Introduction
T-cell lymphomas (TCL) are rare hematolymphoid
malignancies with poor overall survival. A major subtype of TCL,
angioimmunoblastic T-cell lymphoma (AITL), has been the focal point
of numerous studies. AITL is characterized by unique clinical,
morphologic, immunophenotypic and molecular features. AITL
possesses a characteristic background of increased vascularity
along with morphologically atypical T-cells that present with a
follicular helper T-cell immunophenotype (1-3).
In recent years, broad genetic profiling of AITL
reported that it often harbors mutations in ras homolog family
member A (RHOA; G17V), tet methylcytosine dioxygenase 2
(TET2), DNA methyltransferase 3 alpha (DNMT3A) and
isocitrate dehydrogenase [NADP(+)] 2 (IDH2) (4-9).
For example, RHOA G17V mutations have been the focus of
intense research. RHOA G17V mutations are believed to be
highly associated with the most classic cases of AITL (10) and are found in 50-80% of AITL
cases (7). In addition,
RHOA mutated cases are believed to be characterized by
increased microvascular density and to express a high number of
follicular helper T-cell markers (10).
To better understand the pathophysiology of AITL,
the present study performed targeted deep sequencing on 18 tissues
samples from 10 patients with AITL, which were biopsied at the time
of diagnosis. Both targeted DNA sequencing and RNA-sequencing,
including single nucleotide polymorphisms (SNPs) and
insertions/deletions (indel) analysis, translocation analysis, and
gene expression as well as pathway analysis were performed on AITL
cases. Computational image segmentation and analysis from
haematoxylin and eosin (H&E) stained sections were completed in
order to quantify and differentiate morphological parameters
between the RHOA mutated and RHOA non-mutated cases.
In addition, this analysis was coupled to the outcomes of patients
to compare overall survival.
Materials and methods
Patient cohort
In the present study, 10 cases of AITL were selected
from the archives (July 2004-October 2018) of the Department of
Pathology, Stanford University Medical Center (Stanford, CA) where
adequate tissues were available and diagnoses could be confirmed.
Patient medical record charts, clinical and laboratory data,
treatment data and slides [formalin fixed paraffin embedded (FFPE)
lymph node tissues, clinically stained] were reviewed, and
diagnoses were confirmed by RO, JK, AB and RW according to the 2016
World Health Organization (WHO) (11,12) criteria. In total, 18 tissue
samples were analyzed. H&E stained slides were generated by
staining FFPE tissues slices of 4-µm thickness.
Auto-staining was performed on a Leica Autostainer XL according to
manufacturer's instructions (Leica Microsystems, Inc.). This study
received ethical approval from Stanford University's Institutional
Review Board (reference no. IRB-22359).
Targeted DNA deep sequencing
Targeted sequencing was performed as previously
described (13). Briefly, DNA was
extracted from FFPE lymph node tissues using the DNA Storm FFPE DNA
Extraction kit (Cell Data Sciences). Quality and quantity of
extracted nucleic acids was assessed by Qubit (Thermo Fisher
Scientific, Inc.) and the 2100 Bioanalyzer (Agilent Technologies,
Inc.). For targeted next-generation sequencing (NGS), our Heme
Malignancy Evaluation and Infectious Disease panel (HeME-ID;
Table SI) was used, which
targets 354 genes mutated in hematolymphoid diseases and allows
identification of point mutations, insertion/deletions (indels) and
translocations, as well as 13 viruses and bacteria associated with
hematolymphoid diseases. DNA (150 ng) was used to prepare the DNA
library using the SureSelectXT HS enrichment kit (Agilent
Technologies, Inc.). A 100 base-pair paired end high-throughput
sequencing was performed on a HiSeq4000 platform (Illumina, Inc.)
at an average depth of 1000 fold. For downstream processing of the
output files, Genome Analysis Toolkit (GATK; https://gatk.broadinstitute.org/hc/en-us) best
practices for alignment, single-nucleotide variant and structural
variant analysis were followed (14), BWA-MEM algorithm for alignment was
used and further analysis was performed using GATK (version 4.0;
https://gatk.broadinstitute.org/hc/en-us), Varscan2
(version v2.3.8) (15), and
SNNPET (Agilent Technologies, Inc.). For variant calling, SureCall
(version 4.1; Agilent Technologies, Inc.) was used and mutations
were analyzed with a variant allele frequency at ≥2% in single
sample analysis mode, which was justified by a high read depth and
the use of molecular barcodes in the SureselectXT HS
kit. Filters were applied following the mutation caller's
recommendations. In order to call a mutation, a 200× read coverage
per base, a minimum coverage in forward and reverse direction and a
maximum allele frequency of 40% were required and a minimum
Combined Annotation Dependent Depletion (CADD) score of 20. The
same filters were applied for small indels analysis. For
annotation, SureCall and Seattleseq (University of Washington,
Seattle; version 9.10) were used. For further curation of the
variants, Exome Aggregation Consortium (16), Clinvar (17), 1000 genomes (18), Exome Variant Server (19) and Varsome (20) were used. For further downstream
analysis, the MutationalPatterns (21) Package from R was used [mutational
signatures analysis using COSMIC signatures, enrichment/depletion
analysis of promoter regions, CTCF (CCCTC-binding factor) bindings
sites and promoter flanking regions], and Pathway Analysis was done
with EnrichR (22), Gene Set
Enrichment Analysis (23,24) and ConsensusPathDB-human (25). Evaluation of microorganisms was
performed using the subtraction method as previously described for
shotgun metagenomic sequencing (26,27). For viruses, results were
interpreted by percent coverage of the targeted regions and average
depth. Samples were classified as follows: Negative, equivocal and
positive. A positive result required all three targeted regions of
a sample to have a minimum coverage of 75% and an average depth of
at least 5. An equivocal result indicated that all three targeted
regions had a 10-75% coverage with a minimum average read depth of
1. A negative result indicated all other scenarios.
RNA-sequencing and data analysis
RNA was extracted from FFPE lymph node tissues using
the RNA Storm FFPE DNA Extraction kit (Cell Data Sciences). Quality
and quantity of extracted nucleic acids was assessed by Qubit
analysis and 2100 Bioanalyzer. RNA (200 ng) was used to prepare RNA
libraries with the SureSelectXT RNA Direct kit (Agilent
Technologies, Inc.; with Sureselect Exome V6 + UTR, capture
library) for strand-specific sequencing libraries. A 150 base-pair
paired end whole transcriptome sequencing was performed on a
high-throughput sequencing HiSeq4000 platform with a mean coverage
of 300 million reads. For downstream processing of our output
files, Hisat2 (version 2.1.0) (28) was used for alignment and HT-Seq
(version 0.11.1) (29) was used
for generation of the count files. Gene expression analysis was
performed using RStudio (version 3.5.3) (30). Exploratory analysis [Principal
Component Analysis (PCA) and heatmap with hierarchical clustering
by Euclidean distance] was performed using ClustVis (31). For differential gene expression,
the DeSeq and EdgeR packages were used. The immune cell composition
was analyzed using CIBERSORT (https://ciber-sort.stanford.edu/), which is a tool
that uses the constellation of expressed genes, based on known
signatures, in order to refer to the suspected immune cell
composition within the analyzed sample (32).
Statistical analysis
Statistical analysis was performed using R (Cran;
version 3.6.0; https://cran.r-project.org/) and RStudio (RStudio
Inc., version 1.2.1335; https://rstudio.com/). Student's t-test and
Mann-Whitney U test were performed to evaluate differences between
the mutational burden of RHOA positive and negative cases, the
average CADD score of RHOA positive and negative cases, the
mutational burden of EBV positive and negative cases and the
average CADD score of EBV positive and negative cases.
χ2 test and Kaplan Meier Survival analysis were also
used. Statistical significance was determined with the Log-rank. A
two-sided binomial test was used to compare the two categories of
RHOA positive and negative cases with regards to expected vs.
observed mutational burdens in defined genomic locations. P<0.05
was considered to indicate a statistically significant
difference.
Computational digital image analysis
H&E images of lymph node sections were obtained
using a conventional slide scanner (Leica Aperio AT2 slide scanner;
Leica Microsystems, Inc.). The H&E images were patched at an
optical zoom of ×40, uniform width of 68 pixels and height of 57
pixels. MATLAB program (version 2019b; https://www.mathworks.com/) was used for image
analysis wherein the cells of interest were segmented based on
their pixel intensities (33).
The unsharp filter in the Image Processing Toolbox was used to
improve the edge contours and contrast among the different cells of
interest. Based on the input pixels, cells were clustered using
k-means clustering (34,35). For an input image I(x,y), the
distance between each cluster center,
cI(I=1:n),
and the corresponding data points is denoted by the equation 1:
This iterative algorithm seeks to minimize the sum
of all distances (d) between all the data points in a particular
cluster to its cluster centroid and is calculated using the
equation 2:
Once the necessary cluster that best matched the
target cell body was identified, the respective image was
transformed into its corresponding grayscale format. Finally, the
regionprops function from MATLAB was used to extract eight
parameters, including circularity, area (in pixel2),
major axis, minor axis, eccentricity, equivalent diameter [
], solidity (measure of the indentations on a cell surface) and
perimeter (in pixels). Since it has been reported that cancer cells
exhibit an increased variability in roundness, elongation and
differences in nuclear shape (36,37), cells were classified based on
these parameters. The resulting datasets of RHOA mutation
positive and RHOA mutation negative samples were compared
using Principal Component Analysis (PCA) in R studio [R version
3.5.1 (2018-07-02); https://rstudio.com/] to quantify the deviation
between them and identify the most variable parameters between the
samples.
Results
Patient cohort
Tissue biopsies from 10 patients with AITL were
selected in the present study. All cases were reviewed by RO, JK,
AB and RW and classified according to the 2016 revised WHO
classification. The 10 patients consisted of four women and six men
(average age, 60 years; age range, 39-78 years). Five patients were
positive for Epstein-Barr virus (EBV) infected cells
following analysis by in situ hybridization (ISH). Five
patients were RHOA mutation positive and five patients were
RHOA mutation negative. Treatment regimens are presented in
Table I. Among the 10 patients,
only one (case 9) did not relapse following initial therapy.
 | Table IClinicopathological characteristics
of the 10 patients with angioimmunoblastic T-cell lymphoma. |
Table I
Clinicopathological characteristics
of the 10 patients with angioimmunoblastic T-cell lymphoma.
| Case number | Age, years | Sex | Histology | RHOA status | Treatment | Time to relapse,
days | Survival, days |
|---|
| Case 1 | 73 | Female | AITL+BCP | RHOA+ | Prednisone,
Chlorambucil, Hydroxyurea, Rituximab | 1,364 | 3,449 |
| Case 2 | 70 | Male | AITL+BCP | RHOA− | 6 cycles R-CHOP
+GCSF | 310 | 1,177 |
| Case 3 | 47 | Male | AITL+BCP | RHOA− | 1 cycle R-CHOP | 715 | 725 |
| Case 4 | 49 | Male | AITL | RHOA− | NA | NA | NA |
| Case 5 | 78 | Female | AITL | RHOA+ | Prednisone | 42 | 42 |
| Case 6 | 59 | Male | AITL | RHOA− | 8 cycles CHOP | 181 | 181 |
| Case 7 | 72 | Male | AITL | RHOA+ | Etanercept
treatment | 1,005 | 1,005 |
| Case 8 | 45 | Male | AITL | RHOA+ | 6 cycles CHOP +
Vincristine vs. Brentuximab, auto-HCT | 1,733 | 1,733 |
| Case 9 | 68 | Female | AITL | RHOA+ | Steroids, 1 dose
Cytoxan, Romiplostim + 2 doses IVIG, cyclosporine treatment,
prednisone + IVIG continuously | No relapse | 1,839 |
| Case 10 | 39 | Female | AITL | RHOA− | SGN-35-014: 4
cycles CHOP + Vincristine vs. Brentuximab, 3 cycles
ICE+Brentuximab, auto-HCT | 127 | 264 |
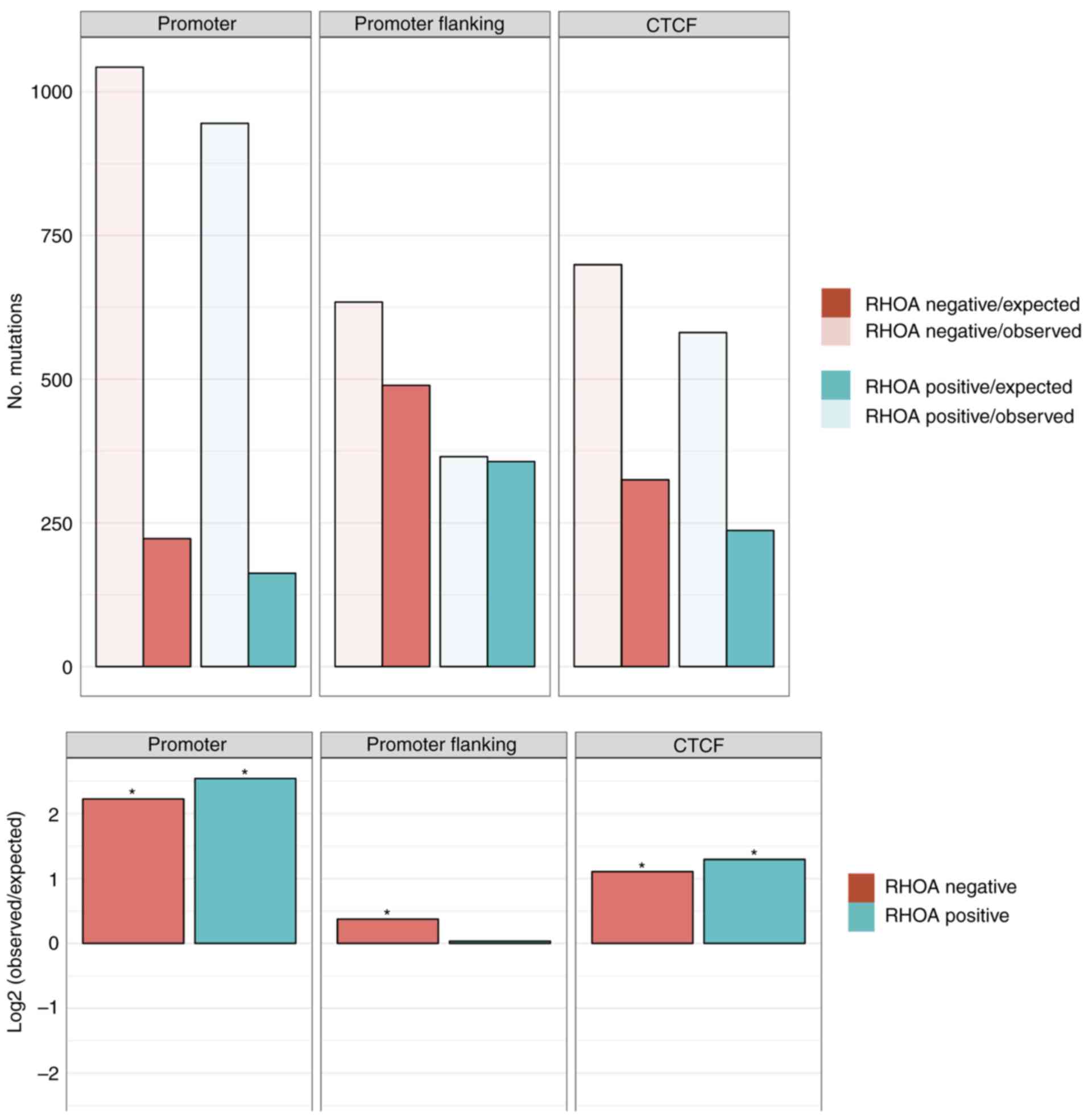
DNA mutational analysis demonstrates
differential mutational profiles in RHOA positive cases vs. RHOA
negative cases
Targeted DNA deep sequencing was performed on all
cases to understand the pathophysiology of AITL. The overall
mutational burden ranged from 0-14. RHOA mutation positive
cases showed a higher mutational burden (5.2 mutations/case) vs.
RHOA mutation negative cases (1.8 mutations/case) with
mutations generally showing more deleterious changes in RHOA
mutation positive cases (average CADD score of 24.9 vs. 21.8). The
mutational profile in promoter regions and CTCF binding sites were
also investigated and the results demonstrated that the mutational
burden was higher than expected in these regions, especially for
RHOA mutation positive cases vs. the RHOA mutation
negative cases (Fig. 1).
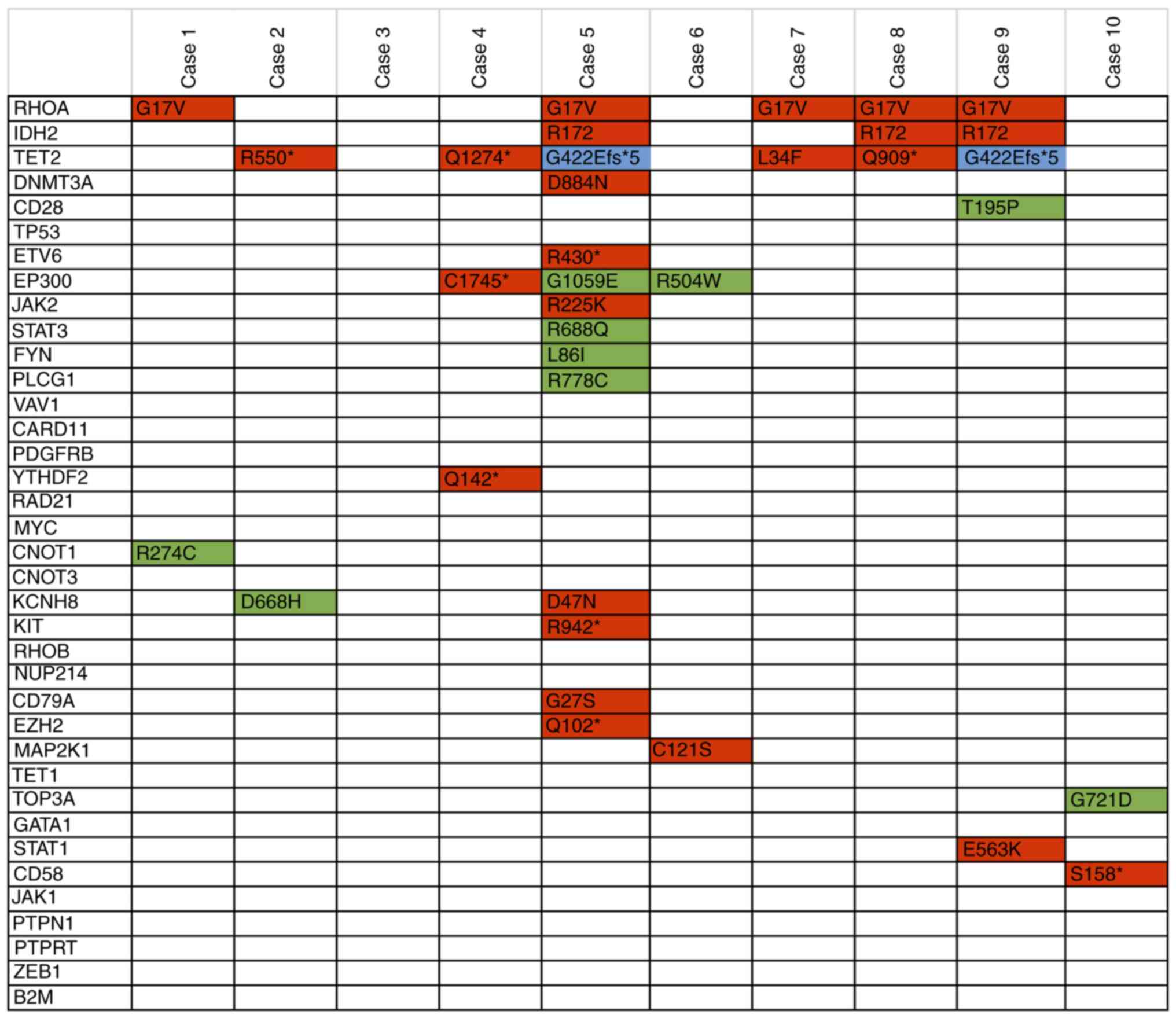
Recurrent IDH2, TET2 and DNMT3A mutations have also been
reported in AITL cases. We identified IDH2 R172 mutations in
three of our cases (cases 5, 8 and 9). Two of these cases also
showed the recurrent TET2 G422Efs*5 mutation (Fig. 2). The results demonstrated that
TET2 was mutated in four additional samples. In addition,
other genes known to be mutated in TCLs were mutated in our cases,
and CD28 was mutated in one case (case 9) and DNMT3A
was mutated in another case (case 5). Furthermore, ETV6, EP300,
STAT3, JAK2, FYN and PLCG1 genes were mutated in the
samples analyzed in the present study. The HeME-ID Panel can
evaluate common breakpoints and translocations, including for vav
guanine nucleotide exchange factor 1 (VAV1; Table SI). No translocations were
identified in the samples (data not shown). Finally, the absolute
contribution of the COSMIC signatures (version 2), which are
cancer-type specific signatures (liver, lung, stomach, B-cell
lymphomas), was also investigated in the AITL samples with the
Mutational Patterns package in R. However, no major contribution of
a mutational signature in the AITL cases was identified. The
mutational signature amongst all cases was the same with a majority
of C>T and T>C base exchanges independent from RHOA mutation
status or tissue type (Fig.
S1).
EBV infection in AITL tissues
Five out of 10 patients had EBV infected cells by
Epstein-Barr encoding region ISH (Table II). EBV and 13 other
microorganisms as well as 354 human genetic mutations were targeted
by using our targeted panel and sequencing approach. We performed
targeted deep sequencing with our HeME-ID panel on our tissue
samples directly and detected EBV in 4 cases as positive and 2
cases as equivocal (Table SII).
All matched bone marrow samples were negative for EBV infection
based on our criteria, apart from case 10 which had equivocal EBV
infection of the bone marrow. Table
II represents the viral findings from immunohistochemistry
compared with results from NGS. No difference in the gene
expression pattern was observed between EBV positive and EBV
negative cases. The genomic analysis showed a higher mutational
burden in EBV negative cases (average 8.25 vs. average 4 in EBV
positive); however, a higher CADD score in mutated genes in EBV
positive cases was observed (average CADD score 27.11 vs. average
CADD score 17.7 in EBV negative). Survival analysis showed no
difference between EBV infected and non-infected cases (Fig. S2).
| Table IIOverview of EBV in AITL cases. |
Table II
Overview of EBV in AITL cases.
| Case | Tissue | EBV infected cells
(by EBER ISH) | EBV NGS |
|---|
| Case 1 | LN | Negative | Negative |
| Case 2 | LN | Positive | Equivocal |
| Case 3 | LN | Negative | Negative |
| BM | | Negative |
| Case 4 | LN | Positive | Positive |
| BM | Negative | Negative |
| Case 5 | LN | Negative | Negative |
| Case 6 | LN | N/A | Negative |
| BM | | Negative |
| Case 7 | LN | Positive | Positive |
| BM | | Negative |
| Case 8 | LN | Positive | Positive |
| BM | | Negative |
| Case 9 | LN | Negative | Equivocal |
| Case 10 | LN | Positive | Positive |
| BM | N/A | Negative |
| BM | Positive | Equivocal |
| BM | Negative | Negative |
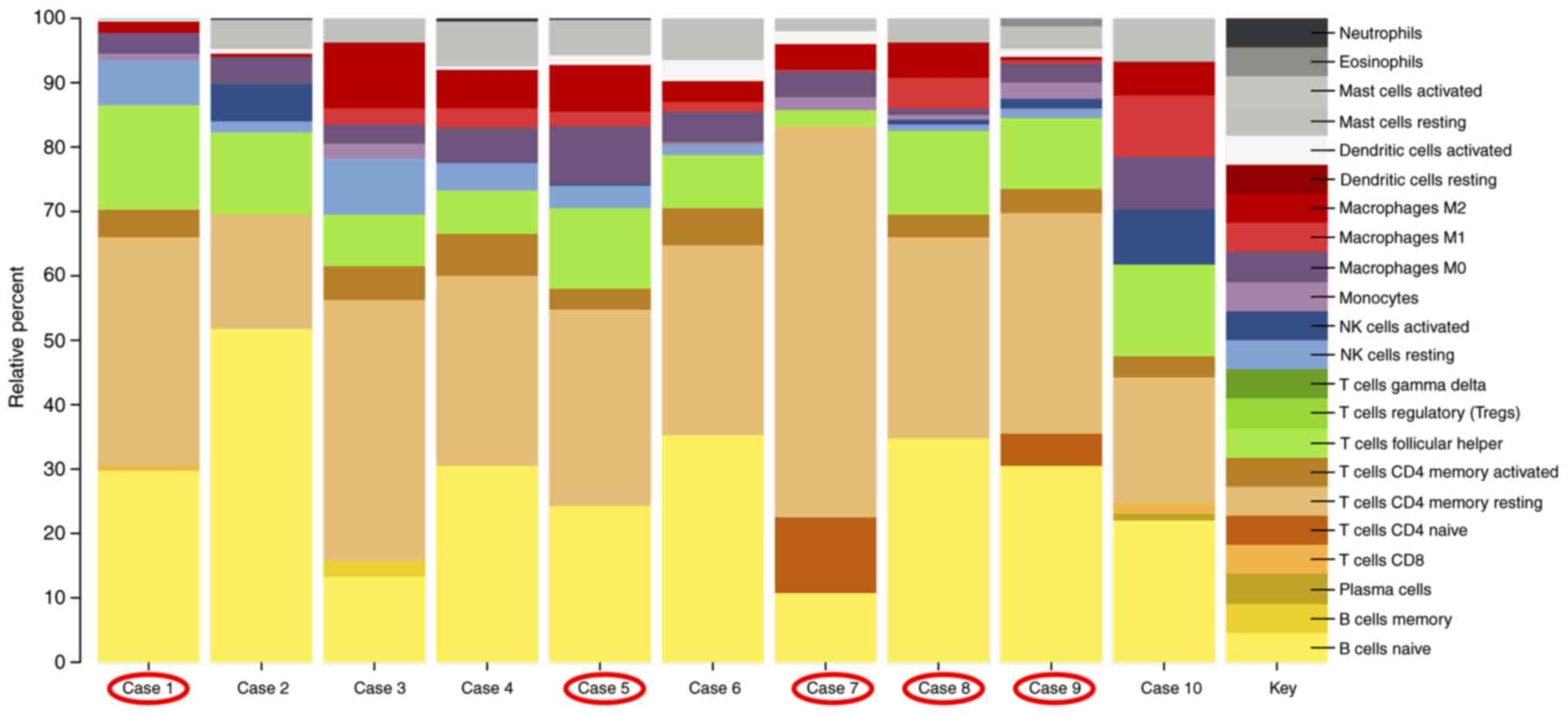
RNA expression profiling identifies an
altered immunologic environment in all AITL cases
Investigation of the associated immune system based
on the gene expression profile of the AITL cases demonstrated that
the majority of infiltrating immune cells consisted of naïve
B-cells (yellow, Fig. 3) and
resting memory CD4 T-cells (beige, Fig. 3). The third largest component in
our AITL cases were the T-follicular helper cells (green, Fig. 3). Fig. 3 is a representation of the
contribution of the immune cell expression patterns analyzed by the
CIBERSORT pipeline. RHOA positive cases are circled in red.
There is no difference in the immunologic background of RHOA
mutation positive compared with RHOA mutation negative
cases.
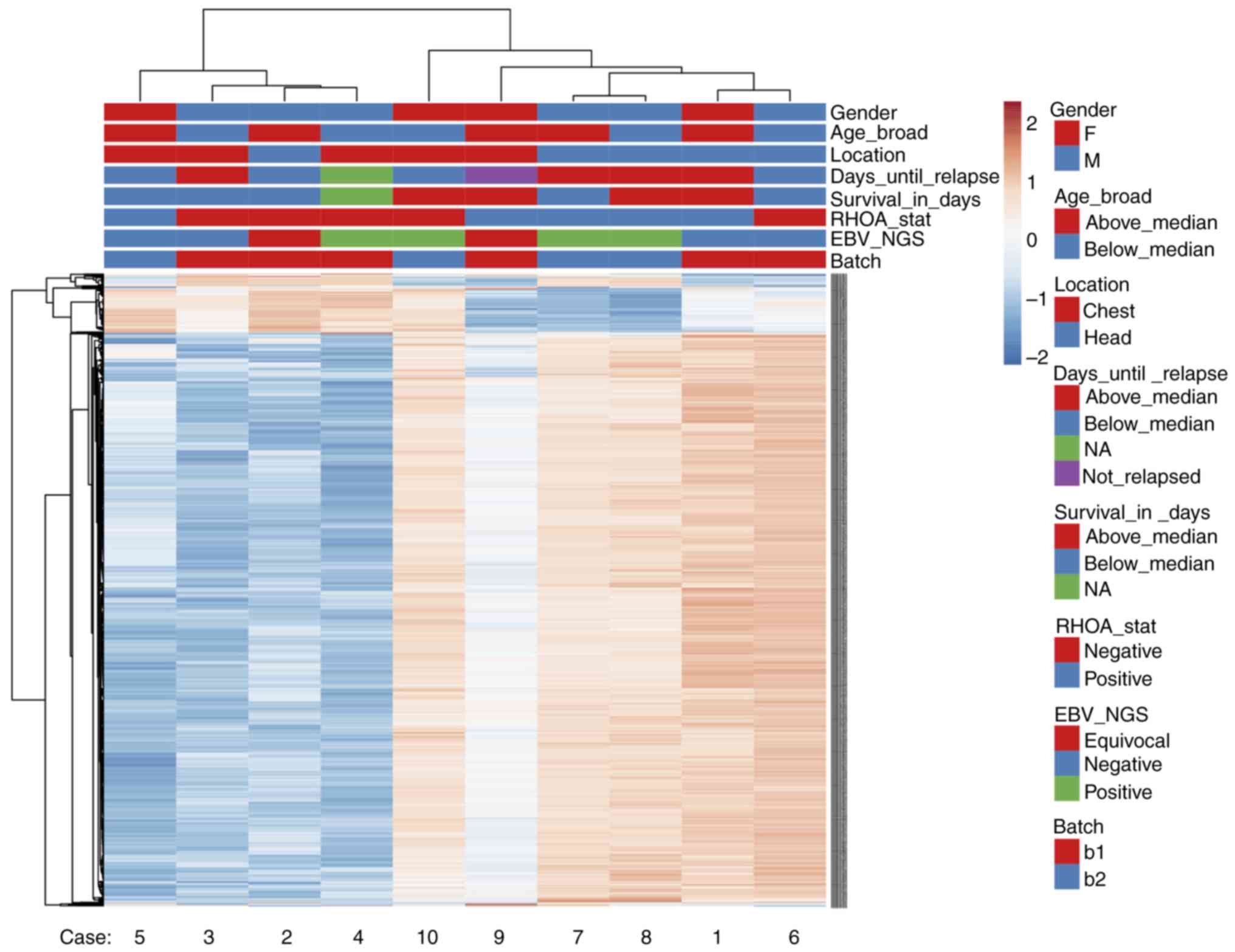
AITL cases form two groups independent
from RHOA mutational status or EBV infection status based on gene
expression analysis
Following gene expression analysis, AITL cases were
separated into two clusters. Fig.
4 represents a heatmap of the gene expression in our AITL
samples. Hierarchical clustering was performed based on Euclidean
distance. The results demonstrated that AITL samples were separated
into two groups: Cases 3, 4, 5 and 6 in one group, and cases 1, 2,
7, 8 and 9 are in another group. Case 10 seemed to be an outlier
with different expression patterns compared with the other samples.
PCA was also performed on RHOA mutational status (Fig. S3) and the results confirmed
similar sample grouping. However, no association to any of our
tested conditions, such as RHOA status, EBV infection status or
gender and age, was observed.
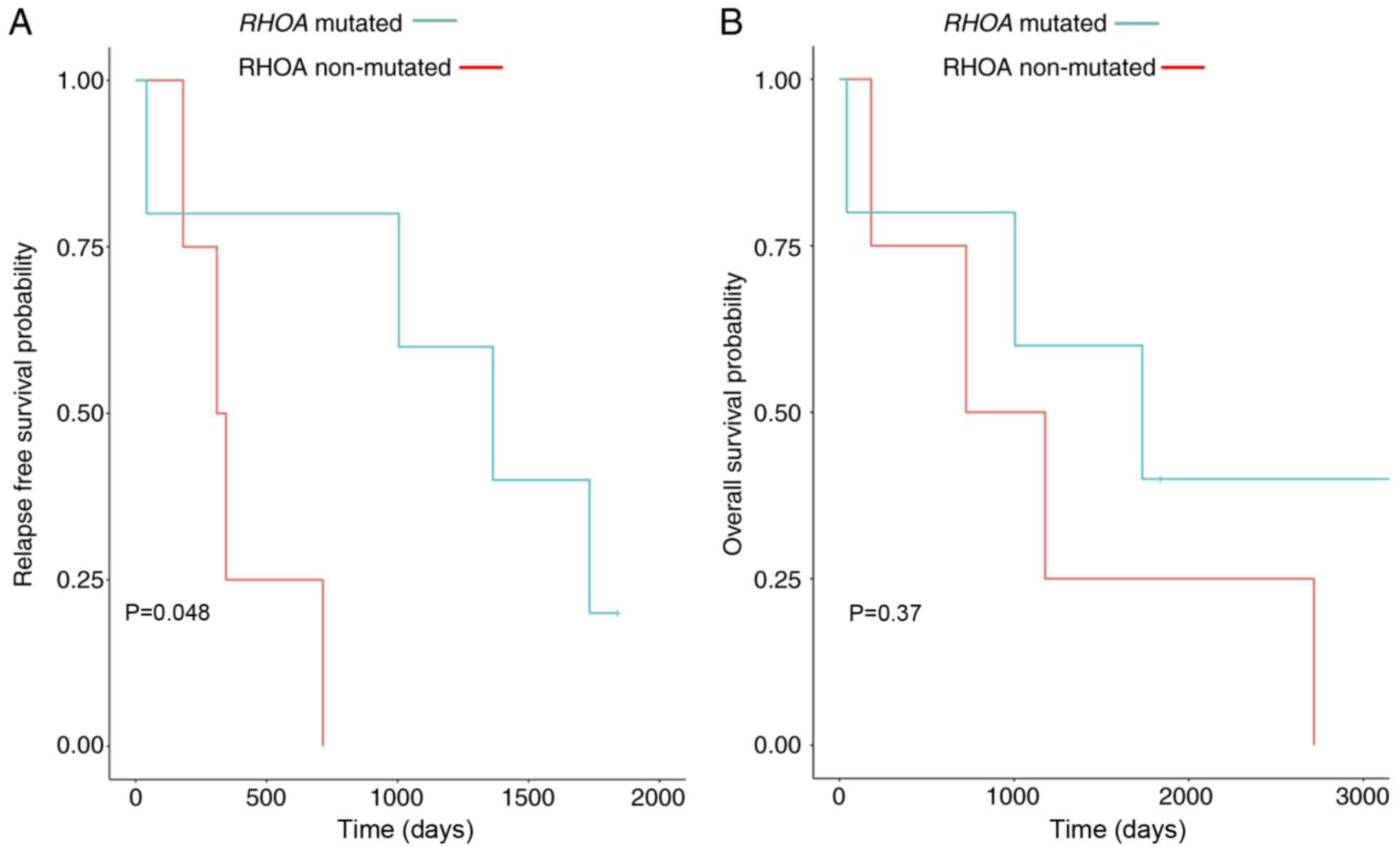
RHOA G17V mutated cases have better
overall survival than RHOA mutation negative cases
Although treatments were different in our study
group (Table I), the results from
overall survival and relapse-free survival demonstrated that the
RHOA mutation negative cases had a shorter relapse-free
survival and showed a trend towards shorter overall survival
(Fig. 5). Furthermore, we also
performed Kaplan Meier analysis for EBV infection status of all
cases, but no significant association between outcomes and EBV
infection was identified (data not shown).
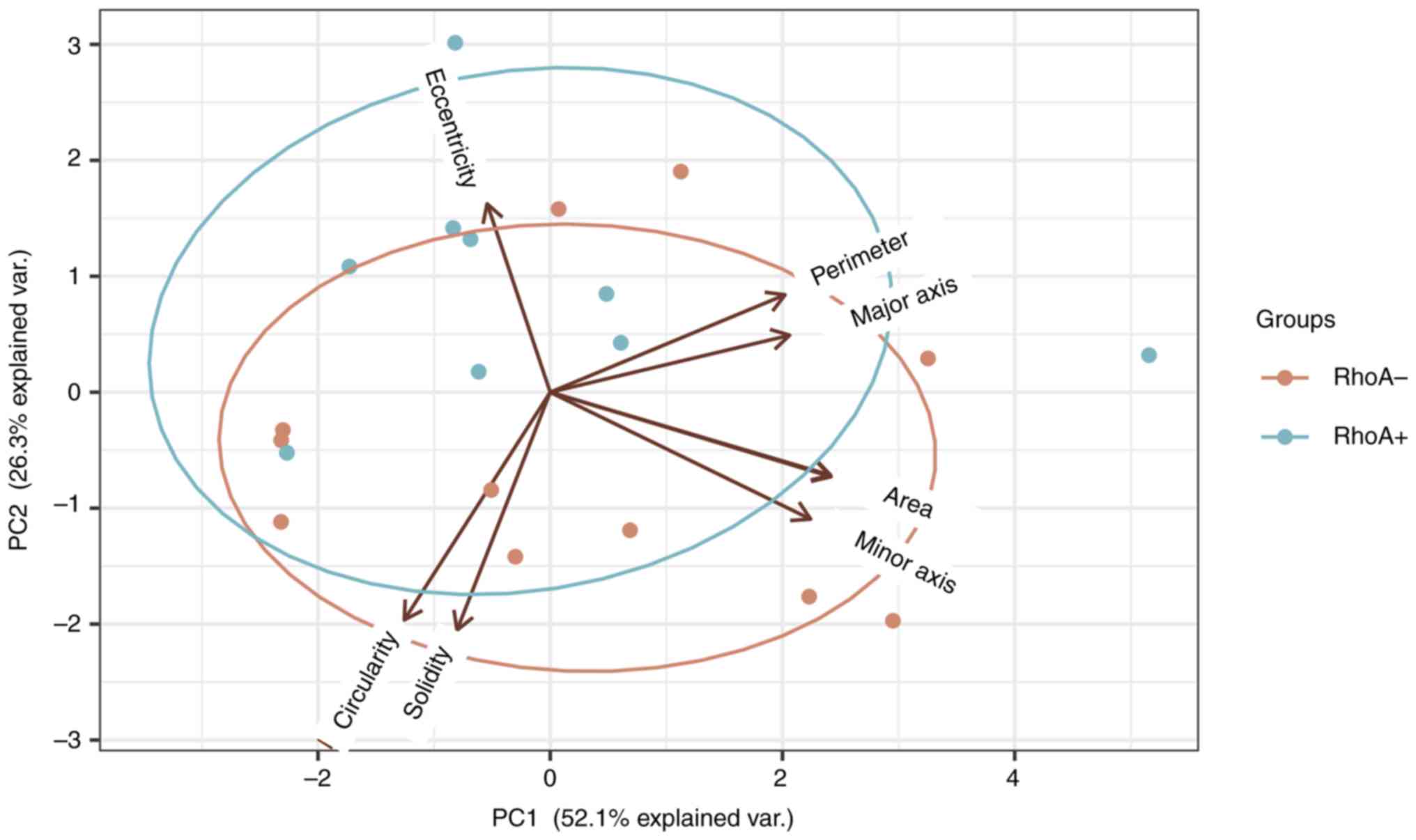
Computational single-cell image analysis
separates neoplastic T-cells in RHOA mutated cases from neoplastic
T-cells in RHOA wild type cases
It has been reported that RHOA mutations can
affect tumorigenesis, in particular cell motility due to cellular
processes associated with the formation of actin fibers and myosin
activation (38). Subsequently,
cell morphology of RHOA mutation positive samples and
RHOA mutation negative samples was evaluated.
The results from PCA of samples revealed that
although the two subtypes, RHOA mutation positive and
RHOA mutation negative, showed a maximum variance of ~53%
along the first principal component (Figs. 6 and 7), there was an increased variation in
the eccentricity of samples with RHOA mutation positive
cells (0.73) and RHOA mutation negative cells (0.74). This
indicated that RHOA mutation positive cells may be more
circular compared with RHOA mutation negative cells, which
is explained by the reduced actin formation. Increased variation
between these subtypes may be observed by increasing the total
sample size and we hypothesize that greater variance may also be
observed with respect to circularity (36,37).
Discussion
In the present study, 10 cases of AITL were reported
and compared for RHOA mutational status. Genomic analysis of
the bone marrow samples for six of the cases was also performed.
Although the present study was limited in size, only a few studies
have compared RHOA mutation positive cases side-by-side with
RHOA negative cases (10,39,40). The present study provided a
broader genomic analysis coupled with computational digital image
analysis.
Recurrent RHOA G17V mutations have been
previously identified in >50% of AITL cases, and mutations in
epigenetic regulators, including IDH2, TET2 and
DNMT3A, have also been described (41,42). The present study also identified
the frequently found mutations in AITL. The results also identified
IDH2 and TET2 mutations in association with the
RHOA G17V mutation. The role of RHOA G17V mutations
in combination with IDH2 and TET2 within the
pathogenesis of AITL has been previously studied as promoting
T-follicular helper cell differentiation and as an important
genetic hit for T-cell lymphomagenesis (8,9,38,43). Other mutations that frequently
occur in AITL, such as DNMT3A, VAV1, PLCG1,
STAT3, JAK2 and FYN, are typically observed
independently from RHOA mutations (40). A previous study by Abate et
al (44) also identified
recurrent VAV1 translocations in peripheral TCL (PTCL). Our
HeME-ID Panel also covered the break-point region of VAV1
translocations; however, the present study did not identify any
VAV1 translocation in our cases. These differences could be
due to the small sample size of AITL cases compared with the 152
PTCL samples (of which 60 were AITL samples) evaluated in the study
by Abate et al.
Overall, RHOA mutation positive cases showed
a more damaging mutational burden with an average CADD score of
24.9 compared with 21.8 for the RHOA negative cases,
suggesting that other genetic mutations in RHOA mutated
cases may be more damaging. This could indicate that RHOA
mutations may be more oncogenic and result in increased mutations
due to increased DNA damage. Alternatively, it is possible that
cases with RHOA mutations require more damaging co-mutations
in order for oncogenesis to take form. Further in vitro and
in vivo investigation is therefore required to assess these
possibilities.
The present study also assessed samples for COSMIC
signatures seen in B-cell lymphomas and solid tumors (Fig. S4). Some broad COSMIC signatures
that are seen in most tumors were also present in our samples, and
no COSMIC signature was unique to RHOA mutational status. In
addition, a COSMIC T-cell lymphoma tumor signature was not present
in the COSMIC database. Generating such a signature by using
multiple T-cell lymphoma subtypes may be required.
Although the mutational burden was higher in the
RHOA positive cases (8.2) compared with the RHOA
negative cases (2.4), which was consistent with a previous study by
Sakata-Yanagimoto et al (7), the present study reported a higher
mutational burden in promoter regions and CTCF binding sites for
the RHOA negative group compared with the RHOA
positive group. Promoter regions and CTCF binding sites serve
crucial roles in the regulation of gene transcription and
expression (45,46). In the present study, RHOA
negative cases seemed to present with a more heterogenous
mutational landscape, which makes them difficult to describe and
classify. These mutations may affect common pathways that
ultimately lead to an AITL phenotype, even in the absence of a
RHOA mutation. This dysregulation of yet unidentified
pathways may also happen in other non-coding areas. In order to
identify the promoter regions or CTCF binding sites regulating the
oncogenesis in the RHOA negative or positive cases, CHIP-Seq
or functional studies would be required in the future.
AITL is often associated with EBV infected cells and
EBV is identified primarily in non-T-cells (the B-cells) (47); however, only a few studies
evaluated the role of EBV in AITL pathology. In the present study,
six out of 10 patients had a positive EBV infection status
following NGS and ISH analysis. When looking at the gene expression
of our cases with regards to EBV infected cells, there was no
difference in the gene expression profile between positive and
negative cases. After comparison of the mutational profiles of our
cases, a higher mutational burden was observed in the EBV negative
cases. However, the average CADD score was higher in the EBV
positive cases, suggesting that an EBV infection may contribute to
a more deleterious mutational scenery. Previous studies suggested
that a contribution to T-cell lymphomagenesis is excluded as there
was no virus found in the neoplastic T-cells (48,49); however, it was reported that an
EBV infection in AITL cases could lead to histologic progression of
these cases (49). A previous
study investigating 270 cases of AITL reported that in young
patients with AITL, an EBV infection was associated with a
significantly improved prognosis (50). Kaplan-Meier Survival analysis was
performed with regards to EBV infection, and the results
demonstrated no improved overall survival for EBV positive cases.
Our results do not confirm these previous findings of Eladl et
al (50) but our sample size
is small. Another study by Hoffman et al (51) assessed the association between EBV
infection and diffuse large B-cell lymphoma (DLBCL) occurring in
PTCL, and reported that patients with PTCL and DLBCL frequently
have EBV infected B-cells, suggesting an important role of EBV in
B-cell transformation.
The immunologic environment of AITL is characterized
by a massive infiltration of inflammatory cells (52). An analysis of immune cell
infiltration within our AITL cases based on gene expression
profiling was performed. The results demonstrated that the greatest
contributors to the gene expression profiles were naïve B-cells,
resting memory CD4 T-cells and T-follicular helper cells. Amongst
the RHOA positive cases, the portion of naïve B-cells and
T-follicular helper cells seemed to be elevated compared with other
cases. Previous studies investigating the gene expression profiles
of AITL reported an overexpression of T-follicular helper cells in
AITL, since these are the neoplastic infiltrates, and of vascular
endothelial growth factor (53,54). Nguyen et al (55) identified mutations specific to
both T-cell and B-cells within nodal T-cell lymphomas.
Although the present study only included 10 patients
with AITL, we were interested in assessing statistics associated
with the prognosis of patients with AITL. The relapse-free survival
and overall survival were evaluated and compared by RHOA
mutational status. The results demonstrated that RHOA
positive cases had a better overall survival and relapse-free
survival (P-value=0.048), although these cases tend to have an
increased mutational burden. However, improved outcomes may be due
to improved immune system surveillance in the setting of more tumor
antigenic stimulation. Further studies are needed to assess this
possibility.
Single cell imaging analysis demonstrated that
RHOA mutation positive cells may be more circular than
RHOA mutation negative cells, which, based on the known
function of RHOA, could be a consequence of altered actin dynamics.
This difference was not significant, but an increased variation
between these subtypes may be observed by increasing the total
sample size, and a greater variance may also be detected with
respect to circularity (36,37). However, it is important to
acknowledge that the mutational status of RHOA in the
individual cells was unclear, since they were selected randomly. A
single cell NGS study is therefore required to confirm the absolute
mutational status in these cells.
In conclusion, the results from the present study
demonstrate the differences and similarities of RHOA mutated
and non-mutated AITL, identified both increased burden and
deleterious mutations in coding and non-coding (promoter and
transcription binding site) regions of RHOA mutated AITLs,
highlighted differences in the immune system infiltrate, as well as
a specific single cell morphologic manifestation in cases of AITL
(increased circularity). Further studies are needed to investigate
the immunologic environment in patients with AITL in the context of
understanding RHOA mutations and their cell biologic
responses that will be important future avenues of study.
Supplementary Data
Acknowledgments
Not applicable.
Funding
This study was supported through grant funding
(grant no. 1187454) from Agilent Technologies.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
RO, AB, MKS, RW, BP and ER designed the research.
RSO and AB wrote the paper. DJ and AB performed analyses. RSO, AB,
DJ, KS and JK analyzed data and edited the paper. NS and AB
performed research. ER provided expertise. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics approval from
Stanford University's Institutional Review Board (approval no.
IRB-22359).
Patient consent for publication
Not applicable.
Competing interests
This work was supported by funding from Agilent
Technologies to RO, and ER is an employee of Agilent Technologies.
Other authors declare that they have no competing interest.
References
|
1
|
Attygalle AD, Kyriakou C, Dupuis J, Grogg
KL, Diss TC, Wotherspoon AC, Chuang SS, Cabeçadas J, Isaacson PG,
Du MQ, et al: Histologic evolution of angioimmunoblastic T-cell
lymphoma in consecutive biopsies: Clinical correlation. Am J Surg
Pathol. 31:1077–1088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dogan A, Atygalle AD and Kyriakou C:
Angioimmunoblastic T-cell lymphoma. Br J Haematol. 121:681–691.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Attygalle A, Al-Jehani R, Diss TC, Munson
P, Liu H, Du MQ, Isaacson PG and Dogan A: Neoplastic T cells in
angioimmunoblastic T-cell lymphoma express CD10. Blood. 99:627–633.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manso R, Sánchez-Beato M, Monsalvo S,
Gómez S, Cereceda L, Llamas P, Rojo F, Mollejo M, Menárguez J,
Alves J, et al: The RHOA G17V gene mutation occurs frequently in
peripheral T-cell lymphoma and is associated with a characteristic
molecular signature. Blood. 123:2893–2894. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cairns RA, Iqbal J, Lemonnier F, Kucuk C,
de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et
al: IDH2 mutations are frequent in angioimmunoblastic T-cell
lymphoma. Blood. 119:1901–1903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Enami T, Yoshida K, Shiraishi Y, Ishii R,
Miyake Y, Muto H, Tsuyama N, Okuno Y, Sakata S, Kamada Y, et al:
Somatic G17V Rhoa mutation specifies angioimmunoblastic T-Cell
lymphoma. Blood. 122:8152013. View Article : Google Scholar
|
|
7
|
Sakata-Yanagimoto M, Enami T, Yoshida K,
Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A,
Okuno Y, et al: Somatic RHOA mutation in angioimmunoblastic T cell
lymphoma. Nat Genet. 46:171–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujisawa M, Sakata-Yanagimoto M, Nishizawa
S, Komori D, Gershon P, Kiryu M, Tanzima S, Fukumoto K, Enami T,
Muratani M, et al: Activation of RHOA-VAV1 signaling in
angioimmunoblastic T-cell lymphoma. Leukemia. 32:694–702. 2018.
View Article : Google Scholar
|
|
9
|
Cortes JR, Ambesi-Impiombato A, Couronné
L, Quinn SA, Kim CS, da Silva Almeida AC, West Z, Belver L, Martin
MS, Scourzic L, et al: RHOA G17V induces T follicular helper cell
specification and promotes lymphomagenesis. Cancer Cell.
33:259–273, e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ondrejka SL, Grzywacz B, Bodo J, Makishima
H, Polprasert C, Said JW, Przychodzen B, Maciejewski JP and His ED:
Angioimmunoblastic T-cell lymphomas with the RHOA p. Gly17Val
mutation have classic clinical and pathologic features. Am J Surg
Pathol. 40:335–341. 2016. View Article : Google Scholar
|
|
11
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2391. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H and Thiele J: WHO Classification of Tumours of
Haematopoietic and Lymphoid Tissues. 4th edition. International
Agency for Research in Cancer (IARC); Lyon: 2017
|
|
13
|
Kumar J, Butzmann A, Wu S, Easly S,
Zehnder JL, Warnke RA, Bangs CD, Jangam D, Cherry A, Lau J, et al:
Indolent in situ B-cell neoplasms with MYC rearrangements show
somatic mutations in MYC and TNFRSF14 by next-generation
sequencing. Am J Surg Pathol. 43:1720–1725. 2019.PubMed/NCBI
|
|
14
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A
MapReduceframework for analyzing next-generation DNA sequencing
data. Genome Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan2: Somatic mutation and copy number alteration discovery in
cancerby exome sequencing. Genome Res. 22:568–576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rehm HL, Berg JS, Brooks LD, Bustamante
CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL,
Nussbaum RL, et al: ClinGen-the clinical genome resource. N Engl J
Med. 372:2235–2242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
1000 Genomes Project Consortium; Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Exome Variant Server. http:////evs.gs.washington.edu/EVS/urisimple//evs.gs.washington.edu/EVS/Accessed
February 16 2020.
|
|
20
|
Kopanos C and Tsiolkas V: VarSome: The
human genomic variant search engine. Bioinformatics. 35:1978–1980.
2019. View Article : Google Scholar :
|
|
21
|
Blokzijl F, Janssen R, Van Boxtel R and
Cuppen E: MutationalPatterns: Comprehensive genome-wide analysis of
mutational processes. Genome Med. 10:332018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z,
Meirelles GV, Clark NR and Ma'ayan A: Enrichr: Interactive and
collaborative HTML5 gene list enrichment analysis tool. BMC
Bioinformatics. 14:1282013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kamburov A, Stelzl U, Lehrach H and Herwig
R: The ConsensusPathDB interaction database: 2013 update. Nucleic
Acids Res. 41(Database Issue): D793–D800. 2013. View Article : Google Scholar :
|
|
26
|
Gyarmati P, Kjellander C, Aust C, Song Y,
Öhrmalm L and Giske CG: Metagenomic analysis of bloodstream
infections in patients with acute leukemia and therapy-induced
neutropenia. Sci Rep. 21:235322016. View Article : Google Scholar
|
|
27
|
Baheti S, Tang X, O'Brien DR, Chia N,
Roberts LR, Nelson H, Boughey JC, Wang L, Goetz MP, Kocher JA and
Kalari KR: HGT-ID: An efficient and sensitive workflow to detect
human-viral insertion sites using next-generation sequencing data.
BMC Bioinformatics. 19:2712018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim D, Paggi JM, Park C, Bennett C and
Salzberg SL: Graph-based genome alignment and genotyping with
HISAT2 and HISAT-genotype. Nat Biotechnol. 37:907–915. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anders S, Pyl PT and Huber W: HTSeq-a
Python frame-work to work with high-throughoutput sequencing data.
Bioinformatics. 31:166–169. 2015. View Article : Google Scholar
|
|
30
|
Integrated Development for R. Version
125001. Mode: desktop. RStudio, PBC; Boston, MA: 2020, http://www.rstudio.com/.
Accessed February 16 2020.
|
|
31
|
Metsalu T and Vilo J: ClustVis: A web tool
visualizing clustering of multivariate data using Prinicpal
Component Analysis and heatmap. Nucleic Acids Res. 43(W1):
W566–W570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jangam D, Sridhar K, Butzmann A,
Sambhagati P, Plowey ED and Ohgami RS: TBL1XR1 Mutations in primary
marginal zone lymphomas of occular adnexa are associated with
unique morphometric phenotypes. Curr Eye Res. Jun 19–2020.Epub
ahead of print. View Article : Google Scholar
|
|
34
|
Wong MA and Hartigan JA: Algorithm AS 136:
A K-means clustering algorithm. J R Stat Soc Ser C. 28:100–108.
1979.
|
|
35
|
Fouad S, Randell D, Galton A, Mehanna H
and Landini G: Unsupervised morphological segmentation of tissue
compartments in histopathological images. PLoS One.
12:e01887172017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hart M, Lauer J, Selig M, Hanak M, Walters
B and Rolauffs B: Shaping the cell and the future: Recent
advancements in biophysical aspects relevant to regenerative
medicine. J Funct Morphol Kinesiol. 3:22017. View Article : Google Scholar
|
|
37
|
Lyons SM, Alizadeh E, Mannheimer J,
Schuamberg K, Castle J, Schroder B, Turk P, Thamm D and Prasad A:
Changes in cell shape are correlated with metastatic potential in
murine and human osteosarcomas. Biol Open. 5:289–299. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chiba S, Enami T, Ogawa S and
Sakata-Yanagimoto M: G17V RHOA: Genetic evidence of GTP-unbound
RHOA playing a role in tumorigenesis in T cells. Small GTPases.
6:100–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nagao R, Kikuti YY, Carreras J, Kikuchi T,
Miyaoka M, Matsushita H, Kojima M, Ando K, Sakata-Yanagimoto M,
Chiba S and Nakamura N: Clinicopathologic analysis of
angioimmunoblastic T-cell lymphoma with or without RHOA G17V
mutation using formalin-fixed paraffin-embedded sections. Am J Surg
Pathol. 40:1041–1050. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Willemsen M, Hamid MA, Winkens B and Zur
Hausen A: Mutational heterogeneity of angioimmunoblastic T-cell
lymphoma indicates distinct lymphomagenic pathways. Blood Cancer J.
8:62018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoo HY, Sung MK, Lee SH, Kim S, Lee H,
Park S, Kim SC, Lee B, Rho K, Lee JE, et al: A recurrent
inactivating mutation in RHOA GTPase in angioimmunoblastic T cell
lymphoma. Nat Genet. 46:371–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang M, Zhang S, Chuang SS, Ashton-Key M,
Ochoa E, Bolli N, Vassiliou G, Gao Z and Du MQ: Angioimmunoblastic
T cell lymphoma: Novel molecular insights by mutation profiling.
Oncotarget. 8:17763–17770. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ng SY, Brown L, Stevenson K, deSouza T,
Aster JC, Louissaint A Jr and Weinstock DM: RhoA G17V is sufficient
to induce autoimmunity and promotes T-cell lymphomagenesis in mice.
Blood. 132:935–947. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Abate F, Da Silva-Almeida AC, Zairis S,
Zairis S, Robles-Valero J, Couronne L, Khiabanian H, Quinn SA, Kim
MY, Laginestra MA, et al: Activating mutations and trans-locations
in the guanine exchange factor VAV1 in peripheral T-cell lymphomas.
Proc Natl Acad Sci USA. 114:764–769. 2017. View Article : Google Scholar
|
|
45
|
Cornish AJ, Hoang PH, Dobbins SE, Law PJ,
Chubb D, Orlando G and Houlston RS: Identification of recurrent
non-coding mutations in B-cell lymphoma using Hi-C. Blood Adv.
3:21–32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang X, Zhang Y, Ba Z, Kyritsis N,
Casellas R and Alt FW: Fundamental roles of chromatin loop
extrusion in antibody class switching. Nature. 575:385–389. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Weiss LM, Jaffe ES, Liu XF, Chen YY,
Shibata D and Medeiros LJ: Detection and localization of
Epstein-Barr viral genomes in angioimmunoblastic lymphadenopathy
and angioimmunoblastic lymphadenopathy-like lymphoma. Blood.
79:1789–1795. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Khan G, Norton AJ and Slavin G:
Epstein-Barr virus in angioimmunoblastic T-cell lymphomas.
Histopathology. 22:145–149. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou Y, Attygalle AD, Chuang SS, Diss T,
Ye H, Liu H, Hamoudi RA, Munson P, Bacon CM, Dogan A and Du MQ:
Angioimmunoblastic T-cell lymphoma: Histological progression
associates with EBV and HHV6B viral load. Br J Haematol. 138:44–53.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Eladl AE, Shimada K, Suzuki Y, Takahara T,
Kato S, Kohno K, Elsayed AA, Wu CC, Tokunaga T, Kinoshita T, et al:
EBV status has prognostic implication among young patients with
angioimmunoblastic T-cell lymphoma. Cancer Med. 9:678–688. 2020.
View Article : Google Scholar
|
|
51
|
Hoffmann JC, Chisholm KM, Cherry A, Chen
J, Arber DA, Natkunam Y, Warnke RA and Ohgami RS: An analysis of
MYC and EBV in diffuse large B-cell lymphomas associated with
angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma
not otherwise specified. Hum Pathol. 48:9–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bal M, Gujral S, Gandhi J, Shet T, Epari S
and Subramanian PG: Angioimmunoblastic T-cell lymphoma: A critical
analysis of clinical, morphologic and immunophenotypic features.
Indian J Pathol Microbiol. 53:640–645. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
De Leval L, Rickman DS, Thielen C, Reynies
Ad, Huang YL, Delsol G, Lamant L, Leroy K, Brière J, Molina T, et
al: The gene expression profile of nodal peripheral T-cell lymphoma
demonstrates a molecular link between angioimmunoblastic T-cell
lymphoma (AITL) and follicular helper T (TFH) cells. Blood.
109:4952–4963. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Piccaluga PP, Agostinelli C, Califano A,
Carbone A, Fantoni L, Ferrari S, Gazzola A, Gloghini A, Righi S,
Rossi M, et al: Gene expression analysis of angioimmunoblastic
lymphoma indicates derivation from T follicular helper cells and
vascular endothelial growth factor deregulation. Cancer Res.
67:10703–10713. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nguyen TB, Sakata-Yanagimoto M, Asabe Y,
Matsubara D, Kano J, Yoshida K, Shiraishi Y, Chiba K, Tanaka H,
Miyano S, et al: Identification of cell-type-specific mutations in
nodal T-cell lymphomas. Blood Cancer J. 7:e5162017. View Article : Google Scholar : PubMed/NCBI
|