. sc-32233, Santa Cruz Biotechnology,
Inc.) was used as the loading control. After washing in PBS/ 0.1%
Tween, the appropriate secondary antibodies were added for 1 h at
room temperature, as anti-rabbit (diluted 1:5,000; rabbit conjugate
horseradish peroxidase, cat. no. GtxRb-003-DHRPX; Microtech) or
anti-mouse (diluted 1:4,000, mouse conjugate horseradish
peroxidase, cat. no. GtxMu-033-DHRPX; Microtech). The
immunoreactive protein bands were detected using enhanced
chemiluminescence immunoassay reagents and blot imaging (LAS 4000;
GE Healthcare), densitometry was performed using ImageJ, 1.52t
(Wayne Rasband).
Flow cytometric analysis
The H9c2 cells were cultured at 4.5×105
cells/well in 6-well plates and allowed to grow for 24 h. They were
then treated as described above to determine the CAT, MnSOD, p-IKB
and nitrotyrosine levels. The H9c2 cells were collected using a
scraper, treated with fixing buffer (4% formaldehyde, 0.1%
NaN3, 2% FBS, in PBS) for 20 min, and then permeabilized
using a fixation and permeabilization buffer (fixing buffer
containing 0.1% Triton X) for 30 min. Subsequently, these cells
were incubated with the anti-CAT (diluted 1:250; cat. no.
sc-271803; Santa Cruz Biotechnology, Inc.), anti-MnSOD (diluted
1:250; cat. no. sc-137254; Santa Cruz Biotechnology, Inc.),
anti-p-IKB and anti-nitrotyrosine antibodies (both diluted 1:250;
cat. no. sc-137254 and sc-32757 respectively; Santa Cruz
Biotechnologies) for 1 h at 4°C, as required, followed by the
respective anti-rabbit or anti-mouse FITC antibodies as the
secondary antibodies (diluted both 1:5,000; cat. nos. A120-208F and
A90-146F, respectively; Bethyl Laboratories), for 1 h at 4°C. The
cells were collected and analyzed by FACS (FACSscan; BD
Biosciences) and the data obtained were processed using the
CellQuest software, version 5.2.1. These data are shown as
percentages of positive cells.
Analysis of apoptosis
The H9c2 cells were plated at 4.5×105
cells/well in 6-well plates and allowed to grow for 24 h, and then
treated as described above. In order to estimate specifically the
pro-apoptotic effect of iNOS and its products in Doxo-treated
cells, in other experiments, the H9c2 cells were treated with
L-NAME (1 µM), a specific iNOS inhibitor, administered 30
min prior to Doxo treatment. The H9c2 cells were washed twice with
PBS, and incubated in 500 µl 0.1% Triton X-100, 0.1% sodium
citrate, and 50 µg/ml propidium iodide, at 4°C for 30 min in
the dark. The propidium-iodide-stained cells were analyzed by FACS,
using CellQuest software, version 5.2.1. These data are expressed
as the percentages of cells in the hypodiploid region.
Measurement of nitrite/nitrate (NOx)
concentrations
NOx concentrations provide a surrogate marker for NO
generation, and this was measured for the cell medium of the H9c2
cells, as previously described (25). The cell medium was incubated with
0.1 U/ml nitrate reductase, 1 mM NADPH, and 50 µM
flavin adenine dinucleotide, at 37°C. After 15 min, the
samples were incubated with 100 U/ml lactate dehydrogenase and 10
mM sodium pyruvate, for 5 min. The total NOx concentrations in the
samples were measured using the Griess reaction, with the addition
of 100 µl Griess reagent (0.1% naphthyl ethylenediamide
dihydrochloride in H2O, 1% sulfanilamide in 5% conc.
H2PO4; 1:1) to 100 µl samples, carried
out in triplicate. The optical densities at 550 nm
(OD550) were measured in a micro-plate reader (Titertek;
Dasit). Total NOx concentrations (µm) were calculated from
the standard curve with sodium nitrate.
Statistical analysis
The data are presented as the means ± standard error
of the mean (SEM) for at least 3 independent experiments, with each
performed in duplicate. The RT-qPCR data are presented as the means
± SD for at least 3 independent experiments, each performed in
triplicate. Statistical analysis was performed using one-way ANOVA,
with multiple comparisons using Bonferroni's tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibition of Cx43 translocation to the
mitochondria alters H9c2 cell morphology
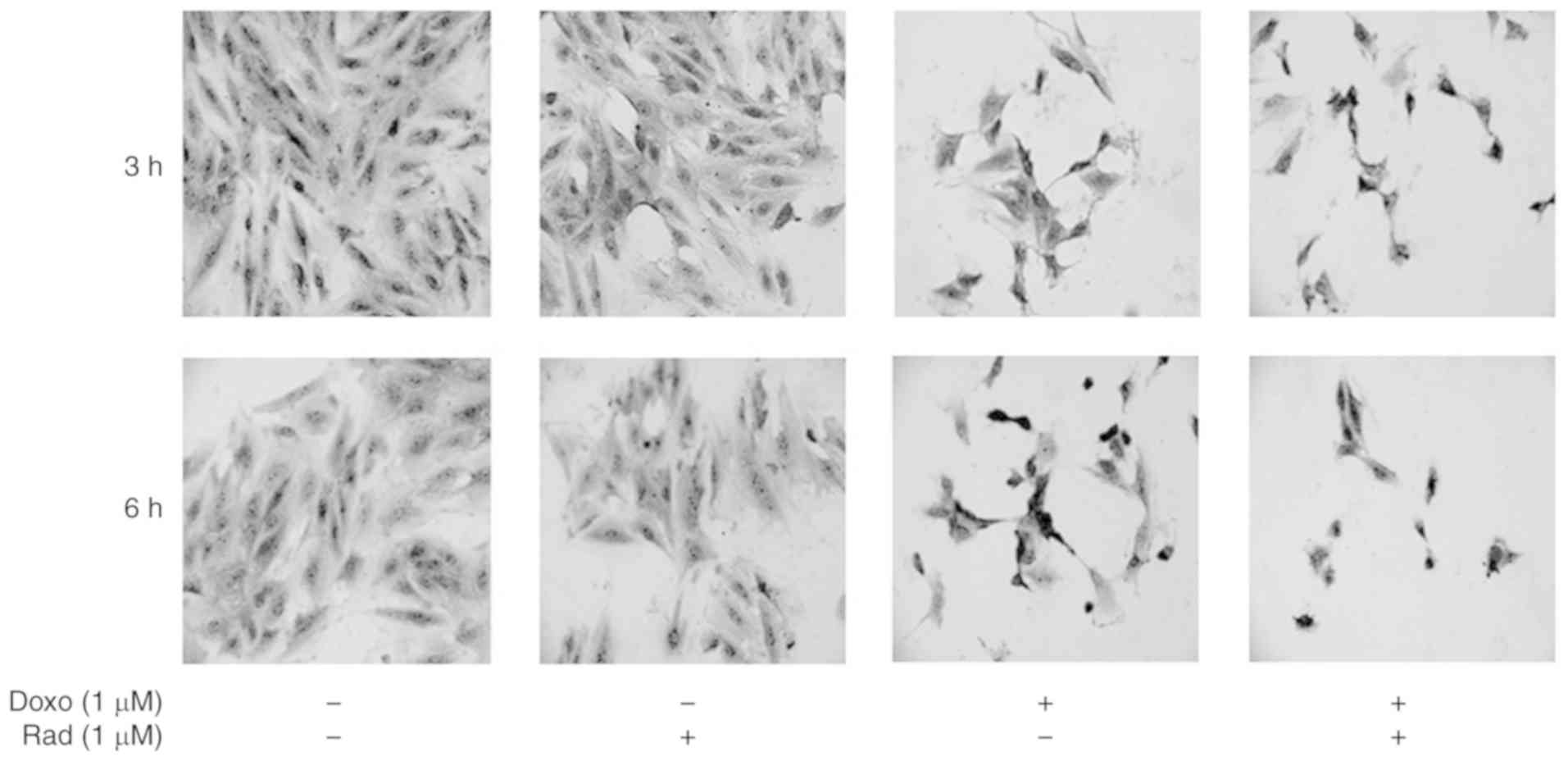
Morphological modifications of the H9c2 cells
treated with Doxo alone, with radicicol alone, or both in
combination were evaluated using toluidine blue staining. As
depicted in Fig. 1, the untreated
control cells exhibited a characteristic spindle shape, with the
nuclei clearly visible as round and central. There were also a
number of nucleoli and an abundant cytoplasm component at both 3
and 6 h of incubation. Treatment with radicicol alone, which
inhibits Cx43 translocation to the mitochondria, did not alter the
morphology of these cells at 3 or 6 h of treatment. For the H9c2
cells treated with Doxo for 3 h, the nuclei acquired an atypical,
wrinkled appearance, and the cytoplasm component appeared reduced.
These cells lost their cylindrical shape, and acquired an irregular
shape. Moreover, following 6 h of Doxo treatment, nuclear and
cytoplasmic involution changes were observed. Indeed, the untreated
control cells were cylindrical, in contact, with roundish nuclei
and evident nucleoli and abundant cytoplasm component. The treated
cells, instead, exhibited a thin form, and the nuclei exhibited
atypical and wrinkled aspects. Radicicol pre-treatment increased
these Doxo-induced morphological changes at both 3 and 6 h of
treatment.
Inhibition of Cx43 translocation to the
mitochondria increases the Doxo effects on ROS signaling and cell
death
The administration of 1 µM Doxo for 3 and 6 h
induced an increase in mitochondrial Cx43 expression, which was
inhibited in the radicicol pre-treated cells (Fig. 2A). The inhibition of Cx43
translocation to the mitochondria was increased significantly under
Doxo-induced mitochondrial ROS production and mitochondrial
membrane depolarization (P<0.05; Fig. 2B and C). In particular, ROS
formation was determined as the mitochondrial superoxide formation
with MitoSOX red, which once targeted to the mitochondria, was
oxidized by superoxide and exhibited red fluorescence that was
proportional to the amount of enzyme present (Fig. 2B). Mitochondrial membrane
depolarization was measured using FACSscan with the fluorescent
dye, TMRE. Due to its relative negative charge, TMRE is normally
stored in active mitochondria, whereas mitochondria with a
decreased membrane potential, and depolarized or inactive
mitochondria, fail to sequester TMRE (Fig. 2C). Aerobic organisms have
developed efficient defense systems of enzymatic and non-enzymatic
antioxidants to ameliorate and cope with injury from oxidative
damage and to maintain redox homeostasis, such as MnSOD and
catalase (26). In the present
study, to evaluate the effects of Doxo administration on MnSOD and
catalase, the H9c2 cells were treated as described above, and MnSOD
and catalase gene and protein expression were evaluated by RT-qPCR
and cytofluorimetric techniques, respectively.
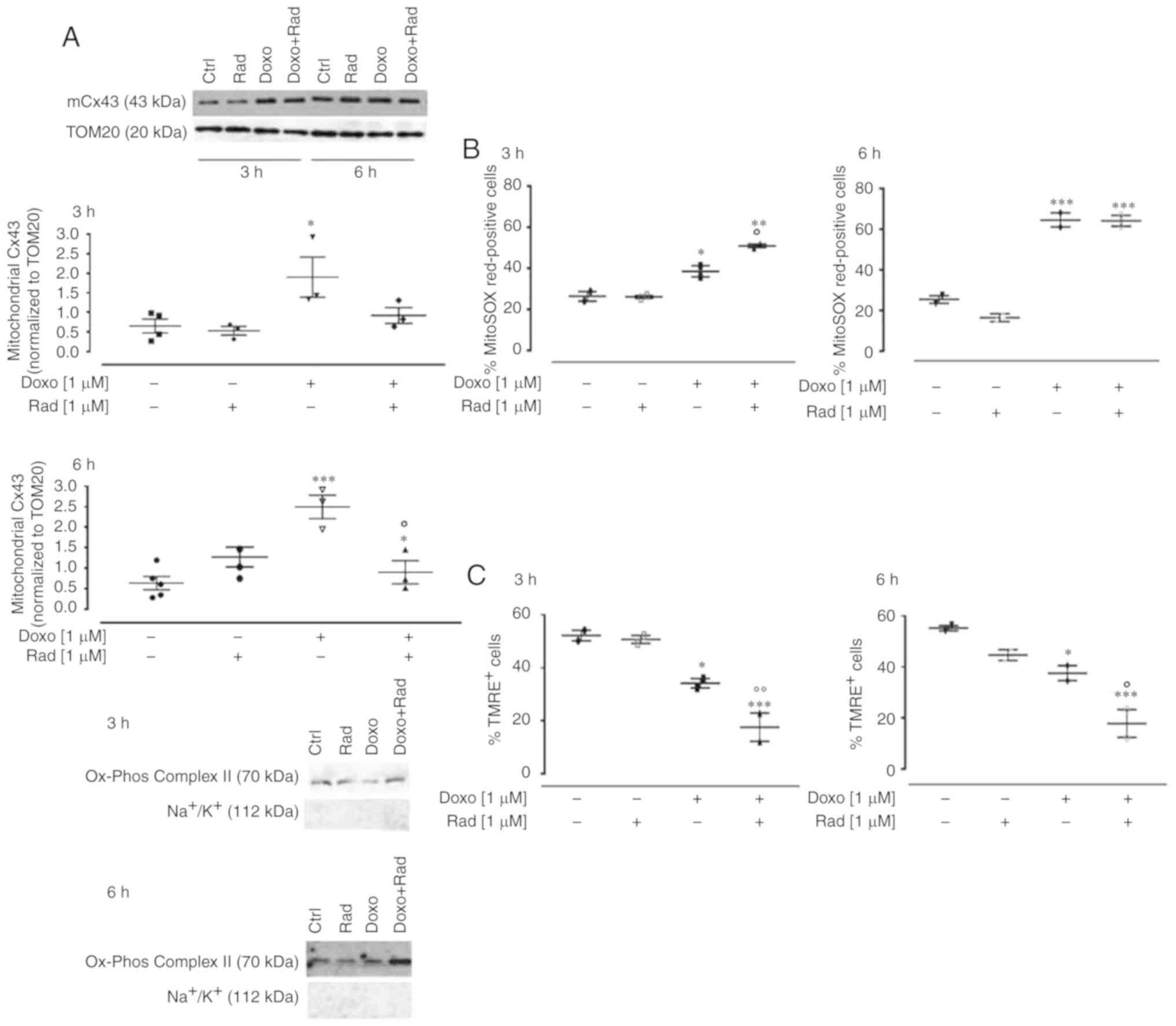 | Figure 2Effects of treatments with Doxo alone
and in combination with radicicol on (A) mitochondrial Cx43
expression, (B) mitochondrial ROS production, and (C) mitochondrial
membrane potential of H9c2 cells. Doxo (1 µM) was
administered for 3 and 6 h, and where indicated, radicicol (1
µM) was administered 30 min prior to Doxo. (A) Mitochondrial
Cx43 expression was detected by western blot analysis. TOM20
protein expression was used as the loading control. Inset, top
panel, representative western blot for
Na+/K+ATPase and Ox-phos complex II is shown
as markers of mitochondria and sarcolemma, respectively, to
demonstrate the purity of the mitochondrial extracts. (B)
Mitochondrial superoxide production evaluated using MitoSOX red in
the H9c2 cells by flow cytometry, as the percentages of MitoSOX
Red-positive cells. (C) Mitochondrial membrane potential was
evaluated by flow cytometry analysis with tetramethylrhodamine
ethyl ester (TMRE), a cell permeant, positively charged, red-orange
dye that penetrates cells and accumulates in the mitochondria in
inverse proportion to the membrane potential. A low percentage of
TMRE-positive cells indicates that the TMRE dye was not trapped
within the mitochondrial membranes, due to its depolarization. Data
are the means ± SEM of fluorescence intensity, from at least 3
independent experiments, each performed in duplicate.
*P<0.05, **P<0.005,
***P<0.001 vs. control; °P<0.05, °°P<0.005 vs.
cells treated with Doxo under the same conditions (one-way ANOVA
and multiple comparisons by Bonferroni's test). Doxo, doxorubicin;
Rad, radicicol. |
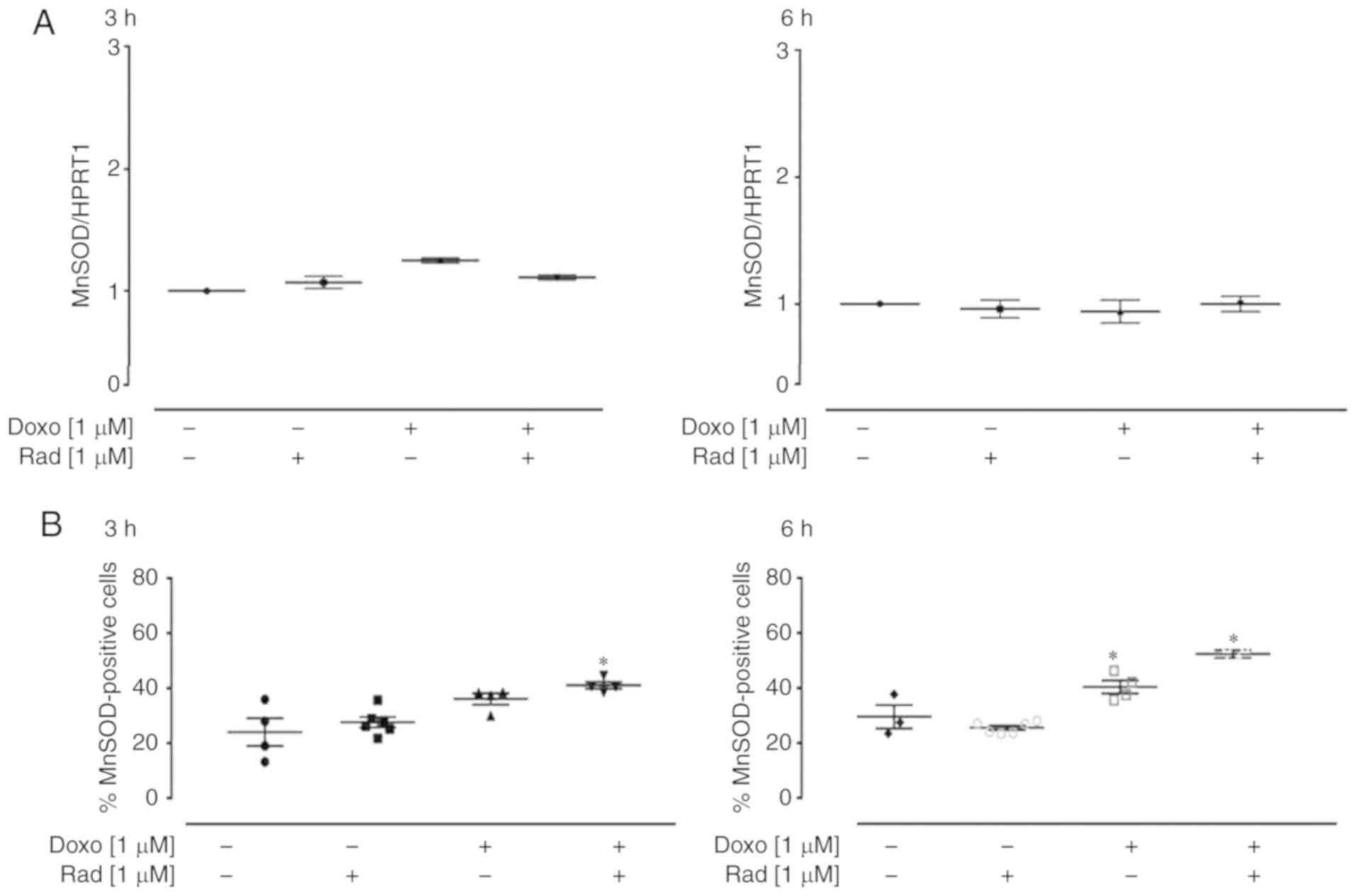
The enzyme MnSOD belongs to the class of
oxidoreductases, and it catalyzes the dismutation of superoxide
anion into H2O2 and molecular oxygen,
according to the following reaction: 2O2− +
2H+ ↔ O2 + H2O2.
Following treatment with 1 µM Doxo for 3 h, a small, and
non-significant increase in MnSOD expression was induced,
which was not observed after 6 h of treatment (Fig. 3A). The cytofluorimetric analysis
revealed that Doxo treatment significantly increased MnSOD
expression (P<0.05; Fig. 3B),
after 6 h of treatment. Furthermore, in the H9c2 cells pre-treated
with radicicol, at both experimental times, MnSOD expression was
higher than that in the cells treated with Doxo alone, as well as
showing a significant increase compared to the untreated control
cells (P<0.05; Fig. 3B).
In the H9c2 cells treated with Doxo and radicicol,
an increase in CAT expression was observed after 3 h, and,
in particular, a significant additive effect (P<0.05) was
observed following co-treatment for 3 h. On the contrary, following
co-treatment for 6 h, CAT expression was significantly
down-regulated (P<0.005; Fig.
4A). Agarose gel electrophoresis was used to monitor the PCR
products, which confirmed the data obtained (Fig. 4B). The results of FACS analysis
supported the RT-qPCR data, with a significant increase observed in
CAT expression following treatment for 3 and 6 h (P<0.05).
Indeed, as shown in Fig. 4C, the
inhibition of Cx43 translocation to the mitochondria significantly
increased Doxo-induced CAT expression at 3 h (P<0.05), while no
significant differences were observed between the cells treated
with Doxo alone and those treated with Doxo and radicicol for 6
h.
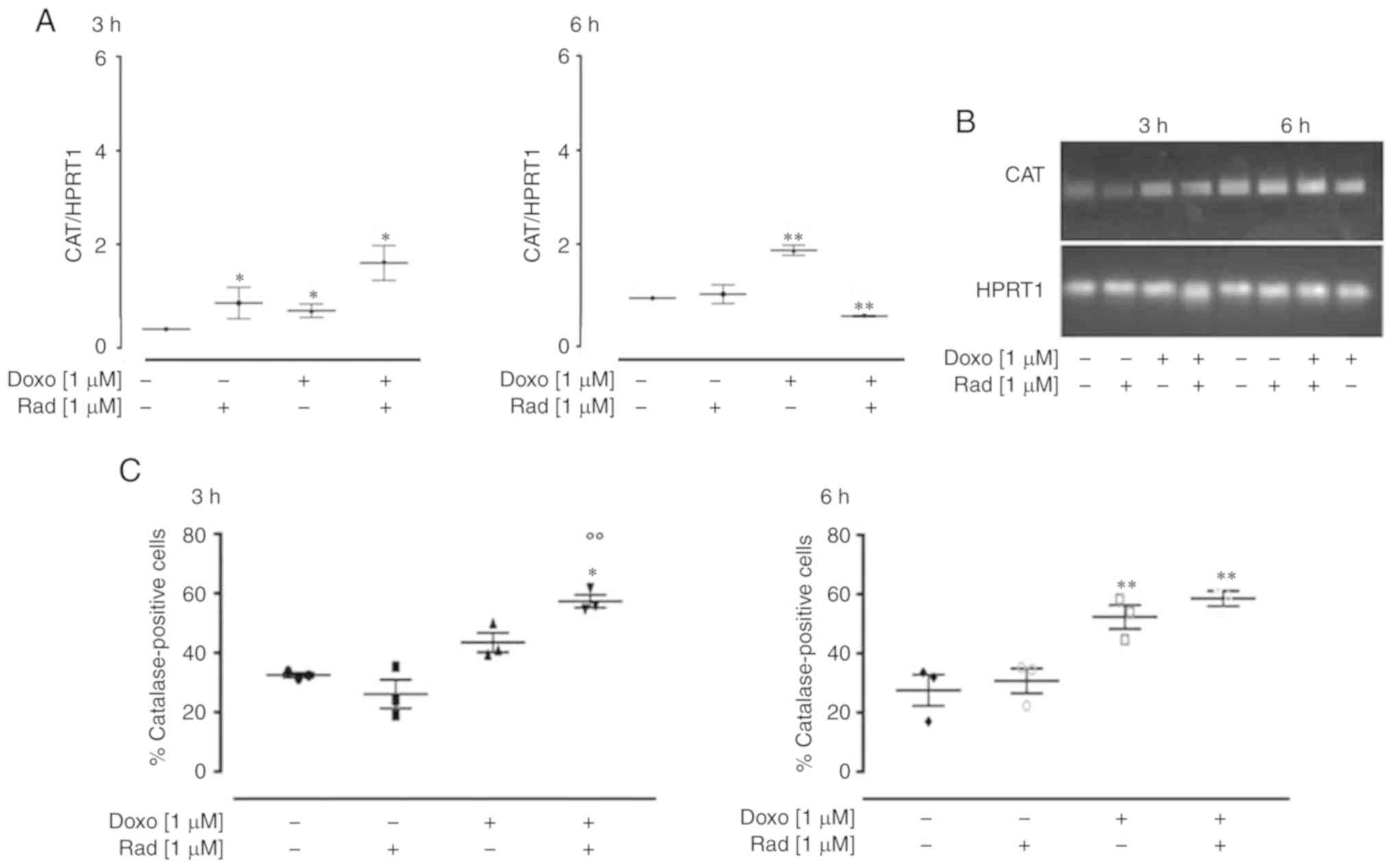 | Figure 4Effects of treatments with Doxo alone
and in combination with radicicol on catalase (A and B) gene and
(C) protein expression in the H9c2 cells. Doxo (1 µM) was
administered for 3 and 6 h, and where indicated, radicicol (1
µM) was administered 30 min prior to Doxo. (A) CAT mRNA
levels analyzed by RT-qPCR, with the data normalized to the HPRT1
gene, The data are presented as the means ± SD for at least 3
independent experiments, which each performed in triplicate. (B)
Representative agarose gel of RT-qPCR products of the data shown in
(A). (C) Catalase levels in the cytosol, using flow cytometry for
the H9c2 cells. Data are means ± SEM for the percentages of
catalase-positive cells, from at least 3 independent experiments,
each performed in duplicate. *P<0.05,
**P<0.005 vs. control, °°P<0.005 vs. cells treated
with Doxo under the same conditions (one-way ANOVA and multiple
comparisons by Bonferroni's test). Doxo, doxorubicin; Rad,
radicicol. |
ROS are considered to be responsible for the
apoptotic effects of Doxo (14).
Indeed, if a large amount of O2− is produced
within the mitochondria, this cannot pass through the mitochondrial
membrane, and can thus damage the mitochondrial DNA (27), which then induces the release of
cytochrome c from the mitochondria (28). In the present study, treatment
with Doxo induced an increase in the apoptotic response,
significantly (P<0.005) after 6 h of treatment, as shown by the
hyplodiploid nuclei in Fig. 5A.
Indeed, it is known that Doxo induces apoptosis through caspase
activation via the mitochondrial pathway in cardiomyocytes
(29). In the Doxo-treated cells
at both treatment times, western blot analysis revealed an increase
in caspase 9 expression (Fig.
5B), and a concomitant decrease in procaspase 3 levels
(Fig. 5C). The inhibition of Cx43
translocation to the mitochondria promoted by radicicol enhanced
all the pro-apoptotic effects induced by Doxo (Fig. 5).
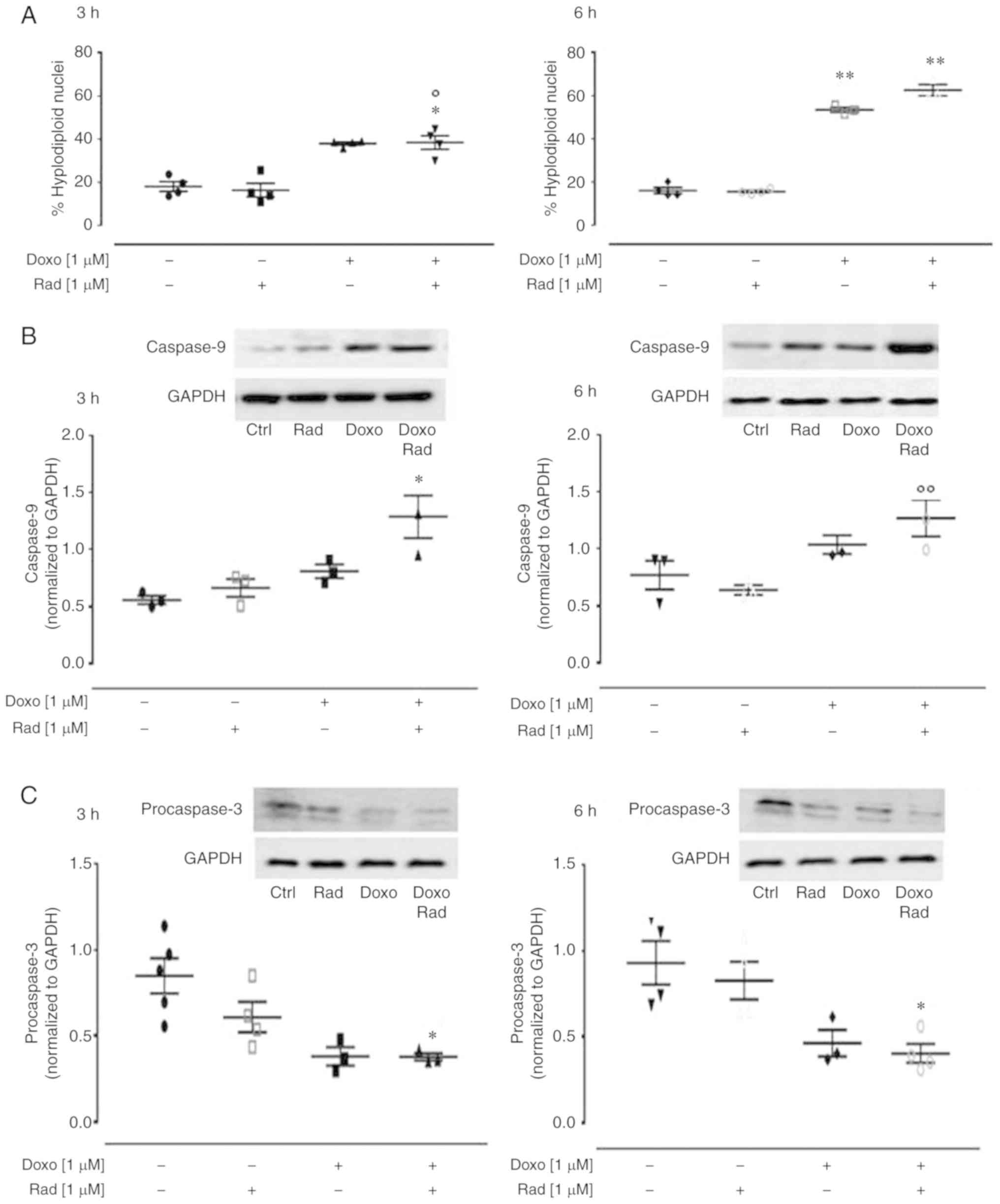 | Figure 5Effects of treatment with Doxo alone
and in combination with radicicol on the apoptotic pathway of the
H9c2 cells. Doxo (1 µM) was administered for 3 and 6 h, and
where indicated, radicicol (1 µM) was administered 30 min
prior to Doxo. (A) The H9c2 cells were stained by propidium iodide
and fluorescence of individual nuclei was measured by flow
cytometry. Data are the means ± SEM for the percentages of
hyplodiploid nuclei, from at least 3 independent experiments, each
performed in duplicate. (B and C) Caspase 9 (B) and procaspase 3
(C) expression detected by western blot analysis, with Gapdh
expression as the loading control. Insets: Representative western
blots. Data are the means ± SEM, from at least 3 independent
experiments, each performed in duplicate. *P<0.05,
**P<0.005, vs. control; °P<0.05 and °°P<0.005
vs. cells treated with Doxo under the same conditions (one-way
ANOVA and multiple comparisons by Bonferroni's test). Doxo,
doxorubicin; Rad, radicicol. |
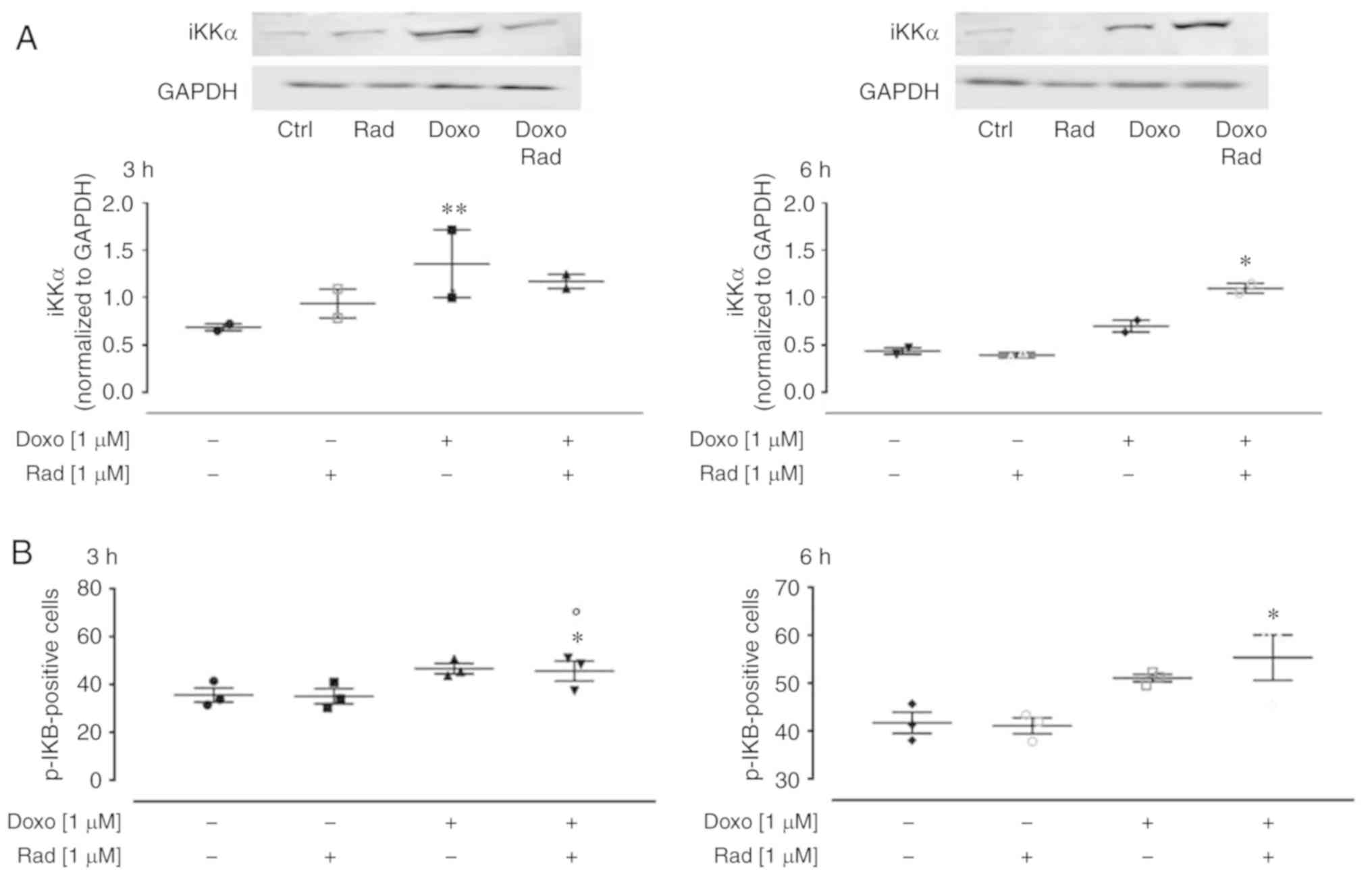
Inhibition of Cx43 translocation to the
mitochondria increases Doxo-induced nitrosative stress
Doxo-derived ROS have been reported to activate the
NF-κB signaling pathway, which results in an imbalance between the
pro-apoptotic and anti-apoptotic proteins (30). NF-κB is a key orchestrator of
inflammation, as it induces expression of the main inflammatory
cytokines, adhesion molecules, and NO synthase (31). Usually, the NF-κB proteins are
bound to and inhibited by the IκB proteins. Various external
stimuli activate the iKK complex (such as iKKβ, iKKα and NEMO),
which phosphorylates the IκB proteins, and thus free NF-κB to
translocate to the nucleus (32).
In the present study, western blot analysis revealed that Doxo
treatment significantly enhanced iKKα expression (P<0.005;
Fig. 6A) at 3 h of treatment, and
in agreement with the canonical NF-κB pathway, there was an
increase in the levels of phosphorylated IKB, as detected by flow
cytometry (Fig. 6B). The
inhibition of Cx43 translocation to the mitochondria activated the
NF-κB signaling pathway from following 3 h co-treatment with Doxo
(Fig. 6A).
The translocation of NF-κB to the nucleus induces
the transcription of iNOS. Thus, the present study investigated
iNOS expression in this experimental model. iNOS is not normally
expressed in cardiomyocytes, but it can be activated under
intracellular stress conditions. Therefore, due to its generation
of free radicals, iNOS represents an indirect marker of
intracellular stress. In the present study, indeed, in both the
untreated control and Doxo-treated H9c2 cells, there was a low
iNOS expression, as shown in Fig. 7A. However, following 6 h of
co-treatment with Doxo plus radicicol, there was a significant
increase in iNOS expression (P<0.005). Western blot
analysis revealed that following 6 h of treatment, there was
anincrease in iNOS expression in the Doxo-treated H9c2 cells, and a
significant (P<0.05) increase in the radicicol-pre-treated cells
(Fig. 7B). The quantitative
protein analysis of iNOS by western blot analysis confirmed the
data from RT-qPCR.
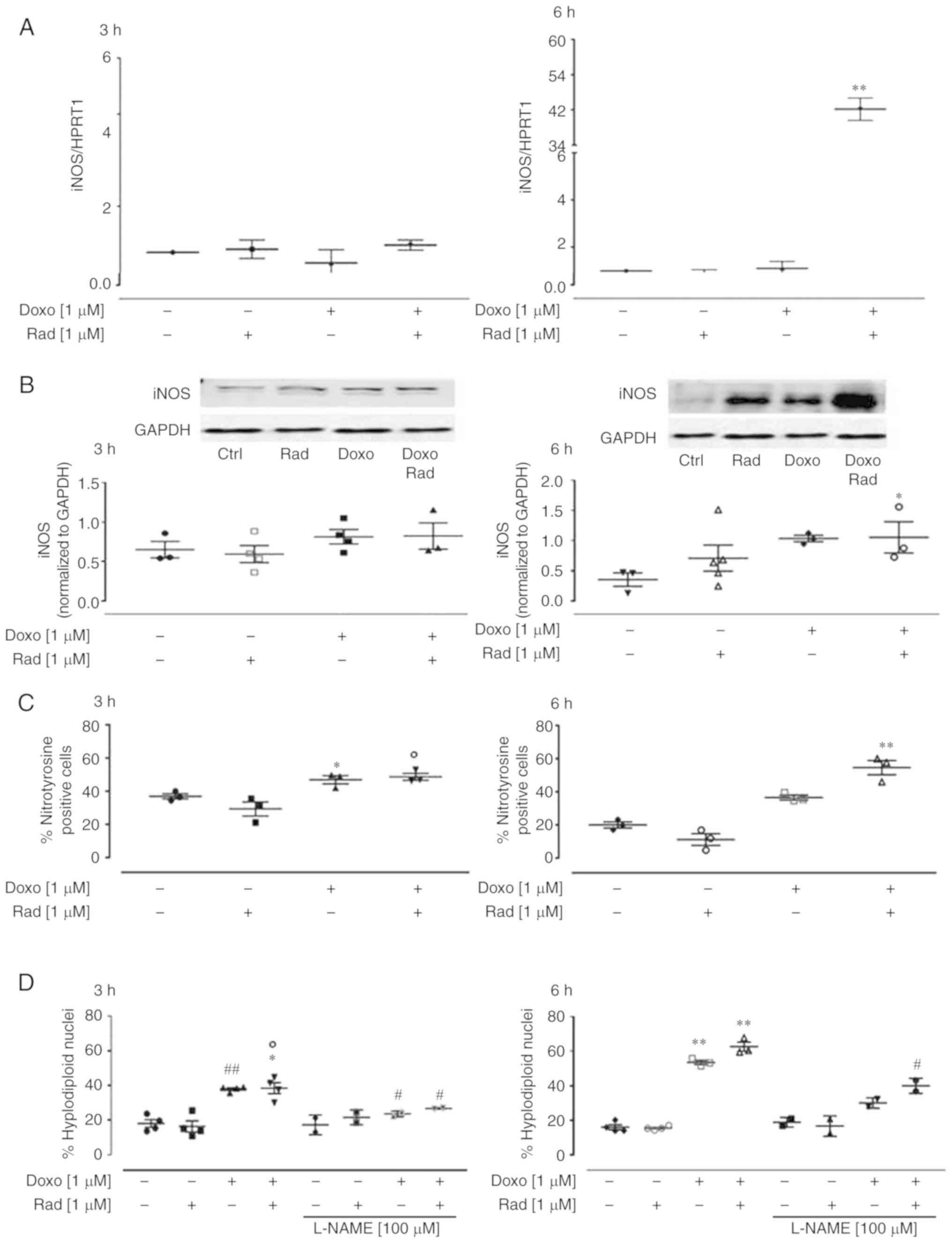 | Figure 7Effects of treatment with Doxo alone
and in combination with radicicol on iNOS (A) gene and (B) protein
expression, and (C) nitrotyrosine formation, and with the (D)
addition of L-NAME for apoptosis, of the H9c2 cells. Doxo (1
µM) was administered for 3 or 6 h, and where indicated,
radicicol (1 µM) was administered 30 min prior to Doxo. (A)
iNOS mRNA levels analyzed by RT-qPCR, with the data normalized to
the HPRT1 gene The data are presented as the means ± SD for at
least 3 independent experiments, which each performed in
triplicate. (B) iNOS expression was detected by western blot
analysis, with Gapdh expression as the loading control. Insets:
Representative western blots. (C) Nitrotyrosine levels using flow
cytometry of the H9c2 cells. Data are the means ± SEM for the
percentages of nitrotyrosine-positive cells, from at least 3
independent experiments, each performed in duplicate. (D) Effects
of L-NAME on apoptosis induced under the same conditions. Data are
the means ± SEM for the percentages of hypodiploid nuclei, from at
least 3 independent experiments, each performed in duplicate.
*P<0.05, **P<0.005, vs. control;
°P<0.05 vs. cells treated with Doxo under the same experimental
conditions; and #P<0.05 and ##P<0.005,
for cells co-treated with Doxo and L-NAME vs. Doxo (one-way ANOVA
and multiple comparisons by Bonferroni's test). Doxo, doxorubicin;
Rad, radicicol. |
Nitrite release represents an indirect indicator of
NO production, and this was measured in the medium of the H9c2
cells to determine the enzymatic activity of iNOS. However, the
nitrite levels were not quantifiable under the current experimental
conditions (data not shown). As NO rapidly reacts in the presence
of high O2− levels to form peroxynitrite,
which can then induce nitration of the aromatic side-chains of
tyrosine in proteins, the levels of nitrotyrosine were investigated
in the H9c2 cells treated as described above. Fig. 7C illustrates the data from the
cytofluorimetric analysis that revealed an increase in
nitrotyrosine levels in the Doxo-treated H9c2 cells. Indeed, the
inhibition of Cx43 translocation to the mitochondria significantly
(P<0.005) enhanced the nitrotyrosine levels following 6 h of
treatment.
Finally, to determine the contribution of iNOS and
its products to Doxo-induced apoptosis, L-NAME, a specific iNOS
inhibitor, was used (33). The
data obtained by FACS indicated that L-NAME significantly decreased
Doxo-induced apoptosis (P<0.05) after 3 h of treatment. The
inhibition of Cx43 translocation to the mitochondria reduced the
protective effects of L-NAME on Doxo-induced apoptosis,
particularly after 6 h of treatment (Fig. 7D).
Discussion
The survival of cancer patients has significantly
increased over the past 20 years. However, to obtain these
benefits, a large price has to be paid in terms of the side-effects
associated with the use of intensive anticancer treatments. In
particular, chronic cardiotoxicity can compromise the clinical
efficacy of chemotherapies, which will affect the survival and
quality of the life of patients, regardless of the oncological
prognosis. Indeed, cardiotoxicity is the main limitation to the
clinical use of Doxo. The proposed mechanisms for Doxo-induced
cardiotoxicity are complex, and they involve increased
oxidative/nitrosative stress and the activation of downstream
effector pathways (15). As the
involvement of Cx43 in Doxo-induced mitochondrial dysfunction and
oxidative stress was recently demonstrated (9), the present study was designed to
determine whether mitochondrial Cx43 is also involved in
Doxo-induced nitrosative stress. For this purpose, H9c2 cells were
treated with 1 µM Doxo for 3 and 6 h, in both the absence
and presence of radicicol, an Hsp90 inhibitor. Indeed, although it
is well known that Hsp90 is involved in a number of other cellular
processes (22), radicicol is
widely used as a pharmacological tool to block the Doxo-induced
translocation of Cx43 to the mitochondria (4,9),
just as another Hsp90 inhibitor has been used to study the role of
mitochondrial Cx43 in cardioprotection (35).
In a previous study, the authors reported data
obtained by FACS on the effects of Doxo on mitochondrial ROS
production and on mitochondrial membrane depolarization (10). These data were repeated in the
present study as part of the background to the present the
investigation (as shown in Fig.
2), to confirm that the experimental conditions were suitable
to reproduce the cardio-toxic effects of Doxo and its consequences
on mitochondrial Cx43 expression. The deleterious effects of Doxo
on the H9c2 cells treated for 3 and 6 h were also confirmed using
Toluidine blue staining. The morphological variations observed
following these treatments are in agreement with those of another
study that described atypical and wrinkled aspects of the nuclei of
the damaged cells (34). Once the
increase in intracellular ROS was confirmed herein through MitoSox
and TMRE analyses, the various antioxidant systems that were
activated in response to this were investigated. Two antioxidant
enzymes were evaluated: MnSOD and CAT, which are known to cooperate
in the defense mechanisms against free radicals. Interesting data
emerged herein regarding these two enzymes. The expression of
MnSOD was shown to increase following treatment with Doxo
for 3 h, which represents the nuclear response that was aimed at
intensifying the production of MnSOD, to reduce the high ROS
concentrations detected by the intracellular sensing systems.
Conversely, following 6 h of Doxo treatment, no changes in
MnSOD expression were observed. CAT expression
analysis revealed a similar trend. Indeed, after 3 h of Doxo
treatment, the expression of CAT was enhanced, with an
evident additive effect observed with co-treatment with radicicol,
while at 6 h of Doxo treatment, it was also diminished. However,
the data obtained by FACS demonstrated that the expression of both
the MnSOD and CAT proteins remained high, even in the cells treated
with Doxo for 6 h.
The apparent contrast between the data obtained by
FACS on the expression of these enzymes, where there was a
time-dependent increase in MnSOD, with the data obtained by
RT-qPCR, can be explained by consideration that the cellular
concentrations of proteins usually correlate with the abundances of
their corresponding mRNAs, although not necessarily strongly.
Further data addressing this issue indicate that in almost every
organism, the transcript abundance only partially predicts the
protein levels, which suggests that other modes of regulation need
to be invoked to explain how proteins levels are indeed set within
cells (37).
Moreover, as Vogel and Marcotte previously reported
(36), in mammalian cells, mRNAs
are produced at much lower rates than the proteins are; similarly,
the mRNAs are less stable than the proteins. This condition may be
complicated by the post-transcriptional mechanisms involved in
turning mRNAs into proteins. In addition, the data of the present
study can be explained by the activation of programmed cell death,
which was strongly initiated following 6 h of Doxo treatment,
compared with 3 h. Indeed, the activation of the apoptotic pathway
would inhibit the nuclear response in terms of the modulation of
gene expression, by shifting the cellular energies towards the
activation of these 'controlled' death mechanisms, hence reducing
mRNA production.
It is well known that Doxo also induces the NF-κB
signaling pathway (37). The data
of the present study demonstrated that Doxo induced an increase in
iKKα and p-IKB expression, thus indicating the activation of NF-κB
in the current experimental model. This activation of NF-κB results
in an increase in iNOS gene and protein expression. Nitrite and
nitrate release into the medium of these treated H9c2 cells as
indicators of NO production was not evaluable herein, although
elevated levels of nitrotyrosine were shown. Indeed, in agreement
with previous reports, it was found that the NO produced by iNOS
will rapidly react with H2O2 or
O2− generated by the mitochondria, to form
the highly reactive and harmful peroxynitrite (21,38), which then finally induces
nitrotyrosine formation (28,39). Furthermore, and for the first
time, to the best of our knowledge, it was demonstrated herein that
the inhibition of Cx43 translocation to the mitochondria, and the
consequent increase in mitochondrial superoxide release,
significantly increases the deleterious effects of Doxo-induced
iNOS overexpression. Indeed, the inhibition of iNOS alone in the
presence of L-NAME did not completely block Doxo-induced
apoptosis.
In conclusion, the present study supports the
concept that Doxo induces both oxidative and nitrosative stress,
and that these are closely related to each other. However, the
present study we focused mainly on the role of the mitochondria. It
was hypothesized that mitochondrial damage appears at an earlier
stage, as indicated by the compensatory mechanisms implemented by
the cells (e.g., mitoCx43 overexpression, increased MnSOD
and CAT gene expression). However, if the increase in
mitochondrial ROS production cannot be counteracted, this will also
amplify the damage produced by activation of the NF-κB-mediated
pathway. This is in agreement with the findings of previous studies
that have reported that mitochondrial super-oxide generation occurs
within minutes of Doxo administration, whereas cell death becomes
evident only at a later stage, when significant amounts of
peroxynitrite have been generated (15). Indeed, the present study
demonstrated that the mechanisms that further increase the
mitochondrial superoxide generation (e.g., the inhibition of Cx43
translocation to the mitochondria) can significantly accelerate the
occurrence of cell death.
Further studies are scheduled to determine whether
the oxidative stress observed in cells in which Cx43 translocation
to the mitochondria is blocked can be rescued using antioxidants,
as the protective effects of antioxidants against Doxo-induced
toxicity have been reported (40).
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by the University of
Salerno, through a FARB 2018 grant to APo.
Availability of data and materials
All data used during the current study are available
from the corresponding author on reasonable request.
Authors' contributions
APo and GM conceived and designed the experiments.
MP, BP and MCDM performed the experiments. MCDM and SM analyzed the
data and revised the manuscript. APi and RM conceived the
experiments and revised the manuscript. MP, APo and GM wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Michela P, Velia V, Aldo P and Ada P: Role
of connexin43 in cardiovascular diseases. Eur J Pharmacol.
768:71–76. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fontes MS, van Veen TA, de Bakker JM and
van Rijen HV: Functional consequences of abnormal Cx43 expression
in the heart. Biochim Biophys Acta. 1818:2020–2029. 2012.
View Article : Google Scholar
|
|
4
|
Ruiz-Meana M, Rodríguez-Sinovas A,
Cabestrero A, Boengler K, Heusch G and Garcia-Dorado D:
Mitochondrial connexin43 as a new player in the pathophysiology of
myocardial ischaemia-reperfusion injury. Cardiovasc Res.
77:325–333. 2008. View Article : Google Scholar
|
|
5
|
Boengler K, Stahlhofen S, van de Sand A,
Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G and Schulz R:
Presence of connexin 43 in subsarcolemmal, but not in
interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol.
104:141–147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heinzel FR, Luo Y, Li X, Boengler K,
Buechert A, García-Dorado D, Di Lisa F, Schulz R and Heusch G:
Impairment of diazoxide-induced formation of reactive oxygen
species and loss of cardioprotection in connexin 43 deficient mice.
Circ Res. 97:583–586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rehling P, Brandner K and Pfanner N:
Mitochondrial import and the twin-pore translocase. Nat Rev Mol
Cell Biol. 5:519–530. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu G, Haider HKH, Porollo A and Ashraf M:
Mitochondria-specific transgenic overexpression of connexin-43
simulates preconditioning-induced cytoprotection of stem cells.
Cardiovasc Res. 88:277–286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pecoraro M, Sorrentino R, Franceschelli S,
Del Pizzo M, Pinto A and Popolo A: Doxorubicin-mediated
cardiotoxicity: Role of mitochondrial connexin 43. Cardiovasc
Toxicol. 15:366–376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pecoraro M, Ciccarelli M, Fiordelisi A,
Iaccarino G, Pinto A and Popolo A: Diazoxide improves mitochondrial
connexin 43 expression in a mouse model of doxorubicin-induced
cardiotox-icity. Int J Mol Sci. 19:7572018. View Article : Google Scholar
|
|
11
|
Kondru SK, Potnuri AG, Allakonda L and
Konduri P: Histamine 2 receptor antagonism elicits protection
against doxorubicin-induced cardiotoxicity in rodent model. Mol
Cell Biochem. 441:77–88. 2018. View Article : Google Scholar
|
|
12
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berthiaume JM and Wallace KB: Persistent
alterations to the gene expression profile of the heart subsequent
to chronic doxorubicin treatment. Cardiovasc Toxicol. 7:178–191.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Angsutararux P, Luanpitpong S and
Issaragrisil S: Chemotherapy-induced cardiotoxicity: Overview of
the roles of oxidative stress. Oxid Med Cell Longev.
2015:7956022015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mukhopadhyay P, Rajesh M, Bátkai S,
Kashiwaya Y, Haskó G, Liaudet L, Szabó C, Pacher P and Am J: Role
of superoxide nitric oxide and peroxynitrite in doxorubicin-induced
cell death in vivo and in vitro. Physiol Heart Circ Physiol.
296:H1466–H1483. 2009. View Article : Google Scholar
|
|
16
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Z, Lin S, Wu W, Tan H, Wang Z, Cheng C,
Lu L and Zhang X: Ghrelin prevents doxorubicin-induced
cardiotoxicity through TNF-alpha/NF-kappaB pathways and
mitochondrial protective mechanisms. Toxicology. 247:133–138. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shirazi LF, Bissett J, Romeo F and Jawahar
LM: Role of Inflammation in heart failure. Curr Atheroscl Rep.
19:272017. View Article : Google Scholar
|
|
20
|
Bartekova M, Radosinska J, Jelemensky M
and Dhalla NS: Role of cytokines and inflammation in heart function
during health and disease. Heart Failure Rev. 23:733–758. 2018.
View Article : Google Scholar
|
|
21
|
Sanskriti K, Gurinder BS and Madhu K:
Nitric oxide synthase and diabetic cardiomyopathy. Nitric Oxide.
43:29–34. 2014. View Article : Google Scholar
|
|
22
|
Schulte TW, Akinaga S, Soga S, Sullivan W,
Stensgard B, Toft D and Neckers LM: Antibiotic radicicol binds to
the N-terminal domain of Hsp90 and shares important biologic
activities with geldanamycin. Cell Stress Chaper. 3:100–108. 1998.
View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Boengler K, Dodoni G, Rodriguez-Sinovas A,
Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C,
Garcia-Dorado D, Di Lisa F, et al: Connexin 43 in cardiomyocyte
mitochondria and its increase by ischemic preconditioning.
Cardiovasc Res. 67:234–244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Di Iorio B, Marzocco S, Di Micco L, Adesso
S, De Blasio A, Autore G, Sirico ML, Fazeli G and Heidland A:
High-tone external muscle stimulation in patients with acute kidney
injury (AKI): Beneficial effects on NO metabolism asymmetric
dimethylarginine and endothelin-1. Clin Nephrol. 82:304–312. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miao L and St Clair DK: Regulation of
superoxide dismutase genes: Implications in disease. Free Radic
Biol Med. 47:344–356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wallace DC: Mitochondrial defects in
cardiomyopathy and neuromuscular disease. Am Heart J. 139:S70–S85.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pecoraro M, Del Pizzo M, Marzocco S,
Sorrentino R, Ciccarelli M, Iaccarino G, Pinto A and Popolo A:
Inflammatory mediators in a short-time mouse model of
doxorubicin-induced cardiotoxicity. Toxicol Appl Pharmacol.
293:44–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ueno M, Kakinuma Y, Yuhki K, Murakoshi N,
Iemitsu M, Miyauchi T and Yamaguchi I: Doxorubicin induces
apoptosis by activation of caspase-3 in cultured cardiomyocytes in
Vitro and rat cardiac ventricles in Vivo. J Pharmacol Sci.
101:151–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghosh J, Das J, Manna P and Sil PC: The
protective role of arjunolic acid against doxorubicin induced
intracellular ROS dependent JNK-p38 and p53-mediated cardiac
apoptosis. Biomaterials. 32:4857–4866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Del Prete A, Allavena P, Santoro G,
Fumarulo R, Corsi MM and Mantovani A: Molecular pathways in
cancer-related inflammation. Biochem Med (Zagreb). 21:264–275.
2011. View Article : Google Scholar
|
|
32
|
Perkins ND: Post-translational
modifications regulating the activity and function of the nuclear
factor κB pathway. Oncogene. 25:6717–6730. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marzocco S, Adesso S, Alilou M, Stuppner H
and Schwaiger S: Anti-inflammatory and anti-oxidant potential of
the root extract and constituents of doronicum austriacum.
Molecules. 22:10032017. View Article : Google Scholar
|
|
34
|
Witek P, Korga A, Burdan F, Ostrowska M,
Nosowska B, Iwan M and Dudka J: The effect of a number of H9C2 rat
cardiomyocytes passage on repeatability of cytotoxicity study
results. Cytotechnology. 68:2407–2415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tu RH, Li QJ, Huang Z, He Y, Meng JJ,
Zheng HL, Zeng ZY and Zhong GQ: Novel functional role of heat shock
protein 90 in mitochondrial connexin 43-mediated hypoxic
postconditioning. Cell Physiol Biochem. 44:982–997. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vogel C and Marcotte EM: Insights into the
regulation of protein abundance from proteomic and transcriptomic
analyses. Nat Rev Genet. 13:2227–2232. 2012. View Article : Google Scholar
|
|
37
|
Wang J, Ma L, Tang X, Zhang X, Qiao Y, Shi
Y, Xu Y, Wang Z, Yu Y and Sun F: Doxorubicin induces apoptosis by
targeting Madcam1 and AKT and inhibiting protein translation
initiation in hepatocellular carcinoma cells. Oncotarget.
6:24075–24091. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Balligand JL and Cannon PJ: Nitric oxide
synthases and cardiac muscle. Autocrine and paracrine influences.
Arterioscler Thromb Vasc Biol. 17:1846–1858. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ahsan H: 3-Nitrotyrosine: A biomarker of
nitrogen free radical species modified proteins in systemic
autoimmunogenic conditions. Hum Immunol. 74:1392–1399. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ye M, Zhang L, Yan Y and Lin H:
Punicalagin protects H9c2 cardiomyocytes from doxorubicin-induced
toxicity through activation of Nrf2/HO-1 signaling. Biosci Rep.
39:BSR201902292019. View Article : Google Scholar : PubMed/NCBI
|