Introduction
Urolithiasis is a type of solid mass that occurs in
all parts of the urinary system. It is a disease with a complex
etiology and is associated with a high incidence and recurrence
rates (1). In recent years, the
incidence of urolithiasis has increased each year, thereby placing
a heavy burden on the global healthcare system (2). The formation of urinary calculi is a
complex process, which includes urinary supersaturation, crystal
nucleation, crystal growth and aggregation, and crystal attachment
to renal tubular epithelial cells (3). The influence of various factors
leads to the difference in the composition of calculi in patients
with urinary calculi, among which the proportion of calcium oxalate
stones and the mixed stone of calcium oxalate and calcium phosphate
stones accounts for approximately 70%. Calcium oxalate (CaOx) is an
important component of oxalate calculi, and the supersaturated
state of CaOx in urine is a major risk factor for urolithiasis
(4). This aspect prompted us to
conduct research on CaOx-induced development of uroli-thiasis.
Ferroptosis is a novel type of regulatory cell death
caused by iron-dependent lipid peroxidation (5). Lipid peroxidation occurs due to the
depletion of glutathione in cells. Moreover, Fe2+ is
transported at an increased rate in cells. A combined effect of
these two factors causes the accumulation of reactive oxygen
species (ROS) in cells, eventually leading to ferrop-tosis. p53 is
a classic tumor suppressor gene (6). XCT is a light chain subunit of the
cystine/glutamate transport system, which can promote the uptake of
cystine and the biosynthesis of glutathione, and can produce a
variety of antioxidants in cells, thus protecting the cells from
oxidative stress (7). Glutathione
peroxidase 4 (GPX4) is a subtype of glutathione peroxidase and the
only one that can detoxify lipid peroxidase (8,9).
During ferroptosis, p53 induces a decrease in the transcription
efficiency of XCT via a transcription-dependent mechanism, which
leads to a decrease in intracellular cystine intake and a
corresponding decrease in the content of synthetic cysteine. As the
rate-limiting precursor of GSH synthesis, the decrease in cysteine
level directly leads to a decrease in GSH synthesis level, which
indirectly affects the activity level of GPX4 in cells and finally
leads to lipid peroxidation (6-9).
Long-chain acyl-CoA synthetases (ACSL4) is a member of the Acyl-CoA
synthetase long-chain family (10), which can esterify arachi-donoyl
and adrenoyl into phosphatidylethanolamine in cells.
Phosphatidylethanolamine, as the only esterified oxygenation center
in endoplasmic reticulum (ER) septum, plays an impor-tant role in
ferroptosis (11). The overload
of Fe2+ in cells is also a necessary factor for the
induction of ferroptosis. Fe3+ can combine with
transferrin (TF) in plasma. TF then combines with transferrin
receptor (TRC) on the cell membrane, carrying Fe3+,
which is ingested by the receptor cells in the form of endocytosis.
The TRC-TF complex entering the cell is acidified, which alters the
conformation of the TF-TRC complex protein and promotes the release
of iron. Finally, the released Fe3+ is reduced to
Fe2+ by ferrireductases of the STEAP protein family
(12,13).
According to previous studies, CaOx crystal-induced
autophagy, endoplasmic reticulum (ER) stress (ERS) and
epithelial-mesenchymal transition (EMT) play important roles in the
formation of urolithiasis (14-17). Previous research has also
confirmed that ferroptosis is closely associated with autophagy,
ERS and EMT (18-20). It is understood that ferrop-tosis
is caused by a type of Fe2+ overload-dependent lipid
ROS. Related studies have reported that the exposure of renal
tubular epithelial cells to high concentrations of CaOx crystals
can produce excessive ROS, leading to inflammation and injury to
the surrounding tissue (21).
Thus, it can be considered that ferroptosis is associated with the
development of urolithiasis. Furthermore, during the formation of
urolithiasis, renal tubular epithelial cells and renal tissue can
be injured by autophagy, which is induced by ROS. Bound iron is
stable in cells and does not affect their normal physiological
function. Autophagy can degrade iron storage proteins in cells,
leading to excessive release of free iron and accumulation of ROS,
thereby acti-vating the process of ferroptosis (12). These research findings indicate
that ferroptosis is inextricably linked to a variety of causative
factors for urolithiasis, which provides further reason to suggest
the aforementioned scientific hypothesis. However, with regard to
the current research status, although ferroptosis has been reported
to be a causative factor of renal diseases, its role in the
development of urolithiasis is unclear (18,22). Therefore, as ferroptosis is
considered to be closely associated with multiple causes of
urolithiasis, the role of ferroptosis in the formation of
urolithiasis needs to be understood. The present study
preliminarily explored the role of ferroptosis in the development
of urolithiasis.
Materials and methods
Main reagents and antibodies
CaOx (cat. no. 455997-5G) was purchased from
Sigma-Aldrich; Merck KGaA. Erastin (cat. no. S7242) and
Ferrostain-1 (cat. no. S7243) were ordered from Selleck Chemicals
Co., Ltd. The CCK-8 kit (cat. no. CK04) was obtained from Dojindo
Laboratories Co., Ltd. DAPI (cat. no. P0131), the mitochondrial
membrane potential assay kit (cat. no. C2006) and the TUNEL
staining kit (cat. no. C1086) were purchased from Beyotime
Institute of Biotechnology, Inc. The lactate dehydrogenase (LDH)
assay kit (cat. no. A020-2-2), the reduced glutathione (GSH) assay
kit (cat. no. A006-2-1), the superoxide dismutase (SOD) assay kit
(cat. no. A001-3-2), the total antioxidant capacity (T-AOC) assay
kit (cat. no. A015-2-1) and the cell malondialdehyde (MDA) assay
kit (cat. no. A003-4-1) were purchased from Nanjing Jiancheng
Bioengineering Institute Co., Ltd. The MitoSOX Red Mitochondrial
Superoxide Indicator (cat. no. 40778ES50) was obtained from Yeasen
Biotech Co., Ltd. The catalase activity assay kit (cat. no. BC0205)
and penicillin-strepto-mycin solution (cat. no. P1400) were
obtained from Beijing Solarbio Science & Technology Co., Ltd.
The neutrophil gela-tinase-associated lipocalin (NGAL; cat. no.
CSB-E09409r) and kidney injury molecule-1 (KIM-1; cat. no.
CSB-E08808r) ELISA kits were purchased from Cusabio Technology,
LLC. Anti-GAPDH (cat. no. 10494-1-AP), anti-p53 (cat. no.
10442-1-AP), anti-GPX4 (cat. no. 14432-1-AP), anti-TF (cat. no.
17435-1-AP) and anti-TRC (cat. no. 1004-2-AP) anti-bodies were
ordered from Proteintech Group, Inc. The iron assay kit (cat. no.
ab83366), and anti-XCT (cat. no. ab37185) and anti-ACSL4 (cat. no.
ab155282) antibodies were obtained from Abcam. DMEM/F12 (cat. no.
C10010500BT) and fetal bovine serum (cat. no. 10270-106) were
purchased from Thermo Fisher Scientific, Inc.
Cells, cell culture and exposure
Human proximal tubular cells (HK-2 cells) were
obtained from the Cell Bank of the Chinese Academy of Sciences,
maintained in complete DMEM/F12 (supplemented with 10% fetal bovine
serum and 100 U/ml penicillin/streptomycin) and cultured at 37°C in
an incubator with 5% CO2. CaOx crystals were added to
the serum-free DMEM/F12 to prepare the CaOx intervention solution.
When cell confluence reached 80%, the cells were exposed to 0
(nega-tive control; NC), 1, 2 and 4 mM CaOx intervention solution
for 24 h prior to analysis. The CCK-8, LDH and MDA assays were then
performed, and their results were obtained using a microplate
reader, as described below. Morphological changes in the cells were
observed using an inverted microscope, and the protein levels of
p53, XCT, ACSL4, GPX4, TF and TRC were examined by western blot
analysis. Subsequently, the HK-2 cells were treated with erastin
(agonist of ferroptosis, 10 µM) and ferrostatin-1 (inhibitor
of ferroptosis, 8 µM) with or without CaOx intervention
solution (2 mM) for 24 h.
Animal experiments
All animal experiment protocols were approved by the
Animal Care Committee of Wuhan University (Wuhan, China) and the
Laboratory Animal Welfare and Ethics Committee of Renmin Hospital
of Wuhan University. A total of 32 male adult Sprague-Dawley rats
(weight, 180-220 g; age, 6 weeks) were used in the experiments. Rat
models of urolithiasis were established by the administration of
0.75% ethylene glycol (EG) in the drinking water for 4 weeks. The
rats were randomly divided into 4 groups (n=8 per group) as
follows: i) The NC group; ii) urolithiasis model group; iii)
nephrolithiasis model treated with erastin (10 mg/kg/day) group;
and iv) nephrolithiasis model treated with ferrostatin-1 (4
mg/kg/day) group. In the latter 2 groups, the rats were treated
with erastin and ferrostatin-1, respectively, by an
intra-peritoneal injection.
Cell morphology
HK-2 cells were exposed to 0, 1, 2 and 4 mM CaOx for
24 h. Morphological changes and cell confluence were then observed
using an inverted microscope (IX51; Olympus Corporation) at ×200
magnification.
Cell activity assay
Cell activity was measured using a CCK-8 assay kit.
The appropriate amount of CCK-8 reagent was added to the cells in
strict accordance with the manufacturer's instructions. After
complete mixing, the cells were placed in an incubator (37°C, 5%
CO2) for 2.5 h. The optical density (OD) value was
measured at 450 nm using a microplate reader (VICTOR Nivo;
PerkinElmer Corporation).
LDH analysis
LDH is a very stable enzyme involved in energy
metabolism of the body. The change in the quality and quantity of
LDH directly affects the energy metabolism of the body. A
pathological state of tissues and organs leads to the release of
LDH into the blood (23). In the
present study, the LDH levels were measured using an LDH assay kit.
The super-natant of the cell culture medium of each group was
collected, and subsequent analyses were performed in strict
accordance with the requirements of the supplier's manual. The OD
value was measured at 450 nm using a microplate reader.
Cell biochemistry assay
GSH, CAT, T-AOC, SOD and MDA are classic indicators
for measuring the capacity of cells to resist antioxidant and
oxidative stress (24-27). In the present study, the levels of
GSH were measured using the reduced glutathione assay kit. The
levels of CAT were measured using the catalase activity assay kit.
The degrees of T-AOC were detected using the total antioxidant
capacity assay kit. The levels of SOD were measured using the
superoxide dismutase assay kit. The levels of lipid peroxidation
were measured using the cell malondialdehyde assay kit. Following
treatment, the cells were homogenized (reagents provided with the
kits) and centrifuged (15,00 g × 10 min, 25°C) to remove the
precipitate. The supernatant was collected and the assay kit
reagents were added, and the assays were performed in strict
accordance with the respective manufacturer's instructions. The OD
value was measured using a microplate reader.
Mitochondrial injury assay
Changes in mitochondrial membrane potential reflect
mitochondrial injury. In the present study, the degree of
mitochondrial injury was deter-mined using the mitochondrial
membrane potential assay kit with JC-1. All reagents were prepared
in advance according to the instructions. Following treatment, the
supernatant was removed, and HK-2 cells were washed once with PBS;
the cells were then collected in an Eppendorf tube. In accordance
with the manufacturer's instructions, an appropriate amount (1 ml)
of JC-1 staining reagent was added, and the cells were incubated
for 20 min at 37°C in 5% CO2. Following incubation [HBSS
medium (containing Ca2+, Mg2+) was used to
prepare a 5 µM probe working solution; an appropriate amount
of probe working solution was then added and incubated at 37°C for
10 min in the dark], the cells were washed with JC-1 buffer and
centrifuged at 600 × g and at 4°C for 3 min. This process was
repeated twice. Finally, the cells were resuspended in JC-1 buffer
for flow cytometric analysis by using a flow cytometer (BD
FACSCalibur, BD Biosciences). Flow cytometry was performed at an
excitation wavelength of 488 nm, an emission wavelength FL1
(EM=525±20 nm), FL2 (EM=585±20 nm). FlowJo (8.8.2, BD Biosciences)
software was used to analyze the data.
ROS analysis
The levels of ROS were detected using the MitoSOX
Red Mitochondrial Superoxide Indicator. Once the probe enters the
mitochondria, it can be oxidized by superoxide to produce red
fluorescence. HBSS medium (containing Ca2+,
Mg2+) was used to prepare a 5 µM probe working
solution; an appropriate amount of probe working solution was then
added and incubated at 37°C for 10 min in the dark. Following
incubation, the working solution was removed, and the cells were
washed twice with buffer solution. Finally, the outbreak degree of
ROS in each group was observed with an inverted fluorescence
microscope (IX1; Olympus Corporation) at ×200 magnification.
Western blot analysis
Both HK-2 cells and rat kidney tissues were
completely lysed using cold RIPA buffer, and proteins were
extracted from the lysed cell/tissue suspension. Protein was
quantified by BCA assay (cat. no. PC0020, Beijing Solarbio Science
& Technology Co., Ltd), and the same amount (30 µg) of
proteins were separated using 10-12% SDS-PAGE gel and then
transferred to PVDF (0.45-µM) membranes. Subsequently, the
PVDF membranes was blocked at room temperature using 5% skim milk.
The membranes were incubated with p53, XCT, GPX4, ACSL4, TF and TRC
antibodies (1:1,000) overnight at 4°C. The membranes were then
washed with TBST buffer and incubated with anti-rabbit IgG (H+L)
secondary antibodies (cat. no. 5366S, Cell Signaling Technology,
Inc.; 1:20,000) for 1 h at room temperature. Finally, the relative
expression levels of each protein were observed by an Odyssey dual
color infrared laser imager (Odyssey Infrared Imaging System,
LI-COR Corporation), and data were analyzed using Odyssey software
(Odyssey Version 3.0.29, LI-COR Corporation).
Immunofluorescence assay
The HK-2 cells were seeded on coverslips; following
treatment as described above, the medium was removed, and HK-2
cells were washed twice with cold PBS. The cells were then fixed
with 4% paraformalde-hyde (cat. no. P0099; Beyotime Institute of
Biotechnology, Inc.) for 15 min and washed twice with cold PBS. The
cells were blocked with 5% BSA (cat. no. ST023; Beyotime Institute
of Biotechnology, Inc.) at room temperature and washed twice with
cold PBS. The cells on coverslips were incubated with XCT, GPx4,
ACSl4, TF and TRC antibodies (1:100) at 4°C for 24 h. This was
followed by incubation with FITC- or Cy3-conjugated secondary
antibodies (Servicebio; cat. nos. GB22303 and GB21303,
respectively) at room temperature for 1 h. Finally, the coverslips
were blocked with the antifade mounting medium with DAPI. The
fluorescence intensity was measured using an automatic fluorescence
micro-scope (BX51; Olympus Corporation) at ×200 magnification.
Histological analysis
The degrees of renal histological injury were
determined by Von Kossa staining and the renal tubule
histopathological score. After 4 weeks of treatment, the rats were
exposed to carbon dioxide (30% of CO2) and sacrificed by
decapitation, then their kidneys were then removed, fixed with 4%
paraformaldehyde and embedded in paraffin. The tissue paraffin
block was sliced and the slices were immersed in xylene for 30 min,
followed by dewaxing in an ethanol gradient solution. A silver
nitrate solution was then dropped on the slices and irradiated
under an ultraviolet lamp for 10 min. The excess silver nitrate
solution was washed away with distilled water, a hyposulfite
solution was added to the slices, and the slices were placed at
room temperature for 2 min. Finally, the cell nuclei were
counterstained (hematoxylin, 10 min, 25°C; eosin, 40 sec, 25°C)
with an H&E staining solution (cat. no. G1120, Beijing Solarbio
Science & Technology Co., Ltd.). Scoring standards for the
degree of renal tubule injury were as follows: 1 point for marked
renal tubular expan-sion and flattening of cells; 1 point for brush
border injury and 2 points for brush border loss; 2 points for
cellular cast formation, and 1 point for necrotic cells and cell
desquamation (non-casted formation or cell debris). All slices were
observed under an automatic microscope (BX51; Olympus Corporation)
at ×400 magnification.
In addition, following treatment, venous blood
samples and 24-h urine samples of rats were collected. The blood
samples were placed in an automatic clinical chemistry analyzer to
detect serum creatinine (CRE) and blood urea nitrogen (BUN) levels.
The 24-h urine samples were subjected to NGAL and KIM-1 analyses
using the NGAL and KIM-1 ELISA assay kits. Samples and kits were
used in strict accordance with the manufacturer's instructions, and
the NGAL and KIM-1 levels were determined using a microplate reader
(VICTOR Nivo; PerkinElmer Corporation).
TUNEL assay
The preparation of cell coverslips and tissue slices
was consistent with that of the previous experiment. After the
tissue slices were prepared, proteinase K was used to strengthen
the binding between the kit probe and the broken DNA. The samples
were then mixed with the TUNEL staining kit reagent and incubated
at 37°C for 1 h. Finally, the samples were blocked with the
antifade mounting medium with DAPI. The positive rate of TUNEL
staining was observed using an automatic fluorescence microscope
(BX51; Olympus Corporation) at ×200 magnification.
Fe2+ assay
The levels of intracellular Fe2+ were
detected using the iron assay kit. Following treatment, both HK-2
cells and rat kidney tissues were homogenized in an appropriate
amount of iron assay buffer. The samples and kits were used in
strict accordance with the manufacturer's instructions, and the
absorbance was measured at 593 nm using a microplate reader.
Statistical analysis
All data from the experiments are expressed as the
means ± standard error of the mean of ≥3 independent experiments.
All statistical analyses were performed using GraphPad Prism
software version 6.0 (GraphPad Software, Inc.). Significant
differences among multiple groups were analyzed with one-way
analysis of variance followed by the Bonferroni's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
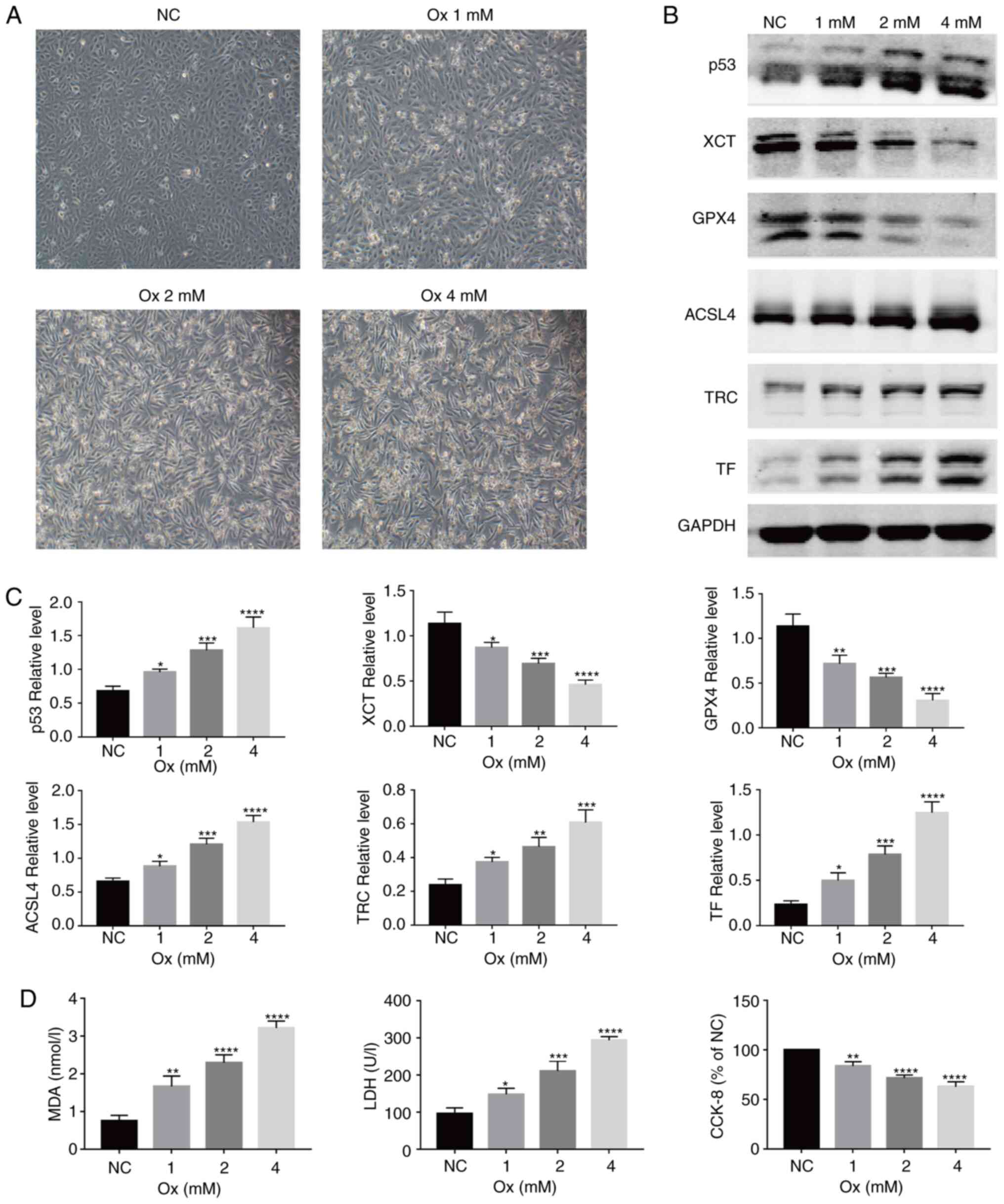
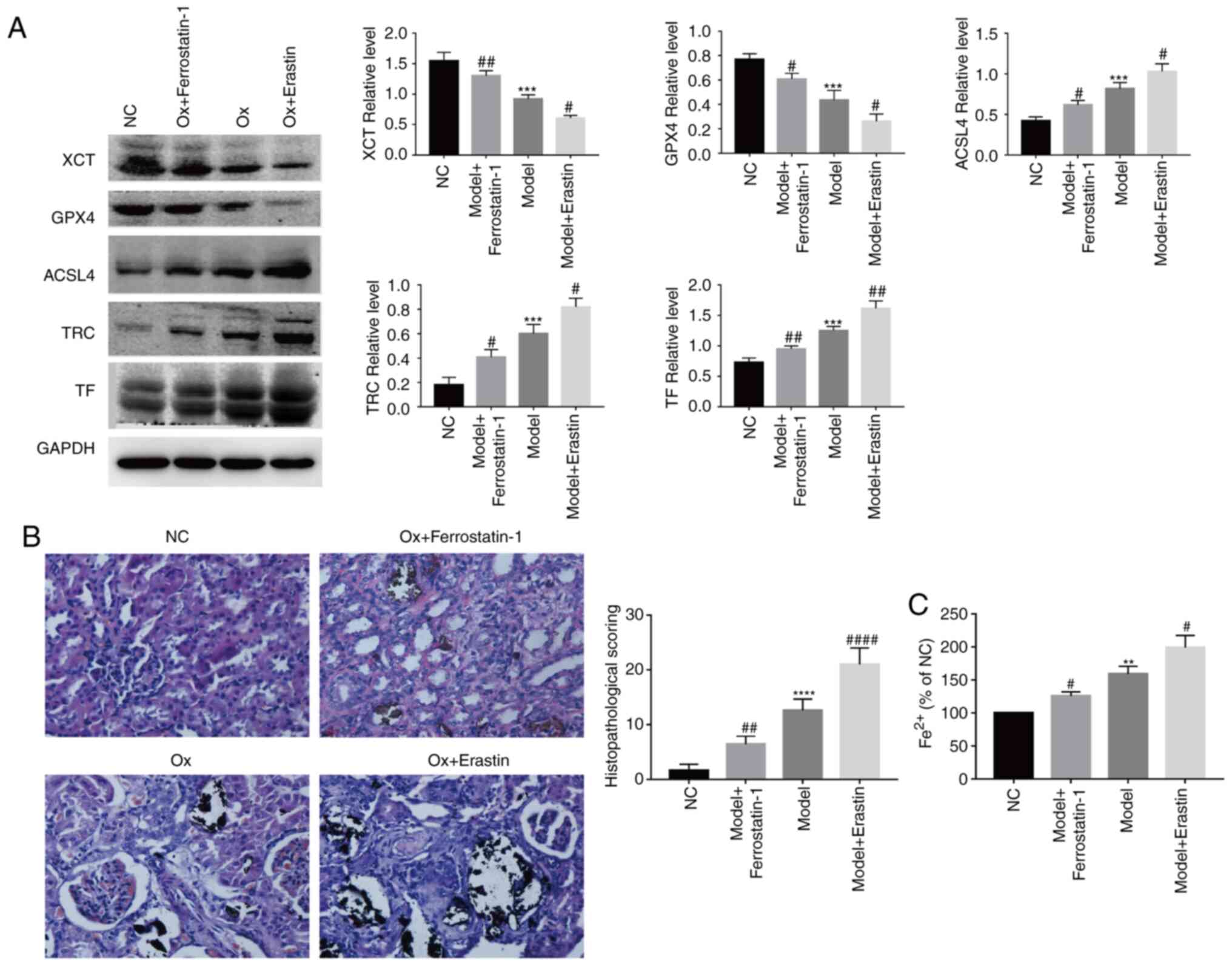
CaOx activates ferroptosis in HK-2 cells
in a concentration-dependent manner
With the increase in the concentration of CaOx, the
morphology of the HK-2 cells gradually changed from an original
oval shape to a long spindle-like shape, and the cell confluency
decreased (Fig. 1A). To examine
the role of CaOx-mediated ferroptosis in the formation of
urolithiasis, the relative expression levels of ferroptosis-related
proteins were detected in the HK-2 cells following exposure to
CaOx. It was identified that with the increase in the CaOx
concentration, compared with the NC group, the relative expression
of the ferroptosis agonist proteins, p53, ACSL4, TF and TRC,
increased, while that of the ferroptosis inhibitory proteins, XCT
and GPX4, significantly decreased (Fig. 1B and C). Changes in the MDA level
reflect the changes in intracellular lipid peroxidation levels;
compared with the NC group, the MDA levels were significantly
increased in the treatment groups (Fig. 1D, left panel). In terms of changes
in protein expres-sion and lipid peroxidation, CaOx activated
ferroptosis in the HK-2 cells. However, compared with the NC group,
the levels of LDH released were significantly increased (Fig. 1D, middle panel), and cell
viability was markedly decreased (Fig. 1D, right panel). On the basis of
these results, it could be deduced that a certain concentration of
CaOx induces the occurrence of ferroptosis, and with the occurrence
of ferroptosis, the damage inflicted on HK-2 cells gradually became
severe, and the degree of damage increased in a
concentration-dependent manner.
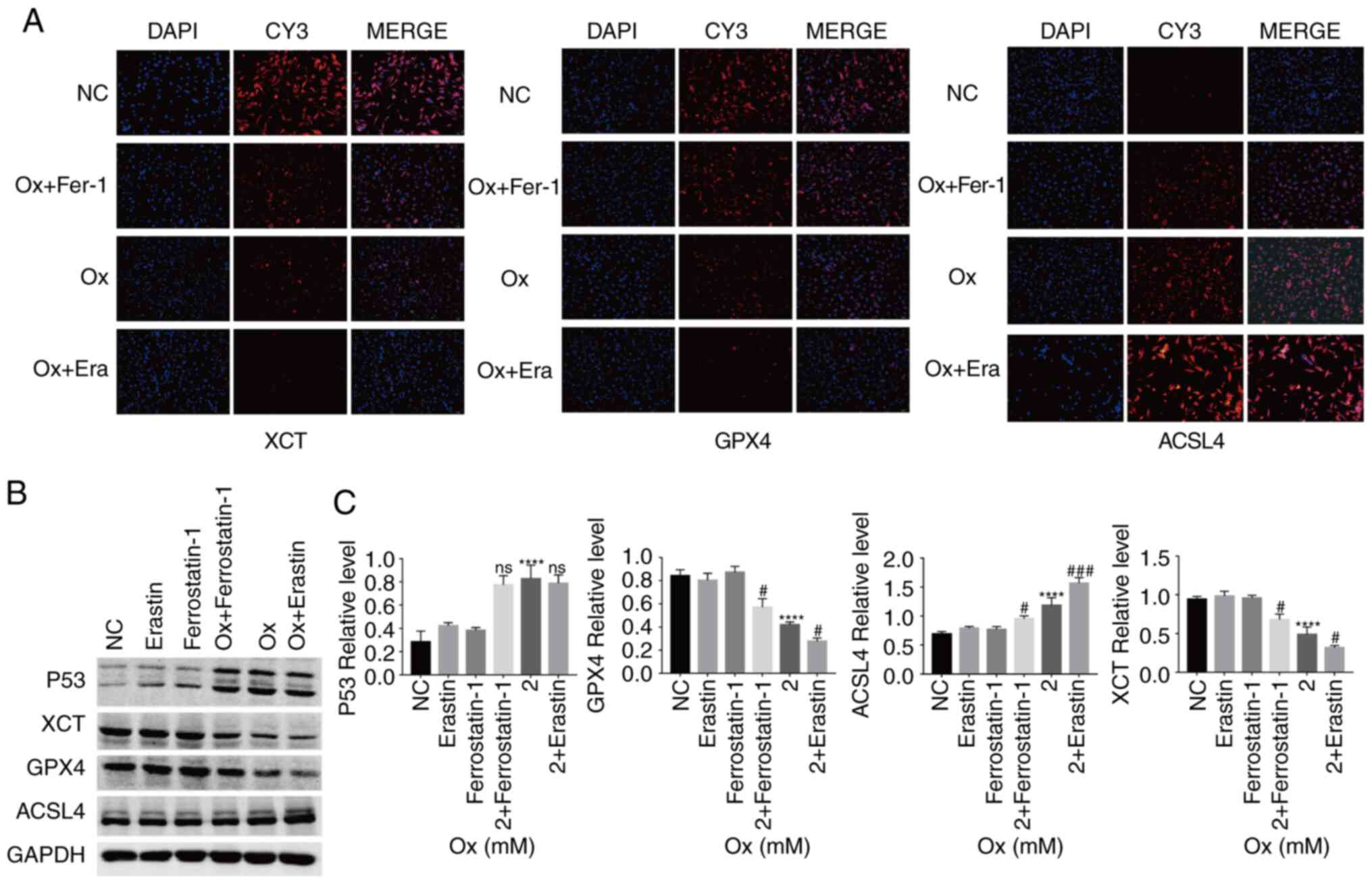
CaOx-induced ferroptosis decreases the
resistance of HK-2 cells to oxidative stress
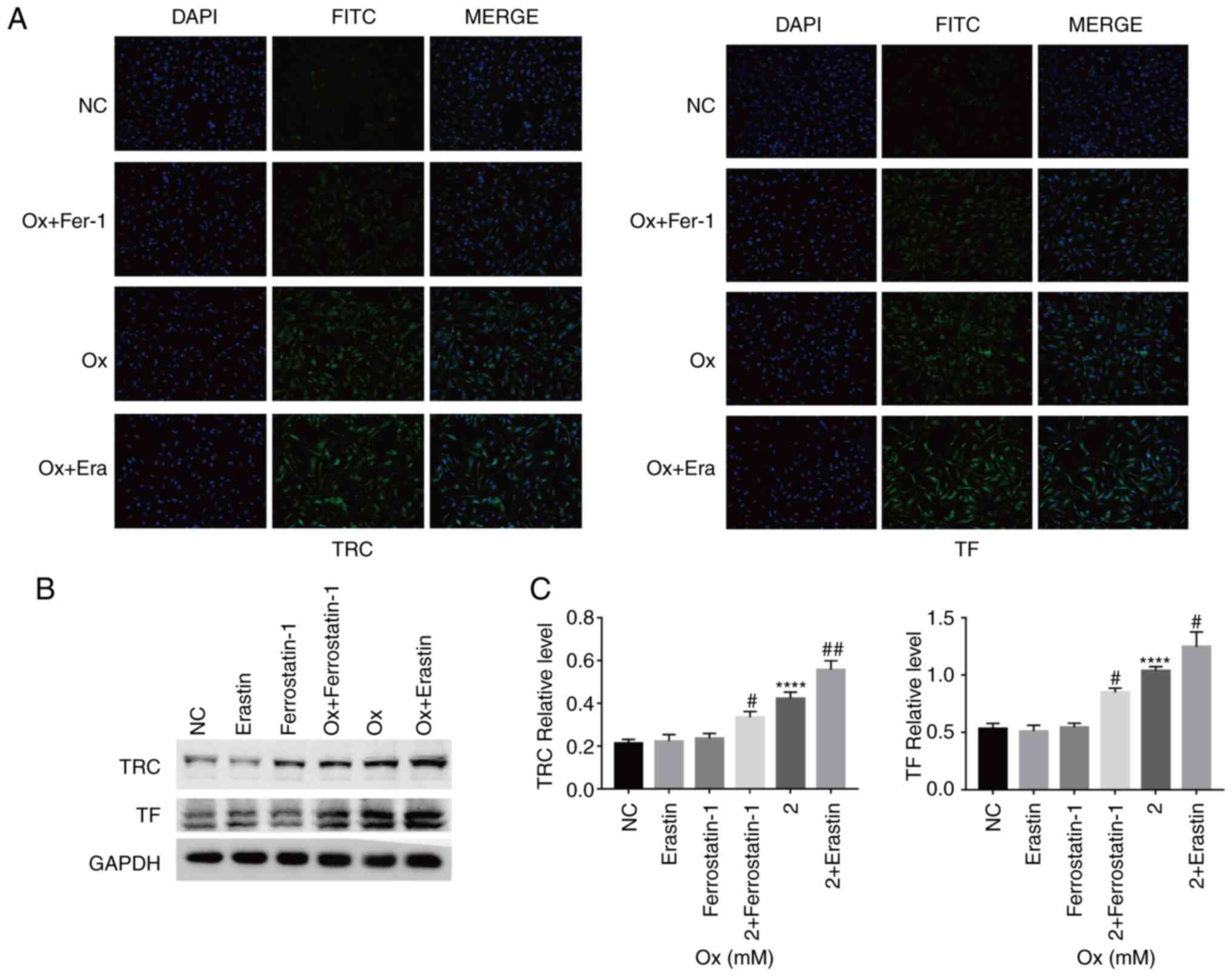
Immunofluorescence detection of the relative
fluorescence intensity of ACSL4, TF, TRC, XCT and GPX4 was then
performed. Compared with the CaOx group, treatment with the
agonist, erastin, increased the relative expression of ACSL4, TF
and TRC, and reduced that of XCT and GPX4. Treatment with the
inhibitor, ferrostatin-1, increased the relative expression of XCT
and GPX4, and reduced that of ACSL4, TF and TRC (Figs. 2A and 3A). Western blot analysis revealed
similar results as those obtained with immunofluorescence assay.
However, the p53 protein level was not markedly affected by erastin
and ferrostatin-1 treatment alone (Figs. 2B and C, and 3B and C). On the basis of the results of
protein level analyses, the effectiveness of erastin and
ferrostatin-1 in regulating the degree of ferroptosis occurrence in
the HK-2 cells under a high-concentration CaOx environment was
fully demonstrated. Under the condition wherein the degree of
ferroptosis could be controlled in the CaOx environment, it was
identified that the levels of intracellular CAT, T-AOC, SOD and GSH
decreased with the increase in ferroptosis, and vice versa
(Fig. 4A). The results obtained
from the aforementioned experiments confirmed that erastin and
ferrostatin-1 regulated the degree of ferroptosis in HK-2 cells in
a high-concentration CaOx environment. Intracellular
Fe2+ overload and upregulation of lipid peroxidation are
necessary conditions for ferroptosis. The agonist, erastin,
significantly increased the intracellular Fe2+ and MDA
concentrations, while the inhibitor, ferrostatin-1, notably
decreased the intracellular Fe2+ and MDA concentrations
(Fig. 4B and C). These results
revealed that ferroptosis induced by high concentrations of CaOx
could reduce the effectiveness of antioxidants and antioxidant
enzymes in cells, thereby affecting the ability of the cells to
resist oxida-tive stress in response to external stimuli.
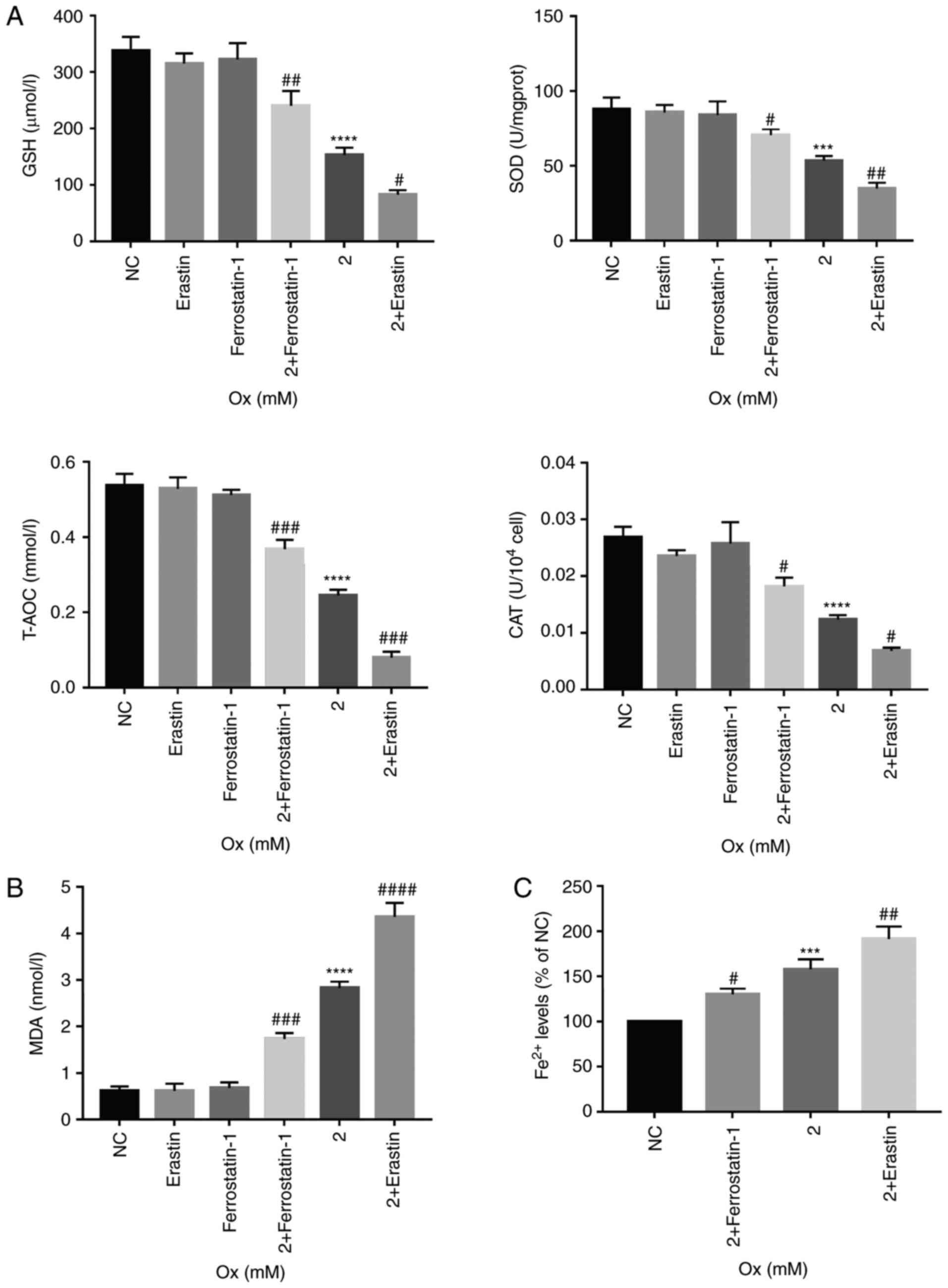 | Figure 4Changes in antioxidant capacity,
lipid peroxidation and concentration of Fe2+ in HK-2
cells induced by ferroptosis. HK-2 cells were treated with erastin
(10 µM) and ferrostatin-1 (8 µM) with or without CaOx
intervention solution (2 mM) for 24 h. (A) Quantification analysis
of antioxidant markers in HK-2 cells. Data are presented as the
means ± SEM (n=3). (B) Quantification analysis of lipid
peroxidation levels in HK-2 cells. Data are presented as the means
± SEM (n=3). (C) Quantification analysis of Fe2+ levels in HK-2
cells. Data are presented as the means ± SEM (n=3). CaOx/Ox,
calcium oxalate; GSH, glutathione; SOD, superoxide dismutase;
T-AOC, total antioxidant capacity; CAT, catalase; MDA,
malondialdehyde. ***P<0.001,
****P<0.0001 vs. the control (NC) group;
#P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001 vs. the Ox
group. |
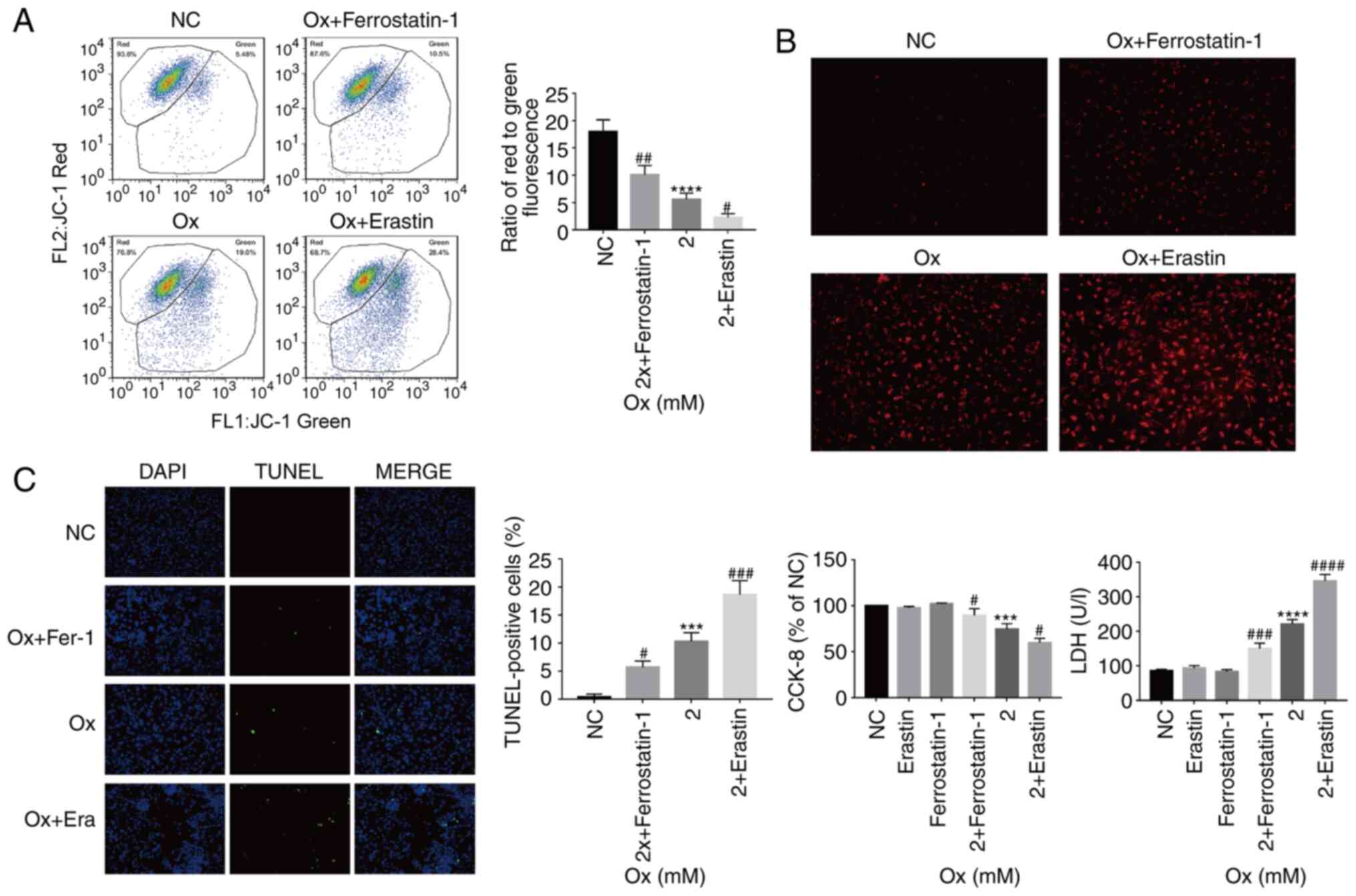
Changes in mitochondria and ROS in HK-2
cells
The results of the above-mentioned analyses
demonstrated that with the increase in the degree of ferroptosis,
the overall resistance of HK-2 cells to oxidation weakened, and the
degree of lipid peroxidation increased in the high-concentration
CaOx envi-ronment. By analyzing the fluorescence ratio by JC-1
assay, it was identified that as the degree of ferroptosis
increased, the mitochondrial membrane potential significantly
decreased, and vice versa (Fig.
5A). Furthermore, by measuring the intensity of the MitoSox
probe signal in the cell, it was demon-strated that with the
increase in the degree of ferroptosis, the levels of ROS increased
significantly and vice versa (Fig.
5B). From these results, it was considered that the gradual
increase in ferroptosis leads to the weakening of the overall
antioxidant capacity of cells. The mitochondria are important
subcellular structures that regulate the ROS levels. The
experiments of the present study demonstrated that the mitochondria
were damaged with the strengthening of external stimuli, and when
the mitochondria were severely damaged, they lost their ability to
regulate ROS, which further led to an increase in ROS generation in
cells.
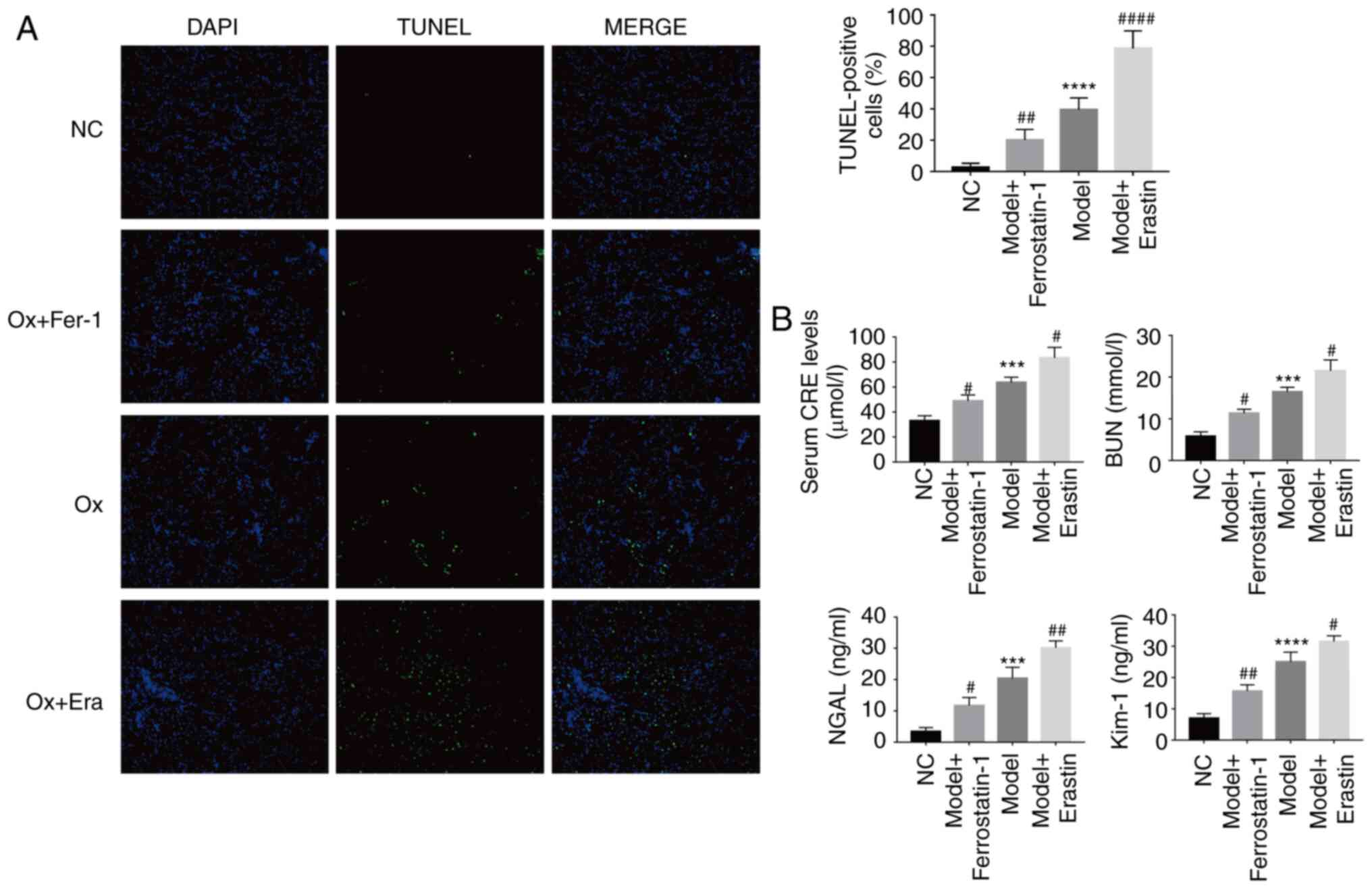
Ferroptosis promotes injury to HK-2 cells
under a high concentration of CaOx
TUNEL staining was used to observe cell death in the
different treatment groups. It was identified that as the degree of
ferroptosis increased, the proportion of TUNEL-positive cells
significantly increased, suggesting that ferroptosis promoted HK-2
cell death under a high concentration of CaOx (Fig. 5C). The present study also examined
cell viability and LDH release levels. With the increase in the
degree of ferroptosis, the LDH release levels significantly
increased and cell viability significantly decreased, and vice
versa (Fig. 5C). From these
results, it could be inferred that CaOx-induced ferroptosis, as
observed in the previous series of biochemical reactions, reduced
the ability of cells to resist oxidation, thereby resulting in
Fe2+ overload, mitochondrial damage and an increase in
lipid peroxidation, which then induced an intracellular ROS
outbreak, eventually causing severe cell injury.
Regulating the degree of ferroptosis in a
rat model
By using a rat model, the results of the in
vitro experiments were confirmed in terms of
ferroptosis-related proteins. Compared with the urolithiasis model
group, the ferroptosis agonist, erastin, significantly increased
the relative expression of ACSL4, TF and TRC, and reduced that of
XCT and GPX4. The ferroptosis inhibitor, ferrostatin-1,
significantly increased the relative expression of XCT and GPX4,
and reduced that of ACSL4, TF and TRC (Fig. 6A). These results were similar to
those obtained in the in vitro experiments.
Crystal deposition and pathological
changes in renal tissue
Von Kossa staining was used to evaluate crystal
deposition and pathological changes in renal tissue. Von
Kossa-stained slices from the NC group exhibited a normal
histological morphology and no pathological changes. Compared with
the NC group, crystal formation in the urolithiasis model group was
signifi-cantly increased, and renal tubules were dilated and
vacuolated, leading to glomerular degeneration. The ferroptosis
agonist, erastin, exacerbated the aforementioned pathological
changes, while the ferroptosis inhibitor, ferrostatin-1, alleviated
these pathological changes (Fig.
6B). The Von Kossa-stained slices were scored for the
histological analysis of the renal tubules, and the same results
were obtained as those for Von Kossa staining (Fig. 6B). These results demonstrated that
ferroptosis aggravated renal crystal deposition and pathological
changes in the rat model of urolithiasis.
Regulating the degree of Fe2+
in renal tissue
With the increase in the degree of ferroptosis, the
levels of Fe2+ in the renal tissue of rats was
significantly increased, and vice versa (Fig. 6C). These results were similar to
those obtained with the in vitro experiments.
Renal cell death detection
By using TUNEL staining, the effect of ferroptosis
on renal cell death in the rat urolithiasis model was investigated.
Compared with the NC group, the number of TUNEL-positive cells was
significantly increased in the urolithiasis model group; the
ferroptosis agonist, erastin, further increased the number of
TUNEL-positive cells, while the ferroptosis inhibitor,
ferrostatin-1, markedly reduced the number of TUNEL-positive cells
(Fig. 7A). From these results, it
could be suggested that ferroptosis aggravated renal cell death in
the rat model of urolithiasis.
Changes in renal function in the rat
model
Compared with the NC group, the levels of CRE and
BUN in the urolithiasis model group were significantly increased.
The ferroptosis agonist, erastin, reduced the clearance rate of CRE
and BUN, while the ferroptosis inhibitor, ferrostatin-1,
significantly improved the clearance rate of CRE and BUN (Fig. 7B). Furthermore, compared with the
NC group, the levels of the renal tubular injury markers, NGAL and
KIM-1, were significantly increased in the urolithiasis model
group. The ferroptosis agonist, erastin, further increased the
levels of NGAL and KIM-1, while the ferroptosis inhibitor,
ferrostatin-1, significantly downregulated the levels of NGAL and
KIM-1 (Fig. 7B), which was
consistent with the results noted for CRE and BUN. These results
indi-cate that ferroptosis can further damage the renal function of
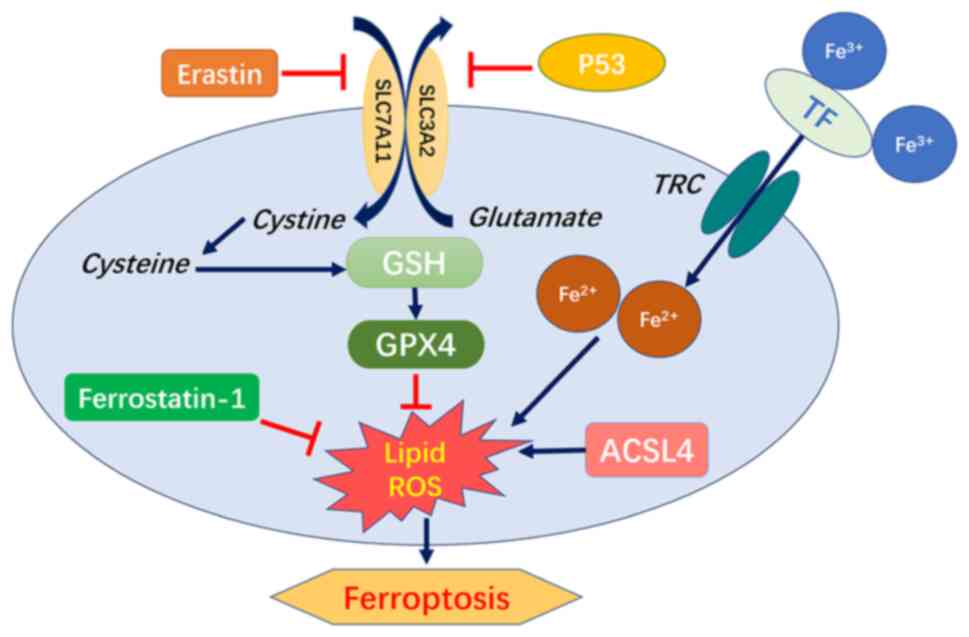
rats with urolithiasis. Based on the above-mentioned results, it
was recognized that ferroptosis is a phenotype caused by multiple
signaling pathways and multiple factors. The level of antioxidants
in the microenvironment and the expression of ferroptosis-related
proteins all have a marked impact on the degree of ferroptosis
(Fig. 8).
Discussion
Ferroptosis, as a regulated cell death phenomenon,
was first reported in 2012, and is a type of iron-dependent lipid
peroxidation (5). With the
advancements in research, ferroptosis has been reported to play a
role in the development of blood diseases, neurological diseases,
respiratory diseases, cardiovascular diseases, reproductive system
diseases and tumors (28-32). However, the related reports of the
role of ferroptosis in urinary system diseases have mainly focused
on renal failure and renal tumors (18,22), and to date, there is no relevant
report available on the topic of urolithiasis, at least to the best
of our knowledge. Through in vivo and in vitro
experiments, the present study, for the first time, to the best of
our knowledge, provided strong evidence of the important role of
ferroptosis in the formation of urolithiasis induced by high
concentrations of CaOx. The results of the in vitro
experiments demonstrated that as the concentration of CaOx
increased, the viability of the HK-2 cells gradually decreased, the
cell morphology gradually changed from an oval to a long
spindle-like shape, and the levels of LDH release and lipid
peroxidation increased significantly. Moreover, with the increase
in the CaOx concentration, the relative expres-sion levels of
ferroptosis-related signaling pathway proteins (p53, ACSL4, TF, and
TRC) significantly increased, while the relative expression levels
of XCT and GPX4 decreased. Thus, it was observed that cell injury
and the degree of ferroptosis exhibits a positive association with
the increase in the CaOx concentration. A high concentration of
CaOx led to markedly increased ferroptosis and injury in the HK-2
cells. With the accumulation of Fe2+ and the increase in
lipid peroxidation, the mitochondria were damaged, leading to the
gradual weakening of the overall antioxidant capacity of cells and
the rapid release of ROS, which eventually resulted in oxidative
stress injury (33,34). Therefore, the present study wished
to determine the mutual association between ferroptosis and
high-concentration CaOx-induced urolithiasis from the aspects of
Fe2+ levels, cell antioxidant capacity, lipid
peroxidation levels, mitochondrial damage, ROS accumulation levels
and cell death.
To further investigate the association between
ferrop-tosis and injury to HK-2 cells induced by exposure to high
concentrations of CaOx, the classic agonist, erastin, and the
ferroptosis inhibitor, ferrostatin-1, were used (33). It was iden-tified that erastin
significantly increased the relative expression of the
ferroptosis-promoting signaling pathway proteins, ACSL4, TF and
TRC, and reduced the relative expression of the
ferroptosis-inhibiting signaling pathway proteins, XCT and GPX4. As
was expected, ferrostatin-1 induced the exact opposite effect. It
was also identified that the relative expres-sion of was is not
affected by erastin and ferrostatin-1. Thus, it was demonstrated
that the regulation of ferroptosis by erastin and ferrostatin-1
began with the cystine/glutamate transport system. As regards cell
biochemical markers, in the uroli-thiasis model, erastin
significantly reduced the antioxidant capacity of HK-2 cells,
leading to an increase in the levels of mitochondrial membrane
potential, lipid peroxidation and ROS. At the same time, the
concentration of intracellular Fe2+ markedly increased,
as well as the degree of cell damage, and vice versa (23-27,33-35). From the aforementioned evidence,
it was concluded that a high concentration of CaOx could induce
ferroptosis, and the increase in ferroptosis levels aggravated
lipid peroxidation levels and mitochondrial injury, leading to an
overload of ROS in cells and eventually increasing the degree of
cell injury. By reducing the degree of ferroptosis, the damage and
death of HK-2 cells caused by the aforementioned effects could be
reduced. Studies have demonstrated that a high CaOx concentration
in urine and crystal deposition could induce the production of ROS
and damage renal epithelial cells (36,37), and that this damage to renal
tubular epithe-lial cells plays a vital role in the formation of
CaOx kidney stones (38). From
the aforementioned results and the findings of the in vitro
experiments, it was concluded that ferroptosis promoted the
formation of urolithiasis.
In the rat model of urolithiasis, the same result as
that obtained in the in vitro experiment was observed. By
performing Von Kossa staining, it was identified that with the
increase in the degree of ferroptosis, the deposition of crystals
in the kidneys significantly increased, and the pathological
fraction of renal tubules markedly increased. Serum CRE and BUN are
classic clinical diagnostic markers for investigating renal
function, while NGAL and KIM-1 are more widely used as clinical
diagnostic indicators of early renal function (39). The present study found that with
the increase in the degree of ferroptosis, renal function in the
rat model of urolithiasis was significantly reduced, and the number
of TUNEL-positive cells in renal tissues was notably increased.
This phenomenon was suppressed by ferroptosis inhibitor. These data
once again confirmed the promoting effect of ferroptosis in the
formation of urolithiasis
Through a series of in vivo and in
vitro experiments, the present study inferred that when exposed
to a high concentration of CaOx, crystal deposition occurred as
CaOx was in a supersaturated state, leading to the occurrence of
cell-crystal reactions, which inhibited the transcriptional
regulation of the cystine/glutamate transport system. Furthermore,
the intracellular transport of cystine was affected, leading to the
inhibition of intracellular GSH synthesis and downregulation of
intracellular activity of GPX4, eventually resulting in lipid
peroxidation. On the other hand, the cell-crystal response
activated the function of the TRC-TF complex, causing a large
amount of iron ions to be transported into cells, and then through
the redox reaction, resulting in an excess of intracellular
Fe2+. The combined effects of these factors caused lipid
peroxidation and mitochondrial injury, and then triggered the burst
of ROS, resulting in cell injury, which eventually produced initial
conditions for the formation of urolithiasis. In the
ferroptosis-lipid peroxidation-ROS injury mode, the damage to renal
tubular epithelial cells induced a more severe state of crystal
deposition, which triggered a positive feedback cell-crystal injury
reaction, and eventu-ally led to the formation of kidney stones.
Therefore, the formation of ferroptosis-induced urolithiasis is a
complex injury mode.
The present study demonstrated that ferroptosis
plays an important role in the formation of urolithiasis; however,
the formation of urolithiasis is a complex pathological process,
and ferroptosis does not play the only role in the pathological
process alone. In some previous studies, it was reported that a
high concentration of CaOx crystals induced an excessive burst of
autophagy and ERS (14-16), which led to the subsequent
oxidative stress reaction and eventually induced the formation of
kidney stones. As an important component of urolithiasis, whether
ferroptosis has a crosstalk with autophagy and ERS, and whether it
affects the formation of urolithiasis through a synergistic effect,
has not been reported in the field of urolithiasis, at least to the
best of our knowledge. However, in other related studies, a close
interaction between ferroptosis, autophagy and ERS has been shown
(18,20). Some researchers have considered
that autophagy was a form of autophagy cell death (12). Mechanistically, autophagy promotes
ferroptosis via a form of cargo-specific autophagy known as
ferrotinophagy. The upregulation of autophagy levels can cause the
overexpres-sion of the NCOA4 gene, and NCOA4 can cause iron storage
as a cargo receptor. The autophagy degradation of protein ferritin
induces the overload of intracellular Fe2+ and causes
ferroptosis (40). On the other
hand, it has been demonstrated that beclin-1 can not only affect
the regulation of autophagy as an autophagy regulator, but also
affect the intake of cystine by combining with XCT to form the
beclin-1/XCT complex, which leads to the inhibition of GPx4
synthesis and the increase of lipid peroxidation level (41). Finally, the Beclin1-XCT signaling
pathway can promote ferroptosis. At the same time, ATF4 is a
critical mediator of metabolic and oxidative homeostasis and cell
survival; as a key regulator of the p-ERK signaling pathway in the
ERS process, it plays a positive regulatory role on ERS. On the
other hand, ATF4 can inhibit the cystine transport function of XCT
via the ATF4/CHOP/CHAC1 signaling pathway, which will further
hinder the synthesis of GSH and decrease the expression of GPX4,
which eventually leads to ferroptosis (42). Therefore, there is a complex
crosstalk between ferroptosis, autophagy and ERS, which can be
regulated by multiple signaling pathways, and there is a common
target among multiple signaling pathways, which can achieve
multi-level regulation by regulating a common target. This makes it
more practical to study the interaction mechanism of ferroptosis,
autophagy and ERS in the formation of kidney stones. On the basis
of the present study, this is also the research direction of our
group. Based on the experimental results and discussions presented
in the current study, we have reason to consider that ferroptosis
plays an important role in the formation of urolithiasis, and it
has been reported that ferroptosis is different from irreversible
injury caused by apoptosis. Ferroptosis is reversible (43), which has made research on
ferroptosis and its relation to the formation of urolithiasis to
have a very promising future, thus bringing a new direction for the
treatment of urolithiasis.
Funding
The present study was supported by the National
Natural Science Funds of China (grant no. 31600785 and 82070723),
the Fundamental Research Funds for the Central Universities of
China (grant no. 2042016kf0097) and the Applied Basic Research
Programs of Wuhan Science and Technology Bureau (grant no.
2017060201010203).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZH designed the study. ZH, BL and QS performed the
experiments and drafted the manuscript. BL, JL and YX participated
in data analysis. WL, CS and SY were involved in the discus-sion
and interpretation of the results. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal experiment protocols were approved by the
Animal Care Committee of Wuhan University (Wuhan, China) and the
Laboratory Animal Welfare and Ethics Committee of Renmin Hospital
of Wuhan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Sorokin I, Mamoulakis C, Miyazawa K,
Rodgers A, Talati J and Lotan Y: Epidemiology of stone disease
across the world. World J Urol. 35:1301–1320. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scales CD Jr, Tasian GE, Schwaderer AL,
Goldfarb DS, Star RA and Kirkali Z: Urinary stone disease:
Advancing knowledge, patient care, and population health. Clin J Am
Soc Nephrol. 11:1305–1312. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsujihata M: Mechanism of calcium oxalate
renal stone formation and renal tubular cell injury. Int J Urol.
15:115–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amato M, Lusini ML and Nelli F:
Epidemiology of nephroli-thiasis today. Urol Int. 72(Suppl 1):
S1–S5. 2004. View Article : Google Scholar
|
|
5
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J,
Zhong M, Yuan H, Zhang L, Billiar TR, et al: The tumor suppressor
p53 limits ferrop-tosis by blocking DPP4 activity. Cell Rep.
20:1692–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid trans-porter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018. View Article : Google Scholar
|
|
8
|
Forcina GC and Dixon SJ: GPX4 at the
crossroads of lipid homeostasis and ferroptosis. Proteomics.
19:e18003112019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Homma T, Kobayashi S, Sato H and Fujii J:
Edaravone, a free radical scavenger, protects against ferroptotic
cell death in vitro. Exp Cell Res. 384:1115922019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar
|
|
12
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ohgami RS, Campagna DR, McDonald A and
Fleming MD: The steap proteins are metalloreductases. Blood.
108:1388–1394. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Li D, He Z, Liu Q, Wu J, Guan X,
Tao Z and Deng Y: Inhibition of autophagy-attenuated calcium
oxalate crystal-induced renal tubular epithelial cell injury in
vivo and in vitro. Oncotarget. 9:4571–4582. 2017. View Article : Google Scholar
|
|
15
|
Liu Y, Liu Q, Wang X, He Z, Li D, Guan X,
Tao Z and Deng Y: Inhibition of autophagy attenuated ethylene
glycol induced crys-tals deposition and renal injury in a rat model
of nephrolithiasis. Kidney Blood Press Res. 43:246–255. 2018.
View Article : Google Scholar
|
|
16
|
Kang J, Sun Y, Deng Y, Liu Q, Li D, Liu Y,
Guan X, Tao Z and Wang X: Autophagy-endoplasmic reticulum stress
inhibition mechanism of superoxide dismutase in the formation of
calcium oxalate kidney stones. Biomed Pharmacother. 121:1096492020.
View Article : Google Scholar
|
|
17
|
Wang XF, Zhang BH, Lu XQ and Wang RQ:
Gastrin-releasing peptide receptor gene silencing inhibits the
development of the epithelial-mesenchymal transition and formation
of a calcium oxalate crystal in renal tubular epithelial cells in
mice with kidney stones via the PI3K/Akt signaling pathway. J Cell
Physiol. 234:1567–1577. 2019. View Article : Google Scholar
|
|
18
|
Kong Z, Liu R and Cheng Y: Artesunate
alleviates liver fibrosis by regulating ferroptosis signaling
pathway. Biomed Pharmacother. 109:2043–2053. 2019. View Article : Google Scholar
|
|
19
|
Gao M, Deng J, Liu F, Fan A, Wang Y, Wu H,
Ding D, Kong D, Wang Z, Peer D and Zhao Y: Triggered ferroptotic
polymer micelles for reversing multidrug resistance to
chemotherapy. Biomaterials. 223:1194862019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park EJ, Park YJ, Lee SJ, Lee K and Yoon
C: Whole cigarette smoke condensates induce ferroptosis in human
bronchial epithelial cells. Toxicol Lett. 303:55–66. 2019.
View Article : Google Scholar
|
|
21
|
Khan SR: Reactive oxygen species as the
molecular modula-tors of calcium oxalate kidney stone formation:
Evidence from clinical and experimental investigations. J Urol.
189:803–811. 2013. View Article : Google Scholar
|
|
22
|
Miess H, Dankworth B, Gouw AM, Rosenfeldt
M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher
DW, et al: The glutathione redox system is essential to prevent
ferroptosis caused by impaired lipid metabolism in clear cell renal
cell carci-noma. Oncogene. 37:5435–5450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jurisic V, Radenkovic S and Konjevic G:
The actual role of LDH as tumor marker, biochemical and clinical
aspects. Adv Exp Med Biol. 867:115–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krych-Madej J and Gebicka L: Interactions
of nitrite with catalase: Enzyme activity and reaction kinetics
studies. J Inorg Biochem. 171:10–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Owen JB and Butterfield DA: Measurement of
oxidized/reduced glutathione ratio. Methods Mol Biol. 648:269–277.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carillon J, Rouanet JM, Cristol JP and
Brion R: Superoxide dismutase administration, a potential therapy
against oxidative stress related diseases: Several routes of
supplementation and proposal of an original mechanism of action.
Pharm Res. 30:2718–2728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Del Rio D, Stewart AJ and Pellegrini N: A
review of recent studies on malondialdehyde as toxic molecule and
biological marker of oxidative stress. Nutr Metab Cardiovasc Dis.
15:316–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang F, Lv H, Zhao B, Zhou L, Wang S, Luo
J, Liu J and Shang P: Iron and leukemia: New insights for future
treatments. J Exp Clin Cancer Res. 38:4062019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Masaldan S, Belaidi AA, Ayton S and Bush
AI: Cellular senescence and iron dyshomeostasis in Alzheimer's
disease. Pharmaceuticals (Basel). 12:E932019. View Article : Google Scholar
|
|
30
|
Alvarez SW, Sviderskiy VO, Terzi EM,
Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K
and Possemato R: NFS1 undergoes positive selection in lung tumours
and protects cells from ferroptosis. Nature. 551:639–643. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stamenkovic A, Pierce GN and Ravandi A:
Phospholipid oxidation products in ferroptotic myocardial cell
death. Am J Physiol Heart Circ Physiol. 317:H156–H163. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ng SW, Norwitz SG and Norwitz ER: The
impact of iron overload and ferroptosis on reproductive disorders
in humans: Implications for preeclampsia. Int J Mol Sci.
20:32832019. View Article : Google Scholar :
|
|
33
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxi-dation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar
|
|
34
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thévenod F: Iron and its role in cancer
defense: A double-edged sword. Met Ions Life Sci. View Article : Google Scholar
|
|
36
|
Li CY, Deng YL and Sun BH: Taurine
protected kidney from oxidative injury through mitochondrial-linked
pathway in a rat model of nephrolithiasis. Urol Res. 37:211–220.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li CY, Deng YL and Sun BH: Effects of
apocynin and losartan treatment on renal oxidative stress in a rat
model of calcium oxalate nephrolithiasis. Int Urol Nephrol.
41:823–833. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang JM, Yao XQ, Tan J and Wang FX:
Renal epithelial cell injury and its promoting role in formation of
calcium oxalate monohydrate. J Biol Inorg Chem. 16:405–416. 2011.
View Article : Google Scholar
|
|
39
|
Kandur Y, Gonen S, Fidan K and
Soylemezoglu O: Evaluation of urinary KIM-1, NGAL, and IL-18 levels
in determining early renal injury in pediatric cases with
hypercalciuria and/or renal calculi. Clin Nephrol. 86:62–69. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kang R, Zhu S, Zeh HJ, Klionsky DJ and
Tang D: BECN1 is a new driver of ferroptosis. Autophagy.
14:2173–2175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang N, Zeng GZ, Yin JL and Bian ZX:
Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects
ferroptosis in Burkitt's lymphoma. Biochem Biophys Res Commun.
519:533–539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang HM and Tang HL: Cell recovery by
reversal of ferroptosis. Biol Open. 8:bio0431822019. View Article : Google Scholar : PubMed/NCBI
|