Atrial fibrillation (AF) decreases the quality of
life of patients whilst also presenting as a financial burden, due
to its severe complications (1).
Although significant progress has been made in the treatment
options that are available for management of AF, such as drugs and
catheter ablation, there are complications associated with these
strategies, and they are hampered by low long-term success rates.
An improved understanding of the fundamental mechanisms underlying
the development of AF and subsequent atrial remodelling may
facilitate the development of novel and more effective therapeutic
approaches for AF treatment. However, the mechanisms underlying AF
are complex, and include structural and electrical remodelling,
autonomic nervous system dysfunction (2) and dysregulated calcium
homeostasis/handing (3). Atrial
structural remodelling is the key factor linking all the AF-related
mechanisms, and atrial fibrosis is the most prominent feature of
atrial structural remodelling (4), but an in-depth understanding of the
molecular mechanisms underlying this process has not yet been fully
elucidated. For this reason, in the present review, the body of
knowledge regarding AF pathophysiology, as well as the involvement
of atrial fibrosis in the initiation and perpetuation of AF, were
reviewed, and the available fibrosis-guided approaches for
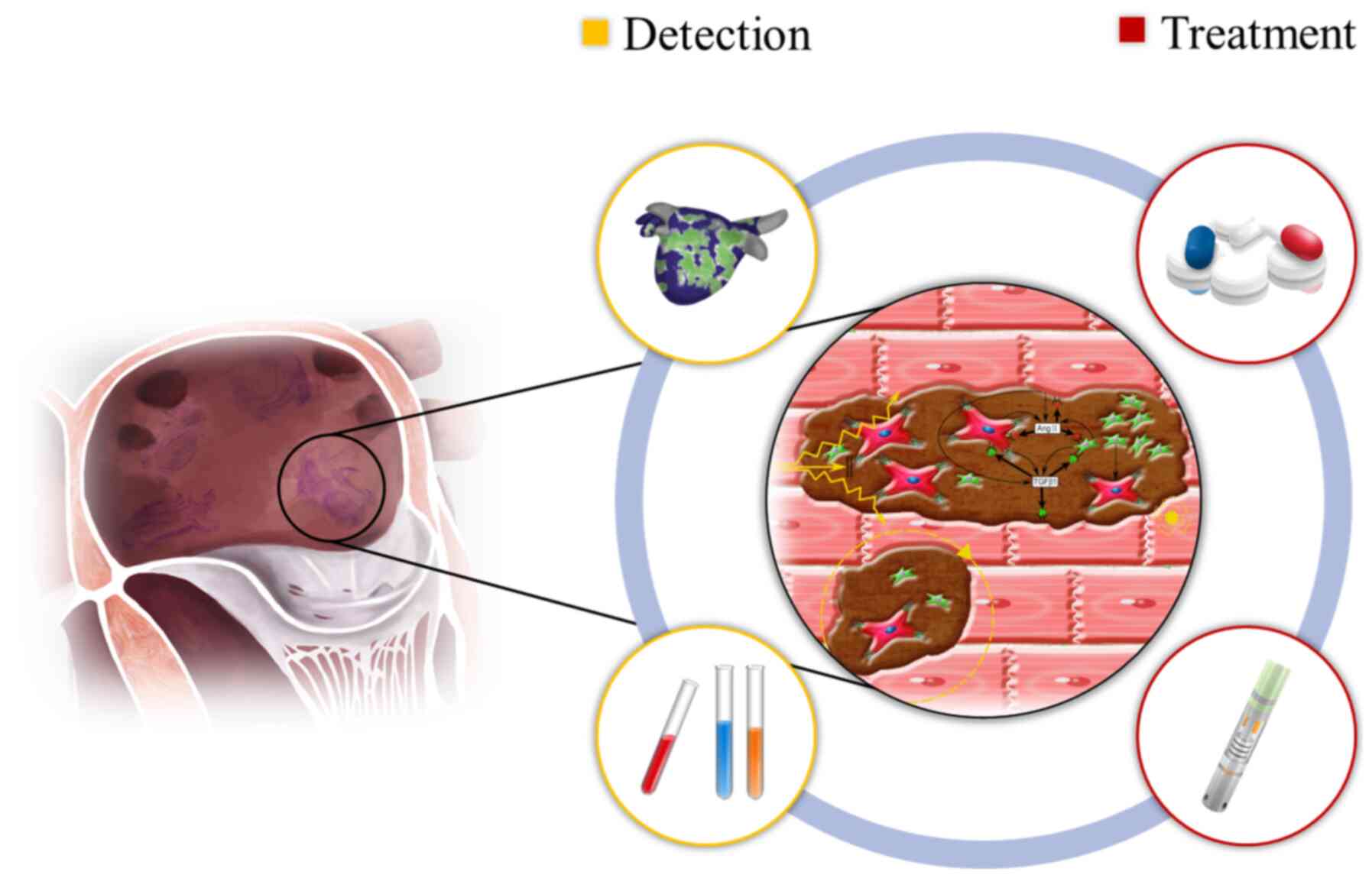
prevention and management of AF are discussed (Fig. 1).
As mentioned above, atrial fibrosis is an hallmark
of atrial structural remodelling, characterized by the aberrant
activation, proliferation and differentiation of fibroblasts, and
subsequent excessive synthesis and irregular deposition of
extracellular matrix (ECM) proteins, which have been identified as
substrates of AF, and are involved in the initiation and
perpetuation of AF (5). Atrial
fibrosis can be divided into two types, reactive and reparative
fibrosis (6,7). Reactive fibrosis is a response to
cardiac inflammation or pressure overload, and can be divided into
perivascular and interstitial fibrosis (8). Reparative fibrosis occurs due to the
loss of cardiomyocytes, with myocardial infarction being the most
cause (8). Various pro-fibrotic
stimulants activate fibroblasts to proliferate and differentiate
into secretory myofibroblasts, often accompanied by the
upregulation of matrix metalloproteinases (MMPs) and downregulation
of tissue inhibitors of metalloproteinases (TIMPs). These
abnormalities result in an imbalance in ECM deposition and
degradation in the intervascular space and myocardial interstitium,
ultimately altering the cardiac ultrastructure (8,9).
The primary benefit of fibrosis is to maintain the integrity of the
heart. However, these collagen-based scars can form barriers to
electrical conduction and separate the well-connected syncytium,
thereby directly interfering with conduction (10). In addition to physical uncoupling,
the membrane of fibroblasts and myofibroblasts can fuse with that
of cardiomyocytes to form gap junctions via connexins 40, 43 and 45
(Cx40, Cx43 and Cx45) (11,12). Despite the passive
electrophysiological qualities of fibroblasts and myofibroblasts,
they have a lower membrane potential than atrial resting potential
and can act as an electrical source during their resting phase and
as a sink during their activation, thereby reducing the conduction
speed and maximum level of depolarization of action potentials
(13). It has also been reported
that cross-linked collagen between cardiomyocyte bundles forms a
thick insulating layer that increases longitudinal conduction
velocity, which is also associated with the occurrence of AF
(14). When sufficient
fibroblasts/myofibroblasts-cardiomyocytes interactions are formed,
the arrhythmogenic mechanisms are fulfilled (15). Pathological coupling escalates the
spontaneous depolarization during phase 4, and this favours
triggered activity (15).
Anatomical barriers decrease conduction velocity and increase
conduction heterogeneity, as well as the dispersion of
refractoriness, which favours re-entry (13). The interactions between triggered
activity and arrhythmogenic substrates allows for the occurrence
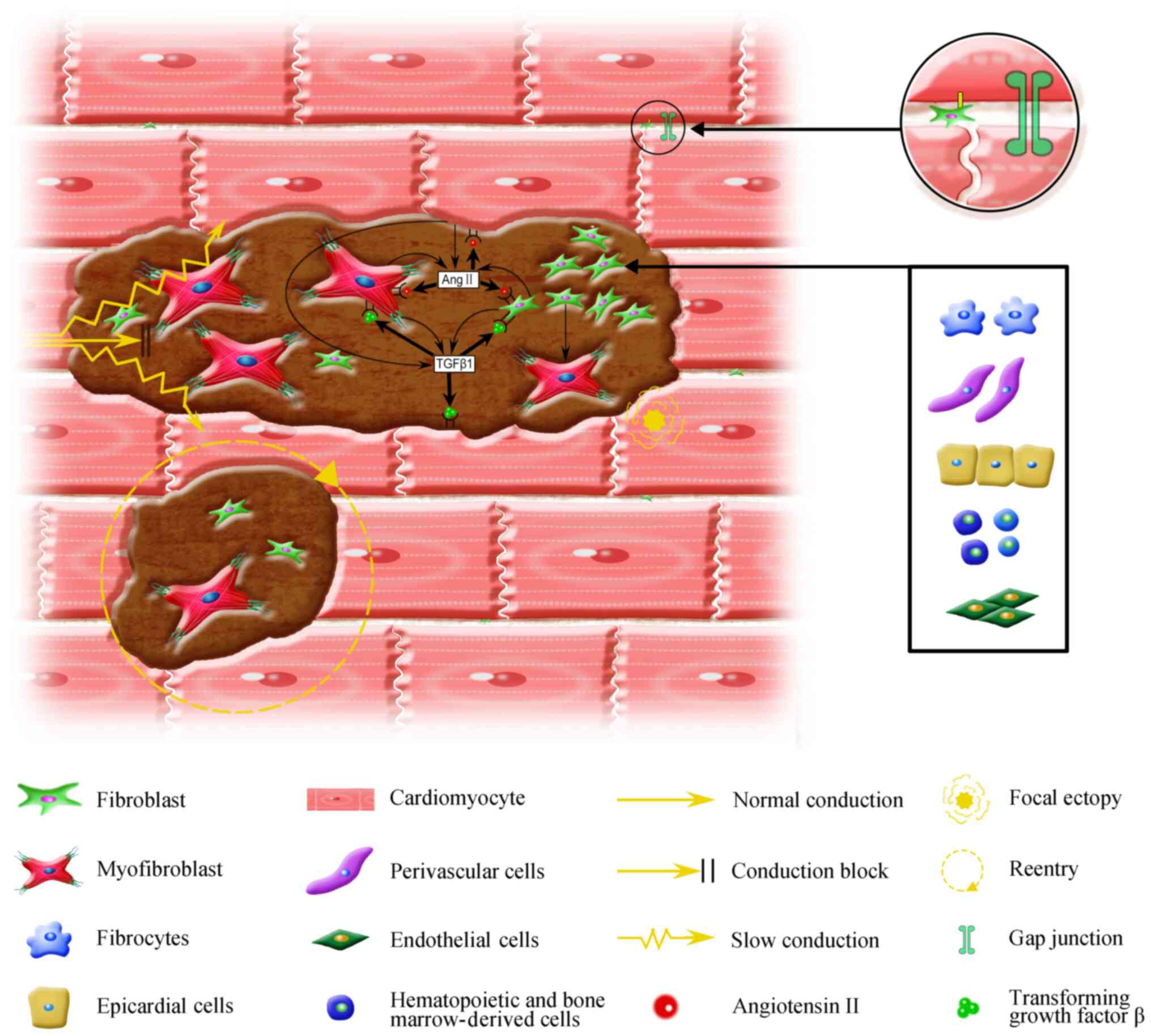
and perpetuation of AF (Fig.
2).
In total, four types of cells, namely endothelial
cells, cardiomyocytes, fibroblasts and smooth muscle cells, make up
a large proportion of cardiac cells (16). Fibroblasts are the second largest
population of non-myocyte cells in the heart, accounting for ~10%
of cardiac cells, and are the primary source of ECM (17). Cardiomyocytes are predominant in
volume, and are the primary constituents of the heart (18). The distribution of cardiac
fibroblasts in the atrium is higher than that in the ventricles,
and the responses of atrial fibroblasts to pro-fibrotic stimuli are
different from those of ventricular fibroblasts, which may account
for the difference in the degree of fibrosis between atriums and
ventricles under similar pathological conditions (19,20).
Cardiac fibroblasts are flat, spindle-shaped cells
that are generally considered to have a mesenchymal origin, and
they determine the homeostasis of ECM (21). During the development of the
heart, most cardiac fibroblasts are differentiated from
epicardium-derived cells (22).
The rest are derived from the endocardium and the neural crest,
which are primarily located in the interventricular septum and
right atrium, respectively (Fig.
2) (23,24). Under homeostatic conditions,
fibroblasts remain dormant. Apart from the activation and
proliferation of resident fibroblasts, several cell linages, such
as endothelial cells, bone marrow progenitor cells, circulating
fibrocytes and monocytes, can differentiate into fibroblasts when
activated by pathological stimulants, thus, markedly increasing the
number of cardiac fibroblasts (25,26). Activated fibroblasts then
synthesize not only a variety of ECM proteins, but also proteolytic
enzymes that modify these proteins and can differentiate into
myofibroblasts, which are contractile cells with a more potent
ability to synthesize more ECM proteins (27). This differentiation causes
disequilibrium in the synthesis and degradation of ECM proteins,
ultimately leading to an arrhythmogenic atrial substrate (28).
The ECM not only acts as a scaffold for all cells in
the heart, but it is also involved in regulating cardiac function
and mediating extracellular signal transmission (29). In addition to collagens,
proteoglycans, glycoprotein and other proteins (such as MMPs and
TIMPs) are necessary components of the ECM (30). There are also non-glycosylated
proteins and soluble components within the extracellular space,
such as dermatopontin, transforming growth factor β (TGF-β) and
interleukins (ILs), which are involved in the regulation of ECM
remodelling (31). Of these,
collagens (primarily types I and III) are the predominant
constituents of the cardiac ECM (5). The synthesis of collagens starts
when their progenitors, pro-collagens, are cleaved by procollagen
C-terminal proteinase and procollagen N-terminal proteinase at the
C- and N-terminal domains to form mature collagen molecules
(9). The final step is
self-assembly and cross-linking of mature collagen molecules form
collagen fibres. In this enzymatic process, some proteolytic
products, such as N-terminal pro-peptide of procollagen type III
and C-terminal pro-peptide of procollagen type I, are released into
the blood and can be used as biomarkers to assess cardiac fibrosis
and evaluate AF recurrence (32-34). The process of ECM protein
synthesis is a dynamic and balanced process under the fine
regulation of proteolytic enzymes and their inhibitors (9). Amongst these enzymes, the most
important are the MMP family members of which there are >25.
They can not only degrade almost all ECM proteins, but also
cytokines and growth factors, amongst other molecules, which
affects the synthesis of ECM (35). Increased expression of MMP-9 has
been observed in the atrial tissue and blood serum of patients with
AF, and the MMP-9 levels appear to be associated with the stage of
AF (36,37). In addition, it was found that
serum MMP-9 levels can also be used as an independent factor to
predict the recurrence of AF following catheter ablation (38). A previous meta-analysis
demonstrated that the enhanced MMP-1 mRNA expression and decreased
serum TIMP-2 levels may act as predictive markers for the incidence
of AF (39). In addition, MMP-2
was also shown to be associated with an increased risk of AF, and
may be used to identify patients that are most likely to benefit
from rhythmic control strategies (40).
The past few years have witnessed an impressive
growth in the number of studies studying the signalling pathways
involved in atrial fibrosis, but the specific mechanism remains
poorly understood. However, some effective therapeutic options that
target atrial fibrosis could not have been developed without taking
into account the complex signalling pathways. The key factors and
mechanisms leading to progressive atrial fibrosis are discussed
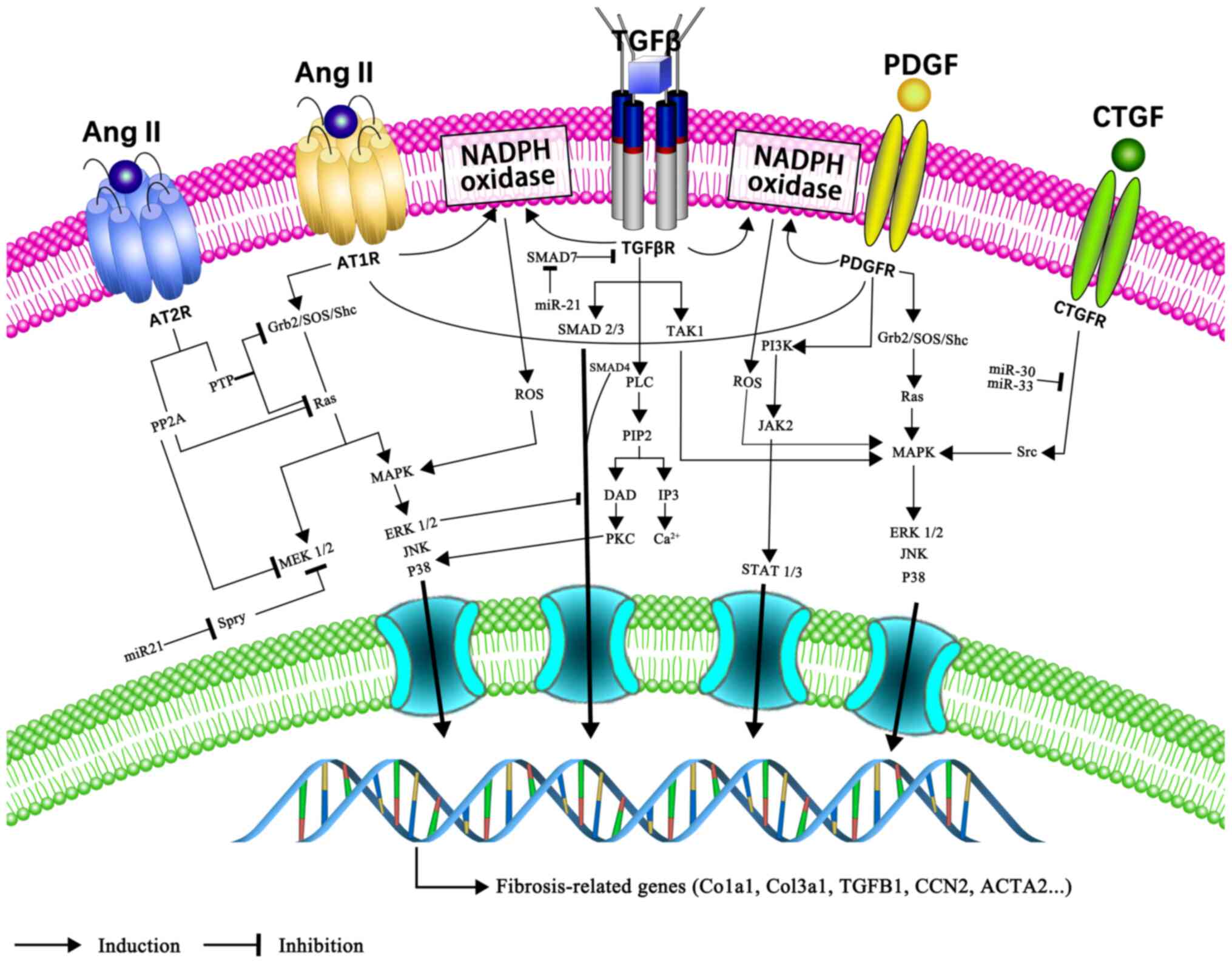
below (Fig. 3).
TGF-β is one of the most potent pro-fibrotic growth
factors, with >30 family members, including TGF-β1-3,
of which TGF-β1 is the predominant member (41). TGF-β1 promotes the
synthesis of collagen fibres by cardiac fibroblasts and their
differentiation into myofibroblasts via the typical Smad-dependent
and non-canonical Smad-independent pathways (42). In the canonical Smad-dependent
pathway, TGF-β binds to two types of serine/threonine kinase
receptors [type I TGFβ receptor (TβRI)/activin receptor-like kinase
5 and TβRII], which together form a Smad2/3/4 complex that
subsequently leads to Smad protein-mediated signal transduction
(43,44). Smad7, an inhibitory Smad,
antagonizes the TGF-β/Smad signalling pathway (44). Non-canonical pathways include the
mitogen-activated protein kinases (MAPKs)/TGF-β1/tumour
necrosis factor (TNF) receptor associated factor 6/TGF-β-activated
kinase 1, TGF-β1/cluster of differentiation
(CD)44/signal transducer and activator of transcription 3 (STAT3)
and angiotensin II (Ang II)/TGF-β/Ras homolog family member A
(RhoA)/Rho-kinase (ROCK) signalling pathways (45-47). The thrombospondin-1/TGF-β/MMP-9
axis is also involved in atrial fibrosis in patients with AF
(48).
Atrial myofibril loss was higher in patients with AF
compared with those with sinus rhythm. In an electrical stimulation
experiment of cultured HL-1 atrial myocytes, Yeh et al
(49) demonstrated that
Nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase-mediated oxidative stress may account for
tachycardia-induced myofibril degradation. They also reported
increased levels of p-Smad3 in a tachypacing model, and confirmed
there was crosstalk between the two signalling pathways in
tachypacing-stimulated reactive oxygen species (ROS)
production.
The RAAS is a system involving the
pathophysiological involvement of multiple organs including the
heart, kidney and lungs (50).
Ang II is a major mediator of this system and serves an important
role in atrial fibrosis. Ang II exerts pro-fibrotic effects by
binding to its type 1 receptor (AT1-R), a member of the
G-protein-coupled receptor superfamily. G protein activation
stimulates phospholipase C (PLC) to generate
inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3
mediates the increase of Ca2+ levels in the cytoplasm.
Intracellular Ca2+ overload promotes fibroblast
proliferation and differentiation (51). DAG activates protein kinase C,
which in turn activates extracellular-signal-regulated kinases
(ERKs). In addition, acting as a potent NADPH oxidase activator,
Ang II induces ROS overproduction, which, in-turn, activates
multiple downstream second messengers, including MAPK, nuclear
factor-κB and cytokines (52,53). Through the activation of the MAPK
signalling pathway, Ang II promotes the secretion of
TGF-β1. TGF-β1 reciprocally upregulates the
density of AT1-R and the expression of connective tissue
growth factor (CTGF), thereby further promoting fibrosis (54). Conversely, the stimulation of Ang
II type 2 receptor (AT2-R) constrains the pro-fibrotic
effects of AT1-R (55). Several studies have confirmed that
the blockade of Ang II by Ang-converting enzyme inhibitors (ACEIs)
or Ang receptor blockers (ARBs) reduces atrial fibrosis (56,57). Aldosterone is the end product of
RAAS, and its role in AF pathophysiology has proven very valuable.
By binding to the mineralocorticoid receptor (MR), aldosterone
serves its pro-fibrotic roles via the MAPK intracellular signalling
pathway in HL-1 atrial myocytes (58,59). Furthermore, there is crosstalk
between the MR/AT1-R and MAPK signalling pathway,
suggesting that the combined blocking of MR and AT1-R
can prevent the occurrence of AF (59).
The pro-fibrotic effect of inflammation is generally
attributed to oxidative stress, which promotes the initiation and
perpetuation of AF by activating the MAPK signalling pathway
(62). Mitochondria and NADPH
oxidase are hypothesized to be the major sources of ROS, which is a
second messenger that activates downstream signals. Amongst other
things, uncoupled nitric oxide (NO) synthase and xanthine oxidase
are also sources of ROS (63).
Ang II promotes ROS production, and both are involved in aberrant
Ca2+ handling, increasing the cytosolic Ca2+
concentration (64,65). Intracellular Ca2+
overload further aggravates electrical remodelling by
downregulating the L-type Ca2+-current (66). In addition, microRNA
(miRNA/miR)-26 is also downregulated by the activation of the
Ca2+-calcineurin-nuclear factor of activated T-cells
signalling pathway, promoting the expression of
KCNJ2/IK1 in both cardiomyocytes and fibroblasts.
Treatments targeting the upstream inflammatory cascade can decrease
the inflammatory response and oxidative stress, and alleviate
atrial structural and electrical remodelling, which further
elucidates the mechanisms underlying this disease (67).
Adipose, particularly epicardial adipose tissue
(EAT), is strongly associated with the initiation, duration and
recurrence of AF. With regard to the pathological mechanism of EAT
by which it promotes the occurrence and development of atrial
fibrosis, considerable evidence has consistently confirmed its role
in local inflammation. Abe et al (68,69) evaluated the levels of
cytokines/chemokines in a specimen from human left atrial
appendage. The results showed that the expression levels of IL-1,
IL-6, IL-10 and TNF-α in EAT increased, consistent with a previous
result from Mazurek et al (68,69). In addition, adipokines secreted by
EAT are another mechanism underlying fibrosis. Activin A, an
adipokine belonging to the TGF-β superfamily, has the ability to
initiate atrial fibrosis (70).
In addition, CTGF, a fibrotic cytokine that functions via the
TGF-β1/Smad pathway, has been shown to be upregulated in
EAT and is strongly associated with AF (71,72).
Significant non-invasive technological advances have
opened up more possibilities for the characterization and
quantification of focal and diffuse left ventricular (LV)
myocardial fibrosis in patients with AF, which have provided
evidence that the cardiac pro-fibrotic microenvironment in AF is
unlikely to be strictly limited to the atria (82,83). Late gadolinium enhanced cardiac
magnetic resonance (LGE-CMR) imaging is an established technique
for the evaluation of focal myocardial scars on the basis of the
different abilities of healthy myocardium and areas of fibrotic
tissue to clear gadolinium (84).
With regard to diffuse myocardial fibrosis, gadolinium contrast may
be evenly retained throughout the diffusely fibrotic myocardium,
and the signal intensity of diffusely fibrotic areas may be nearly
isointense, as compared with that of normal tissue. Diffuse
interstitial fibrosis is challenging to distinguish using
conventional delayed enhancement (DE)-CMR (85-87). With the development of novel
contrast-enhanced T1 mapping techniques, diffuse
myocardial fibrosis may be detected through a quantitative measure
of the myocardial T1 relaxation times (86,87). Ling et al (88) used myocardial T1
mapping in patients with AF to detect diffuse myocardial fibrosis
of the LV. They showed that LV fibrosis could be detected and
quantified by T1 mapping in patients with AF and HF
concurrently. Of note, several studies have shown that diffuse
ventricular fibrosis measured by T1 mapping on CMR predicts the
success of catheter ablation for AF, although the mechanism behind
this association is not clear (89,90).
There may be some possible explanations for the
association between AF and the presence of diffuse LV fibrosis. For
example, arrhythmia-mediated cardiomyopathy may predispose patients
to diffuse interstitial fibrosis (91). Ventricular fibrotic changes are
more extensive in patients with AF compared to those with sinus
rhythm (82,92). Data from an animal study suggested
that a rapid ventricular response from AF could result in a
decrease in ventricular function, and an increase in ventricular
and atrial fibrosis (93). In
addition, the restoration of the sinus rhythm with catheter
ablation is accompanied by significant improvements in reverse
cardiac remodelling and ventricular function (94). Fibrotic cardiomyopathy has been
suggested to predispose patients to diffuse interstitial fibrosis
development. A plethora of non-cardiac factors have been shown to
contribute to fibrosis in AF, including obesity, systemic
inflammation, metabolic syndrome, thyrotoxicosis and obstructive
sleep apnoea, which could ultimately affect the myocardium
(95). Obstructive and central
sleep apnoea leads to myocardial hypertrophy and diastolic
dysfunction, thus further potentiating the development of HF in
patients with AF (96,97). Obesity in AF is associated with
diastolic ventricular impairment and myocardial lipidosis (98). Alternatively, the association
between AF and ventricular fibrosis may also be due to other
factors which have yet to be uncovered (99). In summary, ventricular fibrosis in
response to AF may be regulated by multiple mechanisms. Additional
studies focusing on the association between AF and diffuse
myocardial fibrosis are required.
Several common mechanisms are known to contribute to
atrial and ventricular fibrosis in AF, whereas the extent of
fibrosis may vary between the 2 parts of the heart. Transgenic mice
with TGF-β1 exhibited higher TGF-β1 levels in
the atria than in the ventricles under the control of an α-MHC
promoter (100). In this model,
80 pro-fibrotic genes in the atria were overexpressed and only 2
genes in the ventricle were differentially expressed, as shown by
RNA microarray analysis (100).
Similarly, transgenic mice overexpressing ACE exhibited a
hypertrophic and dilated atria with focal atrial fibrosis, but
normal ventricles (101). This
differential chamber-specific fibrotic response to ACE
overexpression could be partly explained by the differential
AT1 receptor expression in the atria and ventricles
(102). It has been shown that
atrial fibroblasts show greater fibrotic and oxidative responses to
TGF-β1 than ventricular fibroblasts (103), indicating that the atria has a
more potent fibrotic response to various stimuli (20). The results of these studies
suggested that the mechanisms involved in the development of atrial
and ventricular fibrosis are different. Further studies are
required to investigate whether other important signalling pathways
contribute to the development of selective fibrosis in the atria,
compared to the ventricles.
There is increasing evidence of an association
between atrial fibrosis and the risk of stroke in patients with AF.
Daccaret et al (104)
identified an association between the percentage of atrial fibrosis
detected on LGE-CMR and a higher CHADS2-score [CHF,
hypertension, age >75 years, diabetes mellitus and stroke or
transient ischemic attack (TIA)], and a history for stroke. Left
atrium fibrosis is a strong predictor of left atrial thrombosis or
cerebrovascular events, particularly stroke or TIA (105,106). Another study by Disertori et
al (107) showed that the
risk of stroke may be independently associated with structural
fibrotic remodelling. Left atrial fibrosis is also associated with
an increased risk of cryptogenic stroke (108). Even in patients without AF,
embolic stroke of an undetermined source has been found to be
correlated to atrial fibrosis (109). Spronk et al showed that
hypercoagulability in itself may stimulate fibroblasts and increase
fibrosis. It was revealed that anticoagulation therapy may prevent
thromboembolic events, partly through influencing the substrate by
reducing the degree of fibrosis (110). In combination, these studies
provided quantitative evidence that the risk of stroke in patients
with AF may be associated with the severity of the LA fibrosis.
However, there is a paucity of data on the pathophysiological link
and molecular mechanisms between atrial fibrosis and
thromboembolism. Atrial fibrosis, one of several markers of an
AF-prone atrial substrate, promotes the re-entry of electrical
current by increasing heterogeneity of conduction in the atria,
which ultimately impairs atrial contractility, and reduces ejection
fraction and flow velocity (111). It thus causes increased platelet
aggregation, which further enhances the milieu of intra-atrial
stasis (111).
Endothelium/endocardial tissue not only forms a barrier between
platelets and extracellular matrix, but also secretes factors such
as NO and heparan sulphates to prevent the activation of the
coagulation cascade. Endothelial dysfunction develops as a result
of atrial fibrosis in patients with AF and promotes thrombus
formation (112).
Inflammation and oxidative stress are known to serve
an important pathogenic role in AF, leading to cardiac fibrosis
(113,114). Inflammatory markers, such as
TGF-β1, IL-6 and TNF-α, have been detected in patients with AF and
have been shown to affect the functional stability of myocytes and
endothelial cells, as well as promote atrial fibrosis (53,115). Inflammatory marker levels were
associated with a risk of stroke in patients with chronic AF during
follow-up (116,117). There may be a close interplay
amongst atrial fibrosis, inflammation and oxidative stress, which,
in turn, leads to endothelial and/or endocardial dysfunction and a
pro-thrombotic state; however, further studies are required to
advance from theoretical to pragmatic outcomes.
Stroke in AF appears to be a complex and poorly
understood phenomenon, and the means by which LA fibrosis
predisposes patients to thrombus formation is not completely clear.
LA fibrosis represents a marker of disease, which can improve the
prediction of thromboembolic events in patients with AF.
Conventional antiarrhythmic agent approaches have
limited efficacy and have several adverse effects. Increased
attention has therefore been diverted to upstream therapies with
the use of non-antiarrhythmic drugs targeting substrate development
and modifying risk factors for human AF. Specifically, one of the
most relevant objectives of upstream therapy is the control of the
development and progression of atrial fibrosis, which is a hallmark
of structural remodelling in AF and is considered a substrate for
perpetuation of AF (118-120).
It has become clear that Ang II is a potent stimulator of
pro-fibrotic pathways during AF, and the inhibition of the RAAS by
ACEIs, ARBs and mineralocorticoid receptor antagonists (MRAs) was
shown to reduce the progression of fibrosis (121). Several ACEIs have been shown to
effectively suppress atrial fibrosis and prevent the development of
the AF substrate (122,123). The potential of AT1
receptor blockers for the treatment of fibrosis and AF has been
previously explored. In spontaneously hypertensive rats, valsartan
reduced the degree of myocardial fibrosis (124). Similarly, losartan and
candesartan have been previously shown to suppress atrial
remodelling by inhibiting left atrial fibrosis and improving AF
indices in experimental models (125,126). MRAs also appear to be potential
agents for fibrosis. Lavall et al (58) found that mineralocorticoid
receptor blockers could effectively reduce the incidence of
new-onset AF in patients with systolic heart failure. Eplerenone
treatment has been shown to inhibit the development of atrial
hypertrophy and fibrosis compared with the control group animals
(127). Retrospective analyses
and meta-analyses of databases from clinical trials have suggested
a role of inhibitors of the Ang axis in AF prevention, particularly
in patients with LV hypertrophy and systolic LV dysfunction
(128-132). However, other clinical studies
reported no beneficial effects of Ang blockade treatment on the
incidence of recurrent AF (133,134). These conflicting outcomes may be
partly attributed to the possible interactions or synergistic
effects with other drugs including ACEIs, amiodarone and
β-blockers, and the differences in the baseline parameters of
patients, such as ventricular function, structural substrates and
influence of fibrosis-causing factors. The beneficial effects of
upstream therapies may be due to the prevention of structural
remodelling in both the left atrium and the LV, improved LV
haemodynamics and reduced atrial stretch, and direct or indirect
modulation of ion-channel function and other unknown factors.
There is less evidence in favour of therapies, such
as polyunsaturated omega-3 fatty acids or the inhibitors of
3-hydroxy-3-methylglutaryl-CoA reductase (statins) in the
inhibition of fibrosis and atrial structural remodelling.
Simvastatin attenuated CHF-induced atrial structural remodelling
and AF promotion (135).
Similarly, statin therapy may contribute to the prevention of AF in
the postoperative period of cardiac surgery (136). Omega-3 poly-unsaturated fatty
acids have been found to suppress AF in patients with an evident
structural substrate and presence of atrial remodelling, combined
with high levels of circulating inflammatory biomarkers (137). Treatment of CHF canines with the
antifibrotic drug pirfenidone resulted in significantly reduced
TGF-β1 levels, arrhythmogenic atrial remodelling and AF
vulnerability (138). In
conclusion, these results further highlight the value of upstream
AF prevention therapy.
The potential mechanisms underlying the positive
effects of atrial fibrosis treatment and any fibrosis-related AF in
humans is not well understood. A deeper understanding of these
fundamental mechanisms may assist in identifying novel targets for
pharmacological interventions, which may be even more effective
than conventional antiarrhythmic therapy.
Percutaneous catheter ablation is a widely used and
effective clinical treatment for rhythm control in patients with AF
(139). Circumferential
pulmonary vein isolation (CPVI) alone is an ablation strategy that
is effective in the majority of patients with paroxysmal AF.
However, the frequent need for re-ablation coupled with the lower
long-term success rates are still major limitations of catheter
ablation procedures in the treatment of non-paroxysmal AF (140). AF evolves from a singular rhythm
disturbance to the complex condition that is cardiomyopathy through
arrhythmia substrates (141,142). Studies have reported the
detection of atrial fibrosis using DE-MR imaging (MRI) and
electroanatomic voltage mapping (EAVM) (104,143-146), and suggested that it is an
important predictor of the outcome of AF interventions (146-148).
Substrate modification targeting fibrotic tissue has
been performed for several years using EAVM (149); this procedure has been described
in more detail previously (149). Kottkamp et al (150) described a patient-tailored
ablation strategy termed 'box isolation of fibrotic areas', which
involves the circumferential isolation of substantially affected
fibrotic areas (<0.5 mV), providing a novel selection criterion
for PVI-only ablation in patients with non-paroxysmal AF. Rolf
et al (144) also
demonstrated a tailored substrate modification based on voltage
criteria. Yamaguchi et al (151) described an approach of
homogenizing areas of substantial fibrosis; briefly, the ablation
of all detectable electrograms within the target areas was defined
as an area with bipolar electrograms of <0.5 mV and, in
addition, short linear lesions were created so as to ablate
potential conduction channels. During a follow-up in their study,
absence of AF was notably higher in the low-voltage zone-based
substrate modification group compared with the group that only
underwent PVI (38% vs. 72%). A total of 144/201 patients (74%) who
underwent LA low voltage area-guided AF substrate modification as
an adjunct to PVI during a median follow-up of 3.1 years were free
from recurrence (152).
Similarly, Jadidi et al (153) previously reported that absence
of arrhythmia was higher in the substrate modification approach
group compared with the matched control group that only received
PVI (69% vs. 47%). As compared with the stepwise approach for the
treatment of non-paroxysmal AF, a strategy of selective
electrophysiologically guided atrial substrate modification after
CPVI and cavotricuspid isthmus ablation was found to be more
clinically effective (154).
Voltage mapping as a tool for describing fibrotic changes remains
under investigation and still requires standardization. For
example, the measured voltage depends on the rhythm, various
thresholds of voltage amplitude used to define fibrotic areas, the
contact of the electrode to the tissue, the electrode size and
spacing, the thickness of the atrial myocardium and other variables
(155).
LGE-CMR provides a non-invasive tool for detecting,
quantifying and localizing atrial fibrosis. Jadidi et al
(156) demonstrated that the
large fibrotic substrate detected with LGE-CMR is associated with
the complex fractionated atrial electrogram, proposed as a relevant
phenomenon maintaining AF. Recent data have reported patients being
free of AF recurrence after catheter ablation led to a significant
attenuation of the LA fibrosis burden, as shown by follow-up CMR
studies (157). In contrast to
invasive EAVM during the ablation procedure, LGE-CMR-guided
fibrosis management has improved our understanding of the
individual underlying arrhythmia substrate during the natural
course of human AF. Fochler et al (158) reported that an LGE-MRI
anatomically guided approach for the treatment of recurrent
arrhythmias post-AF ablation is feasible and effective. Similarly,
another study highlights the potential use of the optimal set of
patient-specific targets to ablate fibrotic atrial substrates
(159). The LGE-CMR-guided
assessment may provide novel insights into patient-specific AF
stages and treatment strategies; however, this modality requires
extensive MRI experience, and its reproducibility is still under
intensive investigation (160).
At present, the success rate of non-individualized
substrate modifications of catheter ablation procedures for
patients with persistent and/or long-standing AF is disappointingly
low (161). Completely novel
catheter ablation strategies that are based on the individual
substrates rather than on the 'phenotype' in paroxysmal vs.
non-paroxysmal AF are thus required. The knowledge of the
individual amount and distribution pattern of a patient's AF
fibrotic LA substrate allows for a personalized path to prevention,
monitoring or even targeting arrhythmia substrates in patients with
AF, which need to be confirmed and validated with respect to
efficacy, as well as safety in prospective multicentre randomized
studies.
The prevalence and health burden of AF worldwide
highlights the importance of the development of high-accuracy and
precision therapies aimed at preventing or reversing AF. Clinical
and experimental studies have reported that atrial fibrosis is
closely associated with the occurrence and maintenance of AF. The
development of fibrosis is a highly complex, multifactorial and
patient-specific process, involved in complex neurohumoral,
cellular and molecular interactions. Although a significant
understanding has already been obtained that has led to the
identification of novel targets for fibrotic mechanism-based
therapies, the precise role of fibrosis in AF initiation and
maintenance remains to be determined. There is a wide variation in
the presence, extent and pattern of LA fibrosis. Its use for AF
treatment may assist in designing individually tailored ablation
approached for determining the ablation strategy following
pulmonary vein isolation and high-lights the need for repeated
ablation procedures, which could potentially significantly improve
our understanding of AF and ablation outcomes.
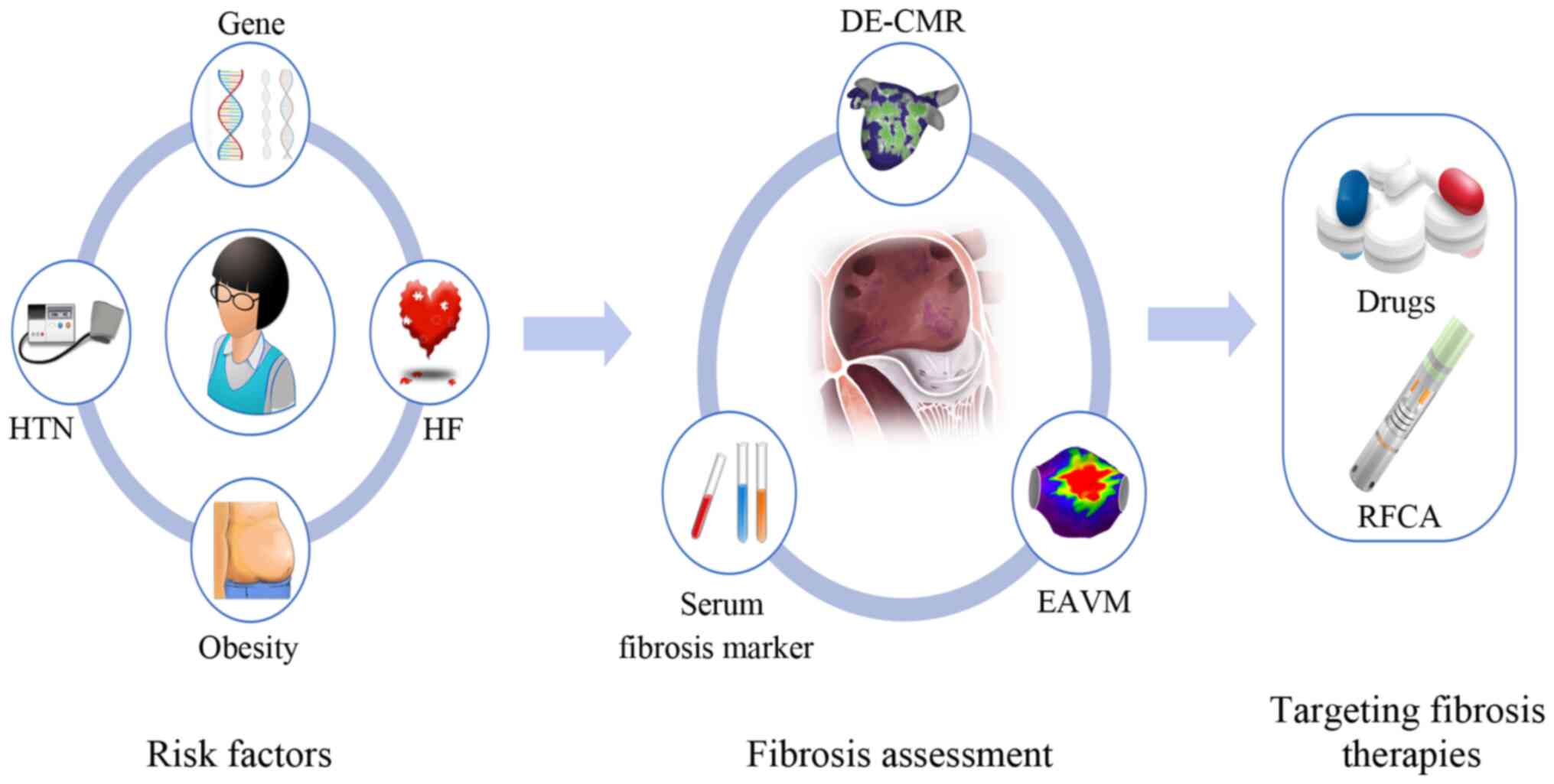
An improved understanding of the roles,
characteristics and mechanisms of fibrosis during AF may facilitate
the identification of new clinical biomarkers, as well as assist in
the development of novel, more effective and patient-tailored
treatment approaches for AF by targeting the fibrotic substrate
(Fig. 4).
This study was supported by Grants from the
National Natural Science Foundation of China (grant no. 81700271),
and the Science and Technology Program of Hebei (grant no.
H2018105054).
Not applicable.
CYL, JRZ, WNH and SNL wrote the manuscript. SNL
critically reviewed the manuscript. All authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
|
1
|
Sanoski CA: Clinical, economic, and
quality of life impact of atrial fibrillation. J Manag Care Pharm.
15(6 Suppl B): S4–S9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen PS, Chen LS, Fishbein MC, Lin SF and
Nattel S: Role of the autonomic nervous system in atrial
fibrillation: Pathophysiology and therapy. Circ Res. 114:1500–1515.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denham NC, Pearman CM, Caldwell JL,
Madders GWP, Eisner DA, Trafford AW and Dibb KM: Calcium in the
pathophysiology of atrial fibrillation and heart failure. Front
Physiol. 9:13802018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pellman J and Sheikh F: Atrial
fibrillation: Mechanisms, therapeutics, and future directions.
Compr Physiol. 5:649–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Polyakova V, Miyagawa S, Szalay Z, Risteli
J and Kostin S: Atrial extracellular matrix remodelling in patients
with atrial fibrillation. J Cell Mol Med. 12:189–208. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nattel S: Molecular and cellular
mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin
Electrophysiol. 3:425–435. 2017. View Article : Google Scholar
|
|
7
|
de Boer RA, De Keulenaer G, Bauersachs J,
Brutsaert D, Cleland JG, Diez J, Du XJ, Ford P, Heinzel FR, Lipson
KE, et al: Towards better definition, quantification and treatment
of fibrosis in heart failure. A scientific roadmap by the committee
of translational research of the heart failure association (HFA) of
the European society of cardiology. Eur J Heart Fail. 21:272–285.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sc. 71:549–574.
2014. View Article : Google Scholar
|
|
9
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rog-Zielinska EA, Norris RA, Kohl P and
Markwald R: The living scar-cardiac fibroblasts and the injured
heart. Trends Mol Med. 22:99–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kohl P and Gourdie RG: Fibroblast-myocyte
electrotonic coupling: Does it occur in native cardiac tissue? J
Mol Cell Cardiol. 70:37–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ongstad E and Kohl P: Fibroblast-myocyte
coupling in the heart: Potential relevance for therapeutic
interventions. J Mol Cell Cardiol. 91:238–246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen TP, Qu Z and Weiss JN: Cardiac
fibrosis and arrhythmogenesis: The road to repair is paved with
perils. J Mol Cell Cardiol. 70:83–91. 2014. View Article : Google Scholar
|
|
14
|
Krul SPJ, Berger WR, Smit NW, van
Amersfoorth SC, Driessen AH, van Boven WJ, Fiolet JW, van Ginneken
AC, van der Wal AC, de Bakker JM, et al: Atrial fibrosis and
conduction slowing in the left atrial appendage of patients
undergoing thoracoscopic surgical pulmonary vein isolation for
atrial fibrillation. Circ Arrhythm Electrophysiol. 8:288–295. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nattel S: Electrical coupling between
cardiomyocytes and fibroblasts: Experimental testing of a
challenging and important concept. Cardiovasc Res. 114:349–352.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perbellini F, Watson SA, Bardi I and
Terracciano CM: Heterocellularity and cellular cross-talk in the
cardiovascular system. Front Cardiovasc Med. 5:1432018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT,
D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA
and Tallquist MD: Revisiting cardiac cellular composition. Circ
Res. 118:400–409. 2016. View Article : Google Scholar :
|
|
18
|
Zhou P and Pu WT: Recounting cardiac
cellular composition. Circ Res. 118:368–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Y, de Castro Brás LE, Toba H, Iyer RP,
Hall ME, Winniford MD, Lange RA, Tyagi SC and Lindsey ML:
Myofibroblasts and the extracellular matrix network in
post-myocardial infarction cardiac remodeling. Pflugers Arch.
466:1113–127. 2014.PubMed/NCBI
|
|
20
|
Burstein B, Libby E, Calderone A and
Nattel S: Differential behaviors of atrial versus ventricular
fibroblasts: A potential role for platelet-derived growth factor in
atrial-ventricular remodeling differences. Circulation.
117:1630–1641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moore-Morris T, Cattaneo P, Puceat M and
Evans SM: Origins of cardiac fibroblasts. J Mol Cell Cardiol.
91:1–5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Krenning G, Zeisberg EM and Kalluri R: The
origin of fibroblasts and mechanism of cardiac fibrosis. J Cell
Physiol. 225:631–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moore-Morris T, Guimarães-Camboa N,
Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu
Y, Dalton ND, Cedenilla M, et al: Resident fibroblast lineages
mediate pressure overload-induced cardiac fibrosis. J Clin Invest.
124:2921–2934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao
P, Subat A, Hojjat A, Kamran P, Müller AM, Volz KS, Tang Z, et al:
Developmental heterogeneity of cardiac fibroblasts does not predict
pathological proliferation and activation. Circ Res. 115:625–635.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lajiness JD and Conway SJ: The dynamic
role of cardiac fibroblasts in development and disease. J
Cardiovasc Transl Res. 5:739–748. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SK, Park JH, Kim JY, Choi JI, Joung B,
Lee MH, Kim SS, Kim YH and Pak HN: High plasma concentrations of
transforming growth factor-β and tissue inhibitor of
metalloproteinase-1: Potential non-invasive predictors for
electroanatomical remodeling of atrium in patients with
non-valvular atrial fibrillation. Circ J. 75:557–564. 2011.
View Article : Google Scholar
|
|
28
|
Goudis CA, Kallergis EM and Vardas PE:
Extracellular matrix alterations in the atria: Insights into the
mechanisms and perpetuation of atrial fibrillation. Europace.
14:623–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frangogiannis NG: The extracellular matrix
in myocardial injury, repair, and remodeling. J Clin Invest.
127:1600–1612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rienks M, Papageorgiou AP, Frangogiannis
NG and Heymans S: Myocardial extracellular matrix: An ever-changing
and diverse entity. Circ Res. 114:872–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu X, Meng L, Shi Q, Liu S, Cui C, Hu S
and Wei Y: Dermatopontin promotes adhesion, spreading and migration
of cardiac fibroblasts in vitro. Matrix Biol. 32:23–31. 2013.
View Article : Google Scholar
|
|
32
|
López B, González A, Ravassa S, Beaumont
J, Moreno MU, San José G, Querejeta R and Díez J: Circulating
biomarkers of myocardial fibrosis: The need for a reappraisal. J Am
Coll Cardiol. 65:2449–2456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Richter B, Gwechenberger M, Socas A, Zorn
G, Albinni S, Marx M, Wolf F, Bergler-Klein J, Loewe C, Bieglmayer
C, et al: Time course of markers of tissue repair after ablation of
atrial fibrillation and their relation to left atrial structural
changes and clinical ablation outcome. Int J Cardiol. 152:231–236.
2011. View Article : Google Scholar
|
|
34
|
Kawamura M, Munetsugu Y, Kawasaki S,
Onishi K, Onuma Y, Kikuchi M, Tanno K and Kobayashi Y: Type III
procollagen-N-peptide as a predictor of persistent atrial
fibrillation recurrence after cardioversion. Europace.
14:1719–1725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Spinale FG: Myocardial matrix remodeling
and the matrix metalloproteinases: Influence on cardiac form and
function. Physiol Rev. 87:1285–1342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li M, Yang G, Xie B, Babu K and Huang C:
Changes in matrix metalloproteinase-9 levels during progression of
atrial fibrillation. J Int Med Res. 42:224–230. 2014. View Article : Google Scholar
|
|
37
|
Nakano Y, Niida S, Dote K, Takenaka S,
Hirao H, Miura F, Ishida M, Shingu T, Sueda T, Yoshizumi M and
Chayama K: Matrix metalloproteinase-9 contributes to human atrial
remodeling during atrial fibrillation. J Am Coll Cardiol.
43:818–825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu G, Wang S, Cheng M, Peng B, Liang J,
Huang H, Jiang X, Zhang L, Yang B, Cha Y, et al: The serum matrix
metalloproteinase-9 level is an independent predictor of recurrence
after ablation of persistent atrial fibrillation. Clinics (Sao
Paulo). 71:251–256. 2016. View Article : Google Scholar
|
|
39
|
Liu Y, Xu B, Wu N, Xiang Y, Wu L, Zhang M,
Wang J, Chen X, Li Y and Zhong L: Association of MMPs and TIMPs
with the occurrence of atrial fibrillation: A systematic review and
meta-analysis. Can J Cardiol. 32:803–813. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Molvin J, Jujic A, Melander O, Pareek M,
Råstam L, Lindblad U, Daka B, Leosdottir M, Nilsson P, Olsen M and
Magnusson M: Exploration of pathophysiological pathways for
incident atrial fibrillation using a multiplex proteomic chip. Open
Heart. 7:e0011902020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morikawa M, Derynck R and Miyazono K:
TGF-β and the TGF-β family: Context-dependent roles in cell and
tissue physiology. Cold Spring Harb Perspect Biol. 8:a0218732016.
View Article : Google Scholar
|
|
42
|
Biernacka A, Dobaczewski M and
Frangogiannis NG: TGF-beta signaling in fibrosis. Growth Factors.
29:196–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Euler-Taimor G and Heger J: The complex
pattern of SMAD signaling in the cardiovascular system. Cardiovasc
Res. 69:15–25. 2006. View Article : Google Scholar
|
|
45
|
Zhang D, Chen X, Wang Q, Wu S, Zheng Y and
Liu X: Role of the MAPKs/TGF-β1/TRAF6 signaling pathway in
postoperative atrial fibrillation. PLoS One. 12:e01737592017.
View Article : Google Scholar
|
|
46
|
Chang SH, Yeh YH, Lee JL, Hsu YJ, Kuo CT
and Chen WJ: Transforming growth factor-β-mediated CD44/STAT3
signaling contributes to the development of atrial fibrosis and
fibrillation. Basic Res Cardiol. 112:582017. View Article : Google Scholar
|
|
47
|
Liu LJ, Yao FJ, Lu GH, Xu CG, Xu Z, Tang
K, Cheng YJ, Gao XR and Wu SH: The role of the Rho/ROCK pathway in
Ang II and TGF-β1-induced atrial remodeling. PLoS One.
11:e01616252016. View Article : Google Scholar
|
|
48
|
Yang Z and Wang H: Increased expression of
the TSP-1/TGF-β/MMP-9 axis in atrial fibrillation related to
rheumatic heart disease. Int J Clin Exp Med. 11:5699–5706.
2018.
|
|
49
|
Yeh YH, Kuo CT, Chan TH, Chang GJ, Qi XY,
Tsai F, Nattel S and Chen WJ: Transforming growth factor-β and
oxidative stress mediate tachycardia-induced cellular remodelling
in cultured atrial-derived myocytes. Cardiovasc Res. 91:62–70.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Patel S, Rauf A, Khan H and Abu-Izneid T:
Renin-angiotensinaldosterone (RAAS): The ubiquitous system for
homeostasis and pathologies. Biomed Pharmacother. 94:317–325. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harada M, Luo X, Qi XY, Tadevosyan A,
Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, et al:
Transient receptor potential canonical-3 channel-dependent
fibroblast regulation in atrial fibrillation. Circulation.
126:2051–2064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu L, Geng J, Zhao H, Yun F, Wang X, Yan
S, Ding X, Li W, Wang D, Li J, et al: Valsartan reduced atrial
fibrillation susceptibility by inhibiting atrial parasympathetic
remodeling through MAPKs/neurturin pathway. Cell Physiol Biochem.
36:2039–2050. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Harada M, Van Wagoner DR and Nattel S:
Role of inflammation in atrial fibrillation pathophysiology and
management. Circ J. 79:495–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang
WF and Zhou L: Angiotensin II increases CTGF expression via
MAPKs/TGF-β1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res.
318:2105–2115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nouet S and Nahmias C: Signal transduction
from the angiotensin II AT2 receptor. Trends Endocrinol Metab.
11:1–6. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sakabe M, Fujiki A, Nishida K, Sugao M,
Nagasawa H, Tsuneda T, Mizumaki K and Inoue H: Enalapril prevents
perpetuation of atrial fibrillation by suppressing atrial fibrosis
and over-expression of connexin43 in a canine model of atrial
pacing-induced left ventricular dysfunction. J Cardiovasc
Pharmacol. 43:851–859. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li D, Shinagawa K, Pang L, Leung TK,
Cardin S, Wang Z and Nattel S: Effects of angiotensin-converting
enzyme inhibition on the development of the atrial fibrillation
substrate in dogs with ventricular tachypacing-induced congestive
heart failure. Circulation. 104:2608–2614. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lavall D, Selzer C, Schuster P, Lenski M,
Adam O, Schäfers HJ, Böhm M and Laufs U: The mineralocorticoid
receptor promotes fibrotic remodeling in atrial fibrillation. J
Biol Chem. 289:6656–6668. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tsai CF, Yang SF, Chu HJ and Ueng KC:
Cross-talk between mineralocorticoid receptor/angiotensin II type 1
receptor and mitogen-activated protein kinase pathways underlies
aldosterone-induced atrial fibrotic responses in HL-1
cardiomyocytes. Int J Cardiol. 169:17–28. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bruins P, te Velthuis H, Yazdanbakhsh AP,
Jansen PG, van Hardevelt FW, de Beaumont EM, Wildevuur CR, Eijsman
L, Trouwborst A and Hack CE: Activation of the complement system
during and after cardiopulmonary bypass surgery: Postsurgery
activation involves C-reactive protein and is associated with
postoperative arrhythmia. Circulation. 96:3542–3548. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hu YF, Chen YJ, Lin YJ and Chen SA:
Inflammation and the pathogenesis of atrial fibrillation. Nat Rev
Cardiol. 12:230–243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang CX, Liu Y, Xia WF, Tang YH and Huang
H: Oxidative stress: A possible pathogenesis of atrial
fibrillation. Med Hypotheses. 72:466–467. 2009. View Article : Google Scholar
|
|
63
|
Sovari AA and Dudley SC Jr: Reactive
oxygen species-targeted therapeutic interventions for atrial
fibrillation. Front Physiol. 3:3112012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Görlach A, Bertram K, Hudecova S and
Krizanova O: Calcium and ROS: A mutual interplay. Redox Biol.
6:260–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Youn JY, Zhang J, Zhang Y, Chen H, Liu D,
Ping P, Weiss JN and Cai H: Oxidative stress in atrial
fibrillation: An emerging role of NADPH oxidase. J Mol Cell
Cardiol. 62:72–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nattel S and Harada M: Atrial remodeling
and atrial fibrillation: Recent advances and translational
perspectives. J Am Coll Cardiol. 63:2335–2345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sirish P, Li N, Timofeyev V, Zhang XD,
Wang L, Yang J, Lee KS, Bettaieb A, Ma SM, Lee JH, et al: Molecular
mechanisms and new treatment paradigm for atrial fibrillation. Circ
Arrhythm Electrophysiol. 9:e0037212016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mazurek T, Zhang L, Zalewski A, Mannion
JD, Diehl JT, Arafat H, Sarov-Blat L, O'Brien S, Keiper EA, Johnson
AG, et al: Human epicardial adipose tissue is a source of
inflammatory mediators. Circulation. 108:2460–2466. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Abe I, Teshima Y, Kondo H, Kaku H, Kira S,
Ikebe Y, Saito S, Fukui A, Shinohara T, Yufu K, et al: Association
of fibrotic remodeling and cytokines/chemokines content in
epicardial adipose tissue with atrial myocardial fibrosis in
patients with atrial fibrillation. Heart Rhythm. 15:1717–1727.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Venteclef N, Guglielmi V, Balse E, Gaborit
B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clément K
and Hatem SN: Human epicardial adipose tissue induces fibrosis of
the atrial myocardium through the secretion of adipo-fibrokines.
Eur Heart J. 36:795–805a. 2015. View Article : Google Scholar
|
|
71
|
Wang Q, Wang X, Yin L, Wang J, Shen H, Gao
Y, Min J, Zhang Y and Wang Z: Human epicardial adipose tissue cTGF
expression is an independent risk factor for atrial fibrillation
and highly associated with atrial fibrosis. Sci Rep. 8:35852018.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li Y, Jian Z, Yang ZY, Chen L, Wang XF, Ma
RY and Xiao YB: Increased expression of connective tissue growth
factor and transforming growth factor-beta-1 in atrial myocardium
of patients with chronic atrial fibrillation. Cardiology.
124:233–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liao CH, Akazawa H, Tamagawa M, Ito K,
Yasuda N, Kudo Y, Yamamoto R, Ozasa Y, Fujimoto M, Wang P, et al:
Cardiac mast cells cause atrial fibrillation through
PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J
Clin Invest. 120:242–253. 2010. View Article : Google Scholar
|
|
74
|
Chen Y, Surinkaew S, Naud P, Qi XY, Gillis
MA, Shi YF, Tardif JC, Dobrev D and Nattel S: JAK-STAT signalling
and the atrial fibrillation promoting fibrotic substrate.
Cardiovasc Res. 113:310–320. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tuuminen R, Nykänen AI, Krebs R, Soronen
J, Pajusola K, Keränen MA, Koskinen PK, Alitalo K and Lemström KB:
PDGF-A, -C, and -D but not PDGF-B increase TGF-beta1 and chronic
rejection in rat cardiac allografts. Arterioscler Thromb Vasc Biol.
29:691–698. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li PF, He RH, Shi SB, Li R, Wang QT, Rao
GT and Yang B: Modulation of miR-10a-mediated TGF-β1/Smads
signaling affects atrial fibrillation-induced cardiac fibrosis and
cardiac fibroblast proliferation. Biosci Rep. 39:BSR201819312019.
View Article : Google Scholar
|
|
77
|
Thum T, Gross C, Fiedler J, Fischer T,
Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et
al: MicroRNA-21 contributes to myocardial disease by stimulating
MAP kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Adam O, Löhfelm B, Thum T, Gupta SK, Puhl
SL, Schäfers HJ, Böhm M and Laufs U: Role of miR-21 in the
pathogenesis of atrial fibrosis. Basic Res Cardiol. 107:2782012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
He X, Zhang K, Gao X, Li L, Tan H, Chen J
and Zhou Y: Rapid atrial pacing induces myocardial fibrosis by
down-regulating Smad7 via microRNA-21 in rabbit. Heart Vessels.
31:1696–1708. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Wang Y, Cai H, Li H, Gao Z and Song K:
Atrial overexpression of microRNA-27b attenuates angiotensin
II-induced atrial fibrosis and fibrillation by targeting ALK5. Hum
Cell. 31:251–260. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen Y, Wakili R, Xiao J, Wu CT, Luo X,
Clauss S, Dawson K, Qi X, Naud P, Shi YF, et al: Detailed
characterization of microRNA changes in a canine heart failure
model: Relationship to arrhythmogenic structural remodeling. J Mol
Cell Cardiol. 77:113–124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shantsila E, Shantsila A, Blann AD and Lip
GY: Left ventricular fibrosis in atrial fibrillation. Am J Cardiol.
111:996–1001. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
White SK, Sado DM, Fontana M, Banypersad
SM, Maestrini V, Flett AS, Piechnik SK, Robson MD, Hausenloy DJ,
Sheikh AM, et al: T1 mapping for myocardial extracellular volume
measurement by CMR: bolus only versus primed infusion technique.
JACC Cardiovasc Imaging. 6:955–962. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Neilan TG, Shah RV, Abbasi SA, Farhad H,
Groarke JD, Dodson JA, Coelho-Filho O, McMullan CJ, Heydari B,
Michaud GF, et al: The incidence, pattern, and prognostic value of
left ventricular myocardial scar by late gadolinium enhancement in
patients with atrial fibrillation. J Am Coll Cardiol. 62:2205–2214.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ambale-Venkatesh B and Lima JA: Cardiac
MRI: A central prognostic tool in myocardial fibrosis. Nat Rev
Cardiol. 12:18–29. 2015. View Article : Google Scholar
|
|
86
|
Flett AS, Hayward MP, Ashworth MT, Hansen
MS, Taylor AM, Elliott PM, McGregor C and Moon JC: Equilibrium
contrast cardiovascular magnetic resonance for the measurement of
diffuse myocardial fibrosis: Preliminary validation in humans.
Circulation. 122:138–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kammerlander AA, Marzluf BA, Zotter-Tufaro
C, Aschauer S, Duca F, Bachmann A, Knechtelsdorfer K, Wiesinger M,
Pfaffenberger S, Greiser A, et al: T1 mapping by CMR imaging: From
histological validation to clinical implication. JACC Cardiovasc
Imaging. 9:14–23. 2016. View Article : Google Scholar
|
|
88
|
Ling LH, Kistler PM, Ellims AH, Iles LM,
Lee G, Hughes GL, Kalman JM, Kaye DM and Taylor AJ: Diffuse
ventricular fibrosis in atrial fibrillation: Noninvasive evaluation
and relationships with aging and systolic dysfunction. J Am Coll
Cardiol. 60:2402–2408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Neilan TG, Mongeon FP, Shah RV,
Coelho-Filho O, Abbasi SA, Dodson JA, McMullan CJ, Heydari B,
Michaud GF, John RM, et al: Myocardial extracellular volume
expansion and the risk of recurrent atrial fibrillation after
pulmonary vein isolation. JACC Cardiovasc Imaging. 7:1–11. 2014.
View Article : Google Scholar
|
|
90
|
McLellan AJ, Ling LH, Azzopardi S, Ellims
AH, Iles LM, Sellenger MA, Morton JB, Kalman JM, Taylor AJ and
Kistler PM: Diffuse ventricular fibrosis measured by T1 mapping on
cardiac MRI predicts success of catheter ablation for atrial
fibrillation. Circ Arrhythm Electrophysiol. 7:834–840. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Guichard JB, Xiong F, Qi XY, L'Heureux N,
Hiram R, Xiao J, Naud P, Tardif JC, Costa AD and Nattel S: Role of
atrial arrhythmia and ventricular response in atrial fibrillation
induced atrial remodeling. Cardiovasc Res. Jan 24–2020.Epub ahead
of print. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Sasaki N, Okumura Y, Watanabe I, Nagashima
K, Sonoda K, Kogawa R, Takahashi K, Iso K, Ohkubo K, Nakai T, et
al: Transthoracic echocardiographic backscatter-based assessment of
left atrial remodeling involving left atrial and ventricular
fibrosis in patients with atrial fibrillation. Int J Cardiol.
176:1064–1066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Avitall B, Bi J, Mykytsey A and Chicos A:
Atrial and ventricular fibrosis induced by atrial fibrillation:
Evidence to support early rhythm control. Heart Rhythm. 5:839–845.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Prabhu S, Costello BT, Taylor AJ, Gutman
SJ, Voskoboinik A, McLellan AJA, Peck KY, Sugumar H, Iles L, Pathik
B, et al: Regression of diffuse ventricular fibrosis following
restoration of sinus rhythm with catheter ablation in patients with
atrial fibrillation and systolic dysfunction: A substudy of the
CAMERA MRI trial. JACC Clin Electrophysiol. 4:999–1007. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Schoonderwoerd BA, Smit MD, Pen L and Van
Gelder IC: New risk factors for atrial fibrillation: Causes of
'not-so-lone atrial fibrillation'. Europace. 10:668–673. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Iwasaki YK, Kato T, Xiong F, Shi YF, Naud
P, Maguy A, Mizuno K, Tardif JC, Comtois P and Nattel S: Atrial
fibrillation promotion with long-term repetitive obstructive sleep
apnea in a rat model. J Am Coll Cardiol. 64:2013–2023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Neilan TG, Farhad H, Dodson JA, Shah RV,
Abbasi SA, Bakker JP, Michaud GF, van der Geest R, Blankstein R,
Steigner M, et al: Effect of sleep apnea and continuous positive
airway pressure on cardiac structure and recurrence of atrial
fibrillation. J Am Heart Assoc. 2:e0004212013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pathak RK, Mahajan R, Lau DH and Sanders
P: The implications of obesity for cardiac arrhythmia mechanisms
and management. Can J Cardiol. 31:203–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wijesurendra RS and Casadei B: Atrial
fibrillation: Effects beyond the atrium? Cardiovasc Res.
105:238–247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rahmutula D, Marcus GM, Wilson EE, Ding
CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ and Olgin JE:
Molecular basis of selective atrial fibrosis due to overexpression
of transforming growth factor-β1. Cardiovasc Res. 99:769–779. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xiao HD, Fuchs S, Campbell DJ, Lewis W,
Dudley SC Jr, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR
and Bernstein KE: Mice with cardiac-restricted
angiotensin-converting enzyme (ACE) have atrial enlargement,
cardiac arrhythmia, and sudden death. Am J Pathol. 165:1019–1032.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bauer P, regitz-zagrosek V, Kallisch H,
Linz W, Schoelkens B, Hildebrandt AG and Fleck E: Myocardial
angiotensin receptor type 1 gene expression in a rat model of
cardiac volume overload. Basic Res Cardiol. 92:139–146. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yeh YH, Kuo CT, Chang GJ, Qi XY, Nattel S
and Chen WJ: Nicotinamide adenine dinucleotide phosphate oxidase 4
mediates the differential responsiveness of atrial versus
ventricular fibroblasts to transforming growth factor-β. Circ
Arrhythm Electrophysiol. 6:790–798. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Daccarett M, Badger TJ, Akoum N, Burgon
NS, Mahnkopf C, Vergara G, Kholmovski E, McGann CJ, Parker D,
Brachmann J, et al: Association of left atrial fibrosis detected by
delayed-enhancement magnetic resonance imaging and the risk of
stroke in patients with atrial fibrillation. J Am Coll Cardiol.
57:831–838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Akoum N, Fernandez G, Wilson B, Mcgann C,
Kholmovski E and Marrouche N: Association of atrial fibrosis
quantified using LGE-MRI with atrial appendage thrombus and
spontaneous contrast on transesophageal echocardiography in
patients with atrial fibrillation. J Cardiovasc Electrophysiol.
24:1104–1109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
King JB, Azadani PN, Suksaranjit P, Bress
AP, Witt DM, Han FT, Chelu MG, Silver MA, Biskupiak J, Wilson BD,
et al: Left atrial fibrosis and risk of cerebrovascular and
cardiovascular events in patients with atrial fibrillation. J Am
Coll Cardiol. 70:1311–1321. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Disertori M, Quintarelli S, Grasso M,
Pilotto A, Narula N, Favalli V, Canclini C, Diegoli M, Mazzola S,
Marini M, et al: Autosomal recessive atrial dilated cardiomyopathy
with standstill evolution associated with mutation of natriuretic
peptide precursor A. Circ Cardiovasc Genet. 6:27–36. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Fonseca AC, Alves P, Inácio N, Marto JP,
Viana-Baptista M, Pinho-E-Melo T, Ferro JM and Almeida AG: Patients
with undetermined stroke have increased atrial fibrosis: A cardiac
magnetic resonance imaging study. Stroke. 49:734–737. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Tandon K, Tirschwell D, Longstreth WT Jr,
Smith B and Akoum N: Embolic stroke of undetermined source
correlates to atrial fibrosis without atrial fibrillation.
Neurology. 93:e381–e387. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Spronk HM, De Jong AM, Verheule S, De Boer
HC, Maass AH, Lau DH, Rienstra M, van Hunnik A, Kuiper M, Lumeij S,
et al: Hypercoagulability causes atrial fibrosis and promotes
atrial fibrillation. Eur Heart J. 38:38–50. 2017. View Article : Google Scholar
|
|
111
|
D'Souza A, Butcher KS and Buck BH: The
multiple causes of stroke in atrial fibrillation: Thinking broadly.
Can J Cardiol. 34:1503–1511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Cai H, Li Z, Goette A, Mera F, Honeycutt
C, Feterik K, Wilcox JN, Dudley SC Jr, Harrison DG and Langberg JJ:
Downregulation of endocardial nitric oxide synthase expression and
nitric oxide production in atrial fibrillation: Potential
mechanisms for atrial thrombosis and stroke. Circulation.
106:2854–2858. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Friedrichs K, Klinke A and Baldus S:
Inflammatory pathways underlying Atrial Fibrillation. Trends Mol
Med. 17:556–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Korantzopoulos P, Letsas K, Fragakis N,
Tse G and Liu T: Oxidative stress and atrial fibrillation: An
update. Free Radic Res. 52:1199–1209. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Cao H, Wang J, Xi L, Røe OD, Chen Y and
Wang D: Dysregulated atrial gene expression of
osteoprotegerin/receptor activator of nuclear factor-κB (RANK)/RANK
ligand axis in the development and progression of atrial
fibrillation. Circ J. 75:2781–2788. 2011. View Article : Google Scholar
|
|
116
|
Pinto A, Tuttolomondo A, Casuccio A, Di
Raimondo D, Di Sciacca R, Arnao V and Licata G: Immuno-inflammatory
predictors of stroke at follow-up in patients with chronic
non-valvular atrial fibrillation (NVAF). Clin Sci (Lond).
116:781–789. 2009. View Article : Google Scholar
|
|
117
|
Li J, Solus J, Chen Q, Rho YH, Milne G,
Stein CM and Darbar D: Role of inflammation and oxidative stress in
atrial fibrillation. Heart Rhythm. 7:438–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Calvo D, Filgueiras-Rama D and Jalife J:
Mechanisms and drug development in atrial fibrillation. Pharmacol
Rev. 70:505–525. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sardar MR, Saeed W and Kowey PR:
Antiarrhythmic drug therapy for atrial fibrillation. Heart Fail
Clin. 12:205–221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Burashnikov A and Antzelevitch C: Novel
pharmacological targets for the rhythm control management of atrial
fibrillation. Pharmacol Ther. 132:300–313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Novo G, Guttilla D, Fazio G, Cooper D and
Novo S: The role of the renin-angiotensin system in atrial
fibrillation and the therapeutic effects of ACE-Is and ARBS. Br J
Clin Pharmacol. 66:345–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li Y, Li W, Yang B, Han W, Dong D, Xue J,
Li B, Yang S and Sheng L: Effects of Cilazapril on atrial
electrical, structural and functional remodeling in atrial
fibrillation dogs. J Electrocardiol. 40:100.e1–e6. 2007. View Article : Google Scholar
|
|
123
|
Belluzzi F, Sernesi L, Preti P, Salinaro
F, Fonte ML and Perlini S: Prevention of recurrent lone atrial
fibrillation by the angiotensin-II converting enzyme inhibitor
ramipril in normotensive patients. J Am Coll Cardiol. 53:24–29.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Akashiba A, Ono H, Ono Y, Ishimitsu T and
Matsuoka H: Valsartan improves L-NAME-exacerbated cardiac fibrosis
with TGF-ß inhibition and apoptosis induction in spontaneously
hypertensive rats. J Cardiol. 52:239–246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kumagai K, Nakashima H, Urata H, Gondo N,
Arakawa K and Saku K: Effects of angiotensin II type 1 receptor
antagonist on electrical and structural remodeling in atrial
fibrillation. J Am Coll Cardiol. 41:2197–2204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Nomura M, Kawano T, Nakayasu K and Nakaya
Y: The effects of losartan on signal-averaged P wave in patients
with atrial fibrillation. Int J Cardiol. 126:21–27. 2008.
View Article : Google Scholar
|
|
127
|
Takemoto Y, Ramirez RJ, Kaur K,
Salvador-Montañés O, Ponce-Balbuena D, Ramos-Mondragón R, Ennis SR,
Guerrero-Serna G, Berenfeld O and Jalife J: Eplerenone reduces
atrial fibrillation burden without preventing atrial electrical
remodeling. J Am Coll Cardiol. 70:2893–2905. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Neefs J, van den Berg NW, Limpens J,
Berger WR, Boekholdt SM, Sanders P and de Groot JR: Aldosterone
pathway blockade to prevent atrial fibrillation: A systematic
review and meta-analysis. Int J Cardiol. 231:155–161. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Rienstra M, Hobbelt AH, Alings M, Tijssen
JGP, Smit MD, Brügemann J, Geelhoed B, Tieleman RG, Hillege HL,
Tukkie R, et al: Targeted therapy of underlying conditions improves
sinus rhythm maintenance in patients with persistent atrial
fibrillation: Results of the RACE 3 trial. Eur Heart J.
39:2987–2896. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Schneider MP, Hua TA, Böhm M, Wachtell K,
Kjeldsen SE and Schmieder RE: Prevention of atrial fibrillation by
Renin-Angiotensin system inhibition a meta-analysis. J Am Coll
Cardiol. 55:2299–2307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
European Heart Rhythm Association;
European Association for Cardio-Thoracic Surgery; Camm AJ, Kirchhof
P, Lip GY, Schotten U, Savelieva I, Ernst S, Van Gelder IC,
Al-Attar N, et al: Guidelines for the management of atrial
fibrillation: The task force for the management of atrial
fibrillation of the European society of cardiology (ESC). Eur Heart
J. 31:2369–2429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chaugai S, Meng WY and Ali Sepehry A:
Effects of RAAS blockers on atrial fibrillation prophylaxis: An
updated systematic review and meta-analysis of randomized
controlled trials. J Cardiovasc Pharmacol Ther. 21:388–404. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
GISSI-AF Investigators; Disertori M,
Latini R, Barlera S, Franzosi MG, Staszewsky L, Maggioni AP, Lucci
D, Di Pasquale G and Tognoni G: Valsartan for prevention of
recurrent atrial fibrillation. N Engl J Med. 360:1606–1617. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Tveit A, Grundvold I, Olufsen M, Seljeflot
I, Abdelnoor M, Arnesen H and Smith P: Candesartan in the
prevention of relapsing atrial fibrillation. Int J Cardiol.
120:85–91. 2007. View Article : Google Scholar
|
|
135
|
Shiroshita-Takeshita A, Brundel BJ,
Burstein B, Leung TK, Mitamura H, Ogawa S and Nattel S: Effects of
simvastatin on the development of the atrial fibrillation substrate
in dogs with congestive heart failure. Cardiovasc Res. 74:75–84.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Kuhn EW, Liakopoulos OJ, Stange S, Deppe
AC, Slottosch I, Choi YH and Wahlers T: Preoperative statin therapy
in cardiac surgery: A meta-analysis of 90,000 patients. Eur J
Cardiothorac Surg. 45:17–26. 2014. View Article : Google Scholar
|
|
137
|
Salvador-Montañés O, Gómez-Gallanti A,
Garofalo D, Noujaim SF, Peinado R and Filgueiras-Rama D:
Polyunsaturated Fatty acids in atrial fibrillation: Looking for the
proper candidates. Front Physiol. 3:3702012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Lee KW, Everett TH IV, Rahmutula D, Guerra
JM, Wilson E, Ding C and Olgin JE: Pirfenidone prevents the
development of a vulnerable substrate for atrial fibrillation in a
canine model of heart failure. Circulation. 114:1703–1712. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Anderson JL, Halperin JL, Albert NM,
Bozkurt B, Brindis RG, Curtis LH, DeMets D, Guyton RA, Hochman JS,
Kovacs RJ, et al: Management of patients with atrial fibrillation
(compilation of 2006 ACCF/AHA/ESC and 2011 ACCF/AHA/HRS
recommendations): A report of the American college of
cardiology/American heart association task force on practice
guidelines. J Am Coll Cardiol. 61:1935–1944. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Holmqvist F, Kesek M, Englund A,
Blomström-Lundqvist C, Karlsson LO, Kennebäck G, Poçi D, Samo-Ayou
R, Sigurjónsdóttir R, Ringborn M, et al: A decade of catheter
ablation of cardiac arrhythmias in Sweden: Ablation practices and
outcomes. Eur Heart J. 40:820–830. 2019. View Article : Google Scholar :
|
|
141
|
Kottkamp H: Human atrial fibrillation
substrate: Towards a specific fibrotic atrial cardiomyopathy. Eur
Heart J. 34:2731–2738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Kottkamp H and Schreiber D: The substrate
in 'early persistent' atrial fibrillation: Arrhythmia induced, risk
factor induced, or from a specific fibrotic atrial cardiomyopathy?
JACC Clin Electrophysiol. 2:140–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Oakes RS, Badger TJ, Kholmovski EG, Akoum
N, Burgon NS, Fish EN, Blauer JJ, Rao SN, DiBella EV, Segerson NM,
et al: Detection and quantification of left atrial structural
remodeling with delayed-enhancement magnetic resonance imaging in
patients with atrial fibrillation. Circulation. 119:1758–1767.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Rolf S, Kircher S, Arya A, Eitel C, Sommer
P, Richter S, Gaspar T, Bollmann A, Altmann D, Piedra C, et al:
Tailored atrial substrate modification based on low-voltage areas
in catheter ablation of atrial fibrillation. Circ Arrhythm
Electrophysiol. 7:825–833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kapa S, Desjardins B, Callans DJ,
Marchlinski FE and Dixit S: Contact electroanatomic mapping derived
voltage criteria for characterizing left atrial scar in patients
undergoing ablation for atrial fibrillation. J Cardiovasc
Electrophysiol. 25:1044–1052. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Marrouche NF, Wilber D, Hindricks G, Jais
P, Akoum N, Marchlinski F, Kholmovski E, Burgon N, Hu N, Mont L, et
al: Association of atrial tissue fibrosis identified by delayed
enhancement MRI and atrial fibrillation catheter ablation: The
DECAAF study. JAMA. 311:498–506. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Canpolat U, Oto A, Hazırolan T, Sunman H,
Yorgun H, Şahiner L, Kaya EB and Aytemir K: A prospective DE-MRI
study evaluating the role of TGF-β1 in left atrial fibrosis and
implications for outcomes of cryoballoon-based catheter ablation:
New insights into primary fibrotic atriocardiomyopathy. J
Cardiovasc Electrophysiol. 26:251–259. 2015. View Article : Google Scholar
|
|
148
|
McGann C, Akoum N, Patel A, Kholmovski E,
Revelo P, Damal K, Wilson B, Cates J, Harrison A, Ranjan R, et al:
Atrial fibrillation ablation outcome is predicted by left atrial
remodeling on MRI. Circ Arrhythm Electrophysiol. 7:23–30. 2014.
View Article : Google Scholar :
|
|
149
|
Stiles MK, John B, Wong CX, Kuklik P,
Brooks AG, Lau DH, Dimitri H, Roberts-Thomson KC, Wilson L, De
Sciscio P, et al: Paroxysmal lone atrial fibrillation is associated
with an abnormal atrial substrate: Characterizing the 'second
factor'. J Am Coll Cardiol. 53:1182–1191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kottkamp H, Berg J, Bender R, Rieger A and
Schreiber D: Box isolation of fibrotic areas (BIFA): A
patient-tailored substrate modification approach for ablation of
atrial fibrillation. J Cardiovasc Electrophysiol. 27:22–30. 2016.
View Article : Google Scholar
|
|
151
|
Yamaguchi T, Tsuchiya T, Nakahara S, Fukui
A, Nagamoto Y, Murotani K, Eshima K and Takahashi N: Efficacy of
left atrial voltage-based catheter ablation of persistent atrial
fibrillation. J Cardiovasc Electrophysiol. 27:1055–1063. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Yagishita A, Gimbel JR, DE Oliveira S,
Manyam H, Sparano D, Cakulev I, Mackall J and Arruda M: Long-term
outcome of left atrial voltage-guided substrate ablation during
atrial fibrillation: A novel adjunctive ablation strategy. J
Cardiovasc Electrophysiol. 28:147–155. 2017. View Article : Google Scholar
|
|
153
|
Jadidi AS, Lehrmann H, Keyl C, Sorrel J,
Markstein V, Minners J, Park CI, Denis A, Jaïs P, Hocini M, et al:
Ablation of persistent atrial fibrillation targeting low-voltage
areas with selective activation characteristics. Circ Arrhythm
Electrophysiol. 9:e0029622016.PubMed/NCBI
|
|
154
|
Yang G, Yang B, Wei Y, Zhang F, Ju W, Chen
H, Li M, Gu K, Lin Y, Wang B, et al: Catheter ablation of
nonparoxysmal atrial fibrillation using electrophysiologically
guided substrate modification during sinus rhythm after pulmonary
vein isolation. Circ Arrhythm Electrophysiol. 9:e0033822016.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Kottkamp H, Schreiber D, Moser F and
Rieger A: Therapeutic approaches to atrial fibrillation ablation
targeting atrial fibrosis. JACC Clin Electrophysiol. 3:643–653.
2017. View Article : Google Scholar
|
|
156
|
Jadidi AS, Cochet H, Shah AJ, Kim SJ,
Duncan E, Miyazaki S, Sermesant M, Lehrmann H, Lederlin M, Linton
N, et al: Inverse relationship between fractionated electrograms
and atrial fibrosis in persistent atrial fibrillation: Combined
magnetic resonance imaging and high-density mapping. J Am Coll
Cardiol. 62:802–812. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gal P and Marrouche NF: Magnetic resonance
imaging of atrial fibrosis: Redefining atrial fibrillation to a
syndrome. Eur Heart J. 38:14–19. 2017. View Article : Google Scholar
|
|
158
|
Fochler F, Yamaguchi T, Kheirkahan M,
Kholmovski EG, Morris AK and Marrouche NF: Late gadolinium
enhancement magnetic resonance imaging guided treatment of
post-atrial fibrillation ablation recurrent arrhythmia. Circ
Arrhythm Electrophysiol. 12:e0071742019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Boyle PM, Zghaib T, Zahid S, Ali RL, Deng
D, Franceschi WH, Hakim JB, Murphy MJ, Prakosa A, Zimmerman SL, et
al: Computationally guided personalized targeted ablation of
persistent atrial fibrillation. Nat Biomed Eng. 3:870–879. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Sohns C and Marrouche NF: Atrial
fibrillation and cardiac fibrosis. Eur Heart J. 41:1123–1131. 2020.
View Article : Google Scholar
|
|
161
|
Kottkamp H, Bender R and Berg J: Catheter
ablation of atrial fibrillation: How to modify the substrate? J Am
Coll Cardiol. 65:196–206. 2015. View Article : Google Scholar : PubMed/NCBI
|