Introduction
The health emergency caused by the severe acute
respiratory syndrome coronavirus 2 (SARS-CoV-2 virus), the
etiological agent of COVID-19 disease, represents one of the
greatest health and social challenges ever faced worldwide
(1,2). Since its first outbreak in China in
the late 2019, the characteristics of this epidemic have been
controversial for different reasons: i) very limited information
available on both the nature of the virus and its clinical
manifestations; ii) no existing health protocol proven effective in
containing or monitoring the spread of this infection (3-5).
According to the World Health Organization (WHO), the current gold
standard method for the diagnosis of SARS-CoV-2 infection is based
on the reverse transcription-quantitative polymerase chain reaction
(RT-qPCR). Since the first cases recorded in China, WHO has
indicated various portions of the SARS-CoV-2 sequence as possible
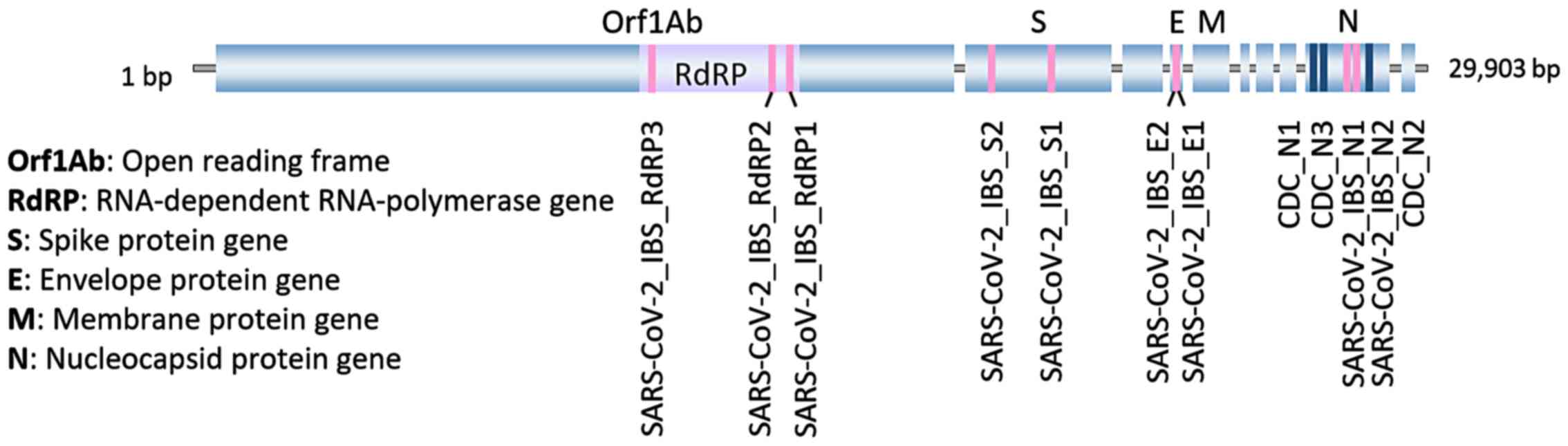
targets for RT-qPCR. The currently most used gene targets are
Orf1Ab, Nucleocapsid protein gene and Spike Protein, used for
single- or multiplex RT-qPCR (6-8)
(Fig. 1).
All the RT-qPCR-based methods analyze SARS-CoV-2
nucleic acids starting from rhino-pharyngeal swab samples obtained
from subjects with suspected COVID-19 infection. However, the
sensitivity of such technique may be very low (depending on the
platform used, sample impurities, low amount of viral cDNA, etc.)
leading to a high percentage of false-negative results and failing
to assess the viral load during the follow-up of quarantined
patients (9,10).
The reasons behind the low sensitivity of RT-qPCR in
detecting SARS-CoV-2 cDNA are not only related to the above
mentioned reasons but depend also on the standardization of the
pre-analytical phases of sampling and extraction of the
rhino-pharyngeal swab. Indeed, significant variations in the
detection of SARS-CoV-2 were related to the different swab and
maintenance buffer used, as well as to the extraction and
amplification kits (one-step or two-step) adopted or the quality of
the RNA extracted (11,12). All the limitations of
RT-qPCR-based approaches suggest that the improvement of the
current diagnostic and follow-up strategies is mandatory to cope
effectively with the COVID-19 emergency.
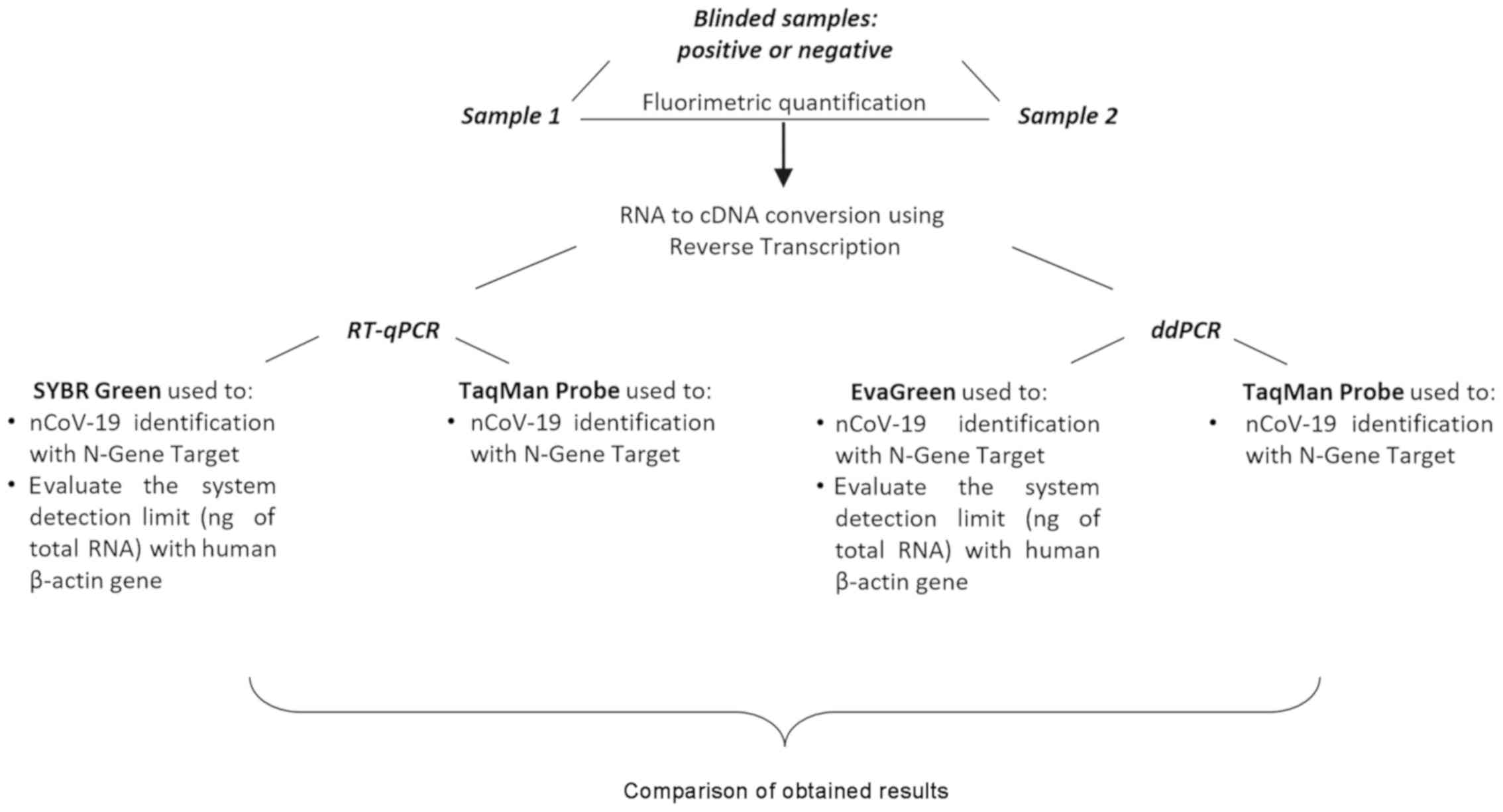
On these bases, the aim of the study was to propose
a novel high-sensitive method for the effective detection of
SARS-CoV-2 in patients with low viral load. For this purpose, the
sensitivity of RT-qPCR SYBR-Green and probe technologies was
compared with the sensitivity of droplet digital PCR (ddPCR)
EvaGreen and probe systems by analyzing swab samples obtained from
two patients negative and positive for COVID-19 infection,
respectively. In this way, the accuracy of both methods in
recognizing as positive COVID-19 patients with low viral load was
assessed (Fig. 2).
In particular, we have chosen to use both DNA
intercalant chemistries and TaqMan-based methods to evaluate the
detection limits and the sensitivity of RT-qPCR and ddPCR.
As regards the ddPCR, different studies have
demonstrated the higher sensitivity and robustness of this method
compared to other molecular techniques, including RT-qPCR (13,14). In particular, ddPCR technology is
based on the absolute quantification of targets using the
principles of dilution and partition of the reaction mix in 20,000
nanodroplets obtained by using oil-water emulsion. This methodology
improves the accuracy and detection of targets in a low-cost and
high-sensitive PCR approach (15). Currently, ddPCR is effectively
used for the absolute quantification of viral load, for the
analysis of circulating DNA, gene and microRNA expression and
analysis of gene copy number variation (16-19).
Materials and methods
Samples included in the study
Two blinded RNA samples extracted from a negative
and a positive rhino-pharyngeal swabs, were included in the study.
Positive and negative SARS-CoV-2 results were preliminary tested
with a commercial platform (Allplex Seegene-Arrow).
RNA samples and reverse
transcription
The concentration of total RNA was determined by
using a fluorometric assay. The RNA quantity was tested by
Qubit® 3.0 Fluorometer (cat. no. Q33216; Life
Technologies; Thermo Fisher Scientific, Inc.) using Qubit RNA HS
Assay kit (250 pg/µl and 100 ng/µl). RT was performed
using 15 ng of total RNA, RNase H reverse transcriptase, and random
primer hexamers (Superscript II; Thermo Fisher Scientific,
Inc.).
RT-qPCR
For the detection of SARS-CoV-2, the CDC-validated
2019-nCoV_N1 primers and probe were used (Table I) (20). The same primers with or without
probe were used for the RT-qPCR performed with SYBR-Green and
TaqMan probe, respectively.
 | Table IThe primers used for qPCR. |
Table I
The primers used for qPCR.
| 2019-novel
coronavirus (2019-nCov) real-time rRT-PCR panel primers and
probes |
|---|
| Name | Oligonucleotide
sequence (5′→3′) | Label | Working
concentration |
|---|
| 2019-nCov_N1-F |
5′-GACCCCAAAATCAGCGAAAT-3′ | None | 20 µM |
| 2019-nCov_N1-R |
5′-TCTGGTTACTGCCAGTTGAATCTG-3′ | None | 20 µM |
| 2019-nCov_N1-P |
5′-FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1-3′ | FAM, BHQ-1 | 5 µM |
The dilution of cDNA samples used for RT-qPCR
analysis (LightCycler®480 System; Roche Molecular
Systems, Inc.) were 1:1 (1.87 ng), 1:10 (0.1875 ng), 1:20 (0.09375
ng), 1:50 (0.0375 ng), 1:100 (0.01875 ng). PCR efficiency, melting
curve analysis and expression rate were calculated using the Light
Cycler® 480 Software (Roche).
TaqMan RT-qPCR analysis was performed used
QuantiNova™ Probe PCR kit (cat. no. 208252; Qiagen) following the
manufacturer's procedure and the following thermal cycle: PCR
initial activation step for 2 min at 95°C; two step-cycling:
denaturation for 5 sec at 95°C, combined annealing/extension
annealing for 5 sec at 60°C; for 45 cycles.
SYBR-Green RT-qPCR analysis was performed using
QuantiTect Syber-Green PCR kit (cat. no. 204145; Qiagen) following
the manufacturer's procedure and the following thermal cycle
conditions: PCR initial activation step for 15 min at 95°C; 3
step-cycling: denaturation for 15 sec at 94°C, annealing for 30 sec
at 60°C, extension for 30 sec at 72°C; for 45 cycles (21). Human β-actin gene (QuantiTect
Primer Assays, Hs_ACTB_2_SG, QT01680476; Qiagen) were used to
overcome SYBR-Green system detection limits.
For the two types of RT-qPCR, the negative control
consisted of a reaction in absence of cDNA and indicated as NTC (no
template control). All the reactions were run in triplicate.
ddPCR amplification
The cDNA previously obtained from the swab samples
was amplified by using both EvaGreen and Probe ddPCR-based method.
Briefly, for EvaGreen ddPCR the reaction mix was prepared by using
11 µl of 2X QX200™ ddPCR™ EvaGreen Supermix (cat. no.
1864034; Bio-Rad Laboratories, Inc.), 0.385 µl of 10
µM Fwd/Rev primer mix, 5.615 µl of RNase and DNase
free-water and 5 µl of cDNA in order to obtain a final
volume of 22 µl.
For the Probe ddPCR, the reaction mix was prepared
by using 11 µl of 2X ddPCR Supermix for Probes (no dUTP)
(cat. no. 1863024; Bio-Rad Laboratories, Inc.), 0.198 µl of
100 µM 2019-nCoV_N1 gene forward and reverse primers (final
concentration 900 nM), 0.055 µl of 100 µM
2019-nCoV_N1 gene TaqMan probe (final concentration 250 nM), 5.5
µl of RNase and DNase free-water and 5 µl of cDNA in
order to obtain a final volume of 22 µl. cDNA was used
undiluted and diluted 1:10 in order to assess the sensitivity of
the ddPCR proposed methods.
Twenty microliters of the reaction mix was used to
generate droplets with the QX200 droplet generator (Bio- Rad
Laboratories, Inc.). After generation, the droplets were
transferred into a 96-well plate, sealed and amplified in a C1000
Thermal Cycler (Bio-Rad Laboratories, Inc.) under the following
thermal conditions:
EvaGreen ddPCR-polymerase activation at 95°C for 10
min, 40 cycles of amplification at 94°C for 30 sec (denaturation)
and 60°C for 1 min (annealing), droplets stabilization at 98°C for
10 min followed by an infinite hold at 4°C. A ramp rate of 2°C/sec
was used among the steps of the amplification;
Probe ddPCR-polymerase activation at 95°C for 10
min, 40 cycles of amplification at 94°C for 30 sec (denaturation)
and 60°C for 1 min (annealing), droplets stabilization at 98°C for
10 min followed by an infinite hold at 4°C. A ramp rate of 2°C/sec
was used among the steps of the amplification. After amplification,
positive and negative droplets were read in the QX200 Droplet
Reader (Bio-Rad Laboratories, Inc.). All the experiments were
performed in triplicate.
Sequencing of SARS-CoV-2-positive ddPCR
droplets
To confirm that the positive signals obtained with
ddPCR were relative to SARS-CoV-2 sequence amplification, the cDNA
amplified by using 2019-nCoV_N1 primers and contained in ddPCR
positive droplets, were extracted and sequenced as follows.
Briefly, the ddPCR reaction mix was prepared for the COVID-19
positive sample as previously described and dispensed into ten
different wells. After ddPCR amplification, three wells were read
with the QX200 Droplet Reader and 8-wells were pooled in a 1.5 ml
tube for the extraction of amplified cDNA. The bottom oil phase was
pipeted-out and 160 µl of TE buffer and 560 µl of
chloroform were added to isolate amplified cDNA. After vortexing (1
min), the samples were centrifuged at 15.500 × g for 10 min to
separate the aqueous phase containing cDNA from the chloroform. The
obtained cDNA was then quantified by fluorimeter Qubit dsDNA BR
Assay kit (cat. no. 32850; Invitrogen; Thermo Fisher Scientific,
Inc.). Then, 5 ng of product was sequenced on a SeqStudio Genetic
Analyzer (Thermo Fisher Scientific, Inc.) using the Applied
Biosystems BigDye terminator cycle sequencing 3.1v (cat. no.
4337455; Thermo Fisher Scientific, Inc.) as previously described
(22). The obtained sequence was
compared with the reference sequence 'MT077125 severe acute
respiratory syndrome coronavirus 2 isolated
SARS-CoV-2/human/ITA/INMI1/2020 (complete genome sequence release
date: April 11, 2020)' by using the BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results
Assessment of RT-qPCR sensitivity and
detection of SARS-CoV-2 N1 gene
Samples included in the study were tested by using
SYBR-Green technology for the detection and quantification of the
β-actin housekeeping gene, as well as for the detection of the
SARS-CoV-2 N gene. The results confirmed the high sensitivity of
RT-qPCR in detecting β-actin human mRNA diluted at different
concentrations from 1.87 to 0.0187 ng (Table SI).
Results obtained with the β-actin gene highlighted
how in the extracted RNA there were some PCR inhibitors. Indeed,
both undiluted samples showed higher Ct values (27.77 and 30.43 for
sample 1 and sample 2, respectively) compared with the 10-fold
dilution, where better Ct values were obtained (25.78 and 29.89 for
sample 1 1:10 and sample 2 1:10, respectively), as a consequence of
the low concentration of inhibitors.
For the detection of the SARS-CoV-2 N gene, only the
1:1 and 1:10 dilutions were used. After amplification, the same Ct
values were obtained for all the samples and dilutions assessed
demonstrating that SYBR-Green RT-qPCR was not sensitive enough for
detecting positive swab samples with low viral load (Table II).
| Table IIAverage of SARS-CoV-2 N gene Ct
values obtained by using SYBR-Green and TaqMan RT-qPCR. |
Table II
Average of SARS-CoV-2 N gene Ct
values obtained by using SYBR-Green and TaqMan RT-qPCR.
| ID sample | SYBR-Green RT-qPCR
| TaqMan RT-qPCR
| [RNA] |
|---|
| Ct | Ct average | Ct | Ct average |
|---|
| Sample 1 | 36.54 | | ND | | 1.87 ng |
| 34.78 | 35.64 | ND | ND | |
| 35.6 | | ND | | |
| Sample 1 1:10 | 33.04 | | ND | | 0.187 ng |
| 34.32 | 34.10 | ND | ND | |
| 34.95 | | ND | | |
| Sample 2 | 33.94 | | 36.93 | | 1.87 ng |
| 34.53 | 34.46 | 36.29 | 36.59 | |
| 34.91 | | 36.54 | | |
| Sample 2 1:10 | 32.8 | | ND | | 0.187 ng |
| 34.84 | 33.86 | ND | ND | |
| 33.95 | | ND | | |
| NTC
2019-nCoV_N1 | - | | - | | |
| - | - | - | - | |
| - | | - | | |
Different results were obtained through the analysis
of the SARS-CoV-2 N gene performed by using the 2019-nCoV_N1 TaqMan
probe, validated by the CDC. Using TaqMan RT-qPCR it was possible
to discriminate between the two samples, thus detecting sample 2 as
positive. However, positive signals were obtained only for the
undiluted sample 2 with a very late Ct value (36.61), while no
signals were obtained in the same sample diluted 10-fold or in
sample 1 (Table II).
ddPCR EvaGreen and Probe systems
effectively detect low amount of SARS-CoV-2 N1 gene
As reported for SYBR-Green RT-qPCR, also ddPCR
EvaGreen chemistry was first used for the absolute quantification
of the SARS-CoV-2 N gene and for the detection of β-actin used as
reference gene. The analysis of β-actin concentration in the two
blinded swab samples showed that sample 1 had a higher
concentration of β-actin compared to sample 2. In particular,
β-actin concentration varied from 247 copies/µl in the
undiluted sample 1 to 0.9 copies/µl in the same sample
diluted 1,000-fold. Noteworthy, at the undiluted concentration the
ddPCR system was saturated with positive droplets resulting in an
underestimation of the actual concentration of the sample, while
the absolute quantification of the diluted samples reflected the
serial dilutions performed. The β-actin absolute quantification of
sample 2 revealed that the overall amount of cDNA was approximately
25-fold lower compared to sample 1 (β-actin concentration of 86
copies/µl and 3.5 copies/µl in the 1:10 diluted
sample 1 and sample 2, respectively) ranging from 36.1
copies/µl in the undiluted sample 2 to 0.07 copies/µl
in the same samples diluted 500-fold (detection limit set at
0.00374 ng) (Fig. S1).
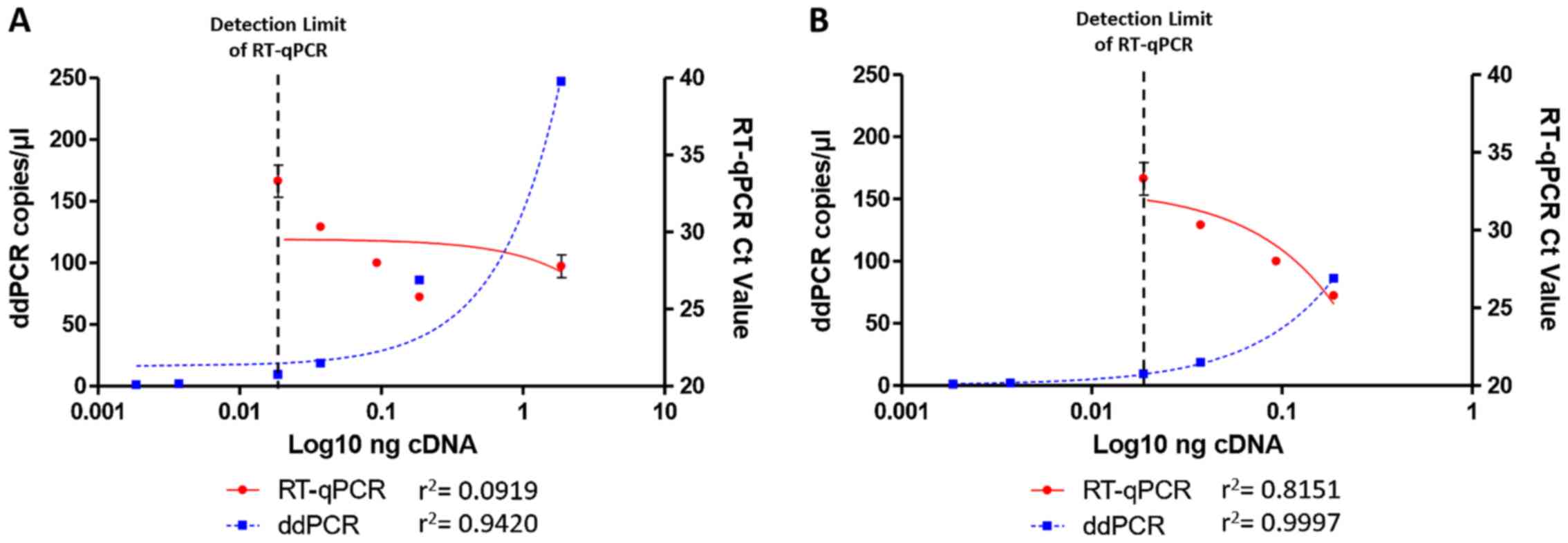
Taking into account the β-actin results obtained by
using both RT-qPCR and ddPCR, it was observed that ddPCR has a
greater accuracy and robustness compared to RT-qPCR. In particular,
linear regression analysis revealed that ddPCR is less susceptible
to the inhibitory action of PCR interferers (r2=0.9420
for ddPCR vs. r2=0.0919 for RT-qPCR) (Fig. 3A). In addition, not considering
the inhibited undiluted samples both methods increased their
accuracy, however, ddPCR still originated more linear data
(r2=0.9997 for ddPCR vs. r2=0.8151 for
RT-qPCR) (Fig. 3B). Finally, the
comparison of β-actin results demonstrated a lower detection limit
and higher sensitivity for ddPCR compared to RT-qPCR (0.00187 vs.
0.0187 ng) (Fig. 3).
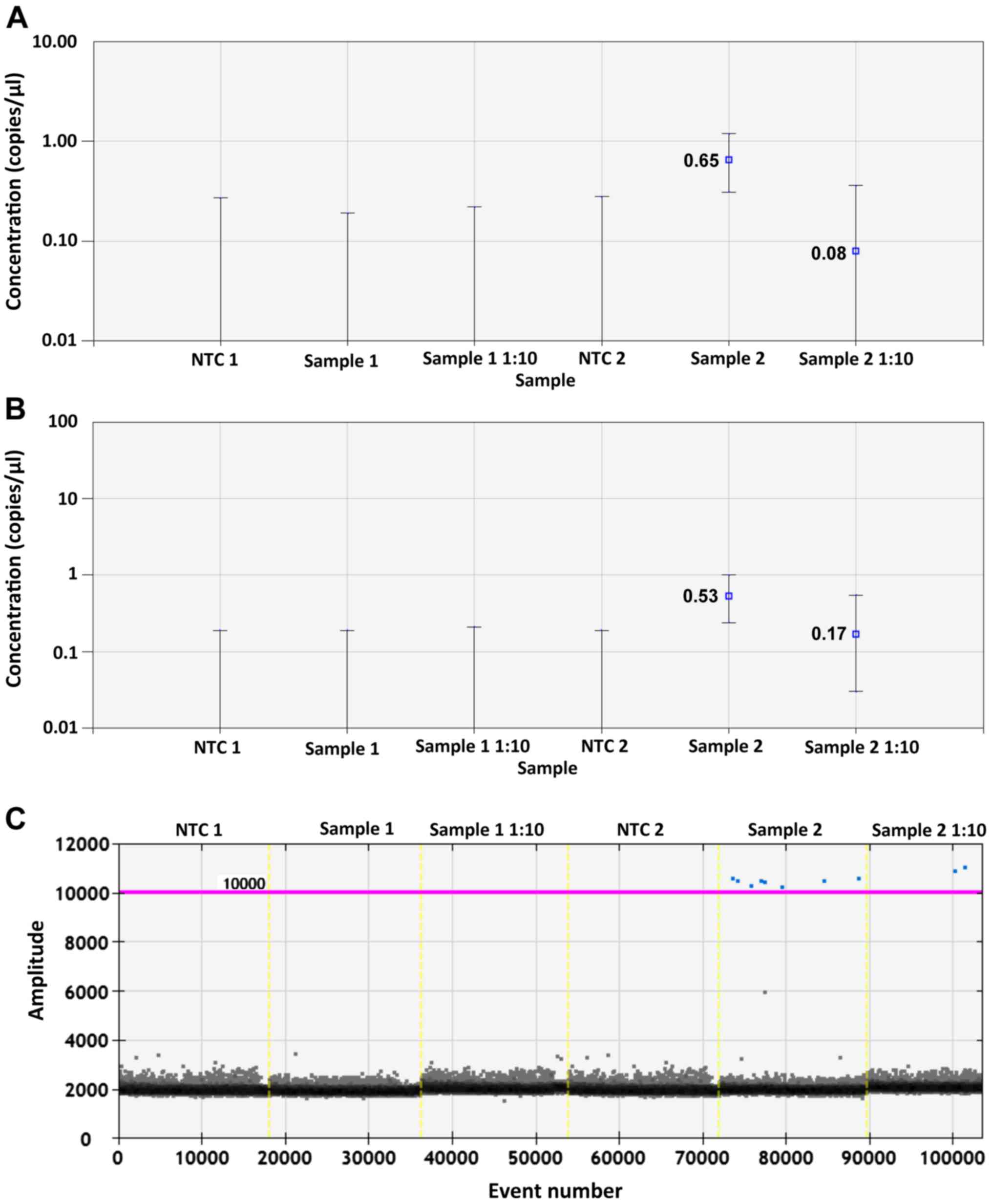
The higher sensitivity of ddPCR allowed the correct
identification of the positive and negative COVID-19 samples.
Despite the lower amount of cDNA in sample 2 compared to sample 1,
the primers specific for the SARS-CoV-2 N gene did not fail in
recognizing sample 1 as negative, and sample 2 as positive.
Therefore, differently from the SYBR-Green and TaqMan RT-qPCR,
EvaGreen ddPCR correctly diagnosed as positive a potentially
false-negative sample due to the low amount of SARS-CoV-2 nucleic
acid. In particular, EvaGreen ddPCR showed positive results in both
undiluted and 10-fold diluted sample 2 detecting 0.65 and 0.08
copies/µl, respectively (Fig.
4A).
The EvaGreen ddPCR results were further confirmed by
using the same 2019-nCoV_N1 TaqMan probe adopted for the RT-qPCR
analysis. Probe ddPCR showed positive results for sample 2
detecting 0.53 and 0.17 copies/µl in the undiluted and
10-fold diluted samples, respectively. Despite the highest
concentration of cDNA, no signals were obtained for the negative
sample 1, thus confirming its negative value (Fig. 4B). In addition, probe ddPCR showed
a more stable signal for the positive droplets that showed an
amplitude greater than 10,000 (Fig.
4C).
Overall, both ddPCR EvaGreen and Probe systems allow
the identification of low amounts of SARS-CoV-2 N gene with great
accuracy and sensitivity. The higher sensitivity of ddPCR allowed
the identification of positive signals not only in the undiluted
sample but also in the 10-fold dilution demonstrating the
usefulness of this method in the diagnosis of COVID-19 positive
patients with very low viral load or in patients not yet in
complete remission and with a minimal residual viral load. In
addition, the detection limit of EvaGreen ddPCR chemistry was
significantly lower compared to that obtained with SYBR-Green and
TaqMan probe RT-qPCR (0.00187 vs. 0.0187 ng, respectively).
Finally, the sequencing of droplets confirmed that
the positive signal was specific for SARS-CoV-2 N gene. In
particular, a perfect match between the amplified fragment and the
reference sequence MT077125 was obtained (Fig. S2).
Discussion
Since the new SARS-CoV-2 emerged, researchers around
the world have tried to develop highly sensitive molecular
techniques in order to effectively diagnose positive COVID-19
subjects and to keep them in quarantine in order to reduce the
number of infections. According to the WHO and the Center for
Disease Control and Prevention (CDC), the gold standard method for
the diagnosis is represented by RT-qPCR. Different molecular
technologies have been developed (23,24), however, many studies have reported
low sensitivity and specificity rates for some of these methods
(25,26). Therefore, it is necessary to
build-up novel robust methodologies ensuring high sensitivity and
specificity rates suitable not only for diagnostic purposes but
also for the follow-up of patients and for the monitoring of the
viral load.
Here we compared the sensitivity of RT-qPCR and
ddPCR techniques in identifying COVID-19 positive patients with low
viral load. By analyzing two blinded pharyngeal swabs obtained from
two patients tested, respectively, positive and negative for
COVID-19, we compared the two methods to establish the advantages
and pitfalls in assessing a potentially critical false-negative
sample. By using SYBR-Green RT-qPCR and WHO/CDC-approved primers
for the SARS-CoV-2 N gene, we demonstrated that, although
high-sensitive (detection limit of β-actin housekeeping gene of
0.0187 ng), RT-qPCR it is not sensitive enough to recognize as
positive samples with low viral load. In parallel, the RT-qPCR
performed by using the WHO/CDC-approved 2019-nCoV-N1 TaqMan probe
showed that TaqMan chemistry allows the identification of the
positive sample only at the undiluted concentration, only at a very
late Ct value (36.59). Notably, according to the CDC-approved
COVID-19 diagnostic panel (CDC 2019-nCoV Real-Time RT-PCR
Diagnostic Panel cat. no. 2019-nCoVEUA-01) a Ct value of 36.59 it
is not considered valid to formulate a diagnosis of COVID-19
positivity. Therefore, the RT-qPCR result obtained here should be
further confirmed before considering the sample as positive.
On the other hand, ddPCR allowed us to certainly
diagnose the tested sample as positive at the undiluted cDNA
concentration and at a 10-fold dilution using both EvaGreen and
Probe ddPCR chemistry with a detection limit for the β-actin
housekeeping gene of more than 0.00187 ng. Therefore, we
demonstrated the higher diagnostic potential of ddPCR compared to
RT-qPCR. In addition, the robustness of the ddPCR approach was
further corroborated by the sequencing of positive droplets that
confirmed that the positive signal was related to the amplification
of the SARS-CoV-2 N gene fragment.
These results pave the way for the use of the ddPCR
not only for diagnostic purposes but also for the follow-up of
patients and the frequent monitoring of the disease state. Indeed,
in addition to being more sensitive than RT-qPCR, the ddPCR has
also a comparable time-costing workflow and costs related to the
reagents and analysis. In particular, the pre-analytical phases are
the same in both methods until the cDNA is obtained; also, the
primers and probes used for the last step of the analysis are the
same. Therefore, the only differences between the two technologies
rely on the slightly greater time of the ddPCR necessary to
generate the droplets and read the plate (approximately 2 h more
than the RT-qPCR) and the moderate additional costs related to the
cartridges and consumables necessary for the generation of the
droplets (8-well cartridge, droplet generator gasket, droplet
generation oil). Overall, the ddPCR requires approximately 15% more
time and 5-10% more cost than the RT-qPCR. Despite the timing and
costs associated with the analysis in RT-qPCR being slightly lower,
the higher sensitivity of the ddPCR tip the balance of the
cost/benefit ratio towards the use of the ddPCR.
Results here obtained highlighted another important
advantage of ddPCR over RT-qPCR. Indeed, the ddPCR is less affected
by the interference of any reaction inhibitors thanks to the
microdilutions that are carried out within each droplet and to the
end-point PCR measurement typical of digital amplification systems
(27). Therefore, the ddPCR is
less dependent on the efficiency of PCR amplification compared to
the RT-qPCR. Finally, substantial differences were related to the
output data generated by using the two methodologies. The output
data obtained by ddPCR are expressed as end-point absolute
quantification of SARS-CoV-2 N gene copies. Therefore, these data
are more robust and repeatable compared to the data obtained with
RT-qPCR that are expressed as relative Ct values and could vary
significantly depending on the platforms used and the quality of
starting materials (28,29).
In addition, as demonstrated for other viral
infections, ddPCR allows the detection of weak and moderate
increase or reduction of the viral load while no significant
variations were observed in the Ct values obtained by using RT-qPCR
(30-32). Therefore, ddPCR may be used also
to early detect SARS-CoV-2 viral load variation after therapeutic
interventions in order to evaluate the efficacy of the treatments
and adjust drug dose and posology.
In conclusion, overall, this preliminary study has a
great translational impact on the fight against COVID-19 infection.
Indeed, the ddPCR analysis here proposed, will improve the current
diagnostic strategies available and will implement novel follow-up
approaches to monitor the viral load of COVID-19 patients, avoiding
false positive or false-negative results. In addition, due to its
high sensitivity, ddPCR could be used also for the detection of
SARS-CoV-2 in blood and saliva samples, thus improving the
diagnostic procedure currently based on the analysis of
rhino-pharyngeal swab not always executable, especially in
uncooperative or unconscious patients.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was funded by the research grant
PRIN 2017SFBFER from MIUR.
Availability of data and materials
The data generated and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML, GS and SS conceived the work; LF, NM, GG, CIP
and DB performed the experiments and analyzed the data; LF and GG
prepared the figures; LF, NM and DB wrote the manuscript; LF, ML,
GS and SS provided critical revisions; SS acquired funding. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Azienda Ospedaliera Universitaria 'Policlinico-Vittorio Emanuele'
of Catania. Patients signed informed consent before participating
in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chakraborty I and Maity P: COVID-19
outbreak: Migration, effects on society, global environment and
prevention. Sci Total Environ. 728:1388822020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Docea AO, Tsatsakis A, Albulescu D,
Cristea O, Zlatian O, Vinceti M, Moschos SA, Tsoukalas D, Goumenou
M, Drakoulis N, et al: A new threat from an old enemy: Re-emergence
of coronavirus (Review). Int J Mol Med. 45:1631–1643.
2020.PubMed/NCBI
|
|
3
|
Mackenzie JS and Smith DW: COVID-19-A
novel zoonotic disease: A review of the disease, the virus, and
public health measures. Asia Pac J Public Health. May 30–2020.Epub
ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goumenou M, Sarigiannis D, Tsatsakis A,
Anesti O, Docea AO, Petrakis D, Tsoukalas D, Kostoff R, Rakitskii
V, Spandidos DA, et al: COVID-19 in Northern Italy: An integrative
overview of factors possibly influencing the sharp increase of the
outbreak (Review). Mol Med Rep. 22:20–32. 2020.PubMed/NCBI
|
|
5
|
Calina D, Docea AO, Petrakis D, Egorov AM,
Ishmukhametov AA, Gabibov AG, Shtilman MI, Kostoff R, Carvalho F,
Vinceti M, et al: Towards effective COVID-19 vaccines: Updates,
perspectives and challenges (Review). Int J Mol Med. 46:3–16.
2020.PubMed/NCBI
|
|
6
|
Corman VM, Landt O, Kaiser M, Molenkamp R,
Meijer A, Chu DK, Bleicker T, Brünink S, Schneider J, Schmidt ML,
et al: Detection of 2019 novel coronavirus (2019-nCoV) by real-time
RT-PCR. Euro Surveill. 25:20000452020. View Article : Google Scholar :
|
|
7
|
World Health Organization (WHO): All
technical guidance on COVID-19 - select topic from drop down menu.
https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/laboratory-guidance.
|
|
8
|
Won J, Lee S, Park M, Kim TY, Park MG,
Choi BY, Kim D, Chang H, Kim VN and Lee CJ: Development of a
Laboratory-safe and low-cost detection protocol for SARS-CoV-2 of
the Coronavirus Disease 2019 (COVID-19). Exp Neurobiol. 29:107–119.
2020. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang X, Yao H, Xu X, Zhang P, Zhang M,
Shao J, Xiao Y and Wang H: Limits of detection of six approved
RT-PCR kits for the novel SARS-coronavirus-2 (SARS-CoV-2). Clin
Chem. 66:977–979. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tahamtan A and Ardebili A: Real-time
RT-PCR in COVID-19 detection: Issues affecting the results. Expert
Rev Mol Diagn. 20:453–454. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lippi G, Simundic AM and Plebani M:
Potential preanalytical and analytical vulnerabilities in the
laboratory diagnosis of coronavirus disease 2019 (COVID-19). Clin
Chem Lab Med. 58:1070–1076. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang YW, Schmitz JE, Persing DH and
Stratton CW: Laboratory diagnosis of COVID-19: Current issues and
challenges. J Clin Microbiol. 58:e00512–e00520. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor SC, Carbonneau J, Shelton DN and
Boivin G: Optimization of Droplet Digital PCR from RNA and DNA
extracts with direct comparison to RT-qPCR: Clinical implications
for quantification of Oseltamivir-resistant subpopulations. J Virol
Methods. 224:58–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hindson CM, Chevillet JR, Briggs HA,
Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL and Tewari M:
Absolute quantification by droplet digital PCR versus analog
real-time PCR. Nat Methods. 10:1003–1005. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hindson BJ, Ness KD, Masquelier DA,
Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY,
Hiddessen AL, Legler TC, et al: High-throughput droplet digital PCR
system for absolute quantitation of DNA copy number. Anal Chem.
83:8604–8610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Filetti V, Falzone L, Rapisarda V,
Caltabiano R, Eleonora Graziano AC, Ledda C and Loreto C:
Modulation of microRNA expression levels after naturally occurring
asbestiform fibers exposure as a diagnostic biomarker of
mesothelial neoplastic transformation. Ecotoxicol Environ Saf.
198:1106402020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salemi R, Falzone L, Madonna G, Polesel J,
Cinà D, Mallardo D, Ascierto PA, Libra M and Candido S: MMP-9 as a
candidate marker of response to BRAF inhibitors in melanoma
patients with BRAFV600E mutation detected in circulating-free DNA.
Front Pharmacol. 9:8562018. View Article : Google Scholar :
|
|
18
|
Battaglia R, Palini S, Vento ME, La
Ferlita A, Lo Faro MJ, Caroppo E, Borzì P, Falzone L, Barbagallo D,
Ragusa M, et al: Identification of extracellular vesicles and
characterization of miRNA expression profiles in human blastocoel
fluid. Sci Rep. 9:842019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Whale AS, Huggett JF, Cowen S, Speirs V,
Shaw J, Ellison S, Foy CA and Scott DJ: Comparison of microfluidic
digital PCR and conventional quantitative PCR for measuring copy
number variation. Nucleic Acids Res. 40:e822012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Department of Health and Human Services,
Public Health Service, Centres for Disease Control and Prevention
(CDC): 2019-Novel Coronavirus (2019-nCoV) Real-time rRT-PCR Panel.
Primers and Probes. Division of Viral Diseases. CDC; Atlanta, GA:
2020, https://www.who.int/docs/default-source/coronaviruse/uscdcrt-pcr-panel-primer-probes.pdf?sfvrsn=fa29cb4b_2.
Accessed March 15, 2020.
|
|
21
|
Fresta CG, Fidilio A, Lazzarino G, Musso
N, Grasso M, Merlo S, Amorini AM, Bucolo C, Tavazzi B, Lazzarino G,
et al: Modulation of pro-oxidant and pro-inflammatory activities of
M1 macrophages by the natural dipeptide carnosine. Int J Mol Sci.
21:E7762020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Musso N, Caronia FP, Castorina S, Lo Monte
AI, Barresi V and Condorelli DF: Somatic loss of an EXT2 gene
mutation during malignant progression in a patient with hereditary
multiple osteochondromas. Cancer Genet. 208:62–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
World Health Organization (WHO):
Emergencies diseases novel coronavirus 2019. https://www.who.int/.
|
|
24
|
Carter LJ, Garner LV, Smoot JW, Li Y, Zhou
Q, Saveson CJ, Sasso JM, Gregg AC, Soares DJ, Beskid TR, et al:
Assay techniques and test development for COVID-19 diagnosis. ACS
Cent Sci. 6:591–605. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao AT, Tong YX and Zhang S:
False-negative of RT-PCR and prolonged nucleic acid conversion in
COVID-19 Rather than recurrence. J Med Virol. https://doi.org/10.1002/jmv.25855.
|
|
26
|
Li Y, Yao L, Li J, Chen L, Song Y, Cai Z
and Yang C: Stability issues of RT-PCR testing of SARS-CoV-2 for
hospitalized patients clinically diagnosed with COVID-19. J Med
Virol. 92:903–908. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dingle TC, Sedlak RH, Cook L and Jerome
KR: Tolerance of droplet-digital PCR vs real-time quantitative PCR
to inhibitory substances. Clin Chem. 59:1670–1672. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dong L, Wang X, Wang S, Du M, Niu C, Yang
J, Li L, Zhang G, Fu B, Gao Y, et al: Interlaboratory assessment of
droplet digital PCR for quantification of BRAF V600E mutation using
a novel DNA reference material. Talanta. 207:1202932020. View Article : Google Scholar
|
|
29
|
Pinheiro LB, O'Brien H, Druce J, Do H, Kay
P, Daniels M, You J, Burke D, Griffiths K and Emslie KR:
Interlaboratory reproducibility of Droplet Digital Polymerase Chain
Reaction using a new DNA reference material format. Anal Chem.
89:11243–11251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rutsaert S, Bosman K, Trypsteen W, Nijhuis
M and Vandekerckhove L: Digital PCR as a tool to measure HIV
persistence. Retrovirology. 15:162018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yucha RW, Hobbs KS, Hanhauser E, Hogan LE,
Nieves W, Ozen MO, Inci F, York V, Gibson EA, Thanh C, et al:
High-throughput characterization of HIV-1 reservoir reactivation
using a single-cell-in-Droplet PCR assay. EBioMedicine. 20:217–229.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trypsteen W, Kiselinova M, Vandekerckhove
L and De Spiegelaere W: Diagnostic utility of droplet digital PCR
for HIV reservoir quantification. J Virus Erad. 2:162–169.
2016.PubMed/NCBI
|