Introduction
Mechanical ventilation using a ventilator is an
essential means of life support for patients with severe
respiratory failure, such as acute respiratory distress syndrome.
However, excess mechanical ventilation may result in
ventilator-induced lung injury (VILI), which is the most common
complication of mechanical ventilation (1). Several theories, such as barotrauma,
volutrauma, atelectrauma and biotrauma have been proposed and, in
particular, the biotrauma theory has now been expanded upon to
illustrate the mechanisms underlying VILI (1). Amongst several biomolecules in the
biotrauma theory involved in the pathogenesis of VILI, the
Sex-determining gene on the Y chromosome related high mobility
group box (SOX) and focal adhesion kinase (FAK) are gaining
increasing attention as therapeutic targets for the treatment of
lung diseases (2-5).
Several members of the SOX family are involved in
the pathophysiological process of lung injury. Gross et al
(6) found that changes in SOX18
expression were involved in endotoxin-induced acute lung injury.
SOX9 exerts protective effects on lipopolysaccharide-induced lung
fibroblasts, apoptosis and the expression of inflammatory factors
(7). SOX11 has been shown to be
involved in epithelial-mesenchymal interactions and is associated
with lung development and pulmonary injury (8). SOX11 can positively regulate members
of the FAK family, which is a subfamily of non-receptor protein
tyrosine kinases and a key regulator of growth factor receptor- and
integrin-mediated signals, and governs fundamental processes of
cells under physiological and pathophysiological conditions through
its kinase activity and scaffolding function (9,10).
The inhibition of FAK expression can lead to the destruction of the
cell barrier and increase pulmonary vascular permeability of
ischemic lung injury (11).
Conversely, the increase in FAK expression may improve pulmonary
vascular permeability caused by acute injury (12,13), reduce the protein levels in
bronchoalveolar lavage fluid and decrease necrosis and apoptosis of
lung epithelial cells (14). In
our previous studies, it was shown that the expression of SOX11 and
FAK in alveolar epithelial cells in HMV-induced VILI in vivo
was dysregulated in vivo (15) and in vitro (16). These findings suggest that the
dysregulation of SOX11 and FAK serve an important role in the
pathogenesis of VILI.
Shear stress resulting from alveolar overstretching
at high pressures during artificial ventilation is the primary
cause of VILI. Alveolar type 2 epithelial (AT2) cells produce
pulmonary surfactants and participate in cell regeneration, which
plays an important role in maintaining alveolar integrity (17). Therefore, in order to illustrate
the role of AT2 cell injury in the pathogenesis of VILI and the
role of SOX-FAK signals in this process, the present study
investigated the effects of cell stretch (CS), which is used to
simulate the shear stress of alveolar overstretching, on the
biological behaviors of cells, including migration, adhesion,
viability and apoptosis of AT2 cells, and the role of SOX/FAK
signaling in this process, through the overexpression and knockdown
of SOX11. Furthermore, to elucidate the molecular mechanisms
downstream of SOX-FAK signaling, changes in the expression levels
of Akt, caspase-3/8, p65 and matrix metalloproteinase (MMP)7 in AT2
cells following CS were also investigated.
Materials and methods
Ethics statement
The animal experiments were approved by the
Committee of Ethics on Animal Experiments of Hebei Medical
University (approval no. ILAS-PL-2010-004). All efforts were made
to minimize the suffering of animals and the number of the animals
used.
Experimental grouping and protocols
AT2 cells (details provided below) were randomly
divided into 5 groups as follows: i) The sham group, cells were
cultured in normal culture medium for 72 h, corresponding to the
culture time following transfection in the other groups, and then
subjected to the sham treatments for CS; ii) CS group, AT2 cells
were cultured for 72 h first and then underwent CS; iii) SOX11
overexpression group, AT2 cells were transfected with SOX11 plasmid
and underwent CS 72 h following transfection with the SOX11
plasmid; iv) SOX11 plasmid + FAK antagonist group, AT2 cells were
transfected with SOX11 plasmid and treated with 1.5 mM FAK
antagonist (PF562271; cat. no. GC12174, Glpbio Technology, Inc.,
dissolved in DMSO) for 72 h and then subjected to CS; v) SOX11
siRNA group, AT2 cells were subjected to CS at 72 h following
transfection with SOX11 siRNA.
Following CS or sham treatment, the AT2 cells were
collected to evaluate the migratory ability using a wound healing
assay, adhesion ability and viability using cell adhesion and
viability assays. The proportion of apoptotic cells was evaluated
by flow cytometry. Furthermore, the expression levels of SOX11,
FAK, Akt, caspase-3/8, p65 and MMP7 were evaluated at the mRNA
(only SOX11 and FAK) and protein levels by reverse
transcription-quantitative PCR (RT-qPCR) western blot analysis
following CS or sham treatment. To directly link the observed
changes in the molecules with the protective effects of SOX11
against the mechanical injury to AT2 cells, changes in the levels
of these molecules were assessed following CS. However, it is
possible that these molecules may have been already activated and
the activation continued to the time point when CS performed.
AT2 cell isolation and culture and
identification
C57 mice supplied by the Chinese Academy of Medical
Sciences were used in the present study. To isolate and culture AT2
cells, 140 neonatal mice born within 24 h were euthanized by an
intraperitoneal injection of 3% pentobarbital sodium (150 mg/kg),
and immersed in 75% alcohol for 2 min. The lungs were collected and
the trachea, macrovascular tissue and bronchus were removed and
placed in pre-cooled PBS. The lung tissue was cut into
1-mm3-thick sections after fully rinsing with PBS and
then digested with trypsin (1 g/l) for 5 min at 37°C after mixing
well. The cell suspension was then collected and centrifuged at
12,000 × g for 3 min at room temperature. The cell pellet was
re-suspended in DMEM containing 10% FBS to terminate digestion. The
remaining lung tissue was then mixed with 2 ml collagenase I (1 g/l
containing 0.01 g/l DNase I), and then incubated at 37°C for 15
min. The cell suspension was collected and an equivalent volume of
10% FBS were added to terminate the digestion. The cell suspensions
were merged and mixed well with a straw 50 times. The cell
suspension was filtered using a filter 200 mesh screen and
centrifuged at 12,000 × g for 5 min at room temperature twice. The
supernatant was discarded and the precipitate was resuspended in an
equivalent volume of 10% FBS DMEM. Pneumonocyte suspensions were
inoculated in a culture dish coated with mouse IgG and incubated
for 45 min at room temperature. The liquid with non-adherent cells
was collected and plated in another culture dish coated with mouse
IgG at 37°C for 40 min. The above-mentioned process was repeated
twice. Finally, the non-adherent cells were collected and
centrifuged at 12,000 × g for 5 min twice at room temperature. The
supernatant was then discarded. The cell pellets were resuspended
in DMEM with 10% FBS and the cell count was adjusted to
1×109/l and inoculated in 6-well plates. The AT2 cells
were identified by morphological evaluation under a microscope
(BX53, Olympus Corporation), and by immunofluorescence staining
targeting LAMP and ABCA3, which are classical biochemical hallmarks
of AT2 cells (Fig. S1).
The cell culture medium was replaced every 2 days,
and the morphology and growth of the AT2 cells were observed using
an inverted microscope (LX71, Olympus Corporation) every day. The
AT2 cells pre-cultured for 3-5 days were used for experiments.
Immunofluorescence staining
AT2 cells plated on coverslips were rinsed with PBS,
and then fixed with 4% paraformaldehyde for 15 min at room
temperature. The AT2 cells were then washed with PBS and exposed to
0.3% Triton X-100 at 37°C for 20 min. After washing with PBS and
blocking with 10% normal goat serum at 37°C for 30 min, the AT2
cells were incubated with the mixture of primary antibodies against
LAMP derived from rabbit (1:200, cat. no. Bs-1970R, lot no.
A106217651, BIOSS) and ABCA3 derived from rabbit (1:200, cat. no.
Bs-17588R, lot. no. AD101749, BIOSS) overnight at 4°C. After
washing 3 times with PBS, the AT2 cells were co-incubated with
secondary antibodies, including Cy3 goat anti-rabbit IgG (1:100,
cat. no. BA1032, lot no. BST15C09A15132, Wuhan Boster Biological
Technology, Ltd.) at 37°C for 60 min. DAPI was then used for
staining with incubation in the dark for 5 min. Subsequently, the
coverslips were washed with PBS and coverslipped with fluorescent
antifade mounting solution. Images were obtained by an investigator
who was blinded to the experiment groups using a microscope (BX53,
Olympus Corporation).
CS
CS was performed using a Flexercell FX-4000T cell
stretch system equipped with a 25-mm BioFlex Loading station
(Flexcell International). The AT2 cells were mounted in the Strain
Unit of the equipment, and then exposed to CS for 4 h (frequency,
0.5 Hz; time ratio of pull and relaxation, 1:1; amplitude, 20%; 30
cycles per min) to mimic stretch-induced AT2 cell injury (11). The AT2 cells in the sham group
were also placed in the equipment and underwent the same process
apart from the stretching.
SOX11 overexpression and knockdown
A recombinant adenovirus-SOX11 plasmid (Addgene,
Inc.) was purchased. The protocol for overexpression was performed
as previously described (17).
Briefly, mouse SOX11 cDNAs were subcloned into the pCMV-IRES-EGFP
plasmid, and the IRES-eGFP sequence was removed to create
pCMV-SOX11. The adenoviral vector and pCMV-SOX11 were then
transfected into 293T cells (Procell Life Science & Technology
Co., Ltd.), and the recombinant adenovirus-SOX11 was harvested from
the cell lysate using the calcium phosphate precipitation method.
In the present study, the SOX11 plasmid at a concentration of 50 nM
in the medium was transfected into the AT2 cells using poly plus
transfection reagent (Polyplus-transfection SA) according to the
manufacturer's protocol.
SOX11 siRNA and its scrambled probe (Santa Cruz
Biotechnology, Inc.) were also purchased. The sequence of the SOX11
siRNA was 5′-GCT GAC TAC CCC GAC TAC AAG-3′. In the present study,
the SOX11 siRNA or its scrambled probe (40 nM) were transfected
into the AT2 cells using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
RT-qPCR and western blot analysis were used to confirm the efficacy
of transfection after 72 h.
Wound healing assay
The migratory ability of the AT2 cells was evaluated
using a wound healing assay. Following the treatment of each group,
the cells were collected and incubated in 6-well plates for culture
until the cells grew to 80% confluency in DMEM/F12 medium
containing 10% fetal bovine serum (Gemini Bio Products) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
Subsequently, a wound was scratched in the cell monolayer using a
sterile pipette tip, and the plates were washed with PBS to remove
debris. The remaining cells were allowed to grow in the wells in
medium without fetal bovine serum for 72 h. The gap distance was
quantitatively evaluated and imaged at 0 and 72 h after creating
the scratch. An inverted microscope (LX71, Olympus Corporation) and
ImageJ software (ImageJ-Pro Plus 6.0, National Institutes of
Health) were used to observe and calculate the percentage of wound
closure.
Cell adhesion assay
For cell adhesion assay, the AT2 cells were
collected immediately following the corresponding treatments in
each group and seeded in fibronectin pre-coated 96-well plates
(50,000 cells/well), and cultured for 2 h at 37°C in DMEM
supplemented with FBS (Gibco; Thermo Fisher Scientific, Inc.).
Subsequently, the plates were washed with PBS buffer to remove any
unattached cells, and subsequently blocked with 1% BSA for 30 min
at 37°C. The cells were stained for 1 h at 37°C using a WST-1 assay
kit (Roche Diagnostics) according to the manufacturer's protocol.
The absorbance was measured at 690 nm using a multimode plate
reader (BioTek Instruments, Inc.).
Cell invasion experiments
Matrigel stored at -20°C was allowed to melt at 4°C
overnight. Matrigel was diluted with pre-cooled serum-free DMEM
medium to a final concentration of 1 mg/ml, and 100 ml diluted
Matrigel were added to the bottom of the central chamber of a
Transwell insert, and incubated at 37°C until it had polymerized.
For gum reconstruction, 200 ml DMEM were added to each well. After
the cells were trypsinized and centrifuged at 10,000 × g for 5 min
at 4°C to remove the culture medium and washed with PBS, they were
resuspended in serum-free medium and seeded on the upper chamber
(10,000 cells per well) of the Transwell insert. Culture medium
containing 10% FBS was added to the lower chamber and cultured at
37°C. Subsequently, after culturing for 36 h, the liquid in the
upper chamber was discarded, the upper chamber was removed, and a
cotton swab was used to wipe off the cells that had not passed
through the cell membrane. Cells were fixed with 3.7% formaldehyde
for 10 min, stained with crystal violet (cat. no. 548-82-9,
Biotopped) for 20 min at room temperature, and cells were observed
under an inverted microscope (LX71, Olympus Corporation). A total
of 3 fields of view were randomly selected, and the number of cells
that had invaded was calculated using ImageJ software (ImageJ-Pro
Plus 6.0, National Institutes of Health) for statistical analysis.
Transwell chambers were purchased from Corning Co. Matrigel matrix
(5 mg/ml) was purchased from Thermo Fisher Scientific, Inc.
Apoptosis assay
Flow cytometry was used to evaluate the apoptosis of
the AT2 cells. Following the treatments, the AT2 cells were
collected and washed with PBS, and then resuspended in 500
µl binding buffer containing 5 µl Annexin V-FITC and
10 µl propidium iodide (Bio-Rad Laboratories, Inc.).
Following incubation for 30 min in the dark at room temperature,
the apoptotic ratio was measured using a FACScan flow cytometer
(Cyto-Flex Beckman) according to the protocol provided with the
Annexin V/PI kit.
RT-qPCR
The mRNA expression levels of SOX11 and FAK in the
AT2 cells were evaluated by RT-qPCR. Following the treatments, the
AT2 cells were collected and the total RNA was extracted using an
RNeasy kit (Qiagen, Inc.) and cDNA was synthesized from 500 ng
total RNA using SuperScript (Invitrogen; Thermo Fisher Scientific,
Inc.). Semi-quantitative levels of SOX11 and FAK mRNA were measured
using a RT-qPCR system (cat. no. K2622, Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The sequences of
the primers used were as follows: GAPDH forward, 5′-AAT GGA TTT GGA
CGC ATT GGT -3′ and reverse, 5′-TTT GCA CTG GTA CGT GTT GAT -3′;
SOX11 forward, 5′-CGA GCC TGT ACG ACG AAG TG-3′ and reverse, 5′-AAG
CTC AGG TCG AAC ATG AGG -3′; and FAK forward, 5′-CCA TGC CCT CGA
AAA GCT ATG -3′ and reverse, 5′-TGA CGC ATT GTT AAG GCT TCT -3′.
PCR (3 min at 94°C and 35 cycles of 30 sec at 94°C, 30 sec at 58°C
and 45 sec at 72°C) was performed in triplicate. Gene expression
was calculated using the 2-∆∆Cq (18) method and normalized to GAPDH
expression.
Western blot analysis
The protein expression levels of SOX11, FAK, Akt,
caspase-3/8, p65 and MMP7 in the AT2 cells were evaluated by
western blot analysis. Following the treatments, the AT2 cells were
collected and lysed in RIPA lysis buffer containing protease
inhibitor. Equal quantities of protein (20 µg) were loaded
on 8-10% SDS-gels, resolved using SDS-PAGE and electrotransferred
to PVDF membranes (EMD Millipore). β-actin was used as the loading
control. The membranes were then incubated with the primary
antibodies overnight at 4°C. The primary antibodies used were the
following: Rabbit SOX11-polyclonal antibody (Cusabio; Ho926s, cat.
no. CSB-PA899716; 1:3,000); rabbit-FAK-monoclonal antibody (Abcam;
CR280473, cat. no. ab40794; 1:3,000); rabbit caspase-3-monoclonal
antibody (Affinity Biosciences; 3253u12 cat. no. AF6311; 1:2,000);
rabbit caspase-8 monoclonal antibody (Affinity Biosciences; 19u71;
cat. no. AF6442; 1:2,500); mouse Akt monoclonal antibody (Affinity
Biosciences; 19u71, cat. no. AF6261; 1:3,000); mouse
phospho-Akt-monoclonal antibody (Epitomics; Abcam; YE112001, cat.
no. 1085-1; 1:4,000); mouse MMP7-derived monoclonal antibody
(ABclonal Biotech Co., Ltd.; 36490, cat. no. A0695; 1:2,000
dilution); mouse p65 monoclonal antibody (GeneTex; 40020, cat. no.
GTX61793; 1:3,000 dilution) and rabbit â-actin monoclonal antibody
(ABclonal Biotech Co., Ltd.; 9100026001, cat. no. AC026; 1:2,000
dilution). Subsequently, the membranes were incubated with
anti-rabbit IgG (KPL, Inc. cat. no. 074-1506; lot no. 140740;
1:3,000), or anti-mouse IgG (Sino Biological Inc.; cat. no.
H013FE2708; lot no. SSA007; 1:2,500). Signals were visualized using
a Western Bright ECL kit (Bio-Rad Laboratories, Inc.). The
integrative optical density (IOD) of the immunoreactive bands was
measured using an Alpha Innotech AlphaImager 2200 (Alpha Image
2200, Alpha Technologies). The relative quantity of protein was
expressed as the ratio of IOD value of target proteins to
β-actin.
Co-immunoprecipitation (Co-IP)
The transfected AT2 cells were solubilized in RIPA
cell lysis buffer (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with a protease inhibitor cocktail and PMSF on ice for
15 min. Lysates were centrifuged at 14,000 × g for 15 min at 4°C.
To assess the interaction between SOX11 and FAK, the supernatant
was incubated overnight at 4°C with SOX11 antibody (Abcam; cat. no.
ab170916). Subsequently, 40 µl protein A cross-linked to
agarose beads (EMD Millipore; cat. no. IP05) were added, and the
mixture was incubated for 1 h at 4°C with constant rotation. The
beads were washed 6 times with ice-cold lysis buffer prior to the
addition of SDS-PAGE sample buffer to each sample. The bound
proteins were dissociated from the beads by heating at 92°C for 3
min before they were resolved on 10% SDS-gels using SDS-PAGE as
described above. Western blot analysis was used to evaluate the
expression of SOX11 and FAK as described above.
Luciferase assay
FAK promoter (from +23 bp to -2,000 bp, NCBI) was
amplified by PCR, and the SOX11 gene sequence was constructed and
amplified by PCR (Addgene, Inc.). Primers of the FAK promoter were
designed (upstream primer, 5′CGA CGC GTC TGG CTA ATT TTT TTG TAT
TTT TAG TAG AG3′ and downstream primer, 5′CCG CTC GAG CCC GAC ACC
GAC CCG GGC TTC 3′). The FAK promoter was then cloned into a
pGL3-FAK-Promotrer-Luc vector and the SOX11 gene was cloned into a
PCDNA vector plasmid. The vectors were then co-transfected into
293T cells with the vectors mentioned above. The assessment was
divided into 4 groups as follows: i) The vector group, where cells
were transfected with the vector and pRL-TK, which was used as the
internal reference plasmid; ii) the SOX11 + PGL3 group, where cells
were transfected (using Lipofectamine 2000 transfection reagent;
lot no. 2103397, Invitrogen; Thermo Fisher Scientific, Inc.) with
pcDNA-SOX11, pGL3 and pRL-TK; iii) the pcDNA + pGL3-FAKP group,
where cells were transfected with pcDNA, pGL3-FAKP and pRL-TK; and
iv) the pcDNASOX11 + pGL3-FAKP group, where cells transfected with
pcDNASOX11, pGL3-FAKP and pRL-TK. The duration between transfection
and activity measurement was 48 h. There were 4 groups of cells
transfected. A total of 3 repeated experiments were carried out in
each group. In addition, the plasmid of Renilla luciferase
was co-transfected as the control. In order to eliminate the
influence of other factors on the accuracy of experimental results,
Firefly luciferase and Renilla luciferase were detected
twice, and the 2 values were divided. The final luciferase activity
of the 4 groups was obtained by averaging the ratio results of 3
replicates. The luciferase activity was measured using a luciferase
assay system (Promega Corporation). The expression of FAK mRNA was
measured by RT-qPCR as described above.
Statistical analysis
All experiments were repeated 3 times independently,
and data are presented as the means ± the standard error of the
mean. A one-way ANOVA followed by the Holm Bonferroni post hoc test
was used to test differences between groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
CS deteriorates the biological behaviors
and increases the apoptosis of, and results in the downregulation
of SOX11 and FAK expression in AT2 cells
Compared with the sham group, the mechanical
stretching of the AT2 cells induced by CS resulted in a decreased
invasion, migration and adhesion of the AT2 cells (Fig. 1). Flow cytometric analysis also
revealed that the proportion of apoptotic AT2 cells increased
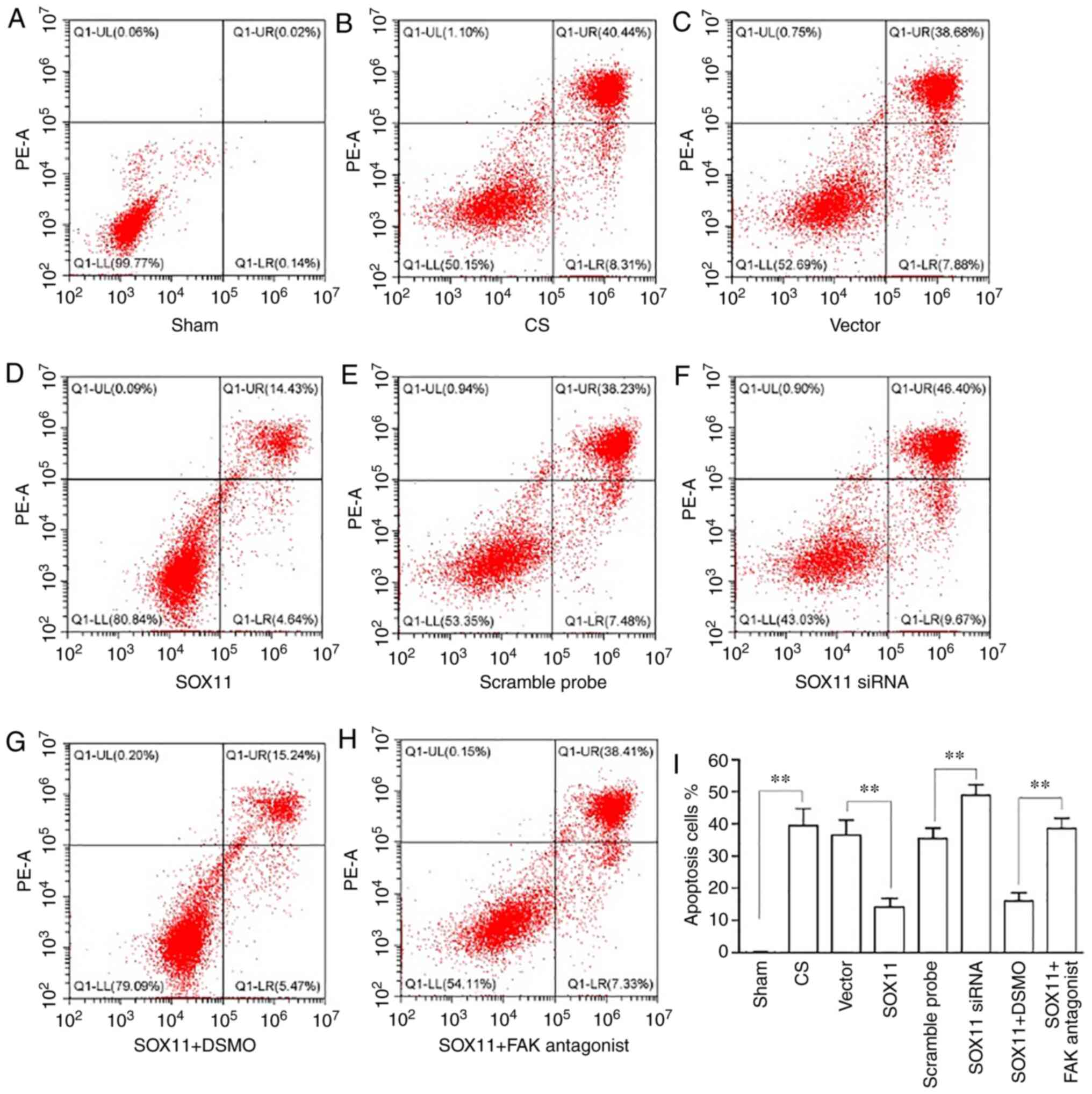
following CS by >2-fold compared with the sham group (Fig. 2). The bar chart shows the
percentages of late apoptotic cells (top right quadrant in the
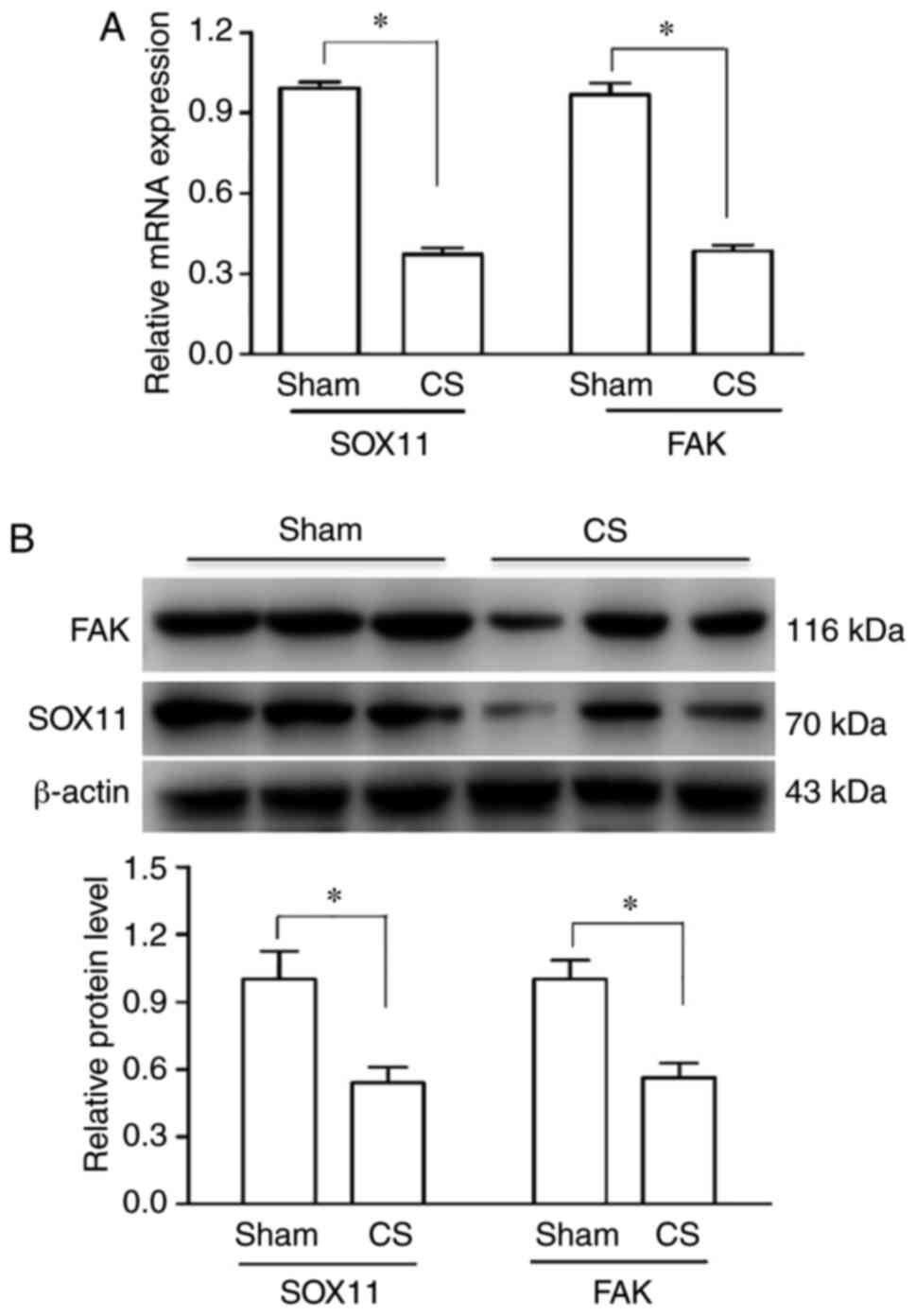
plots). RT-qPCR and western blot analysis revealed that SOX11 and
FAK expression decreased following CS at the mRNA (Fig. 3A) and protein (Fig. 3B) level compared with the sham
group.
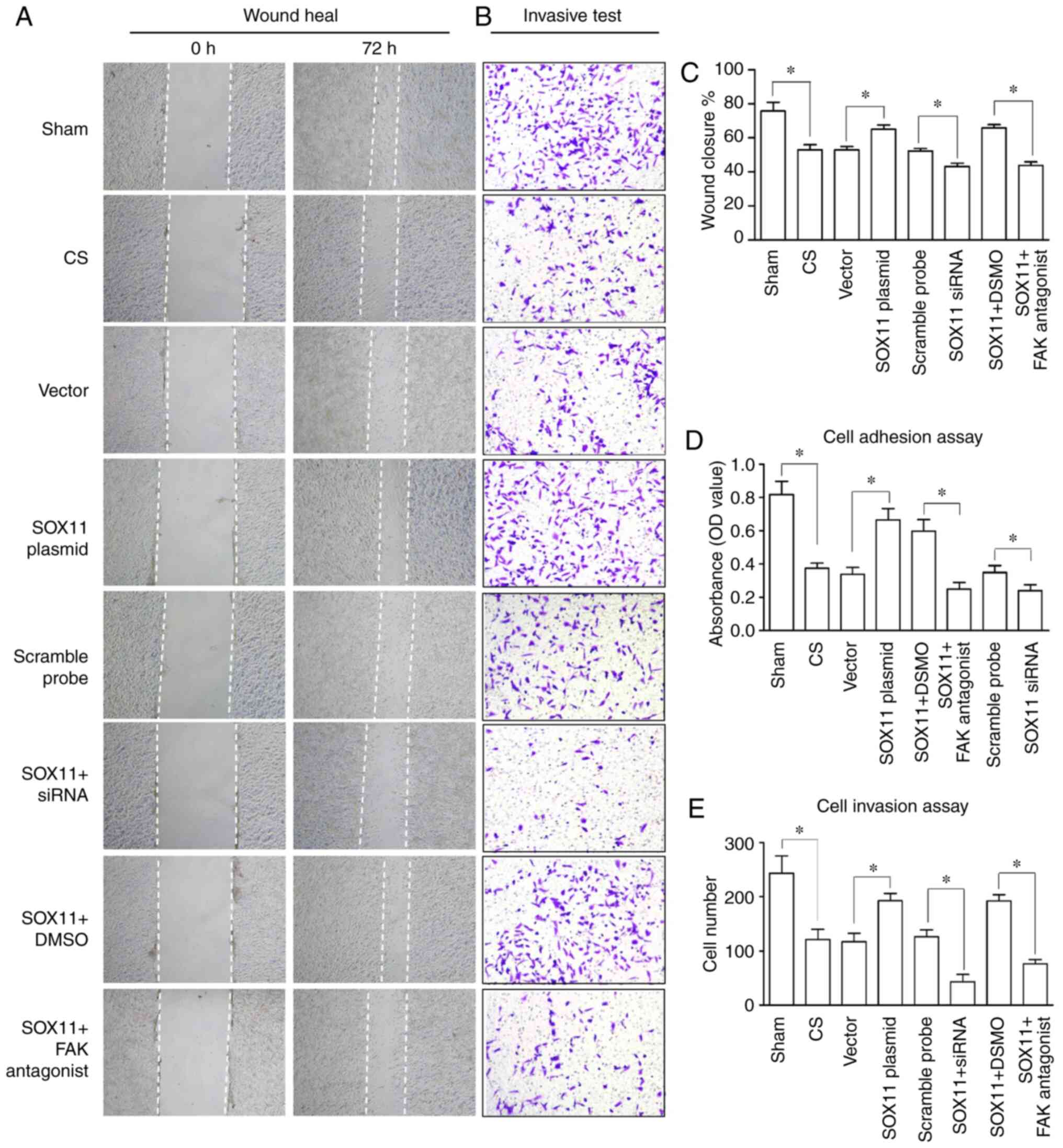 | Figure 1Changes in the biological behaviors
of AT2 cells. (A and C) Wound healing, (B and E) invasion assays
and (D) adhesion assay. The biological behaviors of the AT2 cells
were deteriorated by CS, and the deterioration was reversed
following overexpression of SOX11, or aggravated by knockdown of
SOX11. Furthermore, the effects of SOX11 overexpression were
inhibited by a FAK antagonist. (A) Magnification, x40; (B)
magnification, x100. Data are presented as the means ± standard
error of the mean. *P<0.05. AT2, alveolar type II;
CS, cell stretch; SOX, Sex-determining gene on the Y chromosome
related high mobility group box; FAK, focal adhesion kinase. |
Overexpression of SOX11 attenuates the
effects of CS on AT2 cells
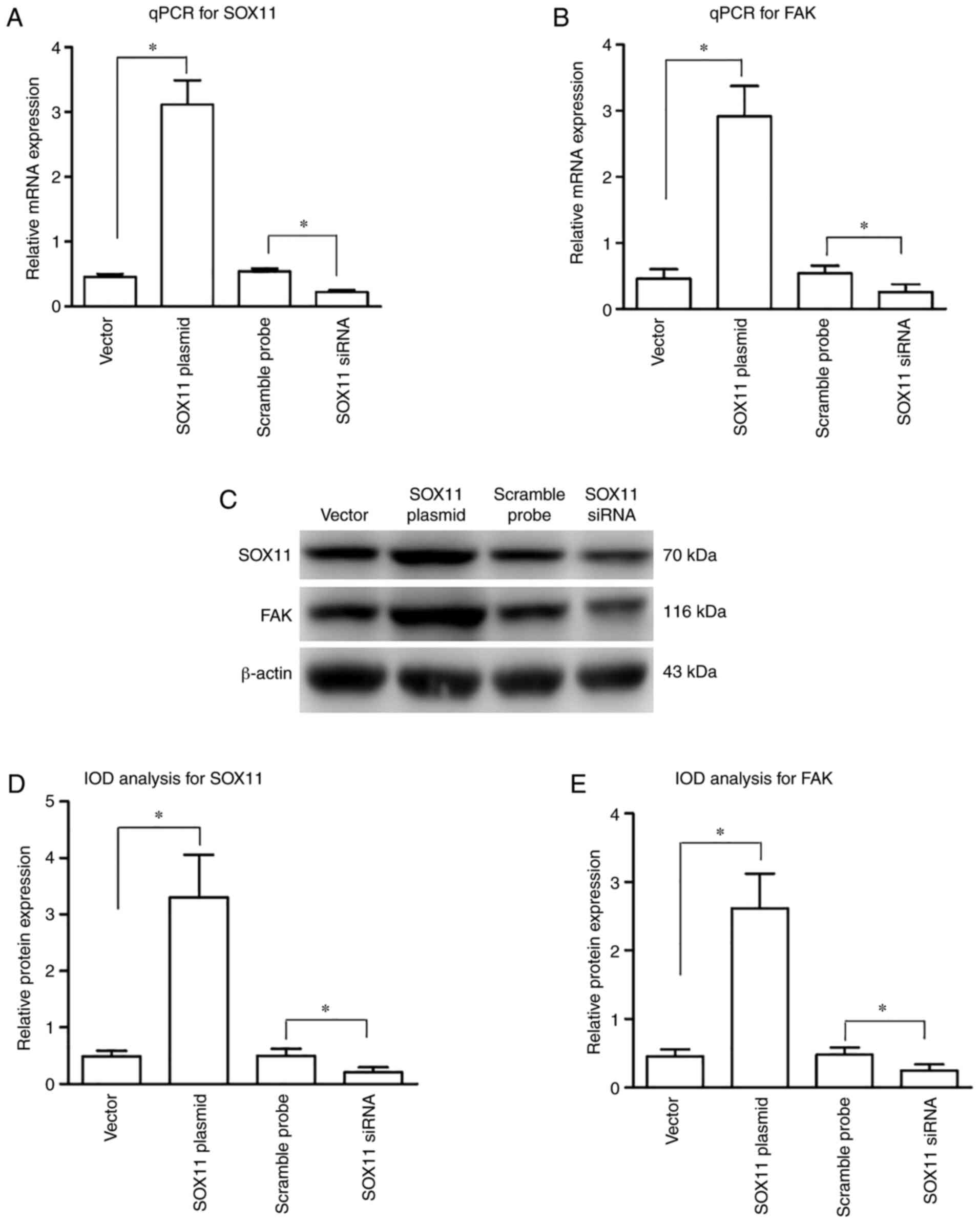
The efficacy of transfection and the effects of
SOX11 overexpression on FAK expression were determined. Compared
with the CS group, the mRNA (Fig. 4A
and B) and protein (Fig. 4C and
D) expression levels of SOX11 and FAK were significantly
increased following transfection with SOX11 overexpression plasmid,
suggesting that plasmid transfection effectively upregulated the
expression of SOX11 and FAK at both the gene and protein levels in
the AT2 cells subjected to CS.
The effects of SOX11 overexpression on the
deterioration of biological behaviors and the increased apoptosis
of AT2 cells induced by CS were then assessed. The CS-induced
decrease in the invasion, migration and adhesion of the AT2 cells
was significantly attenuated following SOX11 overexpression
(Fig. 1). Flow cytometric
analysis indicated that the proportion of apoptotic cells decreased
following SOX11 overexpression (Fig.
2). Furthermore, the protective effects of SOX11
overex-pression against the deterioration of the biological
behaviors and the apoptosis of AT2 cells mentioned above was
prevented by treatment of the cells with a FAK antagonist, which
was manifested by a decrease in invasion, migration and adhesion
(Fig. 1), and an increase in the
apoptotic rate (Fig. 2) of the
AT2 cells in the SOX11 + FAK antagonist group compared with the
SOX11 plasmid transfection group. The negative area
(A/V-PI-) indicates the proportion of normal
AT2 cells. In the present study, the A/V-PI-fluctuated between
43-99% in the different groups (Fig.
2).
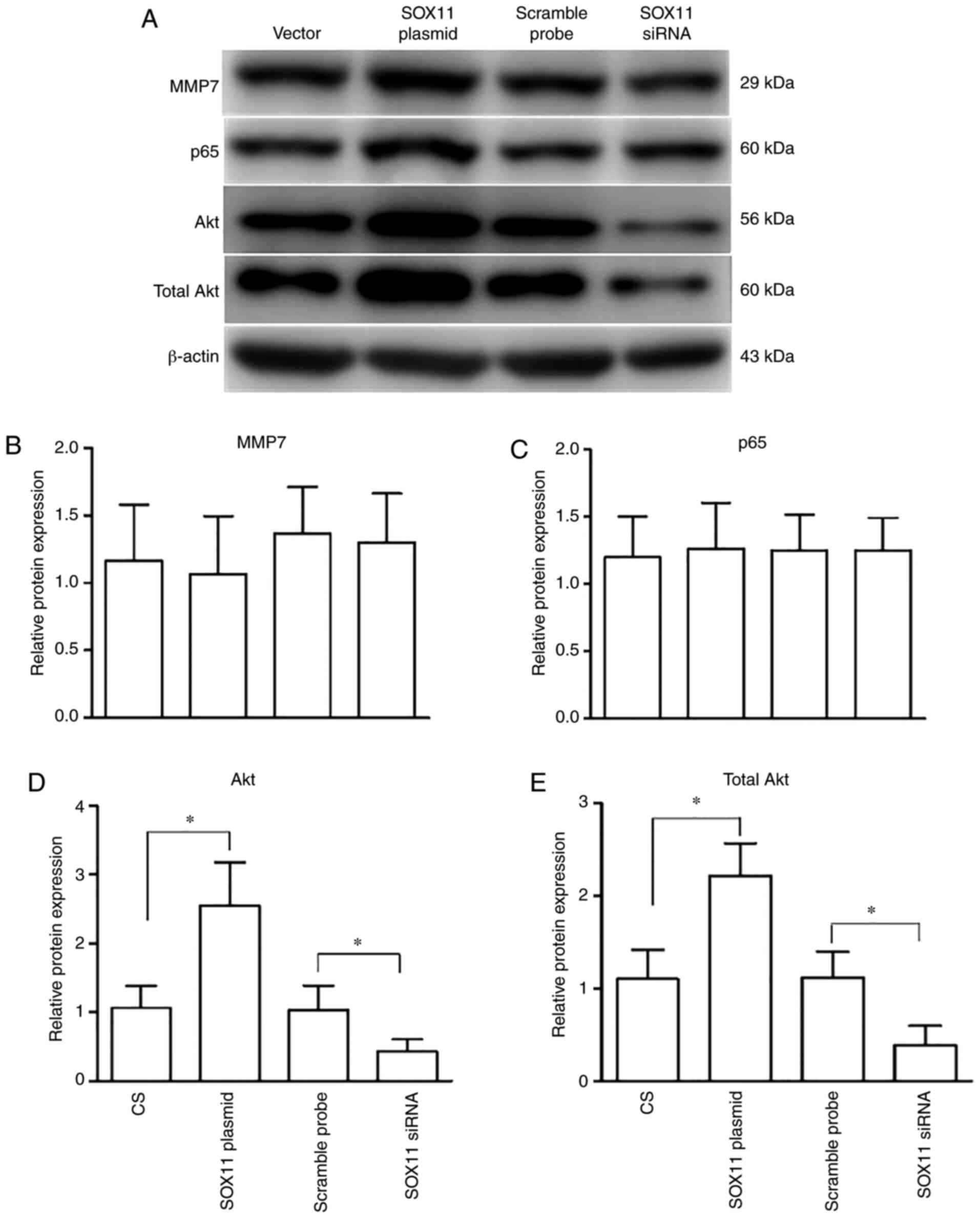
To illustrate the molecular mechanisms downstream of
SOX11 and FAK, the changes in the expression levels of MMP7, p65,
phosphorylated and total Akt, and caspase-3/8 at the protein level
were assessed in the AT2 cells following CS. Western blot analysis
revealed that compared with the CS group, transfection with SOX11
plasmid significantly increased the expressions levels of both
phosphorylated and total Akt, whereas the expression of MMP7 and
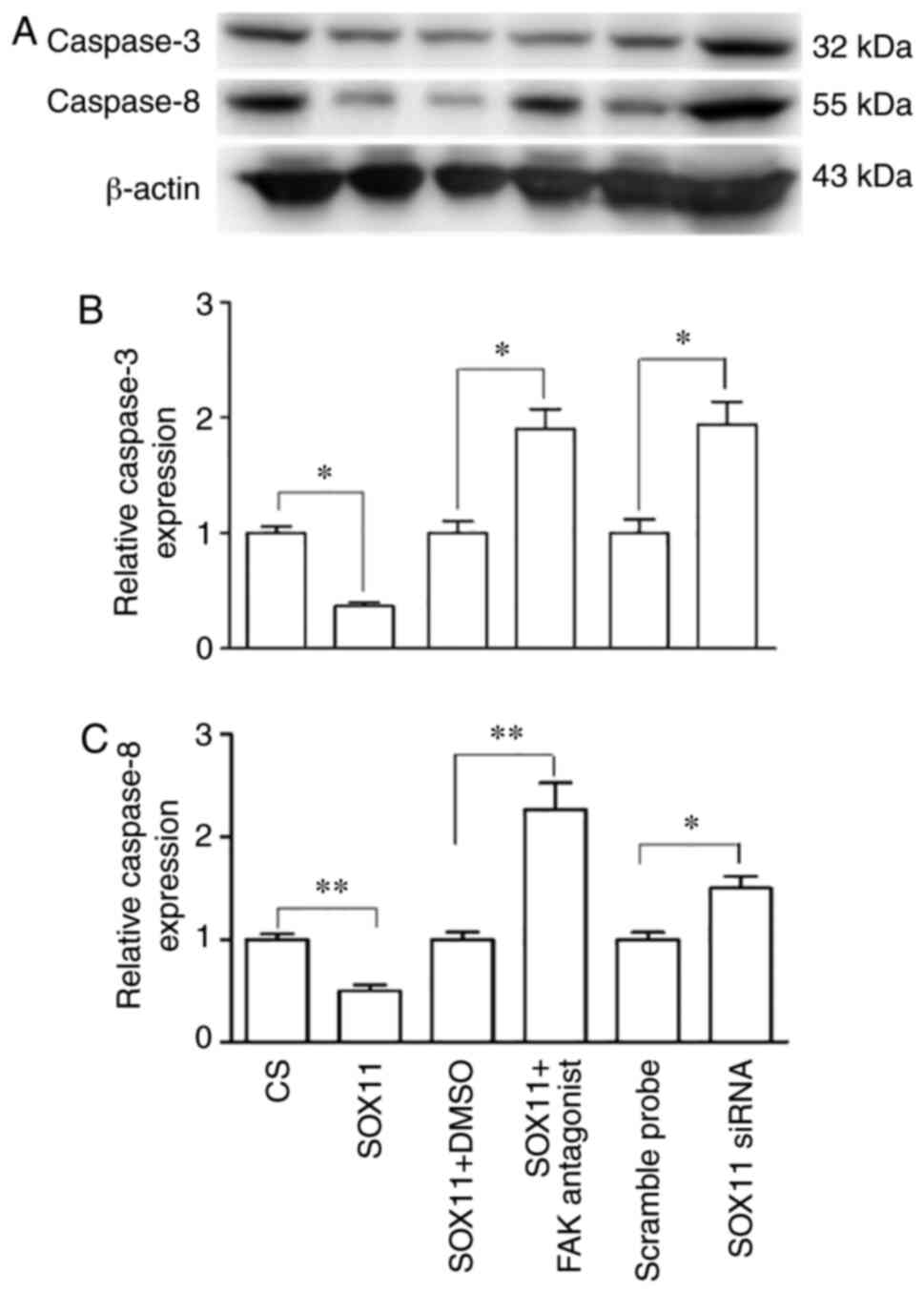
p65 remained unaltered (Fig. 5).
The overexpression of SOX11 resulted in a significant
downregulation of caspase-3/8 expression when compared with the CS
group. Following treatment with a FAK antagonist, the expression of
caspase-3/8 significantly increased (Fig. 6) compared with the SOX11
overexpression group.
Transfection with SOX11 siRNA aggravates
the deterioration of biological behaviors and increases apoptosis
of AT2 cells induced by CS
The efficacy of SOX11 knockdown and the effects on
FAK expression were then determined. Compared with scramble probe
group, the expression levels of SOX11 and FAK at both the mRNA
(Fig. 4A and B) and protein
(Fig. 4C and D) level were
significantly decreased in the SOX11 siRNA group, suggesting that
SOX11 siRNA transfection effectively inhibited the expression of
SOX11 and FAK in the AT2 cells subjected to CS.
Subsequently, the effects of SOX11 knockdown on the
deterioration of biological behaviors and the apoptosis of AT2
cells induced by CS were investigated. It was found that the
invasion, migration and adhesion (Fig. 1) of the AT2 cells were
significantly decreased following the knockdown of SOX11 compared
with the CS group. Flow cytometric analysis indicated that compared
with the cells subjected to CS, the knockdown of SOX11
significantly increased the proportion of apoptotic AT2 cells
induced by CS (Fig. 2).
For the molecules downstream of SOX11 and FAK
mentioned above, western blot analysis showed that knockdown of
SOX11 significantly reduced the expression of both phosphorylated
and total Akt (Fig. 5), and
significantly increased the expression of caspase-3/8 (Fig. 6). This was accompanied by the
deterioration of biological behaviors and an increase in the
apoptosis of AT2 cells induced by CS following the knockdown of
SOX11. The expression levels of MMP7 and p65 remained unaltered
following transfection with SOX11 siRNA (Fig. 5).
SOX11 and FAK interact with each
other
The results of Co-IP revealed that when a SOX11
antibody was incubated with agarose beads with AT2 cell lysates,
SOX11 was detected in the bound protein, and its expression was
markedly upregulated in the SOX11 overexpression group compared
with the control group (Fig. 7A).
Following incubation with a FAK antibody with the bound protein,
FAK bands were also detected in the bound protein. The
above-mentioned results suggested that SOX11 and FAK were both
present in the bound protein (Fig.
7A). Notably, the expression of FAK was markedly increased in
the SOX11 overexpression group compared with the control group
(Fig. 7A). In order to validate
whether an interaction exists between SOX11 and FAK, the AT2 cells
lysates were incubated with a FAK antibody and agarose beads again,
and similar results were observed as that with the SOX11 antibodies
(Fig. 7B). The results of Co-IP
suggested that SOX11 was bound with FAK and regulated the
expression of FAK in AT2 cells.
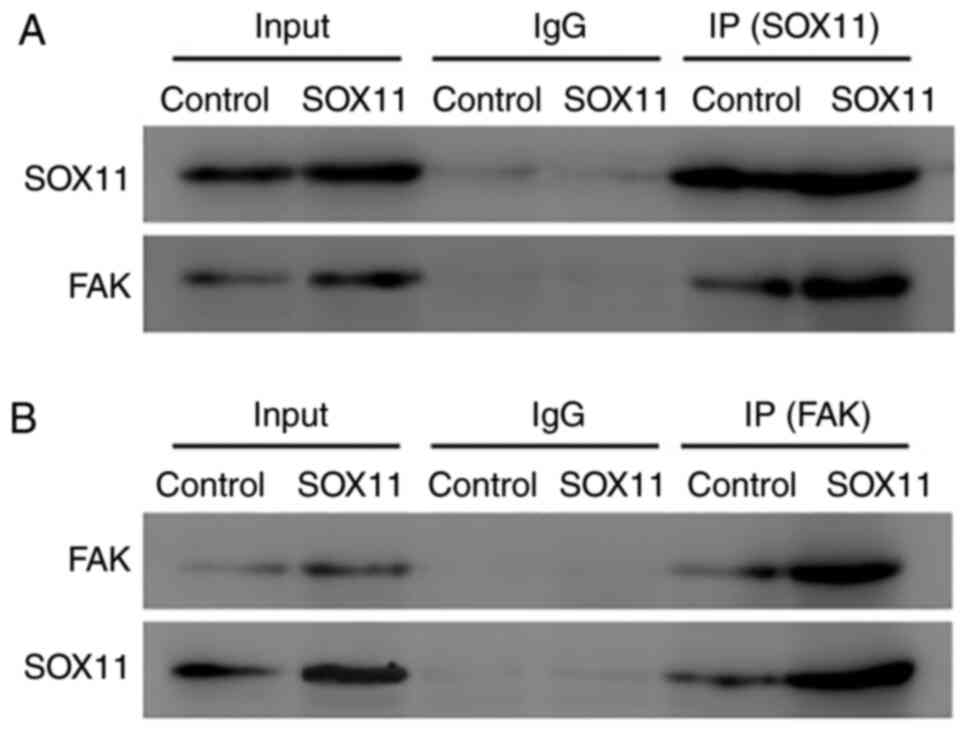 | Figure 7Co-IP showing the interaction between
SOX11 and FAK. CoIP results revealed that when (A) a SOX11 antibody
and agarose beads were incubated with the AT2 cells lysates, SOX11
was detected in the bound protein, and its expression was markedly
upregulated when SOX11 was overexpressed compared with the control
group. When a FAK antibody was incubated with the bound protein,
FAK bands were also detected in the bound protein, and the
expression of FAK was markedly increased in the SOX11
overexpression group compared with the control group, similar to
the expression of SOX11. (B) When AT2 cell lysates were incubated
with FAK antibody and agarose beads, similar results were observed
as that with the SOX11. The results suggested that SOX11 was bound
with FAK and regulated the expression of FAK in AT2 cells. Input,
band from the lysate of AT2 cells; IgG, negative control for the
SOX11 or FAK antibody. AT2, alveolar type II; IP,
immunoprecipitation; SOX, Sex-determining gene on the Y chromosome
related high mobility group box; FAK, focal adhesion kinase. |
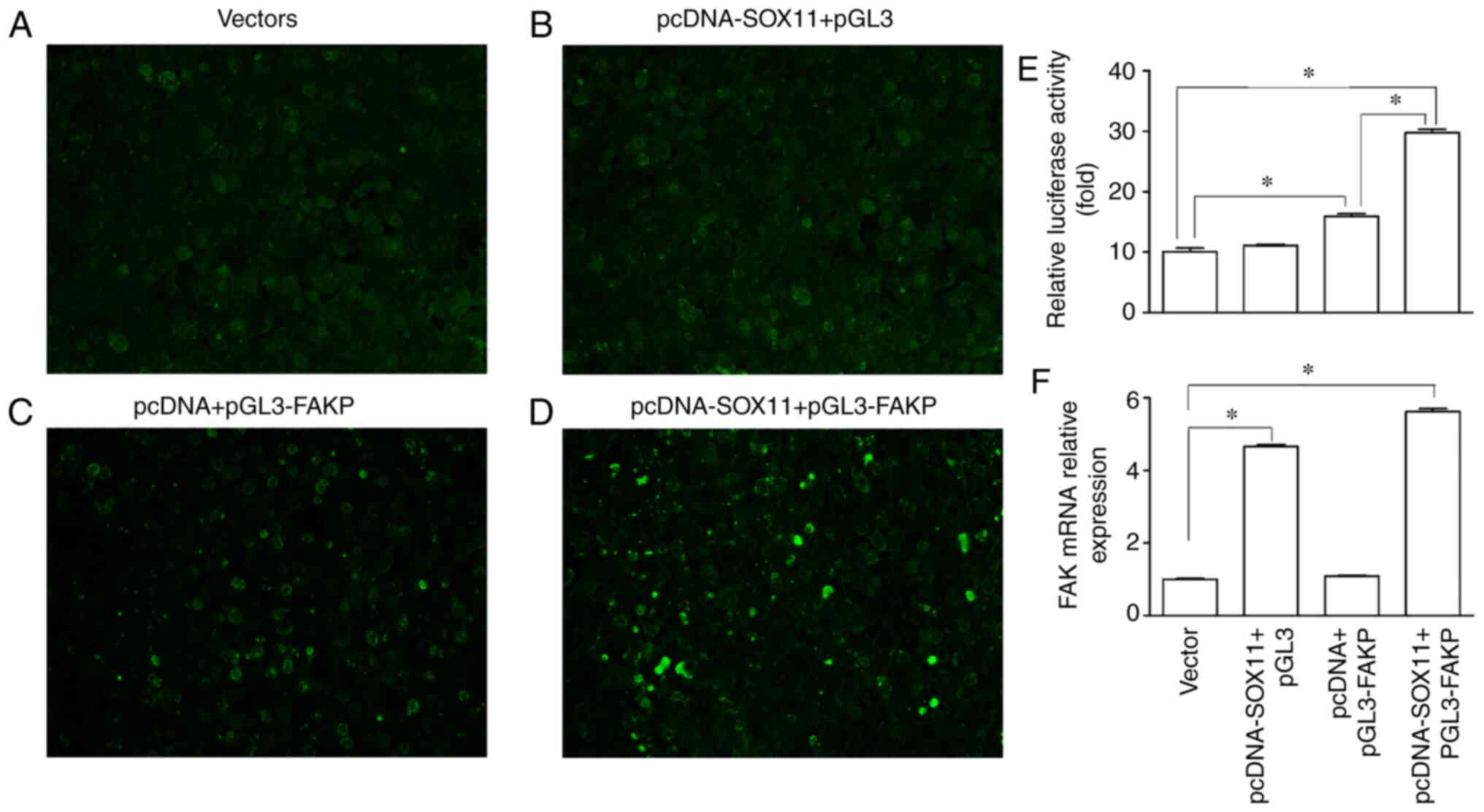
To further confirm the promoting effect of SOX11 on
FAK expression, a luciferase assay was performed using the 293T
cells. It was found that transfection with the pGL3-FAK promoter
increased (Fig. 8C and E), while
transfection with pcDNA SOX11 had no effect (Fig. 8B and E) on the luciferase activity
compared with the vector group (Fig.
8A and E). However, following co-transfection with pcDNA SOX11
and pGL3 FAK promoter, the luciferase activity was further
significantly increased (Fig. 8D and
E) compared with the vector group and pcDNA + pGL3-FAKP group.
RT-qPCR analysis revealed that the mRNA expression of FAK
significantly increased in the pcDNA SOX11 + pGL3 and pcDNA-SOX11 +
pGL3-FAKP groups (Fig. 8F). These
results suggest that SOX11 may enhance the expression of FAK at the
transcriptional level.
Discussion
AT2 cells are the progenitor cells of alveolar
epithelial cells and play an important role in maintaining alveolar
integrity and normal function (19). Under normal conditions, AT2 cells
produce pulmonary surfactant and transform into AT1 cells to repair
lung injury and to prevent severe lung injury (20). An important feature of acute lung
injury is the dysfunction of AT2 cells and the decrease in alveolar
surfactant (21). Mechanical
stimulation, such as periodic mechanical stretching, can produce a
series of adverse effects on AT2 cells, which induces apoptosis of
AT2 cells (22), cell stress
response (23), and even lead to
cell death. Therefore, AT2 cell injury may be an important factor
in the pathogenesis of VILI. A recent study by the authors
demonstrated that impaired SOX11 and FAK signaling in the lungs was
involved in the pathogenesis of VILI induced by hyper-mechanical
ventilation in a mouse model (15). The present study further provides
convincing evidence to indicate that the dysregulation of SOX/FAK
signaling within AT2 cells is involved in the dysfunction of the
biological behaviors and the increased apoptosis of AT2 cells using
CS to directly simulate the shear stress of alveolar
overstretching, a direct factor underlying the pathogenesis of
VILI.
In the present study, it was demonstrated that the
deterioration of biological behaviors and apoptosis of the AT2
cells induced by CS was accompanied by the down-regulation of SOX11
and FAK expression. CS decreased the invasion, migration and
adhesion of the AT2 cells, and increased apoptosis. It has been
reported that mechanical injury to AT2 cells may be assessed based
on their migratory and adhesive abilities, as well as the apoptotic
rate (24,25). The effect of CS on the apoptosis
of AT2 encompasses the entire process of apoptosis, and in
particular, is associated with the activation of the caspase
pathway and the release of cytochrome c in AT2 cells
(22). Additionally, CS has been
shown to decrease the viability of cells and the dysfunction of
cell barrier function (26).
These reports support the findings that the CS test can induce the
deterioration of invasive, migratory and adhesive abilities, and
increase the apoptosis of AT2 cells. Notably, it was found in the
present study that the expression of SOX11 and FAK at both the mRNA
and protein expression level was downregulated in the AT2 cells
that underwent CS, and this was accompanied by the deterioration of
biological behaviors and increased apoptosis. As a member of the
group C of SOX factors, SOX11 can regulate tissue development and
remodeling, including lung development (8). The knockout of SOX11 in mice induces
defects of neurogenesis and lung function (3,27).
FAK is known as a non-receptor tyrosine kinase. The activation of
FAK can facilitate cell survival and attachment in culture, whereas
the degradation of FAK promotes cell detachment and mobility
(28). Desai et al
(29) suggested that
hyper-mechanical ventilation induced lung injury and decreased the
levels of FAK phosphorylation (29). Ding et al (5) demonstrated a role of FAK signaling
in determining the fate of lung epithelial cells. Therefore, the
downregulation of SOX11 and FAK expression in the AT2 cells
accompanied by AT2 mechanical injury in the present study indicates
a clear possibility that the downregulation of SOX11 and FAK
following CS may have been responsible for the pathogenesis of AT2
mechanical injury induced by CS.
Secondly, to further determine the role of SOX11 in
CS-induced AT2 cell impairment, the effects of the overexpression
and knockdown of SOX11 on the CS-induced impairments of the
biological behaviors and apoptosis of AT2 cells were determined. It
was found that following SOX11 overexpression, the effects of CS
were attenuated. The knockdown of SOX11 aggravated the
deterioration in the biological behaviors and further increased the
apoptosis of AT2 cells induced by CS. These findings suggest that
SOX11 dysregulation participates in CS-induced AT2 cell injury.
Researchers have demonstrated the mutually
coordinated association between SOX11 and FAK in certain diseases.
For example, in mantle lymphoma, the overexpression of SOX11 has
been shown to increase the differentiation, proliferation and
invasiveness of tumor cell through the FAK pathway (9). In pulmonary fibrosis, the expression
of SOX11 and FAK has been shown to be increased, and the two
proteins are primarily involved in pulmonary interstitial
remodeling (30). In the present
study, the expression of both SOX11 and FAK were downregulated
simultaneously in the AT2 cells following CS. The overexpression of
SOX11 increased FAK expression, whereas the knockdown of SOX11
decreased FAK expression both at mRNA and protein level.
Furthermore, the attenuation of the deterioration of biological
behaviors and the reduction of the apoptosis of AT2 cells following
the overexpression of SOX11 was blocked by a FAK antagonist. Based
on the above-mentioned results, it is thus suggested that FAK is a
downstream molecule of SOX11, and is involved in the mechanisms
through which SOX11 reduces mechanical injury of AT2 cells induced
by CS.
Akt, also known as protein kinase B, is a
serine/threo-nine protein kinase and plays a principle role in the
survival and growth of cells. Studies have demonstrated the
protective role of Akt in lung injury. For example, Akt activation
has been shown to exhibit a protective function in oxidant-induced
lung injury when adenoviral gene transfer was used (31). In addition, the activating of the
expression of P13K/Akt using dexmedetomidine has been shown to
significantly reduce caspase-3/9 expression, and reduce lung injury
and apoptosis following post-cardiopulmonary bypass (CPB) (32). In the present study, in addition
to the effect on the expression of FAK, the overexpression of SOX11
significantly upregulated Akt expression, and the knockdown of
SOX11 decreased Akt expression, including both the phosphorylated
and total protein levels. FAK acts upstream of Akt-mediated
signaling (33,34), and FAK/PI3/Akt is a classical
signaling pathway in cells (34).
Thus, it was hypothesized that SOX11 can upregulate the expression
of total and phosphorylated Akt via the FAK pathway. Similar
findings have been reported previously. For example, Ji et
al (34) demonstrated that
FAK upregulate the expression of both total and phosphorylated Akt.
Choi et al (35) revealed
that drug-mediated intervention reduced the expression of both
total and phosphorylated Akt. These studies support the hypothesis
of the present study. As regards the determination of whether the
upregulated levels of total Akt occurred through the regulation of
gene transcription, at the protein level, or both, it is necessary
to perform RT-qPCR to assess this. However, in the present study, a
SOX11/FAK/Akt signaling pathway was hypothesized to mediate the
protective effects on AT2 cells following CS. Further studies are
required to investigate the effects of the inhibition of FAK on Akt
expression and activity, and the effects of the inhibition of Akt
on the SOX-mediated effects on mechanical injury to AT2 cells
induced by CS, to provide additional support of the results of the
present study.
As regards the mechanisms underlying the decreased
apoptosis of AT2 cells following the overexpression of SOX11, the
overexpression of SOX11 inhibited, whereas the silencing of SOX11
increased the expression of caspase-3/8, respectively. Furthermore,
the inhibition of caspase-3/8 following the overexpression of SOX11
was prevented by a FAK antagonist. Caspase-3 is a key protease
involved in Fas-mediated apoptosis, and has been reported to act
downstream of caspase-8 (36,37). It has been demonstrated that FAK
regulates apoptosis through the caspase pathway in alveolar
epithelial cell injury (14).
Zhang et al (38) also
obtained similar results, demonstrating that the apoptosis of
cardiac myocytes was also regulated via a FAK/Akt and caspase
pathway (38). Thus, the
inhibition of the caspase-3/8 pathway may be responsible for the
anti-apoptotic effects induced by the overexpression of SOX11 and
FAK following CS of AT2 cells in the present study.
A recent study by the authors demonstrated that the
downregulation of SOX11 and FAK participated in the development of
VILI in a mouse model (15). The
present study assessed the biological behaviors and apoptosis of
AT2 cells using a CS test, which directly mimics the effects of
shear stress on cells, further demonstrating the role of activation
of SOX11 and FAK in mechanical injury to AT2 cells, and the related
molecular mechanisms involved in the process. Taken together, it
can be concluded that the mechanical injury to AT2 cells, which
results from overstretching during artificial ventilation using a
ventilator may be an important factor in the pathogenesis of VILI.
The downregulated expression of SOX11, FAK and Akt and may increase
the apoptosis of AT2 cells. The upregulation of SOX11 and FAK may
be a potential therapeutic target for the prevention and treatment
of VILI in clinical practice.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by TianPu funding
project (grant no. UF201414).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MF, TX and LL performed the experiments, performed
the data analysis and writing the manuscript. NL participated in
data analysis and figure editing. WL, MF, SF and JG conceived,
designed the study, and contributed to data analysis. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The ARRIVE guidelines for care and use of animals
were adhered to in the present study. The animal experiments were
approved by the Committee of Ethics on Animal Experiments of Hebei
Medical University (approval no. ILAS-PL-2010-004). All efforts
were made to minimize the suffering of animals and the number of
the animals used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen L, Xia HF, Shang Y and Yao SL:
Molecular mechanisms of ventilator-induced lung injury. Chin Med J
(Engl). 131:1225–1231. 2018. View Article : Google Scholar
|
|
2
|
Gadi J, Jung SH, Lee MJ, Jami A, Ruthala
K, Kim KM, Cho NH, Jung HS, Kim CH and Lim SK: The transcription
factor protein Sox11 enhances early osteoblast differentiation by
facilitating proliferation and the survival of mesenchymal and
osteoblast progenitors. J Biol Chem. 288:25400–25413. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitamura Y, Nunomura S, Nanri Y, Arima K,
Yoshihara T, Komiya K, Fukuda S, Takatori H, Nakajima H, Furue M
and Izuhara K: Hierarchical control of interleukin 13 (IL-13)
signals in lung fibroblasts by STAT6 and SOX11. J Biol Chem.
293:14646–14658. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castillo SD, Matheu A, Mariani N,
Carretero J, Lopez-Rios F, Lovell-Badge R and Sanchez-Cespedes M:
Novel transcriptional targets of the SRY-HMG box transcription
factor SOX4 link its expression to the development of small cell
lung cancer. Cancer Res. 72:176–186. 2012. View Article : Google Scholar
|
|
5
|
Ding Q, Subramanian I, Luchhardt TR, Che
P, Waghray M, Zhao XK, Bone N, Kurundkar AR, Hecher L, Hu M, et al:
Focal adhesion kinase signaling determines the fate of lung
epithelial cells in response to TGF-β. Am J Physiol Lung Cell Mol
Physiol. 312:L926–L935. 2017. View Article : Google Scholar
|
|
6
|
Gross CM, Kellner M, Wang T, Lu Q, Sun X,
Zemskov EA, Noonepalle S, Kangath A, Kumar S, Gonzalez-Garay M, et
al: LPS-induced acute lung injury involves NF-κB-mediated
down-regulation of SOX18. Am J Respir Cell Mol Biol. 58:614–624.
2018. View Article : Google Scholar :
|
|
7
|
Zhu Z, Dai J, Liao Y and Wang T: SOX9
protects against human lung fibroblast cell apoptosis induced by
LPS through activation of the AKT/GSK3β pathway.
Biochemistry(Mosc). 82:606–612. 2017.
|
|
8
|
Sock E, Retting SD, Enderich J, Bösl MR,
Tamm ER and Wegner M: Gene targeting reveals a widespread role for
the high-mobility-group transcription factor Sox11 in tissue
remodeling. Mol Cell Biol. 24:6635–6644. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Balsas P, Palomero J, Eguileor Á,
Rodriguez ML, Vegliante MC, Planas-Rigol E, Sureda-Gómez M, Cid MC,
Campo E and Amador V: SOX11 promotes tumor protective
microenvironment interactions through CXCR4 and FAK regulation in
mantle cell lymphoma. Blood. 130:501–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee BY, Timpson P, Horvath LG and Daly RJ:
FAK signaling in human cancer as a target for therapeutics.
Pharmacol Ther. 146:132–149. 2015. View Article : Google Scholar
|
|
11
|
Yang S, Yip R, Polena S, Gricius J, Desai
KJ, Sharma M, Ruby C, Gintautas J and Jerome H: Ischemia induced
focal adhesion kinase cleavage in rat lung. Proc West Pharmacol
Soc. 47:57–62. 2004.
|
|
12
|
Chen Q, Yi B, Ma J, Ning J, Wu L, Ma D, Lu
K and Gu J: α2-adrenoreceptor modulated FAK pathway induced by
dexmedetomidine attenuates pulmonary microvascular
hyper-permeability following kidney injury. Oncotarget.
7:55990–56001. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Infusino GA and Jacobson JR: Endothelial
FAK as a therapeutic target in disease. Microvasc Res. 83:89–96.
2012. View Article : Google Scholar
|
|
14
|
Wheaton AK, Agarwal M, Jia S and Kim KK:
Lung epithelial cell focal adhesion kinase signaling inhibits lung
injury and fibrosis. Am J Physiol Cell Mol Physiol. 312:L722–L730.
2017. View Article : Google Scholar
|
|
15
|
Fang M, Fan S, Yao X, Liu N, Gao J, Wang
Z, Xu T, Xian X and Li W: Transfection of Sox11 plasmid alleviates
ventilator-induced lung injury via Sox11 and FAK. Biochem Biophys
Res Commun. 512:182–188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fang M, Liu N, Yao X, Xu T and Wang Z:
Enhancement of FAK alleviates ventilator-induced alveolar
epithelial cell injury. Sci Rep. 10:4192020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hide T, Takezaki T, Nakatani Y, Nakamura
H, Kuratsu J and Kondo T: Sox11 prevents tumorigenesis of
glioma-initiating cells by inducing neuronal differentiation.
Cancer Res. 69:7953–7959. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Castranova V, Rabovsky J, Tucher JH and
Miles PR: The alveolar type II epithelial cell: A multifunctional
pneumocyte. Toxicol Appl Pharmacol. 93:472–483. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jansing NL, McClendon J, Henson PM, Tuder
RM, Hyde DM and Zemans RL: Unbiased quantitation of alveolar type
II to alveolar type I cell transdifferentiation during repair after
lung injury in mice. Am J Respir Cell Mol Biol. 57:519–526. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hallman M, Spragg R, Harrell JH, Moser KM
and Gluck L: Evidence of lung surfactant abnormality in respiratory
failure. Study of bronchoalveolar lavage phospholipids, surface
activity, phospholipase activity, band plasma myoinositol. J Clin
Invest. 70:673–683. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Edwards YS, Sutherland LM, Power JH,
Nicholas TE and Murray AW: Cyclic stretch induces both apoptosis
and secretion in rat alveolar type II cells. FEBS Lett.
448:127–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vlahakis NE and Hubmayr RD: Invited
review: Plasma membrane stress failure in alveolar epithelial
cells. J Appl Physiol 1985. 89:2490–2497. 2000.PubMed/NCBI
|
|
24
|
Tschumperlin DJ, Oswari J and Margulies
AS: Deformation-induced injury of alveolar epithelial cells. Effect
of frequency, duration, and amplitude. Am J Respir Crit Care Med.
162:357–362. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tschumperlin DJ and Margulies SS:
Equibiaxial deformation-induced injury of alveolar epithelial cells
in vitro. Am J Physiol. 275:L1173–L1183. 1988.
|
|
26
|
Birukov KG, Jacobson JR, Flores AA, Ye SQ,
Birukova AA, Verin AD and Garcia JG: Magnitude-dependent regulation
of pulmonary endothelial cell barrier function by cyclic stretch.
Am J Physiol Lung Cell Mol Physiol. 285:L785–L797. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo Y, Liu S, Zhang X, Wang L, Zhang X,
Hao A, Han A and Yang J: Sox11 promotes endogenous neurogenesis and
locomotor recovery in mice spinal cord injury. Biochem Biophys Res
Commun. 446:830–835. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oliveira-Ferrer L, Hauschild J, Fiedler W,
Bokemeyer C, Nippgen J, Celik I and Schuch G: Cilengitide induces
cellular detachment and apoptosis in endothelial and glioma cells
mediated by inhibition of FAK/src/AKT pathway. J Exp Clin Caner
Res. 27:862008. View Article : Google Scholar
|
|
29
|
Desai LP, White SR and Waters CM:
Mechanical stretch decreases FAK phosphorylation and reduces cell
migration through loss of JIP3-induced JNK phosphorylation in
airway epithelial cells. Am J Physiol Lung Cell Mol Physiol.
297:L520–L529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Skubitz KM and Skubitz AP: Gene expression
in aggressive fibromatosis. J Lab Clin Med. 143:89–98. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmed NN, Grimes A, Bellacosa TO, Chan TO
and Tsichlis PN: Transduction of interleukin-2 antiapoptotic and
proliferative signals via Akt protein kinase. Proc Natl Acad Sci
USA. 94:3627–3632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Dou X, Li D, He M, Han M and Zhang
H: Dexmedetomidine ameliorates post-CPB lung injury in rats by
activating the P13K/Akt pathway. J Invest Surg. 33:576–583. 2020.
View Article : Google Scholar
|
|
33
|
Wang Z, Wang Z, Li G, Wu H, Sun K, Chen J,
Feng Y, Chen C, Cai S, Xu J and He Y: CXCL1 from tumor-associated
lymphatic endothelial cells drives gastric cancer cell into
lymphatic system via activating integrin β1/FAK/AKT signaling.
Cancer Lett. 385:28–38. 2017. View Article : Google Scholar
|
|
34
|
Ji Y, Wang Z, Li Z, Huang N, Chen H, Li B
and Hui B: Silencing IGF-II impairs C-myc and N-ras expressions of
SMMC-7721 cells via suppressing FAK/PI3K/Akt signaling pathway.
Cytokine. 90:44–53. 2017. View Article : Google Scholar
|
|
35
|
Choi AR, Kim JH and Yoon S: Sensitization
of cancer cells through reduction of total Akt and downregulation
of salinomycin-induced pAkt, pGSk3β, pTSC2, and p4EBP1 by
cotreatment with MK-2206. Biomed Res Int. 2014:2967602014.
View Article : Google Scholar
|
|
36
|
Schlegel J, Peters I, Orrenius S, Miller
DK, Thornberry NA, Yamin TT and Nicholson DW: CPP32/apopain is a
key interleukin 1 beta converting enzyme-like protease involved in
Fas-mediated apoptosis. J Biol Chem. 271:1841–1844. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rieber M and Rieber MS: Correspondence re:
S. Fulda et al, Betulinic acid triggers (Apo1/Fas)- and
p53-indepedent apoptosis via activation of caspases in
neuroctodernal tumors. Cancer Res. 58:5876–5877. 1998.PubMed/NCBI
|
|
38
|
Zhang R, Li L, Yuan L and Zhao M: Hypoxic
preconditioning protects cardiomyocytes against
hypoxia/reoxygenation-induced cell apoptosis via sphingosine kinase
2 and FAK/AKT pathway. Exp Mol Pathol. 100:51–58. 2016. View Article : Google Scholar
|