1. Introduction
Ischemia is the disruption of blood flow to tissues,
which also results in the reduction of delivery of oxygen and
metabolites to cells (1).
Pathological ischemia in the heart results in oxidative damage,
apoptosis and cardiomyocyte death, which is termed myocardial
infarction (MI) (2). The standard
approach for the management of a patient with MI is to restore
blood flow to the ischemic tissue as quickly as possible. However,
this restoration of oxygenated blood causes extensive tissue
damage, termed ischemia-reperfusion (IR) injury or
hypoxia-reoxygenation injury (3,4),
which may result in morbidity and mortality in the cardiac
intervention following a heart attack or stroke (5,6),
as well as in other pathological situations, such as acute kidney
injury, muscle injury, organ transplantation, hypovolemic shock and
elective surgery (5,7). MI followed by reperfusion is the
major stressor leading to chronic heart failure (8). However, reducing the irreversible
ischemic damage due to MI and the inevitable IR injury that occurs
during the reperfusion of ischemic tissues remains a challenge for
cardiac therapy.
The mitochondria occupy >30% of the cardiomyocyte
volume, provide 95% of adenosine triphosphate (ATP) generated
through oxidative phosphorylation (OXPHOS) for the beating of the
heart, and serve as the metabolic hub for the citric acid cycle and
fatty acid β-oxidation (3,8,9).
IR damages the mitochondrial respiratory chain, and accompanied by
ATP depletion, results in the accumulation of mitochondrial
reactive oxygen species (ROS; mtROS; ROS produced by mitochondria),
and oxidative damage on mitochondrial DNA (mtDNA) or proteins,
ultimately leading to necrotic and apoptotic myocardial cell death.
The extent of mitochondrial damage is a key determinant in the
progression of myocardial IR injury towards to heart failure
(10).
2. Molecular mechanisms underlying IR
injury
In the normoxic heart (Fig. 1A), glucose and fatty acids are
oxidized through glycolysis, β-oxidation and the tricarboxylic acid
(TCA) cycle, respectively. The electrons from the final metabolites
nicotinamide adenine dinucleotide reduced form (NADH;
NAD+ + H+ + 2e− ↔ NADH) and flavin
adenine dinucleotide hydroquinone form (FADH2 ; FAD +
2H+ + 2e− ↔ FADH2) reduce oxygen
(O2) into water through the electron transport chain in
the mitochondrial cristae membrane and generate ATP through OXPHOS
driven by the protonmotive force (∆p) (11). During the conventional forward
electron transport, 2e− are transferred from NADH to
reduce coenzyme Q (CoQ, ubiquinone) into CoQH2 (the
fully reduced form of CoQ, ubiquinol), following pumping of 4
protons (4H+) into the mitochondrial intermembrane space
to maintain the ∆p for the later ATP synthesis. If complex I is
inhibited by the CoQ inhibitor rotenone, the flavin mononucleotide
(FMN) can accept the electrons from NADH and reduce O2
to generate superoxide (O2−) at complex I
(5,12).
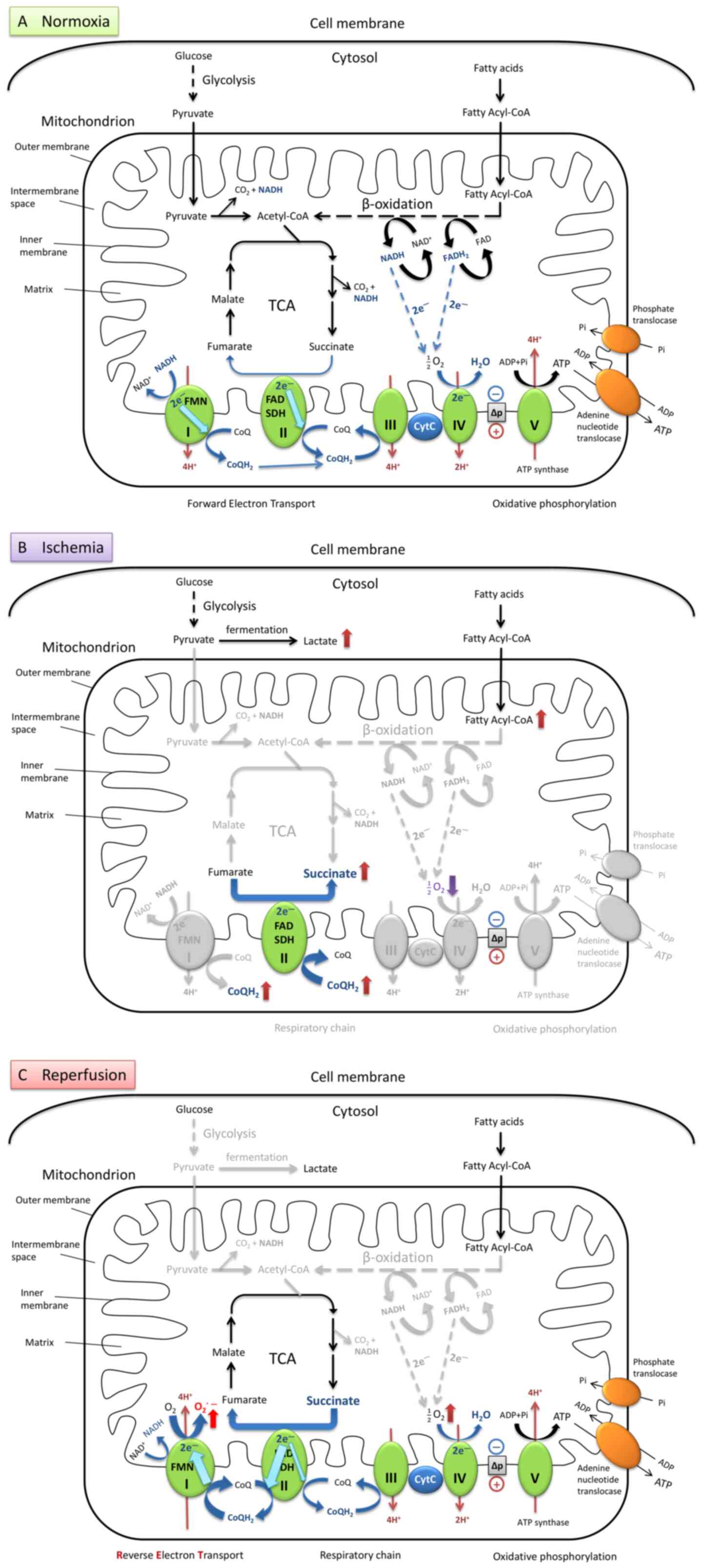 | Figure 1Brief illustration of metabolism via
aerobic respiration under normal conditions and the mechanism of
mtROS production during ischemia-reper-fusion in heart cells. (A)
Under normal oxygen levels, metabolism is balanced by a series of
hydrogen and electron transfer reactions in the respiratory chain.
(B) During ischemia, the oxygen levels are dramatically decreased,
and electron transport flow is diminished. Ubiquinone (CoQ) accepts
an electron and transforms into ubiquinol (CoQH2) at
complex I. SDH catalyzes the reverse reaction by utilizing the
increased levels of CoQH2 to reduce fumarate to
succinate. This results in the accumulation of succinate which
serves as the final electron sink instead of oxygen. (C) When
reperfusion occurs, and oxygen supply is restored, the succinate is
rapidly oxidized into fumarate by forward catalysis by SDH. Due to
the slow reestablishment of ATP synthesis, the electrons from the
excess flux of CoQH2 are forced backward through FMN in
complex I. Production of superoxide is significantly increased,
generated from oxygen reduction, driven by RET together with the
high proton-motive force. mtROS, mitochondrial reactive oxygen
species; TCA, tricarboxylic acid cycle; I, complex I; FMN, flavin
mononucleotide; II, complex II; SDH, succinate dehydrogenase; FAD,
flavin adenine dinucleotide; III, complex III; CytC, cytochrome
c; IV, complex IV; V, complex V (also termed ATP
synthase). |
Under hypoxic conditions (Fig. 1B), the levels of the metabolites
lactate and succinate are significantly increased in ischemic
tissues. The accumulation of succinate is a primary metabolic
signature of ischemia (13,14), and is considered to be a
consequence of the inhibition of the TCA cycle during ischemia or
anoxia. However, recent studies have proposed that it may also be
due to the reversal of succinate dehydrogenase (SDH), driven by the
electrons transported from the accumulated CoQH2 to
fumarate (14,15). Moreover, succinate cannot be
further converted into succinyl-coenzyme A (succinyl-CoA) by
succinyl-CoA synthetase due to the depletion of GTP and CoA during
ischemia (13,14).
During the reperfusion process (Fig. 1C), the accumulated succinate is
rapidly re-oxidized by the SDH in the mitochondrial respiratory
chain complex II, resulting in a burst of superoxide production
from complex I by reverse electron transport (RET). A
CoQH2 pool and a high ∆p are required to drive RET. The
accumulation of CoQH2 is formed during hypoxia and the
high ∆p is restored from proton pumping by complexes III and IV
within seconds of reoxygenation. RET occurs when electrons are
forced to flow backward from a reduced CoQ pool through complex I
and onto the FMN to drive superoxide formation. The superoxide
generated at complex I by RET upon reperfusion is considered as the
major source of ROS during IR injury (5,14,15). In addition to the classic theory
described above, there are also other sources of mtROS which
contribute to IR injury, which have been elucidated in recent
years, such as the ROS formed by monoamine oxidases,
p66Shc protein and NADH oxidases (16,17).
ROS include free superoxide anions
(O2˙−), hydroxyl radicals (OH·), non-radical
hydrogen peroxide (H2O2) and peroxynitrite
(ONOO−) (18).
Usually, the superoxide anion generated in the mitochondrial matrix
can be converted by the manganese-dependent superoxide dismutase
(MnSOD, also known as SOD2) into H2O2, and
then further detoxified by glutathione peroxidase and thioredoxin
enzymes into H2O. The superoxide anion
(O2˙−) diffuses into the cytoplasm and is
transformed by copper-zinc-dependent SOD (CuZnSOD, also known as
SOD1) into H2O2 (19,20). When the production of ROS exceeds
the defensive capacity of cellular antioxidant systems, the
excessive ROS cause functional and structural damage to the cells
(21).
Cardiomyocyte oxidative damage is the major
consequence of cardiac IR injury; however, some damage can be
attributed to circulating platelets. During cardiac ischemia,
activated platelets induce thrombi formation and occlusion of
microcirculation, which causes secondary ischemia injury after
reperfusion (4,22).
3. Alterations in mitochondrial metabolism
during cardiac IR injury
Oxidative stress
Restoring blood flow and the reoxygenation of the
cardiac tissue following myocardial ischemia generates excessive
quantities of ROS. The accumulation of ROS damages cellular
components (such as proteins, lipid and DNA oxidation), increases
the susceptibility of mitochondria and cells, and accelerates aging
and cell death (3,12,21,23). For example, toxic ROS can damage
mitochondrial respiratory complex I via the oxidation of thiols,
which in turn results in the production of excess ROS during
reperfusion (6). The phospholipid
cardiolipin, which is required for the mitochondrial inner
membrane, and the activities of complexes III and IV in the
respiratory chain, contains a high percentage of unsaturated fatty
acids and is sensitive to peroxidation by ROS (24). Cardiolipin is also essential in
the biochemical functions of several mitochondrial proteins, such
as mitochondrial creatine kinase (mitCK) (24). mitCK plays an important role in
the creatine-phosphocreatine energy transferring system (25). The mitCK system has been shown to
be damaged by cardiac ischemia and oxidative stress (26). The burst of ROS from mitochondria
during IR not only causes direct acute damage in cells or tissues,
but also initiates the long-term inflammatory response following
reperfusion or persistent pathologies such as in chronic heart
diseases (7,27).
Carbon stress
During IR, activated acyl-CoA accumulates in the
mitochondrial matrix due to a defective β-oxidation pathway. The
non-enzymatic acylation of lysine residues by acyl-CoA causes
matrix protein dysfunction and aggregation (28-30).
Depletion of NAD+
A damaged mitochondrial respiratory chain also
causes the depletion of NAD+. Since NAD+ is
required for ATP synthesis and also for the deacylation of Lys by
sirtuins, NAD+ depletion disrupts the pathways for
bioenergetics and also the pathways altered by carbon stress
(30,31).
Bioenergetic insufficiency (ATP)
Mitochondria generate ATP through OXPHOS within the
electron transport chain at the mitochondrial inner membrane. The
lack of oxygen and respiratory substrates impairs OXPHOS during
ischemia (3). IR injury markedly
increases mitochondrial permeability, and dissipates electron and
proton gradients. Furthermore, mitochondrial electron transport
chain subunits lose their integrity and are degraded under
conditions of IR. OXPHOS is markedly diminished in the abnormal
mitochondria, leading to bioenergetic insufficiency during MI
(23).
Formation of mitochondrial permeability
transition pore (mPTP)
mPTP is a large conductance channel in the inner
membrane, which is activated by Ca2+ accumulation
(32,33). Ischemic oxidative damage results
in the marked reduction of mitochondrial membrane potential (∆Ψm),
mitochondrial Ca2+ bursts and the opening of the
non-selective mPTP in the inner membrane. Accompanied by the
release of matrix proteins and mtDNA into the cytosol, mPTP opening
increases colloid-osmotic pressure in the matrix, induces the
innate immune response, and initiates the activation of proteases
and lipases, leading to mitochondrial swelling and cardiomyocyte
death through necrosis (34,35). The excessive activation of the
mitochondrial fission protein, dynamin-related protein 1 (Drp1),
also facilitates mPTP opening and accelerates cell death. The
genetic or pharmacological inhibition of Drp1 has been shown to
decrease the frequency of transient opening of mPTP during cardiac
IR and reduce the infarct size following MI (8,36,37).
Calcium overload
During ischemia, ATP depletion and acidosis caused
by the increasing lactate levels drive cytosolic Na+
accumulation through the plasma membrane
Na+/H+ exchanger, which, in turn leads to an
excessive exchange of Ca2+ via the
Na+/Ca2+ exchanger (15). The transport of Ca2+
between sarcoplasmic/endoplasmic reticulum and mitochondria enables
cardiac excitation-contraction coupling (38). However, Ca2+ uptake by
the sarcoplasmic reticulum Ca2+-ATPase is inhibited due
to ATP depletion during ischemia. The rapid restoration of the ∆Ψm
by the subsequent reperfusion drives the uptake of Ca2+
into the mitochondria via the mitochondrial calcium uniporter
(15). Therefore, Ca2+
overload in the mitochondria is one of the hallmarks of IR injury
of the heart (39). During the
reperfusion phase, the accumulation of cytosolic Ca2+
activates calcineurin (CaN) to dephosphorylate Drp1 at
Ser637, initiating mitochondrial fission and
cardiomyocyte apoptosis (40).
4. Mitochondrial dynamics in response to IR
stress
Biogenesis
There are a large number of transcription factors
and regulatory proteins to enhance mitochondrial biogenesis, such
as nuclear respiratory factors 1 and 2, transcription factor A
mitochondrial, peroxisome proliferator-activated receptors (PPARs),
estrogen-related receptors, cAMP response element-binding proteins
and Forkhead box-O. Transcription factors can be regulated by
coactivators and corepressors, such as PPARγ coactivator-1α
(PGC-1α) and nuclear receptor corepressor 1. PGC-1α can be
activated by post translational modifications, such as through
phosphorylation by AMP-activated protein kinase (AMPK) and
deacetylation by Sirtuin 1 (Sirt1) (41). Under oxygen insufficiency, an
energetic stress results in the downregulation of mitochondrial
biogenesis by the hypoxia-inducible factor-mediated suppression of
PGC-1α (42).
Fission and fusion
Continuous mitochondrial fission and fusion enables
a dynamic distribution of mitochondrial proteins and mtDNA through
the cellular mitochondrial network (23). Mitochondrial fission involves
division into two daughter mitochondria, one with a markedly higher
mitochondrial membrane potential (∆Ψm) and fusion affinity than the
other one. Subsequently, the active mitochondrion is further
regenerated and the defective one is targeted for degradation via
mitophagy to maintain mitochondrial homeostasis (3,8,43).
Fission and fusion proteins impact mitochondrial morphology and
respiration, therefore, a balanced dynamic is essential to adjust
mitochondrial metabolism to meet the energy demands in
cardiomyocytes (8,44,45). The alteration of proteins involved
in mitochondrial fission and fusion is tightly associated with the
pathophysiology of several cardiac diseases, such as IR stress and
heart failure (10,46).
The mitochondrial fission protein, Drp1, regulates
mitochondrial dynamics on the mitochondrial outer membrane
(47). Drp1 acts with Bcl-2
family proteins (such as the pro-apoptotic proteins Bax and Bak) to
accelerate mitochondrial fragmentation and apoptosis (48). The phosphorylation at
Ser637 residue of Drp1 by protein kinase A (PKA)
inhibits its GTPase activity and its translocation to the
mitochondria (49). Drp1 is
enriched in the heart and its inhibition exhibits protective
effects during myocardial IR by preventing excessive fission at the
beginning of reperfusion (36,40). The overexpression of the cardiac
proto-oncogene Ser/Thr kinase, Pim-1, causes the downregulation of
Drp1 and an increase in the levels of Ser637
phosphorylation, and also protects against cardiomyocyte ischemia
(37). Drp1 can be reactivated
via the dephosphorylation of Ser637 by the
calmodulin-dependent kinase CaN (50). Under normoxic conditions, the
actions of PKA and CaN on Drp1 are well regulated by mitochondrial
scaffolding protein the A-kinase anchoring protein 1 (AKAP1)
(50). However, in ischemic
myocytes, AKAP1 is ubiquitinated by the hypoxia-induced
E3-ubiquitin ligase Siah2 and rapidly degraded via the
ubiquitin-proteasome system, which impairs PKA-mediated Drp1
inhibition and initiates mitochondrial fission and cardiomyocyte
apoptosis (39,50). In addition, cardiac IR injury may
be reduced when the transient suppression of Drp1 has been achieved
by small molecular disruptors or by shRNA/siRNA-mediated knockdown
(36,40,51). Chronic Drp1 deficiency, such as
the heterozygous knockout (Drp1+/−), results in
enlarged infarct sizes after IR stress (52).
The mitochondrial fission factor (Mff) is important
for Drp1 recruitment in mitochondria during the process of fission
(53). The inhibition of Mff has
been shown to enhance mitochondrial membrane potential (∆Ψm) and
decrease apoptosis under hypoxia/reoxygenation (54).
The dynamin-related GTPases mitofusin (Mfn) 1 and 2
are responsible for the fusion of the mitochondrial outer membrane
between two mitochondria (19),
whereas, the optic atrophy protein 1 (OPA1) mediates mitochondrial
inner membrane fusion via regulation of the cristae structure
(8,19). The down-regulation of Mfn1 under
conditions of ROS-induced stress during IR disturbs mitochondrial
fusion and induces cardiomyocyte apoptosis (55). Conversely, Mfn2 is upregulated in
response to the ROS burst during IR and induces the apoptosis of
cardiomyocytes by inhibiting Akt (also known as protein kinase B)
and activating caspase-9 (39).
In the heart, Mfn2 is involved in the regulation of mitophagy,
altering mitochondrial morphology and forming the
ER/SR-mitochondrial contact sites (8). OPA1 plays a protective role in IR
injury. Opa1+/− mice showed larger infarct sizes
compared with wild type, and Opa1 overexpression results in
decreased damage in cardiac function (17). Bax not only directly interacts
with Mfn2 to physiologically regulate its assembly and distribution
in the normal heart, but also co-localizes with Drp1 to facilitate
mitochondrial fission during early apoptosis (39). The Bcl-2 family proteins, Bax, Bak
and Bcl2-interacting protein 3 (BNIP3), promote OPA1 cleavage and
mitochondrial cristae remodeling following reperfusion, which in
turn exacerbate IR injury (39,56).
Degradation and mitophagy
Mitochondrial dynamics facilitate mitochondrial
quality control, which targets aberrant mitochondrial proteins or
entirely damaged mitochondria for degradation to maintain
myocardial mitochondrial homeostasis (57). The ubiquitin-proteasome system
(UPS) and the autophagy-lysosomal system, termed mitophagy, are the
major proteolytic pathways (58).
The clearance of misfolded proteins usually occurs via UPS, whereas
the entire damaged mitochondrion is degraded selectively by
mitophagy (59). There are
interactions and crosstalk between UPS and mitophagy in several
regards, for example, the inhibition of proteasome induces the
activation of mitophagy (58,60).
Defective mitochondrial proteins are labeled with
ubiquitin proteins by a series of E1, E2 and E3 enzymes and
targeted into the proteasome for elimination (61). Impaired UPS proteolytic function
has been observed in the heart tissue of patients with
cardiomyopathy and heart failure (62); however, the enhancement of
proteasome-dependent degradation plays a cardioprotective role
against proteinopathy and IR injury in mouse models (63). However, the effects of proteasome
inhibitors remain controversial, which could be both beneficial and
detrimental on cardiac function during myocardial ischemia and
reperfusion (64).
Autophagy is induced by the deprivation of oxygen
and nutrition, and refers to the lysosomal degradation of
unnecessary cellular components, such as protein aggregates, waste
lipids and dysfunctional organelles (4). During selective mitochondrial
autophagy (mitophagy), the dysfunctional mitochondria are tagged
with polyubiquitin, embedded into autophagosomes and delivered to
the lysosome for degradation (23,65).
PTEN-induced putative kinase 1 (PINK1)
and ubiquitin ligase Parkin-mediated mitophagy
Kinase PINK1 is a Ser/Thr protein kinase that
phosphorylates ubiquitin, Parkin and the mitochondrial outer
membrane protein Mfn2. The phosphorylation events activate Parkin
to translocate from the cytosol to the mitochondria (4,66).
Parkin is an E3 ubiquitin ligase that polyubiquitinates the
phosphorylated Mfn2 (67),
in-turn promoting the binding of mitophagy proteins (sequestosome
protein p62/SQSTM1 and autophagy-related protein 8 mammalian
homologue LC3) and the cargo sequestration within the autophagosome
for the subsequent degradation in lysosomes (4,66).
The loss of PINK1 has been shown to increase the
myocar-dial infarct size and generate excess ROS during stimulated
IR injury, whereas the overexpression of PINK1 reduces cardiac cell
death following stimulated IR injury (68). On the one hand, Parkin-deficiency
in the hearts of mice reduces mitophagy, resulting in the
accumulation of dysfunctional mitochondria and enlarged infarcts
following MI (69). However,
excessive Parkin-mediated mitophagy also initiates the induction of
apoptotic pathways in cardiac microcirculation endothelial cells
and exacerbates microvascular injury, as well as cardiac
reperfusion injury (70).
Receptor-mediated mitophagy
The mitophagy receptors, such as BNIP3, Nip3-like
protein X (NIX) (also known as Bcl2-interacting protein 3 like) and
FUN14 domain containing 1 (FUNDC1), localize to the mitochondrial
outer membrane and serve a role in acute IR injury by regulating
mitophagy (4,23,62,65,66). For example, BNIP3 binds to
stabilized PINK1, which facilitates the recruitment of Parkin on
the mitochondrial outer membrane (71). NIX is ubiquitinated by Parkin,
enhancing the recruitment of the autophagy receptor NBR1 and
LC3-coated vesicles to the mitochondria (66). Under hypoxic conditions, the
heart-enriched FUNDC1 interacts with LC3 to recruit autophagosomes
to mitochondria and enhances mitophagy (72). The phosphorylation at
Ser13 of FUNDC1 by casein kinase 2 (CK2) inhibits the
interaction between FUNDC1 and Drp1, ultimately decreasing
mitophagy (66). CK2α has been
reported to be upregulated during IR stress. As a consequence,
FUNDC1 is deactivated and suppresses FUNDC1-mediated mitophagy
(4). Ischemic preconditioning has
been reported to induce extensive FUNCD1- or Parkin-mediated
mitophagy in platelets and reduces secondary ischemic injury
(22). In preclinical studies,
myocardial conditioning interventions, including ischemic pre-,
post-conditioning or remote pre-/per-conditioning, are widely used
strategies to reduce infarct sizes following sustained IR. Several
signaling pathways activated in response to conditioning stimuli
end with the preservation of mitochondrial function (17).
Bcl2-associated athanogene (BAG) family
molecular chaperone regulator 3 (BAG3)-mediated non-canonical
mitophagy
BAG3 is highly expressed in cardiac muscles and
functions as a chaperone to target misfolded proteins for
lysosomal-mediated degradation (73). BAG3-Pro209Leu mutation causes the
dysregulation of autophagy, which is associated with the rapid
progression of restrictive cardiomyopathy in myofibrillary
myopathies (74).
Mitochondrial heterogeneity and
subpopulations
In neonatal cardiomyocytes, the mitochondria are
distributed throughout the cytosol; however, in the adult cardiac
cells, the mitochondria are small, round, hypodynamic and
relatively static. In particular, they are heterogeneous and are
clustered into subsarcolemmal mitochondria (SSM), inter-myofibril
mitochondria (IFM) and perinuclear mitochondria subsets with
distinct morphologies, localizations and functions (8,17,75). These three mitochondrial
subpopulations also possess different sensitivities to pathological
processes, such as IR injury. For example, SSM exhibit greater
oxidative damage and increased reduction in complex I-mediated
respiration than IFM, whereas IFM consume less oxygen than SSM in
complex II following IR (17,76). Therefore, the mitochondrial
defects in ischemic cells are in a heterogeneously distributed
manner amongst distinct subpopulations (76,77). A significant heterogeneity of
redox states, Ca2+ and ∆Ψm has also been shown in
myocardial cells following IR injury (76).
5. Current developments in
mitochondria-targeted agents with cardioprotective effects against
IR injury
The defective mitochondria display several different
types of abnormalities and disturbances in metabolism and
morphology, which are often reciprocally coupled (41) and contribute to acute,
irreversible cell death and potentially, MI (77). The prevalence of ischemic heart
disease is statistically the highest in Europe and North America.
An estimated 8.4 million American adults (≥20 years old) suffered
an MI between 2013 and 2016. The mortality related to MI in 2017
was ~0.1 million in the USA. Additionally, ischemic heart diseases
have a notable financial impact. For example, the arithmetic mean
cost for each discharge due to acute coronary syndrome (ACS) was
$63,578 in 2009. Productivity losses connected to ACS were ~$8,000
(short-term) and $50,000 (long-term) per disability claim between
2007 and 2010 according to medical, pharmacy and disability
insurance data (78). Therefore,
drugs designed to directly act on the mitochondria for IR injury
therapy would have a substantial impact on both medical and
societal costs.
With a growing understanding of the molecular
mechanisms of IR injury, several mitochondrion-targeted agents are
under research and development, which are planned to be used for
cardiac IR injury treatment (Table
I). These agents have been designed to improve mitochondrial
function via different underlying strategies, for example: i)
Decreasing the accumulation of succinate during ischemia; ii)
reducing succinate oxidation following reperfusion; iii) inhibiting
RET at complex I; iv) counteracting the superoxide production; v)
reducing ROS damage; vi) increasing NAD+ levels; vii)
blocking the mPTP; viii) maintaining Ca2+ homeostasis;
and ix) regulating mitochondrial dynamics and quality control.
 | Table IResearch and development status of
the current mitochondria-targeted agents with cardioprotective
effects against ischemia-reperfusion injury. |
Table I
Research and development status of
the current mitochondria-targeted agents with cardioprotective
effects against ischemia-reperfusion injury.
| Development
stage |
Mitochondria-targeted agent |
|---|
| Discovery: Natural
source, high- throughput screening and drug design | MitoGSH (96) |
| Pre-clinical:
Tested in cells, isolated tissues and animals to determine
efficacy, toxicity and pharmacokinetic properties | Dimethyl malonate
(80), chloramphenicol succinate
(83), MitoSNO (84), amobarbital (85), S1QELs (86), SkQ (88), Euk-8 (97), CMX-2043 (α-lipoic acid) (98), NIM811 (105), sanglifehrin A (106), 5-aminoimidazole-4-carboxamide
ribonucleotide (115,116), resveratrol (123), mitochondrial division
inhibitor-1 (125), P110
(51), rapamycin (126), miR-499 (127) |
| Clinical trials:
Tested in humans | |
| Phase I | Bendavia
(NCT01572909), nicotinamide mononucleotide (UMIN000021309) |
| Phase II | MitoQ
(NCT00433108), rosiglitazone (NCT00064727) |
| Phase III | Coenzynme
Q10 (ISRCTN94506234), cyclosporine A (NCT01650662),
bezafibrate (https://www.acc.org/Latest-in-Cardiology/Clinical-Trials/2013/04/13/21/02/BIP),
pioglitazone (NCT00091949), metformin (NCT01217307) |
| Marketing,
approved | None |
Inhibitors of respiratory complex II
(SDH)
Malonate is a competitive inhibitor of SDH, reducing
succinate accumulation and oxidation during myocardial IR injury.
Dimethyl malonate is a cell membrane-permeable form of malonate and
its protective effect against IR injury have been investigated in
purified mitochondria, mouse hearts, and in murine and rabbit
models of stroke (14,79,80).
Chloramphenicol succinate (CAPS) is a prodrug of
chloramphenicol (CAP), which is already approved for clinical use
as a human antibiotic, but with a risk of hemotoxicity, including
reversible and irreversible anemia. CAP-induced reversible anemia
with reticulocytopenia, often in conjunction with leucopenia and
thrombocytopenia, develops with the increasing dose of CAP during
treatment. Daily dose levels of CAPS between 500-3,500 mg/kg have
been observed to induce significant reversible myelotoxicity in
different mouse strains. CAP-induced irreversible anemia is a
non-dose-related aplastic anemia with a severe pancytopenia, which
is fatal and unpredictable with an incidence of 20-40 cases per
million following therapies (81). CAPS can be hydrolyzed by esterase
or oxidized by SDH into succinate and CAP, indicating that CAPS may
be a competitive substrate of SDH. The active metabolite, CAP, in
turn inhibits SDH reductive function, underlying the potential
myelotoxicity induced by CAPS (82). The cardioprotective effect of CAPS
has been shown in a pig model of MI, where reduced infarct sizes
were observed following pre-ischemic in vivo administration
of CAPS, as well as in the delayed treatment, where CAPS was
administered prior to reperfusion. CAPS may also induce autophagy,
indicated by the upregulated specific autophagy markers Beclin-1
and microtubule-associated protein light chain (LC3-II). However,
the specific molecular mechanisms underlying its action remain
unclear (83).
Inhibitors of respiratory complex I
MitoSNO is a mitochondria-targeted S-nitrosating
agent, temporarily inhibiting complex I by reversible S-nitrosation
of ND3 Cys39 residue during ischemia. The capability of
MitoSNO as a potential drug has been assessed through incubation of
mitochondria from rat and mouse hearts, ex vivo heart
treatment and in vivo administration in mice models of MI
(84).
Amobarbital is also a rapidly reversible inhibitor
of complex I, which can improve mitochondrial function and protect
cell survival during cardiac IR injury possibly via reducing the
release of apoptosis inducing factor from the mitochondrial
intermembrane space into cytosol. The protective effects of
amobarbital against cardiac IR damage has been assessed in buffer
perfused rats and Harlequin (Hq) mouse hearts (85).
Following high-throughput chemical screening and
validation, performed by Brand et al (86), two families of the cell-permeant
compounds synthesized from commercial companies were identified as
effective and selective suppressors of ubiquinone (Q)-binding site
of respiratory complex I (site IQ). Suppressors of site
IQ electron leak (S1QELs), S1QELs 1.1-1.6 and S1QELs
2.2-2.4, can inhibit ROS production at complex I during reverse
electron transport without altering basal respiration or the
resting OXPHOS. S1QEL1.1 is an outstanding candidate, which
strongly suppresses tunicamycin-triggered endoplasmic reticulum
stress and caspase signaling pathways that leads to apoptosis in
cardiomyocyte H9c2 cells and protects Langendorff-perfused mouse
heart models of IR injury from endogenous oxidative damage,
including enhancement of cardiac function in post-ischemic recovery
and the decrease in infarct size. This highlights S1QEL1.1 as a
novel therapeutic agent which can be used at the time of
reperfusion. The suppression of ROS generation and oxidative stress
signaling by S1QELs have also been assessed in isolated rat
skeletal muscle mitochondria, 293 cells, primary astrocytes and
live Drosophila melanogaster (86).
Mitochondria-permeable or -targeted
antioxidants
Coenzyme Q10 (CoQ10) is an
endogenous lipid, with an oxidized-form (ubiquinone) and fully
reduced-form (ubiquinol), which is the most common form of CoQ in
humans. In addition to its primary role in electron- and
proton-transport in mitochondria for production of ATP (87), CoQ10 is also a
mitochondria-permeable antioxidant (88). CoQ10 deficiency is
associated with several diseases, such as cardiovascular diseases,
muscular dystrophy and neurodegenerative disorders (87). Administration of CoQ10
as a supplement along with standard clinical therapy shows positive
effects in patients with hypertension (87) and heart failure (89). Higher plasma CoQ10
concentrations in patients with MI lead to improved left
ventricular function. An intravenous CoQ10 injection in
rat models of coronary artery blockage has been shown to reduce
cardiomyocyte necrosis and limit post-infarction hypertrophy of the
left ventricle (87). A
significant improvement in patients with chronic heart failure was
shown in the Q-SYMBIO study (a randomized double-blind trial of 420
international patients) (90).
MitoQ is a bioactive ubiquinone covalent compound
linked with a lipophilic cation triphenylphosphonium (TPP), which
is reduced into the antioxidant ubiquinol in the mitochondria, and
protects against oxidative injury. MitoQ is a promising
mitochondria-targeted antioxidant, which has shown positive results
in an intravenous and oral toxicity study in mice, in long-term
oral administration studies in mice and rats, and in human Phase I
and II trials (no significant difference in the NCT00329056 study
and a potential benefit in the NCT00433108 study) (91,92).
10-(6′-Plastoquinonyl) decyltriphenyl-phosphonium
(SkQ) is a lipophilic cation attached to a plastoquinone, which is
a mitochondria-targeted antioxidant that protects cells from
age-related diseases and ROS-burst-mediated acute diseases, such as
IR. SkQ has not yet been approved by the US Food and Drug
Administration as the safety and the clinical application of SkQ
requires further study (88).
Bendavia (also known as SS31 and MTP-131) is a
Szeto-Schiller (SS) peptide that can penetrate the mitochondria and
ameliorate IR injury by interacting with the phospholipid
cardiolipin and protecting mitochondrial cristae membranes
(93). Positive results have been
indicated in animal studies (93,94); however, Bendavia was unsuccessful
in the EMBRACE STEMI study (Phase 2a trial). Therefore, MTP-131 is
considered a potential pharmacological compound that protects
mitochondria, but has not yet successfully translated into clinical
use for the patients with ST-elevation MI (95).
MitoGSH is a group of synthetic forms of reduced
glutathione (GSH) tagged with SS peptides or TPP ions that can
target the mitochondria. MitoGSH is a class of novel drugs that may
be highly valuable compounds for treatment of heart diseases by
effectively restoring mitochondrial GSH levels. However, the
limitations of TPP and SS peptides should be taken into
consideration (96).
Euk-8 is a SOD/catalase mimetic antioxidant that is
capable of potently scavenging oxyradicals. The positive
cardiac-protective effects of Euk-8 include the reduction of both
oxidative stress and pressure overload-triggered heart failure in
the X-linked Hq mutant and WT mice (97).
α-lipoic acid is an important
mitochondria-permeable antioxidant (88). α-lipoic acid is a natural dithiol
that exerts protective effects against IR injury in several organs
(98) and is used as a treatment
of diabetic neuropathy and eye-disorders clinically, but has
several side effects (88).
CMX-2043 [N-((R)-1,2-dithiolane-3-pentanoyl)-L-glutamyl-L-alanine]
is a novel analogue of α-lipoic acid, which has been shown to
achieve efficacious reduction of infarct damage in a rat model of
myocardial IR injury (98).
Precursors of NAD+
Nicotinamide riboside (NR) and nicotinamide
mononucleotide (NMN) are the natural precursors of NAD+.
The supplementation of NR or NMN can increase cellular
NAD+ levels, without any noticeable side-effects
(99,100). Bioactive NMN is synthesized from
nicotinamide, a phosphate group and a ribonucleoside by
nicotinamide phosphoribosyltransferase (100). Both NR and NMN have been shown
to exert beneficial effects on cardio-vascular functions under IR
conditions. The administration of NMN has been shown to exhibit
timing-dependent patterns on the reduction of infarct size (before
ischemia or before reperfusion) (100). Several clinical studies
regarding the safety and effects of NR on human physiology are
currently underway in Europe and the USA (NCT03432871, NCT03423342
and NCT02812238). NMN is available on the market as a dietary
supplement. A Phase I human clinical study for NMN has commenced in
Japan and has evaluated the safety and the bioavailability of NMN
in humans, with the aim of developing NMN into an anti-aging
nutraceutical (99,101).
Inhibitors of mPTP
Cyclosporine A (CsA) is widely used as an
immunosuppressive drug following organ transplantation and
autoimmune disorders clinically, due to its ability to suppress the
transcription of cytokines in activated T cells via blocking the
CaN/nuclear factor of activated T cells pathway (95,102). However, CsA is not only an
inhibitor of cytosolic CaN, but also an inhibitor of mPTP, binding
to mitochondrial cis-transprolyl isomerase
cyclophilin D (CyD) (95). CyD is
a positive regulator of mPTP with a chaperone localized in the
mitochondrial matrix (3,8). The cardioprotective effects of CsA
have been assessed in several species of animals using reperfused
MI models, with inconsistent results in reduction of infarct size
(95). Patients with ST-elevation
MI (STEMI) administered CsA have exhibited promising results in a
Phase II trial; however, CsA was unsuccessful in the Phase III
CIRCUS and CYCLE trials (95,103,104).
NIM811 is a CsA derivative without
immunosuppressive capabilities, which can also specifically inhibit
mPTP opening in the mitochondria from the reperfused myocardium
(95). Adult male New Zealand
white rabbits injected with NIM811 at the time of reperfusion have
been shown to exhibit preserved OXPHOS in the heart mitochondria,
reduced infarct sizes and reduced myocardial damage (105).
Sanglifehrin A (SfA) is a specific inhibitor of
cyclophilin D, similar to CsA, although it cannot suppress CaN
activity. The specific inhibition of CyD by SfA protects against
cardiac IR injury by limiting mPTP opening (3,106). The beneficial effects of SfA
have been examined in MI female rat hearts. Significantly improved
cardiac function was only shown in reperfused hearts treated with
one intravenous bolus of SfA in the acute phase (2 days) of post-MI
remodeling (106).
PPAR agonists for the activation of
mitochondrial biogenesis
Fibrates have been discovered to activate PPARs,
particularly PPARα, which regulate glucose and lipid metabolism in
the heart. Fibrates have been widely used as a class of
hypolipidemic drugs for treatment of heart attacks and strokes
(107,108). Bezafibrate (marketed as Bezalip)
is a fibrate drug widely used for the treatment of metabolic
syndrome and hyperlipidemias. Bezafibrate is an agonist of PPARα
and partially activates PPARγ and PPARδ, upregulating mitochondrial
biogenesis by increasing PGC-1α expression (109). A small clinical trial including
patients with acute STEMI and hyperfibrinogenemia showed
bezafibrate treatment resulted in lower fibrinogen levels and lower
frequency of major cardiovascular events compared with conventional
therapy (110). The clinical
trial Bezafibrate Infarction Prevention (BIP) study with 16 years
of follow-up has shown that bezafibrate treatment reduces the risk
of cardiac death and the non-fatal MI as well as mortality in
patients with coronary arterial diseases (107).
Thiazolidinediones (TZDs) are structurally and
pharmaco-logically similar to fibrates and more specifically act on
PPARγ. TZD (also known as glitazone) is a class of hypoglycemic
drugs used for treatment of type 2 diabetes clinically and protects
against cardiovascular diseases. Pioglitazone and rosiglitazone are
TZDs, which are also PPARγ agonists and are used as anti-diabetic
drugs (108). With the risk of
bias and uncertain adverse events, pioglitazone was shown to also
reduce non-fatal MI and fatal or non-fatal stroke in four clinical
studies (IRIS, NCT00091949; J-SPIRIT, UMIN000013499; Kernan et
al (111); and PROactive,
NCT00174993) (112).
Rosiglitazone has exhibited opposite results in pre-clinical and
clinical trials. Rosiglitazone administration to pig and rat with
IR-affected hearts has been shown to significantly decrease the
infarct size, but without improvement of cardiac mitochondrial
function (113). From the data
pooled from 42 clinical studies, a significant increase in MI and
severe adverse cardiovascular causes were observed with the use of
rosiglitazone for patients with type 2 diabetes (114). Only one phase II clinical trial
(NCT00064727) has been completed to investigate the therapeutic
efficiency of rosiglitazone in patients with congestive heart
failure (107).
Activators of mitochondrial
biogenesis
5-Aminoimidazole-4-carboxamide ribonucleotide
(AICAR) is an AMPK agonist, activating PGC-1α by mimicking AMP
(109). Serum-starved rat
cardiomyocytes stimulated with AICAR have exhibited the full
activation of AMPK downstream targets, such as Sirt1 and PGC-1α,
which may contribute to mitochondrial homeostasis and
cardioprotective effects against ischemic stress (115). Dogs treated with AICAR during
cardiopulmonary bypass surgery followed by reperfusion have
exhibited an improved recovery of myocardial glucose uptake and
utilization (116).
Metformin is also an AMPK activator which exerts
protective effects against cardiac ischemia, MI and heart failure.
Additionally, metformin is a hypoglycemic drug widely used for the
treatment of diabetes. Metformin has been proposed to reduce
myocardial IR injury via increasing PGC-1α levels and mitochondrial
biogenesis (117). Even though
both preclinical and clinical studies (such as the DIGAMI 2 trial)
have suggested the cardioprotective effects of metformin against IR
injury (118-121), 4 months of metformin treatment
in the GIPS-III trial did not result in improved left ventricular
function in patients without diabetes presenting with STEMI. The
2-year follow-up results of the GIPS-III trail also revealed no
beneficial long-term effects (122).
Resveratrol is a natural polyphenol (stilbenoid)
found in red wine, that functions as a Sirt1 and PGC-1α activator,
and an inhibitor of mitochondrial ATPase (109). The protective effects of
resveratrol have been shown in animal models of cardiovascular,
neurodegenerative and metabolic disorders. Five clinical trials of
resveratrol for treatment of pre-diabetes and pulmonary diseases
are currently in progress (NCT02502253; NCT02565979; NCT03762096;
NCT02245932; NCT03819517). However, resveratrol has limitations in
human therapeutic application due to its low potency and poor
pharmacokinetics. Resveratrol and several other polyphenols are
often used as dietary antioxidants (123).
Inhibitors of mitochondrial fission
Mitochondrial division inhibitor-1 (Mdivi-1) is a
derivative of quinazolinone, which can inhibit mitochondrial
fission by decreasing Drp1 activity, thereby reducing short term
ischemic stress and myocardial IR injury. Mdivi-1 has been shown to
exert cardioprotective effects in pre-treated cardiomyocytes (HL-1
cells), primary isolated neonatal mice ventricular myocytes, hearts
from Langendorff perfusion in a rat model of IR, a murine coronary
artery ligation model and a pressure overload mouse model with
induced heart failure (40,124,125).
P110 is a small peptide (7-amino acids) which
blocks the contact between Drp1 and mitochondrial fission 1
protein, ameliorating MI and long-term heart remodeling. P110
peptide has been demonstrated to inhibit excessive mitochondrial
fission in primary neonatal rat cardiomyocytes, decreasing the
infarct size and improving cardiac function in an ex vivo
Langendorff rat heart model and an in vivo rat model of MI
(51).
Regulator of mitophagy
Rapamycin has been found to promote mitophagy by
inhibiting the mammalian target of rapamycin (mTOR) pathway and
reducing infarct size in MI and cardiomyocyte apoptosis. As an
allosteric inhibitor of mTOR complex I (mTORC1), rapamycin plays a
role in the suppression of cell growth and division, and has been
used as an immunosuppressant drug, or for the treatment of cancer
and in-stent restenosis in clinical practice. Rapamycin has also
been shown to reduce cardiomyocyte hypertrophy in a mouse model of
chronic ischemic injury and to inhibit cardiomyocyte apoptosis
in vitro (angiotensin II-induced myocyte apoptosis model)
and in vivo (MI-induced chronic heart failure rat model)
(126).
MicroRNA (miRNA/miR) regulation
miR-499 is an intronic miRNA identified to be
involved in inhibiting apoptosis and MI induced by ischemia.
miR-499 suppresses the expression of CaN and reduces cell apoptosis
following cardiac ischemia, protecting against the MI. miR-499
transgenic mice were previously created, and when they were
intravenously injected with the antagomir of miR-499 following IR
induction via surgery, they were used for the in vivo
evaluation of cardiac function, and hearts harvested from these
mice were used for histological and western blot analysis (127).
6. Future perspectives
Limiting early mtROS production in IR
injury
Superoxide production driven by succinate oxidation
upon reperfusion through RET at complex I in the mitochondrial
respiratory chain is considered the major source of ROS in IR
injury (5). In vivo
experiments have indicated that the succinate accumulated during
ischemia undergoes rapid oxidation and returns to normoxic levels
within 5 min following reperfusion (14). Therefore, it could be an effective
therapeutic strategy for the treatment of IR injury by inhibiting
RET at complex I for at least 5 min, until the succinate is no
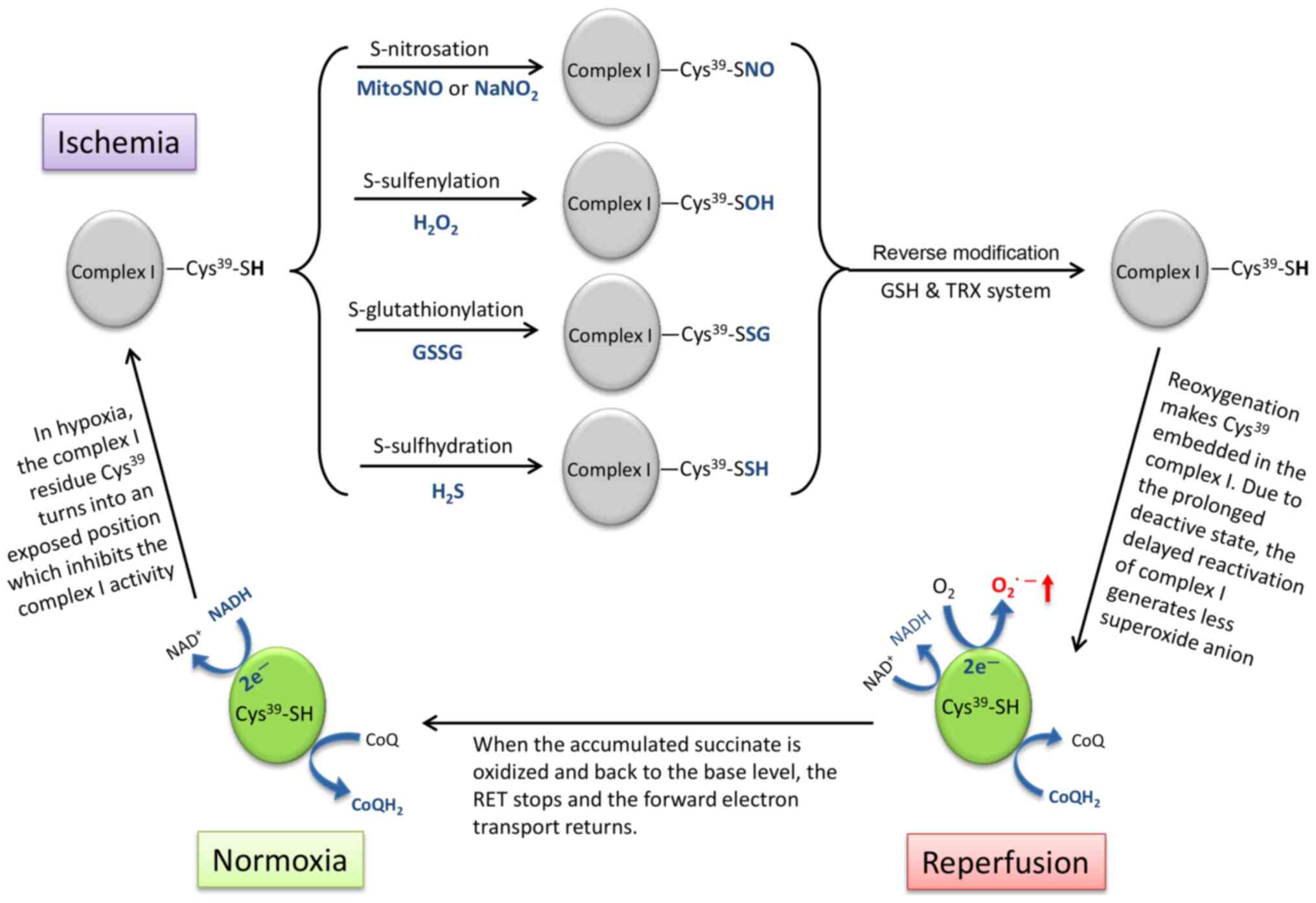
longer functional as an electron sink (Fig. 2).
The electron transport chain complex I undergoes a
structural change, which is termed 'active/deactive transition'
during IR (128).
Cys39 in complex I is the critical switch of this
conformational transition (Fig.
2), which is exposed in the deactive state of complex I under
ischemic conditions and embeds in the reactivated complex I upon
reperfusion (5,129). Several thiol-reactive agents can
be used to lock complex I in the deactive state by thiol
modification on Cys39, including S-nitrosation (-SH into
-SNO) by S-nitrosothiols (MitoSNO) or sodium nitrite
(NaNO2) (84,130), S-sulfenylation (-SH into -SOH)
by H2O2 (131), S-glutathionylation (-SH into
-SSG) by glutathione disulfide (GSSG) (132) and S-sulfhydration/persulfidation
(-SH into -SSH) by hydrogen sulfide (H2S) (133). These post-translational
modifications can later be reversed by the internal mitochondrial
GSH and thioredoxin systems (84).
However, the appropriate onset and duration of
administration of thiol-reactive agents remains uncertain,
otherwise this intervention based on the structural interconversion
of complex I may be an ideal therapeutic method for preventing the
burst of ROS production via RET and to return complex I activity in
time.
Mitochondrial quality control
A poor quality of mitochondrial network generates
and accumulates excess ROS (35).
The burst of mtROS results in a series of downstream oxidative
damages in cardiomyocytes after acute MI or during cardiac
senescence (134,135), which may lead to sudden cardiac
death or heart failure (136).
Mitophagy plays an important role in maintaining
mitochondrial quality control, particularly in adult
cardio-myocytes (43,137). However, cardiac mitophagy plays
both beneficial and detrimental roles in response to IR stress
(22,66,138). On the one hand, the clearance of
the defective mitochondria via mitophagy may reduce the risk of ROS
release and cell apoptosis, which exerts protective effects by
decreasing myocardial oxidative stress and infarct size following
MI (23,137,138). During IR, additional
mitochondrial mass causes excess oxygen consumption and ATP
depletion, which exacerbates the energetic stress. When energy is
insufficient, active mitophagy can enhance cell survival (19,23). However, excessive mitophagy causes
a drastic loss of mitochondrial mass and ATP supply in
cardio-myocytes, leading to cell death and heart failure (54,66). When mitophagy is insufficient, the
accumulation of damaged mitochondria and mtROS weakens the
adaptiveness of cardiomyocytes towards cellular stress and
apoptosis (4,66). Therefore, intervention with
properly modulated mitophagy can serve as an ideal, yet difficult
to achieve, therapeutic method for cardiac IR stress and other
diseases (19,66,138). Both mitophagy and mitochondrial
biogenesis are upregulated following heart surgery (139). Thus, it is essential to maintain
a balance between mitochondrial degradation and biogenesis to
maintain mitochondrial homeostasis and myocardial protection under
stress (138,140).
Attention must be paid to mitophagy, which is a
complex process and intersects with several other cellular events.
Some mediators in mitophagy signaling pathways, such as mTOR and
AMPK, also play roles in mitochondrial biogenesis (19,66), intrinsic fatty acid metabolism
(141), glucose-modulated amino
acid sensing and utilization (142), endothelium-dependent
vasodilation in vessels (143),
vascular smooth muscle cell proliferation and migration (144), suggesting that the manipulation
of these may cause a series of unexpected side-effects.
Gap between pre-clinical studies and
clinical practice of putative infarct-sparing drugs
As discussed above in Chapter 5 entitled 'Current
developments in mitochondria-targeted agents with cardioprotective
effects against IR injury', even though several agents have shown
promising cardioprotective effects in pre-clinical studies, notable
clinical trials in the past have highlighted the limitations or
translational failures. The administration of certain autacoids,
such as adenosine, bradykinin and opioids, can mimic the
cardioprotective state of ischemic pre-conditioning, triggering
several signaling pathways and finally inhibiting the formation of
mPTP. As adjunctive therapies, a common limitation of these
pre-conditional mimetics is the prerequisite administration before
lethal ischemia, which makes them suitable for the planned cardiac
surgeries but not effective in the clinical practice of acute MI.
Two large-scale clinical trials, AMISTAD I and II, which evaluated
the effects of adenosine during acute STEMI therapy, exhibited
restrictive infarct size-reduction and a number of adverse events,
such as hypotension, bradycardia, heart block and ventricular
tachycardia, especially in the adenosine patients with nonanterior
MI (145).
Bradykinin, as the vasodilatory peptide, plays a
role in decreasing blood pressure and increasing vascular
permeability (146). Bradykinin
signaling deficiency may be responsible for the vascular
contraction and the high epithelial Na+ reabsorption
observed in patients with hypertension and cardiovascular diseases.
Bradykinin signaling has also been demonstrated to reduce
inflammation and endothelial apoptosis in animal models of MI and
cardiac IR injury (147).
However, an increasing bradykinin concentration either inherited or
acquired, which is a main mediator of hyper-permeability of
capillaries and accumulation of local edema fluid, results in a
rare disease termed bradykinin-mediated angioedema (146,148).
Opioids, such as morphine have been widely and
frequently used as analgesics for the patients with STEMI (149). First of all, STEMI patients
often have intense chest pain and anxiety in the acute phase of the
treatment. As a traditional analgesia, opioids are effective in
symptomatic relief. In addition, the reduction of pain and stress
also results in vasodilation, an increase in blood flow, a decrease
in arterial blood pressure and heart rate, equilibrating the supply
and demand of oxygen and nutrients in cardiomyocytes (149,150). However, increased severe
side-effects of opioids in STEMI patients have been revealed in
recent clinical studies. For example, the low myocardial
contractility, hypotension and fainting due to morphine-induced
peripheral vasodilation are the main hemodynamic adverse effects
for the patients with ventricular failure. A long-term use of
opioids raises the risk for respiratory and gastrointestinal
adverse effects, such as suppression of breathing, gut motility and
absorption (150). Therefore,
opioid administration may disrupt the absorption of certain
essential oral medicaments, such as platelet inhibitors, and
consequently delay the action of antiplatelet agents and prolong
the platelet reactivity in the treatment of STEMI (149).
One of the major reasons for the limited clinical
success is incomplete or insufficient pre-clinical investigation
that does not offer accurate, reproducible and systematic
examination of results from multiple animal models before
large-scale clinical trials. The multicenter CAESAR (Consortium for
PreclinicAl AssESsment of CARdioprotective Therapies) initiative
supported by the National Heart Lung and Blood Institute was
created to improve translation of cardioprotective therapies into
clinical use (151). CAESAR
promotes integrated, standardized, solid and rigorous preclinical
evaluations to examine potential drugs or combined therapies in
centralized laboratories.
7. Conclusions
In conclusion, MI is one of the leading causes of
mortality and economic burden on health systems. Cardiac IR injury
following MI is due to the oxidative damage from mtROS, which leads
to severe mitochondrial damage and cardiomyocyte death. Therefore,
the mitochondria are strongly recommended to be considered as a
therapeutic target to prevent cardiac IR injury. Several promising
mitochondria-targeted agents are under research and development
with significant focus on protecting against cardiac IR injury by
counteracting the changes in mitochondrial metabolism and/or
morphology during IR. However, none of these have exhibited
sufficient efficacy and safety for clinical use. For future
studies, some theoretical and practical points are suggested to be
taken into consideration: Limiting ROS production by the reversible
and temporary interference on the complex I structure is an
interesting novel potential strategy. In addition, the side-effects
and limitations of drug use should be thoroughly examined. A
beneficial short-term outcome could lead to undesirable long-term
outcomes over time, such as mitophagy, which is a double-edged
sword for the mitochondrial quality and cell survival. Finally, an
innovative multicenter approach is suggested, such as that
advocated by CAESAR, in order to improve the translation of drugs
from the bench to the bedside.
Funding
The present study was funded by grants from the
National Natural Science Foundation of China (nos. 81702785 and
81802822) and supported by the postdoctoral programme of Qingdao
University.
Availability of data and materials
Not applicable.
Authors' contributions
WM conceived the idea for the review, drafted and
revised the manuscript. DM contributed to the revision, the
provision of some of the data and databases, and also contributed
to the proofreading and language correction of the manuscript. WM
and XA performed the literature search and prepared the figures. YL
critically discussed and revised the manuscript, and provided some
updates on the literature and clinical trials. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Carlucci A, Adornetto A, Scorziello A,
Viggiano D, Foca M, Cuomo O, Annunziato L, Gottesman M and
Feliciello A: Proteolysis of AKAP121 regulates mitochondrial
activity during cellular hypoxia and brain ischaemia. EMBO J.
27:1073–1084. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haidarali S, Patil CR, Ojha S, Mohanraj R,
Arya DS and Goyal SN: Targeting apoptotic pathways in myocardial
infarction: Attenuated by phytochemicals. Cardiovasc Hematol Agents
Med Chem. 12:72–85. 2014. View Article : Google Scholar
|
|
3
|
Kuznetsov AV, Javadov S, Margreiter R,
Grimm M, Hagenbuchner J and Ausserlechner MJ: The role of
mitochon-dria in the mechanisms of cardiac ischemia-reperfusion
injury. Antioxidants (Basel). 8:4542019. View Article : Google Scholar
|
|
4
|
Yang M, Linn BS, Zhang Y and Ren J:
Mitophagy and mitochondrial integrity in cardiac
ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis.
1865:2293–2302. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chouchani ET, Pell VR, James AM, Work LM,
Saeb-Parsy K, Frezza C, Krieg T and Murphy MP: A unifying mechanism
for mitochondrial superoxide production during
ischemia-reperfu-sion injury. Cell Metab. 23:254–263. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lesnefsky EJ, Chen Q, Tandler B and Hoppel
CL: Mitochondrial dysfunction and myocardial ischemia-reperfusion:
Implications for novel therapies. Annu Rev Pharmacol Toxicol.
57:535–565. 2017. View Article : Google Scholar
|
|
7
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang W, Fernandez-Sanz C and Sheu SS:
Regulation of mitochondrial bioenergetics by the non-canonical
roles of mitochondrial dynamics proteins in the heart. Biochim
Biophys Acta Mol Basis Dis. 1864:1991–2001. 2018. View Article : Google Scholar
|
|
9
|
Spinelli JB and Haigis MC: The
multifaceted contributions of mitochondria to cellular metabolism.
Nat Cell Biol. 20:745–754. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Niemann B, Schwarzer M and Rohrbach S:
Heart and mitochondria: Pathophysiology and implications for
cardiac surgeons. Thorac Cardiovasc Surg. 66:11–19. 2018.
View Article : Google Scholar
|
|
11
|
Bender DA: Oxidative phosphorylation.
Encyclopedia of food sciences and nutrition. Caballero B: 2nd
edition. Academic Press; Oxford; pp. 4295–4301. 2003, View Article : Google Scholar
|
|
12
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
13
|
Guzy RD, Sharma B, Bell E, Chandel NS and
Schumacker PT: Loss of the SdhB, but Not the SdhA, subunit of
complex II triggers reactive oxygen species-dependent
hypoxia-inducible factor activation and tumorigenesis. Mol Cell
Biol. 28:718–731. 2008. View Article : Google Scholar :
|
|
14
|
Chouchani ET, Pell VR, Gaude E,
Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord
ENJ, Smith AC, et al: Ischaemic accumulation of succinate controls
reperfusion injury through mitochondrial ROS. Nature. 515:431–435.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pell VR, Chouchani ET, Frezza C, Murphy MP
and Krieg T: Succinate metabolism: A new therapeutic target for
myocardial reperfusion injury. Cardiovasc Res. 111:134–141. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866:1657682020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boengler K, Lochnit G and Schulz R:
Mitochondria 'THE' target of myocardial conditioning. Am J Physiol
Heart Circ Physiol. 315:H1215–H1231. 2018. View Article : Google Scholar
|
|
18
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marzetti E, Csiszar A, Dutta D, Balagopal
G, Calvani R and Leeuwenburgh C: Role of mitochondrial dysfunction
and altered autophagy in cardiovascular aging and disease: From
mechanisms to therapeutics. Am J Physiol Heart Circ Physiol.
305:H459–H476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nordberg J and Arner ES: Reactive oxygen
species, antioxidants, and the mammalian thioredoxin system. Free
Radic Biol Med. 31:1287–1312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Judge S and Leeuwenburgh C: Cardiac
mitochondrial bioenergetics, oxidative stress, and aging. Am J
Physiol Cell Physiol. 292:C1983–C1992. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Chen C, Wang J, Liu L, He Y and
Chen Q: Mitophagy in cardiomyocytes and in platelets: A major
mechanism of cardioprotection against ischemia/reperfusion injury.
Physiology (Bethesda). 33:86–98. 2018.
|
|
23
|
Tahrir FG, Langford D, Amini S, Mohseni
Ahooyi T and Khalili K: Mitochondrial quality control in cardiac
cells: Mechanisms and role in cardiac cell injury and disease. J
Cell Physiol. 234:8122–8133. 2019. View Article : Google Scholar :
|
|
24
|
Paradies G, Petrosillo G, Paradies V and
Ruggiero FM: Role of cardiolipin peroxidation and Ca2+ in
mitochondrial dysfunction and disease. Cell Calcium. 45:643–650.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kay L, Nicolay K, Wieringa B, Saks V and
Wallimann T: Direct evidence for the control of mitochondrial
respiration by mitochondrial creatine kinase in oxidative muscle
cells in situ. J Biol Chem. 275:6937–6944. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dolder M, Wendt S and Wallimann T:
Mitochondrial creatine kinase in contact sites: Interaction with
porin and adenine nucleotide translocase, role in permeability
transition and sensitivity to oxidative damage. Biol Signals
Recept. 10:93–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arslan F, de Kleijn DP and Pasterkamp G:
Innate immune signaling in cardiac ischemia. Nat Rev Cardiol.
8:292–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
James AM, Hoogewijs K, Logan A, Hall AR,
Ding S, Fearnley IM and Murphy MP: Non-enzymatic N-acetylation of
lysine residues by AcetylCoA often occurs via a proximal
S-acetylated thiol intermediate sensitive to glyoxalase II. Cell
Rep. 18:2105–2112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wagner GR, Bhatt DP, O'Connell TM,
Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva
OR, Stevens RD, et al: A class of reactive Acyl-CoA species reveals
the non-enzymatic origins of protein acylation. Cell Metab.
25:823–837.e8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wagner GR and Hirschey MD: Nonenzymatic
protein acylation as a carbon stress regulated by sirtuin
deacylases. Mol Cell. 54:5–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ,
Lee WY, Oh KW, Park SW and Park CY: Nicotinamide improves glucose
metabolism and affects the hepatic NAD-sirtuin pathway in a rodent
model of obesity and type 2 diabetes. J Nutr Biochem. 25:66–72.
2014. View Article : Google Scholar
|
|
32
|
Carraro M and Bernardi P: Calcium and
reactive oxygen species in regulation of the mitochondrial
permeability transition and of programmed cell death in yeast. Cell
Calcium. 60:102–107. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giorgio V, Guo L, Bassot C, Petronilli V
and Bernardi P: Calcium and regulation of the mitochondrial
permeability transition. Cell Calcium. 70:56–63. 2018. View Article : Google Scholar
|
|
34
|
Nakagawa T, Shimizu S, Watanabe T,
Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T and Tsujimoto Y:
Cyclophilin D-dependent mitochondrial permeability transition
regulates some necrotic but not apoptotic cell death. Nature.
434:652–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martin JL, Gruszczyk AV, Beach TE, Murphy
MP and Saeb-Parsy K: Mitochondrial mechanisms and therapeutics in
ischaemia reperfusion injury. Pediatr Nephrol. 34:1167–1174. 2019.
View Article : Google Scholar :
|
|
36
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Din S, Mason M, Volkers M, Johnson B,
Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, et
al: Pim-1 preserves mitochondrial morphology by inhibiting
dynamin-related protein 1 translocation. Proc Natl Acad Sci USA.
110:5969–5974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Balaban RS: Cardiac energy metabolism
homeostasis: Role of cytosolic calcium. J Mol Cell Cardiol.
34:1259–1271. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nan J, Zhu W, Rahman MS, Liu M, Li D, Su
S, Zhang N, Hu X, Yu H, Gupta MP and Wang J: Molecular regulation
of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta
Mol Cell Res. 1864:1260–1273. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sharp WW, Fang YH, Han M, Zhang HJ, Hong
Z, Banathy A, Morrow E, Ryan JJ and Archer SL: Dynamin-related
protein 1 (Drp1)-mediated diastolic dysfunction in myocardial
ischemia-reperfusion injury: Therapeutic benefits of Drp1
inhibition to reduce mitochondrial fission. FASEB J. 28:316–326.
2014. View Article : Google Scholar :
|
|
41
|
Murphy MP and Hartley RC: Mitochondria as
a therapeutic target for common pathologies. Nat Rev Drug Discov.
17:865–886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thomas LW and Ashcroft M: Exploring the
molecular interface between hypoxia-inducible factor signalling and
mitochondria. Cell Mol Life Sci. 76:1759–1777. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Song M, Mihara K, Chen Y, Scorrano L and
Dorn GW II: Mitochondrial fission and fusion factors reciprocally
orchestrate mitophagic culling in mouse hearts and cultured
fibroblasts. Cell Metab. 21:273–286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen Y, Liu Y and Dorn GW II:
Mitochondrial fusion is essential for organelle function and
cardiac homeostasis. Circ Res. 109:1327–1331. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Song M, Gong G, Burelle Y, Gustafsson AB,
Kitsis RN, Matkovich SJ and Dorn GW II: Interdependence of
Parkin-Mediated mitophagy and mitochondrial fission in adult mouse
hearts. Circ Res. 117:346–351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sabbah HN: Targeting the mitochondria in
heart failure: A trans-lational perspective. JACC Basic Transl Sci.
5:88–106. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ikeda Y, Shirakabe A, Brady C, Zablocki D,
Ohishi M and Sadoshima J: Molecular mechanisms mediating
mitochondrial dynamics and mitophagy and their functional roles in
the cardio-vascular system. J Mol Cell Cardiol. 78:116–122. 2015.
View Article : Google Scholar
|
|
48
|
Große L, Wurm CA, Bruser C, Neumann D,
Jans DC and Jakobs S: Bax assembles into large ring-like structures
remodeling the mitochondrial outer membrane in apoptosis. EMBO J.
35:402–413. 2016. View Article : Google Scholar
|
|
49
|
Kim H, Scimia MC, Wilkinson D, Trelles RD,
Wood MR, Bowtell D, Dillin A, Mercola M and Ronai ZeA: Fine-tuning
of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial
adaptation to hypoxia. Mol Cell. 44:532–544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Marin W: A-kinase anchoring protein 1
(AKAP1) and its role in some cardiovascular diseases. J Mol Cell
Cardiol. 138:99–109. 2020. View Article : Google Scholar
|
|
51
|
Disatnik MH, Ferreira JC, Campos JC, Gomes
KS, Dourado PM, Qi X and Mochly-Rosen D: Acute inhibition of
excessive mitochondrial fission after myocardial infarction
prevents long-term cardiac dysfunction. J Am Heart Assoc.
2:e0004612013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ikeda Y, Shirakabe A, Maejima Y, Zhai P,
Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et
al: Endogenous Drp1 mediates mitochondrial autophagy and protects
the heart against energy stress. Circ Res. 116:264–278. 2015.
View Article : Google Scholar
|
|
53
|
Otera H, Wang C, Cleland MM, Setoguchi K,
Yokota S, Youle RJ and Mihara K: Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells. J Cell Biol. 191:1141–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar
|
|
55
|
Li J, Li Y, Jiao J, Wang J, Li Y, Qin D
and Li P: Mitofusin 1 is negatively regulated by microRNA 140 in
cardiomyocyte apoptosis. Mol Cell Biol. 34:1788–1799. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jiang X, Jiang H, Shen Z and Wang X:
Activation of mitochon-drial protease OMA1 by Bax and Bak promotes
cytochrome c release during apoptosis. Proc Natl Acad Sci USA.
111:14782–14787. 2014. View Article : Google Scholar
|
|
57
|
Chistiakov DA, Shkurat TP, Melnichenko AA,
Grechko AV and Orekhov AN: The role of mitochondrial dysfunction in
cardio-vascular disease: A brief review. Ann Med. 50:121–127. 2018.
View Article : Google Scholar
|
|
58
|
Minoia M, Boncoraglio A, Vinet J, Morelli
FF, Brunsting JF, Poletti A, Krom S, Reits E, Kampinga HH and Carra
S: BAG3 induces the sequestration of proteasomal clients into
cytoplasmic puncta: Implications for a proteasome-to-autophagy
switch. Autophagy. 10:1603–1621. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hammerling BC and Gustafsson AB:
Mitochondrial quality control in the myocardium: Cooperation
between protein degradation and mitophagy. J Mol Cell Cardiol.
75:122–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kocaturk NM and Gozuacik D: Crosstalk
between mammalian autophagy and the Ubiquitin-Proteasome system.
Front Cell Dev Biol. 6:1282018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Escobar-Henriques M, Altin S and Brave FD:
Interplay between the ubiquitin proteasome system and mitochondria
for protein homeostasis. Curr Issues Mol Biol. 35:35–58. 2020.
View Article : Google Scholar
|
|
62
|
Nishida K and Otsu K: Sterile inflammation
and degradation systems in heart failure. Circ J. 81:622–628. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li J, Horak KM, Su H, Sanbe A, Robbins J
and Wang X: Enhancement of proteasomal function protects against
cardiac proteinopathy and ischemia/reperfusion injury in mice. J
Clin Invest. 121:3689–3700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu X and Kem DC: Proteasome inhibition
during myocardial infarction. Cardiovasc Res. 85:312–320. 2010.
View Article : Google Scholar
|
|
65
|
Zhou H and Toan S: Pathological roles of
mitochondrial oxidative stress and mitochondrial dynamics in
cardiac microvascular Ischemia/Reperfusion injury. Biomolecules.
10:852020. View Article : Google Scholar :
|
|
66
|
Morales PE, Arias-Duran C, Avalos-Guajardo
Y, Aedo G, Verdejo HE, Parra V and Lavandero S: Emerging role of
mitophagy in cardiovascular physiology and pathology. Mol Aspects
Med. 71:1008222020. View Article : Google Scholar
|
|
67
|
Chen Y and Dorn GW II:
PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling
damaged mitochondria. Science. 340:471–475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Siddall HK, Yellon DM, Ong SB, Mukherjee
UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson
SM, et al: Loss of PINK1 increases the heart's vulnerability to
ischemia-reperfusion injury. PLoS One. 8:e624002013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kubli DA, Zhang X, Lee Y, Hanna RA,
Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN and
Gustafsson AB: Parkin protein deficiency exacerbates cardiac injury
and reduces survival following myocardial infarction. J Biol Chem.
288:915–926. 2013. View Article : Google Scholar :
|
|
70
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar
|
|
71
|
Zhang T, Xue L, Li L, Tang C, Wan Z, Wang
R, Tan J, Tan Y, Han H, Tian R, et al: BNIP3 protein suppresses
PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol
Chem. 291:21616–21629. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wu W, Lin C, Wu K, Jiang L, Wang X, Li W,
Zhuang H, Zhang X, Chen H, Li S, et al: FUNDC1 regulates
mitochondrial dynamics at the ER-mitochondrial contact site under
hypoxic conditions. EMBO J. 35:1368–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tahrir FG, Knezevic T, Gupta MK, Gordon J,
Cheung JY, Feldman AM and Khalili K: Evidence for the role of BAG3
in mitochondrial quality control in cardiomyocytes. J Cell Physiol.
232:797–805. 2017. View Article : Google Scholar :
|
|
74
|
Schänzer A, Rupp S, Graf S, Zengeler D,
Jux C, Akinturk H, Gulatz L, Mazhari N, Acker T, Van Coster R, et
al: Dysregulated autophagy in restrictive cardiomyopathy due to
Pro209Leu mutation in BAG3. Mol Genet Metab. 123:388–399. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ong SB, Kalkhoran SB, Hernandez-Resendiz
S, Samangouei P, Ong SG and Hausenloy DJ: Mitochondrial-shaping
proteins in cardiac health and disease-the long and the short of
it! Cardiovasc Drugs Ther. 31:87–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kuznetsov AV and Margreiter R:
Heterogeneity of mitochondria and mitochondrial function within
cells as another level of mitochondrial complexity. Int J Mol Sci.
10:1911–1929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Geng Y, Hu Y, Wang H, Shi S, Shi J and Qiu
Z: Deficiency of interfibrillar mitochondria in post-acute
myocardial infarction heart failure. Pak J Pharm Sci. 30:1089–1094.
2017.PubMed/NCBI
|
|
78
|
Virani SS, Alonso A, Benjamin EJ,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Delling FN, et al: Heart disease and stroke
statistics-2020 update: A report from the American heart
association. Circulation. 141:e139–e596. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhu SC, Chen C, Wu YN, Ahmed M, Kitmitto
A, Greenstein AS, Kim SJ, Shao YF and Zhang YH: Cardiac complex II
activity is enhanced by fat and mediates greater mitochondrial
oxygen consumption following hypoxic re-oxygenation. Pflugers Arch.
472:367–374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kohlhauer M, Pell VR, Burger N, Spiroski
AM, Gruszczyk A, Mulvey JF, Mottahedin A, Costa ASH, Frezza C,
Ghaleh B, et al: Protection against cardiac ischemia-reperfusion
injury by hypothermia and by inhibition of succinate accumulation
and oxidation is additive. Basic Res Cardiol. 114:182019.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Turton JA, Fagg R, Sones WR, Williams TC
and Andrews CM: Characterization of the myelotoxicity of
chloramphenicol succinate in the B6C3F1 mouse. Int J Exp Pathol.
87:101–112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ambekar CS, Lee JS, Cheung BM, Chan LC,
Liang R and Kumana CR: Chloramphenicol succinate, a competitive
substrate and inhibitor of succinate dehydrogenase: Possible reason
for its toxicity. Toxicol In Vitro. 18:441–447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sala-Mercado JA, Wider J, Undyala VV,
Jahania S, Yoo W, Mentzer RM Jr, Gottlieb RA and Przyklenk K:
Profound cardio-protection with chloramphenicol succinate in the
swine model of myocardial ischemia-reperfusion injury. Circulation.
122(11 Suppl): S179–S184. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chouchani ET, Methner C, Nadtochiy SM,
Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley
KS, et al: Cardioprotection by S-nitrosation of a cysteine switch
on mitochondrial complex I. Nat Med. 19:753–759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Xu A, Szczepanek K, Hu Y, Lesnefsky EJ and
Chen Q: Cardioprotection by modulation of mitochondrial respiration
during ischemia-reperfusion: Role of apoptosis-inducing factor.
Biochem Biophys Res Commun. 435:627–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Brand MD, Goncalves RL, Orr AL, Vargas L,
Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT,
et al: Suppressors of superoxide-H2O2
production at site I(Q) of mitochondrial complex I protect against
stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab.
24:582–592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zozina VI, Covantev S, Goroshko OA,
Krasnykh LM and Kukes VG: Coenzyme Q10 in cardiovascular and
metabolic diseases: Current state of the problem. Curr Cardiol Rev.
14:164–174. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhang ZW, Xu XC, Liu T and Yuan S:
Mitochondrion-permeable antioxidants to treat ROS-Burst-mediated
acute diseases. Oxid Med Cell Longev. 2016:68595232016.
|
|
89
|
Di Lorenzo A, Iannuzzo G, Parlato A, Cuomo
G, Testa C, Coppola M, D'Ambrosio G, Oliviero DA, Sarullo S, Vitale
G, et al: Clinical evidence for Q10 coenzyme supple-mentation in
heart failure: From energetics to functional improvement. J Clin
Med. 9:12662020. View Article : Google Scholar
|
|
90
|
Mortensen AL, Rosenfeldt F and Filipiak
KJ: Effect of coen-zyme Q10 in Europeans with chronic heart
failure: A sub-group analysis of the Q-SYMBIO randomized
double-blind trial. Cardiol J. 26:147–156. 2019.
|
|
91
|
Reily C, Mitchell T, Chacko BK, Benavides
G, Murphy MP and Darley-Usmar V: Mitochondrially targeted compounds
and their impact on cellular bioenergetics. Redox Biol. 1:86–93.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Smith RA and Murphy MP: Animal and human
studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y
Acad Sci. 1201:96–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lee FY, Shao PL, Wallace CG, Chua S, Sung
PH, Ko SF, Chai HT, Chung SY, Chen KH, Lu HI, et al: Combined
therapy with SS31 and mitochondria mitigates myocardial
ischemia-reperfusion injury in rats. Int J Mol Sci. 19:27822018.
View Article : Google Scholar :
|
|
94
|
Kloner RA, Hale SL, Dai W, Gorman RC,
Shuto T, Koomalsingh KJ, Gorman JH III, Sloan RC, Frasier CR,
Watson CA, et al: Reduction of ischemia/reperfusion injury with
bendavia, a mitochondria-targeting cytoprotective peptide. J Am
Heart Assoc. 1:e0016442012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Botker HE, Cabrera-Fuentes HA, Ruiz-Meana
M, Heusch G and Ovize M: Translational issues for mitoprotective
agents as adjunct to reperfusion therapy in patients with
ST-segment elevation myocardial infarction. J Cell Mol Med.
24:2717–2729. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mailloux RJ: Application of
mitochondria-targeted pharmaceuticals for the treatment of heart
disease. Curr Pharm Des. 22:4763–4779. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
van Empel VP, Bertrand AT, van Oort RJ,
van der Nagel R, Engelen M, van Rijen HV, Doevendans PA, Crijns HJ,
Ackerman SL, Sluiter W and De Windt LJ: EUK-8, a superoxide
dismutase and catalase mimetic, reduces cardiac oxidative stress
and ameliorates pressure overload-induced heart failure in the
harlequin mouse mutant. J Am Coll Cardiol. 48:824–832. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Baguisi A, Casale RA, Kates SA, Lader AS,
Stewart K and Beeuwkes R III: CMX-2043 efficacy in a rat model of
cardiac ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther.
21:563–569. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tsubota K: The first human clinical study
for NMN has started in Japan. NPJ Aging Mech Dis. 2:160212016.
View Article : Google Scholar
|
|
100
|
Hong W, Mo F, Zhang Z, Huang M and Wei X:
Nicotinamide mononucleotide: A promising molecule for therapy of
diverse diseases by targeting NAD+ metabolism. Front Cell Dev Biol.
8:2462020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Irie J, Inagaki E, Fujita M, Nakaya H,
Mitsuishi M, Yamaguchi S, Yamashita K, Shigaki S, Ono T, Yukioka H,
et al: Effect of oral administration of nicotinamide mononucleotide
on clinical parameters and nicotinamide metabolite levels in
healthy Japanese men. Endocr J. 67:153–160. 2020. View Article : Google Scholar
|
|
102
|
Bendickova K, Tidu F and Fric J:
Calcineurin-NFAT signalling in myeloid leucocytes: New prospects
and pitfalls in immuno-suppressive therapy. EMBO Mol Med.
9:990–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Ottani F, Latini R, Staszewsky L, La
Vecchia L, Locuratolo N, Sicuro M, Masson S, Barlera S, Milani V,
Lombardi M, et al: Cyclosporine a in reperfused myocardial
infarction: The multicenter, controlled, open-label CYCLE trial. J
Am Coll Cardiol. 67:365–374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ikeda G, Matoba T, Nakano Y, Nagaoka K,
Ishikita A, Nakano K, Funamoto D, Sunagawa K and Egashira K:
Nanoparticle-mediated targeting of cyclosporine a enhances
cardioprotection against ischemia-reperfusion injury through
inhibition of mitochondrial permeability transition pore opening.
Sci Rep. 6:204672016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jahandiez V, Cour M, Bochaton T, Abrial M,
Loufouat J, Gharib A, Varennes A, Ovize M and Argaud L: Fast
therapeutic hypothermia prevents post-cardiac arrest syndrome
through cyclophilin D-mediated mitochondrial permeability
transition inhibition. Basic Res Cardiol. 112:352017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Parodi-Rullán RM, Soto-Prado J, Vega-Lugo
J, Chapa-Dubocq X, Díaz-Cordero SI and Javadov S: Divergent effects
of cyclophilin-D inhibition on the female rat heart: Acute versus
chronic post-myocardial infarction. Cell Physiol Biochem.
50:288–303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Khuchua Z, Glukhov AI, Strauss AW and
Javadov S: Elucidating the beneficial role of PPAR agonists in
cardiac diseases. Int J Mol Sci. 19:34642018. View Article : Google Scholar :
|
|
108
|
Lalloyer F and Staels B: Fibrates,
glitazones, and peroxisome proliferator-activated receptors.
Arterioscler Thromb Vasc Biol. 30:894–899. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Viscomi C, Bottani E, Civiletto G, Cerutti
R, Moggio M, Fagiolari G, Schon EA, Lamperti C and Zeviani M: In
vivo correction of COX deficiency by activation of the AMPK/PGC-1α
axis. Cell Metab. 14:80–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Madrid-Miller A, Moreno-Ruiz LA,
Borrayo-Sánchez G, Almeida-Gutiér rez E, Martínez-Gómez DF and
Jáuregui-Aguilar R: Ipact of bezafibrate treatment in patients with
hyperfibrinogenemia and ST-elevation acute myocardial infarction: A
randomized clinical trial. Cir Cir. 78:229–237. 2010.PubMed/NCBI
|
|
111
|
Kernan WN, Inzucchi SE, Viscoli CM, Brass
LM, Bravata DM, Shulman GI, McVeety JC and Horwitz RI: Pioglitazone
improves insulin sensitivity among nondiabetic patients with a
recent transient ischemic attack or ischemic stroke. Stroke.
34:1431–1436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu J and Wang LN: Peroxisome
proliferator-activated receptor gamma agonists for preventing
recurrent stroke and other vascular events in people with stroke or
transient ischaemic attack. Cochrane Database Syst Rev.
12:CD0106932017.PubMed/NCBI
|
|
113
|
Palee S, Weerateerangkul P, Surinkeaw S,
Chattipakorn S and Chattipakorn N: Effect of rosiglitazone on
cardiac electrophysi-ology, infarct size and mitochondrial function
in ischaemia and reperfusion of swine and rat heart. Exp Physiol.
96:778–789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nissen SE and Wolski K: Effect of
rosiglitazone on the risk of myocardial infarction and death from
cardiovascular causes. N Engl J Med. 356:2457–2471. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sunaga H, Koitabashi N, Iso T, Matsui H,
Obokata M, Kawakami R, Murakami M, Yokoyama T and Kurabayashi M:
Activation of cardiac AMPK-FGF21 feed-forward loop in acute
myocardial infarction: Role of adrenergic overdrive and lipolysis
byproducts. Sci Rep. 9:118412019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang DS, Liang GY, Liu DX, Yu J and Wang
F: Role of phosphorylated AMP-activated protein kinase (AMPK) in
myocardial insulin resistance after myocardial ischemia-reperfusion
during cardiopulmonary bypass surgery in dogs. Med Sci Monit.
25:4149–4158. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhou J, Massey S, Story D and Li L:
Metformin: An old drug with new applications. Int J Mol Sci.
19:28632018. View Article : Google Scholar :
|
|
118
|
Chin JT, Troke JJ, Kimura N, Itoh S, Wang
X, Palmer OP, Robbins RC and Fischbein MP: A novel cardioprotective
agent in cardiac transplantation: Metformin activation of
AMP-activated protein kinase decreases acute ischemia-reperfusion
injury and chronic rejection. Yale J Biol Med. 84:423–432.
2011.PubMed/NCBI
|
|
119
|
Palee S, Higgins L, Leech T, Chattipakorn
SC and Chattipakorn N: Acute metformin treatment provides
cardioprotection via improved mitochondrial function in cardiac
ischemia / reperfusion injury. Biomed Pharmacother. 130:1106042020.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Holman RR, Paul SK, Bethel MA, Matthews DR
and Neil HA: 10-Year follow-up of intensive glucose control in type
2 diabetes. N Engl J Med. 359:1577–1589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mellbin LG, Malmberg K, Norhammar A, Wedel
H and Rydé L; DIGAMI 2 Investigators: The impact of glucose
lowering treatment on long-term prognosis in patients with type 2
diabetes and myocardial infarction: A report from the DIGAMI 2
trial. Eur Heart J. 29:166–176. 2008. View Article : Google Scholar
|
|
122
|
Hartman MHT, Prins JKB, Schurer RAJ,
Lipsic E, Lexis CPH, van der Horst-Schrivers ANA, van Veldhuisen
DJ, van der Horst ICC and van der Harst P: Two-year follow-up of 4
months metformin treatment vs. placebo in ST-elevation myocardial
infarction: Data from the GIPS-III RCT. Clin Res Cardiol.
106:939–946. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Whitaker RM, Corum D, Beeson CC and
Schnellmann RG: Mitochondrial biogenesis as a pharmacological
target: A new approach to acute and chronic diseases. Annu Rev
Pharmacol Toxicol. 56:229–249. 2016. View Article : Google Scholar
|
|
124
|
Kim H, Lee JY, Park KJ, Kim WH and Roh GS:
A mitochondrial division inhibitor, Mdivi-1, inhibits mitochondrial
fragmentation and attenuates kainic acid-induced hippocampal cell
death. BMC Neurosci. 17:332016. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Veeranki S and Tyagi SC: Mdivi-1 induced
acute changes in the angiogenic profile after ischemia-reperfusion
injury in female mice. Physiol Rep. 5:e132982017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Gao G, Chen W, Yan M, Liu J, Luo H, Wang C
and Yang P: Rapamycin regulates the balance between cardiomyocyte
apoptosis and autophagy in chronic heart failure by inhibiting mTOR
signaling. Int J Mol Med. 45:195–209. 2020.
|
|
127
|
Wang JX, Jiao JQ, Li Q, Long B, Wang K,
Liu JP, Li YR and Li PF: miR-499 regulates mitochondrial dynamics
by targeting calcineurin and dynamin-related protein-1. Nat Med.
17:71–78. 2011. View Article : Google Scholar
|
|
128
|
Babot M, Birch A, Labarbuta P and Galkin
A: Characterisation of the active/de-active transition of
mitochondrial complex I. Biochim Biophys Acta. 1837:1083–1092.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Gorenkova N, Robinson E, Grieve DJ and
Galkin A: Conformational change of mitochondrial complex I
increases ROS sensitivity during ischemia. Antioxid Redox Signal.
19:1459–1468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Chouchani ET, James AM, Methner C, Pell
VR, Prime TA, Erickson BK, Forkink M, Lau GY, Bright TP, Menger KE,
et al: Identification and quantification of protein S-nitrosation
by nitrite in the mouse heart during ischemia. J Biol Chem.
292:14486–14495. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Pan J and Carroll KS: Light-mediated
sulfenic acid generation from photocaged cysteine sulfoxide. Org
Lett. 17:6014–6017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ukuwela AA, Bush AI, Wedd AG and Xiao Z:
Reduction potentials of protein disulfides and catalysis of
glutathionylation and deglutathionylation by glutaredoxin enzymes.
Biochem J. 474:3799–3815. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kolluru GK, Shen X and Kevil CG: Reactive
sulfur species: A new redox player in cardiovascular
pathophysiology. Arterioscler Thromb Vasc Biol. 40:874–884. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Davalli P, Mitic T, Caporali A, Lauriola A
and D'Arca D: ROS, cell senescence, and novel molecular mechanisms
in aging and age-related diseases. Oxid Med Cell Longev.
2016:35651272016. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shahzad S, Hasan A, Faizy AF, Mateen S,
Fatima N and Moin S: Elevated DNA damage, oxidative stress, and
impaired response defense system inflicted in patients with
myocardial infarction. Clin Appl Thromb Hemost. 24:780–789. 2018.
View Article : Google Scholar
|
|
136
|
Dey S, DeMazumder D, Sidor A, Foster DB
and O'Rourke B: Mitochondrial ROS drive sudden cardiac death and
chronic proteome remodeling in heart failure. Circ Res.
123:356–371. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Gottlieb RA and Thomas A: Mitophagy and
mitochondrial quality control mechanisms in the heart. Curr
Pathobiol Rep. 5:161–169. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Guan R, Zou W, Dai X, Yu X, Liu H, Chen Q
and Teng W: Mitophagy, a potential therapeutic target for stroke. J
Biomed Sci. 25:872018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Andres AM, Tucker KC, Thomas A, Taylor DJ,
Sengstock D, Jahania SM, Dabir R, Pourpirali S, Brown JA, Westbrook
DG, et al: Mitophagy and mitochondrial biogenesis in atrial tissue
of patients undergoing heart surgery with cardio-pulmonary bypass.
JCI Insight. 2:e893032017. View Article : Google Scholar
|
|
140
|
Moyzis AG, Sadoshima J and Gustafsson AB:
Mending a broken heart: The role of mitophagy in cardioprotection.
Am J Physiol Heart Circ Physiol. 308:H183–H192. 2015. View Article : Google Scholar :
|
|
141
|
Ding S, Wu D, Lu Q, Qian L, Ding Y, Liu G,
Jia X, Zhang Y, Xiao W and Gong W: Angiopoietin-like 4 deficiency
upregulates macrophage function through the dysregulation of
cell-intrinsic fatty acid metabolism. Am J Cancer Res. 10:595–609.
2020.PubMed/NCBI
|
|
142
|
Cai J, Wang D, Zhao FQ, Liang S and Liu J:
AMPK-mTOR pathway is involved in glucose-modulated amino acid
sensing and utilization in the mammary glands of lactating goats. J
Anim Sci Biotechnol. 11:322020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Mackenzie RM, Salt IP, Miller WH, Logan A,
Ibrahim HA, Degasperi A, Dymott JA, Hamilton CA, Murphy MP, Delles
C and Dominiczak AF: Mitochondrial reactive oxygen species enhance
AMP-activated protein kinase activation in the endothelium of
patients with coronary artery disease and diabetes. Clin Sci
(Lond). 124:403–411. 2013. View Article : Google Scholar
|
|
144
|
Zhao Y, Shang F, Shi W, Zhang J, Liu X, Li
B, Hu X and Wang L: Angiotensin II receptor type 1 antagonists
modulate vascular smooth muscle cell proliferation and migration
via AMPK/mTOR. Cardiology. 143:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Gerczuk PZ and Kloner RA: An update on
cardioprotection: A review of the latest adjunctive therapies to
limit myocardial infarction size in clinical trials. J Am Coll
Cardiol. 59:969–978. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Obtułowicz K: Bradykinin-mediated
angioedema. Pol Arch Med Wewn. 126:76–85. 2016.
|
|
147
|
Taddei S and Bortolotto L: Unraveling the
pivotal role of bradykinin in ACE inhibitor activity. Am J
Cardiovasc Drugs. 16:309–321. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lepelley M, Bernardeau C, Defendi F,
Crochet J, Mallaret M and Bouillet L: Update on bradykinin-mediated
angioedema in 2020. Therapie. 75:195–205. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Koh JQS, Fernando H, Peter K and Stub D:
Opioids and ST elevation myocardial infarction: A systematic
review. Heart Lung Circ. 28:697–706. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Tavenier AH, Hermanides RS, Ottervanger
JP, Ter Horst PGJ, Kedhi E and van 't Hof AWJ: Risks of opioids in
ST-elevation myocardial infarction: A review. Drug Saf.
41:1303–1308. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Jones SP, Tang XL, Guo Y, Steenbergen C,
Lefer DJ, Kukreja RC, Kong M, Li Q, Bhushan S, Zhu X, et al: The
NHLBI-sponsored consortium for preclinicAl assESsment of
cARdioprotective therapies (CAESAR): A new paradigm for rigorous,
accurate, and reproducible evaluation of putative infarct-sparing
interventions in mice, rabbits, and pigs. Circ Res. 116:572–586.
2015. View Article : Google Scholar :
|