Introduction
Epilepsy is one of the most common clinical
neurological diseases, with a high incidence. The prevalence of
epilepsy worldwide is approximately 6.38-7.60% (1,2).
The pathogenesis of epilepsy is not very clear, although studies
have confirmed that the normal function of brain cells requires a
stable neurovascular unit, suggesting that vascular endothelial
dysfunction may be related to its pathogenesis (3,4).
Endothelial monocyte-activating polypeptide II (EMAP
II), which is derived from its precursor aminoacyl-tRNA
synthetase-interacting multi-functional protein 1 (AIMP1), has a
molecular weight of approximately 22 kDa. It can promote cell
apoptosis, inhibit tumor angiogenesis and mediate inflammation
(5). EMAP II has a wide range of
biological associations with endothelial cells. It can stimulate
the expression of P-selectin and E-selectin, promote the release of
von Willebrand factor (vWF) in endothelial cells, and induce the
production of tissue factors on the surface of endothelial cells,
activating pro-coagulant activity. In addition, it can promote the
secretion of tumor necrosis factor (TNF)-α and interleukin (IL)-8
by monocytes, regulate the expression of TNF-α receptor 1, and
enhance the sensitivity of endothelial cells to TNF-α-induced
apoptosis (5,6). The majority of research into EMAP II
has focused on its roles in tumors, diabetes, athero-sclerosis,
chronic myocardial infarction and lung injury (7-10).
Studies have found that EMAP II is a sensitive
marker of neurotoxic injury in a variety of central nervous system
diseases, such as autoimmune encephalomyelitis and traumatic brain
injury (11,12). Mice with genetic defects in the
EMAP II precursor, AIMP1, can develop degenerative diseases of the
motor neuron axons or muscle atrophy and exhibit movement
dysfunction (13). Patients with
the homozygous deletion of the AIMP1 gene exhibit developmental
delays, refractory epilepsy, or poor myelination of the brain
(14). However, research into the
relationship between EMAP II and epilepsy is rare.
AIMP1 is enzymatically converted into EMAP II under
hypoxic conditions or in response to certain chemotherapeutic drugs
or apoptosis (15-17). Knies et al (15), through in vitro
experiments, confirmed that EMAP II can be detected in the cultures
of apoptotic cells, but not in those of necrotic cells, suggesting
that the enzymolysis and the release of EMAP II may be associated
with apoptosis. Some researchers have found that caspase 3, which
has an apoptosis-promoting function, can significantly increase
EMAP II expression in the lungs, with caspase inhibitors reducing
EMAP II expression (18). Barnett
et al (16) found that the
induction of apoptosis or the necrosis of prostate cancer cells
allowed AIMP1 to be released into the extracellular space and
cleaved to form EMAP II. These experiments suggest that apoptotic
cells can produce EMAP II and that EMAP II can itself promote cell
apoptosis.
The CD31 molecule is also known as platelet
endothelial cell adhesion molecule-1 (PECAM-1). Intracranial CD31
is mainly found at the tight junction between endothelial cells,
and its immunohistochemical detection can be used to prove the
presence of endothelial cells and evaluate angiogenesis.
Angiogenesis is often associated with the development of epilepsy.
Clinically, compared with non-epileptic patients, the vascular
density in the hippocampal tissue of patients with chronic
refractory temporal lobe epilepsy is significantly increased, is
positively associated with the frequency of epileptic seizures, and
is unrelated to the etiology and degree of neuronal loss (19). In cultured hippocampal brain
slices, following the epileptic discharge induced by kainic acid,
the numbers of cells exhibiting laminin and rat endothelial cell
antigen (RECA-1) immunofluorescence are significantly increased,
the vascular density and branching are augmented, and the tight
junction protein, zonula occludens 1 (ZO-1), is downregulated
(20).
Neurons in the hippocampus have been reported to be
hypoxic several seconds before, during, and after the seizure,
particularly the pyramidal cell layer of the CA1 region (21). In addition, extensive neuronal
apoptosis and pyroptosis have been identified to be involved in
epileptogenesis (22). Therefore,
it was hypothesized that the expression of EMAP II in the rat brain
may become altered following status epilepticus (SE). Accordingly,
the present study examined the changes in the expression of EMAP II
and the tight junction proteins, ZO-1 and occluding, as well as in
the numbers of CD31-positive microvascular endothelial cells in the
rat hippocampus following SE.
Materials and methods
Animals and groups
A total of 240 healthy male Sprague-Dawley rats, 21
days old and weighing 45-65 g, were used in the present study. The
rats were kept at a room temperature of 22-24°C and under a 12
a.m./12 p.m. light/dark cycle. All animal experiments were approved
by the Ethics Committee of Shengjing Hospital Affiliated to China
Medical University (no. 2016PS201K).
All the rats were randomly divided into 2 groups as
follows: i) The control group (n=120), which received an
intraperitoneal injection of saline; and ii) the SE group (n=120),
which received an intraperitoneal injection of lithium
chloride-pilocarpine. The time of SE appearance in the experimental
group was considered 0 h. In total of 6 time points, were selected
for both groups: 2 and 24 h, and 3, 7, 14 and 21 days. A total of
20 rats were selected at each time point.
Establishment of animal models of SE
According to the model described in the previous
studies (23,24), the rats in the model group were
intraperitoneally injected with lithium chloride solution [3 mEq/kg
(127 mg/kg), Sigma-Aldrich; Merck KGaA]. Subsequently, 18-20 h
later, the rats in the model group were intraperitoneally injected
with pilocarpine solution (30 mg/kg, Sigma-Aldrich; Merck KGaA); at
30 min prior to the pilocarpine administration, the rats were
intraperitoneally injected with bromide scopolamine solution (1
mg/kg, Sigma-Aldrich; Merck KGaA) to antagonize the peripheral
cholinergic reaction induced by pilocarpine. Level IV-V Racine
seizures, according to the rat grade evaluation standard (25), were considered to indicate a
successful model. If no seizures occurred after the first dose of
pilocarpine, an additional injection of pilocarpine (10 mg/kg dose)
was administered every 30 min until the rats developed level IV-V
Racine seizure attacks. The maximum dose of pilocarpine
intraperitoneally administered to each rat was not >60 mg/kg.
The rats in the control group were administered the same amount of
normal saline via intraperitoneal injection. To reduce the
mortality rate of the epileptic rats, diazepam solution (10 mg/kg)
was intraperitoneally injected into the rats after the SE lasted 30
min to terminate the SE attack as described in a previous study
(24). The weights of the rats in
each group were measured before their drug treatment and sampling.
Samples from the 2 groups were collected at 6 time points after
modeling. Rats were gas-anesthetized by inhaling 5% isoflurane
mixed with 50/50% oxygen/nitrogen. When the limbs were fixed and
the tail pinched, there was no significant limb response, the rats
were subjected to abdominal aorta bleeding until cardiac arrest,
followed by sampling. At different time points following SE, the
intact brain tissues from 6 rats were obtained and fixed in 4%
paraformaldehyde for morphological analysis, while hippocampal
tissues from another 6 rats were collected and stored in a freezer
at −80°C for biological analysis.
Electroencephalogram recording of rats
with SE
At 3 days before modeling, all the rats were
administered 5% isoflurane mixed with 50/50% oxygen/nitrogen as an
inducible anesthesia, and the isoflurane concentration was
subsequently reduced to 2% to maintain the anesthesia. Their heads
were fixed in the stereoscopic locator in the prone position to
fully expose the anterior fontanelle. The cross-point of the
anterior fontanelle of the rats was selected as the zero point with
reference to the 5th edition of Paxinos & Watson brain
stereolocation map (26), and
then 2 points 4-mm posterior and 2-mm lateral to the anterior
fontanelle were selected. The electrode needles were inserted to a
depth of approximately 4 mm under the skull and were fixed by
tissue glue. The rats were placed in a shield and the
electroencephalogram (EEG) was used before and after model
establishment.
Immunofluorescence staining
Brain tissues fixed in 4% paraformaldehyde were
embedded in paraffin and then cut into 4-µm-thick sections.
Brain sections were dewaxed with xylene, dehydrated with a gradient
of alcohol, and then subjected to a microwave antigen retrieval
method (for EMAP II, the sections were heated with 0.01 mol/l
sodium citrate buffer, pH 6.0, and for CD31 they were heated with
Tris-EDTA buffer, pH 9.0). The sections were incubated with normal
goat serum to reduce non-specific staining and then with primary
antibody at 4°C overnight. The primary antibodies comprised rabbit
anti-rat EMAP II polyclonal antibody (1:100, 11091-1-AP,
ProteinTech Group, Inc.) and mouse anti-rat CD31 monoclonal
antibody (1:200, ab64543, Abcam). According to the primary antibody
source, goat anti-rabbit polyclonal secondary antibody (1:200,
ab150077, Abcam) or goat anti-mouse polyclonal secondary antibody
(1:200, ab150115, Abcam) were added the following day in a wet box
in the dark at room temperature. DAPI (C1005, Biyuntian Institute
of Biotechnology) was added for 5 min in the dark at room
temperature. The sections were then observed under a microscope
(Nikon Eclipse 80i; Nikon Corporation). The control group was
incubated with phosphate-buffered saline (PBS) instead of primary
antibodies. To quantify the EMAP II- and CD31-positive cells, 10
microscopic fields of the CA1, CA3 and dentate gyrus (DG) regions
were randomly observed at ×200 magnification. Image Pro Plus 6.0
image analysis software was used to determine the optical density
value.
Western blot analysis
Hippocampal tissue was incubated in RIPA lysis
buffer (PMSF, 100:1; P0013B, ST505, Biyuntian Institute of
Biotechnology), homogenized under ultrasound and then pyrolysed at
4°C for 30 min. The tissue was then centrifuged at 4°C for 15 min
at 18,800 × g. The protein concentration of the supernatant was
determined with the BCA protein assay kit (P0010S, Biyuntian
Institute of Biotechnology). The supernatant was mixed with 5X
buffer and denatured in a water bath at 100°C. Sample proteins in
each subgroup were adjusted to the same volume and concentration
according to the measured protein concentration. Proteins (40
µg) were then separated using various concentrations (10%
for EMAP II; 6% for ZO-1 and occludin) of Bis-Tris sodium dodecyl
sulfate-polyacrylamide electrophoresis gel (SDS-PAGE; Beyotime
Institute of Biotechnology), according to the molecular weight of
the target protein. The separated proteins were then transferred to
a PVDF membrane. The PVDF membrane was then placed in 5% skim milk
and blocked for 2 h at room temperature prior to incubation with
primary antibodies at 4°C overnight. The primary antibodies used
included mouse anti-rat EMAP II monoclonal antibody (1:1,000,
ab15693, Abcam), rabbit anti-rat ZO-1 polyclonal antibody (1:500,
WL03419, Biological Technology Co., Ltd.), rabbit anti-rat occludin
polyclonal antibody (1:500, WL03419, Biological Technology Co.,
Ltd.) and rabbit anti-rat alpha-tubulin antibody (1:10,000,112-1-24
AP, ProteinTech Group, Inc.). The following day, the membrane was
incubated for 2 h with goat anti-rabbit secondary antibody
(1:7,000, sa00001-2, ProteinTech Group, Inc.) or goat anti-mouse
secondary antibody (1:7,000, sa00001-1, ProteinTech Group, Inc.).
Protein bands were visualized using ECL (NCI5079; Thermo Fisher
Scientific, Inc.) and imaged using an electrophoresis gel imaging
system. ImageJ software (version 1.4.3.67) was used to analyze the
gray values of the western blot results, and the gray values of the
target proteins were normalized to that of α-tubulin.
Statistical analysis
SPSS 22.0 statistical software was used to analyze
the data. The data are presented as the means ± standard deviation,
and an independent sample t-test was used to compare the data
between the 2 groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Behavioral changes in SE rats
Following the establishment of the model, peripheral
cholinergic reactions, such as erect hair and salivation were
observed after 13.4±2.1 min in the rats in the SE group
administered pilocarpine. Level IV-V Racine seizures developed in
75.6% of rats in the SE group. After the convulsion lasted for 30
min, diazepam solution was intraperitoneally injected to terminate
the SE attack. Within 24 h, the rats still had repeated
convulsions, with an average of 5.6±1.2 convulsions per hour. The
24-h mortality rate of the rats was approximately 10.4%.
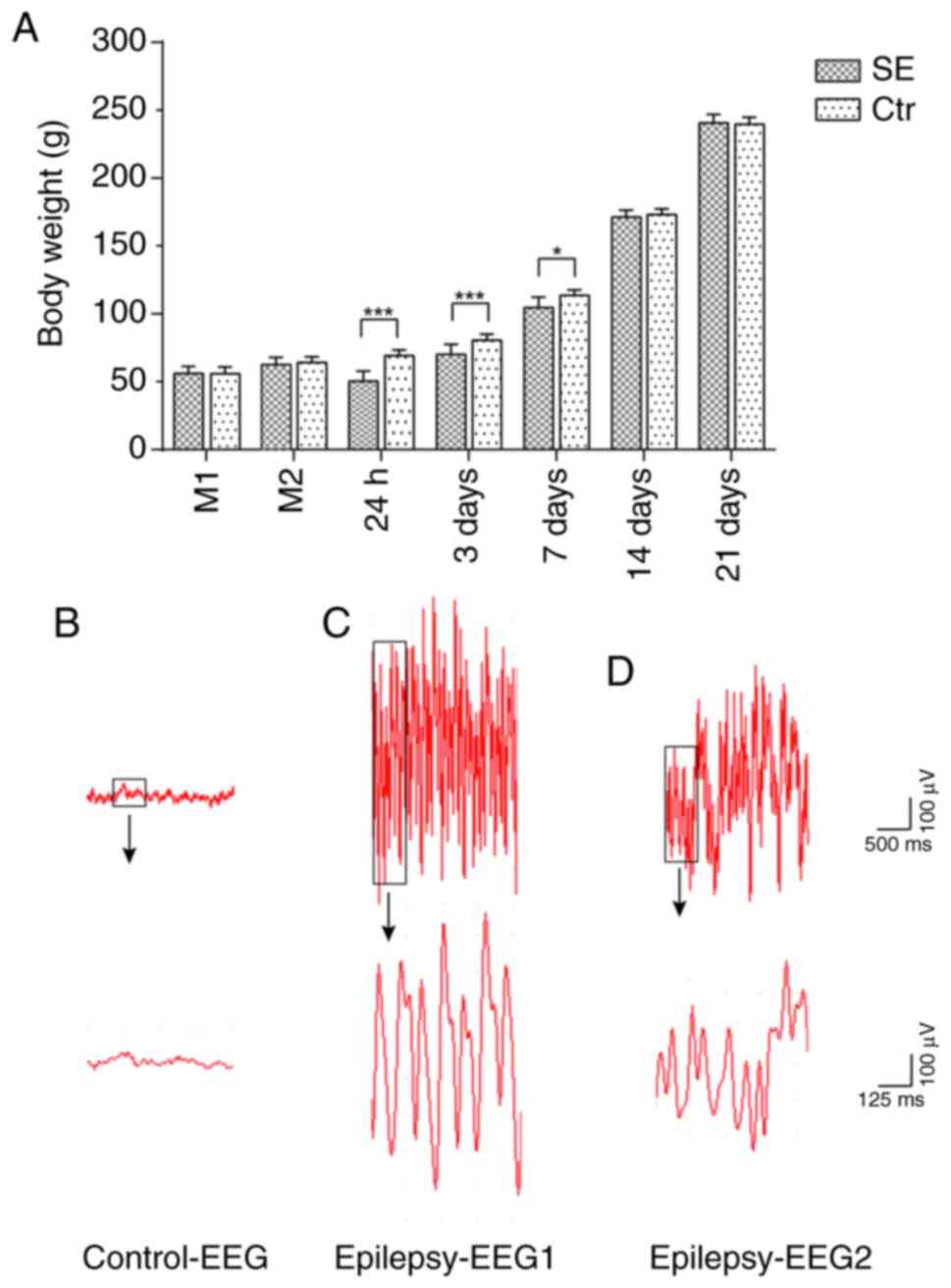
Body weight changes in rats with SE
Prior to model establishment, no significant
differences in body weight were observed between the rats in the
control and SE groups. Following the administration of lithium
chloride, no significant differences in body weight were observed
between the 2 groups. Following the administration of pilocarpine,
the rats in the control group gained weight regularly at 5-12 g per
day. By contrast, the rats in the SE group exhibited significant
weight loss 24 h after SE compared with the controls with a maximum
of 27.0% body weight loss and then began to gain weight. At 3 days,
their body weight returned to the pre-model weight, although it was
still significantly lower than that of the control group. At 7
days, there was still a difference in body weight between the 2
groups. Subsequently, the weights of the rats in the SE group
recovered, and no significant difference in the weights of the rats
were observed between the 2 groups at 14 days (Fig. 1A).
EEG monitoring of rats with SE
Following model establishment, the EEG of control
group rats revealed a basic rhythm of α and θ waves of a
low-to-middle amplitude of approximately 50 µV. The EEG of
the SE group revealed high-amplitude spike wave emission with a
frequency of approximately 10-20 Hz (Fig. 1B-D).
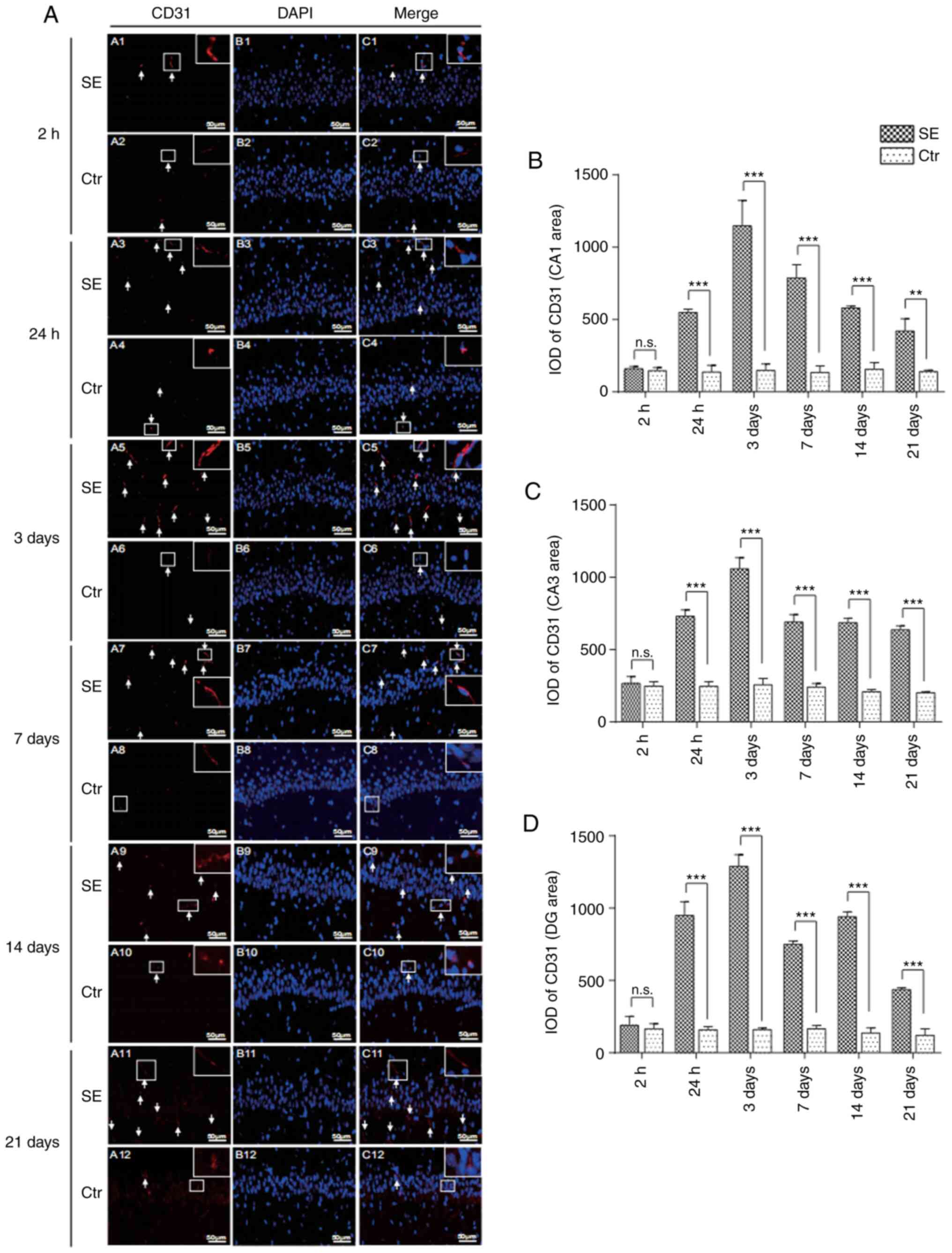
Changes in microvascular endothelial
cells in the hippocampus of rats with SE
Compared with the control group at the same time
point, the number of endothelial cells with positive CD31 staining
in the hippocampal CA1, CA3 and DG areas began to increase
significantly from 24 h in the SE group, with the optical density
being significantly higher in the SE group than in the control
group (P<0.001). The number of CD31-positive endothelial cells
peaked in the SE group at 3 days, and the optical density value was
significantly higher in the SE group than in the control group
(P<0.001). At 21 days, the number of CD31-positive endothelial
cells was significantly higher in the SE group than in the control
group at the same time point (Figs.
2 and S1).
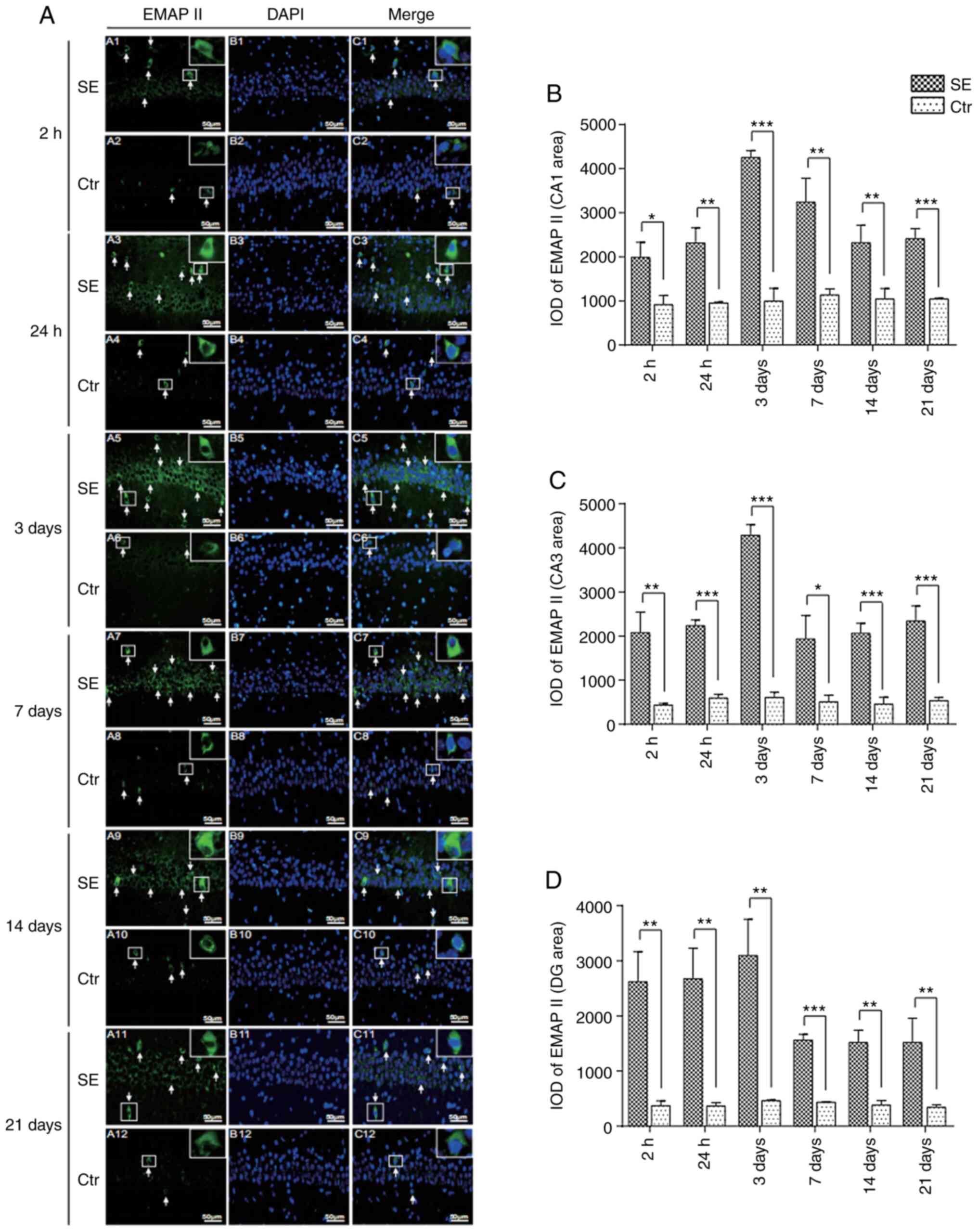
Changes in the expression of EMAP II in
the hippocampus of rats with SE
EMAP II was expressed at low levels in the
hippocampal CA1, CA3 and DG areas of the control group. However,
the numbers of EMAP II-positive cells were higher in the SE group
at 2 h. In addition, the optical density value was higher in the SE
group than in the control group (P<0.05, CA1 area; P<0.01,
CA3 and DG areas). The numbers of EMAP II-positive cells peaked on
day 3, and the optical density value of the SE group was
significantly higher than that of the control group (P<0.001,
CA1 and CA3 areas; P<0.01, DG area). The expression then
decreased, although the numbers of EMAP II-positive cells were
still significantly higher at 21 days in the SE group than in the
control group (Figs. 3 and
S2).
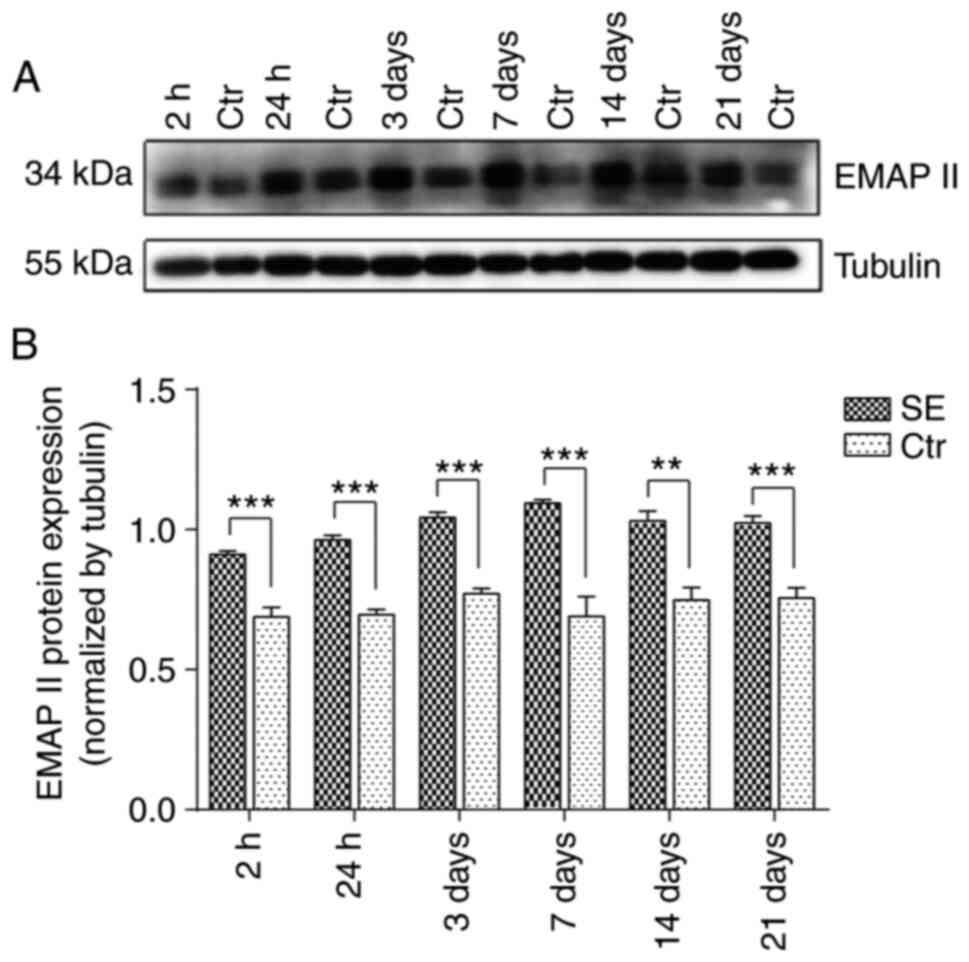
According to the results of western blot analysis,
EMAP II expression was not altered significantly over time in the
control group. However, the EMAP II protein expression levels were
significantly higher in the SE group at the same time points vs.
the control group. EMAP II protein expression began to gradually
increase after the seizures began, peaking at 7 days (P<0.001)
and then gradually declining. Nonetheless, the levels were still
significantly higher at 21 days than those in the control group
(Fig. 4).
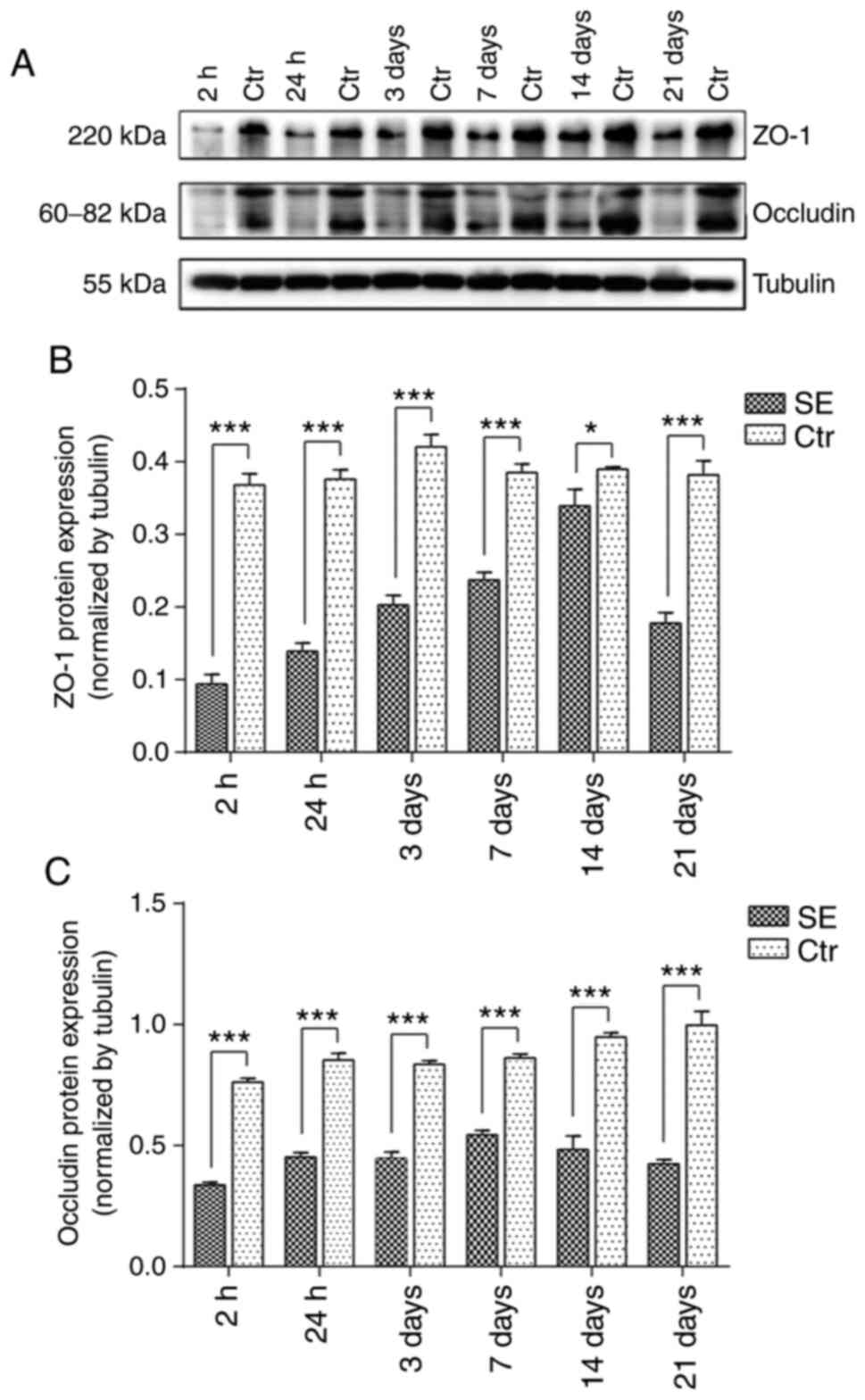
Changes in the expression of ZO-1 and
occludin in the hippocampal region of rats with SE
Western blot analysis revealed that, compared with
the control group at the same time points, the protein expression
of ZO-1 was significantly decreased at each time point in the SE
group. ZO-1 protein expression was most evidently decreased at 2 h
after SE and then gradually increased until 14 days, at which point
the expression of ZO-1 protein was still lower than that of the
control group. Compared with the control group at the same time
point, the protein expression of occludin was significantly
decreased in the SE group at each time point. At 2 h following SE,
occludin protein expression was at its lowest level, but then
gradually increased before decreasing again (Fig. 5).
Discussion
In the present study, an animal model of epilepsy
was established via an intraperitoneal injection of lithium
chlorine-pilocarpine, with EEG monitoring confirming that epileptic
spikes were discharged in the experimental rats. Within 24 h, the
rats still had frequent convulsions, and a small number of rats
died, with a 24-h mortality rate of approximately 10.4%, which was
similar to that in a previous study (24). We also found that the weights of
the rats had decreased significantly by 24 h before gradually
recovering. After 7 days, the weights of the rats began to recover
and they exhibited the same weight as the rats of the same age in
the control group at 14 days after model establishment. As the
durations of convulsions in the experimental group included in the
present study all exceeded 30 min, the current model can be used to
study SE and may better reflect the degree of brain injury in
rats.
Hypoxia and brain ischemic injury are always
involved in the process of epilepsy (21,27). Long-term seizures always cause
brain hypoxia, and hypoxia can inflict a direct toxic injury on
endothelial cells, as well as secondary blood-brain barrier (BBB)
damage. Such secondary damage includes abnormal mitochondrial
metabolism in endothelial cells, a thickening of the basal
membrane, compensatory endothelial cell proliferation, and the
presence of abnormal tight junctions (28,29).
Angiogenesis and the destruction of the BBB are
closely related to the occurrence and development of epilepsy. In
the rat model of SE induced by pilocarpine, cerebral vessels
exhibit varying degrees of morphology, with changes in, for
example, distribution, density, length, angiogenesis, BBB opening,
and cerebral blood flow and velocity (28). Recently, it was found that the
expression level of occludin and ZO-1 in the neocortical
microvessels of patients with drug-resistant temporal lobe epilepsy
was significantly decreased compared with autopsy samples (30). In the present study, vascular
endothelial cells were labeled through CD31 immunofluorescence and
it was found that the numbers of vascular endothelial cells in each
part of the rat hippocampus were significantly increased at 24 h
following SE and that the numbers of vascular endothelial cells
continued to be higher than that of the control group 21 days after
SE. At the same time, it was also found that the expression levels
of ZO-1 and occludin proteins in the SE group were significantly
decreased at each time point, which is consistent with previous
reports (20,30,31). Tight junctions are important for
the composition of the BBB. The increased vascular density
following SE is accompanied by decreased expression of tight
junction proteins, which indicates pathological angiogenesis. The
downregulation of tight junction proteins further confirmed the
increase in BBB permeability following SE, which would lead to the
abnormal exchange of substances inside and outside blood vessels.
This would make the distribution of neurotransmitters in the brain
abnormal and aggravate the occurrence and development of epilepsy
(28).
EMAP II is a type of tissue factor that can induce
endothelial cells to exert coagulant activity. A number of animal
and clinical studies have found that EMAP II is highly expressed in
endocrine organs, particularly neuroendocrine organs (17,32). In addition, cerebral ischemia
injury can induce EMAP II in brain tissue (33). Brabeck et al (34) found that the EMAP II-positive cell
number was proportional to the severity of the brain damage induced
by trimethyltin. Schikorski et al (12) found, through the analysis of nerve
repair, that EMAP II expression was significantly higher within 24
h, suggesting that EMAP II plays an important role in the process
of tissue regeneration. The present study found that the
hippocampal EMAP II expression of the SE group began to
significantly increase at 2 h after SE compared with the normal
control group, continuing until 21 days. As reported in previous
studies, epileptogenesis is related to hypoxia and apoptosis
(21,22), and the conversion of AIMP1 to EMAP
II is enhanced under hypoxic conditions and apoptosis (15-17). The present study demonstrated that
the expression of EMAP II increased after SE, suggesting that EMAP
II maybe related to epileptogenesis.
In the present study, EMAP II expression was
significantly increased at all time points following SE, while the
expression of ZO-1 and occludin proteins decreased. It has been
reported that a low expression of EMAP II could selectively
increase blood-tumor barrier (BTB) permeability via a transcellular
pathway through the RhoA/Rho kinase signaling pathway (35). EMAP II has also been reported to
decrease the levels of tight junction-related proteins, ZO-1 and
occluding, through the RhoA/ROCK and PKC signaling pathways in rat
brain microvascular endothelial cells (BMECs) (36,37). Based on these findings, it was
hypothesized speculate that endogenously increased EMAP II may also
decrease ZO-1 and occludin proteins to change the tight junction
integrity of ECs following SE.
Park et al (38) found that the angiogenesis
regulatory function of the EMAP II precursor protein AIMP1 was
dependent on its concentration. At low concentrations, AIMP1
mediates angiogenesis, whereas, at high concentrations, AIMP1 leads
to apoptosis of vascular endothelial cells and angiogenesis
inhibition. Their results revealed that AIMP1 may inhibit
angiogenesis through the C-terminal hydrolysate EMAP II.
Clinically, owing to EMAP II inhibiting the growth of blood
vessels, it is widely applied in the treatment of tumors at
relatively high concentrations (39,40). The present study demonstrated that
the increased expression of EMAP II was accompanied by increased
numbers of CD31-positive microvascular endothelial cells. It was
hypothesized that endogenous EMAP II expression may be present at a
relatively low concentration, which can promote angiogenesis
following SE. Limitations of the present study include the absence
of immunofluorescence analysis of ZO-1 and occludin, and the
absence of detailed mechanism through which EMAP II alters
angiogenesis and tight junction integrity following SE. Research is
currently ongoing to explore the mechanisms exogenous EMAP II
influences the progression of SE.
In conclusion, the expression of EMAP II in the rat
hippocampus was found to be upregulated in the model of SE, which
may promote angiogenesis with an increased number of CD31-positive
microvascular endothelial cells. Moreover, an increase in
endogenous EMAP II may also alter the tight junction integrity of
BMECs, with a decreased expression of ZO-1 and occludin. However,
further studies are required to reveal the exact mechanisms
underlying the effects of EMAP II in the model of epilepsy.
Supplementary Data
Funding
The present study was supported by the National Key
Research and Development Program of China (grant no.
2016YFC1306203).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL, WM and HW were involved in the conception and
design of the study and the drafting of the manuscript. CL and WM
were involved in the analysis and interpretation of the data. YZ
assisted in setting up the protocols. CL and WM performed all the
experiments and acquired all data. All authors read and approved
the manuscript.
Ethics approval and consent to
participate
The China Medical University Institutional Animal
Care and Use Committee (No. 2016PS201K) approved all
procedures.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Thurman DJ, Begley CE, Carpio A, Helmers
S, Hesdorffer DC, Mu J, Touré K, Parko KL and Newton CR: The
primary prevention of epilepsy: A report of the prevention task
force of the international league against epilepsy. Epilepsia.
59:905–914. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiest KM, Sauro KM, Wiebe S, Patten SB,
Kwon CS, Dykeman J, Pringsheim T, Lorenzetti DL and Jetté N:
Prevalence and incidence of epilepsy: A systematic review and
meta-analysis of international studies. Neurology. 88:296–303.
2017. View Article : Google Scholar :
|
|
3
|
Bertini G, Bramanti P, Constantin G,
Pellitteri M, Radu BM, Radu M and Fabene PF: New players in the
neurovascular unit: Insights from experimental and clinical
epilepsy. Neurochem Int. 63:652–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takemiya T and Yamagata K: Intercellular
signaling pathway among Endothelia, astrocytes and neurons in
excitatory neuronal damage. Int J Mol Sci. 14:8345–8357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mogylnytska LA: Endothelial
monocyte-activating polypeptide-II: Properties, functions, and
pathogenetic significance. Fiziol Zh. 61:102–111. 2015.In
Ukrainian. View Article : Google Scholar
|
|
6
|
Kao J, Houck K, Fan Y, Haehnel I, Libutti
SK, Kayton ML, Grikscheit T, Chabot J, Nowygrod R, Greenberg S, et
al: Characterization of a novel tumor-derived cytokine.
Endothelial-monocyte activating polypeptide II. J Biol Chem.
269:25106–25119. 1994.PubMed/NCBI
|
|
7
|
Yuan C, Yan L, Solanki P, Vatner SF,
Vatner DE and Schwarz MA: Blockade of EMAP II protects cardiac
function after chronic myocardial infarction by inducing
angiogenesis. J Mol Cell Cardiol. 79:224–231. 2015. View Article : Google Scholar :
|
|
8
|
Lal CV and Schwarz MA: Vascular mediators
in chronic lung disease of infancy: Role of endothelial monocyte
activating poly-peptide II (EMAP II). Birth Defects Res A Clin Mol
Teratol. 100:180–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adly AAM, Ismail EA, Tawfik LM, Ebeid FSE
and Hassan AAS: Endothelial monocyte activating polypeptide II in
children and adolescents with type 1 diabetes mellitus: Relation to
micro-vascular complications. Cytokine. 76:156–162. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Liu XB, Liu YH, Xue YX, Wang P, Liu
LB, Liu J, Yao YL and Ma J: Roles of serine/threonine phosphatases
in low-dose endothelial monocyte-activating polypeptide-II-induced
opening of blood-tumor barrier. J Mol Neurosci. 57:11–20. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma J, Meng F, Li S, Liu L, Zhao L, Liu Y,
Hu Y, Li Z, Yao Y, Xi Z, et al: Autophagy induction by endothelial
monocyte activating polypeptide II contributes to the inhibition of
malignant biological behaviors by the combination of EMAP II with
rapamycin in human glioblastoma. Front Mol Neurosci. 8:742015.
View Article : Google Scholar
|
|
12
|
Schikorski D, Cuvillier-Hot V,
Boidin-Wichlacz C, Slomianny C, Salzet M and Tasiemski A:
Deciphering the immune function and regulation by a TLR of the
cytokine EMAPII in the lesioned central nervous system using a
leech model. J Immunol. 183:7119–7128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu X, Liu Y, Yin Y, Shao A, Zhang B, Kim
S and Zhou J: MSC p43 required for axonal development in motor
neurons. Proc Natl Acad Sci USA. 106:15944–15949. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Armstrong L, Biancheri R, Shyr C, Rossi A,
Sinclair G, Ross CJ, Tarailo-Graovac M, Wasserman WW and van
Karnebeek CD: AIMP1 deficiency presents as a cortical
neurodegenerative disease with infantile onset. Neurogenetics.
5:157–159. 2014. View Article : Google Scholar
|
|
15
|
Knies UE, Behrensdorf HA, Mitchell CA,
Deutsch U, Risau W, Drexler HC and Clauss M: Regulation of
endothelial monocyte-activating polypeptide II release by
apoptosis. Proc Natl Acad Sci USA. 95:12322–12327. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barnett G, Jakobsen AM, Tas M, Rice K,
Carmichael J and Murray JC: Prostate adenocarcinoma cells release
the novel proinflammatory polypeptide EMAP-II in response to
stress. Cancer Res. 60:2850–2857. 2000.PubMed/NCBI
|
|
17
|
Murray JC, Barnett G, Tas M, Jakobsen A,
Brown J, Powe D and Clelland C: Immunohistochemical analysis of
endothelial-monocyte-activating polypeptide-II expression in vivo.
Am J Pathol. 157:2045–2053. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clauss M, Voswinckel R, Rajashekhar G,
Sigua NL, Fehrenbach H, Rush NI, Schweitzer KS, Yildirim AÖ,
Kamocki K, Fisher AJ, et al: Lung endothelial monocyte-activating
protein 2 is a mediator of cigarette smoke-induced emphysema in
mice. J Clin Invest. 121:2470–2479. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rigau V, Morin M, Rousset MC, de Bock F,
Lebrun A, Coubes P, Picot MC, Baldy-Moulinier M, Bockaert J,
Crespel A, et al: Angiogenesis is associated with blood-brain
barrier permeability in temporal lobe epilepsy. Brain.
130:1942–1956. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morin-Brureau M, Lebrun A, Rousset MC,
Fagni L, Bockaert J, de Bock F and Lerner-Natoli M: Epileptiform
activity induces vascular remodeling and zonula occludens 1
downregulation in organotypic hippocampal cultures: Role of VEGF
signaling pathways. J Neurosci. 31:10677–10688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ingram J, Zhang CF, Cressman JR, Hazra A,
Wei Y, Koo YE, Žiburkus J, Kopelman R, Xu J and Schiff SJ: Oxygen
and seizure dynamics: I. Experiments J Neurophysiol. 112:205–212.
2014. View Article : Google Scholar
|
|
22
|
Wu Q and Wang H: The spatiotemporal
expression changes of CB2R in the hippocampus of rats following
pilocarpine-induced status epilepticus. Epilepsy Res. 148:8–16.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Curia G, Longo D, Biagini G, Jones RS and
Avoli M: The pilocarpine model of temporal lobe epilepsy. J
Neurosci Methods. 172:143–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Glien M, Brandt C, Potschka H, Voigt H,
Ebert U and Löscher W: Repeated low-dose treatment of rats with
pilocarpine: Low mortality but high proportion of rats developing
epilepsy. Epilepsy Res. 46:111–119. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Racine RJ, Burnham WM and Gartner JG:
First trial motor seizures triggered by amygdaloid stimulation in
the rat. Electroencephalogr Clin Neurophysiol. 35:487–494. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paxinos G and Watson C: The Rat Brain in
Stereotaxic Coordinates - the New Coronal Set. 5th edition.
Academic Press; New York: 2005
|
|
27
|
Li J, Jiang G, Chen Y, Chen L, Li Z, Wang
Z and Wang X: Altered expression of hypoxia-Inducible factor-1α
participates in the epileptogenesis in animal models. Synapse.
68:402–409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ndode-Ekane XE, Hayward N, Gröhn O and
Pitkänen A: Vascular changes in epilepsy: Functional consequences
and association with network plasticity in pilocarpine-induced
experimental epilepsy. Neuroscience. 166:312–332. 2010. View Article : Google Scholar
|
|
29
|
Friedman A and Heinemann U: Role of
blood-brain barrier dysfunction in epileptogenesis. Jasper's Basic
Mechanisms of the Epilepsies [Internet]. 4th edition. Noebels JL,
Avoli M, Rogawski MA, Olsen RW and Delgado-Escueta AV: National
Center for Biotechnology Information (US); Bethesda, MD, USA: pp.
1–12. 2012
|
|
30
|
Castañeda-Cabral JL, Colunga-Durán A,
Ureña-Guerrero ME, Beas-Zárate C, Nuñez-Lumbreras MLA,
Orozco-Suárez S, Alonso-Vanegas M, Guevara-Guzmán R, Deli MA,
Valle-Dorado MG, et al: Expression of VEGF- and tight
junction-related proteins in the neocortical microvasculature of
patients with drug-resistant temporal lobe epilepsy. Microvasc Res.
132:1040592020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim JY, Ko AR, Hyun HW and Kang TC: ETB
receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1
protein degradation following status epilepticus. Neuroscience.
304:355–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Z, Liu YH, Xue YX, Xie H and Liu LB:
Role of ATP synthase alpha subunit in low-dose endothelial
monocyte-activating poly-peptide-II-induced opening of the
blood-tumor barrier. J Neurol Sci. 300:52–58. 2011. View Article : Google Scholar
|
|
33
|
Liao YL, Zhang ZY, Liu JW, Schluesener HJ,
Zhang ZR and Wu YZ: Lesional expression of EMAPII in
macro-phages/microglia following cerebral ischemia in rats. Int J
Neurosci. 121:58–64. 2011. View Article : Google Scholar
|
|
34
|
Brabeck C, Michetti F, Geloso MC, Corvino
V, Goezalan F, Meyermann R and Schluesener HJ: Expression of
EMAP-II by activated monocytes/microglial cells in different
regions of the rat hippocampus after trimethyltin-induced brain
damage. Exp Neuro. 177:241–246. 2002. View Article : Google Scholar
|
|
35
|
Li Z, Liu YH, Liu XB, Xue YX, Wang P and
Liu LB: Low-dose endothelial monocyte-activating polypeptide-II
increases permeability of blood-tumor barrier via a
PKC-ζ/PP2A-dependent signaling mechanism. Exp Cell Res.
331:257–266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Z, Liu YH, Xue YX, Liu LB and Xie H:
Mechanisms for endothelial monocyte-activating
polypeptide-II-induced opening of the blood-tumor barrier. J Mol
Neurosci. 47:408–417. 2012. View Article : Google Scholar
|
|
37
|
Xie H, Xue YX, Liu LB, Liu YH and Wang P:
Role of RhoA/ROCK signaling in endothelial-monocyte-activating
polypeptide II opening of the blood-tumor barrier: Role of
RhoA/ROCK signaling in EMAP II opening of the BTB. J Mol Neurosci.
46:666–676. 2012. View Article : Google Scholar
|
|
38
|
Park SG, Kang YS, Ahn YH, Lee SH, Kim KR,
Kim KW, Koh GY, Ko YG and Kim S: Dose-dependent biphasic activity
of tRNA synthetase-associating factor, p43, in angiogenesis. J Biol
Chem. 277:45243–45248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schwarz RE, Awasthi N, Konduri S, Caldwell
L, Cafasso D and Schwarz MA: Antitumor effects of EMAP II against
pancreatic cancer through inhibition of fibronectin-dependent
proliferation. Cancer Biol Ther. 9:632–639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Awasthi N, Schwarz MA, Verma V, Cappiello
C and Schwarz RE: Endothelial monocyte activating polypeptide II
interferes with VEGF-induced proangiogenic signaling. Lab Invest.
89:38–46. 2009. View Article : Google Scholar
|