In 1972, the Austrian pathologist J.F. Kerr, in
cooperation with his Scottish colleagues A.H. Wyllie and A.R.
Currie, introduced the concept of 'apoptosis' (after the ancient
Greek ἀπόπτωσις-leaf fall) to describe a morphologically
stereotypical form of cell death characterized by cytoplasmic
volume depletion, chromatin condensation and margination, shrinkage
of the nucleus (pyknosis), fragmentation of the nucleus
(karyorhexis), blebbing of the membranes and formation of discrete
apoptotic bodies with an undamaged cell membrane (1,2).
According to the contemporary biochemical
classification of Nomenclature Committee on Cell Death, apoptosis
is considered to be one of the morphological signs typical for
different types of regulated cell death (RCD) (3). One of the forms of RCD is intrinsic
apoptosis. Intrinsic apoptosis, initiated by the cell itself in
response to intracellular damage, is also known as mitochondrial
apoptosis, as the mitochondria performs the key role in this
process (4). The trigger event is
the increase in mitochondrial outer membrane permeabilization
(MOMP) and release of proteins that are normally sequestered
between the two mitochondrial membranes (5,6).
The MOMP and thus the entire process of intrinsic apoptosis is
regulated by members of the BCL2 protein family that are embedded
in the outer membrane (6,7).
BCL2 is an acronym for B-cell lymphoma/leukemia-2.
As its name suggests, the gene expressing BCL2 was for the first
time found in B-cell malignant neoplasms. This acronym is also used
for the designation of the entire family of homological proteins
(8). Different proteins of this
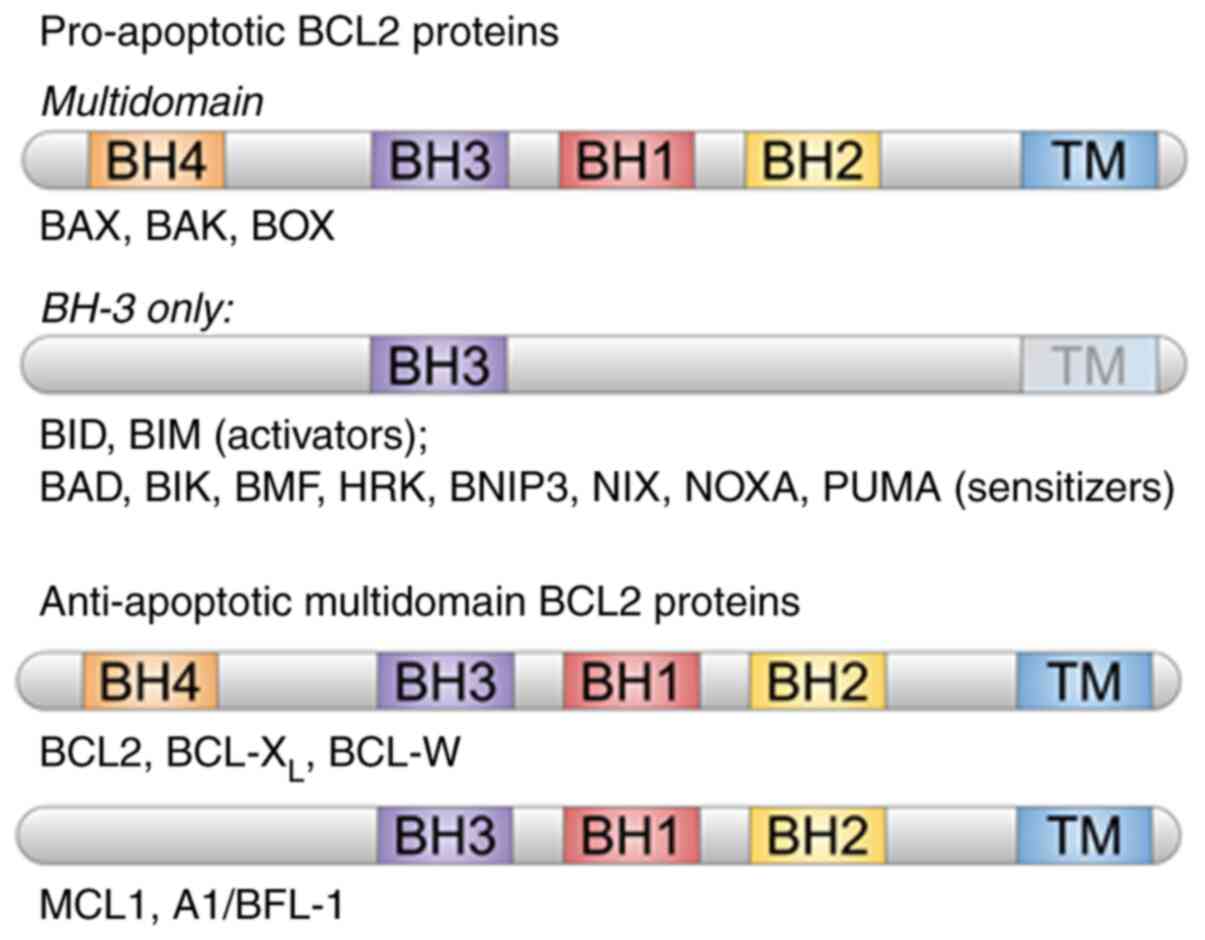
family contain BCL2 homology domains (BH: BH1, BH2, BH3 and BH4)
(Fig. 1) (9) and can be divided into two groups:
Pro-apoptotic and anti-apoptotic. Pro-apoptotic proteins include
BCL2-associated X protein (BAX), (BCL2 antagonist/killer (BAK),
BCL2-related ovarian killer, BH3 interacting domain death agonist,
BCL2-associated agonist of cell death, BCL2-interacting killer,
BCL2-interacting mediator of cell death), BCL2-modifying factor,
activator of apoptosis harakiri, BCL2-interacting protein 3
(ANIP3), NIX (BNIP3-like), phorbol-12-myristate-13-acetate-induced
protein 1 (NOXA) and p53 upregulated modulator of apoptosis (PUMA).
Meanwhile, anti-apoptotic proteins include BCL2, BCL2 X-linked
protein (BCL-XL), myeloid cell leukemia 1 and BCL-w and
A1/BFL-1 (10-12). The BCL2 family proteins are
capable of interacting with each other, whereby their different
partnerships result in different outcome of the cell fate (9). In response to apoptosis stimulation,
BAX and BAK proteins are exposed to oligomerization on the
mitochondrial outer membrane (13,14). This process is blocked by BCL2
protein, which inhibits mitochondrial permeabilization and cell
death by interacting with BAX and BAK (9,15).
Enhanced expression of Bcl2 may increase cell resistance to
apoptosis in cells, such as tumor cells. The BCL2/BAX ratio is a
type of 'rheostat' regulating cell death depending on the balance
between BCL2 and BAX in cells (16).
Cardiomyocyte apoptosis is a well-known key process
during the development of ischemia (17) During apoptosis inhibition, the
BCL2/BAX ratio is increased, which contributes to cardiomyocyte
survival in the peri-infarct area (18). Previous investigations revealed a
significant role of abnormal Bcl2 expression in
cardiomyocyte apoptosis modulation in MI/RI, as its expression rate
has a direct effect on cardiomyocyte apoptosis and cardiac function
(19,20).
The key object in the clinical treatment of MI/RI
based on molecular mechanisms of the injury progression is
decreasing the rate of cardiomyocyte apoptosis. BCL2 is the key
protein in the entire BCL2 family that is responsible for the
anti-apoptotic process and promotion of cell survival. Therefore,
the present review focused mainly on this protein and aimed to
investigate how it is regulated during MI/RI and how this can be
exploited for clinical use. The present review also assessed
possible ways of using BCL2 as a target for pharmacological
correction.
Cardiovascular diseases are the main cause of death
world-wide. In 2016, 85% of cases resulted from myocardial
infarction or cerebral stroke (21,22). Coronary heart disease is the main
cause of death and disabilities (23). Myocardial infarction is tissue
necrosis following acute ischemia, which is characterized by
absolute insufficiency of coronary blood circulation (24).
Ischemia is a complex pathological mechanism
resulting from a decrease in the local blood flow in a tissue or
organ (25). Ischemia occurs
commonly in the myocardium due to occlusion of the coronary
arteries responsible for myocardial perfusion (25). The heart is a constantly
contracting organ, requiring a high rate of metabolic activity,
which makes it extremely susceptible to any disorders of oxygen
supply. Under normal conditions, mitochondria consume oxygen and
generate ATP. A decrease in oxygen supply leads to the inhibition
of mitochondrial oxidative phosphorylation and, consequentially,
the switch from aerobic to anaerobic metabolism (26). Anaerobic glycolysis causes a
reduction of intracellular pH (26). The combination of enhanced sodium
and calcium influx into cells, due to Na+-H+
and Na+-Ca2+ exchange, correspondingly
increases acidity and intracellular calcium levels (27). Moreover, a rapid elevation in
intracellular Ca2+ leads to a pathological increase in
mitochondrial permeability transition; however, a reduction of
intracellular pH inhibits this process (27). Disordered ion homeostasis is
followed by osmotic gradient formation, which is accompanied by
water inflow into the cell with a subsequent swelling and
disturbance in intracellular ion balance (28). If blood supply is not properly
restored after ischemia, the absence of sufficient ATP levels and
high levels of Ca2+ lead to myocyte atrophy and
eventually apoptosis and necrosis (28). The activation of caspase-3 and
maximal activity of pro-apoptotic proteins BAX, Noxa and PUMA are
observed on the 1st day post-coronary artery occlusion; however,
anti-apoptotic proteins BCL2 and BCL-XL remain relatively
unchanged, which indicated that the pro-apoptotic pathways are
activated rapidly in MI/RI while cell protective pathways remain
inactive (29).
Reperfusion of the stunned myocardium during
percutaneous coronary intervention is necessary to minimize
myocardial damage. For patients with myocardial infarction
accompanied by elevation of ST-segment, the timely reperfusion of
the myocardium using either thrombolytic therapy or primary
percutaneous coronary intervention, is the most effective method of
treatment to restrict the size of infarction area, support systolic
function and reduce manifestations of heart failure (30). Reperfusion therapy of coronary
insufficiency after myocardial infarction is also the most
effective method to save cardiomyocytes suffering from hypoxia,
support cardiac function and save patients' lives (31). Reperfusion is the most significant
method to prevent tissue death after ischemia. However, restoration
of blood flow can paradoxically lead to MI/RI, characterized by
metabolic disturbances, local inflammatory response, cell death and
a consequent cardiac remodeling and dysfunction, contributing to
adverse cardiac events after myocardial ischemia (25,32,33). Although reperfusion is necessary
for the restoration of oxygen and nutrient influx, which supports
cellular metabolism, it may paradoxically cause consequent
pathological processes aggravating tissue damage (34,35). MI/RI may exacerbate structural and
functional disturbances of the myocardium and cause a strong effect
on the restoration of cardiac function after recurrent reperfusion
(35-37).
The phenomenon of paradoxical aggravation after
oxygen flux restoration was described for the first time >50
years ago when it was shown that reperfusion caused several
pathological changes in heart exposed to coronary occlusion
(26). MI/RI is associated with
different pathophysiological mechanisms, including calcium
overload, production of oxygen free radicals, endothelial
dysfunction, immune response, mitochondrial dysfunction,
cardiomyocyte apoptosis and autophagy and platelet aggregation
(38-41). During this process, apoptosis is
the main pathological mechanism, which plays a critical role in
cardiac remodeling after myocardial infarction (42).
Cardiomyocyte apoptosis and necrosis caused by MI/RI
are the most critical pathological processes in cases of cardiac
dysfunction after previous myocardial infarction (43). Myocardial necrosis is
predominantly observed at the late stages of MI/RI while cell
apoptosis is observed throughout the whole process (43). Apoptosis is one of the most
important mechanisms of MI/RI and it has a considerable effect on
the degree of damage and consequently on the prognosis of heart
failure development (42). Thus,
effective inhibition of apoptosis caused by MI/RI is one of the
important lines of research and it is of great importance for
cardiac function improvement after myocardial infarction and for
preventing myocardial remodeling.
Two main types of protein activity regulation are
known: Fast regulation via post-translational modification (usually
phosphorylation/dephosphorylation) and slow regulation via gene
expression regulation. BCL2 was shown to have three basic sites of
phosphorylation (T69, S70 and S87) which results in changes in its
anti-apoptotic activity (11).
The modulating role of BCL2 phosphorylation remains to be fully
elucidated, moreover, there are contradicting facts described in
literature which can derive from the feasibility of single
phosphorylation of different amino acids or triple phosphorylation
of all three amino acids in the structure of BCL2 (10,46,53). Moreover, BCL2 phosphorylation of
the same type in normal and cancer cells can lead to different
effects. An attempt to clarify these contradictions was undertaken
by Song et al (53), who
managed to build a mathematical model for BCL2 phosphorylation in
different types of cancer cells and revealed that the turning point
was 50% triple phosphorylation (T69, S70 and S87) that switched
BCL2 from apoptotic to anti-apoptotic action.
The limitation of this conclusion is that it can
only be reliably applied to cancer cells.
Several kinases that have BCL2 as a target for
phosphorylation are well described in literature: Protein kinase C
α, JNK, p38/MAPK, ERK and pyruvate kinase isoform M2 (PKM2).
Dephosphorylation of phosphorylated (p)-BCL2 is performed by
protein phosphatase A2. BCL2 phosphorylation mediated by JNK,
p38/MAPK and PKM2 was shown to occur in cardiomyocytes. JNK and
p38/MAPK inactivate BCL2 by phosphorylating and inducing apoptosis,
causing cardiomyocyte injury after ischemia and during oxidative
stress (54,55). By contrast, PKM2 phosphorylates
BCL2 with the aid of heat shock protein 90 to prevent its
degradation, thus enhancing its stability and promoting its
anti-apoptotic properties (56).
Several publications link the degree between BCL2 triple
phosphorylation with the crosstalk between autophagy and apoptosis
(57-59). This switch point is feasible due
to different affinities of BCL2 and p-BCL2 to beclin-1 as the main
autophagy inducer (57). Thus,
phosphorylation of BCL2 leads to the dissociation of beclin-1 from
the BCL2-beclin-1 complex with consequent phosphorylation of
beclin-1 and the formation of an active PI3K III complex and
autophagy induction (57). The
lower degree of BCL2 phosphorylation resulted in autophagy
induction, while more extensive BCL2 phosphorylation reduced its
affinity to BAX, causing its dissociation and thus resulting in
apoptosis induction (58).
Other mechanisms of BCL2 regulation involve gene
expression and result in changes in BCL2 intracellular levels.
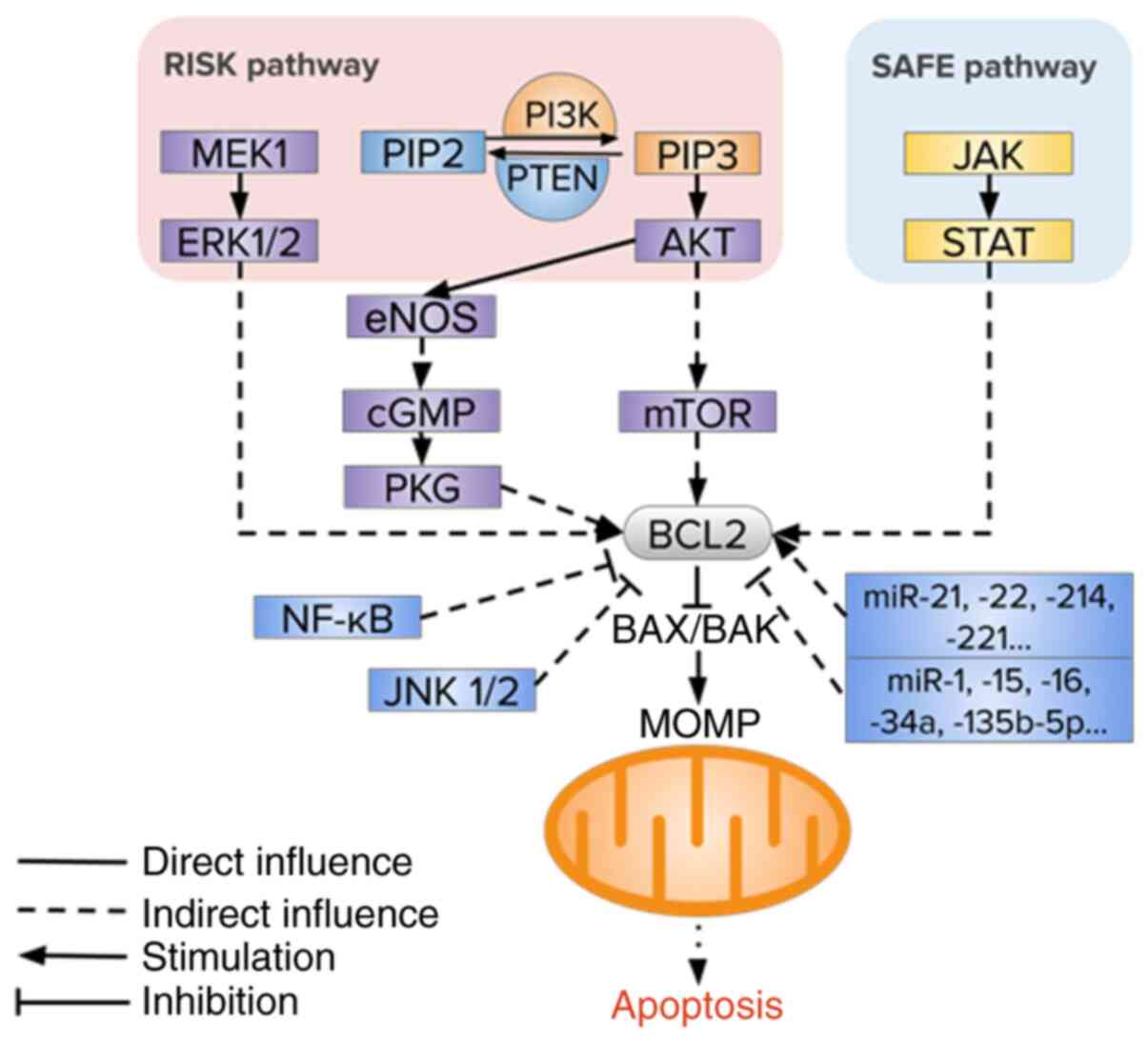
Several signaling pathways are known to regulate the rate of
intrinsic apoptosis including PI3K/AKT and MEK1-ERK1/2, endothelial
nitric oxide synthase (eNOS), PTEN and JAK2/STAT3 (59-65) (Fig.
2).
The reperfusion injury salvage kinase (RISK) pathway
was described for the first time by Schulman et al (59) in 2002, while they were studying
the mechanisms underlying the cardioprotective effect caused by
urocortin. The RISK pathway is a combination of two parallel
cascades: PI3K/AKT and MEK1/ERK1/2. The pathways were analyzed in
detail in a series of subsequent pharmacological experiments in
which the protective effect of several interventions was blocked by
a simultaneous administration of PI3K and ERK inhibitors at
different times (60). In the
broadest term, RISK refers to the group of pro-survival protein
kinases responsible for cardio-protection via specific activation
during reperfusion.
The PI3K/AKT/mTOR signaling pathway is an important
regulatory mechanism for protein synthesis and is closely
associated with intracellular oxidation and reduction in the
mitochondria (61). It was found
that stress in vitro and in vivo may lead to an
increase in the rate of tyrosine receptor phosphorylation which
activates PI3K, indirectly stimulating AKT phosphorylation,
increasing the rate of p-mTOR and activating the expression of the
anti-apoptotic factor Bcl2 (61,62). It was also shown that the levels
of PI3K, p-AKT and p-mTOR in rat myocardial cells after
MI/RI were significantly lower compared with the controls (62). Following MI/RI, expression levels
of caspase-3 and Bax were significantly increased in
myocardial cells whereas Bcl2 expression significantly
decreased (63).
The conformation of PI3K can be changed and
activated by the action of growth factors and mitogens, which
convert phosphatidylinositoldiphospate 2 (PIP2) into
phosphatidylino-sitoltriphospate 3 (PIP3) (63). Several studies demonstrated that
the PI3K/AKT signaling pathway may facilitate cell apoptosis in
case of MI/RI by influencing the BCL2/BAX ratio (64,65). Zhang et al (66) showed that the PI3K/AKT/mTOR
signaling pathway is inhibited in the cardiomyocytes of rats with
myocardial infarction, which leads to significant activation of
cardiomyocyte apoptosis.
A significant role in the regulation of the PI3K/AKT
signaling pathway in MI/RI belongs to PTEN which dephosphorylates
PIP3 back into PIP2, thus inhibiting the PI3K/AKT signaling pathway
(Fig. 2). This protein plays an
important role in apoptosis (67). Nevertheless, only a few studies
evaluated the role of PTEN in MI/RI experimental models. In
particular, it was shown that PTEN inhibition protected the
myocardium from MI/RI by activating the PI3K/AKT/eNOS/ERK pathway,
which is one of the variants of pro-apoptotic pathway induction
(67). An increase of PTEN levels
may suppress the activity of the PI3K/AKT signaling pathway, which
may cause myocardial cell apoptosis during MI/RI (68). It was also shown that expression
of PTEN and BAX levels in myocardial cells in the MI/RI group were
markedly higher compared with sham-operated animals, but
phosphorylation of AKT and BCL2 levels were significantly lower
(69).
The anti-apoptotic effects of nitrogen oxide (NO)
mediated-cGMP/protein kinase G (PKG) signaling can be associated
with increased synthesis of anti-apoptotic BCL2 and inhibition of
MPTP formation (77,78). Moreover, NO and natriuretic
peptides may prevent cardiomyocyte apoptosis via cGMP/PKG-dependent
inhibition of intracellular calcium overload (79).
The JAK/STAT signaling pathway is a key component of
the survivor activating factor enhancement (SAFE) pathway, which
can transmit cell signals from the plasmalemma to the nucleus,
providing regulation of gene expression (80-85). The JAK/STAT pathway plays an
important role in different mechanisms in the myocardium, including
apoptosis (81,86), MI/RI (87,88), preconditioning (89) and postconditioning (90,91). In 2009, Lecour (92) showed that in addition to the RISK
pathway, SAFE can be an alternative pathway mediating signaling
activated by post-conditioning. The JAK/STAT pathway consists of
the family of receptor-associated cytosol tyrosine kinases, which
phosphorylate tyrosine (93).
Phosphorylation and activation of signal transducer and activator
of transcription (STAT) in response to ischemic preconditioning
(IPC) contribute to cardioprotection by means of signaling cascades
and inhibition of pro-apoptotic factors (94). STAT3 is a central component of
cardioprotection (95,96). Subsequent studies showed that the
JAK2/STAT3 signaling pathway takes part in the anti-apoptotic
effect of preconditioning, which is realized by increasing the
synthesis of anti-apoptotic BCL2 and suppressing the pro-apoptotic
protein BAX (90,97).
The inhibition of pathways that increase the
BCL2/Bax ratio and enhancement of pathways leading to its lowering
is typically observed in MI/RI, which is associated with hypoxic
conditions (84). In vitro
MI/RI modeling in cardiac myoblasts revealed an increase in BCL2
protein levels accompanied by an increase in p-PI3K and p-AKT
levels after antioxidant treatment (94). Cell survival was also increased
while the expression of pro-apoptotic BAX was downregulated
(98). These results supported
the idea that hypoxia-induced oxidative stress acts as a main
downregulatory factor for BCL2 and BCL2-family controlled intrinsic
apoptosis.
Cardiac ankyrin repeat protein (CARP), a
transcription co-factor regulating gene expression in
cardiomyocytes, inhibits apoptosis induced by MI/RI increasing
Bcl2 gene expression (104). CARP is linked with the promotor
site of the gene Bcl2 through formation of a complex with
transcription factor GATA-4 which regulates transcription and
enhances cardioprotection (104).
Hyperlipidemia can stimulate the activation of
cardio-myocyte apoptosis in MI/RI. Immunocytochemical analysis
revealed an increase in the expression of pro-apoptotic Bax
and inhibition anti-apoptotic Bcl2 expression in the
myocardium of rats exposed to a hypercholesterol diet (105). These results are in agreement
with the data obtained by Guo et al (106) and Kuo et al (107). In this model, the levels of
pro-apoptotic proteins BAK and BAX are significantly increased,
which is a sign of induction of intrinsic apoptosis (108). Hypercholesterinemia is
associated with an increase in the BCL2/BAX ratio in the myocardium
which leads to the aggravation of myocardial damage after its
reperfusion due to the activation of cardiomyocyte apoptosis rate
(107). It was also shown in the
experiments in Oryctolagus (rabbits) that Bcl2
expression is increased in the myocardium during
hypercholesterolemia by 50% compared with the controls (109). In Oryctolagus with
hypercholesterolemia and myocardial ischemia, a marked reduction of
Bcl2 expression and similar degree of the increase in
Bax expression were observed (109).
For example, miR-1 is predominantly expressed in
cardiac myocytes and closely associated with MI/RI in rats as its
levels inversely correlate with BCL2 protein synthesis in
cardiomyocytes in MI/RI (119).
Mice studies also showed that enhancement of miR-135b-5p expression
in MI/RI leads to activation of the JAK2/STAT3 signaling pathway,
Bax expression and Bcl2 inhibition (120). Hullinger et al (121) demonstrated that miR-15b, a
member of the miR-15 family, aggravated myocardial damage caused by
MI/RI via affecting BCL2. miR-16 expression is activated during
MI/RI and has an inhibiting effect on Bcl2 expression, which
contributed to the enhancement of cardiomyocyte apoptosis after
MI/RI (122). Inhibition of
miR-16 expression may suppress cardiomyocyte apoptosis after MI/RI,
resulting in a reduction of infarction area (122). miR-221 is involved in the
pathogenesis of MI/RI by regulating the PTEN/AKT signaling pathway,
along with Bax and Bcl2 expression (123-125). Expression of Bcl2 and
microtubule-associated proteins 1A/1B light chain 3B II in
cardiomyocytes of newly born rats is significantly decreased, which
is accompanied by enhanced expression of miR-497 in
anoxia-reoxygenation (126).
Another study revealed the cardioprotective role of mir-21 in MI/RI
via the activation of the PTEN/AKT signaling pathway and BCL2
(127). miRNA-22 may inhibit
cardiomyocyte apoptosis by inhibiting p53 acetylation and
decreasing the levels of pro-apoptotic genes Bax and
p21 by affecting one of its targets-cAMP response
element-binding protein (128-130). miR-214 reduced myocardial damage
caused by MI/RI via the PI3K/AKT signaling pathway, accompanied by
a decrease in BAX levels and an increase in BCL2 levels (131). miR-34a, activated in rats with
MI/RI, repressed Bcl2 in vivo and in vitro (132).
The regulation of BCL2-dependent apoptosis in MI/RI
is quite versatile and depends on a large number of factors,
including activation of emergency genetic programs, changes in
metabolic processes and the involvement of additional signaling
pathways protecting the myocardium from the negative effects of
hypoxia. The ability to influence these mechanisms makes it
possible to reduce cardiomyocyte damage, also via induction of
BCL2.
Various forms of cell death may occur during acute
MI/RI including necrosis, apoptosis, autophagy, necroptosis and
pyroptosis, which may influence the terminal size of the myocardial
infarction area after MI/RI (3).
This may be used as a new target for cardioprotection, which may
include the activation of endogenous cardioprotective signaling
pathways: Cascade NO/cGMP/PKG, RISK and SAFE pathways,
mitochondrial morphology, cardiomyocyte apoptosis and others
(77-79).
Cardiomyocytes of adult humans are characterized by
an extremely limited regeneration capacity (133). As a result, there is a
continuous process of renewal and reparation of cells mediated by
different mechanisms, including apoptosis (134).
In the 1990s, studies focused on the role of
different types of cell death in cardioprotection after MI/RI
(135). Pro-apoptotic proteins
were the main subjects of research at the time, where they were
considered to be new targets in MI/RI (135). This was based on a hypothesis
suggesting a possibility of saving viable cardiomyocytes when the
signaling pathway of regulated cell death was potentially
interrupted (135). For example,
caspase inhibition during reperfusion restricted the size of the
myocardial infarction area in animal models (136). Besides preventing cell death by
inhibition of pro-apoptotic caspases, the focus was also given to
the use of growth factors that prevented apoptotic processes via
activation of proteins contributing to cell survival, such as
kinases responsible for the survival associated with PI3K and
ERK1/2 activation. This method was suggested to be protective
against MI/RI (137,138).
However, there are still no effective methods for
prevention of MI/RI in patients with myocardial infarction
(139). Previous attempts to
perform cardioprotective treatment of MI/RI (antioxidants, calcium
blockers and anti-inflammatory drugs) were not successful (140). The advantages of growth factors
(137,138) was restricted because the
signaling pathways they were involved in lead simultaneously to
activation of apoptosis and induction of fibrosis (141).
Regardless of the success in the research of
cardioprotective methods on animals, their use in clinical practice
still present with severe difficulties (145-147). Some pharmaceutical approaches
faced just little success, and although the suggested methods of
ischemic conditioning seem promising, their effects may be minor
and, in some cases, even controversial (148). Differences between preclinical
models of transient myocardial ischemia and coronary heart disease
with specific characteristics in patients including age,
concomitant diseases and drug therapy may help explain the
difficulties in introducing the potential cardioprotective
techniques into clinical practice (149).
Numerous different methods of cardioprotective
therapy of MI/RI have been suggested in the past three decades
(150). These approaches are
commonly based on the controlled use of short-term ischemia and
reperfusion (ischemic conditioning), pharmacotherapy or
physiotherapy including hypothermia or electric stimulation of
nerve terminals (30,140).
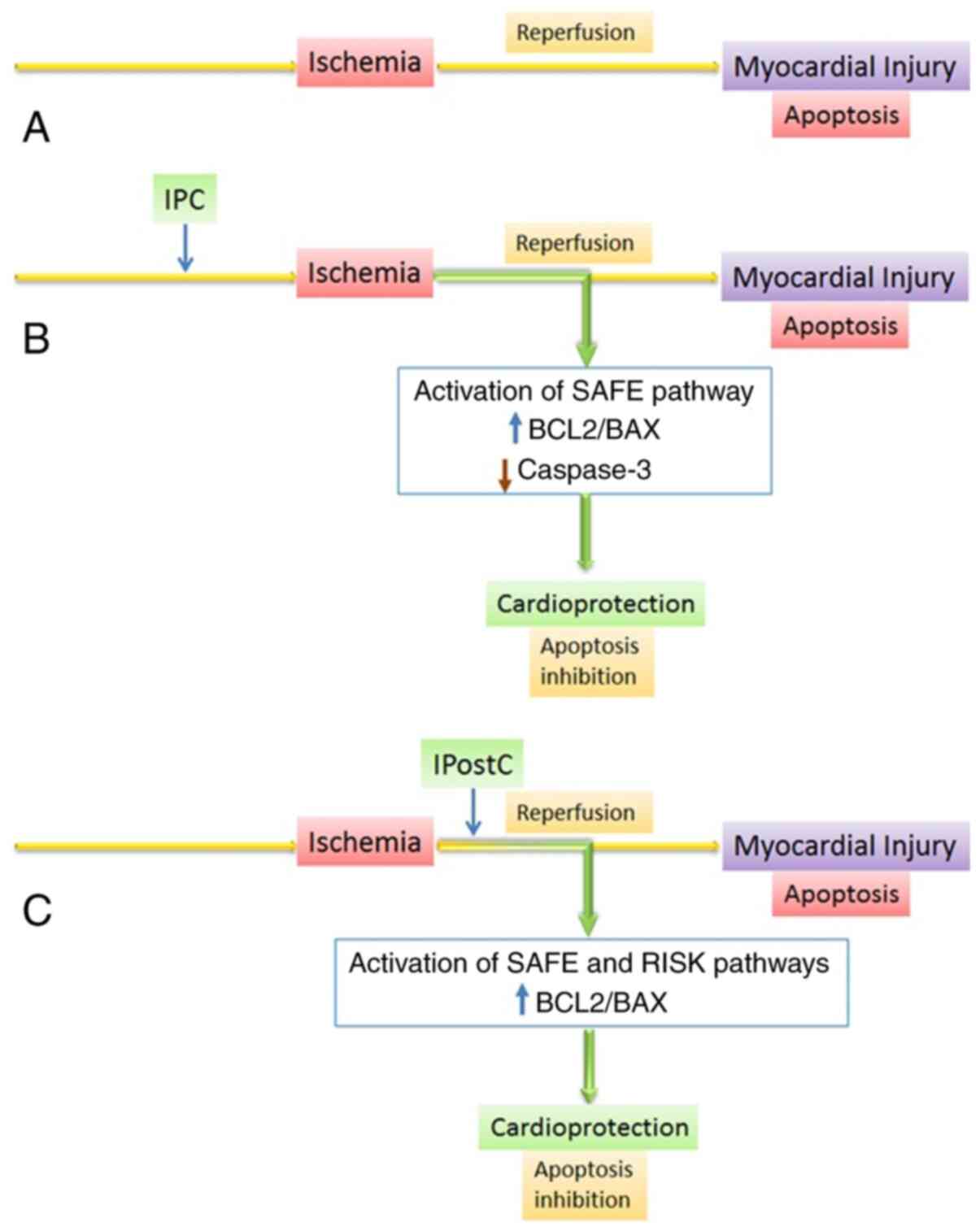
Therapeutic methods of MI/RI based on ischemic
conditioning include local IPC and ischemic post-conditioning
(IPostC) as well as remote ischemic conditioning (140), which delays pH restoration,
prevent NOS decomposition and consequent formation of reactive
forms of oxygen and nitrogen and also increase the content of PKG,
a component of the RISK pathway, and cause enhancement of the SAFE
pathway in reperfused cardiomyocytes (151-153). As aforementioned, all these
factors regulate BCL2 in MI/RI, indicating cardio-protective
effects of ischemic conditioning due to inhibition of BCL2-family
dependent apoptosis (Fig. 3). The
following part of the review details the exploration of the
mechanisms underlying these strategies of MI/RI therapy.
The potential of IPC is inevitably restricted by
the necessity to use it before ischemia, which is of great
difficulty for patients with myocardial infarction (158). However, this method initiated a
number of subsequent studies, which have brought considerable
success in understanding the mechanisms underlying MI/RI and IPC as
a result of the potential development of cardioprotective therapy
(159).
The cardioprotective effect of IPC is evidenced by
a decrease in the size of the myocardial infarction area and a
reduction in the number of apoptotic cardiomyocytes (157). Activation of the JAK2/STAT3
signaling pathway in response to IPC contributed to
cardioprotection via signaling cascades responsible for the
inhibition of pro-apoptotic factors (160). Early phase of IPC enhanced
JAK/STAT signal transduction by activation of STAT3, which is
nearly neutralized by AG490, a JAK2 inhibitor (161). Constitutive deletion of STAT3
stimulated apoptosis, increased the size of infarction area and
caused a reduction in cardioprotective effects after
pharmacological preconditioning (162).
Studies showed that IPC increased the activity of
cyclo-oxygenase-2 and inducible NOS 24 h after intervention, which
depends on transcriptional regulation via the JAK/STAT signaling
pathway (163,164). Taken together, these
observations lead to the conclusion that IPC activated the SAFE
pathway (Figs. 2 and 3).
Restoration of myocardial blood circulation caused
by postconditioning improved the contractile function of the
myocardium and also restricts the size of infarction area, which is
confirmed by a lower serum concentration of creatine kinase (CK)
and the activity of lactate dehydrogenase compared with the data
obtained after MI/RI without previous postconditioning (167,168).
The effectiveness of IPostC as a method of
myocardial protection from MI/RI was also confirmed in several
other studies. IPostC does not only decrease the size of the
infarction area (143,167) but also limits cardiomyocyte
apoptosis after reperfusion. Budhram-Mahadeo et al (29) showed that IPostC stimulated BCL2
synthesis and inhibited BAX production. Another study demonstrated
the ability of IPostC, similar to IPC, to restrict cardiomyocyte
apoptosis after reperfusion via the SAFE pathway (169). IPostC activated STAT3 after
reperfusion, and a JAK2 inhibitor (AG490) suppressed the
anti-apoptotic effects of IPostC (170). The anti-apoptotic effects of the
JAK2-STAT3 signaling pathway were demonstrated in several studies
performed on tumors (171).
Several genes encoding proteins mediating apoptosis, such as
Bcl2 and Bcl-xl, were identified as target genes for
STAT3 (170,171). Notably, an increase in BCL2
levels is typical for the period between the 2nd and 24th h after
reperfusion in IPostC (166).
IPostC might inhibit cardiomyocyte apoptosis during long-term
reperfusion via regulation of anti-apoptotic factors such as BCL2
(167). A long-term
anti-apoptotic effect of IPostC may be associated with an increase
in BCL2 levels 24 h after reperfusion, which is controlled by
JAK2/STAT3 (167). Moreover, the
PI3K/AKT signaling pathway, regulated by JAK2 signaling, is
necessary for cardioprotection of IPostC (169).
An increase in the expression of AKT and BCL2
proteins is accompanied by inhibition of BAX synthesis, which is a
sign of activation of the PI3K/AKT signaling pathway and inhibition
of cardiomyocyte apoptosis (44).
Activation of this pathway, as the main component of the RISK
pathway, prevented cardiomyocyte apoptosis, protected the
myocardium from MI/RI and plays a critical role in IPostC effects
(172-174). Goodman et al (175) demonstrated that JAK/STAT
signaling may contribute to the initiation of RISK signal
transduction via activation of PI3K/AKT, and JAK/STAT signaling
alone, without subsequent activation of RISK, is not sufficient for
cardioprotection after IPostC. Other studies showed that JAK2
signaling regulated the activation of the PI3K/AKT pathway after
IPostC (169). Blocking the
PI3K/AKT pathway decreased the cardioprotective effects of IPostC
at every timepoint (169).
Activation of the JAK2/STAT3/BCL2 pathway without activation of the
PI3K/AKT pathway may be insufficient for apoptosis limitation
(169).
In recent years, there is an increasing interest in
studying the pharmacological methods of cardioprotection (150). The ultimate objectives of
cardioprotection strategies include molecular targets mainly
involved in signaling pathways of regulated cell death such as ion
channels, proteases, reactive oxygen species, contractile elements
or components of MPTP (141). As
a rule, these strategies are based on existing medicines and they
rarely undergo pre-clinical trials (140). The only exclusion is
cyclosporine A, which is targeted at MPTP. However, cyclosporine A
showed controversial results and failed in clinical trials
(140).
Although pharmacotherapy is not commonly included
in cardioprotective strategies, several investigations have shown
that a number of medicines are capable of cushioning the effects of
MI/RI (176,177,179-182,186-190). The present review briefly
reviews those that promote cell survival and reduce apoptosis by
affecting the BCL2/Bax ratio through specific signaling pathways.
Most of the agents provide pleiotropic effects and activate several
pathways simultaneously, leading to an increase of BCL2 expression
(179-182,186-190). The comparative data on these
medicines is summarized in Table
I.
Metformin, which is widely used for the treatment
of carbohydrate metabolism disorders, inhibits apoptosis in culture
(H9c2 cells) and rat cardiomyocytes following injury caused by
hypoxia-reoxygenation or ischemia-reoxygenation by increasing the
BCL2/BAX ratio with the involvement of metalloreductase STEAP4
(177). These results in
vitro and in vivo affirmed the hypothetical effects of
metformin on MI/RI produced by cellular apoptosis inhibition. The
molecular mechanisms of this anti-apoptotic function of metformin
are still poorly understood, though it was earlier reported that
they include activation of AMPK (178). AMPK is considered to be a key
molecule for cardioprotection based on the modulation of several
signaling pathways involved in glucose metabolism and energy
homeostasis (179). AMPKs are
proteins that promote cell protection in ischemic conditions as the
AMP/ATP ratio indicates intercellular energetic status and is
increased in ischemic tissues (191).
The antidepressant escitalopram was shown to
suppress cardiomyocyte apoptosis in patients with previous
myocardial infarction compared with the controls, which was
accompanied by a decrease in the BAX/BCL2 ratio (181).
Preliminary administration of Ilexsaponin A
increased the levels of anti-apoptotic protein BCL2 and decreased
pro-apoptotic protein BAX. These results confirmed that Ilexsaponin
could suppress cardiomyocyte apoptosis in MI/RI, being a new
potential cardioprotective agent which may be used for MI/RI
treatment (182).
Preliminary introduction of Salvianolic acid (10,
20 or 30 mg/kg/day) effectively decreased myocardial synthesis of
BAX and caspase-3 and increased BCL2 levels (50).
Inhaled administration of sevoflurane (halogenated
anesthetic) inhibited BAX expression and enhanced Bcl2
expression in mice, which was mediated by suppression of
miRNA-135b-5p, whereby drug prevented MI/RI by activating the
JAK2/STAT3 signaling pathway (120).
Postconditioning with dexmedetomidine
(high-selective agonist of α2-adrenoreceptors), which is widely
used in anesthesiology and resuscitation, significantly increased
the BCL2/BAX ratio in the rat myocardium with modeled diabetes
mellitus and MI/RI via the PI3K/AKT/GSK-3β signaling pathway
(183).
Rapamycin (an inhibitor of mTOR) is used for
coating coronary stents containing special drugs to prevent
in-stent restenosis after coronary angioplasty (184,185). Rapamycin induces unique
cardioprotective signal transduction that includes phosphorylation
of ERK, STAT3, eNOS and GSK-3β in association with increased
BCL2/BAX ratio (184).
JAK2/STAT3 signal transduction plays a critical role in
cardioprotection induced by rapamycin, which is associated with an
increase in BCL2/BAX (185).
BCL2 expression was enhanced after STAT3 activation via
ERK-dependent phosphorylation caused by rapamycin administration
(186). Introduction of
rapamycin before reperfusion is a promising method that might be
capable of considerable restriction of the myocardial infarction
area and inhibition of cardiomyocyte apoptosis after MI/RI via
signaling pathways involving MAP kinases and PI3K/AKT (187).
Interestingly, a study showed an evident role of
melatonin in cardioprotection through the enhancement of
Bcl-xl and Bcl2 expression and inhibition of
Bax gene expression by reduction of oxidative stress via the
activation of the NAD-dependent protein deacetylase sirtuin-3
(SIRT3) signaling pathway (188-190). SIRT3 is localized in the
mitochondria and regulates several mitochondrial metabolic pathways
(192). Moreover, during MI/RI
and type 1 diabetes, melatonin significantly inhibited apoptosis by
suppression of caspase-3 and BAX production, cleavage of caspase-3
and an increase in BCL2 levels (192). These effects were also inhibited
by a specific blocker of AMPK signal transduction (compound C)
which determines that this signaling pathway plays a key role in
the cardioprotective action of melatonin (192). Firstly, it was demonstrated that
melatonin treatment is a potential strategy for prevention of MI/RI
injury in cases of type 1 diabetes mellitus as it could enhance
mitochondrial biogenesis and support normal functions of the
mitochondria (192). Secondly,
it was also shown that the AMPK/peroxisome proliferator-activated
receptor γ coactivator 1α/SIRT3 signaling pathway played a key role
in the cardioprotective action of melatonin (193). Melatonin also showed a strong
protective effect via Notch1/Hes1 signal transduction in a
receptor-dependent manner (193). The PTEN/AKT signaling pathway is
a key consequent mediator of BCL2 expression enhancement in rats
(in vivo) and cultivated H9C2 cardiomyocytes (in
vitro) (194).
It is important to note that the mechanisms of
metabolic cardioprotection of most preparations have been poorly
investigated to date (177,180-184). The data of different randomized
controlled trials often do not prove the effectiveness of the
suggested methods (180-184). Clinical data is available for
metformin, rapamycin, dexmedetomidine, berberine and sevoflurane,
but sufficient evidence of effective cardioprotection is still
missing (195). Considerable
appending of new theoretical data is required that would include
information concerning molecular and cellular mechanisms which this
therapy would be targeted at.
In recent years, focus on apoptosis has become a
promising direction in the research of cardiovascular pathology
since there is an opportunity to control this process and to
protect the functional reserve of the myocardium. The studies
mentioned in this review have demonstrated a number of effective
methods for inhibiting cell apoptosis. Conclusions based on these
results, unfortunately, did not lead to a final solution to the
problem of prevention and treatment of MI/RI. There is still a lack
of data to recommend or to introduce these results into clinical
practice. This is predominantly explained by the fact that there is
no consensus for common biological and pathogenetic significance of
BCL2 associated processes: Is it cardioprotective or only a
pathological mechanism leading to cardiomyocyte death and
aggravation of myocardial degradation?
Proteins of the BCL2 family play main roles in
intrinsic apoptosis, and regulation of their activity allows
significantly reduced cell death. In addition to the influence of
BCL2 protein on apoptosis development, it is worth paying attention
to its non-apoptotic functions in MI/RI development. For example,
BCL2 regulation features mitochondrial, nuclear and endoplasmic
reticulum processes (including calcium homeostasis) and glucose and
lipid metabolism (196-198).
The preservation of functionally active
cardiomyocytes is a priority in the development of new algorithms
for MI/RI treatment. A wider research of BCL2 integration into
cellular processes in MI/RI is likely to result in building a more
complete signaling network that can be targeted at for preventing
reperfusion injury of cardiomyocytes.
The publication has been prepared with the support
of the 'RUDN University Program 5-100'.
Not applicable.
AYK and MLB conceptualized the study; AYK and MLB
wrote the original draft; AYK, MLB, EVN, SPS and EA participated in
writing and edited the review; EVN and APS prepared the figures;
SMS edited the manuscript. All authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
We thank Dr Bianca K. Verlinden (Department of
Biochemistry, Centre for Sustainable Malaria Control, Faculty of
Natural and Agricultural Sciences, University of Pretoria,
Pretoria, South Africa) for assisting in English language text
editing.
|
1
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rogalińska M: Alterations in cell nuclei
during apoptosis. Cell Mol Biol Lett. 7:995–1018. 2002.
|
|
3
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the Nomenclature Committee on Cell Death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kalkavan H and Green DR: MOMP, cell
suicide as a BCL-2 family business. Cell Death Differ. 25:46–55.
2018. View Article : Google Scholar
|
|
6
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar
|
|
7
|
Galluzzi L, Kepp O and Kroemer G:
Mitochondrial regulation of cell death: A phylogenetically
conserved control. Microb Cell. 3:101–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moldoveanu T, Follis AV, Kriwacki RW and
Green DR: Many players in BCL-2 family affairs. Trends Biochem Sci.
39:101–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shamas-Din A, Kale J, Leber B and Andrews
DW: Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb
Perspect Biol. 5:a0087142013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hardwick JM, Chen Y and Jonas EA:
Multipolar functions of BCL-2 proteins link energetics to
apoptosis. Trends Cell Biol. 22:318–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pihán P, Carreras-Sureda A and Hetz C:
BCL-2 family: Integrating stress responses at the ER to control
cell demise. Cell Death Differ. 24:1478–1487. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J and Ney PA: Role of BNIP3 and NIX
in cell death, autophagy, and mitophagy. Cell Death Differ.
16:939–946. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zamorano S, Rojas-Rivera D, Lisbona F,
Parra V, Court FA, Villegas R, Cheng EH, Korsmeyer SJ, Lavandero S
and Hetz C: A BAX/BAK and cyclophilin D-independent intrinsic
apoptosis pathway. PLoS One. 7:e377822012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kilbride SM and Prehn JH: Central roles of
apoptotic proteins in mitochondrial function. Oncogene.
32:2703–2711. 2013. View Article : Google Scholar
|
|
15
|
Parsons M and Green D: Mitochondria in
cell death. Essays Biochem. 47:99–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: BCL2/Bax: A rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
17
|
Abbate A, Bussani R, Amin MS, Vetrovec GW
and Baldi A: Acute myocardial infarction and heart failure: Role of
apoptosis. Int J Biochem Cell Biol. 38:1834–1840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahmad F, Lal H, Zhou J, Vagnozzi RJ, Yu
JE, Shang X, Woodgett JR, Gao E and Force T: Cardiomyocyte-specific
deletion of Gsk3α mitigates post-myocardial infarction remodeling,
contractile dysfunction, and heart failure. J Am Coll Cardiol.
64:696–706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chinda K, Sanit J, Chattipakorn S and
Chattipakorn N: Dipeptidyl peptidase-4 inhibitor reduces infarct
size and preserves cardiac function via mitochondrial protection in
ischaemia-reperfusion rat heart. Diab Vasc Dis Res. 11:75–83. 2014.
View Article : Google Scholar
|
|
20
|
Gao CK, Liu H, Cui CJ, Liang ZG, Yao H and
Tian Y: Roles of MicroRNA-195 in cardiomyocyte apoptosis induced by
myocardial ischemia-reperfusion injury. J Genet. 95:99–108. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
World Health Organization (WHO): World
Health Statistics 2019: Monitoring health for the SDGs. WHO;
Geneva: 2019
|
|
22
|
Rajaleid K, Janszky I and Hallqvist J:
Small birth size, adult over-weight, and risk of acute myocardial
infraction. Epidemiology. 22:138–147. 2011. View Article : Google Scholar
|
|
23
|
Minamino T: Cardioprotection from
ischemia/reperfusion injury: Basic and translational research. Circ
J. 76:1074–1082. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Z, Lu J, Luo Y, Li S and Chen M: High
association between human circulating microRNA-497 and acute
myocardial infarction. ScientificWorldJournal.
2014:9318452014.PubMed/NCBI
|
|
25
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
27
|
Bak MI and Ingwall JS: Contribution of
Na+/H+ exchange to Na+ overload in the ischemic hypertrophied
hyperthyroid rat heart. Cardiovasc Res. 57:1004–1014. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Budhram-Mahadeo V, Fujita R, Bitsi S,
Sicard P and Heads R: Co-expression of POU4F2/Brn-3b with p53 may
be important for controlling expression of pro-apoptotic genes in
cardiomyocytes following ischaemic/hypoxic insults. Cell Death Dis.
5:e15032014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fröhlich GM, Meier P, White SK, Yellon DM
and Hausenloy DJ: Myocardial reperfusion injury: Looking beyond
primary PCI. Eur Heart J. 34:1714–1722. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen S, Hua F, Lu J, Jiang Y, Tang Y, Tao
L, Zou B and Wu Q: Effect of dexmedetomidine on myocardial
ischemia-reperfusion injury. Int J Clin Exp Med. 8:21166–21172.
2015.
|
|
32
|
Moens AL, Claeys MJ, Timmermans JP and
Vrints CJ: Myocardial ischemia/reperfusion-injury, a clinical view
on a complex pathophysiological process. Int J Cardiol.
100:179–190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu LF, Liang Z, Lv ZR, Liu XH, Bai J,
Chen J, Chen C and Wang Y: MicroRNA-15a/b are up-regulated in
response to myocardial ischemia/reperfusion injury. J Geriatr
Cardiol. 9:28–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown DI and Griendling KK: Regulation of
signal transduction by reactive oxygen species in the
cardiovascular system. Circ Res. 116:531–549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Ischemia/reperfusion. Compr Physiol. 7:113–170. 2016.
View Article : Google Scholar
|
|
36
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017:70183932017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Ha T, Zou J, Ren D, Liu L, Zhang
X, Kalbfleisch J, Gao X, Williams D and Li C: MicroRNA-125b
protects against myocardial ischaemia/reperfusion injury via
targeting p53-medi-ated apoptotic signalling and TRAF6. Cardiovasc
Res. 102:385–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
LeBaron TW, Kura B, Kalocayova B,
Tribulova N and Slezak J: A new approach for the prevention and
treatment of cardiovascular disorders. Molecular hydrogen
significantly reduces the effects of oxidative stress. Molecules.
24:20762019. View Article : Google Scholar :
|
|
39
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Radomski MW, Palmer RM and Moncada S:
Endogenous nitric oxide inhibits human platelet adhesion to
vascular endothelium. Lancet. 2:1057–1058. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xia Y and Zweier JL: Substrate control of
free radical generation from xanthine oxidase in the postischemic
heart. J Biol Chem. 270:18797–18803. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang WP, Zong QF, Gao Q, Yu Y, Gu XY,
Wang Y, Li ZH and Ge M: Effects of endomorphin-1 postconditioning
on myocardial ischemia/reperfusion injury and myocardial cell
apoptosis in a rat model. Mol Med Rep. 14:3992–3998. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu L, Zhang G, Liang Z, Liu X, Li T, Fan
J, Bai J and Wang Y: MicroRNA-15b enhances
hypoxia/reoxygenation-induced apoptosis of cardiomyocytes via a
mitochondrial apoptotic pathway. Apoptosis. 19:19–29. 2014.
View Article : Google Scholar
|
|
44
|
Li CM, Shen SW, Wang T and Zhang XH:
Myocardial ischemic post-conditioning attenuates ischemia
reperfusion injury via PTEN/Akt signal pathway. Int J Clin Exp Med.
8:15801–15807. 2015.PubMed/NCBI
|
|
45
|
Liou SF, Ke HJ, Hsu JH, Liang JC, Lin HH,
Chen IJ and Yeh JL: San-Huang-Xie-Xin-Tang prevents rat hearts from
ischemia/reperfusion-induced apoptosis through eNOS and MAPK
pathways. Evid Based Complement Alternat Med. 2011:9150512011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Q and Lesnefsky EJ: Blockade of
electron transport during ischemia preserves bcl-2 and inhibits
opening of the mitochondrial permeability transition pore. FEBS
Lett. 585:921–926. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen Q, Xu H, Xu A, Ross T, Bowler E, Hu Y
and Lesnefsky EJ: Inhibition of Bcl-2 sensitizes mitochondrial
permeability transition pore (MPTP) opening in ischemia-damaged
mitochondria. PLoS One. 10:e01188342015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gustafsson AB and Gottlieb RA: Bcl-2
family members and apoptosis, taken to heart. Am J Physiol Cell
Physiol. 292:C45–C51. 2007. View Article : Google Scholar
|
|
49
|
Murphy E, Imahashi K and Steenbergen C:
Bcl-2 regulation of mitochondrial energetics. Trends Cardiovasc
Med. 15:283–290. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qiao Z and Xu Y: Salvianolic acid b
alleviating myocardium injury in ischemia reperfusion rats. Afr J
Tradit Complement Altern Med. 13:157–161. 2016. View Article : Google Scholar
|
|
51
|
Zhang HY, McPherson BC, Liu H, Baman TS,
Rock P and Yao Z: H(2)O(2) opens mitochondrial K(ATP) channels and
inhibits GABA receptors via protein kinase C-epsilon in
cardiomyocytes. Am J Physiol Heart Circ Physiol. 282:H1395–H1403.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meng G, Wang J, Xiao Y, Bai W, Xie L, Shan
L, Moore PK and Ji Y: GYY4137 protects against myocardial ischemia
and reperfusion injury by attenuating oxidative stress and
apoptosis in rats. J Biomed Res. 29:203–213. 2015.PubMed/NCBI
|
|
53
|
Song T, Wang P, Yu X, Wang A, Chai G, Fan
Y and Zhang Z: Systems analysis of phosphorylation-regulated Bcl-2
interactions establishes a model to reconcile the controversy over
the significance of Bcl-2 phosphorylation. Br J Pharmacol.
176:491–504. 2019. View Article : Google Scholar
|
|
54
|
Markou T, Dowling AA, Kelly T and Lazou A:
Regulation of Bcl-2 phosphorylation in response to oxidative stress
in cardiac myocytes. Free Radic Res. 43:809–816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Syeda MZ, Fasae MB, Yue E, Ishimwe AP,
Jiang Y, Du Z, Yang B and Bai Y: Anthocyanidin attenuates
myocardial ischemia induced injury via inhibition of ROS-JNK-BCL2
pathway: New mechanism of anthocyanidin action. Phytother Res.
33:3129–3139. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Z, Deng X, Liu Y, Liu Y, Sun L and
Chen F: PKM2, function and expression and regulation. Cell Biosci.
9:522019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Menon MB and Dhamija S: Beclin 1
Phosphorylation-at the center of autophagy regulation. Front Cell
Dev Biol. 6:1372018. View Article : Google Scholar
|
|
58
|
Wei Y, Sinha S and Levine B: Dual role of
JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis
regulation. Autophagy. 4:949–951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schulman D, Latchman DS and Yellon DM:
Urocortin protects the heart from reperfusion injury via
upregulation of p42/p44 MAPK signaling pathway. Am J Physiol Heart
Circ Physiol. 283:H1481–H1488. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hausenloy DJ, Tsang A, Mocanu MM and
Yellon DM: Ischemic preconditioning protects by activating
prosurvival kinases at reperfusion. Am J Physiol Heart Circ
Physiol. 288:H971–H976. 2005. View Article : Google Scholar
|
|
61
|
Carter AN, Born HA, Levine AT, Dao AT,
Zhao AJ, Lee WL and Anderson AE: Wortmannin attenuates
seizure-induced hyperactive PI3K/Akt/mTOR signaling, impaired
memory, and spine dysmorphology in rats. eNeuro. 4. pp.
ENEURO.0354–16.2017. 2017, View Article : Google Scholar
|
|
62
|
Very N, Vercoutter-Edouart AS, Lefebvre T,
Hardivillé S and El Yazidi-Belkoura I: Cross-dysregulation of
O-GlcNAcylation and PI3K/AKT/mTOR axis in human chronic diseases.
Front Endocrinol (Lausanne). 9:6022018. View Article : Google Scholar
|
|
63
|
Chi Y, Ma Q, Ding XQ, Qin X, Wang C and
Zhang J: Research on protective mechanism of ibuprofen in
myocardial ischemia-reperfusion injury in rats through the
PI3K/Akt/mTOR signaling pathway. Eur Rev Med Pharmacol Sci.
23:4465–4473. 2019.PubMed/NCBI
|
|
64
|
Liang K, Ye Y, Wang Y, Zhang J and Li C:
Formononetin mediates neuroprotection against cerebral
ischemia/reperfusion in rats via downregulation of the Bax/BCL2
ratio and upregulation PI3K/Akt signaling pathway. J Neurol Sci.
344:100–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yu LN, Yu J, Zhang FJ, Yang MJ, Ding TT,
Wang JK, He W, Fang T, Chen G and Yan M: Sevoflurane
postconditioning reduces myocardial reperfusion injury in rat
isolated hearts via activation of PI3K/Akt signaling and modulation
of Bcl-2 family proteins. J Zhejiang Univ Sci B. 11:661–672. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang J, Wang C, Yu S, Luo Z, Chen Y, Liu
Q, Hua F, Xu G and Yu P: Sevoflurane postconditioning protects rat
hearts against ischemia-reperfusion injury via the activation of
PI3K/AKT/mTOR signaling. Sci Rep. 4:73172014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Keyes KT, Xu J, Long B, Zhang C, Hu Z and
Ye Y: Pharmacological inhibition of PTEN limits myocardial infarct
size and improves left ventricular function postinfarction. Am J
Physiol Heart Circ Physiol. 298:H1198–H1208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zu L, Zheng X, Wang B, Parajuli N,
Steenbergen C, Becker LC and Cai ZP: Ischemic preconditioning
attenuates mitochondrial localization of PTEN induced by
ischemia-reperfusion. Am J Physiol Heart Circ Physiol.
300:H2177–H2186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhu YB, Ding N, Yi HL and Li ZQ: The
expression of overexpressed PTEN enhanced IR-induced apoptosis of
myocardial cells. Eur Rev Med Pharmacol Sci. 23:4406–4413.
2019.PubMed/NCBI
|
|
70
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hernández-Reséndiz S, Roldán FJ, Correa F,
Martínez-Abundis E, Osorio-Valencia G, Ruíz-de-Jesús O,
Alexánderson-Rosas E, Vigueras RM, Franco M and Zazueta C:
Postconditioning protects against reperfusion injury in
hypertensive dilated cardiomyopathy by activating MEK/ERK1/2
signaling. J Card Fail. 19:135–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Holderfield M, Deuker MM, McCormick F and
McMahon M: Targeting RAF kinases for cancer therapy: BRAF-mutated
melanoma and beyond. Nat Rev Cancer. 14:455–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Balmanno K and Cook SJ: Tumour cell
survival signalling by the ERK1/2 pathway. Cell Death Differ.
16:368–377. 2009. View Article : Google Scholar
|
|
74
|
Zhou QL, Teng F, Zhang YS, Sun Q, Cao YX
and Meng GW: FPR1 gene silencing suppresses cardiomyocyte apoptosis
and ventricular remodeling in rats with ischemia/reperfusion injury
through the inhibition of MAPK signaling pathway. Exp Cell Res.
370:506–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sun G, Ye N, Dai D, Chen Y, Li C and Sun
Y: The protective role of the TOPK/PBK pathway in myocardial
ischemia/reperfusion and H2O2-induced injury
in H9C2 cardiomyocytes. Int J Mol Sci. 17:2672016. View Article : Google Scholar
|
|
76
|
Sun MH, Chen XC, Han M, Yang YN, Gao XM,
Ma X, Huang Y, Li XM, Gai MT, Liu F, et al: Cardioprotective
effects of constitutively active MEK1 against
H2O2-induced apoptosis and autophagy in
cardiomyocytes via the ERK1/2 signaling pathway. Biochem Biophys
Res Commun. 512:125–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lee ML, Sulistyowati E, Hsu JH, Huang BY,
Dai ZK, Wu BN, Chao YY and Yeh JL: KMUP-1 ameliorates
ischemia-induced cardiomyocyte apoptosis through the
NO−cGMP−MAPK signaling pathways. Molecules.
24:13762019. View Article : Google Scholar
|
|
78
|
Razavi HM, Hamilton JA and Feng Q:
Modulation of apoptosis by nitric oxide: Implications in myocardial
ischemia and heart failure. Pharmacol Ther. 106:147–162. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Burley DS, Ferdinandy P and Baxter GF:
Cyclic GMP and protein kinase-G in myocardial
ischaemia-reperfusion: Opportunities and obstacles for survival
signaling. Br J Pharmacol. 152:855–869. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shvedova M, Anfinogenova Y,
Atochina-Vasserman EN, Schepetkin IA and Atochin DN: c-Jun
N-Terminal Kinases (JNKs) in myocardial and cerebral
ischemia/reperfusion injury. Front Pharmacol. 9:7152018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang W, Zhang Y, Ding K, Zhang H, Zhao Q,
Liu Z and Xu Y: Involvement of JNK1/2-NF-κBp65 in the regulation of
HMGB2 in myocardial ischemia/reperfusion-induced apoptosis in human
AC16 cardiomyocytes. Biomed Pharmacother. 106:1063–1071. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang Z, Huang H, He W, Kong B, Hu H, Fan
Y, Liao J, Wang L, Mei Y, Liu W, et al: Regulator of G-protein
signaling 5 protects cardiomyocytes against apoptosis during in
vitro cardiac ischemia-reperfusion in mice by inhibiting both
JNK1/2 and P38 signaling pathways. Biochem Biophys Res Commun.
473:551–557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen Q, Xu T, Li D, Pan D, Wu P, Luo Y, Ma
Y and Liu Y: JNK/PI3K/Akt signaling pathway is involved in
myocardial ischemia/reperfusion injury in diabetic rats: Effects of
salvianolic acid A intervention. Am J Transl Res. 8:2534–2548.
2016.PubMed/NCBI
|
|
84
|
Frias MA and Montessuit C: JAK-STAT
signaling and myocardial glucose metabolism. JAKSTAT.
2:e264582013.
|
|
85
|
Harhous Z, Booz GW, Ovize M, Bidaux G and
Kurdi M: An update on the multifaceted roles of STAT3 in the heart.
Front Cardiovasc Med. 6:1502019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang WY, Zhang QL and Xu MJ: Effects of
propofol on myocardial ischemia reperfusion injury through
inhibiting the JAK/STAT pathway. Eur Rev Med Pharmacol Sci.
23:6339–6345. 2019.PubMed/NCBI
|
|
87
|
Bolli R, Dawn B and Xuan YT: Emerging role
of the JAK-STAT pathway as a mechanism of protection against
ischemia/reperfusion injury. J Mol Cell Cardiol. 33:1893–1896.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu Y, Che G, Di Z, Sun W, Tian J and Ren
M: Calycosin-7-O- β-D-glucoside attenuates myocardial
ischemia-reperfusion injury by activating JAK2/STAT3 signaling
pathway via the regulation of IL-10 secretion in mice. Mol Cell
Biochem. 463:175–187. 2020. View Article : Google Scholar
|
|
89
|
Bolli R, Dawn B and Xuan YT: Role of the
JAK-STAT pathway in protection against myocardial
ischemia/reperfusion injury. Trends Cardiovasc Med. 13:72–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Luan HF, Zhao ZB, Zhao QH, Zhu P, Xiu MY
and Ji Y: Hydrogen sulfide postconditioning protects isolated rat
hearts against ischemia and reperfusion injury mediated by the
JAK2/STAT3 survival pathway. Braz J Med Biol Res. 45:898–905. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Boengler K, Buechert A, Heinen Y, Roeskes
C, Hilfiker-Kleiner D, Heusch G and Schulz R: Cardioprotection by
ischemic post-conditioning is lost in aged and STAT3-deficient
mice. Circ Res. 102:131–135. 2008. View Article : Google Scholar
|
|
92
|
Lecour S: Activation of the protective
Survivor Activating Factor Enhancement (SAFE) pathway against
reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell
Cardiol. 47:32–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Myers MG Jr: Cell biology. Moonlighting in
mitochondria. Science. 323:723–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Suleman N, Somers S, Smith R, Opie LH and
Lecour SC: Dual activation of STAT-3 and Akt is required during the
trigger phase of ischaemic preconditioning. Cardiovasc Res.
79:127–133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Boengler K, Hilfiker-Kleiner D, Drexler H,
Heusch G and Schulz R: The myocardial JAK/STAT pathway: From
protection to failure. Pharmacol Ther. 120:172–185. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bolli R, Stein AB, Guo Y, Wang OL, Rokosh
G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y and Xuan YT: A
murine model of inducible, cardiac-specific deletion of STAT3: Its
use to determine the role of STAT3 in the upregulation of
cardioprotective proteins by ischemic preconditioning. J Mol Cell
Cardiol. 50:589–597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hattori R, Maulik N, Otani H, Zhu L,
Cordis G, Engelman RM, Siddiqui MA and Das DK: Role of STAT3 in
ischemic preconditioning. J Mol Cell Cardiol. 33:1929–1936. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shen P, Chen J and Pan M: The protective
effects of total paeony glycoside on ischemia/reperfusion injury in
H9C2 cells via inhibition of the PI3K/Akt signaling pathway. Mol
Med Rep. 18:3332–3340. 2018.PubMed/NCBI
|
|
99
|
Koeppen M, Lee JW, Seo SW, Brodsky KS,
Kreth S, Yang IV, Buttrick PM, Eckle T and Eltzschig HK:
Hypoxia-inducible factor 2-alpha-dependent induction of
amphiregulin dampens myocardial ischemia-reperfusion injury. Nat
Commun. 9:8162018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li T, Yu J, Chen R, Wu J, Fei J, Bo Q, Xue
L and Li D: Mycophenolate mofetil attenuates myocardial
ischemia-reperfusion injury via regulation of the TLR4/NF-κB
signaling pathway. Pharmazie. 69:850–855. 2014.
|
|
101
|
Lin J, Wang H, Li J, Wang Q, Zhang S, Feng
N, Fan R and Pei J: κ-Opioid receptor stimulation modulates
TLR4/NF-κB signaling in the rat heart subjected to
ischemia-reperfusion. Cytokine. 61:842–848. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li J, Xie C, Zhuang J, Li H, Yao Y, Shao C
and Wang H: Resveratrol attenuates inflammation in the rat heart
subjected to ischemia-reperfusion: Role of the TLR4/NF-κB signaling
pathway. Mol Med Rep. 11:1120–1126. 2015.
|
|
103
|
Gao Y, Song G, Cao YJ, Yan KP, Li B, Zhu
XF, Wang YP, Xing ZY, Cui L, Wang XX and Zhu MJ: The Guizhi Gancao
Decoction attenuates myocardial ischemia-reperfusion injury by
suppressing inflammation and cardiomyocyte apoptosis. Evid Based
Complement Alternat Med. 2019:19474652019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang N, Ye F, Zhu W, Hu D, Xiao C, Nan J,
Su S, Wang Y, Liu M, Gao K, et al: Cardiac ankyrin repeat protein
attenuates cardiomyocyte apoptosis by upregulation of Bcl-2
expression. Biochim Biophys Acta. 1863:3040–3049. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ibrahim A: Inhibition of α-SMA, Bax and
increase of BCL2 expression in myocardiocytes as response to
chitosan administration to hypercholesterolemic rats. World J Pharm
Pharmac Sci. 5:164–176. 2016.
|
|
106
|
Guo J, Li HZ, Wang LC, Zhang WH, Li GW,
Xing WJ, Wang R and Xu CQ: Increased expression of calcium-sensing
receptors in atherosclerosis confers hypersensitivity to acute
myocardial infarction in rats. Mol Cell Biochem. 366:345–354. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kuo WW, Hsu TC, Chain MH, Lai CH, Wang WH,
Tsai FJ, Tsai CH, Wu CH, Huang CY and Tzang BS: Attenuated cardiac
mitochondrial-dependent apoptotic effects by li-fu formula in
hamsters fed with a hypercholesterol diet. Evid Based Complement
Alternat Med. 2011:5303452011. View Article : Google Scholar :
|
|
108
|
Latif N, Khan MA, Birks E, O'Farrell A,
Westbrook J, Dunn MJ and Yacoub MH: Upregulation of the Bcl-2
family of proteins in end stage heart failure. J Am Coll Cardiol.
35:1769–1777. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang TD, Chen WJ, Su SS, Lo SC, Lin WW and
Lee YT: Increased cardiomyocyte apoptosis following ischemia and
reperfusion in diet-induced hypercholesterolemia: Relation to Bcl-2
and Bax proteins and caspase-3 activity. Lipids. 37:385–394. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ye Y, Perez-Polo JR, Qian J and Birnbaum
Y: The role of microRNA in modulating myocardial
ischemia-reperfusion injury. Physiol Genomics. 43:534–542. 2011.
View Article : Google Scholar
|
|
111
|
Tang R, Long T, Lui KO, Chen Y and Huang
ZP: A roadmap for fixing the heart: RNA regulatory networks in
cardiac disease. Mol Ther Nucleic Acids. 20:673–686. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kukreja RC, Yin C and Salloum FN:
MicroRNAs: New players in cardiac injury and protection. Mol
Pharmacol. 80:558–564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Çakmak HA and Demir M: MicroRNA and
cardiovascular diseases. Balkan Med J. 37:60–71. 2020.PubMed/NCBI
|
|
114
|
Peterson SM, Thompson JA, Ufkin ML,
Sathyanarayana P, Liaw L and Congdon CB: Common features of
microRNA target prediction tools. Front Genet. 5:232014. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Cao L, Wang J and Wang PQ: MiR-326 is a
diagnostic biomarker and regulates cell survival and apoptosis by
targeting Bcl-2 in osteosarcoma. Biomed Pharmacother. 84:828–835.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cheng Y, Tan N, Yang J, Liu X, Cao X, He
P, Dong X, Qin S and Zhang C: A translational study of circulating
cell-free microRNA-1 in acute myocardial infarction. Clin Sci
(Lond). 119:87–95. 2010. View Article : Google Scholar
|
|
117
|
Dehaini H, Awada H, El-Yazbi A, Zouein FA,
Issa K, Eid AA, Ibrahim M, Badran A, Baydoun E, Pintus G and Eid
AH: MicroRNAs as potential pharmaco-targets in ischemia-reperfusion
injury compounded by diabetes. Cells. 8:1522019. View Article : Google Scholar :
|
|
118
|
Yan H, Li Y, Wang C, Zhang Y, Liu C, Zhou
K and Hua Y: Contrary microRNA expression pattern between fetal and
adult cardiac remodeling: Therapeutic value for heart failure.
Cardiovasc Toxicol. 17:267–276. 2017. View Article : Google Scholar
|
|
119
|
Tang Y, Zheng J, Sun Y, Wu Z, Liu Z and
Huang G: MicroRNA-1 regulates cardiomyocyte apoptosis by targeting
Bcl-2. Int Heart J. 50:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Xie XJ, Fan DM, Xi K, Chen YW, Qi PW, Li
QH, Fang L and Ma LG: Suppression of microRNA-135b-5p protects
against myocardial ischemia/reperfusion injury by activating
JAK2/STAT3 signaling pathway in mice during sevoflurane anesthesia.
Biosci Rep. 37:BSR201701862017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hullinger TG, Montgomery RL, Seto AG,
Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C,
Latimer PA, et al: Inhibition of miR-15 protects against cardiac
ischemic injury. Circ Res. 110:71–81. 2012. View Article : Google Scholar :
|
|
122
|
Liu X, Nie J and Li C: Targeted regulation
of Bcl 2 by miR-16 for cardiomyocyte apoptosis after cardiac
infarction. Int J Clin Exp Pathol. 10:4626–4632. 2017.
|
|
123
|
Yang W, Yang Y, Xia L, Yang Y, Wang F,
Song M, Chen X, Liu J, Song Y, Zhao Y and Yang C: MiR-221 promotes
Capan-2 pancreatic ductal adenocarcinoma cells proliferation by
targeting PTEN-Akt. Cell Physiol Biochem. 38:2366–2374. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ye Z, Hao R, Cai Y, Wang X and Huang G:
Knockdown of miR-221 promotes the cisplatin-inducing apoptosis by
targeting the BIM-Bax/Bak axis in breast cancer. Tumour Biol.
37:4509–4515. 2016. View Article : Google Scholar
|
|
125
|
Kong QR, Ji DM, Li FR, Sun HY and Wang QX:
MicroRNA-221 promotes myocardial apoptosis caused by myocardial
ischemia-reperfusion by down-regulating PTEN. Eur Rev Med Pharmacol
Sci. 23:3967–3975. 2019.PubMed/NCBI
|
|
126
|
Li X, Zeng Z, Li Q, Xu Q, Xie J, Hao H,
Luo G, Liao W, Bin J, Huang X and Liao Y: Inhibition of
microRNA-497 ameliorates anoxia/reoxygenation injury in
cardiomyocytes by suppressing cell apoptosis and enhancing
autophagy. Oncotarget. 6:18829–18844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yang Q, Yang K and Li A: microRNA-21
protects against ischemia-reperfusion and
hypoxia-reperfusion-induced cardiocyte apoptosis via the
phosphatase and tensin homolog/Akt-dependent mechanism. Mol Med
Rep. 9:2213–2220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fan ZX and Yang J: The role of microRNAs
in regulating myocardial ischemia reperfusion injury. Saudi Med J.
36:787–793. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yang J, Chen L, Yang J, Ding J, Li S, Wu
H, Zhang J, Fan Z, Dong W and Li X: MicroRNA-22 targeting CBP
protects against myocardial ischemia-reperfusion injury through
anti-apoptosis in rats. Mol Biol Rep. 41:555–561. 2014. View Article : Google Scholar
|
|
130
|
Yang J, Fan Z, Yang J, Ding J, Yang C and
Chen L: microRNA-22 attenuates myocardial ischemia-reperfusion
injury via an anti-inflammatory mechanism in rats. Exp Ther Med.
12:3249–3255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Liu S, Yang Y, Song YQ, Geng J and Chen
QL: Protective effects of N(2)-L-alanyl-L-glutamine mediated by the
JAK2/STAT3 signaling pathway on myocardial ischemia reperfusion.
Mol Med Rep. 17:5102–5108. 2018.PubMed/NCBI
|
|
132
|
Xu D, Li H, Zhao Y and Wang C:
Downregulation of miR-34 a attenuates myocardial
ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis.
Int J Clin Exp Pathol. 10:3865–3875. 2017.
|
|
133
|
Kikuchi K and Poss KD: Cardiac
regenerative capacity and mechanisms. Annu Rev Cell Dev Biol.
28:719–741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Biala AK and Kirshenbaum LA: The interplay
between cell death signaling pathways in the heart. Trends
Cardiovasc Med. 24:325–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
McCully JD, Wakiyama H, Hsieh YJ, Jones M
and Levitsky S: Differential contribution of necrosis and apoptosis
in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ
Physiol. 286:H1923–H1935. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Mocanu MM, Baxter GF and Yellon DM:
Caspase inhibition and limitation of myocardial infarct size:
Protection against lethal reperfusion injury. Br J Pharmacol.
130:197–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Baxter G, Mocanu M, Brar B, Latchman D and
Yellon D: Cardioprotective effects of transforming growth
factor-beta1 during early reoxygenation or reperfusion are mediated
by p42/p44 MAPK. J Cardiovasc Pharmacol. 38:930–939. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yellon DM and Baxter GF: Reperfusion
injury revisited: Is there a role for growth factor signaling in
limiting lethal reperfusion injury? Trends Cardiovasc Med.
9:245–249. 1999. View Article : Google Scholar
|
|
139
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Soares ROS, Losada DM, Jordani MC, Évora P
and Castro-E-Silva O: Ischemia/reperfusion injury revisited: An
overview of the latest pharmacological strategies. Int J Mol Sci.
20:50342019. View Article : Google Scholar :
|
|
141
|
Euler G: Good and bad sides of
TGFβ-signaling in myocardial infarction. Front Physiol. 6:662015.
View Article : Google Scholar
|
|
142
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Mykytenko J, Kerendi F, Reeves JG, Kin H,
Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J and Zhao ZQ:
Long-term inhibition of myocardial infarction by postconditioning
during reperfusion. Basic Res Cardiol. 102:90–100. 2007. View Article : Google Scholar
|
|
144
|
Argaud L, Gateau-Roesch O, Raisky O,
Loufouat J, Robert D and Ovize M: Postconditioning inhibits
mitochondrial permeability transition. Circulation. 111:194–197.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Pagliaro P, Femminò S, Popara J and Penna
C: Mitochondria in cardiac postconditioning. Front Physiol.
9:2872018. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Heusch G: Critical issues for the
translation of cardioprotection. Circ Res. 120:1477–1486. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Heusch G: Cardioprotection research must
leave its comfort zone. Eur Heart J. 39:3393–3395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Hausenloy DJ, Botker HE, Engstrom T,
Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM and
Garcia-Dorado D: Targeting reperfusion injury in patients with
ST-segment elevation myocardial infarction: Trials and
tribulations. Eur Heart J. 38:935–941. 2017.
|
|
149
|
Ferdinandy P, Hausenloy DJ, Heusch G,
Baxter GF and Schulz R: Interaction of risk factors, comorbidities,
and comedications with ischemia/reperfusion injury and
cardioprotection by preconditioning, postconditioning, and remote
conditioning. Pharmacol Rev. 66:1142–1174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Hausenloy DJ, Garcia-Dorado D, Bøtker HE,
Davidson SM, Downey J, Engel FB, Jennings R, Lecour S, Leor J,
Madonna R, et al: Novel targets and future strategies for acute
cardioprotection: Position paper of the European Society of
Cardiology Working Group on Cellular Biology of the Heart.
Cardiovasc Res. 113:564–585. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Inserte J, Hernando V, Vilardosa Ú, Abad
E, Poncelas-Nozal M and Garcia/Dorado D: Activation of cGMP/protein
kinase G pathway in postconditioned myocardium depends on reduced
oxidative Stress and preserved endothelial nitric oxide synthase
coupling. J Am Heart Assoc. 2:e0059752013. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kleinbongard P, Skyschally A and Heusch G:
Cardioprotection by remote ischemic conditioning and its signal
transduction. Pflugers Arch. 469:159–181. 2017. View Article : Google Scholar
|
|
153
|
Heusch G: Molecular basis of
cardioprotection: Signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wever KE, Hooijmans CR, Riksen NP,
Sterenborg TB, Sena ES, Ritskes-Hoitinga M and Warlé MC:
Determinants of the efficacy of cardiac ischemic preconditioning: A
systematic review and meta-analysis of animal studies. PLoS One.
10:e01420212015. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Wever KE, Menting TP, Rovers M, van der
Vliet JA, Rongen GA, Masereeuw R, Ritskes-Hoitinga M, Hooijmans CR
and Warlé M: Ischemic preconditioning in the animal kidney, a
systematic review and meta-analysis. PLoS One. 7:e322962012.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Yellon DM, Alkhulaifi AM and Pugsley WB:
Preconditioning the human myocardium. Lancet. 342:276–277. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Rossello X and Yellon DM: The RISK pathway
and beyond. Basic Res Cardiol. 113:22017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Hausenloy DJ, Barrabes JA, Bøtker HE,
Davidson SM, Di Lisa F, Downey J, Engstrom T, Ferdinandy P,
Carbrera-Fuentes HA, Heusch G, et al: Ischaemic conditioning and
targeting reperfusion injury: A 30 year voyage of discovery. Basic
Res Cardiol. 111:702016. View Article : Google Scholar
|
|
160
|
Sun XJ and Mao JR: Role of Janus kinase
2/signal transducer and activator of transcription 3 signaling
pathway in cardioprotection of exercise preconditioning. Eur Rev
Med Pharmacol Sci. 22:4975–4986. 2018.PubMed/NCBI
|
|
161
|
Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou
J, Zhang J, Jin Z and Yi D: The effects of curcumin post-treatment
against myocardial ischemia and reperfusion by activation of the
JAK2/STAT3 signaling pathway. Basic Res Cardiol. 107:2632012.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Goodman MD, Koch SE, Afzal MR and Butler
KL: STAT subtype specificity and ischemic preconditioning in mice:
Is STAT-3 enough? Am J Physiol Heart Circ Physiol. 300:H522–H526.
2011. View Article : Google Scholar :
|
|
163
|
Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein
AB, Hunt G, Wu WJ, Tan W and Bolli R: IL-6 plays an obligatory role
in late preconditioning via JAK-STAT signaling and upregulation of
iNOS and COX-2. Cardiovasc Res. 64:61–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G,
Messing RO and Bolli R: Role of the protein kinase
C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the
activation of signal transducers and activators of transcription 1
and 3 and induction of cyclooxygenase-2 after ischemic
preconditioning. Circulation. 112:1971–1978. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Chen TI, Shen YJ, Wang IC and Yang KT:
Short-term exercise provides left ventricular myocardial protection
against intermit-tent hypoxia-induced apoptosis in rats. Eur J Appl
Physiol. 111:1939–1950. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F,
Wang NP, Guyton RA and Vinten-Johansen J: Inhibition of myocardial
injury by ischemic postconditioning during reperfusion: Comparison
with ischemic preconditioning. Am J Physiol Heart Circ Physiol.
285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Thibault H, Piot C, Staat P, Bontemps L,
Sportouch C, Rioufol G, Cung TT, Bonnefoy E, Angoulvant D, Aupetit
JF, et al: Long-term benefit of postconditioning. Circulation.
117:1037–1044. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhao CM, Yang XJ, Yang JH, Cheng XJ, Zhao
X, Zhou BY, Xu SD and Wang HF: Effect of ischaemic postconditioning
on recovery of left ventricular contractile function after acute
myocardial infarction. J Int Med Res. 40:1082–1088. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Tian Y, Zhang W, Xia D, Modi P, Liang D
and Wei M: Postconditioning inhibits myocardial apoptosis during
prolonged reperfusion via a AK2-STAT3-BCL2 pathway. J Biomed Sci.
18:532011. View Article : Google Scholar
|
|
170
|
Shyu WC, Lin SZ, Chiang MF, Chen DC, Su
CY, Wang HJ, Liu RS, Tsai CH and Li H: Secretoneurin promotes
neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway
in murine models of stroke. J Clin Invest. 118:133–148. 2008.
View Article : Google Scholar
|
|
171
|
Siddiquee K, Zhang S, Guida WC, Blaskovich
MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence
NJ, et al: Selective chemical probe inhibitor of Stat3, identified
through structure-based virtual screening, induces antitumor
activity. Proc Natl Acad Sci USA. 104:7391–7396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Hausenloy DJ, Tsang A and Yellon DM: The
reperfusion injury salvage kinase pathway: A common target for both
ischemic preconditioning and postconditioning. Trends Cardiovasc
Med. 15:69–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Jin YC, Lee YS, Kim YM, Seo HG, Lee JH,
Kim HJ, Yun-Choi HS and Chang KC:
(S)-1-(alpha-naphthylmethyl)-6,7-di
hydroxy-1,2,3,4-tetrahydroisoquinoline (CKD712) reduces rat
myocardial apoptosis against ischemia and reperfusion injury by
activation of phosphatidylinositol 3-kinase/Akt signaling and
anti-inflammatory action in vivo. J Pharmacol Exp Ther.
330:440–448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Takahama H, Minamino T, Hirata A, Ogai A,
Asanuma H, Fujita M, Wa keno M, Tsukamoto O, Okada K, Komamura K,
et al: Granulocyte colony-stimulating factor mediates
cardioprotection against ischemia/reperfusion injury via
phosphatidylinositol-3-kinase/Akt pathway in canine hearts.
Cardiovasc Drugs Ther. 20:159–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Goodman MD, Koch SE, Fuller-Bicer GA and
Butler KL: Regulating RISK: A role for JAK-STAT signaling in
postconditioning? Am J Physiol Heart Circ Physiol. 295:H1649–H1656.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Wu N, Zhang X, Jia P and Jia D:
Hypercholesterolemia aggravates myocardial ischemia reperfusion
injury via activating endoplasmic reticulum stress-mediated
apoptosis. Exp Mol Pathol. 99:449–454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Luo T, Zeng X, Yang W and Zhang Y:
Treatment with metformin prevents myocardial ischemia-reperfusion
injury via STEAP4 signaling pathway. Anatol J Cardiol. 21:261–271.
2019.PubMed/NCBI
|
|
178
|
Hu M, Ye P, Liao H, Chen M and Yang F:
Metformin protects H9C2 cardiomyocytes from high-glucose and
hypoxia/reoxygenation injury via inhibition of reactive oxygen
species generation and inflammatory responses: Role of AMPK and
JNK. J Diabetes Res. 2016:29619542016. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Wang Y, Liu J, Ma A and Chen Y:
Cardioprotective effect of berberine against myocardial
ischemia/reperfusion injury via attenuating mitochondrial
dysfunction and apoptosis. Int J Clin Exp Med. 8:14513–14519.
2015.PubMed/NCBI
|
|
181
|
Wang Y, Zhang H, Chai F, Liu X and Berk M:
The effects of escitalopram on myocardial apoptosis and the
expression of Bax and BCL2 during myocardial ischemia/reperfusion
in a model of rats with depression. BMC Psychiatry. 14:3492014.
View Article : Google Scholar
|
|
182
|
Zhang SW, Liu Y, Wang F, Qiang J, Liu P,
Zhang J and Xu JW: Ilexsaponin A attenuates
ischemia-reperfusion-induced myocardial injury through
anti-apoptotic pathway. PLoS One. 12:e01709842017. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Cheng X, Hu J, Wang Y, Ye H, Li X, Gao Q
and Li Z: Effects of dexmedetomidine postconditioning on myocardial
ischemia/reperfusion injury in diabetic rats: Role of the
PI3K/Akt-dependent signaling pathway. J Diabetes Res.
2018:30719592018. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Morris RE: Prevention and treatment of
allograft rejection in vivo by rapamycin: Molecular and cellular
mechanisms of action. Ann N Y Acad Sci. 685:68–72. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Menown IBA, Mamas MA, Cotton JM,
Hildick-Smith D, Eberli FR, Leibundgut G, Tresukosol D, Macaya C,
Copt S, Sadozai Slama S and Stoll HP: First clinical evidence
characterizing safety and efficacy of the new CoCr Biolimus-A9
eluting stent: The Biomatrix Alpha™ registry. Int J Cardiol Heart
Vasc. 26:1004722020.
|
|
186
|
Das A, Salloum FN, Durrant D, Ockaili R
and Kukreja RC: Rapamycin protects against myocardial
ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J
Mol Cell Cardiol. 53:858–869. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Filippone SM, Samidurai A, Roh SK, Cain
CK, He J, Salloum FN, Kukreja RC and Das A: Reperfusion therapy
with rapamycin attenuates myocardial infarction through activation
of AKT and ERK. Oxid Med Cell Longev. 2017:46197202017. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Zhai M, Li B, Duan W, Jing L, Zhang B,
Zhang M, Yu L, Liu Z, Yu B, Ren K, et al: Melatonin ameliorates
myocardial ischemia reperfusion injury through SIRT3-dependent
regulation of oxidative stress and apoptosis. J Pineal Res.
63:2017. View Article : Google Scholar
|
|
189
|
Hsu CP, Zhai P, Yamamoto T, Maejima Y,
Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J:
Silent information regulator 1 protects the heart from
ischemia/reperfusion. Circulation. 122:2170–2182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Yu L, Sun Y, Cheng L, Jin Z, Yang Y, Zhai
M, Pei H, Wang X, Zhang H, Meng Q, et al: Melatonin
receptor-mediated protection against myocardial
ischemia/reperfusion injury: Role of SIRT1. J Pineal Res.
57:228–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Hardi DG: Keeping the home fires burning:
AMP-activated protein kinase. J R Soc Interface. 15:201707742018.
View Article : Google Scholar
|
|
192
|
Zhang M, Zhao Z, Shen M, Zhang Y, Duan J,
Guo Y, Zhang D, Hu J, Lin J, Man W, et al: Polydatin protects
cardiomyocytes against myocardial infarction injury by activating
Sirt3. Biochim Biophys Acta Mol Basis Dis. 1863:1962–1972. 2017.
View Article : Google Scholar
|
|
193
|
Yu L, Gong B, Duan W, Fan C, Zhang J, Li
Z, Xue X, Xu Y, Meng D, Li B, et al: Melatonin ameliorates
myocardial ischemia/reperfusion injury in type 1 diabetic rats by
preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3
signaling. Sci Rep. 7:413372017. View Article : Google Scholar
|
|
194
|
Yu L, Liang H, Lu Z, Zhao G, Zhai M, Yang
Y, Yang J, Yi D, Chen W, Wang X, et al: Membrane receptor-dependent
Notch1/Hes1 activation by melatonin protects against myocardial
ischemia-reperfusion injury: In vivo and in vitro studies. J Pineal
Res. 59:420–433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Heusch G: Myocardial ischaemia-reperfusion
injury and cardio-protection in perspective. Nat Rev Cardiol. Jul
3–2020.Epub ahead of print. View Article : Google Scholar
|
|
196
|
Gross A and Katz SG: Non-apoptotic
functions of BCL-2 family proteins. Cell Death Differ.
24:1348–1358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Chiu WT, Chang HA, Lin YH, Lin YS, Chang
HT, Lin HH, Huang SC, Tang MJ and Shen MR: Bcl-2 regulates
store-operated Ca2+ entry to modulate ER stress-induced
apoptosis. Cell Death Discov. 4:372018. View Article : Google Scholar
|
|
198
|
Bonneau B, Prudent J, Popgeorgiev N and
Gillet G: Non-apoptotic roles of Bcl-2 family: The calcium
connection. Biochim Biophys Acta. 1833:1755–176. 2013. View Article : Google Scholar : PubMed/NCBI
|