Introduction
Lung cancer is one of most common malignancies in
the world, and is major cause of cancer-associated death; it
accounts for 11.6% of the total cancer cases, and the mortality
accounts for ~18.4% of all cancer deaths (1). There are two main subtypes of lung
cancer, small cell lung cancer (SCLC) and non-small cell lung
cancer (NSCLC); NSCLC accounts for >85% of all lung cancer cases
(2). Despite advances in
diagnostic methods, patients with NSCLC are frequently diagnosed in
the first instance with advanced stage lung cancer, which has a
5-year survival rate of 15.9% (3). Elucidating the mechanisms
under-lying the development and progression of NSCLC may result in
improvements in diagnosis and treatment.
Sphingosine kinase (SphK) is a conserved lipid
kinase that phosphorylates sphingosine into sphingosine 1-phosphate
(S1P) (4). Two subtypes of SphKs
are encoded in the human genome, SPHK1 and SPHK2, which regulate
sphingolipid metabolism (5). A
total of five evolutionarily conserved domains have been identified
in all SphKs, and although the amino acid sequences are highly
similar among the subtypes, the functions of the subtypes differ
(6). S1P is a bioactive signaling
molecule regulating cell health and certain diseases (7). Following intracellular formation by
phosphorylation of SphKs, S1P is secreted and binds to its
corresponding receptor (8). A
total of five specific G protein-coupled receptors, S1P receptor
(S1PR)1-5, have been identified in mammalian cells (9). SphK1 has been reported to be highly
expressed in multiple tumor types, such as breast and prostate
cancer (10). Additionally, SphK1
is closely associated with signaling pathways involved in cell
proliferation, migration, metastasis, epithelial-to-mesenchymal
transition (EMT) and other cellular processes (11). High levels of SphK1 expression are
associated with a less favorable outcome in patients with cancer;
for example, increased expression of SphK1 in tumor cells is
significantly associated with shorter survival in patients with
metastatic melanoma (12). Signal
transducer and activator of transcription (STAT) 3 is a
transcription factor with significant roles in the regulation of
multiple cellular activities (13). All STATs share similar domains,
including the N-terminal domain, the central DNA-binding domain and
the classic SRC homology 2 (SH2) domain (14). There are two phosphorylation sites
in STAT3, and phosphorylation of Tyr705 is crucial for the
activation of STAT3; once Tyr705 is phosphorylated by upstream
kinases, STAT3 dimerizes through reciprocal interactions between
the phosphotyrosine-SH2 domains, the dimer translocates to the
nucleus and binds to the promoter regions of its target genes
(15). Interleukin-6 (IL-6) is
the most common factor that activates STAT3 (15). Numerous receptors have also been
reported to activate STAT3, including epidermal growth factor
receptor and vascular endothelial growth factor (16,17). Activation of S1PR1 is considered
to result in persistent activation of STAT3 (18). Using gene expression microarrays,
STAT3 has been demonstrated to regulate ~100 target genes, using a
threshold of a 1.5-fold increase in expression (19). Of note, enhanced expression of
STAT3 target genes was observed in of several different types
cancer. For example, early growth response-1 and JunB
proto-oncogene are immediate response genes that activate the
induction of the cell cycle (20). Cyclin D1 is also modulated by
STAT3 and is considered to promote cell proliferation (21). Another target gene of STAT3 is
matrix metallopeptidase 2 (MMP-2), which is involved in metastasis
of cancer cells (22). Other
factors, including Bcl-2, Bcl-xl and survivin, inhibit apoptosis
(23). Therefore, the activation
of STAT3 is associated with tumor progression.
SphK1 has been reported to be involved in the
pathological processes associated with NSCLC by modulating certain
signaling pathways. For example, SphK1 has been demonstrated to
promote proliferation of NSCLC cells by modulating the downstream
molecules in the PI3K/Akt pathway in vitro (24). SphK1 mediates the inhibitory
effect of transforming growth factor β on EMT in A549 cells
(25). However, the mechanism
underlying Sphk1 function remains to be fully elucidated, and there
are multiple downstream target genes that have not been
explored.
The present study hypothesized that SphK1 may
promote the development of NSCLC and aimed to detect the expression
of SphK1 in NSCLC, as well as to further examine the effects of
SphK1 on NSCLC development in vitro and in vivo. The
underlying molecular mechanism of SphK1 in NSCLC was also
explored.
Materials and methods
Human tissue samples
Human NSCLC and SCLC tissues were obtained from 56
patients with NSCLC and 15 patients with SCLC treated by surgical
resection at the Department of Thoracic surgery of The First
Affiliated Hospital of Xi'an Jiaotong University (Xi'an, China)
between May 2017 and May 2018. Tumor and adjacent non-cancerous
tissues (3 cm away from the tumor) were collected. All procedures
performed in the present study were approved by the Research Ethics
Committee of The First Affiliated Hospital of Xi'an Jiaotong
University, and written informed consent was obtained from all
patients (approval no. 2019-1260). The tissue samples were
immediately frozen in liquid nitrogen until total RNAs or proteins
were extracted. The inclusion criteria for the study participants
were as follows: i) Pathologically or cytologically confirmed NSCLC
and SCLC; ii) resectable stage IA to IIIA iii) age ≥20 and ≤75
years; iv) at least one measurable lesion meeting the Response
evaluation criteria in solid tumors version 1.1 (26); and v) written informed consent was
provided. The exclusion criteria for the study participants were as
follows: i) patients with interstitial lung disease or pulmonary
fibrosis; ii) severe pleural effusion, peritoneal fluid and
pericardial fluid; iii) superior vena cava syndrome; iv) patients
with brain metastasis; v) uncontrollable diabetes mellitus and
hypertension; vi) liver cirrhosis; vii) active heart disease; viii)
pregnancy, possible pregnancy or breastfeeding; ix) tendency to
bleed; and x) unresectable stage IIIB/IVA. The basic information of
the enrolled patients is presented in Table I.
 | Table IBasic characteristics of the enrolled
patients. |
Table I
Basic characteristics of the enrolled
patients.
|
Characteristics | NSCLC | SCLC |
|---|
| Age, years, median
(range) | 53.5 (28-76) | 50.4 (35-71) |
| Sex, n
(male/female) | 30/26 | 8/7 |
| Stage, n (%) | | |
| IA-B | 10 (14.1%) | 2 (2.8%) |
| IIA-B | 29 (40.8%) | 8 (11.3%) |
| IIIA | 17 (23.9%) | 5 (7.1%) |
Cell culture
All cell lines were purchased from The Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences. Normal
bronchial epithelial cells (16HBE), NSCLC cell lines H460 and A549,
and SCLC cell lines H69 and H446 were cultured in DMEM (Gibco;
Thermo Fishers Scientific, Inc.) supplemented with 10% FBS (Thermo
Fisher Scientific, Inc.) in a humidified atmosphere with 5%
CO2 at 37°C.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from the frozen tissues and
cells using the TRIzol® reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
isolated RNA was reverse transcribed into complementary DNA using a
Reverse Transcription kit (Takara Bio, Inc.) at 42°C for 15 min,
and qPCR waas performed using a 20-µl reaction system with
0.5 µl sense and antisense primers, 50 ng RT product and 10
µl 2X SYBR TransStar Green PCR Super Mix in an IQ™ 5
Real-Time PCR Detection System (Thermo Fisher Scientific, Inc.) to
determine the expression levels of the target genes in SCLC and
NSCLC tissues and cell lines. The thermocycling conditions were as
follows: Denaturation at 95°C for 4 min, followed by 40 cycles of
denaturation at 95°C for 20 sec, annealing at 56°C for 30 sec and
extension at 72°C for 32 sec. Relative gene expression was
determined using the 2−ΔΔCT method (27), and GAPDH was used as the internal
control. The sequences of the primers used are listed in Table II.
| Table IIPrimer sequences. |
Table II
Primer sequences.
| Gene | Sequence (5′→
3′) |
|---|
| GAPDH | F:
GACCTGCCGTCTAGAAAAAC |
| R:
TTGAAGTCAGAGGAGACCAC |
| SphK1 | F:
CTGTCACCCATGAACCTGCT |
| R:
TACAGGGAGGTAGGCCAGTC |
| Bcl-xL | F:
AGACCCAGACCTTCCTCTTTCT |
| R:
CCCGGTTGCTCTGAGACATTT |
| Bcl-2 | F:
ATGTGTGTGGAGAGCGTCAACC |
| R:
TGAGCAGAGTCTTCAGAGACAGCC |
| MMP-2 | F:
TACAGGATCATTGGCTACACACC |
| R:
GGTCACATCGCTCCAGACT |
| Cyclin D1 | F:
GTGAAGTTCATTTCCAATCCGC |
| R:
GGGACATCACCCTCACTTAC |
The Cancer Genome Atlas (TCGA) dataset
analysis
The expression of SphK1 in patients with lung
adenocarcinoma and control subjects from TCGA data was analyzed
using an online tool GEPIA2 (http://gepia2.cancer-pku.cn/) (28). According to the website
instructions, the results were analyzed using the 'Boxplot'
function in the 'Expression DIY' section. Data from 483 patients
with lung adenocarcinoma and 59 control subjects were compared.
Chemicals and antibodies
Antibodies against SphK1 (sc-365401), β-actin
(sc-81178) and E-cadherin (sc-8426) were purchased from Santa Cruz
Biotechnology, Inc. Antibodies against SphK2 (ab37977), Ki67
(ab15580), Bcl-2 (ab32124), Bcl-xL (ab32370) and cyclin D1
(ab16663) were purchased from Abcam. MMP-2 (87809S), phospho-
(p-)STAT3 (9145S) and STAT3 (9139S) antibodies were purchased from
Cell Signaling Technology, Inc. C188-9 (S8605) was purchased from
Selleck Chemicals. The 3-diaminobenzidine (DAB) staining kit (cat.
no. CW0125) was purchased from CWBio.
Transfection
The target genes were inserted into the pcDNA3
vector (OBiO Technology Corp., Ltd) to construct the plasmid.
Plasmids containing the transcript for SphK1, SphK1G82D
or an empty vector were purchased from OBiO Technology (Shanghai)
Corp., Ltd. G82D mutation was selected as a negative control as it
led to the loss of catalytical ability of SphK1 (29). A549 cells were transfected with 3
µg plasmids in one well of a 6-well plate using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) at
room temperature when they reached 80% confluence. The cells were
transfected for 36 h prior to use in subsequent experiments.
Colony formation
A549 cells transfected with SphK1,
SphK1G82D or an empty vector were seeded into 6-well
plates (1×103 cells/well) and cultured in complete
medium at 37°C with 5% CO2 for 2 weeks. When colonies
were formed, the cells were fixed with methanol for 10 min,
followed by staining with 0.1% crystal violet (Beijing ComWin
Biotech Co., Ltd.) for 5 min at room temperature. The stained
colonies were counted using an E100 light microscope (Nikon
Corporation). Clusters of ≥50 cells were defined as a colony.
Lentiviral infection
SphK1 was inserted in BamHI/AgeI
restriction sites of the lentiviral expression vector GV358, which
was purchased from OBiO Technology (Shanghai) Corp., Ltd. When A549
cells reached 80% confluence, they were transduced with the
lentivirus using an Envirus™ virus infection enhancer solution
(Engreen Biosystem Co., Ltd.) for 48 h at 37°C before use in
subsequent experiments. The multiplicity of infection was 30. The
cells stably expressing SphK1 were used to establish the xenograft
lung cancer model in nude mice.
Transwell invasion assay
The upper chamber of the Transwell inserts was
pre-coated with Matrigel at room temperature for 1 h. A total of
1×105 A549 cells were added to the upper chamber in 200
µl serum-free medium (Corning, Inc.). A total of 800
µl medium supplemented with 10% FBS was added to the lower
chamber of each well. The cells that had invaded through the
membrane after 24 h were fixed with methanol for 30 min and stained
with 0.1% crystal violet for 20 min at room temperature. The
stained cells were counted using an E100 light microscope (Nikon
Corporation) with ×10 magnification in five randomly chosen
fields.
Western blotting
A549 cells were washed with cold PBS and lysed using
RIPA lysis buffer (Beyotime Institute of Biotechnology, Inc.)
supplemented with protease inhibitors for 13 min on ice.
Subsequently, the cells were centrifuged at 13,000 × g for 15 min
at 4°C. The protein was quantified using a bicinchoninic acid
assay, and 20 µg protein per lane was loaded on a 10% SDS
gel, resolved using SDS-PAGE and transferred to PVDF membranes (EMD
Millipore). The membranes were blocked with 0.5% non-fat milk for 2
h at room temperature and incubated with the SphK1 (1:100), β-actin
(1:1,000), E-cadherin (1:200), SphK2 (1:1,000), Bcl-2 (1:1,000),
Bcl-xL (1:1,000), cyclin D1 (1:1,000), MMP-2 (1:1,000), phospho-
(p-) STAT3 (1:500) and STAT3 (1:1,000) primary antibodies with
gentle agitation at 4°C for 12 h. Subsequently, the membranes were
washed with TBS + 0.1% Tween-20 three times and incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG H&L
(cat. no. as014) or goat anti-mouse IgG H&L (cat. no. as003;
1:5,000; both from ABclonal Biotech Co., Ltd.) antibodies for 2 h
at room temperature, and the signals were visualized using an
enhanced chemiluminescence reagent (Thermo Fisher Scientific,
Inc.). ImageJ version 1.4.7 (National Institutes of Health) was
used for densitometry analysis.
MTT assay
A549 cells were plated in 96-well plates at the
density of 5×104 cells/well. After transfection with
SphK1 or SphK1G82D plasmid and treatment with a STAT3
inhibitor, 5 mg/ml MTT was added to each well for 5 h. The medium
was removed, and 100 µl DMSO was added to each well to
dissolve the formazan crystals. Cell viability was measured at 12,
24, 48 and 72 h after treatment. The absorbance was measured at 490
nm using a SpectraMax M2 microplate reader (Molecular Devices,
LLC).
Immunohistochemistry
Tissues were sectioned into 5-µm slices.
Antigen retrieval was performed using citric acid buffer under high
pressure for 2 min, and tissue sections were rehydrated using a
graded series of ethanol (100, 95, 85 and 75%), deparaffinized and
heated in a microwave for 15 min. To inactivate endogenous
peroxidases, 3% hydrogen peroxide solution was added. Subsequently,
slides were blocked in 5% BSA (Sigma-Aldrich; Merck KGaA) in PBS
for 1 h, after which slides were incubated with the Ki67 antibody
(1:50) overnight in a humidified chamber at 4°C. The following day,
the slides were washed with PBS, and the tissues were incubated
with the horseradish peroxidase-conjugated goat anti-rabbit
antibody (1:200; cat. no. ab205718; Abcam) at room temperature for
2 h. Subsequently, the sections were stained with DAB and incubated
with a streptavidin-biotin complex (P0615; Beyotime Institute of
Biotechnology) for 30 min at 37°C. Finally, the sections were
rinsed with sterile water three times and analyzed using an E100
light microscope (Nikon Corporation) with ×10 magnification in five
randomly chosen fields.
BrdU staining and immunofluorescence
A549 cells were seeded onto coverslips (Thermo
Fisher Scientific, Inc.) and transfected with SphK1 or
SphK1G82D plasmids. Subsequently, the cells were
incubated at room temperature with BrdU (cat. no. ST1056; Beyotime
Institute of Biotechnology) for 1 h and with an anti-BrdU antibody
(cat. no. A1482; ABclonal Biotech Co., Ltd.) according to the
manufacturer's protocol. Fluorescence was observed using a Leica
BMI-6000 confocal microscope with ×10 magnification (Leica
Microsystems, Ltd.).
Tumor xenografts in nude mice
For in vivo experiments, 6-week old male
BALB/c nude mice were purchased from the Shanghai Experimental
Animal Centre of the Chinese Academy of Sciences. Each mouse was
subcutaneously injected in the flank with 1×107 A549
cells stably expressing SphK1 or the control lentivirus in 0.1 ml
PBS. Once tumors were established, mice were intraperitoneally
injected with either DMSO or 100 mg/kg C188-9 five times a week.
Tumors were measured every 3 days using calipers, and the volume
was calculated as follows: Volume=(width2 × length)/2.
Mice in the control and treatment groups (n=5 per group) were
sacrificed by cervical dislocation, and their neoplastic tissues
were dissected for further analysis and weighed. The procedures
performed on mice were approved by the Research Ethics Committee of
The First Affiliated Hospital of Xi'an Jiaotong University
(approval no. 2019-1261).
Statistical analysis
The data are presented as the mean ± standard
deviation of at least three repeats. SPSS 13.0 (SPSS, Inc.) was
used for statistical analysis. Unpaired Student's t-test or one-way
ANOVA followed by Tukey's post hoc test was used to compare the
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression levels of SphK1 and SphK2 are
upregulated in NSCLC tissues and cell lines
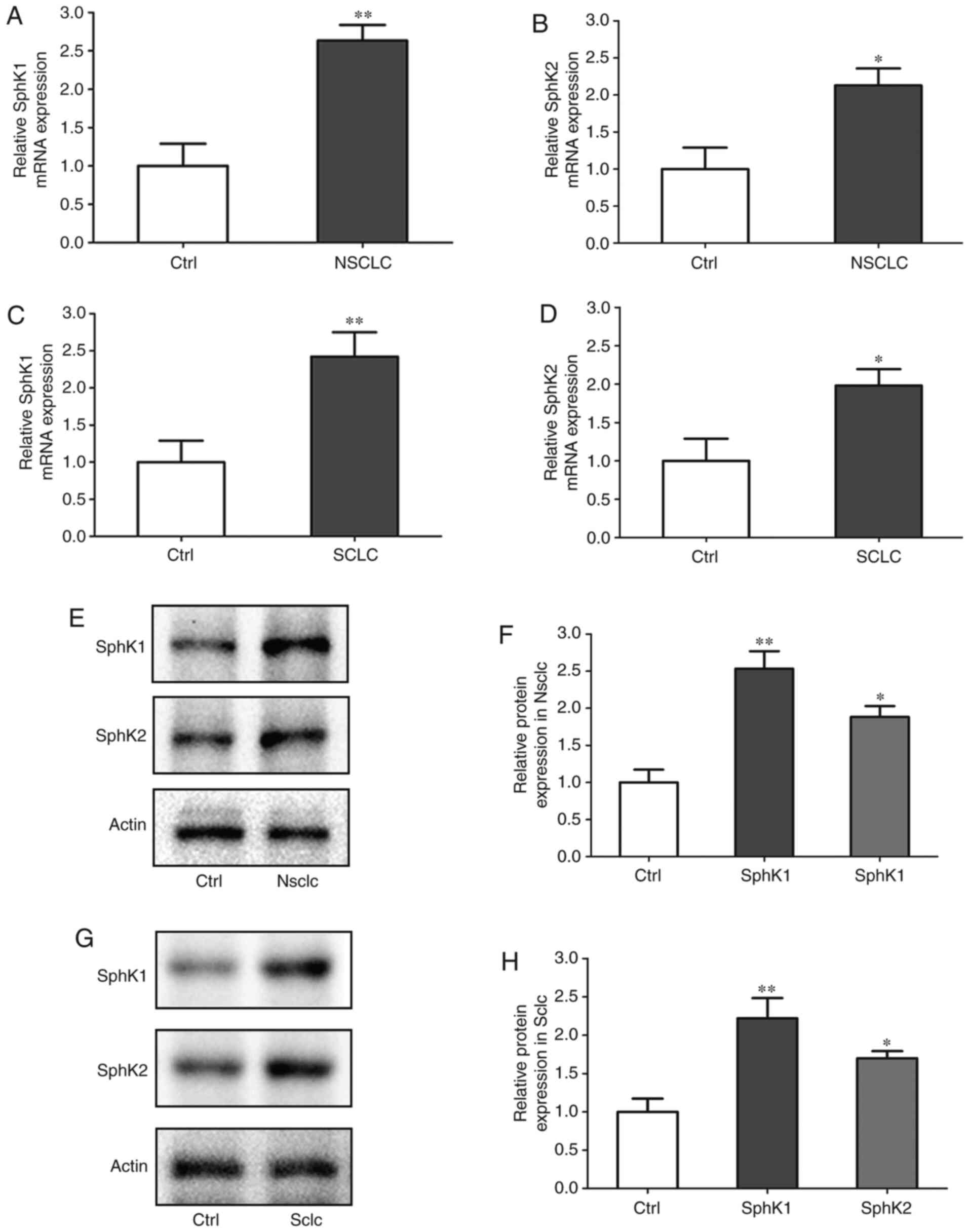
The mRNA expression levels of SphK1 and SphK2 in
clinical NSCLC and SCLC samples were assessed. Compared with the
corresponding normal tissues, SphK1 mRNA expression was 2.63-fold
higher, and SphK2 mRNA expression was 2.21-fold higher in NSCLC. In
SCLC, SphK1 mRNA expression was 2.35-fold higher, and that of SphK2
was 1.92-fold higher compared with the corresponding normal tissues
(Fig. 1A-D). In addition, the
protein expression levels of SphK1 and SphK2 were assessed. Similar
to the mRNA results, the protein expression of SphK1 was 2.51-fold
higher, and that of SphK2 was 1.82-fold higher in NSCLC compared
with the corresponding normal tissues. In SCLC, Sphk1 protein level
was 2.25-fold higher, and that of SphK2 was 1.67-fold higher
compared with the corresponding normal tissues (Fig. 1E-H). Additionally, the mRNA and
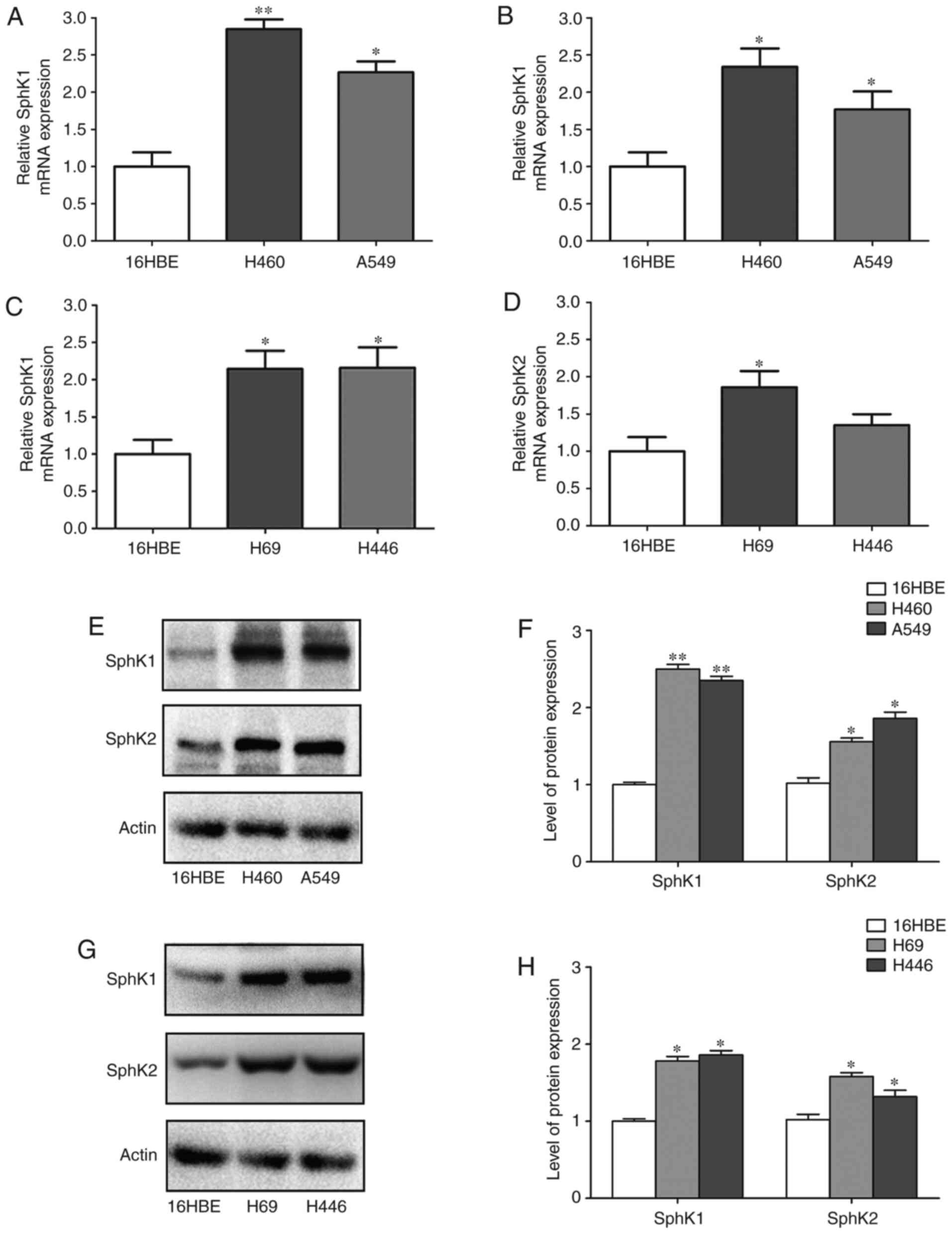
protein expression levels of SphK1 and SphK2 in NSCLC and SCLC cell
lines were compared with those in the 16HBE cells. The results
revealed that SphK1 mRNA expression was 2.78- and 2.21-fold higher
in the H460 and A549 cells, respectively, compared with the16HBE
cells. In addition, SphK2 mRNA expression was 2.35- and 1.73-fold
higher in the H460 and A549 cells, respectively, compared with the
16HBE cells. SphK1 mRNA expression was 2.12- and 2.20-fold higher,
and SphK2 expression was 1.85- and 1.36-fold higher in the H69 and
H446 cells, respectively, compared with the 16HBE cells (Fig. 2A-D). The western blotring result
demonstrated that SphK1 protein expression was 2.58- and 2.35-fold
higher, and that of SphK2 was 1.81- and 1.85-fold higher in the
H460 and A549 cells, respectively, compared with the 16HBE cells.
SphK1 protein expression was 1.52- and 1.78-fold higher, and that
of SphK2 was 1.62- and 1.27-fold higher in the H69 and H446 cells,
respectively, compared with the 16HBE cells (Fig. 2E-H). Therefore, the expression of
SphK1 was further analyzed between patients with lung
adenocarcinoma and healthy controls in The Cancer Genome Atlas
dataset. The result demonstrated that SphK1 expression was higher
in patients with lung adenocarcinoma compared with the control
group (Fig. S1). Thus, based on
these results, the effects and mechanism of SphK1 in NSCLC were
investigated.
SphK1 promotes the proliferation of NSCLC
cells
To examine the role of SphK1 in the progression of
NSCLC, SphK1 or SphK1G82D was overexpressed in A549
cells (30). Western blot-ting
results confirmed the successful transfection of SphK1 (Fig. 3A). An MTT assay was used to
analyze cell proliferation; the results demonstrated that
overexpression of SphK1 in A549 cells increased cell proliferation,
whereas transfection with SphK1G82D reduced cell
proliferation compared with the control group (Fig. 3B). Cell viability was also
measured after different durations (12, 24, 48 and 72 h). The
results suggested that SphK1 induced, whereas SphK1G82D
repressed cell proliferation at 48 and 72 h (Fig. 3C). BrdU staining was used to
verify these results. The BrdU staining results revealed that
overexpression of SphK1 induced proliferation in A549 cells, and
overexpression of SphK1G82D reduced cell proliferation
compared with the control cells (Fig.
3D and E). The colony formation assay was also conducted to
study the cell proliferation; the results demonstrated that SphK1
increased, whereas SphK1G82D impaired the colony
formation ability of A549 cells compared with the control, which
was similar to the MTT and BrdU assay results (Fig. 3F and G).
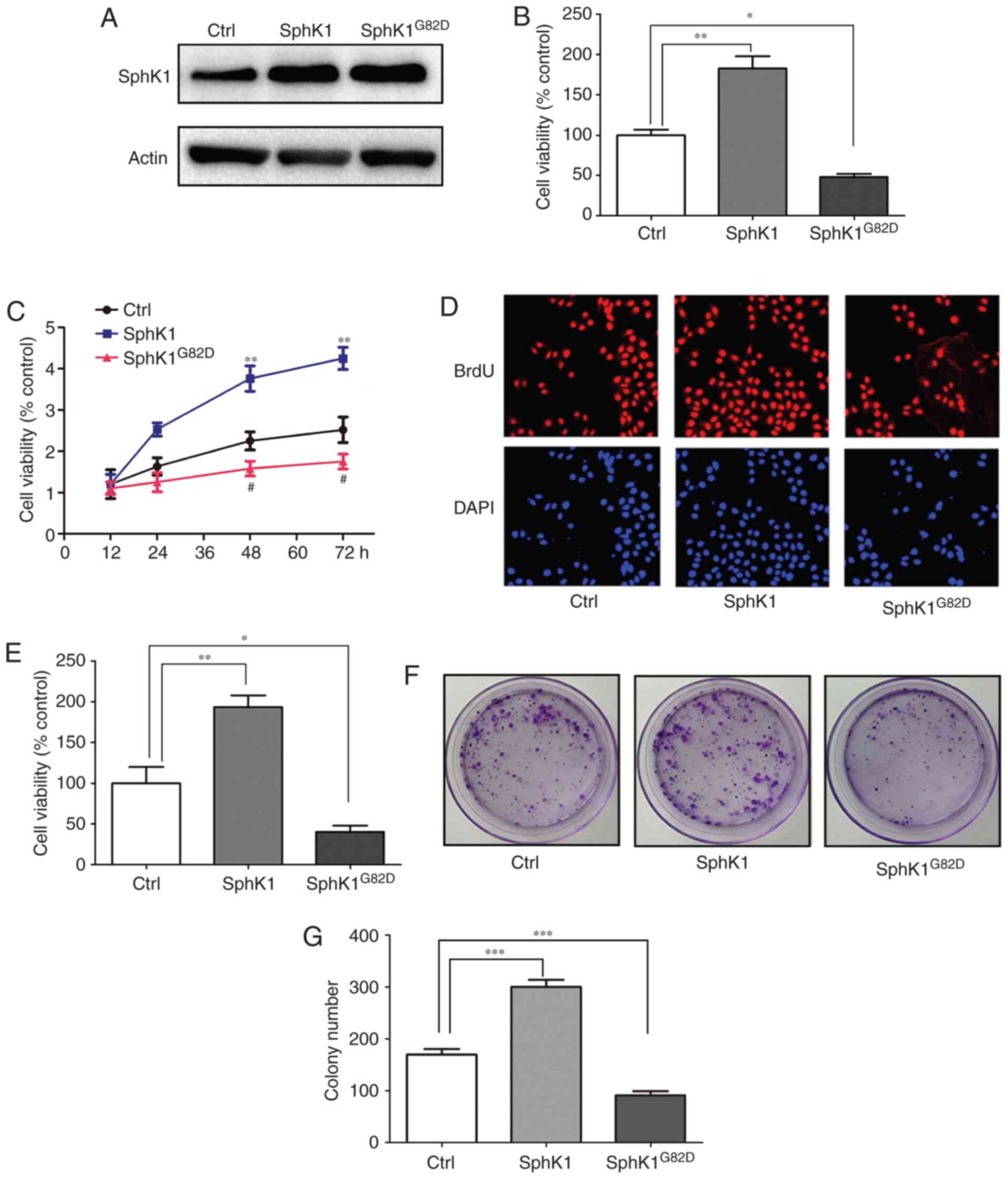 | Figure 3SphK1 promotes the proliferation of
NSCLC cells. (A) Verification of successful transfection of the
SphK1 overexpression vectors in A549 cells. (B) Overexpression of
SphK1 increased the proliferation, whereas transfection with
SphK1G82D reduced the proliferation of A549 cells
compared with the control. (C) Cell viability at different time
points following transfection with SphK1 and SphK1G82D.
**P<0.01, SphK1 group vs. control group;
##P<0.01, SphK1G82D group vs. control
group. (D) The viability of A549 cells was assessed using a BrdU
staining assay; SphK1 overexpression increased cell viability,
whereas SphK1G82D reduced it compared with the control.
(E) The viability of cells increased in the SphK1-transfected
cells, but decreased in SphK1G82D-transfected cells
compared with the control. (F) The colony formation assay of A549
cells following transfection with SphK1 and SphK1G82D.
(G) The colony numbers increased in the SphK1-transfected cells,
but decreased in SphK1G82D-transfected cells compared
with the control. *P<0.05, **P<0.01,
***P<0.001. SphK, sphingosine kinase; NSCLC,
non-small cell lung cancer; Ctrl, control. |
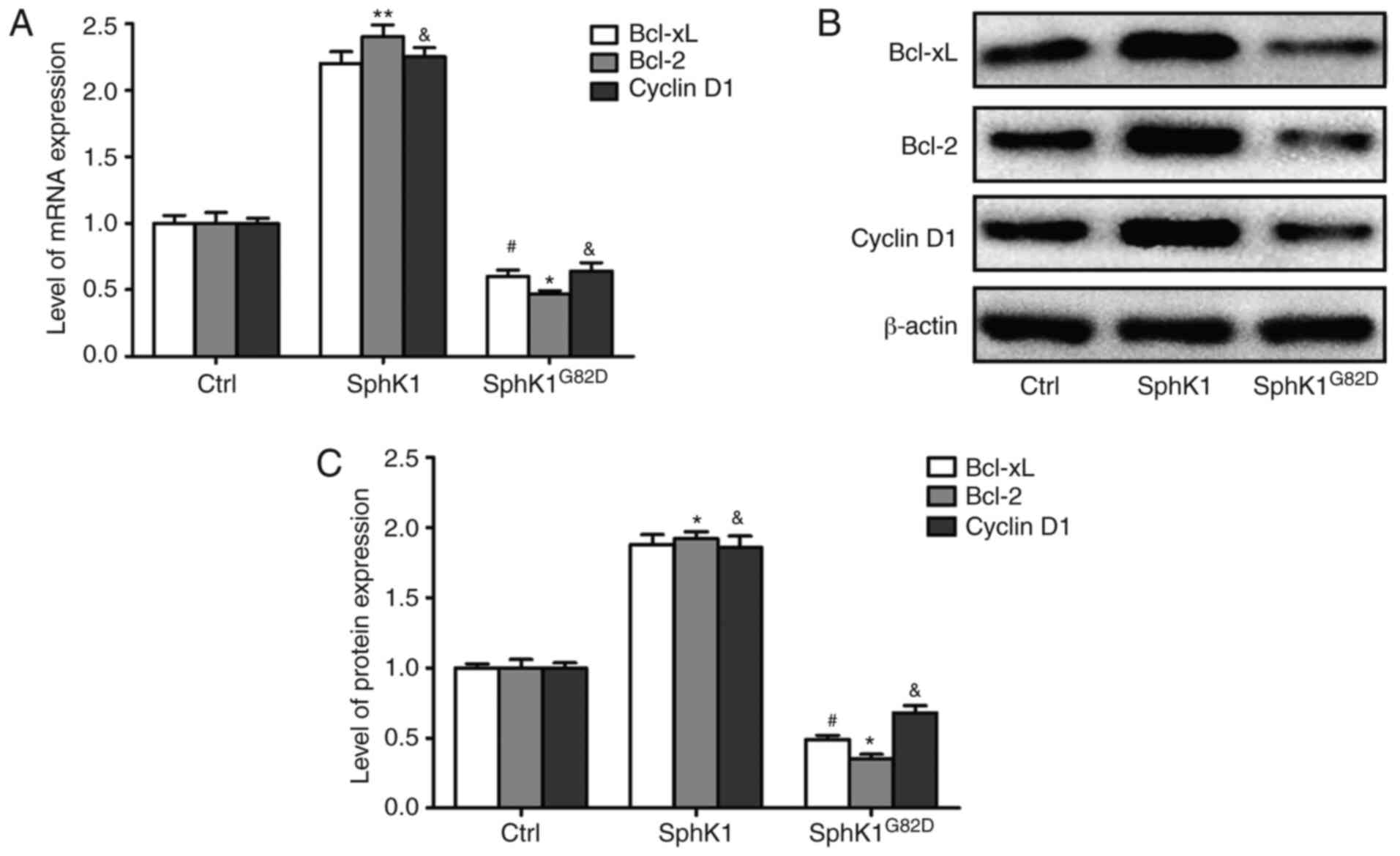
SphK1 modulates the expression of the
apoptosis-related genes
To determine the mechanism underlying the
SphK1-mediated increase in A549 proliferation, the expression
levels of apoptosis-associated genes Bcl-2, Bcl-xL and cyclin D1,
which are required for maintenance of proliferation of cancer cells
and tumorigenesis, were assessed after transfection of SphK1 or
SphK1G82D. The mRNA expression levels of the
antiapoptotic genes were increased in the SphK1 overexpression
group compared with the control cells. SphK1G82D
overexpression resulted in a decrease in mRNA expression levels of
these genes compared with the control cells (Fig. 4A). Additionally, the protein
expression levels of these factors were assessed, and the results
were similar to the mRNA results (Fig. 4B and C). These results suggested
that SphK1 may promote the proliferation of NSCLC cells at least
partly by upregulating the mRNA and protein expression levels of
Bcl-2, Bcl-xL and cyclin D1.
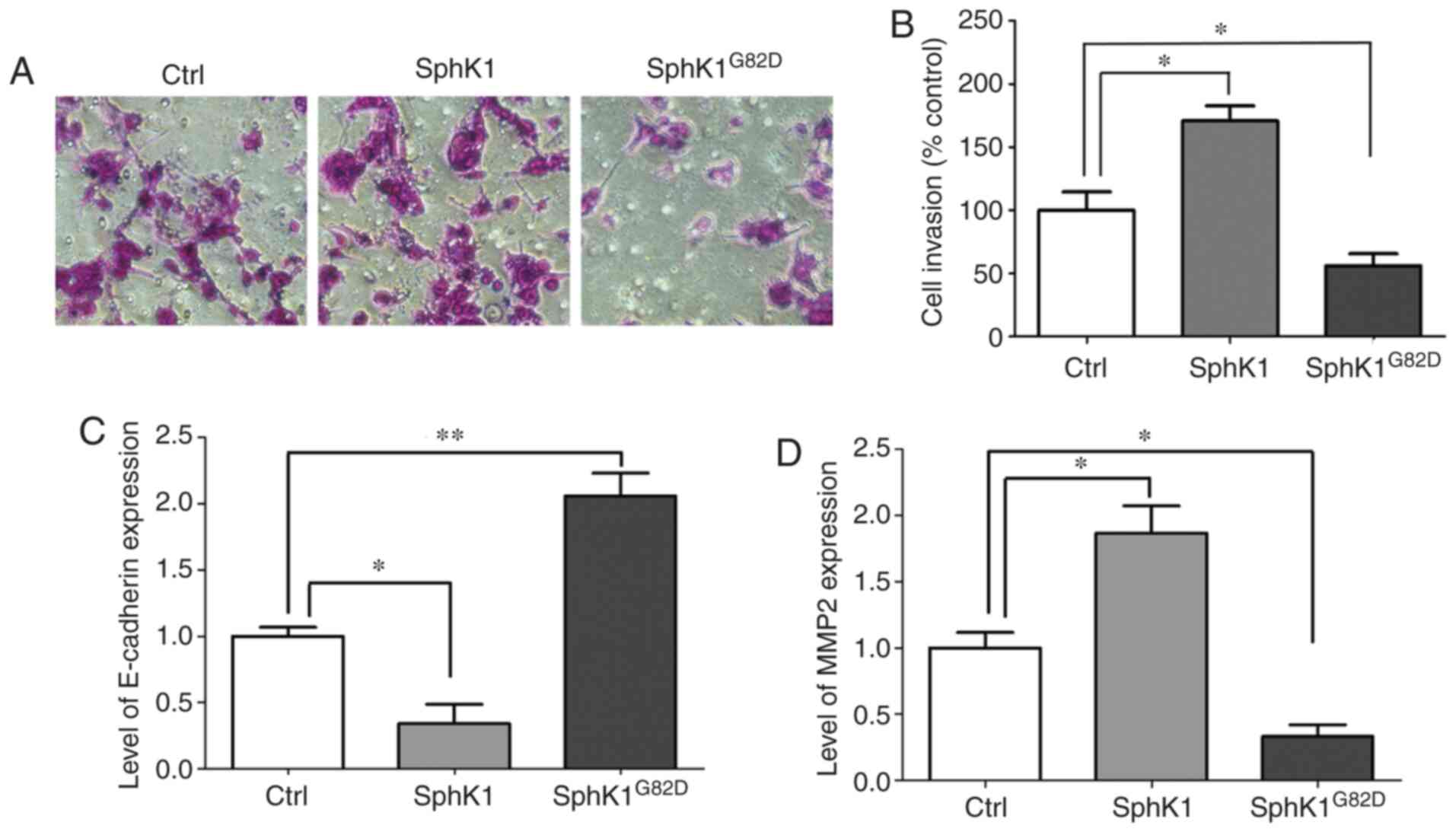
SphK1 promotes the invasion of NSCLC
cells by modulating metastasis-associated genes
To further elucidate the roles of SphK1 in NSCLC,
SphK1 and SphK1G82D were overexpressed in A549 cells,
and the invasive capacity of these cells was assessed. The
Transwell invasion assay results revealed that transfection with
SphK1 increased the cell invasive capacity compared with that of
the control group. Overexpression of SphK1G82D decreased
the invasive capacity of A549 cells compared with the control group
(Fig. 5A and B). E-cadherin and
MMP2 have been reported to be involved in the invasion and
metastasis of multiple types of cancer cells (31). To further examine the role of
SphK1 in NSCLC, the mRNA levels of these two genes were determined.
The mRNA expression levels of E-cadherin were decreased in the
SphK1-transfected cells and increased in the
SphK1G82D-transfected cells compared with the control
cells (Fig. 5C). By contrast, the
mRNA expression levels of MMP2 were upregulated following
transfection with SphK1 and downregulated in the
SphK1G82D-overexpressing cells compared with the control
cells (Fig. 5D).
SphK1 expression increases STAT3 activity
in NSCLC cells
STAT3 has been reported to be a master regulator of
cell proliferation, metastasis, immune evasion and EMT (32). Thus, it was determined whether
STAT3 was activated by SphK1 overexpression. The western blotting
results demonstrated that compared with those in the control cells,
SphK1 overexpression resulted in an increase in p-STAT3 levels,
indicative of STAT3 activation. By contrast, treatment with the
SphK1 inhibitor C188-9 attenuated the activation of STAT3 (Fig. 6A and B). To determine whether
SphK1 regulated the proliferation and invasion of A549 cells
through the activation of STAT3, A549 cells were treated with the
STAT3 inhibitor, C188-9, after transfection with SphK1.
Subsequently, the mRNA expression levels of apoptosis- and
metastasis-associated genes were assessed. The results demonstrated
that the SphK1-induced upregulation of Bcl-2, cyclin D1 and MMP2
mRNA expression levels were reversed by the STAT3 inhibitor C188-9,
whereas treatment with C188-9 exhibited a less prominent effect on
the expression of the aforementioned genes in the control cells
(Fig. 6C-E). MTT and Transwell
invasion assays were performed to examine the role of C188-9 on
SphK1-overexpressing cells. The results revealed that
over-expression of SphK1 increased the proliferation and invasion
of A549 cells compared with the control group, and treatment with
C188-9 significantly suppressed the SphK1-enhanced proliferation
and invasion (Fig. 6F-H).
Together, these results suggested that SphK1 promoted the
progression of NSCLC by activating STAT3.
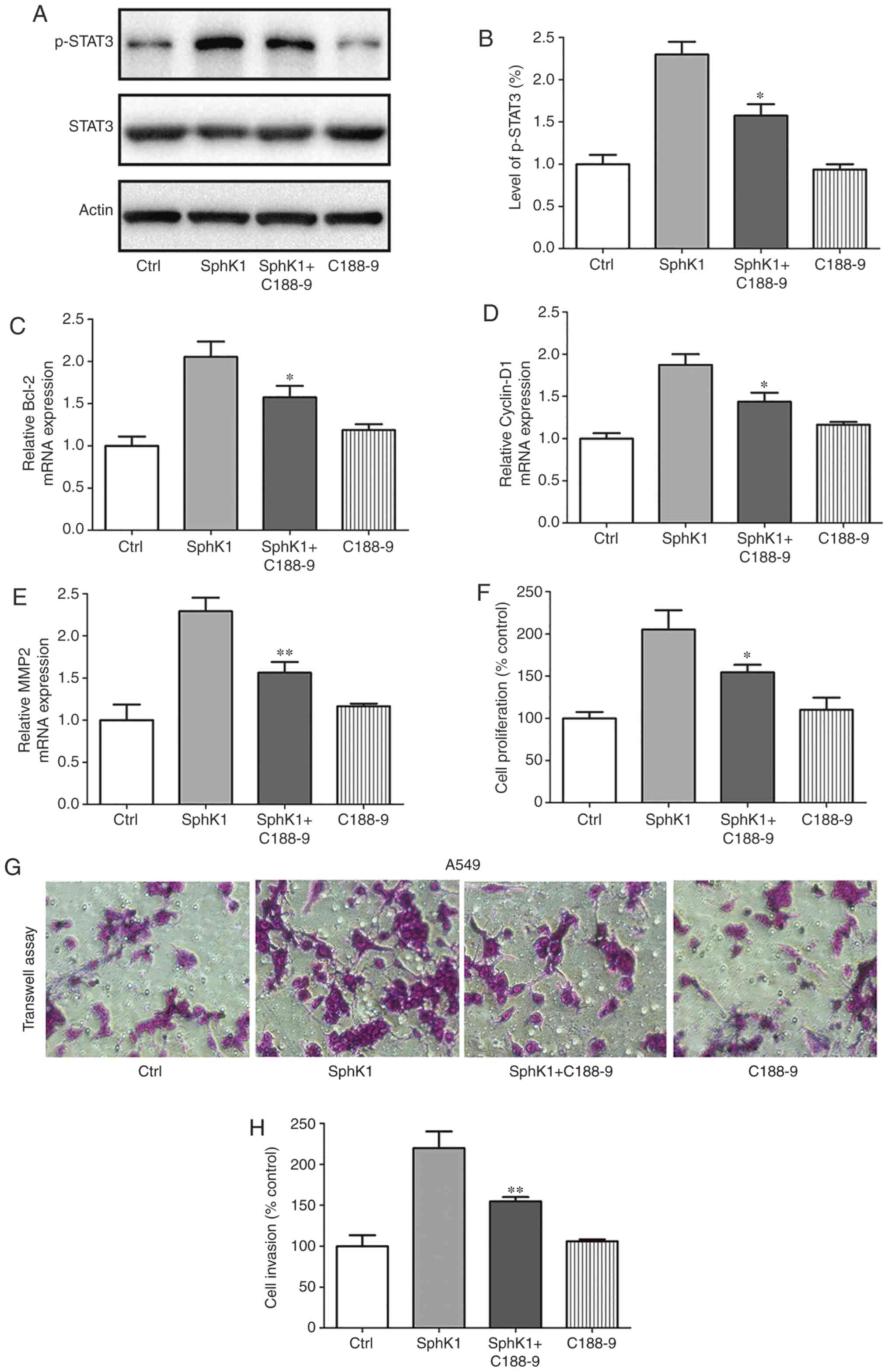 | Figure 6SphK1 regulates the proliferation and
invasion of A549 cells by activating STAT3. (A and B) SphK1
overexpression induced the activation of STAT3, whereas
SphK1G82D transfection attenuated the activation of
STAT3 compared with the control. (C-E) A549 cells were treated with
C188-9 (STAT3 inhibitor) following transfection with SphK1. mRNA
expression levels of (C) Bcl-2, (D) cyclin D1 and (E) MMP2 were
increased after transfection with SphK1 compared with the control
cells, and treatment with C188-9 reversed the increase in Bcl-2,
cyclin D1 and MMP2 mRNA levels. (F) Transfection with SphK1
increased cell viability compared with the control, whereas
treatment with C188-9 reversed this effect. (G and H) Transfection
with SphK1 increased the invasive capacity of A549 cells compared
with the control cells, whereas treatment with C188-9 reversed the
SphK1-induced increase. *P<0.05,
**P<0.01. SphK, sphingosine kinase; MMP, matrix
metalloproteinase STAT3, signal transducer and activator of
transcription 3; Ctrl, control. |
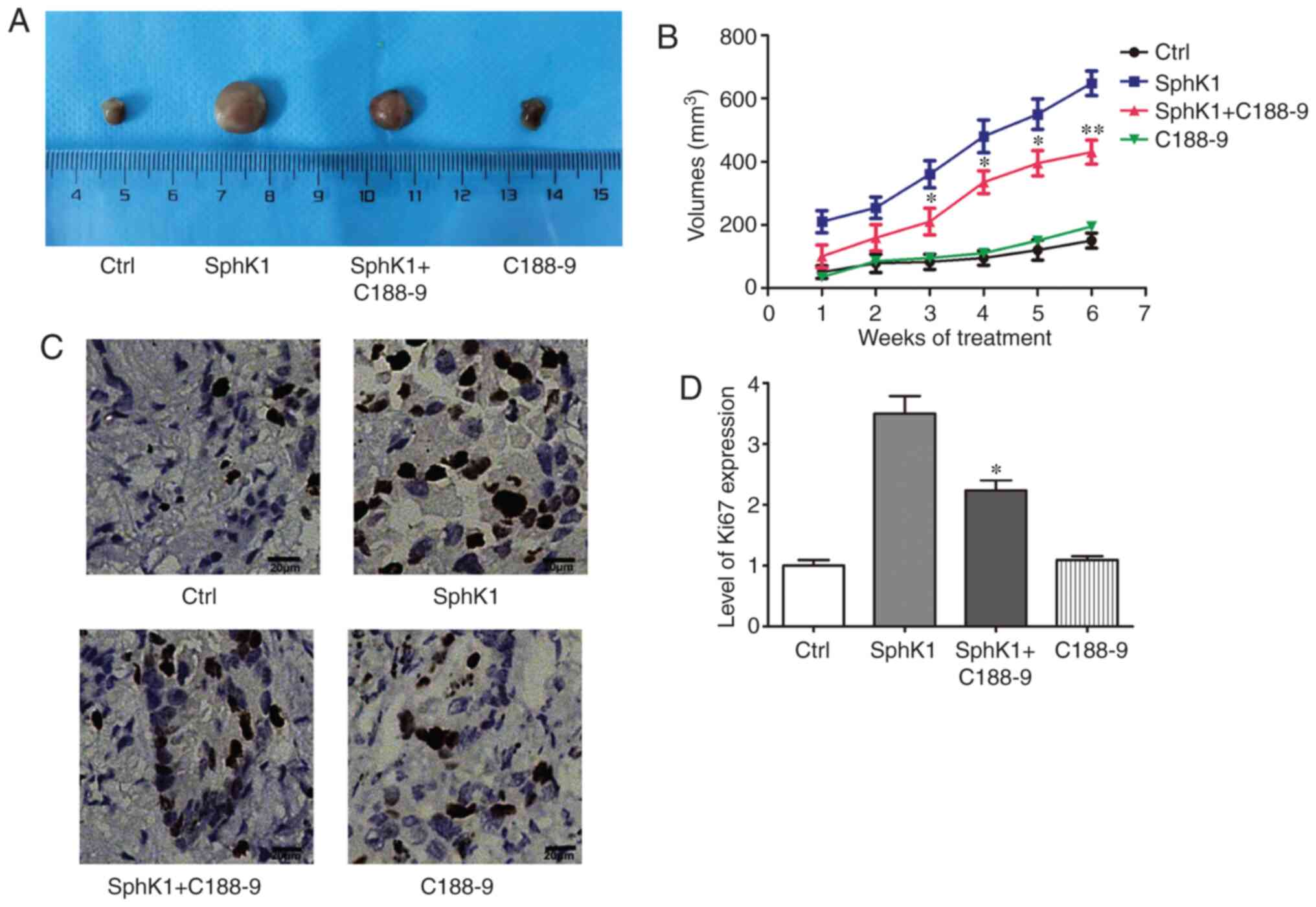
SphK1 promotes NSCLC progression in vivo
by activating STAT3
To further evaluate the effects of SphK1 on
tumorigenesis and explore the underlying mechanism, A549 cells
stably overexpressing SphK1 were used to establish a xenograft lung
cancer model in nude mice (Fig.
S2). The results demonstrated that overexpression of SphK1
resulted in a significant increase in tumor size compared with the
control group; however, treatment with C188-9 reversed the
SphK1-induced increase in tumor size (Fig. 7A and B). Ki67 expression was
analyzed in these tumor samples as an indicator of proliferation in
the tumor. The immunohistochemistry results revealed that Ki67 was
highly expressed in the SphK1 group, and the STAT3 inhibitor
reduced the SphK1-induced increase in Ki67 levels (Fig. 7C and D). Together, these results
suggested that SphK1 promoted tumor growth through activation of
STAT3 in vivo.
Discussion
Despite notable advances in our understanding of
lung cancer, the underlying mechanisms remain to be completely
elucidated. SphK1 is an oncogenic enzyme that participates in cell
proliferation, migration and invasion (33). Upregulated expression of SphK1 has
been observed in gastric, breast and colorectal cancer (34-36). In addition, SphK1 was reported to
be highly expressed in NSCLC cell lines (37). High levels of SphK1 are associated
with tumor progression and unfavorable outcomes in patients with
NSCLC (12). In the present
study, the expression of SphK1 in NSCLC cell lines and tissues was
assessed, and the results demonstrated that both mRNA and protein
expression levels of SphK1 were increased in NSCLC compared with
the control cells and tissues. These results were consistent with
previous studies (38,39). Additionally, overexpression of
SphK1 promoted the proliferation and metastasis of NSCLC cells,
whereas inhibition of SphK1 catalytic activity (overexpression of
SphK1G82D) had the opposite effects. The results of the
present study were consistent with a previous study (40).
SphK1 has been demonstrated to modulate the
expression of EMT-associated genes, promoting the migratory and
metastatic capacity of cancer cells (41). SphK1 also promoted autophagy and
accelerated lysosomal degradation of CDH1/E-cadherin to induce EMT
in hepatoma cells (12). In
addition, SphK1 increased the resistance of breast cancer cells to
chemotherapeutic agents and radiotherapy (42). In the present study,
overexpression of SphK1 increased the expression of antiapoptotic
and metastasis-associated genes cyclin D1, MMP2 and Bcl-2. These
target genes have been demonstrated to affect tumor progression in
gastric and renal cell carcinoma (43,44). The results of the present study
provided novel evidence demonstrating the role of SphK1 in cancer
development.
Expression of STAT3 has been estimated to be
aberrantly increased in >70% of the different types of cancer,
including acute myeloid leukemia, bone, breast cancer and solid
tumors of the bladder (19,45). Modulating the function or
expression of STAT3 has demonstrated the importance of STAT3 in the
development of different types of cancer (46). STAT3 is typically involved in the
regulation of cell proliferation, immunosuppression and apoptosis
resistance (47). As a
transcription factor, STAT3 is hypothesized to function by inducing
the expression of its target genes; for example, STAT3 affects cell
proliferation by modulating transcription of Bcl-xL, cyclin D1 or
MYC (48,49). Through modulation of the
expression of vascular endothelial growth factor receptor, STAT3
regulates angiogenesis (50). In
osteosarcoma, STAT3 regulates the transcription of serine/threonine
kinase 35, which results in increased proliferation and reduced
apoptosis of osteosarcoma cells (51). In breast cancer, STAT3 cross-talks
with the NF-κB signaling pathway by targeting the tumor necrosis
factor receptor superfamily member 1A to induce cancer development
(19). Small-molecule STAT3
inhibitors have been reported to induce apoptosis in acute myeloid
leukemia (46). In NSCLC, STAT3
promotes metastasis via aldoketo reductase family 1 member C1
(52). In the present study,
STAT3 was activated in A549 cells overexpressing SphK1, and
overexpression of SphK1G82D resulted in reduced STAT3
activity.
SphK1 has been demonstrated to modulate and to be
modulated by multiple signaling pathways. SphK1 activates the
PI3K/Akt/NF-κB pathway in NSCLC to enhance cell viability (24). By compromising protein kinase C
activity, SphK1 has been demonstrated to induce the proliferation
and survival in breast, lung and colon carcinoma cells (53-55). S1P, the product of SphK1, is an
important factor involved in the activation of STAT3 in cancer
cells (56). However, the
association between SphK1 and STAT3 remains to be elucidated in
NSCLC. To address this gap in our knowledge, A549 cells where
treated with STAT3 inhibitor following transfection of SphK1 in the
present study. The results revealed that overexpression of SphK1
activated STAT3 compared with the control cells, whereas inhibition
of STAT3 reduced the antiapoptotic role of SphK1 by disturbing the
regulation of target genes.
However, the means by which SphK1 regulates
expression of STAT3 remains unknown. A previous study has
demonstrated that the activation of SphK1 results in increased
production of S1P, which in turn increases the expression of IL-6
and subsequent activation of STAT3 in colitis-associated cancer
(18). In dystrophic muscles, S1P
activates STAT3 by inhibiting Rho GTPase Rac1 in a S1P receptor 2
(S1PR2)-dependent manner (57).
Although it has been reported high SphK1 expression is associated
with poor overall survival and acts as a prognosis predictor in
NSCLC (56), the results of the
present study further confirmed that SphK1 affected the NSCLC cell
proliferation and invasion by modulating apoptosis- and
metastasis-associated genes Bcl-2, Bcl-xL, cyclin D1, E-cadherin
and MMP2. The signaling pathways regulated by SphK1 have also been
investigated in multiple types of cancer. For example, STAT3 was
revealed to be targeted by SphK1 in colorectal cancer,
neuroblastoma, colitis-associated cancer and hepatocellular
carcinoma (58). However, whether
SphK1 regulates the expression of STAT3 has not been reported. It
also remains to be explored whether this pathway may promote
tumorigenesis in NSCLC. The present study demonstrated that SphK1
modulated the activation of STAT3 in NSCLC. In addition, SphK1
promoted the NSCLC development via the STAT3 pathway in
vitro and in vivo. Inhibiting STAT3 may be a potential
strategy to compromise the oncogenic role of SphK1 in NSCLC.
In summary, increased expression of SphK1 was
observed in NSCLC tissues and cells compared with control tissues
and normal lung cells, respectively, and potential target genes
were identified, which may account for the oncogenic effects of
SphK1. Additionally, an SphK1-STAT3 axis was identified as a novel
mechanism underlying the progression of NSCLC. The results of the
present study suggested that inhibition of the SphK1-STAT3 axis may
represent a potential strategy for treatment of patients with
NSCLC.
Supplementary Data
Funding
The present study was supported by the Key Research
and Development Plan Project of Shaanxi Province (grant no.
2019SF-221; Henan, China).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GC and YM designed the study. YM, XX and RK
performed the experiments. CC, SuL and ShL performed the
statistical analysis. YM, XX, XY and LS validated the data. GC, FZ
and YM analyzed the data. XX and YM performed the experiments. GC
curated the data. YM, XX and GC wrote and revised the manuscript.
YM, XX, RK and CC produced the graphs. GC supervised the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All mouse experiments were approved by The
Institutional Animal Care and Use Committee of The First Affiliated
Hospital of Xi'an Jiaotong University (Xi'an, China). All
procedures performed on patient tissues in the present study were
approved by the Research Ethics Committee of The First Affiliated
Hospital of Xi'an Jiaotong University, and written informed consent
was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
IL-6
|
interleukin 6
|
|
NC
|
negative control
|
|
NSCLC
|
non-small cell lung cancer
|
|
SCLC
|
small cell lung cancer
|
|
SH2
|
SRC homology 2
|
|
SphK1
|
sphingosine kinase 1
|
|
S1PR1
|
sphingosine-1-phosphate receptor 1
|
|
SRC
|
homology 2
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zheng H, Zhan Y, Liu S, Lu J, Luo J, Feng
J and Fan S: The roles of tumor-derived exosomes in non-small cell
lung cancer and their clinical implications. J Exp Clin Cancer Res.
37:2262018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hirsch FR, Suda K, Wiens J and Bunn PA Jr:
New and emerging targeted treatments in advanced non-small-cell
lung cancer. Lancet. 388:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu L, Wang Z, Lin Y, Chen Z, Liu H, Chen
Y, Wang N and Song X: Sphingosine kinase 1 enhances the invasion
and migration of non-small cell lung cancer cells via the AKT
pathway. Oncol Rep. 33:1257–1263. 2015. View Article : Google Scholar
|
|
5
|
Maceyka M, Sankala H, Hait NC, Le Stunff
H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, et
al: SphK1 and SphK2, sphingosine kinase isoenzymes with opposing
functions in sphingolipid metabolism. J Biol Chem. 280:37118–37129.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adams DR, Pyne S and Pyne NJ: Sphingosine
kinases: Emerging structure-function insights. Trends Biochem Sci.
41:395–409. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meacci E and Garcia-Gil M: S1P/S1P
receptor signaling in neuromuscolar disorders. Int J Mol Sci.
20:E63642019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sukocheva OA, Furuya H, Ng ML, Friedemann
M, Menschikowski M, Tarasov VV, Chubarev VN, Klochkov SG, Neganova
ME, Mangoni AA, et al: Sphingosine kinase and
sphingosine-1-phosphate receptor signaling pathway in inflammatory
gastrointestinal disease and cancers: A novel therapeutic target.
Pharmacol Ther. 207:1074642020. View Article : Google Scholar
|
|
9
|
Andrieu G, Ledoux A, Branka S, Bocquet M,
Gilhodes J, Walzer T, Kasahara K, Inagaki M, Sabbadini RA,
Cuvillier O and Hatzoglou A: Sphingosine 1-phosphate signaling
through its receptor S1P5 promotes chromosome
segregation and mitotic progression. Sci Signal. 10:eaah40072017.
View Article : Google Scholar
|
|
10
|
Lee CF, Dang A, Hernandez E, Pong RC, Chen
B, Sonavane R, Raj G, Kapur P, Lin HY, Wu SR, et al: Activation of
sphingosine kinase by lipopolysaccharide promotes prostate cancer
cell invasion and metastasis via SphK1/S1PR4/matriptase. Oncogene.
38:5580–5598. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng X, Li W, Ren L, Liu J, Pang X, Chen
X, Kang, Wang J and Du G: The sphingosine
kinase-1/sphingosine-1-phosphate axis in cancer: Potential target
for anticancer therapy. Pharmacol Ther. 195:85–99. 2019. View Article : Google Scholar
|
|
12
|
Liu H, Ma Y, He HW, Zhao WL and Shao RG:
SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal
transition by promoting the autophagy-linked lysosomal degradation
of CDH1/E-cadherin in hepatoma cells. Autophagy. 13:900–913. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hillmer EJ, Zhang H, Li HS and Watowich
SS: STAT3 signaling in immunity. Cytokine Growth Factor Rev.
31:1–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galoczova M, Coates P and Vojtesek B:
STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett.
23:122018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, van Boxel-Dezaire AH, Cheon H,
Yang J and Stark GR: STAT3 activation in response to IL-6 is
prolonged by the binding of IL-6 receptor to EGF receptor. Proc
Natl Acad Sci USA. 110:16975–16980. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao D, Pan C, Sun J, Gilbert C,
Drews-Elger K, Azzam DJ, Picon-Ruiz M, Kim M, Ullmer W, El-Ashry D,
et al: VEGF drives cancer-initiating stem cells through
VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene.
34:3107–3119. 2015. View Article : Google Scholar
|
|
18
|
Liang J, Nagahashi M, Kim EY, Harikumar
KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et
al: Sphingosine-1-phosphate links persistent STAT3 activation,
chronic intestinal inflammation, and development of
colitis-associated cancer. Cancer Cell. 23:107–120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Egusquiaguirre SP, Yeh JE, Walker SR, Liu
S and Frank DA: The STAT3 target gene TNFRSF1A modulates the NF-κB
pathway in breast cancer cells. Neoplasia. 20:489–498. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Horibata S, Rice EJ, Zheng H, Mukai C, Chu
T, Marks BA, Coonrod SA and Danko CG: A bi-stable feedback loop
between GDNF, EGR1, and ERα contribute to endocrine resistant
breast cancer. PLoS One. 13:e01945222018. View Article : Google Scholar
|
|
21
|
Masuda M, Suzui M, Yasumatu R, Nakashima
T, Kuratomi Y, Azuma K, Tomita K, Komiyama S and Weinstein IB:
Constitutive activation of signal transducers and activators of
transcription 3 correlates with cyclin D1 overexpression and may
provide a novel prognostic marker in head and neck squamous cell
carcinoma. Cancer Res. 62:3351–3355. 2002.PubMed/NCBI
|
|
22
|
Xu L, Wang P, Feng X, Tang J, Li L, Zheng
X, Zhang J, Hu Y, Lan T, Yuan K, et al: SETD3 is regulated by a
couple of microRNAs and plays opposing roles in proliferation and
metastasis of hepatocellular carcinoma. Clin Sci (Lond).
133:2085–2105. 2019. View Article : Google Scholar
|
|
23
|
Merino D, Lok SW, Visvader JE and Lindeman
GJ: Targeting BCL-2 to enhance vulnerability to therapy in estrogen
receptor-positive breast cancer. Oncogene. 35:1877–1887. 2016.
View Article : Google Scholar
|
|
24
|
Liu L, Zhou XY, Zhang JQ, Wang GG, He J,
Chen YY, Huang C, Li L and Li SQ: LncRNA HULC promotes non-small
cell lung cancer cell proliferation and inhibits the apoptosis by
up-regulating sphingosine kinase 1 (SPHK1) and its downstream
PI3K/Akt pathway. Eur Rev Med Pharmacol Sci. 22:8722–8730.
2018.PubMed/NCBI
|
|
25
|
Fan Z, Jiang H, Wang Z and Qu J:
Atorvastatin partially inhibits the epithelial-mesenchymal
transition in A549 cells induced by TGF-β1 by attenuating the
upregulation of SphK1. Oncol Rep. 36:1016–1022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schwartz LH, Seymour L, Litiere S, Ford R,
Gwyther S, Mandrekar S, Shankar L, Bogaerts J, Chen A, Dancey J, et
al: RECIST 1.1 Standardisation and disease-specific adaptations:
Perspectives from the RECIST working group. Eur J Cancer.
62:138–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Tang Z, Kang B, Li C, Chen T and Zhang Z:
GEPIA2: An enhanced web server for large-scale expression profiling
and interactive analysis. Nucleic Acids Res. 47(W1): W556–W560.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lan T, Liu W, Xie X, Xu S, Huang K, Peng
J, Shen X, Liu P, Wang L, Xia P and Huang H: Sphingosine kinase-1
pathway mediates high glucose-induced fibronectin expression in
glomerular mesangial cells. Mol Endocrinol. 25:2094–2105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Tang H, Sysol JR, Moreno-Vinasco
L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV,
et al: The sphingosine kinase 1/sphingosine-1-phosphate pathway in
pulmonary arterial hypertension. Am J Respir Crit Care Med.
190:1032–1043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hardy E, Hardy-Sosa A and Fernandez-Patron
C: MMP-2: Is too low as bad as too high in the cardiovascular
system? Am J Physiol Heart Circ Physiol. 315:H1332–H1340. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lo UG, Bao J, Cen J, Yeh HC, Luo J, Tan W
and Hsieh JT: Interferon-induced IFIT5 promotes
epithelial-to-mesenchymal transition leading to renal cancer
invasion. Am J Clin Exp Urol. 7:31–45. 2019.PubMed/NCBI
|
|
33
|
Wu X, Wu Q, Zhou X and Huang J: SphK1
functions down-stream of IGF-1 to modulate IGF-1-induced EMT,
migration and paclitaxel resistance of A549 cells: A preliminary in
vitro study. J Cancer. 10:4264–4269. 2019. View Article : Google Scholar :
|
|
34
|
Yin S, Miao Z, Tan Y, Wang P, Xu X, Zhang
C, Hou W, Huang J and Xu H: SPHK1-induced autophagy in peritoneal
mesothelial cell enhances gastric cancer peritoneal dissemination.
Cancer Med. 8:1731–1743. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Acharya S, Yao J, Li P, Zhang C, Lowery
FJ, Zhang Q, Guo H, Qu J, Yang F, Wistuba II, et al: Sphingosine
kinase 1 signaling promotes metastasis of triple-negative breast
cancer. Cancer Res. 79:4211–4226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen Z, Feng X, Fang Y, Li Y, Li Z, Zhan
Y, Lin M, Li G, Ding Y and Deng H: POTEE drives colorectal cancer
development via regulating SPHK1/p65 signaling. Cell Death Dis.
10:8632019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song L, Xiong H, Li J, Liao W, Wang L, Wu
J and Li M: Sphingosine kinase-1 enhances resistance to apoptosis
through activation of PI3K/Akt/NF-κB pathway in human non-small
cell lung cancer. Clin Cancer Res. 17:1839–1849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang L, Hu H, Deng Y and Bai Y: Role of
SPHK1 regulates multi-drug resistance of small cell lung cancer and
its clinical significance. Zhongguo Fei Ai Za Zhi. 17:769–777.
2014.In Chinese. PubMed/NCBI
|
|
39
|
Zhang G, Zheng H, Zhang G, Cheng R, Lu C,
Guo Y and Zhao G: MicroRNA-338-3p suppresses cell proliferation and
induces apoptosis of non-small-cell lung cancer by targeting
sphingosine kinase 2. Cancer Cell Int. 17:462017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Y, Hu T, Chen T, Yang T, Ren H and Chen
M: Combination treatment of FTY720 and cisplatin exhibits enhanced
antitumour effects on cisplatin-resistant non-small lung cancer
cells. Oncol Rep. 39:565–572. 2018.
|
|
41
|
Xu CY, Liu SQ, Qin MB, Zhuge CF, Qin L,
Qin N, Lai MY and Huang JA: SphK1 modulates cell migration and
EMT-related marker expression by regulating the expression of p-FAK
in colorectal cancer cells. Int J Mol Med. 39:1277–1284. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marvaso G, Barone A, Amodio N, Raimondi L,
Agosti V, Altomare E, Scotti V, Lombardi A, Bianco R, Bianco C, et
al: Sphingosine analog fingolimod (FTY720) increases radiation
sensitivity of human breast cancer cells in vitro. Cancer Biol
Ther. 15:797–805. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen XF, Guo JF, Xu JF, Yin SH and Cao WL:
MiRNA-206 inhibits proliferation of renal clear cell carcinoma by
targeting ZEB2. Eur Rev Med Pharmacol Sci. 23:7826–7834.
2019.PubMed/NCBI
|
|
44
|
Wang Z, Tang T, Wang S, Cai T, Tao H,
Zhang Q, Qi S and Qi Z: Aloin inhibits the proliferation and
migration of gastric cancer cells by regulating NOX2-ROS-Mediated
pro-survival signal pathways. Drug Des Devel Ther. 14:145–155.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li
X and Chang C: Chidamide, a novel histone deacetylase inhibitor,
inhibits the viability of MDS and AML cells by suppressing
JAK2/STAT3 signaling. Am J Transl Res. 8:3169–3178. 2016.PubMed/NCBI
|
|
46
|
Guanizo AC, Fernando CD, Garama DJ and
Gough DJ: STAT3: A multifaceted oncoprotein. Growth Factors.
36:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lewis KM, Bharadwaj U, Eckols TK, Kolosov
M, Kasembeli MM, Fridley C, Siller R and Tweardy DJ: Small-molecule
targeting of signal transducer and activator of transcription
(STAT) 3 to treat non-small cell lung cancer. Lung Cancer.
90:182–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kanda N, Seno H, Konda Y, Marusawa H,
Kanai M, Nakajima T, Kawashima T, Nanakin A, Sawabu T, Uenoyama Y,
et al: STAT3 is constitutively activated and supports cell survival
in association with survivin expression in gastric cancer cells.
Oncogene. 23:4921–4929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kijima T, Niwa H, Steinman RA, Drenning
SD, Gooding WE, Wentzel AL, Xi S and Grandis JR: STAT3 activation
abrogates growth factor dependence and contributes to head and neck
squamous cell carcinoma tumor growth in vivo. Cell Growth Differ.
13:355–362. 2002.PubMed/NCBI
|
|
50
|
Lei Z, Duan H, Zhao T, Zhang Y, Li G, Meng
J, Zhang S and Yan W: PARK2 inhibits osteosarcoma cell growth
through the JAK2/STAT3/VEGF signaling pathway. Cell Death Dis.
9:3752018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu Z, Liu J, Hu S, Zhu Y and Li S:
Serine/Threonine kinase 35, a target gene of STAT3, regulates the
proliferation and apoptosis of osteosarcoma cells. Cell Physiol
Biochem. 45:808–818. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhu H, Chang LL, Yan FJ, Hu Y, Zeng CM,
Zhou TY, Yuan T, Ying MD, Cao J, He QJ and Yang B: AKR1C1 activates
STAT3 to promote the metastasis of non-small cell lung cancer.
Theranostics. 8:676–692. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nazouri AS, Asadpour O, Dabiri S,
Pourseyedi B, Lashkarizadeh MR and Zianalinejad H: High expression
of sphingosine kinase 1 in estrogen and progesterone
receptors-negative breast cancer. Iran J Pathol. 12:218–224. 2017.
View Article : Google Scholar
|
|
54
|
Sankala HM, Hait NC, Paugh SW, Shida D,
Lépine S, Elmore LW, Dent P, Milstien S and Spiegel S: Involvement
of sphingosine kinase 2 in p53-independent induction of p21 by the
chemo-therapeutic drug doxorubicin. Cancer Res. 67:10466–10474.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu SQ, Xu CY, Wu WH, Fu ZH, He SW, Qin MB
and Huang JA: Sphingosine kinase 1 promotes the metastasis of
colorectal cancer by inducing the epithelial-mesenchymal transition
mediated by the FAK/AKT/MMPs axis. Int J Oncol. 54:41–52. 2019.
|
|
56
|
Nagahashi M, Yamada A, Katsuta E, Aoyagi
T, Huang WC, Terracina KP, Hait NC, Allegood JC, Tsuchida J, Yuza
K, et al: Targeting the SphK1/S1P/S1PR1 axis that links obesity,
chronic inflammation, and breast cancer metastasis. Cancer Res.
78:1713–1725. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Loh KC, Leong WI, Carlson ME, Oskouian B,
Kumar A, Fyrst H, Zhang M, Proia RL, Hoffman EP and Saba JD:
Sphingosine-1-phosphate enhances satellite cell activation in
dystrophic muscles through a S1PR2/STAT3 signaling pathway. PLoS
One. 7:e372182012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jin Z, Li H, Hong X, Ying G, Lu X, Zhuang
L and Wu S: TRIM14 promotes colorectal cancer cell migration and
invasion through the SPHK1/STAT3 pathway. Cancer Cell Int.
18:2022018. View Article : Google Scholar : PubMed/NCBI
|