Introduction
As a common complication of cutaneous wound healing,
hypertrophic scar (HTS) results in unsatisfactory appearance or
even permanent loss of normal skin function. Current therapies for
HTS include surgical procedures, anti-inflammatory drugs, cytotoxic
medications, compression therapy and radiation therapy (1,2).
However, these approaches may fail to achieve satisfactory
therapeutic effects, and they have certain limitations. Although
the exact cause for the development of HTS is not yet fully
understood, it is typically hypothesized to result from an abnormal
response of connective tissues to dermal injury, and is
characterized by excessive proliferation of myofibroblasts and
aberrant deposition of extracellular matrix (ECM) components, most
notably collagen (3).
Myofibroblasts are the major source of ECM, including collagen I
(COL I), and the key effector cells of HTS. Excessive proliferation
of myoblasts is characterized by an increased expression of
α-smooth muscle actin (α-SMA) (4,5).
With the development of HTS, myofibroblasts are continuously
hyperactivated and continue to proliferate, leading to excessive
synthesis and deposition of collagen (6). Therefore, inhibiting the excessive
proliferation and secretion of myofibroblasts may become a
potential target for the management of HTS.
Transforming growth factor-β1 (TGF-β1) is considered
to be the most potent fibrogenic cytokine in inducing the
transformation of fibroblasts into myofibroblasts (7,8).
However, given the diversity and complexity of downstream signaling
networks regulated by TGF-β1, the consequences of complete
inhibition of its expression can be difficult to predict.
Therefore, targeting the downstream signaling of TGF-β1 may be a
more practical and promising therapeutic strategy to avoid
potential toxicity caused by TGF-β1 inhibition. TGF-β1 has been
reported to regulate the progression and transition of various
types of tumors by inducing autophagy (9,10).
These findings indicated that autophagy may also serve as a
therapeutic target for TGF-β1-induced tissue fibrosis. As an
evolutionarily conservative process, autophagy facilitates the
degradation of damaged organelles or intracellular components, and
provides cells with nutrients (11,12). Autophagy is a vital process for
the maintenance of cellular homeostasis and cell survival. However,
excessive levels of autophagy may result in autophagic cell death.
It has been reported that dysregulation of autophagy is closely
associated with the pathological process of numerous fibrotic
diseases, including liver, kidney and heart disease, as well as
cystic fibrosis (13-16). Nevertheless, the roles of
autophagy in different fibrotic conditions may be multifaceted and
complex. A number of studies have suggested that autophagy is a
cytoprotective mechanism (17-19). However, some studies have also
indicated that autophagy is involved in promoting fibrosis
(20-22). The actual function of autophagy
may depend on the specific type and stage of the fibrotic disease.
Apart from the aforementioned examples, there is limited
information available regarding the role of autophagy in HTS, and
the precise molecular mechanisms linking autophagy to HTS need to
be further elucidated.
Transcription factor EB (TFEB), a pivotal regulator
of lysosome biogenesis and autophagy (23), has become an ideal target for
regulating autophagy. In the present study, using TGF-β1-treated
human skin fibroblasts as a model system, the aim was to
investigate whether TFEB mediated-autophagy was involved in
fibroblast differentiation and collagen production, and to explore
the underlying molecular mechanism in this process, and thus
provide a novel perspective for the development of strategies for
the treatment of HTS.
Materials and methods
Cell isolation and culture
The present study was approved by the Medical Ethics
Committee of Zhongnan Hospital, Wuhan University (Wuhan, China;
approval no. 2019006). Skin tissues of healthy male patients were
collected during circumcision, primary skin fibroblasts were
disinfected, cut into small pieces, digested using 0.25% dispase II
(cat. no. 11760200; Roche Diagnostics) at 4°C for 24 h, and then
digested using 0.2% collagenase II (cat. no. 17101015; Gibco;
Thermo Fisher Scientific, Inc.) at 37°C for 4 h. The mixture was
vortexed and filtered to obtain a single-cell suspension, which was
then added to the culture bottle, and cultured using DMEM (cat. no.
10567014; Gibco; Thermo Fisher Scientific, Inc.) with 10% calf
serum (cat. no. 16010159; Gibco; Thermo Fisher Scientific, Inc.) at
37°C in a cell culture incubator with 5% CO2, culture
medium was replaced with fresh medium every 3 days. After reaching
~80% confluence, cells were digested with 0.25% trypsin (cat. no.
C0201; Beyotime Institute of Biotechnology) and then passaged.
Fibroblasts in passages 2-4 were used for further experiments.
Determination of TGF-β1 concentration by
lactate dehydrogenase (LDH) release assay
LDH is released upon damage to the plasma membrane
of cells, and LDH concentration in the culture medium can be used
to evaluate the level of cell damage (24). Fibroblasts of the same passage
were seeded onto 24-well plates (5×105 cells/ml), and
were exposed to TGF-β1 (cat. no. AF-100-21C-2; PeproTech, Inc.) at
concentrations ranging between 0 and 80 ng/ml for 48 h.
Subsequently, 200 µl culture medium was taken from each well
and centrifuged at 4°C for 5 min at 300 × g. Next, 20 µl of
each supernatant sample was added to the corresponding well in a
96-well plate, followed by successive addition of 65 µl
potassium pyruvate phosphate buffer and 65 µl NADH solution.
Absorbance measurements were performed using a microplate reader at
340 nm. TGF-β1 concentrations were calculated on the basis of the
release rates of LDH.
Enzyme-linked immunosorbent assay
(ELISA)
The secretion levels of pro-COL Iα by fibroblasts
into the supernatant were detected using an ELISA kit (cat. no.
DY6220-05; R&D Systems, Inc.). Optical density values were
measured at 540 nm.
Western blotting
Fibroblasts were seeded into 6-well plates
(1×106 cells/ml) and treated with TGF-β1 (10 ng/ml) with
or without tauroursodeoxycholic acid (TUDCA; 2.5 µmol/l;
cat. no. T0266; Sigma-Aldrich; Merck KGaA) at 37°C for 48 h.
Fibroblasts were seeded into 6-well plates (1×106
cells/ml) and transfected with 50 nmol/l TFEB-siRNA or NC-siRNA for
48 h, and then treated with or without 10 ng/ml TGF-β1 at 37°C for
48 h. Cells were collected for lysate preparation. RIPA lysis
buffer (cat. no. P0013C; Beyotime Institute of Biotechnology) was
used for protein extraction. The protein concentrations of
fibroblast lysates were measured using the BCA protein assay kit
(cat. no. P0010; Beyotime Institute of Biotechnology). Total
protein (20 µg per lane) was loaded onto 10 or 15% gels and
separated via SDS-PAGE, the separated proteins were then
transferred onto PVDF membranes (cat. no. IPVH08130; EMD
Millipore). Membranes were subsequently blocked with 5% BSA (cat.
no. ST023; Beyotime Institute of Biotechnology) for 1 h at 25°C,
and incubated with corresponding primary antibodies overnight at
4°C, including anti-COL I (1:1,000; cat. no. ab6308; Abcam),
anti-α-SMA (1:2,000; cat. no. ab5694; Abcam),
anti-glucose-regulated proteins 78 (GRP78; 1:1,000; cat. no.
ab32618; Abcam), anti-protein kinase R-like endoplasmic reticulum
kinase (PERK; 1:1,000; cat. no. 3192; Cell Signaling Technology,
Inc.), anti-phosphorylated (p)-PERK (1:1,000; cat. no. 3179; Cell
Signaling Technology, Inc.), anti-activating transcription factor 6
(ATF6; 1:1,000; cat. no. 65880; Cell Signaling Technology, Inc.),
anti-inositol-requiring enzyme-1α (IRE1α; 1:1,000; cat. no. 3294;
Cell Signaling Technology, Inc.), anti-lysosome-associated membrane
protein 1 (LAMP1; 1:1,000; cat. no. ab62562; Abcam), anti-cathepsin
B (CTS B; 1:1,000; cat. no. 31718; Cell Signaling Technology,
Inc.), anti-TFEB (1:200; cat. no. ab220695; Abcam), anti-α subunit
of eukaryotic initiation factor 2 (eIF2α; 1:1,000; cat. no. 5324;
Cell Signaling Technology, Inc.), anti-p-eIF2α (1:2,000; cat. no.
3398; Cell Signaling Technology, Inc.), anti-spliced X-box binding
protein 1 (XBP-1s; 1:2,000; cat. no. 47134; Cell Signaling
Technology, Inc.), anti-cysteinyl aspartate specific proteinase 3
(caspase 3; 1:1,000; cat. no. 14220; Cell Signaling Technology,
Inc.), anti-microtubule associated protein 1 light chain 3 (LC3;
1:1,000; cat. no. L8918; Sigma-Aldrich; Merck KGaA), anti-p62
(1:1,000; cat. no. SAB1406748; Sigma-Aldrich; Merck KGaA),
anti-C/EBP-homologous protein caspase 3 (CHOP; 1:1,000; cat. no.
2895; Cell Signaling Technology, Inc.), anti-β-actin (1:1,000; cat.
no. AF0003; Beyotime Institute of Biotechnology), anti-GAPDH
(1:1,000; cat. no. AF1186; Beyotime Institute of Biotechnology) and
anti-Histone H3 (1:1,000; cat. no. AF0009; Beyotime Institute of
Biotechnology). After washing with TBS with 0.1% Tween-20,
membranes were then incubated with goat anti-rabbit IR dye 800 CW
(1:10,000; cat. no. P/N 926-32211; LI-COR Biosciences) or goat
anti-mouse (1:10,000; cat. no. P/N 926-32210; LI-COR Biosciences)
secondary anti-bodies at room temperature for 2 h. The intensity of
bands was detected using the Odyssey infrared image processing
system (LI-COR Biosciences). The gray values of the specific blots
were analyzed with Image-Pro Plus version 6.0 software (Media
Cybernetics, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (cat. no. 15596026; Invitrogen; Thermo Fisher Scientific,
Inc.), and cDNA synthesis was performed using RevertAid First
Strand cDNA Synthesis kit (cat. no. K1622; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
RT-qPCR was performed using LightCycler® 96 System
(Roche Diagnostics) and FastStart™ Universal SYBR Green Master kit
(cat. no. 4913850001; Roche Diagnostics). The qPCR thermo-cycling
conditions were as follows: 94°C for 3 min, 40 cycles at 95°C for
15 sec, 60°C for 30 sec and 72°C for 30 sec. Relative mRNA levels
were normalized to β-actin and analyzed using the 2−ΔΔCq
method (25). Primer sequences
used in this study were as follows: TFEB forward, 5′-ACC TGT CCG
AGA CCT ATG GG-3′ and reverse, 5′-CGT CCA GAC GCA TAA TGT TGT C-3′;
and β-actin forward, 5′-GAA ATC GTG CGT GAC ATT AAA GAG-3′ and
reverse, 5′-GCG GCA GTG GCC ATC TC-3′.
Terminal deoxynucleotidyl
transferase-mediated biotinylated UTP nick-end labeling (TUNEL)
assay
TUNEL staining was performed to evaluate apoptotic
cell death by using the In SituCell Death Detection kit, TMR
red (cat. no. 12156792910; Roche Diagnostics), according to the
manufacturer's instruction. Cells were fixed on slides using 4%
paraformaldehyde (cat. no. P0098; Beyotime Institute of
Biotechnology) for 30 min and permeabilized with 0.3% Triton X-100
(cat. no. ST795; Beyotime Institute of Biotechnology) for 10 min at
25°C. The slides were washed with PBS, covered with the TUNEL
reaction mixture and incubated for 30 min in the dark. DAPI (cat.
no. C1006; Beyotime Institute of Biotechnology) was added for
nuclei staining for 15 min at room temperature. A total of six cell
slides per group were examined by fluorescence microscopy (LSM780;
Carl Zeiss AG) at ×400 magnification, and the proportion of
TUNEL-positive stained cells vs. total cells from three random
fields of each slice were calculated using Image-Pro Plus 6.0
software (Media Cybernetics, Inc.).
Immunofluorescence staining
Cells were fixed on slides with 2% paraformaldehyde
(cat. no. P0099; Beyotime Institute of Biotechnology) for 15 min
and permeabilized with 0.3% Triton X-100 for 10 min, and then
blocked with 5% BSA (cat. no. ST023; Beyotime Institute of
Biotechnology) for 1 h, all at 25°C. Subsequently, cells were
incubated with the following primary antibodies overnight at 4°C:
Anti-TFEB (1:50; cat. no. ab220695; Abcam), anti-LAMP1 (1:200; cat.
no. ab62562; Abcam), anti-COL I (1:200; cat. no. ab6308; Abcam) and
anti-Ras-related protein Rab-8A (Rab8a; 1:200; cat. no.
ABIN6290618; Beijing 4A Biotech Co., Ltd.). The slides were then
incubated with Alexa Fluor 488-conjugated donkey anti-mouse IgG
(1:500; cat. no. A-21202; Invitrogen; Thermo Fisher Scientific,
Inc.), Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:500;
cat. no. A-31572; Invitrogen; Thermo Fisher Scientific, Inc.) or
Alexa Fluor 647-conjugated donkey anti-goat IgG (1:500; cat. no.
A-21447; Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature for 2 h, counterstained with DAPI (cat. no C1006;
Beyotime Institute of Biotechnology) for 15 min at room
temperature, and then observed under confocal laser scanning
microscope (LSM780; Carl Zeiss AG). Blocking buffer without the
primary anti-bodies was considered the negative control. Image-Pro
Plus 6.0 software (Media Cybernetics, Inc.) was used for
quantitative analysis of fluorescence microscopy images, and the
'LINE PROFILE' function module was used for line tracing analysis
of fluorescence colocalization.
Detection of autophagic flux and small
interfering RNA (siRNA)-induced knockdown
Autophagic flux in fibroblasts was detected using
Autophagy Tandem Sensor RFP-GFP-LC3B kit (cat. no. P36239;
Invitrogen; Thermo Fisher Scientific, Inc.). After TGF-β1 treatment
for 24 h, fibroblasts were treated with mRFP-GFP-LC3-expressing
plasmid for another 24 h. Autophagic flux was quantified by
determining the number of autophagosomes (yellow puncta) and
autolysosomes (red puncta) per cell per µm2.
The following sequences were applied for
siRNA-mediated TFEB knockdown: TFEB siRNA-1 sense, 5′-GAA AGG AGA
CGA AGG UUC AAC AUC A-3′ and antisense, 5′-AUU CGC UCC UAA CCG AGC
CAU UCC C-3′; TFEB siRNA-2 sense, 5′-GGA UCA AGG AGC UGG GAA UUU-3′
and anti-sense, 5-AUU CCC AGC UCC UUG AUC CUU-3′. Fibroblasts were
seeded into 24- or 6-well plates (5.0×105 cells/ml or
1.0×106 cells/ml) and transfected with 50 nmol/l
TFEB-siRNA for 48 h at 37°C using Lipofectamine® RNAiMAX
transfection reagent (cat. no. 13778075; Invitrogen; Thermo Fisher
Scientific, Inc.). Negative control (NC)-siRNA was used as the
control (sense, 5′-UAG CGA CUA AAC ACA UCA AUU-3′ and antisense,
5′-UUG AUG UGU UUA GUC GCU AUU-3′). After transfection with
TFEB-siRNA or NC-siRNA for 48 h, the cells were then treated with
10 ng/ml TGF-β1 for 48 h at 37°C. TFEB-siRNA and siRNA NC were
purchased from BrainVTA (Wuhan) Co., Ltd.
Statistical analysis
GraphPad Prism 6.0 software (GraphPad Software,
Inc.) was used for statistical analyses. Data are expressed as the
mean ± standard deviation and were analyzed using one-way analysis
of variance followed by Tukey's post hoc test. Pearson's
correlation analysis was used to determine the correlation between
protein colocalization. P<0.05 was considered to indicate a
statistically significant difference.
Results
TGF-β1 promotes fibroblastic phenotypic
transformation as well as collagen synthesis and secretion in
fibroblasts in a dose-dependent manner
In the present study, TGF-β1-treated human dermal
fibroblasts were used as a model of HTS pathogenesis. To determine
the optimal concentrations of TGF-β1 for fibroblast treatment, the
effects of different concentrations of TGF-β1 on the integrity and
viability of fibroblasts were investigated. Cells were treated with
0, 2.5, 5, 10, 20, 40 or 80 ng/ml TGF-β1 for 48 h, and the
concentrations of LDH released from fibroblasts were determined.
Treatment with 80 ng/ml TGF-β1 induced a significant increase in
LDH release from fibroblasts, whereas treatment with 40 ng/ml
TGF-β1 produced a small and insignificant increase in LDH release
compared with the control (Fig.
1A). Hence, 2.5-20 ng/ml TGF-β1 was selected for further
evaluation.
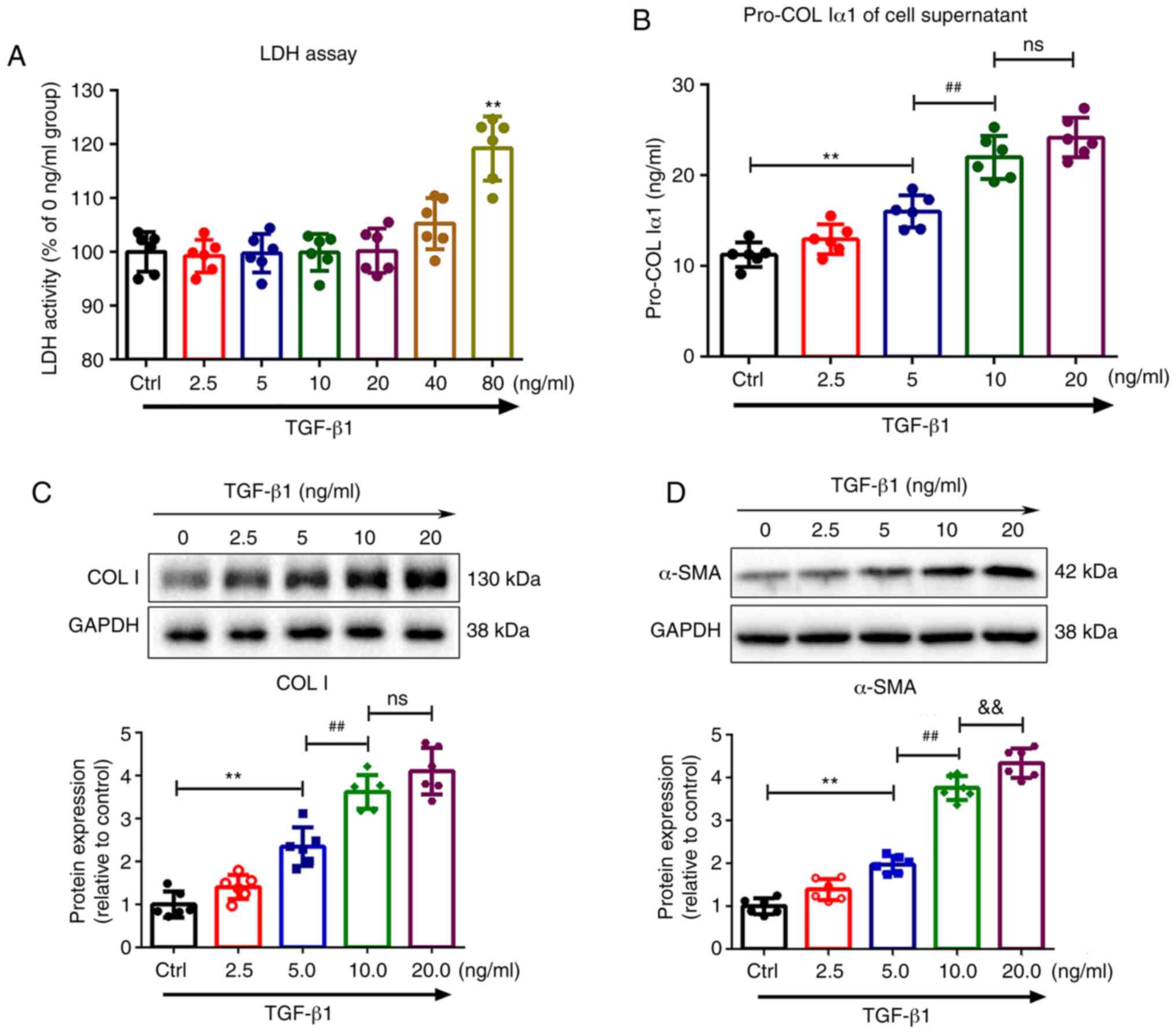 | Figure 1TGF-β1 promotes phenotypic
transformation of fibroblasts, and collagen synthesis and secretion
in fibroblasts. (A) Fibroblasts were treated with 0, 2.5, 5, 10,
20, 40 or 80 ng/ml TGF-β1 for 48 h and LDH release was then
determined. (B) Secretion of pro-COL Iα1 in fibroblasts was
assessed by ELISA. Expression of (C) COL I and (D) α-SMA in
fibroblasts was measured by western blotting after treatment with
0, 2.5, 5, 10 or 20 ng/ml TGF-β1 for 48 h. GAPDH served as a
loading control. Results are presented as the mean ± SD (n=6).
**P<0.01 vs. control group; ##P<0.01
vs. 5 ng/ml TGF-β1 treatment group; &&P<0.01 vs. 10
ng/ml TGF-β1 treatment group. TGF-β1, transforming growth
factor-β1; LDH, lactate dehydrogenase; ELISA, enzyme-linked
immunosorbent assay; COL I, collagen I; α-SMA, α-smooth muscle
actin; ns, not significant. |
The effects of different concentrations of TGF-β1 on
COL I and α-SMA expression levels in the fibroblasts were detected
by western blotting. Basal expression levels of COL I and α-SMA in
fibroblasts were low, but following treatment with TGF-β1, COL I
(Fig. 1C) and α-SMA (Fig. 1D) expression significantly
increased in a dose-dependent manner in cells, which started to
occur at the concentration of 5 ng/ml and reached a maximum at 20
ng/ml. Similar trends were also observed in the secretion levels of
pro-COL Iα1 (Fig. 1B) in the
supernatant. However, there was no difference in COL I expression
or pro-COL Iα1 secretion in fibroblasts treated with 10 and 20
ng/ml TGF-β1. Therefore, 10 ng/ml was used as the optimal
concentration in subsequent experiments.
TGF-β1 triggers autophagy upregulation
through endoplasmic reticulum (ER) stress in fibroblasts
Whether and how TGF-β1 induced autophagy in primary
human dermal fibro-blasts was next evaluated. Western blotting
analysis showed that treatment with 10 ng/ml TGF-β1 led to
increased expression of ER stress-related proteins in fibroblasts
compared with the control group, including GRP78 (1.73±0.30 vs.
1.00±0.24; P<0.01; Fig. 2A),
p-PERK (2.13±0.19 vs. 1.00±0.12; P<0.01; Fig. 2B), ATF6 (1.80±0.22 vs. 1.00±0.16;
P<0.01; Fig. 2C) and IRE1α
(2.38±0.26 vs. 1.00±0.18; P<0.01; Fig. 2D). As shown in Fig. 2E-H, TGF-β1 (10 ng/ml) triggered
autophagy upregulation in fibroblasts compared with the control
group, as evidenced by higher expression levels of LC3 II
(3.24±0.31 vs. 1.00±0.15; P<0.01; Fig. 2E), LAMP1 (2.26±0.19 vs. 1.00±0.24;
P<0.01; Fig. 2G) and CTS B
(1.98±0.12 vs. 1.00±0.24; P<0.01; Fig. 2H), along with reduced expression
of p62 (0.31±0.09 vs. 1.00±0.15; P<0.01; Fig. 2F). Notably, TUDCA, an inhibitor of
ER stress, significantly reversed the upregulation of autophagy
(P<0.05 or P<0.01; Fig.
2A-H), indicating that ER stress is essential for
TGF-β1-induced autophagy.
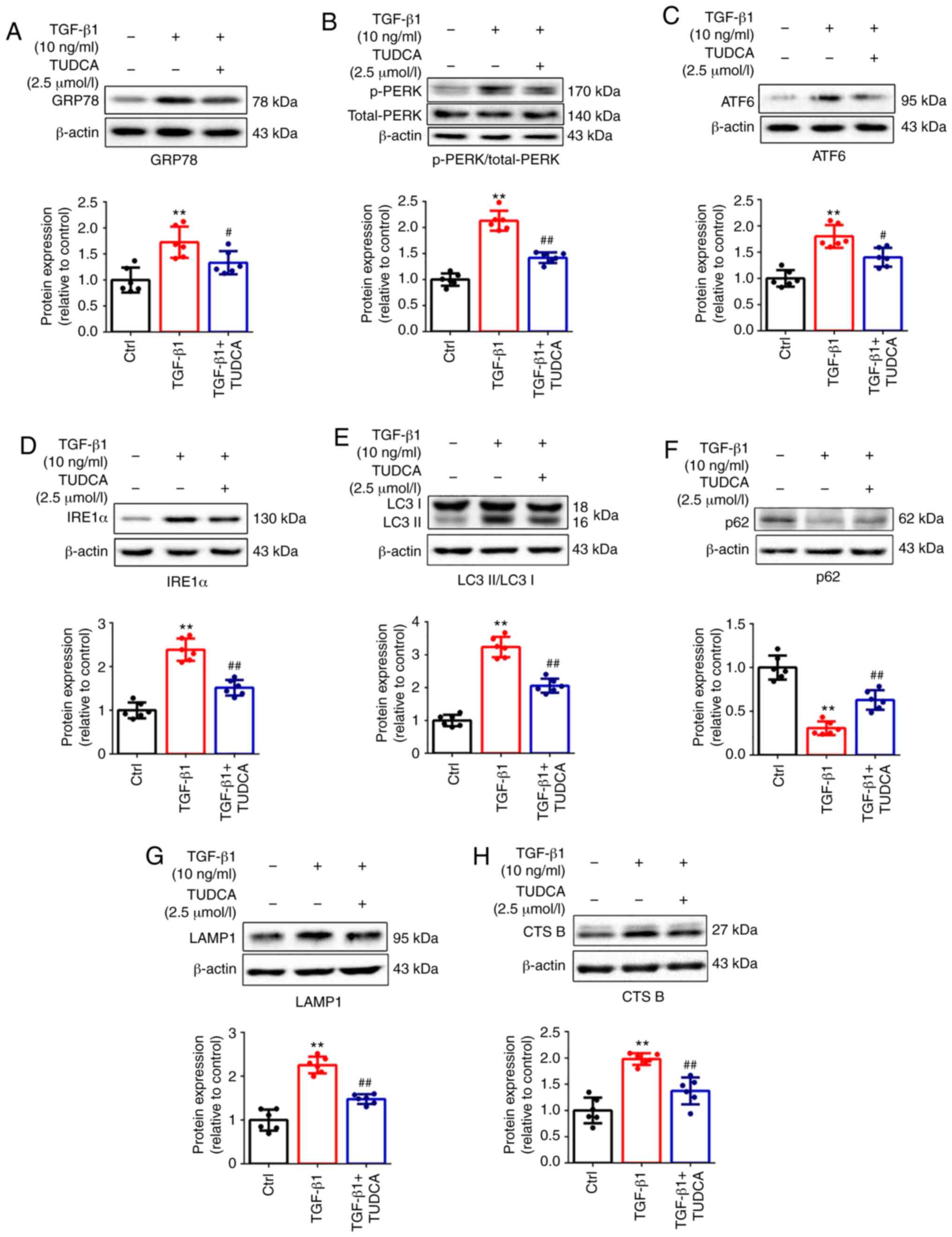 | Figure 2ER stress caused by TGF-β1 triggers
autophagy, which is repressed by TUDCA in fibroblasts. Fibroblasts
were treated with TGF-β1 (10 ng/ml) for 48 h with or without TUDCA
(2.5 µmol/l). Representative immunoblots of cultured
fibroblasts, with detection of (A) GRP78, (B) p-PERK/total-PERK,
(C) ATF6, (D) IRE1α, (E) LC3 II/I, (F) p62, (G) LAMP1 and (H) CTS
B. Results are presented as the mean ± SD (n=6).
**P<0.01 vs. control group; #P<0.05 and
##P<0.01 vs. TGF-β1 treatment group. ER, endoplasmic
reticulum; TGF-β1, transforming growth factor-β1; TUDCA,
tauroursodeoxycholic acid; GRP78, glucose-regulated proteins 78;
p-, phosphorylated; PERK, protein kinase R-like endoplasmic
reticulum kinase; ARF6, activating transcription factor 6; IRE1α,
inositol-requiring enzyme-1α; LC3, microtubule associated protein 1
light chain 3; LAMP1, lysosome-associated membrane protein 1; CTS
B, cathepsin B. |
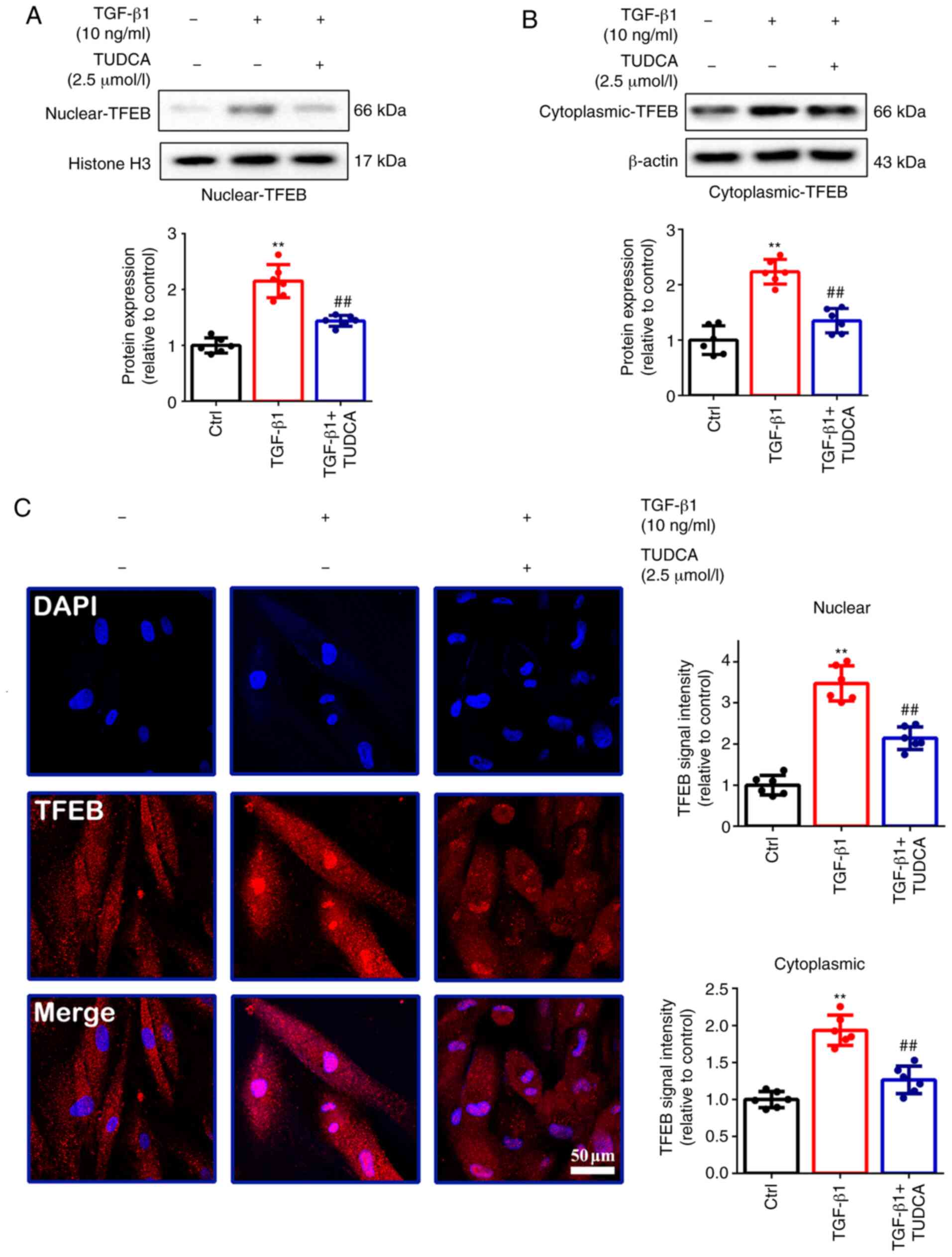
The present study also evaluated the effect of
TGF-β1 on the activation/overexpression of TFEB, which is a central
regulator of lysosomal biogenesis and autophagy (23). TFEB activation/overexpression has
been shown to promote the degradation of autophagy substrates
(23). Western blotting assays
were performed to determine the distributions in the nuclear
(2.15±0.30 vs. 1.00±0.14; P<0.01; Fig. 3A) and cytoplasmic (2.23±0.22 vs.
1.00±0.26; P<0.01; Fig. 3B)
fractions of TFEB, showing that TGF-β1 treatment resulted in
increased protein expression and nuclear translocation of TFEB in
fibro-blasts compared with the control group. TGF-β1 also increased
the fluorescence intensity of TFEB compared with the control group
in the nucleus (3.47±0.43 vs. 1.00±0.24; P<0.01; Fig. 3C) and the cytoplasm (1.93±0.20 vs.
1.00±0.11; P<0.01; Fig. 3C).
TUDCA also reduced the activity of TGF-β1-induced TFEB
transcription (P<0.01; Fig.
3A-C).
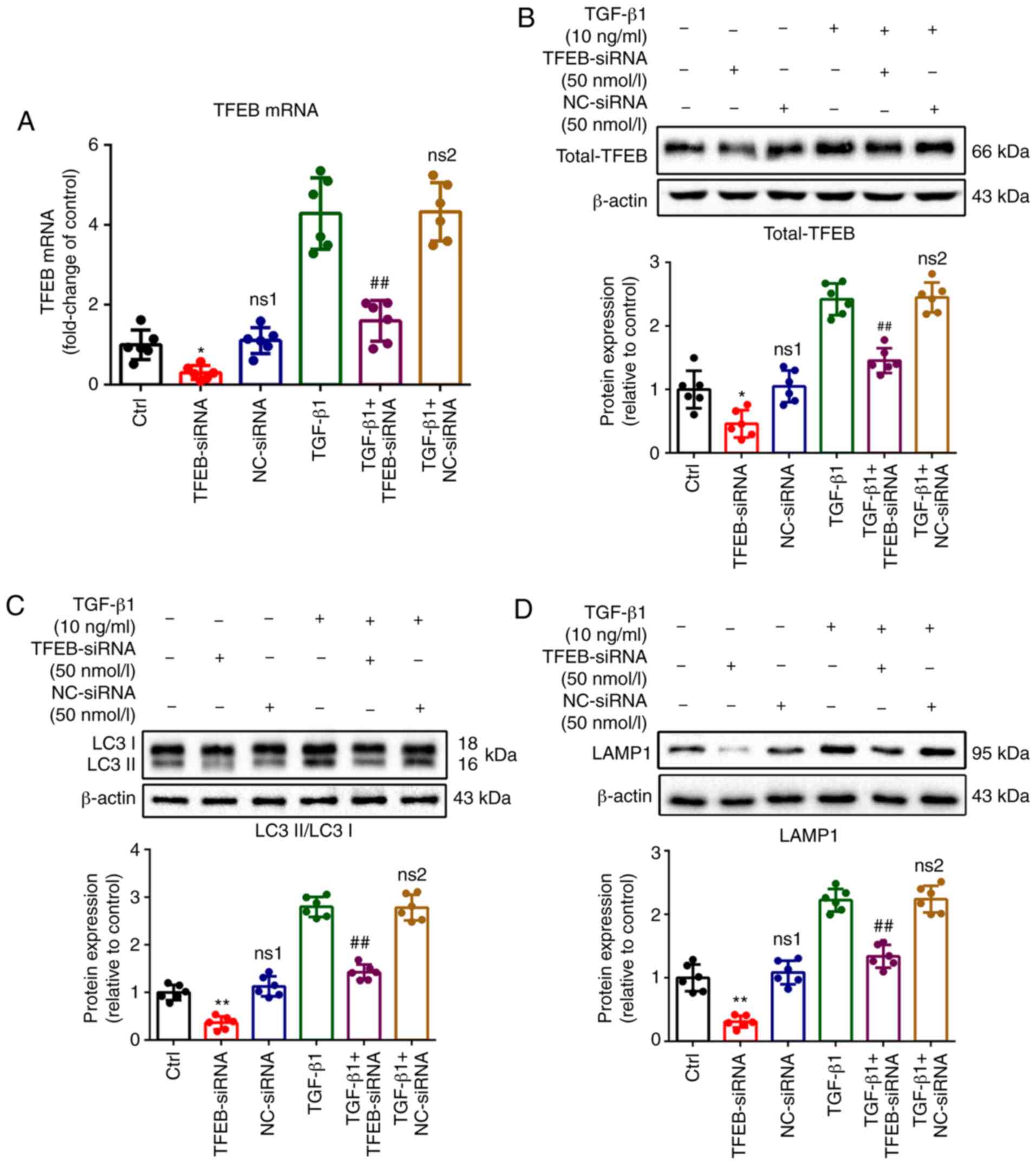
Knockdown of TFEB enhances TGF-β1-induced
ER stress and apoptosis, while it reduces fibroblastic phenotypic
transformation and collagen synthesis in fibroblasts
To further understand whether autophagy plays a
significant role in ER stress, fibroblastic phenotypic
transformation, as well as collagen synthesis and secretion in
fibroblasts, gene knock-down of TFEB was performed using siRNA.
Compared with the control group, efficient knockdown of TFEB was
confirmed by RT-qPCR (0.31±0.17 vs. 1.00±0.36; P<0.05; Fig. 4A) and western blotting (0.46±0.21
vs. 1.00±0.30; P<0.05; Fig.
4B; 1.47±0.34 vs. 2.50±0.44; P<0.01; Fig. S1A). Knockdown of TFEB
significantly reversed the increased expression of LC3 (1.43±0.16
vs. 2.80±0.21; P<0.01; Fig.
4C; 1.47±0.27 vs. 3.02±0.37; P<0.01; Fig. S1B) and LAMP1 (1.34±0.18 vs.
2.22±0.18; P<0.01; Fig. 4D;
1.47±0.25 vs. 2.24±0.34; P<0.01; Fig. S1C) induced by TGF-β1.
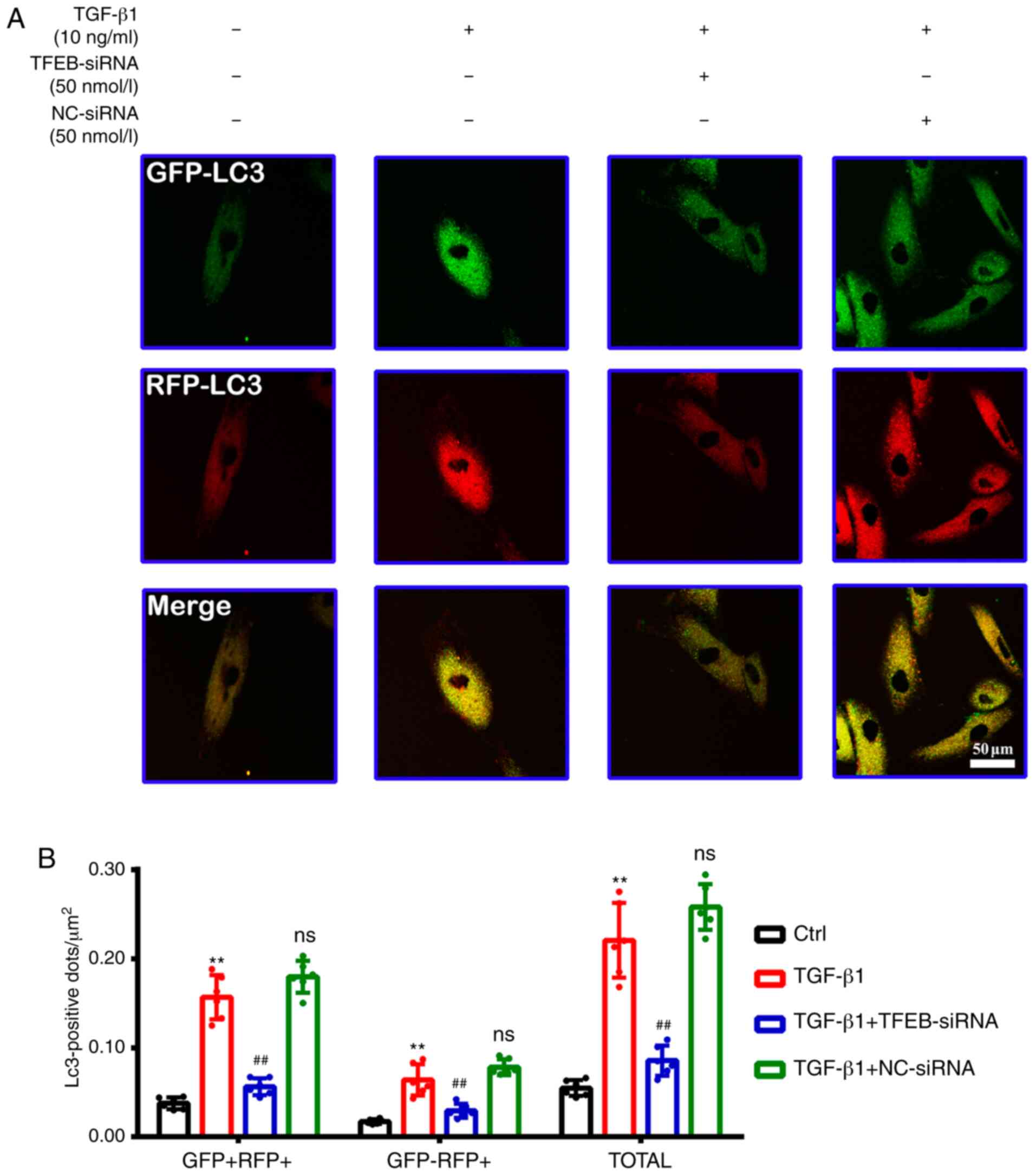
For an improved quantitative assessment of
autophagy, 50 nmol/l TFEB-siRNA or NC-siRNA was used to block the
fusion of lysosomes and autophagosomes, and autophagic flux was
assessed using an RFP-GFP-LC3 plasmid, in which yellow puncta
reflect combined GFP and RFP fluorescence, representing
autophagosomes, while red puncta (RFP only) represent
autolysosomes. The accumulation of both yellow puncta (0.157±0.025
vs. 0.037±0.006; P<0.01; Fig. 5A
and B) and red puncta (0.064±0.018 vs. 0.017±0.003; P<0.01;
Fig. 5A and B) were significantly
increased by TGF-β1 treatment compared with the control. Compared
with the TGF-β1 treatment group, transfection with TFEB-siRNA
significantly decreased the amount of yellow puncta (0.056±0.009
vs. 0.157±0.025; P<0.01; Fig. 5A
and B) and red puncta (0.029±0.008 vs. 0.064±0.018; P<0.01;
Fig. 5A and B); however, no
significant difference was detected between the TGF-β1 group and
TGF-β1 + NC-siRNA group.
Compared with the fibroblasts treated with TGF-β1,
transfection with TFEB-siRNA significantly increased the expression
of p-eIF2α (3.78±0.54 vs. 2.75±0.35; P<0.01; Fig. 6A; 3.51±0.46 vs. 2.80±0.37;
P<0.05; Fig. S2A), XBP-1s
(3.20±0.34 vs.1.93±0.18; P<0.01; Fig. 6B; 3.13±0.41 vs. 1.97±0.38;
P<0.01; Fig. S2B) and CHOP
(2.29±0.41 vs. 1.88±0.20; P<0.05; Fig. 6C; 2.56±0.42 vs. 1.81±0.30;
P<0.01; Fig. S2C), which are
associated with a predisposition to cell death (26), and also increased the levels of
cleaved caspase 3, a marker of apoptosis (2.43±0.30 vs. 1.07±0.18;
P<0.01; Fig. 6E; 2.18±0.19 vs.
1.12±0.20; P<0.01; Fig. S3A)
compared with the TGF-β1 treatment group. TUNEL staining also
demonstrated that knockdown of TFEB induced apoptotic cell death
(32.95±4.11 vs. 7.59±0.80; P<0.01; Fig. 6D) compared with the TGF-β1
treatment group. However, TFEB-siRNA did not induce apoptosis in
quiescent fibroblasts without TGF-β1 treatment (Fig. 6D and E).
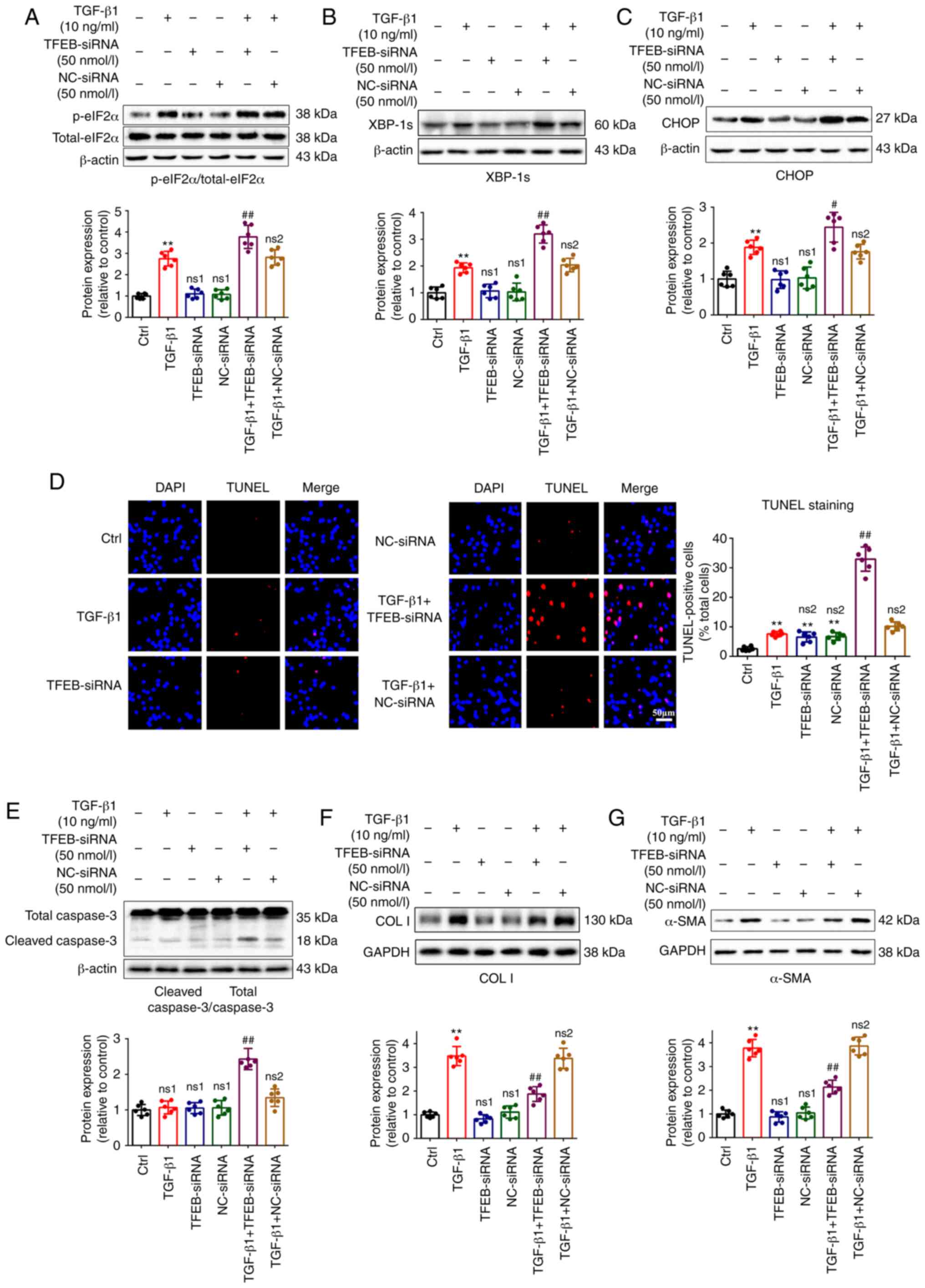 | Figure 6TFEB-siRNA enhances TGF-β1-induced ER
stress and apoptosis, while it reduces the phenotypic
transformation and collagen synthesis in fibro-blasts. Fibroblasts
were transfected with 50 nmol/l TFEB-siRNA or NC-siRNA for 48 h and
then treated with or without 10 ng/ml TGF-β1 for 48 h. The
expression of (A) p-eIF2α/total-eIF2α, (B) XBP-1s, (C) CHOP, (E)
cleaved caspase 3/total caspase 3, (F) COL I and (G) α-SMA in
fibroblasts was determined by western blotting. GAPDH or β-actin
served as a loading control. (D) Apoptotic fibroblasts were
analyzed by TUNEL staining (scale bar, 50 µm). Results are
presented as the mean ± SD (n=6). **P<0.01 vs.
control group; #P<0.05 and ##P<0.01 vs.
TGF-β1 treatment group. TGF-β1, transforming growth factor-β1;
TFEB, transcription factor EB; siRNA, small interfering RNA; NC,
negative control; ER, endoplasmic reticulum; p-, phosphorylated;
eIF2α, α subunit of eukaryotic initiation factor 2; CHOP,
C/EBP-homologous protein caspase 3; XBP-1s, spliced X-box binding
protein 1; caspase 3, cysteinyl aspartate specific proteinase 3;
α-SMA, α smooth muscle actin; COL I, collagen I; TUNEL, terminal
deoxynucleotidyl transferase-mediated biotinylated UTP nick-end
labeling; ns1, not significant compared with control group; ns2,
not significant compared with TGF-β1 treatment group. |
The results also demonstrated that the knockdown of
TFEB significantly reduced the expression levels of COL I
(1.87±0.31 vs. 3.48±0.40; P<0.01; Fig. 6F; 2.07±0.34 vs. 3.39±0.41;
P<0.01; Fig. S3B) and α-SMA
(2.13±0.29 vs. 3.77±0.37; P<0.01; Fig. 6G; 2.24±0.42 vs. 3.99±0.36;
P<0.01; Fig. S3C) compared
with the TGF-β1 treatment group.
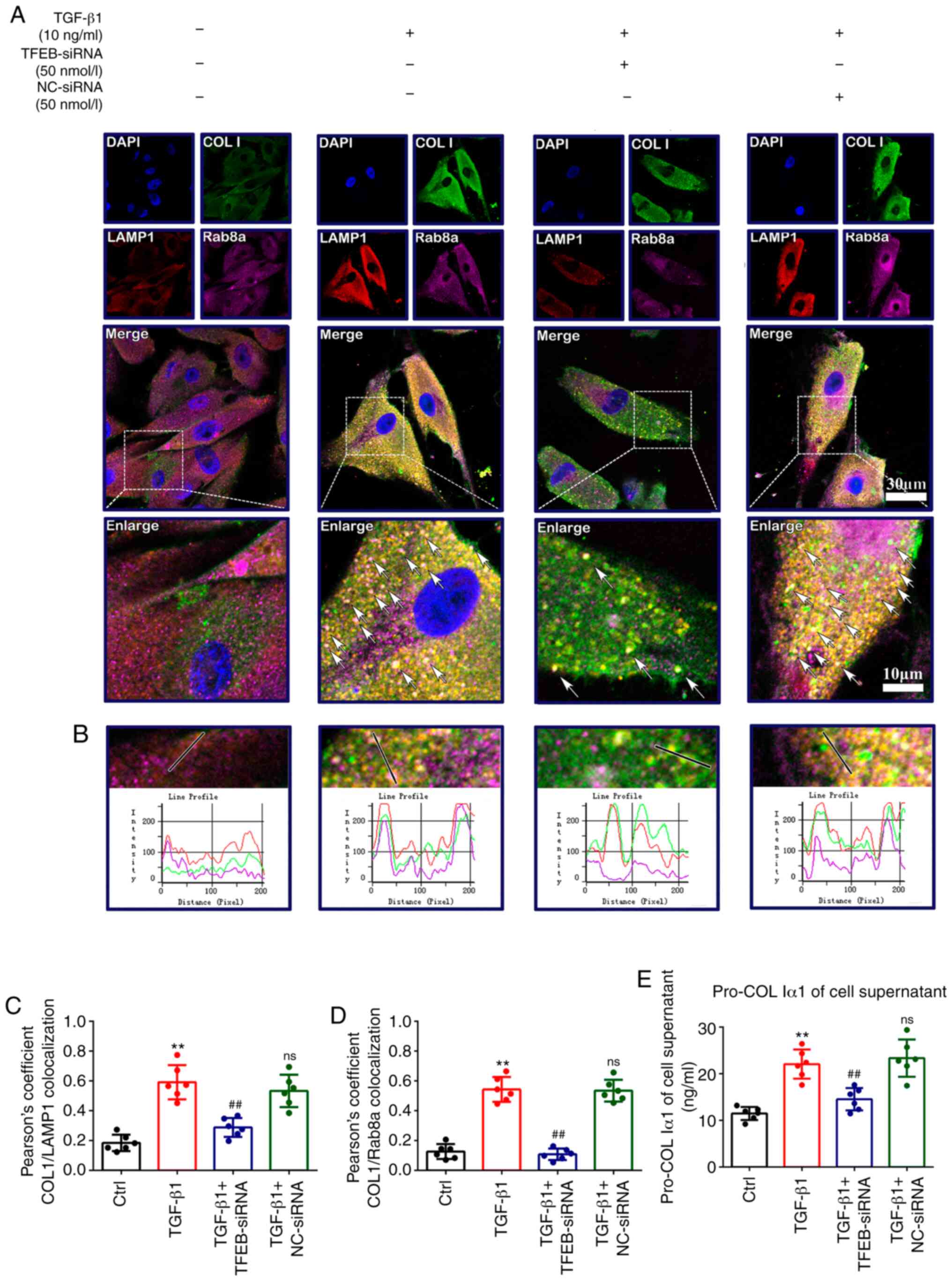
TGF-β1-induced COL I secretion is
associated with an autophagy-based unconventional secretory pathway
and is mediated by Rab8a activity
Since fibroblasts do not contain any evidently large
coat protein complex II (COPII) structures, and collagens are too
large to be incorporated into conventional 80-nm COPII-coated
vesicles (27), the present study
further explored whether autophagy is an alternative secretory
pathway of collagen trafficking. Fibroblasts were transfected with
50 nmol/l TFEB-siRNA or NC-siRNA, and then treated with TGF-β1 (10
ng/ml). Immunofluorescence analysis showed that TGF-β1 increased
the colocalization of COL I with the lysosome marker LAMP1
(0.59±0.12 vs. 0.18±0.05; P<0.01; Fig. 7A-C) and Rab8a (0.54±0.08 vs.
0.13±0.04; P<0.01; Fig. 7A, B and
D), a marker of secretory autophagy vesicles, and also
upregulated the secretion of pro-COL Iα1 in the culture
supernatants (22.07±3.14 vs. 11.49±1.41; P<0.01; Fig. 7E; 21.86±3.25 vs. 11.20±2.10;
P<0.01; Fig. S3D) compared
with the control group. These changes were reversed by TFEB
knockdown [0.29±0.06 vs. 0.59±0.12 (P<0.01; Fig. 7C); 0.10±0.04 vs. 0.54±0.08
(P<0.01; Fig. 7D); 14.54±2.41
vs. 22.07±3.14 (P<0.01; Fig.
7E); 14.57±2.22 vs. 21.86±3.25 (P<0.01; Fig. S3D)]. These data suggested that
COL I may be secreted via the secretory autophagy pathway during
myofibroblast differentiation and proliferation.
Discussion
Myofibroblasts, which are transformed from activated
fibroblasts, can serve as the source of de novo synthesis
and secretion of collagen proteins that connect with the collagen
proteins on the edge of wounded tissues, and promote the edges to
shrink together, thus playing a key role in the early stage of the
wound-healing process (28).
During the remodeling phase of wound-healing, myofibroblasts
undergo a shift from proliferation to apoptosis in order to clear
the wound site (29). However,
under fibrotic conditions, myofibroblasts are continuously
activated, and remain as the fibrotic phenotype, which are
resistant to apoptosis. At present, the underlying molecular
mechanisms during this process have not been fully elucidated.
Autophagy has been demonstrated to be closely associated with the
pathological process of numerous fibrotic diseases. The present
study provided insight into how TGF-β1-induced autophagy leads to
less misfolded collagen in dermal fibroblasts through autophagic
degradation and autophagy-dependent secretion, along with reduced
ER stress and prevention of cell apoptosis.
It is commonly known that TGF-β1 plays a key
modulatory role in the proliferation of fibroblasts and the
accumulation of ECM via the Smad signaling pathway (30). In the present study, western
blotting and ELISA results also confirmed that 10 ng/ml TGF-β1
effectively increased collagen content, not only in fibroblasts,
but also in culture supernatants. Synthesized and folded in the ER,
collagen is composed of three pro-chains that form a triple helix
(31). Despite its relatively
simple structure, the folding of collagen is very difficult due to
its thermodynamic instability, and relies on multiple chaperones,
such as collagen prolyl 4-hydroxylase, GRP78, protein
disulfide-isomerase and heat shock protein 47, which stabilize
native structures and prevent aggregation of unfolded chains
(27,32-35). Unfolded or misfolded proteins are
harmful and must be folded into the appropriate conformation to
perform their cellular functions. TGF-β1 promotes protein synthesis
and increases the demand for the protein-folding capacity of ER,
which disturbs ER homeostasis, resulting in increased ER stress and
activation of a network of signaling pathways, known as the
unfolded protein response (UPR), which is mediated through three
sensors: PERK, IRE1α and ATF6 (36). These proteins dissociate from
GRP78 and then activate downstream signaling pathways, which
suppress protein translation, enhance protein folding and promote
unfolded or misfolded protein degradation (37). The positive effects of TGF-β1 on
ER stress were confirmed in the current study by the upregulated
expression of these ER stress markers. Activation of UPR signaling
is known to induce the ER-associated degradation (ERAD) system,
which mediates the degradation of misfolded collagen molecules from
the ER (38). However, the ERAD
pathway can only degrade collagen monomers, and cannot degrade
collagen trimmers once they are formed (39). The persistence of uncompensated ER
stress eventually induces apoptosis. Thus, there must be other
underlying mechanisms that contribute to the reduction of ER stress
in the context of fibrosis.
Autophagy is an essential lysosome-dependent
degradation pathway that is involved in the breakdown of cellular
organelles and protein complexes too large for proteasomal
degradation (40). Similar to
UPR, autophagy is also associated with both cell survival and death
(41). The present study observed
that the expression levels of autophagy-related proteins and the
key regulator of autophagy, TFEB, were upregulated, which was
consistent with the changes of ER stress marker proteins in
fibroblasts treated with TGF-β1. It was shown in this study that
the ER stress inhibitor TUDCA was also able to downregulate
autophagy, indicating that autophagy may be upregulated by ER
stress in differentiated fibroblasts.
Therefore, autophagy was further downregulated
through knockdown of TFEB to investigate whether autophagy is the
mechanism of the reduction of ER stress and promotion of cell
survival by autophagy under fibrotic conditions. Under normal
conditions, TFEB is phosphorylated and localized in the cytoplasm,
and does not exhibit any function. Under the circumstances of
starvation or oxidation stimulation, TFEB is dephosphorylated,
translocated to the nucleus, and activates the transcription of
genes related to autophagy and lysosome biogenesis (42). TFEB plays a pivotal role in
regulating the process of autophagy (23,43). As expected, in the present study
knockdown of TFEB enhanced the apoptosis of fibro-blasts by
activating the CHOP pathway, as well as reducing the phenotypic
transformation of fibroblasts and the synthesis and secretion of
collagen. CHOP plays an important role in ER stress-induced
apoptosis (44). The
transcription of CHOP is activated via the PERK/eIF2a/ATF4 and
IRE1α/XBP-1s pathways, which transmits to the mitochondria and
finally leads to caspase 3 activation (45). Notably, the downregulation of
autophagy-mediated apoptosis only occurred in activated
fibroblasts, but not in quiescent fibroblasts in the present study.
This finding suggested that knockdown of TFEB aggravated the
pre-existing ER stress response, converting adaptive signaling to
apoptotic signaling. Collectively, these results confirm that
TFEB-mediated autophagy could main-tain the survival and function
of fibroblasts by eliminating misfolded/unfolded proteins. However,
given that the role of autophagy in the pathogenesis of diseases is
multifaceted and complex, whether upregulated autophagy by
overexpression of TFEB could promote fibroblast proliferation or
lead to further autophagic death needs to be further elucidated in
future research.
In general, secretory proteins are released from the
ER to the Golgi apparatus and subsequently into the extracellular
space, and this process is mediated by COPII-coated vesicles
(27). However, fibroblasts do
not contain any evidently large COPII structures, and collagen
molecules are too large to be incorporated into the conventional
80-nm COPII-coated vesicles (27). Thus, an alternative pathway of
trafficking is required for the export of collagen from the ER in
the absence of large carriers, particularly when handling unfolded
and/or misfolded procollagen.
ER exit sites are physically and functionally
associated with autophagosome formation (46). While autophagy was initially
defined as a cellular process that results in isolation of
cytosolic cargoes and their degradation in lysosomes, it has also
been recognized as a contributor to autophagy-dependent secretion
('secretory autophagy'), which bypasses the ER-Golgi complex and
also promotes toxic protein clearance and cell-cell signaling
communications (47-50). Secretory autophagy was initially
identified in studies that investigated unconventional protein
secretion, for example how proteins that are unable to enter the ER
due to lack of signal peptides are released to the extracellular
space (51). However, it has been
revealed that autophagy is also involved in the regulation of
conventional secretion, with different roles in different cell
types (52,53). Secretory autophagy is a tightly
regulated process, but its underlying molecular machinery is far
from elucidated. Evidence has indicated that certain factors are
necessary for secretory autophagy, among which the small GTPase
Rab8a is considered to be a useful available marker as a regulator
of polarized sorting of proteins to the plasma membrane (54). The present study demonstrated that
Rab8a colocalized with COL I and LAMP1 in fibroblasts following
exposure to TGF-β1. Autophagy inhibition by knockdown of TFEB
significantly reduced colocalization of COL I with Rab8a and LAMP1,
and also decreased the secretion level of pro-COL Iα1 in the cell
culture supernatant. These data indicated that apart from
autophagic degradation, COL I in the autolysosome can also be
released to the extracellular space via autophagy-dependent
secretion machinery. Therefore, autophagy-dependent secretion is
also an important cellular clearance mechanism for the maintenance
of cell homeostasis under fibrotic conditions. In addition,
collagen fibrils can only be formed after procollagen is
cross-linked following cleavage of terminal pro-peptides by
procollagen N- and C-proteinases (55). Therefore, it can be speculated
that misfolded or unfolded COL I secreted via autolysosome to the
extracellular environment in the process of secretory autophagy may
be a cause of the faulty assembly and irregular arrangement of
collagen fibers in HTS.
In conclusion, the present study demonstrated that
TFEB-mediated autophagy reduced ER stress, decreased cell apoptosis
and maintained the activated phenotype of fibro-blasts, not only
through degradation of misfolded or unfolded proteins, but also via
release of COL I from the autolysosome to the extracellular
environment via autophagy-dependent secretion machinery. The
results may provide an improved understanding of the roles of
autophagy in the formation of HTS. The inhibition of autophagy and
lysosomal biogenesis may serve as a novel direction for the
development of treatment strategies for HTS.
Supplementary Data
Abbreviations:
|
HTS
|
hypertrophic scar
|
|
TGF-β1
|
transforming growth factor-β1
|
|
TFEB
|
transcription factor EB
|
|
TUDCA
|
tauroursodeoxycholic acid
|
|
GRP78
|
glucose-regulated proteins 78
|
|
p-PERK
|
phosphorylated-protein kinase R-like
endoplasmic reticulum kinase
|
|
ATF6
|
activating transcription factor 6
|
|
IRE1α
|
Inositol-requiring enzyme-1α
|
|
LC3
|
microtubule associated protein 1 light
chain 3
|
|
LAMP1
|
lysosome-associated membrane protein
1
|
|
CTS B
|
cathepsin B
|
|
p-eIF2α
|
phosphorylated α subunit of eukaryotic
initiation factor 2
|
|
CHOP
|
C/EBP-homologous protein caspase 3
|
|
XBP-1s
|
spliced X-box binding protein 1
|
|
caspase 3
|
cysteinyl aspartate specific
proteinase 3
|
|
α-SMA
|
α smooth muscle actin
|
|
COL I
|
collagen I
|
|
ER
|
endoplasmic reticulum
|
|
LDH
|
lactate dehydrogenase
|
|
UPR
|
unfolded protein response
|
Funding
This work was supported by Hubei Natural Science
Foundation (grant no. 2018CKB904).
Availability of data and materials
The datasets used and/or analyzed during this study
are available from the corresponding author upon reasonable
request.
Authors' contributions
LZ and ZL designed and conducted the experiments. LG
and GY participated in designing the experiments and proofreading
the article. SC assisted in data collection and manuscript
preparation. JQ, QL, SW and WZ participated in performing the
experiments. DC performed data analysis. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This research was approved by the Medical Ethics
Committee of Zhongnan Hospital of Wuhan University (Wuhan, China;
approval no. 2019006) and complied with the ethical standards of
the Declaration of Helsinki as well as the relevant national and
international guidelines. Each patient signed an informed consent
for the use of their abandoned skin tissue in the research.
Patient consent for publication
Written informed consent was obtained from each
patient.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Benzaquen M, Collet-Villette AM and
Delaporte E: Combined treatment of hypertrophic and keloid scars
with intralesional injection of corticosteroids and laser-assisted
corticosteroid delivery. Dermatol Ther. 32:e131262019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Del Toro D, Dedhia R and Tollefson TT:
Advances in scar management: Prevention and management of
hypertrophic scars and keloids. Curr Opin Otolaryngol Head Neck
Surg. 24:322–329. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Li Y, Bai X, Li Y, Shi J and Hu
D: Recent advances in hypertrophic scar. Histol Histopathol.
33:27–39. 2018.
|
|
4
|
Gras C, Ratuszny D, Hadamitzky C, Zhang H,
Blasczyk R and Figueiredo C: miR-145 contributes to hypertrophic
scarring of the skin by inducing myofibroblast activity. Mol Med.
21:296–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Darby IA, Zakuan N, Billet F and
Desmoulière A: The myofibro-blast, a key cell in normal and
pathological tissue repair. Cell Mol Life Sci. 73:1145–1157. 2016.
View Article : Google Scholar
|
|
6
|
Hinz B: The role of myofibroblasts in
wound healing. Curr Res Transl Med. 64:171–177. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pakyari M, Farrokhi A, Maharlooei MK and
Ghahary A: Critical role of transforming growth factor beta in
different phases of wound healing. Adv Wound Care (New Rochelle).
2:215–224. 2013. View Article : Google Scholar
|
|
8
|
Zhang Y, Cheng C, Wang S, Xu M, Zhang D
and Zeng W: Knockdown of FOXM1 inhibits activation of keloid
fibro-blasts and extracellular matrix production via inhibition of
TGF-β1/Smad pathway. Life Sci. 232:1166372019. View Article : Google Scholar
|
|
9
|
Alizadeh J, Glogowska A, Thliveris J,
Kalantari F, Shojaei S, Hombach-Klonisch S, Klonisch T and Ghavami
S: Autophagy modulates transforming growth factor beta 1 induced
epithelial to mesenchymal transition in non-small cell lung cancer
cells. Biochim Biophys Acta Mol Cell Res. 1865:749–768. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang C, Xu J, Meng Q, Zhang B, Liu J, Hua
J, Zhang Y, Shi S and Yu X: TGFB1-induced autophagy affects the
pattern of pancreatic cancer progression in distinct ways depending
on SMAD4 status. Autophagy. 16:486–500. 2020. View Article : Google Scholar :
|
|
11
|
Cuomo F, Altucci L and Cobellis G:
Autophagy function and dysfunction: Potential drugs as anti-cancer
therapy. Cancers (Basel). 11:14652019. View Article : Google Scholar
|
|
12
|
Fitzwalter BE and Thorburn A: Recent
insights into cell death and autophagy. FEBS J. 282:4279–4288.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu C, Yang Y, Zhu Y, Lv S and Zhang J: An
intervention target for myocardial fibrosis: Autophagy. Biomed Res
Int. 2018:62159162018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maiuri L and Kroemer G: Autophagy delays
progression of the two most frequent human monogenetic lethal
diseases: Cystic fibrosis and Wilson disease. Aging (Albany NY).
10:3657–3661. 2018. View Article : Google Scholar
|
|
15
|
Tseng YJ, Dong L, Liu YF, Xu N, Ma W, Weng
SQ, Janssen HLA and Wu SD: Role of autophagy in chronic liver
inflammation and fibrosis. Curr Protein Pept Sci. 20:817–822. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao XC, Livingston MJ, Liang XL and Dong
Z: Cell apoptosis and autophagy in renal fibrosis. Adv Exp Med
Biol. 1165:557–584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lodder J, Denaës T, Chobert MN, Wan J,
El-Benna J, Pawlotsky JM, Lotersztajn S and Teixeira-Clerc F:
Macrophage autophagy protects against liver fibrosis in mice.
Autophagy. 11:1280–1292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cabrera S, Maciel M, Herrera I, Nava T,
Vergara F, Gaxiola M, López-Otín C, Selman M and Pardo A: Essential
role for the ATG4B protease and autophagy in bleomycin-induced
pulmonary fibrosis. Autophagy. 11:670–684. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takagaki Y, Lee SM, Dongqing Z, Kitada M,
Kanasaki K and Koya D: Endothelial autophagy deficiency induces
IL6-dependent endothelial mesenchymal transition and organ
fibrosis. Autophagy. 16:1905–1914. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frangou E, Chrysanthopoulou A, Mitsios A,
Kambas K, Arelaki S, Angelidou I, Arampatzioglou A, Gakiopoulou H,
Bertsias GK, Verginis P, et al: REDD1/autophagy pathway promotes
thromboinflammation and fibrosis in human systemic lupus
erythematosus (SLE) through NETs decorated with tissue factor (TF)
and interleukin-17A (IL-17A). Ann Rheum Dis. 78:238–248. 2019.
View Article : Google Scholar :
|
|
21
|
San-Miguel B, Crespo I, Sánchez DI,
González-Fernández B, Ortiz de Urbina JJ, Tuñón MJ and
González-Gallego J: Melatonin inhibits autophagy and endoplasmic
reticulum stress in mice with carbon tetrachloride-induced
fibrosis. J Pineal Res. 59:151–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livingston MJ, Ding HF, Huang S, Hill JA,
Yin XM and Dong Z: Persistent activation of autophagy in kidney
tubular cells promotes renal interstitial fibrosis during
unilateral ureteral obstruction. Autophagy. 12:976–998. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Settembre C, Di Malta C, Polito VA, Garcia
Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D,
Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du F, Zhu L, Qian ZM, Wu XM, Yung WH and
Ke Y: Hyperthermic preconditioning protects astrocytes from
ischemia/reperfusion injury by up-regulation of HIF-1 alpha
expression and binding activity. Biochim Biophys Acta.
1802:1048–1053. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Cybulsky AV: The intersecting roles of
endoplasmic reticulum stress, ubiquitin- proteasome system, and
autophagy in the patho-genesis of proteinuric kidney disease.
Kidney Int. 84:25–33. 2013. View Article : Google Scholar
|
|
27
|
McCaughey J and Stephens DJ: ER-to-golgi
transport: A sizeable problem. Trends Cell Biol. 29:940–953. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schnieder J, Mamazhakypov A, Birnhuber A,
Wilhelm J, Kwapiszewska G, Ruppert C, Markart P, Wujak L, Rubio K,
Barreto G, et al: Loss of LRP1 promotes acquisition of
contractile-myofibroblast phenotype and release of active TGF-β1
from ECM stores. Matrix Biol. 88:69–88. 2020. View Article : Google Scholar
|
|
29
|
Dolivo DM, Larson SA and Dominko T:
Fibroblast growth factor 2 as an antifibrotic: Antagonism of
myofibroblast differentiation and suppression of pro-fibrotic gene
expression. Cytokine Growth Factor Rev. 38:49–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kirkness MW, Lehmann K and Forde NR:
Mechanics and structural stability of the collagen triple helix.
Curr Opin Chem Biol. 53:98–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saito K, Maeda M and Katada T: Regulation
of the Sar1 GTPase cycle is necessary for large cargo secretion
from the endoplasmic reticulum. Front Cell Dev Biol. 5:752017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gjaltema RA and Bank RA: Molecular
insights into prolyl and lysyl hydroxylation of fibrillar collagens
in health and disease. Crit Rev Biochem Mol Biol. 52:74–95. 2017.
View Article : Google Scholar
|
|
34
|
Sorushanova A, Delgado LM, Wu Z, Shologu
N, Kshirsagar A, Raghunath R, Mullen AM, Bayon Y, Pandit A,
Raghunath M and Zeugolis DI: The collagen suprafamily: From
biosynthesis to advanced biomaterial development. Adv Mater.
31:e18016512019. View Article : Google Scholar
|
|
35
|
Ishikawa Y and Bächinger HP: A molecular
ensemble in the rER for procollagen maturation. Biochim Biophys
Acta. 1833:2479–2491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Biol. 21:421–438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shu S, Zhu J, Liu Z, Tang C, Cai J and
Dong Z: Endoplasmic reticulum stress is activated in post-ischemic
kidneys to promote chronic kidney disease. EBioMedicine.
37:269–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kropski JA and Blackwell TS: Endoplasmic
reticulum stress in the pathogenesis of fibrotic disease. J Clin
Invest. 128:64–73. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ishida Y and Nagata K: Autophagy
eliminates a specific species of misfolded procollagen and plays a
protective role in cell survival against ER stress. Autophagy.
5:1217–1219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nabar NR, Shi CS and Kehrl JH: Signaling
by the toll-like receptors induces autophagy through modification
of beclin 1 Molecular Mechanism. Immunology. Hayat MA: Academic
Press; London: pp. 75–84. 2018, View Article : Google Scholar
|
|
41
|
Swart C, Du Toit A and Loos B: Autophagy
and the invisible line between life and death. Eur J Cell Biol.
95:598–610. 2016. View Article : Google Scholar
|
|
42
|
Füllgrabe J, Klionsky DJ and Joseph B: The
return of the nucleus: Transcriptional and epigenetic control of
autophagy. Nat Rev Mol Cell Biol. 15:65–74. 2014. View Article : Google Scholar
|
|
43
|
Napolitano G and Ballabio A: TFEB at a
glance. J Cell Sci. 129:2475–2481. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kandel-Kfir M, Almog T, Shaish A, Shlomai
G, Anafi L, Avivi C, Barshack I, Grosskopf I, Harats D and Kamari
Y: Interleukin-1α deficiency attenuates endoplasmic reticulum
stress-induced liver damage and CHOP expression in mice. J Hepatol.
63:926–933. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
46
|
Sanchez-Wandelmer J, Ktistakis NT and
Reggiori F: ERES: Sites for autophagosome biogenesis and
maturation? J Cell Sci. 128:185–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ponpuak M, Mandell MA, Kimura T, Chauhan
S, Cleyrat C and Deretic V: Secretory autophagy. Curr Opin Cell
Biol. 35:106–116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kimura T, Jia J, Claude-Taupin A, Kumar S,
Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, et al: Cellular
and molecular mechanism for secretory autophagy. Autophagy.
13:1084–1085. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bel S and Hooper LV: Secretory autophagy
of lysozyme in Paneth cells. Autophagy. 14:719–721. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
New J, Arnold L, Ananth M, Alvi S,
Thornton M, Werner L, Tawfik O, Dai H, Shnayder Y, Kakarala K, et
al: Secretory autophagy in cancer-associated fibroblasts promotes
head and neck cancer progression and offers a novel therapeutic
target. Cancer Res. 77:6679–6691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rabouille C: Pathways of unconventional
protein secretion. Trends Cell Biol. 27:230–240. 2017. View Article : Google Scholar
|
|
52
|
Gonzalez CD, Resnik R and Vaccaro MI:
Secretory autophagy and its relevance in metabolic and degenerative
disease. Front Endocrinol (Lausanne). 11:2662020. View Article : Google Scholar
|
|
53
|
Cavalli G and Cenci S: Autophagy and
protein secretion. J Mol Biol. 432:2525–2545. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ejlerskov P, Rasmussen I, Nielsen TT,
Bergström AL, Tohyama Y, Jensen PH and Vilhardt F: Tubulin
polymerization-promoting protein (TPPP/p25α) promotes
unconventional secretion of α-synuclein through exophagy by
impairing autopha-gosome-lysosome fusion. J Biol Chem.
288:17313–17335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Khoshnoodi J, Cartailler JP, Alvares K,
Veis A and Hudson BG: Molecular recognition in the assembly of
collagens: Terminal noncollagenous domains are key recognition
modules in the formation of triple helical protomers. J Biol Chem.
281:38117–38121. 2006. View Article : Google Scholar : PubMed/NCBI
|