Based on GLOBOCAN 2018 statistics, colorectal cancer
(CRC) ranks as the third most frequently detected cancer and the
second prominent cancer-related fatality worldwide despite the
advent of better screening for early detection and therapeutic
advances (1). By the year 2030,
the incidence of CRC is predicted to increase by 60% in developing
countries (2). Adenocarcinomas
constitute >90% of CRC cases developing as a malignant lesion in
glandular epithelial cells of the large intestine comprising of the
colon and rectum (3). The
majority of CRC cases (60-65%) are sporadic (without a family
history of CRC) acquiring somatic mutations and epigenetic
alterations from modifiable risk factors (4). CRC due to hereditary components is
estimated to be approximately 35-40% (5,6)
while family history attributes to approximately 25% of cases
without any disease phenotype (3). CRC is found to be inheritable in 5%
of cases known as hereditary non-polyposis CRC (HNPCC) or familial
adenomatous polyposis (FAP) induced by adenomatous polyposis coli
(APC), MutL homolog 1 (MLH1) and MutS homolog (MSH2) germline
mutation (3). The development of
CRC involves 3 global genetic and epigenetic aberrations: i)
Chromosomal instability (CIN); ii) methylation of CpG island
methylator phenotype (CIMP); and iii) instability of microsatellite
DNA regions (MSI) (7-9). The majority of sporadic cases of CRC
(85%) result from CIN due to structural and numerical alterations,
leading to the loss or gain of chromosomal segments, rearrangements
leading to genetic instability and the loss of heterozygosity
(10). The loss of heterozygosity
(LOH) causes alterations in copy number variations. On the other
hand, CIMP augments alterations in the methylation frequency of CpG
islands in promoter regions of tumor-suppressor genes, rendering
their subdued expression or complete silencing (11,12). Noticeable CRC cases have also been
attributed to the unstable nature of microsatellite DNA (MSI)
causing an alteration in the microsatellite length and are caused
by the loss of DNA mismatch repair gene MLH1 driving
hypermethylation and subsequent gene silencing (13). Alteration in these events results
in the perturbation of tumor-associated genes, leading to changes
in the cell cycle, which ultimately affects different cellular
behaviors viz. cellular invasion, migration, proliferation and
altered cell-to-cell signaling, leading to initiation and
progression to CRC.
Naturally, the progression of CRC results from 4
steps: i) Initiation; ii) promotion; iii) progression and iv)
metastasis (14). In initiation,
irreversible genetic alteration leads to neoplastic transformation.
Promotion involves cell proliferation leading to abnormal growth.
In the progression phase, these genetic/epigenetic aberrations
provide a selective advantage to cells, converting benign cells to
malignant cells, which further progress to gain aggressive
characteristics and metastasis, which is indicative of advanced
disease characteristics with the potential to spread to other
organs of the body through the blood and lymph nodes.
The detection of CRC is defined as stages that
reflect the extent of disease progression. As per the American
Joint Committee on Cancer, the staging of CRC is based on the TNM
(tumor-nodes-metastasis) system (15). Tumor stage (T) characterizes the
extent of tumor infiltration into the bowel wall, nodal stage (N)
refers to local or regional lymph node spread and metastatic spread
(M) defines the presence of distant metastasis. After the TNM
characterization, the disease is assigned into 4 stages (I-IV)
categorizing stages I-II as early and stage III-IV as late-stage
CRC (15).
Alterations in genetic and epigenetic components
lead to the aberrant activation of signaling pathways, a
pre-requisite for the progression from a benign to a malignant
tumor. Crosstalk between signaling pathways further promotes the
disease stage to metastasis, which is the main cause of CRC-related
mortality (16). Despite
advancements being made in early diagnosis and treatments that
include surgery and chemotherapy, significant numbers of patients
with early-stage CRC tend to develop metastasis and thus succumb to
the disease. With the advent of robust next-generation sequencing
techniques, numerous deleterious single nucleotide polymorphisms
(SNPs) and mutations have been identified in genes directly or
indirectly linked with CRC that may be the cause of carcinogenesis
(17). According to Fearon and
Vogelstein (18), a tumor
acquires driver mutations, leading to the dysregulation of
signaling pathways specifically targeting cell growth and
differentiation, leading to colorectal carcinogenesis and further
resulting in the metastatic phenotype. The most prevalent is the
Wnt signaling pathway resulting from the APC mutation and is
regarded as the earliest genetic lesions to induce cell
transformation (19). Thus,
understanding the signaling pathways underlying the
adenoma-carcinoma sequence is essential for the identification of
novel biomarkers for diagnosis and targeted therapeutics for CRC
treatment. The present review article discusses various incidences
and events linked with the development and progression of CRC and
dysregulation in signaling pathways (Wnt, epidermal growth factor
receptor (EGFR), PI3K/AKT, vascular endothelial growth factor
(VEGF), hepatocyte growth factor (HGF)/mesenchymal-epithelial
transition factor (cMET), Notch, Hedgehog, Hippo, NF-E2-related
factor 2 (Nrf2) and immune checkpoint) that can cause malignancy.
An overview of multiple targeted therapeutics that may help
attenuate the course of the disease is also presented.
The acquisition of genetic and epigenetic
aberrations leads to the transformation of normal cells into benign
lesions, which later become malignant. CRC arises as adenocarcinoma
from glandular epithelial cells of the large intestine comprised of
the colon and rectum. The development of CRC may take several years
by establishing the dysregulation of several signaling pathways and
avoiding multiple regulatory routes. Malignant cells arising in the
large intestine constitute CRC. This includes the colon and rectum
and since these include common features, it is grouped and termed
'colorectal cancer'. CRC is the most prevailing cancer of the
gastrointestinal tract. The growth of the majority of CRCs begins
in the innermost colon linings or rectum in the form of polyps. Not
all polyps are cancerous; however, depending on their type, over
time, some polyps can become cancerous. There are 2 main types of
polyps: i) Adenomatous polyps, which are termed 'pre-cancerous' as
they can sometimes develop into cancer; and ii) hyperplastic polyps
(HPs) and inflammatory polyps, which are common, and they are
generally not pre-cancerous. Several other factors can increase the
risk of polyps developing into CRC, such as: If the polyp size
increases by >1 cm, if the number increases by >2, and if
dysplasia occurs following polyp removal.
As has long been considered, CRC develops using the
classical pathway of adenoma to carcinoma route (20). Recently, another alternate pathway
was coined as the serrated pathway. In this pathway, HPs were
regarded as insignificant and only adenomas were responsible for
CRC; accumulating evidence indicates that serrated polyps may form
precursors to CRC, as well as through the serrated neoplasia
pathway (21). Currently,
patients with CRC with several serrated polyps classified as
serrated polyposis syndrome have been demonstrated to have an
increased risk of developing CRC (22). Small tumors are diagnosed within
serrated lesions. It has been suggested that 10-30% of CRC cases
develop from the serrated neoplasia pathway (23). Longacre and Fenoglio-Preiser
described serrated adenomas for the first time (24). Serrated polyps are heterogeneous
lesions histologically marked by glandular serration. Colonic
epithelial cells from crypts display luminal saw-toothed
morphology. In 2010, the WHO classified serrated polyps into 3
groups: i) HPs; ii) sessile serrated adenoma/polyps; and iii)
traditional serrated adenoma (TSA) (25). Three-quarters of serrated polyps
constitute HPs. HPs establish earlier than traditional adenomas;
however, after 50 years, their occurrence does not increase
significantly (26,27). They develop as flat, sessile and
pale lesions of approximately 5 mm in diameter and are
etiologically located at the end of rectal mucosa folds. Sessile
serrated adenoma/polyps (SSA/P) represent 15-20% of all serrated
polyps and these lesions are either flat or slightly elevated
located in the proximal colon (28,29). TSAs are not very common polyps and
constitute up to 5% of serrated polyps. They are found in the
elderly and are located on the left side of the colon (30).
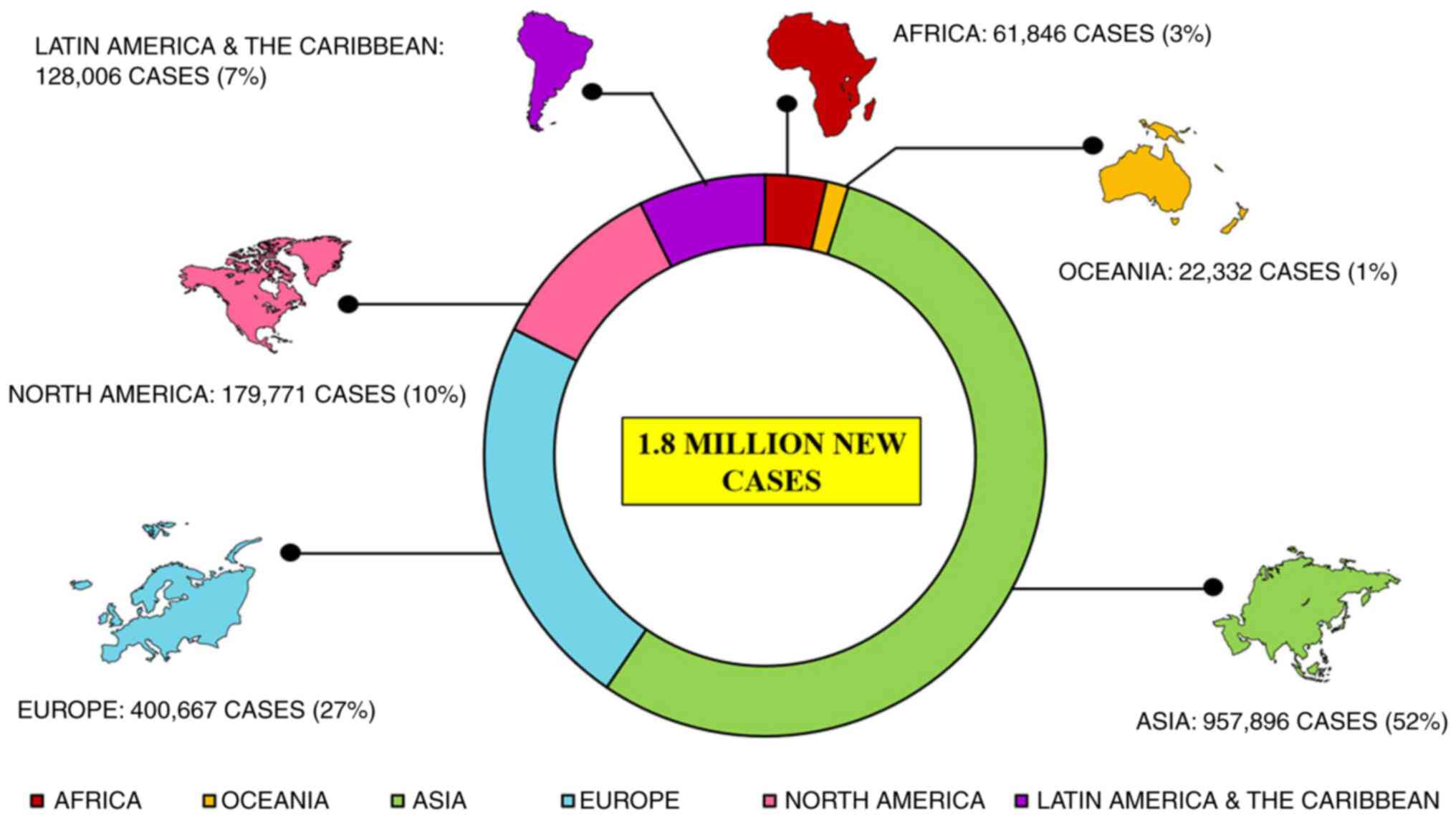
CRC is the third most major type of cancer diagnosed
in both sexes globally. Approximately 1.8 million new cases are
reported annually that account for approximately 10% of all common
cancers investigated globally, leading to approximately 9 million
fatalities in 2018 itself that is 9.2% of all the cases
investigated globally (31), as
per the International Agency for Research on Cancer report 2018.
Reports suggest a large topographical variation in the incidence
and mortality of CRC among several countries worldwide (Fig. 1) (7). CRC is more prevalent in developed
countries than economically transitioning countries (Brazil,
Slovakia and China) where incidence rates have increased; however,
the overall risk of CRC remains low. Incidence rates are decreasing
with a higher human development index (HDI; North America and
Europe), trailing a peak (USA, New Zealand and France), or
increasing (Spain, Italy and Norway). In Saudi Arabia, CRC is the
most common type of cancer in males (19.6%), while the third most
common cancer in females (9.5%), causing highest cancer-related
mortality (32). Several
countries have taken major initiatives, such as screening,
resulting in the early detection of CRC along with better treatment
management that has decreased mortality rates. However, some
countries still need to improve the screening process with a more
effective medical set up, so that CRC can be detected at an early
stage and thus treatment can be improved. Statistics suggest a
spike in CRC growth and mortality rates after 50 years of age. An
estimated 90% of worldwide cases and deaths have been observed
after this age. It is also noteworthy that the incidence rate in
males is 30% higher compared to females, with wider variations for
rectal cancer (60% higher) than for colon cancer (30% higher).
Females also exhibit a lower susceptibility to malignancy overall.
Older females, however, (≥50 years) are more prone to developing
adenomas in the proximal colon than males. Sex inequalities and
lifestyle habits follow differences in exposures to risk factors,
such as smoking and sex hormones, as well as complex interactions
between these factors.
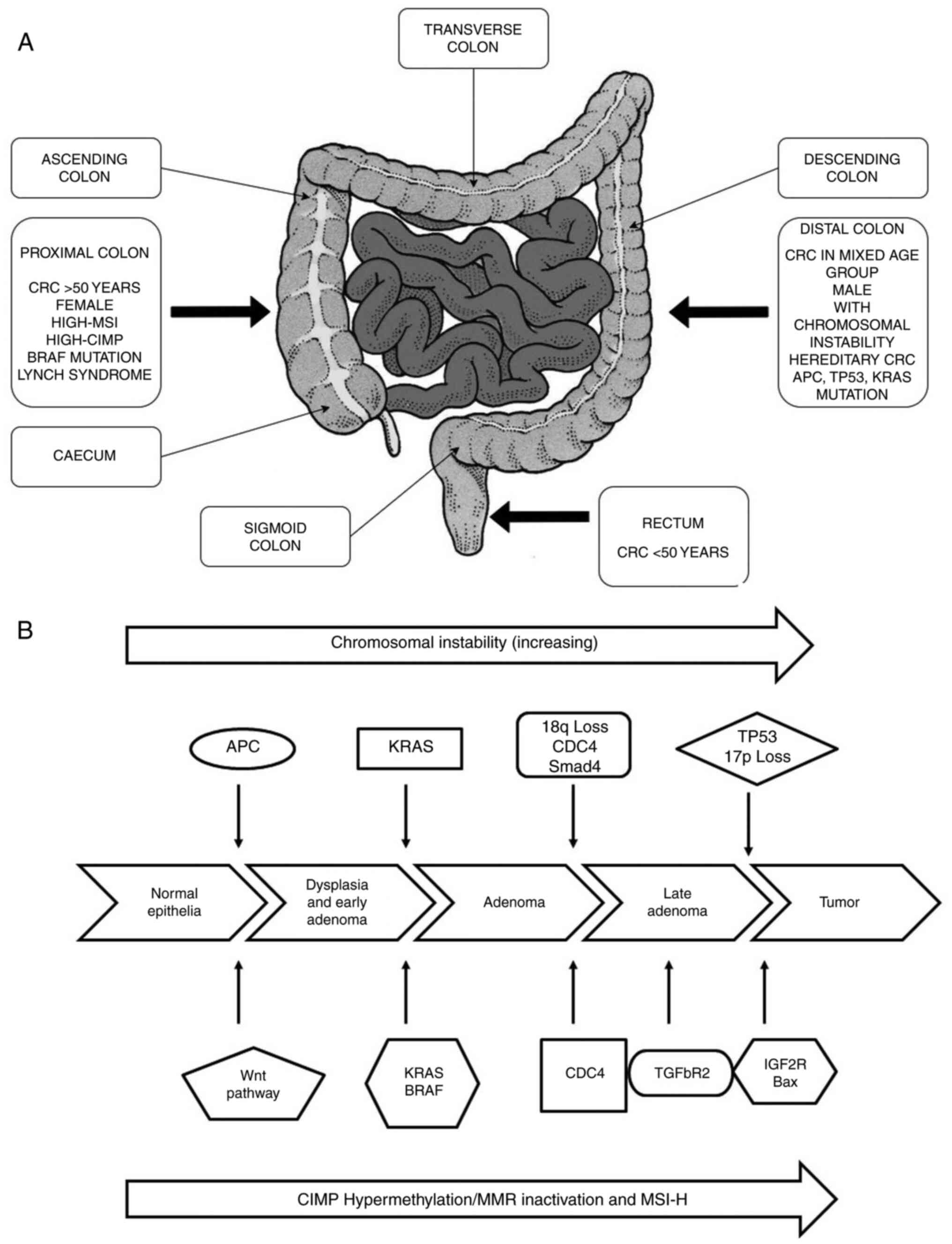
The majority of CRCs are adenocarcinomas, a type of
tumor that contributes to 96% of colon and rectal cancers. These
adenocarcinomas line the inside of the colon and rectal tissues.
Despite its occurrence in the large intestine, CRC is a deeply
heterogeneous disease with subtype variations, causes and clinical
outcomes. Depending on its anatomical site, CRC subtypes have been
divided into 3 segments: Proximal colon, distal colon and rectal
cancer (Fig. 2A) (33). The proximal and distal colon
located within the peritoneal cavity and the rectum lies within the
pelvis. The embryo of the proximal colon usually begins from the
midgut, whereas the distal colon contains segments from the splenic
flexure to the upper anal canal and rectum appears from the
hindgut. These subtypes are comprised of branches of the superior
and inferior mesenteric artery, respectively. Several studies
reveal that CRC subtypes present at different anatomical sites
possess distinct risk factors i.e., smokers are at an increased
risk of proximal colon cancer and rectal cancer (34). Due to etiological heterogeneity of
CRC with tumor locations, major functions, such as nutrient
absorption and fecal storage of the colon and rectum occurs in
distinctly different segments of the large intestine, for example,
sodium and water absorption rates are highest in the cecum and
decrease progressively towards the rectum. This is also due to the
functional variations of different segments of the large intestine.
Demographic factors also contribute to the risk associated with
CRC. The European Prospective Investigation into Cancer cohorts
places females at a higher risk of proximal colon cancer (34%)
compared to males (25%) (34) and
this is age-dependent i.e., increases with age (35% for individuals
<60 years of age to 60% for individuals >70 years of age)
(35). It has also been observed
that subtypes of CRC are widely distributed based on ethnicity:
Proximal colon cancer is more prevalent among Caucasians and
individuals of African origin (USA: Proximal colon cancer accounts
for 44 and 49%; rectal for 29 and 25%; and distal for 27 and 26%
among Caucasians and African-American individuals, respectively)
(36). In Asian countries, such
as Korea, rectal cancer is more prevalent (rectal, 52%; proximal,
22%; and distal, 26%) (37),
whereas, 32-34% of each subtype CRC cases are uniformly shared
among Asian-Pacific Islanders (API) across the 3 colorectal
sections (36).
The majority of CRC cases are sporadic with no
family history or a predisposition to illness in individuals,
although almost 1 out of 3 individuals with a family history of CRC
develop the disease. The average lifetime risk of developing CRC in
most western populations is in the range of 3-5%. However,
individuals with a family history of CRC in first-degree family
members diagnosed at 50-70 years of age are at a double risk; the
risk triples if the relative is <50 years of age at the time of
diagnosis. The risk of developing CRC increases with ≥2 diagnosed
first-degree family members at any stage. There may be several
reasons for the increased risk, such as genetics, environmental
factors, or a combination of both. Approximately 5-10% of the
specific subgroup of individuals who develop CRC symptoms have
inherited gene mutations that cause hereditary cancer syndrome,
rendering them prone to developing the disease. It has been
observed that FAP and HNPCC also known as Lynch syndrome, are the
most prevailing inherited syndromes linked to CRC, accounting for
approximately 2-4% of all CRC cases (38).
HNPCC is the most widespread inherited colorectal
syndrome with an estimated 1 in 3,000 affected individuals in
western populations (39).
Germline mutations in any of the mismatch repair genes, namely
MLH1, MSH2, MSH6, PMS2 and epithelial cell adhesion molecule
(EPCAM) have been reported to be associated with HNPCC. Generally,
tumors harbor MSI (40), which
primarily occurs due to the inability to rectify strand slippage
within repetitive DNA sequences, leading to alterations in the
mononucleotide or dinucleotide repeats, thus also altering the size
and arrangement of microsatellites, which are strewn throughout the
genome (12). This can be
identified by a PCR test and immunohistochemical analysis, which
can pinpoint the loss of expression of the mismatch repair protein
factors (40-43). Individuals affected with
'non-polyposis' tend to have polyps from the defined spectrum,
leading to cancer progression within 2-3 years, as compared to the
8-10 years of the 'normal' community.
FAP is the second major type of hereditary CRC
syndrome, which accounts for >1% of all CRC cases (44). This syndrome affects almost 1 in
11,300-37,600 individuals in the European Union (45) and is caused by hereditary germline
mutation in the APC gene, which regulates the activity of the Wnt
signaling pathway (46). Unlike
patients with HNPCC who develop a few adenomas, patients with FAP
tend to develop numerous adenomas, primarily in the distal colon at
a younger age (44). Among the
numerous adenomas in FAP, one or more adenomas undergo malignant
transformation, virtually increasing the risk of developing CRC to
100% by 40 years of age, unless the colon, or sometimes the rectum,
is not removed (47).
The incidence of EOCRC is increasing in countries,
such as the USA and Canada at an alarming rate, becoming the second
most common type of cancer, and the third most common cause of
cancer-related mortality in individuals <50 years of age
(48). Over the past 4 decades,
the incidence of EOCRC has increased rapidly and by the year 2030,
it is further expected to increase by >140% (49,50). The incidence rates are inversely
associated with age i.e., increasing significantly in younger
individuals and decreasing in older individuals. Up to 13% of cases
of EOCRC have been reported to be linked to a germline mutation in
mismatch repair genes mentioned above, underlying hereditary CRC
syndromes (51). Although
early-onset CRC patients have a higher risk of the prevalence of
hereditary syndrome, approximately half the patients with
early-onset CRC do not have any family history of the disease
(52). Studies have indicated a
prominent difference in pathological characteristics between the
elderly and younger groups of CRC patients (53), demonstrating an increase in EOCRC
at a younger age, but often being detected at a higher stage
(54,55). CRC diagnosed in younger patients
is commonly symptomatic, at a later stage, mucinous and involves
poorly differentiated tumors (56-58). The cancer-specific survival of
younger individuals is markedly higher than older individuals
(53). No statistically
significant difference in the disease-free survival between these 2
groups has been demonstrated over the past 5 years (63.2% in both
the young and elderly group) (53). Between 1992 and 2014, there was a
significant increase in the number of young male patients with CRC
than older patients (53.3% in the young and 49.7% in the older
group). Compared to patients of a screening age (≥50), younger
patients with CRC were mostly of African origin (16.8 vs. 10.4%),
Asian/Pacific Islander (API) (5.6 vs. 2.2%) and Hispanic (13.1 vs.
6.0%). In younger patients with CRC, there are high proportions of
rectal (39.5 vs. 27.7%) and distal colon (43.9 vs. 33.8%) cancers
as compared to elderly patients with a higher percentage of
proximal colon cancers (58.2 vs. 48.2%) (54). However, the reason for EOCRC
remains unclear, although certain risk factors, such as prolonged
exposure to carcinogens and early childhood exposure serve as
critical determinants of risk (31).
Several other physical factors, such as body weight,
age, sex, body mass index (BMI) and lifestyle behaviors, such as
smoking, etc. have also been associated with EOCRC. Weight loss may
be an early symptom of EOCRC (59). Furthermore, females with a BMI of
>30 have a higher risk of developing EOCRC (95% CI, 1.15, 3.25)
compared to those with a normal BMI (60). Low et al suggested that
smoking is not associated with the risk of EOCRC; neither current
nor former smokers are at a risk of developing CRC as compared to
non-smokers (59). Rectal cancer
has a higher chance of developing into EOCRC than colon cancer,
whereas, in late-onset cases, obesity is the major risk factor for
colon cancer (61,62). These differences indicate several
factors associated with EOCRC and further studies are required to
identify the major associated risk factors.
CRC is a heterogeneous disease, resulting from the
continuous accumulation of both genetic and epigenetic alterations
within cells, leading to the transformation from a colorectal
adenoma to a colorectal adenocarcinoma. This transformation is
associated with 3 major pathways of genome instability, namely CIN,
MSI and CIMP, of which the latter 2 fall under the alternate
serrated pathway (Fig. 2B)
(63). The most common is CIN
(chromosomal number and structural alterations) that leads to
karyotyping variability among cells (64). The second most common pathway is
MSI (molecular alteration and hyper-mutable phenotype), which
constitutes approximately 15-20% of CRC (65) caused by defective DNA mismatch
repair system (66). MSI can be
defined as 'alterations in the number of repetitive DNA in
microsatellites (also known as short tandem repeats) throughout the
genome sequence (67). The third
pathway contains a high density of methylated genes termed CIMP
cancers (68). This type of
cancer is mainly located on the proximal side of the colon (up to
40% of the cases) and appears in serrated polyps instead of
adenomas (69).
A number of studies have concluded that the majority
of human tumors are heterogeneous in nature forming a
'mutator-phenotype' due to continuous accumulation of multiple
mutations that occur during cell division in cancer cells that
generally function to maintain genetic stability (70). Both CIN and MSI are well defined
and detected by karyotype and PCR-based analysis, respectively;
however, due to difficulty in predicting random mutations,
particularly point mutations that limit the search for evidence of
a mutator phenotype at single base-levels (70), the 'mutator phenotype' may exhibit
various manifestations, including increased mutation rates and
genetic evolution of cancer cells that propel tumor progression
(71). The mutator-phenotype may
be an attractive target for cancer therapy due to common features
in the majority of cancers (72).
CIN appears in 70% of sporadic CRC cases. CIN causes
an alteration in chromosome number, involving gain or losses of the
whole or a large part of chromosomal aneuploidies, resulting in a
rearrangement of chromosomes (karyotyping) from cell to cell
(73). LOH and an imbalance in
chromosome number (aneuploidy) are the most common characteristics
of CIN. There are several techniques to measure CIN, such as
cytometry, LOH analysis, karyotyping, fluorescent in situ
hybridization (FISH) and a recently developed technique known as
comparative genomic hybridization (CGH). CGH utilizes DNA
microarray or 'chips' that are commonly used to detect copy number
variations (74). In this
advanced CGH microarray technique, cloned DNA fragments with
precise genomic positions are used against the metaphase
chromosomal arrangement in conventional CGH (75). It is not always upfront to
categorize tumors as CIN-positive vs. CIN-negative based on these
different methods and criteria rather different sub-categories of
CIN-high and CIN-low for CIN-positive tumors have been proposed in
several studies (76-78). Moreover, CIN contributing to CRC
tumorigenesis through the aggregation of mutations in specific
oncogenes, including B-Raf proto-oncogene serine/threonine kinase
(BRAF), KRAS proto-oncogene GTPase (KRAS), tumor protein p53 (TP53)
and tumor-suppressor APC gene (12,75). The multistep genetic model
proposal by Fearon and Vogelstein, which is now widely accepted,
examines the different stages of tumor development i.e., from small
adenomas to large adenocarcinomas (18). The model demonstrates the various
events occurring at different stages of tumor development i.e.,
from normal colorectal epithelium to metastatic carcinomas
(79). The first event is the
mutation of APC, transforming normal colorectal epithelium to
adenoma, followed by oncogenic KRAS mutation at early adenomatous
stage and ultimately inactivation of the tumor-suppressor gene TP53
on chromosome 17p and the deletion of chromosome 18q occurring
during the progression to malignancy (80-82). APC is a tumor suppressor gene
located at chromosome 5, which is responsible for familial
adenomatous polyposis constituting approximately 85% of colorectal
cancer cases without a hereditary relationship (83,84). APC and CTNNB1 (β-catenin) are the
most frequently mutated genes in CRC. APC mutation breaks the
association between APC and β-catenin, resulting in a large amount
of β-catenin in the cytoplasm and the overactivation of the Wnt
signaling pathway. Followed by tumor-formation promoting gene
translocation to the nucleus and interacting with other
transcription factors involved in tumorigenesis and invasion
(85). K-RAS is located on
chromosome 12 and is one of the most bulging proto-oncogenes in
colon carcinogenesis. RAS family proteins are involved in signal
transduction. K-RAS activates the mitogen-activated protein kinase
(MAPK) pathway eliciting the nuclear expression of early response
genes. RAF proteins are activated by the GTPase activity of RAS
(86). Thus, K-RAS mutations
result in colon cancer formation in the early adenomatous stage and
contribute to its formation by 37-41% (87,88). The allelic loss in chromosome 18q
is observed in 70% of primary CRC cases in the late-stage adenomas
(89) and exhibits a strong
association with a poor prognosis (90). The inactivation of
tumor-suppressor genes, including deleted in colon cancer (DCC),
SMAD2 and SMAD4 present on the q arm of chromosome 18 due to LOH
plays a significant role in CRC (18,82,91). The SMAD proteins are involved in
TGF-β signaling and in the regulation of genes involved in cell
cycle programming (92). Since
SMAD2 and SMAD4 are located on chromosome 18q, the loss of
chromosome 18q leads to the deregulation of the TGF-β signaling
pathway and contributes to colorectal carcinogenesis (93,94). DCC is localized in the chromosome
band 18q21.2 and is deleted in approximately 70% of the cases
(95). However, no evidence
supports the role of DCC in colorectal tumorigenesis (96). Patients with 18q LOH (70%) have an
increased 5-year survival rate in stage II than those without 18q
LOH (43%), which leads to the analysis of the impact of adjuvant
therapy in stage II (97). There
is an increase in the 5-year survival rate of patients with 18q LOH
receiving adjuvant therapy compared to those without 18q LOH (90
vs. 37%; P=0.01) (97). The TP53
gene located on the short arm of chromosome 17p13.1 consists of 11
exons and 10 introns and is commonly lost in colorectal carcinoma
(20,98). The TP53 mutation is most common in
human cancers with 43.28% in CRC resulting in the loss of tumor
suppressor activity or (gain of function) to support tumor
progression (99). The majority
of mutations occur in exon 5 to 8 (DNA binding domain) (100,101). To date, the majority of TP53
mutations detected in CRC are missense mutations with AT for GC
substitution (102). p53 is
known as the 'guard of the genome' due to its ability to respond to
mutagenic stress, such as DNA-damage and repair, cell cycle arrest
and apoptosis (103). It also
inhibits the development of new blood vessels (angiogenesis)
through the induction of TSP1 (104). However, a mutation in p53 leads
to oligomerization of the wild-type and mutant p53, which can block
the function of TP53, resulting in the loss of DNA binding
specificity (105).
The second most common genomic instability is the
hyper-mutable phenotype (MSI) (106). MSI generally occurs due to
damaged mismatch repair (MMR) along with the slippage of DNA
polymerase which creates a short-term insertion-deletion loop (IDL)
(106). These defects result in
the alteration of the size of the allele as compared to those
detected in the normal cells of the same individual. The DNA MMR
system has several proteins (such as MLH1, MSH2, MSH6 and PMS2)
that repair single base pair mismatch, incorporated into
micro-satellites during DNA synthesis to maintain genomic
stability. MSI CRC mostly occurs in the proximal colon (107). Several studies have examined MSI
a prognostic biomarker for CRC. The Bethesda guidelines proposed
the first panel of MSI markers consisting of 5 microsatellite
markers viz. mononucleotides (BAT25 and BAT26), and dinucleotides
(D2S123, D5S346, and D17S250) to access the status of CRC (108). CRC can be classified based on
the percentage of loci with MSI. In particular, >30% of unstable
markers are classified as CRC with MSI-high (MSI-H), those with
<30% markers exhibiting instability are termed MSI-low (MSI-L),
and markers with no instability are termed microsatellite stable
(MSS) (109). Defects in MMR
genes occur either by mutational inactivation or by epigenetic
silencing of CpG island hyper-methylation of the MLH1 gene promoter
(9).
The results of immunohistochemistry of CRC reveal
the interaction between MMR proteins PMS2 and MSH6 with other
repair factors, such as MLH1 and MSH2, respectively. Therefore, the
inactivation of MSH2 is frequently associated with the loss of the
expression of MSH6, which is highly acceptable in MSH2 germline
mutation. Similarly, MLH1 inactivation is frequently associated
with the loss of expression of PMS2, which may result either from
MLH1 germline mutation or by the epigenetic silencing of CpG island
hyper-methylation of the MLH1 gene promoter. Germ-line mutations of
MSH6 and PMS2 are generally associated with the individual loss of
expression of MSH6 and PMS2 proteins, respectively (110).
CIMP represents a subset of CRC that contains a high
density of hyper-methylated genes, causing transcriptional
silencing within the promoter region, resulting in the loss of gene
expression (111). CIMP
contributes to approximately 30-35% cases of colorectal adenomas,
occurring at an early stage and as a precursor to the serrated
pathway of colorectal tumorigenesis (112-114). CpG island hypermethylation
present in the tumor suppressor gene promoter region results in
gene silencing. The hypermethylation of MLH1 leads to its silencing
and dysregulates MMR (mismatch repair) function (114). CIMP constitutes a prominent
molecular characteristic of the serrated neoplasia in 20-30% of CRC
cases (115,116). CIMP has also been found
histologically in patients with hyperplastic polyposis syndrome,
suggesting that it is an important early event of the serrated
neoplasia pathway (117). The
inactivation of MLH1 by hypermethylation leads to the induction of
MSI-H followed by additional mutations in MSH3, MSH6, Bax,
insulin-like growth factor 2 receptor (IGF2R) and phosphatase and
tensin homolog (PTEN), resulting in the development of dysplasia
and cellular transformation (118,119). Studies suggest that the serrated
pathway is responsible for the rapid development of CRC, as
compared to patients with CRC with Lynch syndrome (120,121).
Risk factors increase the chance of acquiring a
disease. Several factors, such as environment and lifestyle have
been associated with the increased occurrence of CRC (Table I). Smoking, an increased BMI,
intake of red meat, lack of regular physical activity and poor
diets are associated with an increased risk of CRC (122). Various studies show that
approximately 12% of CRC-related deaths are due to cigarette
smoking. Tobacco smoke contains at least 70 chemicals classified as
carcinogens. Smoking is associated with the early onset and distal
location of CRC in males (123).
The relative risk of CRC due to prolonged heavy smoking is 1.18
(95% CI, 1.11-1.25) (124). A
previous study found larger polyps in the colon and rectum of
long-time heavy smokers (123).
Additionally, patients with Lynch syndrome (also known as HNPCC),
who also smoke regularly, are at a higher risk of developing CRC as
compared to former smokers, short-term smokers and light smokers
(125). Smoking is strongly
associated with serrated polyps (relative risk (RR), 2.33 (95% CI,
1.76-3.07), particularly in the left side of the colorectum and a
weak association with adenomas (RR 1.31 (1.08-1.58) (126). Evidence points to the role of
epigenetic modification in smoking-related CRC. Smokers with MSI-H
tumors (RR, 1.99; 95% CI, 1.26-3.14), CIMP-positive tumors (RR,
1.88; 95% CI, 1.22-2.90) and BRAF mutation-positive tumors
(RR, 1.92; 95% CI, 1.22-3.02) are at a higher risk of developing
CRC (127). In a cohort study
from the USA, former smokers that had quit smoking prior to 40
years of age or had quit for ≥30 years, were at no risk of
developing CRC (128). A
previous meta-analysis revealed a significant association of
smoking cessation with improved an overall survival (HR <10
years, 0.78; 95% CI, 0.69-0.88; HR ≥10 years, 0.78; 95% CI,
0.63-0.97) and CRC-specific survival (HR ≥10 years, 0.76; 95% CI,
0.67-0.85) as compared to smokers who had not quit (129).
Several studies have associated alcohol consumption
with an increased risk of developing CRC. Alcoholic beverages
contain reactive metabolites known as acetaldehydes, which can be
carcinogenic and mutagenic and are responsible for
alcohol-dependent carcinogenesis (130). A meta-analysis of 27 cohorts and
34 case-control studies observed that there was a significant
increase in the risk of developing CRC for moderate (2-3 drinks per
day; RR, 1.21; 95% CI, 1.13-1.28) and heavy drinkers (≥4 drinks per
day; RR, 1.52; 95% CI, 1.27-1.81), as compared to non-drinkers
(131). Nevertheless, another
meta-analysis published in 2018 on 14 cohorts in North America,
Europe and Asia revealed a significant increase in the risk of CRC
for light drinkers (≤1 alcoholic drinks per day) as compared with
non-drinkers/occasional drinkers (132). The increased risk of CRC is
generally higher in males than in females, possibly due to the
higher alcohol consumption (133). In 2012, the worldwide incidence
and mortality rates of all cancer cases due to alcohol consumption
were 5.5 and 5.8%, respectively (134). A total of 3.5% of all
cancer-related deaths in the USA are due to alcohol consumption
(135).
Alcohol dehydrogenase is a key metabolic enzyme that
metabolizes ethanol to acetaldehyde, which is then converted into
acetic acid via aldehyde dehydrogenase (ALDH). The polymorphism
ALDH2*2 in ALDH2 leads to an increased circulation of acetaldehyde,
that can reach colonocytes. As compared to other parts of the
world, the ALDH2 variant is very frequent among populations in East
Asian. According to pooled studies in Japan, the relative risk
associated with >45 g/day consumption of alcohol was 2.09 (95%
CI, 1.65, 2.64), but 1.41 (95% CI, 1.16-1.72) in Europe and North
America (133). A meta-analysis
found that obnoxious symptoms of ALDH2 carrier that may be
preventing them from consuming alcohol, thereby, reducing the risk
of CRC by approximately 20%.
Diet is strongly associated with the risk of
developing CRC, with studies showing a 70% risk reduction by a
change to a healthier diet and acquiring healthy food habits
(135). Patients who consume a
high-fat diet, particularly red meat, have been shown to have a
higher risk of developing advanced CRC (136,137). Colon cancer exhibits a stronger
association with the consumption of meat than rectal cancer
(138). The mechanistic link
with the positive association of the consumption of red meat with
CRC is the presence of heme iron in the former (138,139). Meat cooked at a high temperature
produces heterocyclic amines and polycyclic aromatic hydrocarbons,
which are considered to possess carcinogenic properties (138,140). Individuals consuming a diet rich
in calcium (dietary and supplements), fruits, fiber and vegetables
are at a decreased risk of developing CRC (141,142).
Overweight and obese individuals are at a higher
risk of fatality, with this being the fifth leading cause of
cancer-related mortality. Approximately 2.8 million adults die of
obesity-related cancer each year (143). In Europe, approximately 11% of
CRC cases are associated with obesity and being overweight
(143). Researchers have found a
positive association between excess weight and cancer in both
sexes; however, males were found to have a higher risk. This was
attributed to lower testosterone levels in older males as compared
to post-menopausal women with higher estrogen levels (144). Various studies found a
significant positive association between CRC and BMI (145,146); the overall RR for CRC predicted
per 1 kg/m2 of higher BMI was 1.03 (95% CI, 1.02-1.03)
(147). BMI expresses overall
body fat and waist circumference (WC), representing abdominal fat;
studies have reported that WC, more than only the BMI, is strongly
connected to an increased risk of CRC (148,149). Abdominal fat is divided into 2
categories: Visceral adipose tissue (VAT) and subcutaneous adipose
tissue (SAT). VAT secretes higher levels of pro-inflammatory
adipokines (such as TNF) and lower levels of adiponectin (an
insulin-sensitizing hormone) as compared to SAT (150). Visceral obesity is more common
among Asian populations than Caucasian populations for any given
BMI (151). Evidence suggests a
stronger association with obesity in males than in females, colon
cancer over rectal cancer and distal cancer over proximal colon
cancer (152). A previous
meta-analysis reported a link between abdominal obesity and an
increased risk of colorectal adenomas (RR, 1.42; 95% CI, 1.30-1.56)
(153). Another meta-analysis
predicted a higher risk of developing CRC among diabetic patients
(21%; 95% CI, 1.02-1.42) as compared to non-diabetic individuals
(154). Obesity can also cause
hyperinsulinemia and insulin resistance (155) due to the lower expression of
insulin-receptor levels and decreased intracellular insulin
signaling in response to insulin receptor binding (156). This results in an escalated
release of insulin, and lower insulin sensitivity, leading to an
increase in free insulin-like growth factor 1 (IGF1). IGF is
involved in the maintenance of tissue homeostasis, differentiated
phenotype, growth regulation, proliferation, apoptotic imbalance,
angiogenesis, migration, cell adhesion and wound healing (157). The insulin/IGF1 signaling
pathway promotes colorectal carcinogenesis by decreasing apoptosis
and increasing cell proliferation (158). After menopause, adiposity
becomes the main spot for estrogen production in women, protecting
them against susceptibility to CRC (159,160). Thus, cancer caused due to
insulin and IGF1 in overweight/obese elderly women could be
counteracted by the anticancer effects of estrogen (7).
An increased intake of dietary insoluble-fiber
lowers the risk of colorectal epithelium carcinogenesis in the
lumen by increasing fecal bulk, diluting fecal content, and
decreasing transient time (47).
Research has demonstrated a lower risk of developing CRC among
rural Africans compared to Western populations, due to a higher
fiber intake by the former (161). A nested case-control design
predicted the association between the incidence of CRC and dietary
fiber intake, concluding that cereal fiber and whole grains having
a high dietary fiber content were inversely associated with the
risk of CRC (RR for 10 g per day increment, 0.90; 95% CI,
0.83-0.97) as compared to fiber from fruits, vegetables and legumes
(162). In their report, the
World Cancer Research Fund (WCRF) and the American Institute for
Cancer Research (AICR) added whole grains as a possible protective
agent against CRC (163).
CRC is one of the few types of cancers that strongly
suggests the absence of physical activity as a risk factor
(164). It has been demonstrated
that physical activity is inversely related and sedentary
lifestyles are positively associated with the risk of CRC (165). A cohort study reported the
benefits of aerobic exercise against digestive system cancers (of
which CRC contributed to 56%) with optimal levels detected at
approximately 30 metabolic equivalent of task (MET) hours/week (HR,
0.68; 95% CI, 0.56-0.83) (166).
Regardless of the level of physical activity, sedentary activities,
such as prolonged periods of sitting are strongly associated with
an increased risk of CRC. For an increase of 2 h per day of
television watching, the RR was 0.07 (95% CI, 1.05-1.10;
P<0.001) (167). Sedentary
behavior results in weight gain in CRC survivors (168). In 2008, a prospective cohort
study concluded that physical exercise or sports activity >5
times per week was associated with a lower risk of developing colon
cancer among males (P=0.001; RR, 0.79; 95% CI, 0.68-0.91) and
females (P=0.376; RR, 0.85; 95% CI, 0.70-1.04) as compared to very
limited or no activity at all (169).
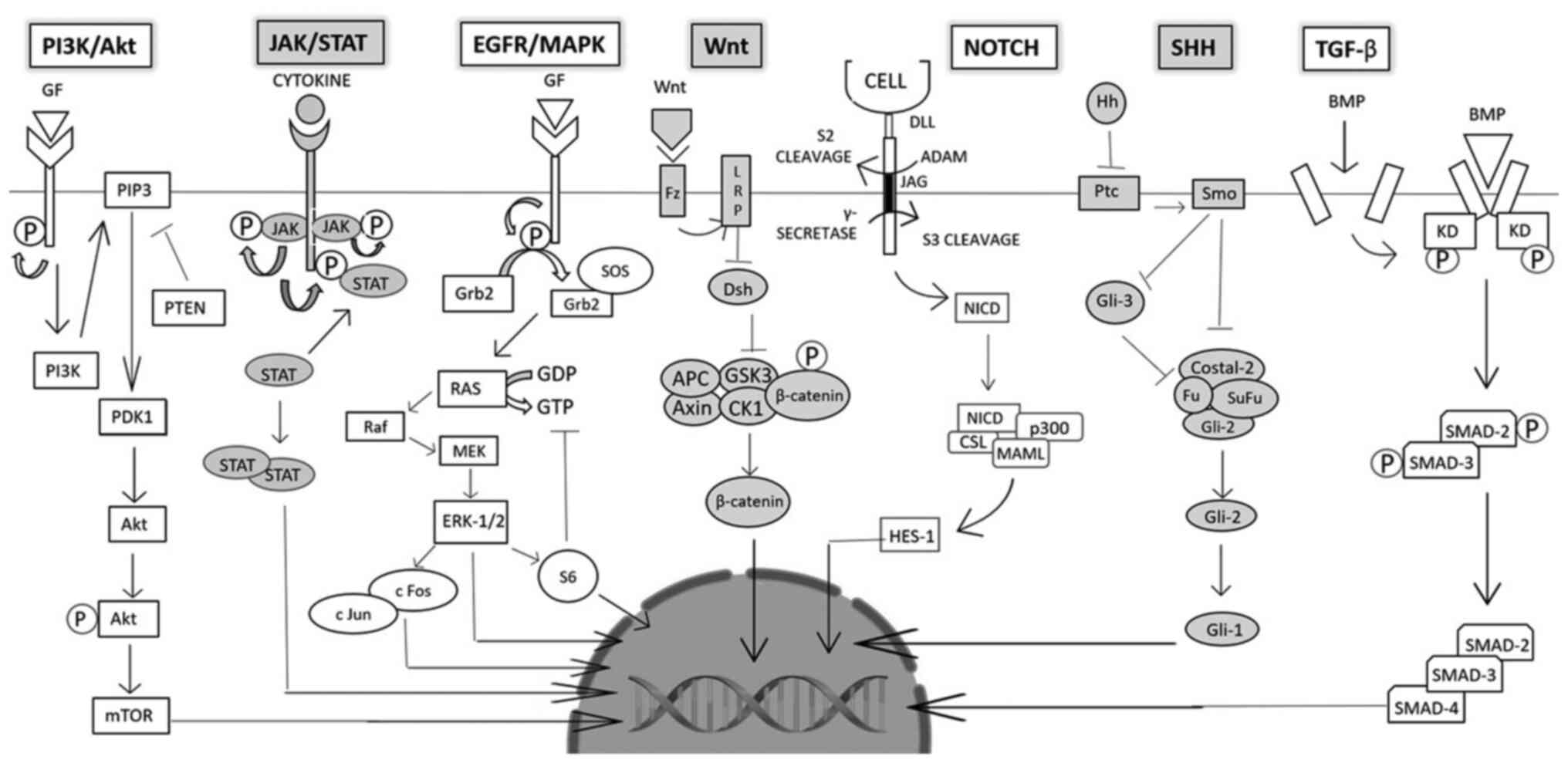
Intestinal epithelial cells renew constantly and are
tightly regulated by several pathways (Fig. 3). Mutations in these pathways can
lead to unchecked growth/delayed or failed apoptosis of epithelial
cells, encouraging tumor formation, survival, angiogenesis and
metastasis. The understanding of these pathways as targets of gene
therapy to combat CRC is underway. While the dysfunction of a few
growth and differentiation pathways may result in CRC,
understanding these mechanisms may help in the prevention of tumor
formation.
The Wnt/β-catenin pathway is highly conserved as it
is essential to embryogenesis. Wnt proteins are growth stimulatory
factors (the attached palmitoleic acid assisting in
protein-binding). These proteins exhibit an abnormal cellular
expression in patients with CRC. There are 19 Wnt genes present in
mammals and all play regulatory roles in several biological and
developmental processes, such as cell fate determination, cell
cycle, proliferation and migration. Membrane surface cell receptors
comprise frizzled (Fz) and low-density lipoprotein (LDL)
receptor-related protein (LRP) complexes at the cell surface. Along
with this, there exists an intracellular complex comprising of
several proteins, such as β-catenin, dishevelled (Dsh), axin,
glycogen synthase kinase-3β (GSK-3) and APC. The protein complex
regulates the level of β-catenin in the cell by proteasomal
degradation. Following phosphorylation and ubiquitination (by
β-trcp) of β-catenin, the transcriptional regulator is degraded by
the cellular proteasome. Upon ligand-binding, the degradation
process is inhibited, leading to the accumulation of active
phosphorylated β-catenin in the cell. The β-catenin then
translocates into the nucleus and induces transcription. Mutations
in the APC gene can lead to colon cancer (reported in 90% of
cases). The overexpression of Wnt is associated with tumorigenic
activity and encourages tumor growth. Mutations in the
Wnt/β-catenin pathway lead to CRC development (170). This pathway also plays an
essential role in tissue regeneration of hair, skin, intestine,
etc. (171). The dysregulation
of the Wnt pathway has been reported in a number of tumors,
including CRC. The hyperactivation of this pathway is imperative
for oncogenesis, leading to CRC development. Targeting
Wnt/β-catenin can be effectively used for the development of small
molecules (172-175).
A catalytic receptor tyrosine kinase (RTK), EGFR, is
present on the cell surface, having an extracellular ligand-binding
domain. EGF acts as a ligand and binds to EGFR, resulting in the
autophosphorylation of the tyrosine residues on the intercellular
side of the transmembrane protein. This offsets a chain of cellular
events; an adaptor molecule Grb-2 interacts with the phosphorylated
tyrosine through its SH2 domain, followed by interaction with the
son of seven-less protein (SOS) through the SH3 domain of Grb-2.
SOS, a guanine nucleotide exchange factor, enables the conversion
of GTP from GDP on the RAS molecule, thereby activating it.
Activation initiation results in a kinase cascade, activating
mitogen-activated protein kinase kinase kinase-Raf (MAPKKK),
mitogen-activated protein kinase kinase-MEK (MAPKK) and MAPK or
extracellular signal-regulated kinase (ERK) in turn through
phosphorylation. ERK regulates cellular events, such as the
proliferation and survival of cells by targeting cytoplasmic or
nuclear substrates. Cytoplasmic substrates include c-fos and c-Jun
(dimerized by MAPK) which enter the nucleus and interact with the
AP-1 motif of the DNA, initiating transcription. ERK also
phosphorylates cytoplasmic substrate, ribosomal S6 kinase (RSK).
The S6 protein can perform one of two functions including, negative
regulation of the SOS molecule (effectively turning 'off' the
signaling pathway by inhibiting the conversion of GTP from GDP) or
entering the nucleus and regulating the CREB transcription factor.
MAPK may also directly regulate the nuclear substrate MYC.
Inactivation of the pathway can also occur through the hydrolysis
of GTP through the GAP protein.
The MAPK pathway engages in various cellular
processes such as growth, proliferation and survival of cells. The
deregulation of the pathway results in the stimulation of growth,
survival, angiogenesis and metastasis of neoplastic cells. The
mutation of the K-Ras gene has been reported in early cancer stages
in almost 40% of CRC cases. Abnormal regulation, amplification,
increased copy number and the overexpression of EGFR promoting MAPK
activation has been reported in cases of CRC and is being studied
as a possible and promising target for treatment (173-175).
The PI3K pathway is associated with cell growth,
proliferation and differentiation. The enzymatic receptor tyrosine
kinase upon ligand-binding, autophosphorylates and activates
phosphatidylinositol 3-kinase (PI3K) that has two subunits: p85 and
p110. PI3K then, in turn, phosphorylates lipid protein,
phosphatidylinositol-4, 5-biphosphate (PIP2) to
phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 signals
proteins, such as 3-phosphoinositide-dependent protein kinase 1
(PDK1) that activates protein kinase B (AKT/PKB) by acting upon its
serine and threonine residues. AKT may be of 3 subtypes (AKT-1,
AKT-2 and AKT-3) depending upon whether it has been encoded by
PKBα, PKBβ, or PKBγ, respectively. AKT targets downstream proteins,
such as mammalian target of rapamycin (mTOR), which is responsible
for cell cycle progression, proliferation, delayed apoptosis,
growth and survival. Phosphatase and tensin homolog protein (PTEN)
downregulate the pathway by dephosphorylating PIP3. PTEN is also a
tumor-suppressing molecule. The aberrant expression of the pathway
(inability to switch-off) results in continuous and unchecked
growth and survival of cells leading to cancer. PI3K consists of 3
classes, of which type class 1A is the most prevalent. The abnormal
expression of PI3K accounts for 30% of human cancers. Overall, it
is shown to serve as an oncogenic factor in the growth and
development of CRC. The overexpression of phosphorylated AKT has
been linked with cell division and the suppression of apoptosis in
70% of patients with CRC, along with the abnormal expression of
PTEN. Akt also targets downstream protein mTOR that has been shown
to favor angiogenesis and growth; research into the use of aspirin
(mTOR inhibitor) has demonstrated that it inhibits CRC progression
(173,175).
Angiogenesis is an essential process for the
formation of blood vessels contributing crucially to cancer
initiation, cell proliferation, and growth, metastasis, and
invasion. Identification of vascular endothelial growth factor
(VEGF-A) and the generation of monoclonal antibodies inhibitor
against VEGF-A led to the direct relationship between new blood
vessel formation and carcinogenesis (176). Various pro-angiogenic and
anti-angiogenic factors regulate angiogenesis like VEGF, FGF,
TGF-α, TGF-β, PDGF, and angiopoietins which are released from the
tumor microenvironment (177-179). The VEGF family of proteins is
comprised of 5 proteins namely, VEGF-A, B, C, D and placental
growth factor (PIGF). These proteins bind to VEGFR: VEGFR1, VEGFR2
and VEGFR3, a type of receptor tyrosine kinases on endothelial
cells. There are 2 non-tyrosine kinase co-receptors, neuropilin-1
(NP-1) and NP-2. The diverse network between VEGF and VEGFR,
VEGF-A, VEGF-B, and PIGF mainly contribute to angiogenesis.
However, VEGF-C and VEGF-D predominantly contribute to lymph
angiogenesis. VEGF-A and VEGF-B prominently bind to endothelial
cells and on some non-endothelial cells via VEGFR-1 and -2
(180). VEGFR-3 is expressed on
endothelial lymphatic cells and bind to VEGF-C and D with increased
affinity (181).
VEGFR-1 belongs to receptor tyrosine kinase family
protein known to be expressed on endothelial cells, inflammatory
cells and tumor cells. VEGFR-1 regulates mainly differentiation and
cell migration of endothelial cells and promotes epithelial cell
differentiation during the early angiogenic event; however, it has
an insignificant role in cell proliferation (182,183). Furthermore, VEGFR-1 activation
mediates the activation of several downstream pathways, such as
PI3K/AKT/MAPK/ERK in inflammatory cells, resulting in the
upregulation of inflammatory cytokine and interleukin (IL)
production, such as TNFα, IL-1β, IL-6 and IL-8, leading to cell
migration. VEGFR-1 function is still unknown and mainly plays a
regulatory role in the angiogenesis process. VEGFR-2 is a 200-230
kDa protein reported in its involvement in vascular formation.
VEGFR-2 is mainly expressed in blood and lymphatic epithelial cells
(183). VEGF-A binds to VEGFR-2
leads to activation of VEGFR-2 resulting in the activation of
several downstream pathways, such as RAS/RAF/ERK/MAPK and PLCγ
which promotes cell growth. The activation of VEGFR-2 also
activates PI3K-AKT signaling, leading to the regulation of cell
death (177-180,184). The binding of VEGF-C and -D to
VEGFR-3 results in lymphatic vessel formation (185,186). Activated VEGFR-3 activates
RAS-MAPK-ERK and PI3K-AKT/PKB pathways leading to differentiation,
proliferation, survival, and migration of lymphatic endothelial
cells (185-187). There is sufficient evidence to
indicate that VEGF levels and VEGFR activity are elevated and
considered to be associated with a poor prognosis in CRC (188). Elevated levels of VEGF are
reported in the early and late advanced stages of CRC (189,190). The interaction between
VEGF-VEGFR is regulated by K-RAS mutation, p53, Cox2 and hypoxia
resulting in cell growth and migration in CRC (190-193). The pro-angiogenic function of
this VEGF/VEGFR complex is critical at the primary site of tumor
enhancing progression and migration and at the metastatic site for
new vessel formation to promote cancer growth and survival.
Targeting this complex with anti-VEGF or anti-VEGFR therapy may
result in the depletion of tumor formation and metastasis.
HGF and cMET play an essential role in
proliferation, survival, drug resistance and metastasis (194-198). HGF is the only ligand known for
MET receptor tyrosine kinase and is secreted from mesenchymal
tissues. An increased expression of HGF in tissue and serum is
related to a poor prognosis in various solid tumors of the breast
and gastrointestinal tumors (199-201). Patients with CRC with advanced
disease symptoms are reported to have higher levels of serum HGF
(202,203). MET belongs to the transmembrane
receptor family known to express in hepatocytes, normal and
malignant epithelial and endothelial cells, neural cells and
hematopoietic cells (204-206). MET has been reported to be
overexpressed in various malignant tumors, such as hepatocellular
carcinoma, lung, breast, thyroid, kidney, gastric cancer and CRC
(207-213). Several studies have demonstrated
elevated levels of MET mRNA and protein in CRC during tumor
progression and metastasis (214-216). HGF binding to MET receptor leads
to the activation of MET signaling, which initiates various
downstream signaling pathways, such as MAPK-ERK, PI3K-AKT, JAK-STAT
and NF-κB, resulting in the regulation of hematopoiesis, wound
healing and organ regeneration (195-200). Aberrant HGF-MET axes are
comprised of gene amplification, overexpression, mutation, and
ligand-mediated auto and paracrine signaling during oncogenesis
(217). Other factors also
modulate the HGF/MET pathway. Recently, it has been reported that a
novel gene metastasis-associated in colon cancer 1 (MACC1) is a
crucial player of HGF-MET signaling and regulates cancer
progression and CRC metastasis (218). Increased levels of MACC1 have
been observed in primary and metastatic CRC tissues. HGF induces
the translocation of MACC1 from the cell membrane into the nucleus
and binds to MET promoter, leading to an increased MET expression.
The MET signaling pathway is also regulated by crosstalk with
receptor tyrosine kinases mainly EGFR. MET and EGFR are both known
to be overexpressed in CRC (219). The individual blocking of MET or
EGFR has little effect on downstream ERK/PI3K activation due to the
compensatory mechanism. Targeting both receptors by combined
therapy results in the abrogation of the downstream pathway
(220-222).
Recent data suggest that targeting
immune-recognition and response may be effective in eradicating
cancer cells. This strategy includes malignant tumors having
different genetic and epigenetic signatures that may be identified
and attacked by the host immune system expressing unique antigens.
This process consists of many steps, such as T cell binding to MHC
molecules presented by antigen-presenting cells (APCs). The next
step involves signals mediated by co-stimulatory or inhibitory
receptors that play a critical role in the T cells activation and
tolerance (223,224). This dual-check mechanism is
essential for avoiding excessive immune response in a normal
scenario and attack diseased cells (225). The process of tumor cells
evading host immune recognition and response is referred to as the
immune escape and has been mentioned in cancer (226). Immune escape results from
immunosuppressive factors, such as TGF-β of Treg cells
and IL-6 regulatory cells, or the loss of immunogenicity by the
inhibition of MHC-1 (227). The
activation of co-inhibitory receptors, also known as immune
checkpoint receptors present on the surface of T cells, leads to
cancer-mediated T cell inactivation. The immune checkpoint
receptors expressed on the surface of T cells comprise of
programmed death-1 (PD-1) and cytotoxic T lymphocyte antigen 4
(CTLA-4). Ligands for these receptors are known as PD-L1 and PD-L2
expressed on cancer, stromal and immune cells (228). Wang et al reported
elevated levels of PD-L1 in metastatic CRC as compared to primary
CRC, allowing its targeting with an immune response (229). High levels of Treg
cells have been found in CRC tissue as compared to adjacent normal
tissue. These Treg cells are known to express PD-1 and
are crucial to the immune response to CRC (230).
The 4 types of Janus kinase proteins (JAKs) include
JAK1-3 and TYK2 that interact with cytokine receptors present in
the colon. Although they are associated with different cytokine
receptors, they have a common mechanism of the intracellular
pathway, including signal transducer and activator of transcription
(STAT) protein. Upon ligand-binding and physiological
transformation, the associated JAKs of the cytokine receptor
autophosphorylate and proceed to phosphorylate specific residues on
the receptors that act as docking sites for STAT proteins.
Associated JAK proteins further phosphorylate these STAT proteins,
causing their dissociation and dimer formation. These dimers
translocate to the nucleus, identifying and binding to gamma
activated sequence (GAS) elements, causing pro-inflammatory gene
expression and transcription and playing a role in the pathogenesis
of inflammatory bowel disease (IBD). Drugs developed to inhibit the
functioning of JAK proteins bind and prevent their phosphorylation,
effectively blocking the pathway. JAK protein can also signal PI3K
protein (Akt pathway) and Ras protein (MAPK pathway) (172).
The TGF-β pathway plays a role in cell growth,
division and adhesion. It also stimulates apoptosis and cellular
differentiation. Its downstream targets include important cell
cycle checkpoint genes (p21, p27 and p15) that trigger growth.
TGF-β receptors occur as transmembrane heterodimers (type 1 and 2)
with kinase domains (KDs) present in the intracellular part. Upon
ligand binding, 2 of these heterodimers come together to form a
complex through receptor dimerization. The KDs are activated
through phosphorylation and further activate SMAD proteins present
in the cytosol. SMAD2 and SMAD3 form phosphorylated heterodimers.
This complex, joined by co-factor SMAD4, forms a heterotrimer. The
heterotrimers then translocate into the nucleus and bind to TGF-β
target genes and initiate transcription. TGF-β is known to function
as a tumor suppressor, normally controlling cell division and the
death of epithelial cells of the colon. CRC cells lose TGF-β,
thereby resisting growth inhibition. However, in the later stages
of CRC, TGF-β expression is increased and influences
epithelia-to-mesenchymal transition (EMT), and as a result,
increases invasion and cell migration thus subduing the normal
cellular immune response. Mutations in SMAD4 have also been
reported in cases of juvenile polyposis (172-175).
The Notch signaling pathway occurs intercellularly
and is highly conserved. Mammals possess 4 types of notch receptors
(Notch 1-4). Ligands are of 2 types, the Jagged protein family (JAG
1 and 2) and the Delta-like protein family (DLL 1, 3, and 4). A
Notch receptor has 3 components, namely the notch extracellular
domain (binds to the ligand), Notch intracellular domain (NICD) and
the transmembrane component. Ligand activation of DLL or JAG
proteins on the 'sending' cell occurs through ubiquitination by a
mind bomb protein (MIB). The activated ligand then binds to the
extracellular component of the notch receptor. A disintegrin and
metalloproteinase (ADAM protease) cleaves the extracellular domain
of the notch receptor (S2 cleavage). Subsequently, another
protease, secretase gamma, cleaves NICD causing it to dissociate
from the transmembrane domain of the receptor (S3 cleavage). NICD,
now free to move in the cytosol, binds to and activates the CSL
transcription factor (suppressor of the hairless) that forms a
complex with co-activators MAML (mastermind-like proteins), and
p300. The complex translocates into the nucleus where the p300 acts
as a histone acetylase, causing the activation of transcription
factors (ex-HES1) and the transcription of notch-target genes
(example-MYC, p21). The downregulation of the pathway may occur by
a ubiquitin ligase, f-box/WD-40 repeat-containing protein 7 (FBW7)
that ubiquitinates NICD causing its proteasomal degradation. The
overexpression of Notch-associated proteins and ligands (JAG, HES1,
NICD, etc.) has been reported in CRC. Cell cycle and apoptotic
regulation of target genes (p21 and PUMA genes) enhance the
severity of CRC through Notch signaling. The pathway also
influences CRC resistance to chemotherapeutic drugs (172-175,231).
The SHH signaling pathway is essential for the
regeneration and differentiation of epithelial cells present in
adult colons. Hedgehog (Hh) ligands produced in the endoplasmic
reticulum of secretory cells are released through a membrane
protein known as Dispatch. In a paracrine-type signaling event,
these Hh molecules bind to Patched (Ptc) protein present in
neighboring cell(s) thereby, inhibiting its function. Upon the
inhibitory action of the Ptc molecule, the smoothened (Smo) protein
molecule (previously inhibited by Ptc) is activated. The Smo
protein present in the primary cilia of the intestine regulates the
action of the 3 intracellular Gli proteins (Gli-1, Gli-2 and
Gli-3). It releases Gli-2 (transcriptional activator) from the
suppressor complex composed of Costal-2, Fused kinase (Fu) and SuFu
(suppressor of fused), thereby, activating it. This Gli-2 then acts
upon and phosphorylates the transcription factor, Gli-1, that
translocates to the nucleus and acts upon SHH-target genes. Smo
protein also acts upon and inhibits the function of Gli-3 (a
transcriptional inhibitor). In CRC tissues, it is noted that the
levels of the proteins SHH, Smo and Gli1 are uncharacteristically
high (175). A previous study
also reported that the subcutaneous transplantation of speckle-type
POZ protein (SPOP) reduced the rate of tumor growth (in BALB/c nude
mice) and increased apoptosis (in HCT116 cells) (232). In CRC, SPOP is shown to degrade
Gli2 by ubiquitinating it (172). The arbitrary regulation of the
signaling pathway, either by a mutation in Ptc (loss of function)
or a mutation in Smo (gain of function), can lead to colon cancer
development (233).
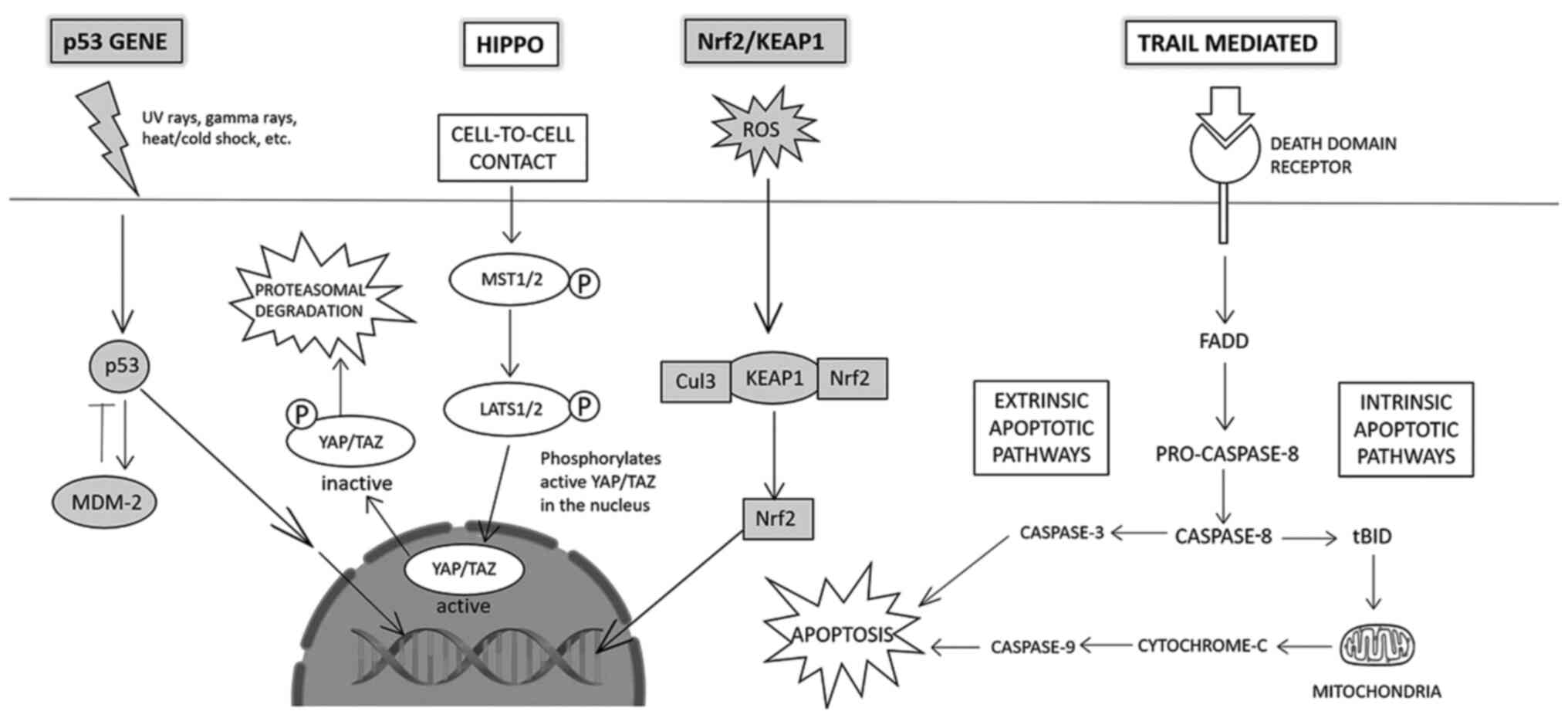
The Hippo signaling pathway controls cell
proliferation, homeostasis and regeneration (Fig. 4). The main transcriptional
regulator of the pathway is Yes-associated protein 1 (YAP). YAP and
its homolog, PDZ-binding domain taffazin (TAZ) regulate the Hippo
pathway. Upon the initiation of the pathway, the activation and
phosphorylation of first, mammalian Ste20-like kinases 1/2
(MST1/2), and subsequently large tumor suppressor 1/2 (LATS1/2)
occurs. LATS1/2, in turn, phosphorylates YAP/TAZ, resulting in its
removal from the nucleus into the cytoplasm where it undergoes
ubiquitin-mediated protein degradation. An abnormally high level of
YAP/TAZ protein has been reported in solid tumors and amplifies the
frequency of tumors (174,234).
Reactive oxygen species (ROS) cause oxidative
damage to cells and are linked to CRC (Fig. 4). In response to the fatal effects
of oxidative stress, the cell releases antioxidant and
detoxification genes, such as NF-E2-related factor 2 (Nrf2). A
Cap-n-collar transcription factor, Nrf2 identifies antioxidant
response elements on target gene promoters and combats free-radical
damage (by carcinogens, inflammation, etc.). Normally, Nrf2 is
confined to the cytosol by Kelch-like ECH associated protein 1
(Keap1). Keap1 acts as a linker protein between Nrf2 and Cul3-based
E3-ubiquitin ligase complex, causing ubiquitination of Nrf2 and its
subsequent proteasomal degradation. Under conditions of oxidative
stress, antioxidant response elements induce separation of Nrf2 and
Keap1, leading to its nuclear transportation. There, Nrf2 undergoes
dimerization and interacts with small musculoaponeurotic
fibrosarcoma (Maf) proteins initiating the attachment of Nrf2 with
antioxidant response elements, initiating transcriptional
activation of target genes (174). The Nrf2/Keap1 pathway can help
regulate the chemopreventive effects of various drugs for the
treatment of CRC (174).
p53 plays an important role in maintaining the
integrity of cellular processes and genetic material. It is a
well-known tumor suppressor and spearheads repair and cellular
apoptosis depending upon the extent of DNA damage (Fig. 4). Secreted in response to external
stress such as UV rays, and hypoxia, p53 has a short half-life in
the cell and is directed by a fool-proof regulatory system. Nicks
or mutations caused due to external stress stimuli result in
activation of the ataxia-telangiectasia mutated (ATM) protein
kinase CHK2, whose absence can delay the action of p53. The
activation of the p53 gene also results in the production of mouse
double minute 2 homolog (MDM-2), an E-3 ubiquitin ligase. MDM-2 is
a negative regulator that degrades the p53 gene by ubiquitinating
it, thus setting up a self-governing 'loop' to maintain the p53
level in the cell. Oncogenic events can lead to the transcription
of the p19 alternate reading frame (ARF) protein that inhibits the
functioning of MDM-2, thus, failing to regulate p53 levels in the
cell. Mutations in the p53 gene and aberrant function lead to a
loss of cellular checkpoints and programmed cell death, thereby
compromising genetic integrity (174). Mutations also encourage EMT, and
the formation of adenomatous polyps, eventually conferring
malignancy, i.e., CRC. It has also been noted that 80% of p53
mutations in CRC cases stemmed from missense mutations, largely in
exons 4-8 (172).
The TRAIL-mediated signaling pathway is a candidate
for anticancer therapy by selectively targeting cancer cells
(Fig. 4). TRAIL receptors span
the cellular membrane and act as a conduit for the extrinsic
apoptotic pathway. TRAIL ligand binds to specific death domain
receptors DR4/DR5 which augments the association of Fas-associated
protein with death domain (FADD), an adaptor protein. FADD
activates pro-caspase 8, initiating the apoptotic pathway (through
recruitment of various caspases initiating the extrinsic, and
cytochrome-c initiating the intrinsic apoptotic pathway) (172).
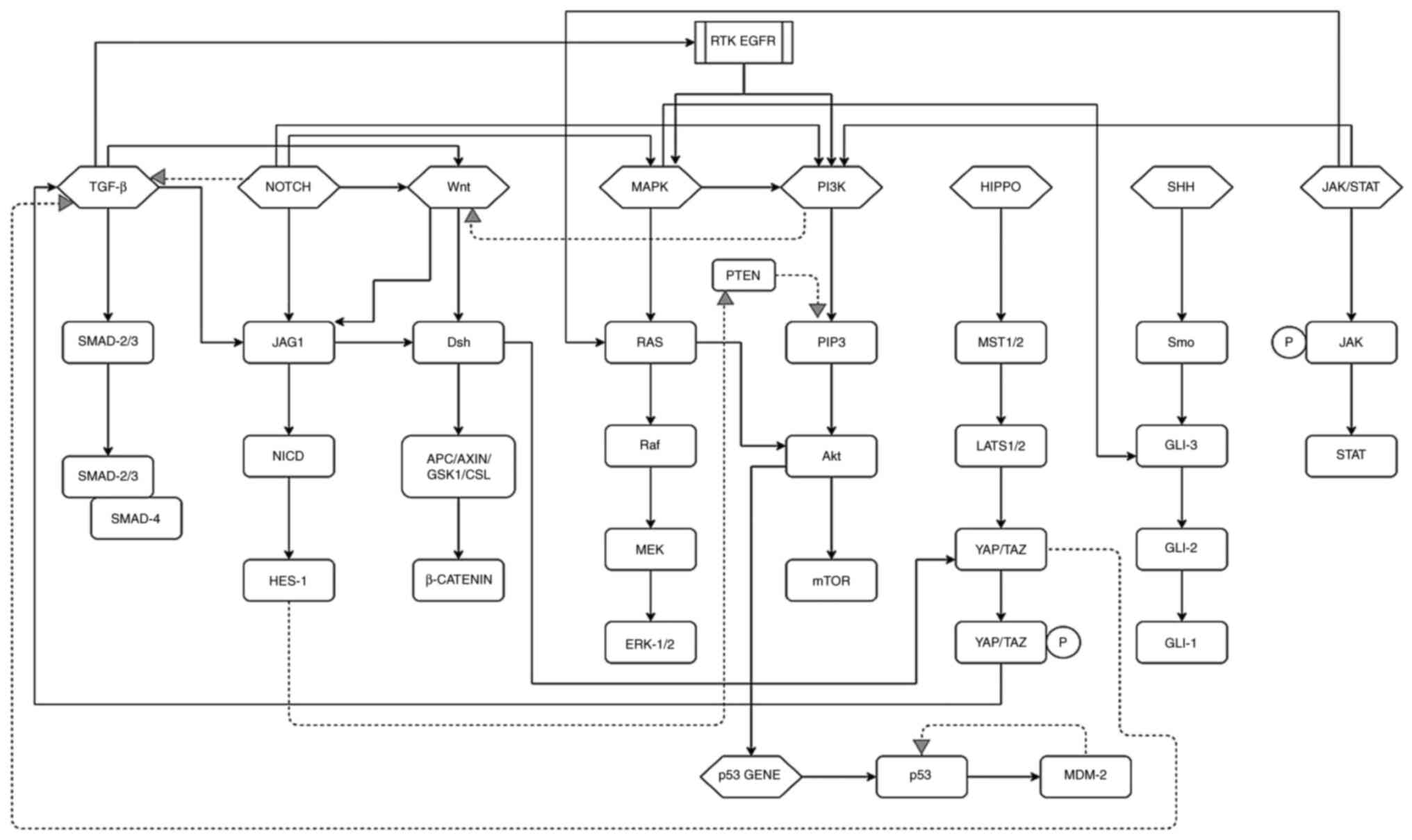
The majority of signaling pathways interact to
maintain cellular homeostasis in healthy cells (Fig. 5). For example, the non-canonical
TGF-β signaling pathway induces the MAPK and PI3K signaling
pathways, both of which otherwise activate upon ligand-binding on
receptor tyrosine kinase (EGFR). TGF-β and its tumor-suppressive
activity can also be inhibited by interaction with the mutant p53
complex. Another example is the JAK/STAT signaling pathway.
Following autophosphorylation, it can activate Ras protein and
ERK-1/2 (MAPK pathway) and AKT in downstream signaling.
The Hippo signaling pathway interacts with the
TGF-β pathway in a positive and negative regulatory manner,
depending upon the circumstances. TAZ protein can bind to the
SMAD2/3-SMAD4 complex and maintain its accumulation in the nucleus
(boosting TGF-β pathway), or phosphorylated TAZ protein can prevent
SMAD 2/SMAD 4 nuclear build-up (repressing TGF-β signaling).
The activation of the MAPK signaling pathway
through RTK can influence Gli protein activity (of the SHH pathway)
in a Smo-independent manner. The co-expression of EGF and Gli
protein activates both pathways and promotes carcinogenic
transformation. Smo-independent and PI3K-dependent Gli factor
activation, occurring due to AKT-associated prevention of
proteasomal degradation of Gli2, is prevalent in colon cancer.
Amplification of Gli1 activity through ribosomal S6 kinases is also
influenced by the PI3K pathway. The non-canonical SHH pathway
promotes CRC cell survival via the Wnt signaling pathway, and EMT
and tumor invasion via the TGF-β signaling pathway (240,241).
5-Fluorouracil (5FU) is the backbone of
chemotherapy with good activity against CRC (242). Folinic acid (FA, leucovorin) was
then also added to the regimen, that achieves a median overall
survival of approximately 8-9 months and has become the standard
treatment for metastatic CRC (mCRC) (243,244). Oxaliplatin (platinum analogue)
and irinotecan (topoisomerase inhibitor) were added to the backbone
of 5FU/FA. Combination chemotherapy has demonstrated a higher
response rate and a better overall survival of 14-18 months, as
well as a progression-free survival (PFS) of 5-8 months. FOLFOX
(5FU/FA and oxaliplatin) or FOLFIRI (5FU/FA and irinotecan)
constitute first-line standard combination chemotherapy (245,246). Both have exhibited approximately
equal clinical responses with different safety profiles. A regimen
containing oxaliplatin gives rise to peripheral neuropathy and
irinotecan results in gastrointestinal toxicity (247,248). The replacement of 5FU/FA with
capecitabine (Xeloda) has been investigated with oxaliplatin
(XELOX) or with irinotecan (XELIRI) resulting in similar efficacy,
as the combination with 5FU/FA (249,250). With the improved screening
efforts, the diagnosis of patients with CRC has improved and the
mortality rate has decreased due to the early detection and the
success of anticancer therapies. Despite the success of current
therapeutics, 40-50% of CRC cases ultimately relapse, leading to
fatality due to metastasis (251). Although the treatment options
for patients with CRC have improved, more effective targeting
agents are required for the treatment of advanced stages of the
disease. Therefore, targeted anticancer therapeutics are warranted
to disrupt the dysregulated signaling pathways of CRC with a better
outcome for patients.
The idea of targeted therapy against cancer has
flourished over the past 2 decades (252,253). Targeted therapies block the
function of certain oncoproteins and downstream pathways using
monoclonal antibodies or small molecules against
receptor/non-receptor tyrosine kinases. Monoclonal antibodies are
the main candidates in targeted therapies that target surface
receptors and membrane-bound factors outside the cancer cells
(253,254). Monoclonal antibodies can
recognize and bind cancer cells directly, regulating downstream
pathways and leading to the inhibition of cell cycle advancement
and subsequent cell death. Immune cells are also targeted by
monoclonal antibodies to manipulate the immune system to attack and
discard cancer cells. Small molecules are robustly developed that
work mostly inside the cells to target receptor and non-receptor
tyrosine kinases, thereby blocking cancer cell growth and inducing
cell death (255). These
targeted treatments lead to the inhibition of the differentiation,
proliferation, invasion and migration of cancer cells. These
therapies also act on the tumor microenvironment, resulting in a
decrease in angiogenesis and rendering immune cells more alert for
stronger surveillance and attack.
Monoclonal antibody against EGFR (cetuximab) was
the first targeted therapy approved by the FDA in 2004 for the
treatment of CRC. In the same year, another monoclonal antibody
targeting VEGF-A (bevacizumab) was approved for the treatment of
CRC (256). Ideal sites for
targeted therapy are present in the dysregulated pathway of
TGF-β/SMAD, Wnt/β-catenin, EGFR, VEGFR/VEGRR, Notch, Hedgehog
activating PI3K/AKT and RAS/RAF pathways (257). The intricate network of
downstream signaling pathways and the crosstalk among these renders
the complete blocking of specific biological interactions complex
and difficult.
The dysregulation of various signaling pathways
leads to CRC initiation, progression and migration. The activation
of signaling pathways leads to the acquisition of a malignant
phenotype. One novel approach could be to use specific inhibitors
targeting these pathways. Small molecule inhibitors-based
anticancer therapeutics provide an excellent opportunity for
scientists to navigate various aspects of cell growth, cell cycle,
cell proliferation to gene expression, and protein-protein
interaction network. The EGFR/MAPK pathway has been targeted by
small molecules and antibodies. Targeting the EGFR pathway and
related factors has been achieved by the use of monoclonal
antibodies, such as anti-EGFR and tyrosine kinase inhibitors (TKIs)
(Table II). Small molecules
targeting EGFR, Ras and Raf are known to interfere with the MAPK
signaling pathway. EGFR antibodies are also known to block MAPK
signaling. For metastatic colorectal cancer, EGFR inhibitors are a
valuable therapeutic option. Monoclonal antibodies for EGFR
(cetuximab or panitumumab) in addition to chemotherapy are
effective for mCRC patients harbouring wild type RAS and BRAF
(258). Previous research has
reported different inhibitors for the targeting of PI3K signaling
(259), namely: i) PI3K
inhibitors; ii) dual inhibitors of PI3K and mTOR; iii) AKT
inhibitors; and iv) mTOR inhibitors. A summary of some of the
inhibitors targeting the PI3K/AKT pathway in CRC is presented in
Table III.
Targeting the TGF-β pathway has been achieved by
specifically targeting the ligand, ligand-receptor complex and the
intracellular levels of TGF-β. Anti-TGF-β therapy holds promise, as
pre-clinical studies and clinical trials have indicated (260). TGF-β synthesis has been blocked
using antisense molecules in CRC. TGF-β-R1 and R2 kinase is known
to be blocked by a small molecule (LY2109761) (261). The Wnt signaling pathway is
targeted by different inhibitors ranging from small molecules,
peptides and antibodies. In total, 4 types of Wnt inhibitors have
been developed based on specific targets: i) Generic; ii) β-catenin
destruction complex; iii) Wnt-receptor complex; and iv)
nuclear-transcription factor complexes (171). It has been found that targeting
notch receptor and notch ligands using siRNA results in more
effective therapeutics (262).
Antibodies against Notch ligands and receptors have been reported
to be effective in blocking the Notch pathway. Anti-Notch-1, -2/3
and anti-DLL4 are in different stages of clinical trials (263) (Table IV).
JAK-STAT pathway inhibition has been achieved using
various approaches. The small molecule inhibitor of this pathway,
such asJAK inhibitors are in clinical trials for gastrointestinal
disorders, including ulcerative colitis (UC) (NCT01959282,
NCT03006068, NCT02914535, NCT02914522, NCT02819635) and Crohn's
disease (CD) (NCT03345836, NCT02782663, NCT03345849, NCT02914600,
NCT02914561) (264). The
anti-IL-6-R antibody is known to inhibit IL-6R function and is in
the preclinical stage of development. A JAK inhibitor (AZD1480) and
STAT3 inhibitors, such as TrichostatinA and bufalin have also been
shown to be successful in the inhibition of the JAK/STAT pathway
(265). The association between
CRC and hedgehog (Hh) signaling remains inconclusive; its
inhibitors have been used in in vitro and in vivo
studies (266). The most common
Hh inhibitor is cyclopamine. Wu et al reported the efficacy
of this inhibitor in the treatment of CRC in vitro (267). It has also been demonstrated
that cyclopamine can inhibit proliferation and induction of
apoptosis in CRC (267,268). The NRF2 pathway is known to act
as a 'double-edged sword', acting as an oncogene and tumor
suppressor in CRC (269).
Inhibitors targeting the NRF2 and Hippo pathways are still in the
initial stage, paving the way for further studies. A combination of
conventional chemotherapeutics with inhibitors of signaling
pathways may enhance patient outcome (270).
The discovery of bevacizumab (anti-VEGF-A) and its
effectiveness against CRC was a milestone in the treatment of solid
tumors by blocking angiogenesis (271). It is a humanized IgG monoclonal
antibody that improves both progression-free survival and overall
survival in mCRC. FDA approved VEGF-targeted bevacizumab for the
treatment of mCRC. Combining bevacizumab with the FOLFOX/FOXFIRI
regimen has been shown to provide a partial significant improvement
in progression-free survival and overall survival (272). Various novel agents have been
reported and few have been endorsed for the treatment of CRC.
Aflibercept is a ligand trap-based VEGFR-1 and -2 extracellular
domain recombinant fusion protein targeting VEGF-A and VEGF-B;
clinical trials have reported that it has a stronger affinity for
VEGF-A (273). A TKI,
regorafenib, targeting VEGFR, FGFR, PDGFR and BRAF has been
approved by the FDA for the treatment of mCRC (274). Another humanized monoclonal
antibody for VEGFR-2, ramucirumab, is an FDA-approved second-line
treatment for mCRC (275)
(Table V).
The HGF/MET pathway is known to be upregulated and
therefore initiatesthe activation of downstream pathways, such as
MAPK/ERK, PI3K/AKT and JAK/STAT in CRC. HGF/MET signaling can be
blocked by antibodies and small molecules using different
mechanisms (202). These small
molecules act by either blocking HGF activation or inhibiting HGF
binding to MET receptors and thereby inhibiting cancer cell growth.
Rilotumumab, a humanized monoclonal antibody for HGF acts by
neutralizing HGF binding to receptors in clinical trials. Various
antibodies targeting MET have been developed that target HGF
binding to MET, resulting in its degradation. DN-30, ABT-700 and
onartuzumab have been studied in solid tumors in clinical trials.
Several selective and non-selective TKIs have been investigated in
clinical trials (276,277) (Table VI).
Targeted therapy for immune checkpoint enhances
immune surveillance and tumor suppression by blocking tumor cell
escape from T cell recognition (278). A phase I trial of immune
checkpoint targeted therapy revealed a significant response in mCRC
(279). Further studies noted
that a small number of patients with mCRC responded to this
therapy, owing to a high mutational burden, exhibiting high levels
of MSI-H and dMMR (MMR deficiency) (280-283). The first PD-1 blocker approved
by the FDA in 2017 was pembrolizumab that exhibited promising
efficacy in MMR-deficient CRC (284). Pembrolizumab is a humanized IgG4
antibody against PD-1 set another milestone in CRC immunotherapy
(285). Another humanized
monoclonal IgG4 antibody for PD-1 (nivolumab) received FDA approval
for the treatment of MSI-H and dMMR mCRC in 2017 (286). Other novel PD-1/PDL-1 inhibitors
have been looking promising in phase I clinical trials of CRC and
other solid tumors (287)
(Table VII).
CRC is a complex and heterogeneous disease,
exhibiting multiple genetic mutations and epigenetic aberrations.
Individuals with predisposing germline mutations may exhibit
somatic mutation accumulation at various stages, resulting in the
cell transformation from normal epithelial cells to malignant and
invasive cancer. Genetic testing for MSI in Lynch syndrome has led
to the targeted surveillance of at-risk family members for the
prevention of CRC. Targeted EGFR and VEGF therapies have already
had a better impact on the management of mCRC. A heterogeneous
disease, such as CRC resulting from the activation of numerous
signaling pathways, is therapeutically challenging, and cannot be
targeted with a single agent. A combination of conventional
therapeutics with novel inhibitors targeting dysregulated pathways
is required for a better outcome. The latest discoveries have
assisted researchers in improving their knowledge of CRC to an
unprecedented level. In the present review, the molecular pathways
and the dysregulation of signaling pathways that play an essential
role in CRC initiation and progression were discussed. More
extensive knowledge of CRC has been acquired, and the efficiency of
the first sign of targeted therapies provides an encouraging
prospect for the future management and development of more
efficient markers for the treatment of CRC. The design of novel
scaffolds, such as TKIs and specific inhibitors/antibodies to
inhibit oncogenes and dysregulated signaling pathways is expanding.
With the discovery of more targeted novel therapeutics, there is
real hope that CRC can be better managed, leading to a lower
disease burden in the future.
No funding was received.
Not applicable.
JKS, RA, AW and SKS conceived, drafted and wrote
the manuscript. OAO and MA were involved in the critical reviewing
of the manuscript. RA and SKS were involved in the revising and
editing of the manuscript. All authors have read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The authors are grateful to the Deanship of
Scientific Research, King Saud University for funding through the
Vice Deanship of Scientific Research Chairs.
|
1
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar
|
|
3
|
Fleming M, Ravula S, Tatishchev SF and
Wang HL: Colorectal carcinoma: Pathologic aspects. J Gastrointest
Oncol. 3:153–173. 2012.PubMed/NCBI
|
|
4
|
Jasperson KW, Tuohy TM, Neklason DW and
Burt RW: Hereditary and familial colon cancer. Gastroenterology.
138:2044–2058. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Graff RE, Möller S, Passarelli MN, Witte
JS, Skytthe A, Christensen K, Tan Q, Adami HO, Czene K, Harris JR,
et al: Familial risk and heritability of colorectal cancer in the
nordic twin study of cancer. Clin Gastroenterol Hepatol.
15:1256–1264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lichtenstein P, Holm NV, Verkasalo PK,
Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A and Hemminki
K: Environmental and heritable factors in the causation of
cancer-analyses of cohorts of twins from Sweden, Denmark, and
Finland. N Engl J Med. 343:78–85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keum N and Giovannucci E: Global burden of
colorectal cancer: Emerging trends, risk factors and prevention
strategies. Nat Rev Gastroenterol Hepatol. 16:713–732. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ogino S and Goel A: Molecular
classification and correlates in colorectal cancer. J Mol Diagn.
10:13–27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bakhoum SF, Silkworth WT, Nardi IK,
Nicholson JM, Compton DA and Cimini D: The mitotic origin of
chromosomal instability. Curr Biol. 24:R148–R149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nazemalhosseini Mojarad E, Kuppen PJK,
Aghdaei HA and Zali MR: The CpG island methylator phenotype (CIMP)
in colorectal cancer. Gastroenterol Hepatol Bed Bench. 6:120–128.
2013.
|
|
12
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pawlik TM, Raut CP and Rodriguez-Bigas MA:
Colorectal carcinogenesis: MSI-H versus MSI-L. Dis Markers.
20:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pitot HC: The molecular biology of
carcinogenesis. Cancer. 72(3 Suppl): S962–S970. 1993. View Article : Google Scholar
|
|
15
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American joint committee on
cancer sixth edition staging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu W, He Y, Wang Y, Li X, Young J,
Ioannidis JPA, Dunlop MG and Theodoratou E: Risk factors and risk
prediction models for colorectal cancer metastasis and recurrence:
An umbrella review of systematic reviews and meta-analyses of
observational studies. BMC Med. 18:1722020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jensen KH, Izarzugaza JMG, Juncker AS,
Hansen RB, Hansen TF, Timshel P, Blondal T, Jensen TS,
Rygaard-Hjalsted E, Mouritzen P, et al: Analysis of a gene panel
for targeted sequencing of colorectal cancer samples. Oncotarget.
9:9043–9060. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
20
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jass JR and Smith M: Sialic acid and
epithelial differentiation in colorectal polyps and cancer-a
morphological, mucin and lectin histochemical study. Pathology.
24:233–242. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dekker E: IJspeert JEG: Serrated pathway:
A paradigm shift in CRC prevention. Gut. 67:1751–1752. 2018.
View Article : Google Scholar
|
|
23
|
East JE, Vieth M and Rex DK: Serrated
lesions in colorectal cancer screening: Detection, resection,
pathology and surveillance. Gut. 64:991–1000. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Longacre TA and Fenoglio-Preiser CM: Mixed
hyperplastic adenomatous polyps/serrated adenomas. A distinct form
of colorectal neoplasia. Am J Surg Pathol. 14:524–537. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Snover DC, Ahnen D, Burt R and Odze R:
Serrated polyps of the colon and rectum and serrated
('hyperplastic') polyposis WHO classification of tumours of the
digestive system. International Agency for Research on Cancer;
Lyon: pp. 160–165. 2010
|
|
26
|
Groff RJ, Nash R and Ahnen DJ:
Significance of serrated polyps of the colon. Curr Gastroenterol
Rep. 10:490–498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aust DE and Baretton GB; Members of the
Working Group GI-Pathology of the German Society of Pathology:
Serrated polyps of the colon and rectum (hyperplastic polyps,
sessile serrated adenomas, traditional serrated adenomas, and mixed
polyps)-proposal for diagnostic criteria. Virchows Arch.
457:291–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Torlakovic E, Skovlund E, Snover DC,
Torlakovic G and Nesland JM: Morphologic reappraisal of serrated
colorectal polyps. Am J Surg Pathol. 27:65–81. 2003. View Article : Google Scholar
|
|
29
|
Goldstein NS, Bhanot P, Odish E and Hunter
S: Hyperplastic-like colon polyps that preceded
microsatellite-unstable adenocarcinomas. Am J Clin Pathol.
119:778–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim KM, Lee EJ, Kim YH, Chang DK and Odze
RD: KRAS mutations in traditional serrated adenomas from Korea
herald an aggressive phenotype. Am J Surg Pathol. 34:667–675. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
International Agency for Research on
Cancer (IARC): Global Cancer Observatory (GLOBOCAN). IARC; Lyon:
2018
|
|
32
|
Ferlay J, Ervik M, Lam F, Colombet M, Mery
L, Piñeros M, Znaor A, Soerjomataram I and Bray F: Global Cancer
Observatory: Cancer Today. International Agency for Research on
Cancer; Lyon: 2018
|
|
33
|
Li FY and Lai MD: Colorectal cancer, one
entity or three. J Zhejiang Univ Sci B. 10:219–229. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murphy N, Ward HA, Jenab M, Rothwell JA,
Boutron-Ruault MC, Carbonnel F, Kvaskoff M, Kaaks R, Kühn T, Boeing
H, et al: Heterogeneity of colorectal cancer risk factors by
anatomical subsite in 10 european countries: A multinational cohort
study. Clin Gastroenterol Hepatol. 17:1323–1331.e6. 2019.
View Article : Google Scholar
|
|
35
|
Yang L, Xiong Z, He W, Xie K, Liu S, Kong
P, Jiang C, Guo G and Xia L: Proximal shift of colorectal cancer
with increasing age in different ethnicities. Cancer Manag Res.
10:2663–2673. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Murphy G, Devesa SS, Cross AJ, Inskip PD,
McGlynn KA and Cook MB: Sex disparities in colorectal cancer
incidence by anatomic subsite, race and age. Int J Cancer.
128:1668–1675. 2011. View Article : Google Scholar :
|
|
37
|
Shin A, Kim KZ, Jung KW, Park S, Won YJ,
Kim J, Kim DY and Oh JH: Increasing trend of colorectal cancer
incidence in Korea,. 1999–2009. Cancer Res Treat. 44:219–226. 2012.
View Article : Google Scholar
|
|
38
|
Rawla P, Sunkara T and Barsouk A:
Epidemiology of colorectal cancer: Incidence, mortality, survival,
and risk factors. Prz Gastroenterol. 14:89–103. 2019.PubMed/NCBI
|
|
39
|
Win AK, Jenkins MA, Dowty JG, Antoniou AC,
Lee A, Giles GG, Buchanan DD, Clendenning M, Rosty C, Ahnen DJ, et
al: Prevalence and penetrance of major genes and polygenes for
colorectal cancer. Cancer Epidemiol Biomarkers Prev. 26:404–412.
2017. View Article : Google Scholar :
|
|
40
|
Aaltonen LA, Peltomäki P, Leach FS,
Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J,
Hamilton SR, et al: Clues to the pathogenesis of familial
colorectal cancer. Science. 260:812–416. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ionov Y, Peinado MA, Malkhosyan S, Shibata
D and Perucho M: Ubiquitous somatic mutations in simple repeated
sequences reveal a new mechanism for colonic carcinogenesis.
Nature. 363:558–561. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thibodeau SN, Bren G and Schaid D:
Microsatellite instability in cancer of the proximal colon.
Science. 260:816–819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hampel H, Frankel WL, Martin E, Arnold M,
Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J,
et al: Screening for the Lynch syndrome (hereditary nonpolyposis
colorectal cancer). N Engl J Med. 352:1851–1860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Patel SG and Ahnen DJ: Familial colon
cancer syndromes: An update of a rapidly evolving field. Curr
Gastroenterol Rep. 14:428–438. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Half E, Bercovich D and Rozen P: Familial
adenomatous polyposis. Orphanet J Rare Dis. 4:222009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vasen HF, Tomlinson I and Castells A:
Clinical management of hereditary colorectal cancer syndromes. Nat
Rev Gastroenterol Hepatol. 12:88–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sir Michael M, Tola A, Tim B and Junshi C:
World Cancer Research Fund/American Institute for Cancer Research.
Food, Nutrition, Physical Activity, and the Prevention of Cancer: A
Global Perspective. AICR; Washington, DC: 2007
|
|
48
|
Hofseth LJ, Hebert JR, Chanda A, Chen H,
Love BL, Pena MM, Murphy EA, Sajish M, Sheth A, Buckhaults PJ and
Berger FG: Early-onset colorectal cancer: Initial clues and current
views. Nat Rev Gastroenterol Hepatol. 17:352–364. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mauri G, Sartore-Bianchi A, Russo AG,
Marsoni S, Bardelli A and Siena S: Early-onset colorectal cancer in
young individuals. Mol Oncol. 13:109–131. 2019. View Article : Google Scholar
|
|
50
|
Bailey CE, Hu CY, You YN, Bednarski BK,
Rodriguez-Bigas MA, Skibber JM, Cantor SB and Chang GJ: Increasing
disparities in the age-related incidences of colon and rectal
cancers in the United States, 1975-2010. JAMA Surg. 150:17–22.
2015. View Article : Google Scholar :
|
|
51
|
Pearlman R, Frankel WL, Swanson B, Zhao W,
Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, et
al: Ohio colorectal cancer prevention initiative study group:
Prevalence and spectrum of germline cancer susceptibility gene
mutations among patients with early-onset colorectal cancer. JAMA
Oncol. 3:464–471. 2017. View Article : Google Scholar :
|
|
52
|
Schellerer VS, Merkel S, Schumann SC,
Schlabrakowski A, Förtsch T, Schildberg C, Hohenberger W and Croner
RS: Despite aggressive histopathology survival is not impaired in
young patients with colorectal cancer: CRC in patients under 50
years of age. Int J Colorectal Dis. 27:71–79. 2012. View Article : Google Scholar
|
|
53
|
Wang MJ, Ping J, Li Y, Adell G, Arbman G,
Nodin B, Meng WJ, Zhang H, Yu YY, Wang C, et al: The prognostic
factors and multiple biomarkers in young patients with colorectal
cancer. Sci Rep. 5:106452015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Crosbie AB, Roche LM, Johnson LM, Pawlish
KS, Paddock LE and Stroup AM: Trends in colorectal cancer incidence
among younger adults-Disparities by age, sex, race, ethnicity, and
subsite. Cancer Med. 7:4077–4086. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ballester V, Rashtak S and Boardman L:
Clinical and molecular features of young-onset colorectal cancer.
World J Gastroenterol. 22:1736–1744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dozois EJ, Boardman LA, Suwanthanma W,
Limburg PJ, Cima RR, Bakken JL, Vierkant RA, Aakre JA and Larson
DW: Young-onset colorectal cancer in patients with no known genetic
predisposition: Can we increase early recognition and improve
outcome? Medicine (Baltimore). 87:259–263. 2008.
|
|
57
|
O'Connell JB, Maggard MA, Liu JH, Etzioni
DA, Livingston EH and Ko CY: Do young colon cancer patients have
worse outcomes? World J Surg. 28:558–562. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Taggarshe D, Rehil N, Sharma S, Flynn JC
and Damadi A: Colorectal cancer: Are the 'young' being overlooked?
Am J Surg. 205:312–316. 2013. View Article : Google Scholar
|
|
59
|
Low EE, Demb J, Liu L, Earles A,
Bustamante R, Williams CD, Provenzale D, Kaltenbach T, Gawron AJ,
Martinez ME and Gupta S: Risk factors for early-onset colorectal
cancer. Gastroenterology. 159:492–501.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu PH, Wu K, Ng K, Zauber AG, Nguyen LH,
Song M, He X, Fuchs CS, Ogino S, Willett WC, et al: Association of
obesity with risk of early-onset colorectal cancer among women.
JAMA Oncol. 5:37–44. 2019. View Article : Google Scholar :
|
|
61
|
Renehan AG, Tyson M, Egger M, Heller RF
and Zwahlen M: Body-mass index and incidence of cancer: A
systematic review and meta-analysis of prospective observational
studies. Lancet. 371:569–578. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dong Y, Zhou J, Zhu Y, Luo L, He T, Hu H,
Liu H, Zhang Y, Luo D, Xu S, et al: Abdominal obesity and
colorectal cancer risk: Systematic review and meta-analysis of
prospective studies. Biosci Rep. 37:BSR201709452017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tejpar S and Van Cutsem E: Molecular and
genetic defects in colorectal tumorigenesis. Best Pract Res Clin
Gastroenterol. 16:171–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instability in colorectal cancers. Nature. 386:623–627.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nojadeh JN, Behrouz Sharif S and Sakhinia
E: Microsatellite instability in colorectal cancer. EXCLI J.
17:159–168. 2018.PubMed/NCBI
|
|
66
|
Vilar E and Gruber SB: Microsatellite
instability in colorectal cancer-the stable evidence. Nat Rev Clin
Oncol. 7:153–162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Geiersbach KB and Samowitz WS:
Microsatellite instability and colorectal cancer. Arch Pathol Lab
Med. 135:1269–1277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lao VV and Grady WM: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kambara T, Simms LA, Whitehall VL, Spring
KJ, Wynter CV, Walsh MD, Barker MA, Arnold S, McGivern A, Matsubara
N, et al: BRAF mutation is associated with DNA methylation in
serrated polyps and cancers of the colorectum. Gut. 53:1137–1144.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Loeb LA, Loeb KR and Anderson JP: Multiple
mutations and cancer. Proc Natl Acad Sci USA. 100:776–781. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Loeb LA: A mutator phenotype in cancer.
Cancer Res. 61:3230–3239. 2001.PubMed/NCBI
|
|
72
|
Prindle MJ, Fox EJ and Loeb LA: The
mutator phenotype in cancer: Molecular mechanisms and targeting
strategies. Curr Drug Targets. 11:1296–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar
|
|
74
|
Le Scouarnec S and Gribble SM:
Characterising chromosome rearrangements: Recent technical advances
in molecular cytogenetics. Heredity (Edinb). 108:75–85. 2012.
View Article : Google Scholar
|
|
75
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Rowan A, Halford S, Gaasenbeek M, Kemp Z,
Sieber O, Volikos E, Douglas E, Fiegler H, Carter N, Talbot I, et
al: Refining molecular analysis in the pathways of colorectal
carcinogenesis. Clin Gastroenterol Hepatol. 3:1115–1123. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Weber JC, Meyer N, Pencreach E, Schneider
A, Guérin E, Neuville A, Stemmer C, Brigand C, Bachellier P, Rohr
S, et al: Allelotyping analyses of synchronous primary and
metastasis CIN colon cancers identified different subtypes. Int J
Cancer. 120:524–532. 2007. View Article : Google Scholar
|
|
78
|
Cheng YW, Pincas H, Bacolod MD, Schemmann
G, Giardina SF, Huang J, Barral S, Idrees K, Khan SA, Zeng Z, et
al: CpG island methylator phenotype associates with low-degree
chromosomal abnormalities in colorectal cancer. Clin Cancer Res.
14:6005–6013. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Grady WM: Epigenetic events in the
colorectum and in colon cancer. Biochem Soc Trans. 33:684–688.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shih IM, Zhou W, Goodman SN, Lengauer C,
Kinzler KW and Vogelstein B: Evidence that genetic instability
occurs at an early stage of colorectal tumorigenesis. Cancer Res.
61:818–822. 2001.PubMed/NCBI
|
|
81
|
Sieber OM, Heinimann K and Tomlinson IP:
Genomic instability-the engine of tumorigenesis? Nat Rev Cancer.
3:701–708. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Thiagalingam S, Lengauer C, Leach FS,
Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton
SR, Kern SE, et al: Evaluation of candidate tumor suppressor genes
on chromosome 18 in colorectal cancers. Nat Genet. 13:343–346.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Powell SM, Zilz N, Beazer-Barclay Y, Bryan
TM, Hamilton SR, Thibodeau SN, Vogelstein B and Kinzler KW: APC
mutations occur early during colorectal tumorigenesis. Nature.
359:235–237. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kinzler KW and Vogelstein B: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Su LK, Vogelstein B and Kinzler KW:
Association of the APC tumor suppressor protein with catenins.
Science. 262:1734–1737. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bokoch GM and Der CJ: Emerging concepts in
the Ras superfamily of GTP-binding proteins. FASEB J. 7:750–759.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Arber N, Shapira I, Ratan J, Stern B,
Hibshoosh H, Moshkowitz M, Gammon M, Fabian I and Halpern Z:
Activation of c-K-ras mutations in human gastrointestinal tumors.
Gastroenterology. 118:1045–1050. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bos JL, Fearon ER, Hamilton SR, Verlaan-de
Vries M, van Boom JH, van der Eb AJ and Vogelstein B: Prevalence of
ras gene mutations in human colorectal cancers. Nature.
327:293–297. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Nguyen HT and Duong HQ: The molecular
characteristics of colorectal cancer: Implications for diagnosis
and therapy. Oncol Lett. 16:9–18. 2018.PubMed/NCBI
|
|
90
|
Tanaka T, Watanabe T, Kazama Y, Tanaka J,
Kanazawa T, Kazama S and Nagawa H: Chromosome 18q deletion and
Smad4 protein inactivation correlate with liver metastasis: A study
matched for T- and N-classification. Br J Cancer. 95:1562–1567.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zauber P, Sabbath-Solitare M, Marotta SP
and Bishop T: Loss of heterozygosity for chromosome 18q and
microsatellite instability are highly consistent across the region
of the DCC and SMAD4 genes in colorectal carcinomas and adenomas. J
Appl Res. 8:142008.
|
|
92
|
Kirley SD, D'Apuzzo M, Lauwers GY,
Graeme-Cook F, Chung DC and Zukerberg LR: The Cables gene on
chromosome 18Q regulates colon cancer progression in vivo. Cancer
Biol Ther. 4:861–863. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jung B, Staudacher JJ and Beauchamp D:
Transforming growth factor β superfamily signaling in development
of colorectal cancer. Gastroenterology. 152:36–52. 2017. View Article : Google Scholar
|
|
94
|
Muzny DM, Bainbridge M, Chang K, Dinh HH,
Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Morgan I, et
al: Comprehensive molecular characterization of human colon and
rectal cancer. Nature. 487:330–337. 2012. View Article : Google Scholar
|
|
95
|
Mehlen P and Fearon ER: Role of the
dependence receptor DCC in colorectal cancer pathogenesis. J Clin
Oncol. 22:3420–3428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hedrick L, Cho KR and Vogelstein B: Cell
adhesion molecules as tumor suppressors. Trends Cell Biol. 3:36–39.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Martínez-López E, Abad A, Font A, Monzó M,
Ojanguren I, Pifarré A, Sánchez JJ, Martín C and Rosell R: Allelic
loss on chromosome 18q as a prognostic marker in stage II
colorectal cancer. Gastroenterology. 114:1180–1187. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Saha MN, Qiu L and Chang H: Targeting p53
by small molecules in hematological malignancies. J Hematol Oncol.
6:232013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Li H, Zhang J, Tong JHM, Chan AWH, Yu J,
Kang W and To KF: Targeting the oncogenic p53 mutants in colorectal
cancer and other solid tumors. Int J Mol Sci. 20:59992019.
View Article : Google Scholar :
|
|
100
|
López I, P Oliveira L, Tucci P,
Alvarez-Valín F, A Coudry R and Marín M: Different mutation
profiles associated to P53 accumulation in colorectal cancer. Gene.
499:81–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Russo A, Bazan V, Iacopetta B, Kerr D,
Soussi T and Gebbia N; TP53-CRC Collaborative Study Group: The TP53
colorectal cancer international collaborative study on the
prognostic and predictive significance of p53 mutation: Influence
of tumor site, type of mutation, and adjuvant treatment. J Clin
Oncol. 23:7518–7528. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sigal A and Rotter V: Oncogenic mutations
of the p53 tumor suppressor: The demons of the guardian of the
genome. Cancer Res. 60:6788–6793. 2000.
|
|
103
|
Teodoro JG, Evans SK and Green MR:
Inhibition of tumor angiogenesis by p53: A new role for the
guardian of the genome. J Mol Med (Berl). 85:1175–1186. 2007.
View Article : Google Scholar
|
|
104
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Howe JR and Guillem JG: The genetics of
colorectal cancer. Surg Clin North Am. 77:175–195. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Boland CR: The molecular biology of
gastrointestinal cancer: Implications for diagnosis and therapy.
Gastrointest Endosc Clin N Am. 18:401–413. vii2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A national cancer institute
workshop on microsatellite instability for cancer detection and
familial predisposition: Development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.PubMed/NCBI
|
|
109
|
Findeisen P, Kloor M, Merx S, Sutter C,
Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W,
et al: T25 repeat in the 3' untranslated region of the CASP2 gene:
a sensitive and specific marker for microsatellite instability in
colorectal cancer. Cancer Res. 65:8072–8078. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lynch HT, Lynch PM, Lanspa SJ, Snyder CL,
Lynch JF and Boland CR: Review of the Lynch syndrome: History,
molecular genetics, screening, differential diagnosis, and
medicolegal ramifications. Clin Genet. 76:1–18. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
van Rijnsoever M, Grieu F, Elsaleh H,
Joseph D and Iacopetta B: Characterisation of colorectal cancers
showing hypermethylation at multiple CpG islands. Gut. 51:797–802.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JPJ: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686.
1999.PubMed/NCBI
|
|
113
|
Jass JR: Serrated adenoma of the
colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol.
2:398–405. 2005.PubMed/NCBI
|
|
114
|
Samowitz WS, Albertsen H, Herrick J, Levin
TR, Sweeney C, Murtaugh MA, Wolff RK and Slattery ML: Evaluation of
a large, population-based sample supports a CpG island methylator
phenotype in colon cancer. Gastroenterology. 129:837–845.
2005.PubMed/NCBI
|
|
115
|
Hawkins N, Norrie M, Cheong K, Mokany E,
Ku SL, Meagher A, O'Connor T and Ward R: CpG island methylation in
sporadic colorectal cancers and its relationship to microsatellite
instability. Gastroenterology. 122:1376–1387. 2002.PubMed/NCBI
|
|
116
|
Chan AOO, Issa JPJ, Morris JS, Hamilton SR
and Rashid A: Concordant CpG island methylation in hyperplastic
polyposis. Am J Pathol. 160:529–536. 2002.PubMed/NCBI
|
|
117
|
Wynter CVA, Walsh MD, Higuchi T, Leggett
BA, Young J and Jass JR: Methylation patterns define two types of
hyperplastic polyp associated with colorectal cancer. Gut.
53:573–580. 2004.PubMed/NCBI
|
|
118
|
Minoo P, Baker K, Goswami R, Chong G,
Foulkes WD, Ruszkiewicz AR, Barker M, Buchanan D, Young J and Jass
JR: Extensive DNA methylation in normal colorectal mucosa in
hyperplastic polyposis. Gut. 55:1467–1474. 2006.PubMed/NCBI
|
|
119
|
Shima K, Morikawa T, Yamauchi M, Kuchiba
A, Imamura Y, Liao X, Meyerhardt JA, Fuchs CS and Ogino S: TGFBR2
and BAX mononucleotide tract mutations, microsatellite instability,
and prognosis in 1072 colorectal cancers. PLoS One.
6:e250622011.PubMed/NCBI
|
|
120
|
Edelstein DL, Axilbund JE, Hylind LM,
Romans K, Griffin CA, Cruz-Correa M and Giardiello FM: Serrated
polyposis: Rapid and relentless development of colorectal
neoplasia. Gut. 62:404–408. 2013.
|
|
121
|
Bettington M, Walker N, Rosty C, Brown I,
Clouston A, McKeone D, Pearson SA, Leggett B and Whitehall V:
Clinicopathological and molecular features of sessile serrated
adenomas with dysplasia or carcinoma. Gut. 66:97–106. 2017.
|
|
122
|
Johnson CM, Wei C, Ensor JE, Smolenski DJ,
Amos CI, Levin B and Berry DA: Meta-analyses of colorectal cancer
risk factors. Cancer Causes Control. 24:1207–1222. 2013.PubMed/NCBI
|
|
123
|
Zisman AL, Nickolov A, Brand RE, Gorchow A
and Roy HK: Associations between the age at diagnosis and location
of colorectal cancer and the use of alcohol and tobacco:
Implications for screening. Arch Intern Med. 166:629–634.
2006.PubMed/NCBI
|
|
124
|
Botteri E, Iodice S, Bagnardi V, Raimondi
S, Lowenfels AB and Maisonneuve P: Smoking and colorectal cancer: A
meta-analysis. JAMA. 300:2765–2778. 2008.PubMed/NCBI
|
|
125
|
Pande M, Lynch PM, Hopper JL, Jenkins MA,
Gallinger S, Haile RW, LeMarchand L, Lindor NM, Campbell PT,
Newcomb PA, et al: Smoking and colorectal cancer in Lynch syndrome:
Results from the colon cancer family registry and the University of
Texas M.D. Anderson cancer center. Clin Cancer Res. 16:1331–1339.
2010.PubMed/NCBI
|
|
126
|
Figueiredo JC, Crockett SD, Snover DC,
Morris CB, McKeown-Eyssen G, Sandler RS, Ahnen DJ, Robertson DJ,
Burke CA, Bresalier RS, et al: Smoking-associated risks of
conventional adenomas and serrated polyps in the colorectum. Cancer
Causes Control. 26:377–386. 2015.
|
|
127
|
Limsui D, Vierkant RA, Tillmans LS, Wang
AH, Weisenberger DJ, Laird PW, Lynch CF, Anderson KE, French AJ,
Haile RW, et al: Cigarette smoking and colorectal cancer risk by
molecularly defined subtypes. J Natl Cancer Inst. 102:1012–1022.
2010.PubMed/NCBI
|
|
128
|
Hannan LM, Jacobs EJ and Thun MJ: The
association between cigarette smoking and risk of colorectal cancer
in a large prospective cohort from the United States. Cancer
Epidemiol Biomarkers Prev. 18:3362–3367. 2009.PubMed/NCBI
|
|
129
|
Ordóñez-Mena JM, Walter V, Schöttker B,
Jenab M, O'Doherty MG, Kee F, Bueno-de-Mesquita B, Peeters PHM,
Stricker BH, Ruiter R, et al: Impact of prediagnostic smoking and
smoking cessation on colorectal cancer prognosis: A meta-analysis
of individual patient data from cohorts within the CHANCES
consortium. Ann Oncol. 29:472–483. 2018.
|
|
130
|
Pöschl G and Seitz HK: Alcohol and cancer.
Alcohol Alcohol. 39:155–165. 2004.PubMed/NCBI
|
|
131
|
Fedirko V, Tramacere I, Bagnardi V, Rota
M, Scotti L, Islami F, Negri E, Straif K, Romieu I, La Vecchia C,
et al: Alcohol drinking and colorectal cancer risk: An overall and
dose-response meta-analysis of published studies. Ann Oncol.
22:1958–1972. 2011.PubMed/NCBI
|
|
132
|
Choi YJ, Myung SK and Lee JH: Light
alcohol drinking and risk of cancer: A meta-analysis of cohort
studies. Cancer Res Treat. 50:474–487. 2018. View Article : Google Scholar :
|
|
133
|
Cho E, Smith-Warner SA, Ritz J, van den
Brandt PA, Colditz GA, Folsom AR, Freudenheim JL, Giovannucci E,
Goldbohm RA, Graham S, et al: Alcohol intake and colorectal cancer:
A pooled analysis of 8 cohort studies. Ann Intern Med. 140:603–613.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Praud D, Rota M, Rehm J, Shield K,
Zatoński W, Hashibe M, La Vecchia C and Boffetta P: Cancer
incidence and mortality attributable to alcohol consumption. Int J
Cancer. 138:1380–1387. 2016. View Article : Google Scholar
|
|
135
|
Nelson DE, Jarman DW, Rehm J, Greenfield
TK, Rey G, Kerr WC, Miller P, Shield KD, Ye Y and Naimi TS:
Alcohol-attributable cancer deaths and years of potential life lost
in the United States. Am J Public Health. 103:641–648. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Butler LM, Sinha R, Millikan RC, Martin
CF, Newman B, Gammon MD, Ammerman AS and Sandler RS: Heterocyclic
amines, meat intake, and association with colon cancer in a
population-based study. Am J Epidemiol. 157:434–445. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Kampman E, Slattery ML, Bigler J, Leppert
M, Samowitz W, Caan BJ and Potter JD: Meat consumption, genetic
susceptibility, and colon cancer risk: A United States multicenter
case-control study. Cancer Epidemiol Biomarkers Prev. 8:15–24.
1999.PubMed/NCBI
|
|
138
|
Santarelli RL, Pierre F and Corpet DE:
Processed meat and colorectal cancer: A review of epidemiologic and
experimental evidence. Nutr Cancer. 60:131–144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kabat GC, Miller AB, Jain M and Rohan TE:
A cohort study of dietary iron and heme iron intake and risk of
colorectal cancer in women. Br J Cancer. 97:118–122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sinha R: An epidemiologic approach to
studying heterocyclic amines. Mutat Res. 506-507:197–204. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Pietinen P, Malila N, Virtanen M, Hartman
TJ, Tangrea JA, Albanes D and Virtamo J: Diet and risk of
colorectal cancer in a cohort of Finnish men. Cancer Causes
Control. 10:387–396. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Terry P, Giovannucci E, Michels KB,
Bergkvist L, Hansen H, Holmberg L and Wolk A: Fruit, vegetables,
dietary fiber, and risk of colorectal cancer. J Natl Cancer Inst.
93:525–533. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Bardou M, Barkun AN and Martel M: Obesity
and colorectal cancer. Gut. 62:933–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Kelly DM and Jones TH: Testosterone and
obesity. Obes Rev. 16:581–606. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Matsuo K, Mizoue T, Tanaka K, Tsuji I,
Sugawara Y, Sasazuki S, Nagata C, Tamakoshi A, Wakai K, Inoue M, et
al: Association between body mass index and the colorectal cancer
risk in Japan: Pooled analysis of population-based cohort studies
in Japan. Ann Oncol. 23:479–490. 2012. View Article : Google Scholar
|
|
146
|
Goh LY and Goh KL: Obesity: An
epidemiological perspective from Asia and its relationship to
gastrointestinal and liver cancers. J Gastroenterol Hepatol.
28(Suppl 4): S54–S58. 2013. View Article : Google Scholar
|
|
147
|
World Cancer Research Fund: Continuous
update project report. Food, nutrition, physical activity, and the
prevention of colorectal cancer. In: WCRF/AICR; 2011
|
|
148
|
Song M, Hu FB, Spiegelman D, Chan AT, Wu
K, Ogino S, Fuchs CS, Willett WC and Giovannucci EL: Long-term
status and change of body fat distribution, and risk of colorectal
cancer: A prospective cohort study. Int J Epidemiol. 45:871–883.
2016. View Article : Google Scholar :
|
|
149
|
Moore LL, Bradlee ML, Singer MR, Splansky
GL, Proctor MH, Ellison RC and Kreger BE: BMI and waist
circumference as predictors of lifetime colon cancer risk in
Framingham study adults. Int J Obes Relat Metab Disord. 28:559–567.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Samaras K, Botelho NK, Chisholm DJ and
Lord RV: Subcutaneous and visceral adipose tissue gene expression
of serum adipokines that predict type 2 diabetes. Obesity (Silver
Spring). 18:884–889. 2010. View Article : Google Scholar
|
|
151
|
Lim U, Ernst T, Buchthal SD, Latch M,
Albright CL, Wilkens LR, Kolonel LN, Murphy SP, Chang L, Novotny R
and Le Marchand L: Asian women have greater abdominal and visceral
adiposity than Caucasian women with similar body mass index. Nutr
Diabetes. 1:e62011. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ma Y, Yang Y, Wang F, Zhang P, Shi C, Zou
Y and Qin H: Obesity and risk of colorectal cancer: A systematic
review of prospective studies. PLoS One. 8:e539162013. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Lee YJ, Myung SK, Cho B, Park BJ, Park JH,
Ju W, Park MS and Choi JH: Adiposity and the risk of colorectal
adenomatous polyps: A meta-analysis. Cancer Causes Control.
22:1021–1035. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Guraya SY: Association of type 2 diabetes
mellitus and the risk of colorectal cancer: A meta-analysis and
systematic review. World J Gastroenterol. 21:6026–6031. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Kahn SE, Hull RL and Utzschneider KM:
Mechanisms linking obesity to insulin resistance and type 2
diabetes. Nature. 444:840–846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Moller DE and Flier JS: Insulin
resistance-mechanisms, syndromes, and implications. N Engl J Med.
325:938–948. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Calle EE and Kaaks R: Overweight, obesity
and cancer: Epidemiological evidence and proposed mechanisms. Nat
Rev Cancer. 4:579–591. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Murphy N, Strickler HD, Stanczyk FZ, Xue
X, Wassertheil-Smoller S, Rohan TE, Ho GY, Anderson GL, Potter JD
and Gunter MJ: A prospective evaluation of endogenous sex hormone
levels and colorectal cancer risk in postmenopausal women. J Natl
Cancer Inst. 107. pp. djv2102015, View Article : Google Scholar
|
|
160
|
Hetemäki N, Savolainen-Peltonen H,
Tikkanen MJ, Wang F, Paatela H, Hämäläinen E, Turpeinen U, Haanpää
M, Vihma V and Mikkola TS: Estrogen metabolism in abdominal
subcutaneous and visceral adipose tissue in postmenopausal women. J
Clin Endocrinol Metab. 102:4588–4595. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Burkitt DP: Epidemiology of cancer of the
colon and rectum. Cancer. 28:3–13. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Aune D, Chan DS, Lau R, Vieira R,
Greenwood DC, Kampman E and Norat T: Dietary fibre, whole grains,
and risk of colorectal cancer: Systematic review and dose-response
meta-analysis of prospective studies. BMJ. 343. pp. d66172011,
View Article : Google Scholar
|
|
163
|
World Cancer Research Fund: Diet,
nutrition, physical activity and cancer: A global perspective: A
summary of the third expert report. World Cancer Research Fund
International. 2018.
|
|
164
|
Rezende LFM, Sá TH, Markozannes G,
Rey-López JP, Lee IM, Tsilidis KK, Ioannidis JPA and Eluf-Neto J:
Physical activity and cancer: An umbrella review of the literature
including 22 major anatomical sites and 770 000 cancer cases. Br J
Sports Med. 52:826–833. 2018. View Article : Google Scholar
|
|
165
|
Morris JS, Bradbury KE, Cross AJ, Gunter
MJ and Murphy N: Physical activity, sedentary behaviour and
colorectal cancer risk in the UK Biobank. Br J Cancer. 118:920–929.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Keum N, Bao Y, Smith-Warner SA, Orav J, Wu
K, Fuchs CS and Giovannucci EL: Association of physical activity by
type and intensity with digestive system cancer risk. JAMA Oncol.
2:1146–1153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Ma P, Yao Y, Sun W, Dai S and Zhou C:
Daily sedentary time and its association with risk for colorectal
cancer in adults: A dose-response meta-analysis of prospective
cohort studies. Medicine (Baltimore). 96:e70492017. View Article : Google Scholar
|
|
168
|
Lynch BM: Sedentary behavior and cancer: A
systematic review of the literature and proposed biological
mechanisms. Cancer Epidemiol Biomarkers Prev. 19:2691–2709. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Howard RA, Freedman DM, Park Y, Hollenbeck
A, Schatzkin A and Leitzmann MF: Physical activity, sedentary
behavior, and the risk of colon and rectal cancer in the NIH-AARP
diet and health study. Cancer Causes Control. 19:939–953. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Jeong WJ, Ro EJ and Choi KY: Interaction
between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer
strategy via degradations of β-catenin and RAS by targeting the
Wnt/β-catenin pathway. NPJ Precis Oncol. 2:52018. View Article : Google Scholar
|
|
171
|
Novellasdemunt L, Antas P and Li VS:
Targeting Wnt signalling in colorectal cancer. A review in the
theme: Cell Signalling: Proteins, pathways and mechanisms. Am J
Physiol Cell Physiol. 309:C511–C521. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Farooqi AA, de la Roche M, Djamgoz MBA and
Siddik ZH: Overview of the oncogenic signalling pathways in
colorectal cancer: Mechanistic insights. Semin Cancer Biol.
58:65–79. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Koveitypour Z, Panahi F, Vakilian M,
Peymani M, Seyed Forootan F, Nasr Esfahani MH and Ghaedi K:
Signaling pathways involved in colorectal cancer progression. Cell
Biosci. 9:972019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Pandurangan AK, Divya T, Kumar K,
Dineshbabu V, Velavan B and Sudhandiran G: Colorectal
carcinogenesis: Insights into the cell death and signal
transduction pathways: A review. World J Gastrointest Oncol.
10:244–259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Tiwari A, Saraf S, Verma A, Panda PK and
Jain SK: Novel targeting approaches and signaling pathways of
colorectal cancer: An insight. World J Gastroenterol. 24:4428–4435.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Folkman J: Tumor angiogenesis: Therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Saharinen P, Eklund L, Pulkki K, Bono P
and Alitalo K: VEGF and angiopoietin signaling in tumor
angiogenesis and metastasis. Trends Mol Med. 17:347–362. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Goel HL and Mercurio AM: VEGF targets the
tumor cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Lee YJ, Karl DL, Maduekwe UN, Rothrock C,
Ryeom S, D'Amore PA and Yoon SS: Differential effects of VEGFR-1
and VEGFR-2 inhibition on tumor metastases based on host organ
environment. The biology of VEGF and its receptors. Cancer Res.
70:8357–8367. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Vaahtomeri K, Karaman S, Mäkinen T and
Alitalo K: Lymphangiogenesis guidance by paracrine and pericellular
factors. Genes Dev. 31:1615–1634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling-in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Takahashi H and Shibuya M: The vascular
endothelial growth factor (VEGF)/VEGF receptor system and its role
under physiological and pathological conditions. Clin Sci (Lond).
109:227–241. 2005. View Article : Google Scholar
|
|
184
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Garnier L, Gkountidi AO and Hugues S:
Tumor-associated lymphatic vessel features and immunomodulatory
functions. Front Immunol. 10:7202019. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Secker GA and Harvey NL: VEGFR signaling
during lymphatic vascular development: From progenitor cells to
functional vessels. Dev Dyn. 244:323–331. 2015. View Article : Google Scholar
|
|
187
|
Cébe-Suarez S, Zehnder-Fjällman A and
Ballmer-Hofer K: The role of VEGF receptors in angiogenesis;
complex partnerships. Cell Mol Life Sci. 63:601–615. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Lopez A, Harada K, Vasilakopoulou M,
Shanbhag N and Ajani JA: Targeting angiogenesis in colorectal
carcinoma. Drugs. 79:63–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Seeber A, Gunsilius E, Gastl G and Pircher
A: Anti-angiogenics: Their value in colorectal cancer therapy.
Oncol Res Treat. 41:188–193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Guba M, Seeliger H, Kleespies A, Jauch KW
and Bruns C: Vascular endothelial growth factor in colorectal
cancer. Int J Colorectal Dis. 19:510–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Jain RK: Antiangiogenesis strategies
revisited: From starving tumors to alleviating hypoxia. Cancer
Cell. 26:605–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Amelio I and Melino G: The p53 family and
the hypoxia-inducible factors (HIFs): Determinants of cancer
progression. Trends Biochem Sci. 40:425–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Tarnawski AS, Ahluwalia A and Jones MK:
Angiogenesis in gastric mucosa: An important component of gastric
erosion and ulcer healing and its impairment in aging. J
Gastroenterol Hepatol. 29(Suppl 4): S112–S23. 2014. View Article : Google Scholar
|
|
194
|
Wang Q, Yang S, Wang K and Sun SY: MET
inhibitors for targeted therapy of EGFR TKI-resistant lung cancer.
J Hematol Oncol. 12:632019. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Cheng F and Guo D: MET in glioma:
Signaling pathways and targeted therapies. J Exp Clin Cancer Res.
38:2702019. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Demkova L and Kucerova L: Role of the
HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol
Cancer. 17:262018. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Lam BQ, Dai L and Qin Z: The role of
HGF/c-MET signaling pathway in lymphoma. J Hematol Oncol.
9:1352016. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Bradley CA, Salto-Tellez M, Laurent-Puig
P, Bardelli A, Rolfo C, Tabernero J, Khawaja HA, Lawler M, Johnston
PG and Van Schaeybroeck S; MErCuRIC consortium: Targeting c-MET in
gastrointestinal tumors: Rationale, opportunities and challenges.
Nat Rev Clin Oncol. 14:562–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Xing F, Liu Y, Sharma S, Wu K, Chan MD, Lo
HW, Carpenter RL, Metheny-Barlow LJ, Zhou X, Qasem SA, et al:
Activation of the c-Met pathway mobilizes an inflammatory network
in the brain microenvironment to promote brain metastasis of breast
cancer. Cancer Res. 76:4970–4980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Ozawa Y, Nakamura Y, Fujishima F, Felizola
SJ, Takeda K, Okamoto H, Ito K, Ishida H, Konno T, Kamei T, et al:
c-Met in esophageal squamous cell carcinoma: An independent
prognostic factor and potential therapeutic target. BMC Cancer.
15:4512015. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Anestis A, Zoi I and Karamouzis MV:
Current advances of targeting HGF/c-Met pathway in gastric cancer.
Ann Transl Med. 6:2472018. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Safaie Qamsari E, Safaei Ghaderi S, Zarei
B, Dorostkar R, Bagheri S, Jadidi-Niaragh F, Somi MH and Yousefi M:
The c-Met receptor: Implication for targeted therapies in
colorectal cancer. Tumor Biol. 39:10104283176991182017. View Article : Google Scholar
|
|
203
|
Otte JM, Schmitz F, Kiehne K, Stechele HU,
Banasiewicz T, Krokowicz P, Nakamura T, Fölsch UR and Herzig K:
Functional expression of HGF and its receptor in human colorectal
cancer. Digestion. 61:237–246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Matsumoto K, Umitsu M, De Silva DM, Roy A
and Bottaro DP: Hepatocyte growth factor/MET in cancer progression
and biomarker discovery. Cancer Sci. 108:296–307. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Bahrami A, Shahidsales S, Khazaei M,
Ghayour-Mobarhan M, Maftouh M, Hassanian SM and Avan A: C-Met as a
potential target for the treatment of gastrointestinal cancer:
Current status and future perspectives. J Cell Physiol.
232:2657–2673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Mo HN and Liu P: Targeting MET in cancer
therapy. Chronic Dis Transl Med. 3:148–153. 2017.PubMed/NCBI
|
|
207
|
Bouattour M, Raymond E, Qin S, Cheng AL,
Stammberger U, Locatelli G and Faivre S: Recent developments of
c-Met as a therapeutic target in hepatocellular carcinoma.
Hepatology. 67:1132–1149. 2018. View Article : Google Scholar :
|
|
208
|
Drilon A, Cappuzzo F, Ou SI and Camidge
DR: Targeting MET in lung cancer: Will expectations finally be MET?
J Thorac Oncol. 12:15–26. 2017. View Article : Google Scholar :
|
|
209
|
Blumenschein GR Jr, Mills GB and
Gonzalez-Angulo AM: Targeting the hepatocyte growth factor-cMET
axis in cancer therapy. J Clin Oncol. 30:3287–3296. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Cabanillas ME, de Souza JA, Geyer S, Wirth
LJ, Menefee ME, Liu SV, Shah K, Wright J and Shah MH: Cabozantinib
as salvage therapy for patients with tyrosine kinase
inhibitor-refractory differentiated thyroid cancer: Results of a
multicenter phase II international thyroid oncology group trial. J
Clin Oncol. 35:3315–3321. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Cancer Genome Atlas Research Network;
Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis
C, Wheeler DA, Murray BA, Schmidt L, et al: Comprehensive molecular
characterization of papillary renal-cell carcinoma. N Engl J Med.
374:135–145. 2016. View Article : Google Scholar
|
|
212
|
Catenacci DVT, Tebbutt NC, Davidenko I,
Murad AM, Al-Batran SE, Ilson DH, Tjulandin S, Gotovkin E,
Karaszewska B, Bondarenko I, et al: Rilotumumab plus epirubicin,
cisplatin, and capecitabine as first-line therapy in advanced
MET-positive gastric or gastro-oesophageal junction cancer
(RILOMET-1): A randomised, double-blind, placebo-controlled, phase
3 trial. Lancet Oncol. 18:1467–1482. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sartore-Bianchi A, Loupakis F, Argilés G
and Prager GW: Challenging chemoresistant metastatic colorectal
cancer: Therapeutic strategies from the clinic and from the
laboratory. Ann Oncol. 27:1456–1466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Gao H, Guan M, Sun Z and Bai C: High c-Met
expression is a negative prognostic marker for colorectal cancer: A
meta-analysis. Tumor Biol. 36:515–520. 2015. View Article : Google Scholar
|
|
215
|
Luo HY and Xu RH: Predictive and
prognostic biomarkers with therapeutic targets in advanced
colorectal cancer. World J Gastroenterol. 20:3858–3874. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Baldus SE, Kort EJ, Schirmacher P, Dienes
HP and Resau JH: Quantification of MET and hepatocyte growth
factor/scatter factor expression in colorectal adenomas, carcinomas
and non-neoplastic epithelia by quantitative laser scanning
microscopy. Int J Oncol. 31:199–204. 2007.PubMed/NCBI
|
|
217
|
Kentsis A, Reed C, Rice KL, Sanda T, Rodig
SJ, Tholouli E, Christie A, Valk PJ, Delwel R, Ngo V, et al:
Autocrine activation of the MET receptor tyrosine kinase in acute
myeloid leukemia. Nat Med. 18:1118–1122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Stein U, Walther W, Arlt F, Schwabe H,
Smith J, Fichtner I, Birchmeier W and Schlag PM: MACC1, a newly
identified key regulator of HGF-MET signaling, predicts colon
cancer metastasis. Nat Med. 15:59–67. 2009. View Article : Google Scholar
|
|
219
|
Parseghian CM, Napolitano S, Loree JM and
Kopetz S: Mechanisms of innate and acquired resistance to anti-EGFR
therapy: A review of current knowledge with a focus on rechallenge
therapies. Clin Cancer Res. 25:6899–6908. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Boccaccio C, Luraghi P and Comoglio PM:
MET-mediated resistance to EGFR inhibitors: An old liaison rooted
in colorectal cancer stem cells. Cancer Res. 74:3647–3651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Van Emburgh BO, Sartore-Bianchi A, Di
Nicolantonio F, Siena S and Bardelli A: Acquired resistance to
EGFR-targeted therapies in colorectal cancer. Mol Oncol.
8:1084–1094. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Viticchiè G and Muller PAJ: c-Met and
other cell surface molecules: Interaction, activation and
functional consequences. Biomedicines. 3:46–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Sharpe AH and Freeman GJ: The B7-CD28
superfamily. Nat Rev Immunol. 2:116–126. 2002.PubMed/NCBI
|
|
224
|
Jelinek T, Mihalyova J, Kascak M, Duras J
and Hajek R: PD-1/PD-L1 inhibitors in haematological malignancies:
Update 2017. Immunology. 152:357–371. 2017.PubMed/NCBI
|
|
225
|
Chen L and Han X: Anti-PD-1/PD-L1 therapy
of human cancer: Past, present, and future. J Clin Invest.
125:3384–3391. 2015.PubMed/NCBI
|
|
226
|
Markman JL and Shiao SL: Impact of the
immune system and immunotherapy in colorectal cancer. J
Gastrointest Oncol. 6:208–223. 2015.PubMed/NCBI
|
|
227
|
Xie YH, Chen YX and Fang JY: Comprehensive
review of targeted therapy for colorectal cancer. Signal Transduct
Target Ther. 5:222020.PubMed/NCBI
|
|
228
|
Pauken KE and Wherry EJ: Overcoming T cell
exhaustion in infection and cancer. Trends Immunol. 36:265–276.
2015.PubMed/NCBI
|
|
229
|
Wang HB, Yao H, Li CS, Liang LX, Zhang Y,
Chen YX, Fang JY and Xu J: Rise of PD-L1 expression during
metastasis of colorectal cancer: Implications for immunotherapy. J
Dig Dis. 18:574–581. 2017.PubMed/NCBI
|
|
230
|
Fujimoto H, Saito Y, Ohuchida K, Kawakami
E, Fujiki S, Watanabe T, Ono R, Kaneko A, Takagi S, Najima Y, et
al: Deregulated mucosal immune surveillance through gut-associated
regulatory T cells and PD-1+ T cells in human colorectal
cancer. J Immunol. 200:3291–3303. 2018.PubMed/NCBI
|
|
231
|
Vinson KE, George DC, Fender AW, Bertrand
FE and Sigounas G: The Notch pathway in colorectal cancer. Int J
Cancer. 138:1835–1842. 2016.
|
|
232
|
Zhi X, Tao J, Zhang L, Tao R, Ma L and Qin
J: Silencing speckle-type POZ protein by promoter hypermethylation
decreases cell apoptosis through upregulating hedgehog signaling
pathway in colorectal cancer. Cell Death Dis.
7:e25692016.PubMed/NCBI
|
|
233
|
Wu C, Zhu X, Liu W, Ruan T and Tao K:
Hedgehog signalling pathway in colorectal cancer: Function,
mechanism, and therapy. Onco Targets Ther. 10:3249–3259. 2017.
|
|
234
|
Han Y: Analysis of the role of the Hippo
pathway in cancer. J Transl Med. 17:1162019.PubMed/NCBI
|
|
235
|
Zhang YE: Non-Smad pathways in TGF-β
signaling. Cell Res. 19:128–139. 2009.
|
|
236
|
Cheruku HR, Mohamedali A, Cantor DI, Tan
SH, Nice EC and Baker MS: Transforming growth factor-β, MAPK and
Wnt signaling interaction in colorectal cancer. EuPA Open
Proteomics. 8:104–115. 2015.
|
|
237
|
Luo K: Signaling cross talk between
TGF-β/Smad and other signaling pathways. Cold Spring Harb Perspect
Biol. 9:a0221372017.
|
|
238
|
Lee Y, Kim NH, Cho ES, Yang JH, Cha YH,
Kang HE, Yun JS, Cho SB, Lee SH, Paclikova P, et al: Dishevelled
has a YAP nuclear export function in a tumor suppressor
context-dependent manner. Nat Commun. 9:23012018.PubMed/NCBI
|
|
239
|
Jing L, Li J, Zhang C, Shang Y and Lin J:
YAP-mediated crosstalk between the Wnt and Hippo signaling pathways
(Review). Mol Med Rep. 22:4101–4106. 2020.
|
|
240
|
Pietrobono S, Gagliardi S and Stecca B:
Non-canonical hedgehog signalling pathway in cancer: Activation of
GLI transcription factors beyond smoothened. Front Genet.
10:5562019.
|
|
241
|
Regan JL, Schumacher D, Staudte S, Steffen
A, Haybaeck J, Keilholz U, Schweiger C, Golob-Schwarzl N, Mumberg
D, Henderson D, et al: Non-canonical hedgehog signaling is a
positive regulator of the WNT pathway and is required for the
survival of colon cancer stem cells. Cell Rep. 21:2813–2828.
2017.PubMed/NCBI
|
|
242
|
Heidelberger C, Chaudhuri NK, Danneberg P,
Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E and
Scheiner J: Fluorinated pyrimidines, a new class of
tumor-inhibitory compounds. Nature. 179:663–666. 1957.PubMed/NCBI
|
|
243
|
Piawah S and Venook AP: Targeted therapy
for colorectal cancer metastases: A review of current methods of
molecularly targeted therapy and the use of tumor biomarkers in the
treatment of metastatic colorectal cancer. Cancer. 125:4139–4147.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Piedbois P, Buyse M, Rustum Y, Machover D,
Erlichman C, Carlson RW, Valone F, Labianca R, Doroshow JH and
Petrelli N: Modulation of fluorouracil by leucovorin in patients
with advanced colorectal cancer: Evidence in terms of response rate
by the advanced colorectal cancer meta-analysis project. J Clin
Oncol. 10:896–903. 1992. View Article : Google Scholar
|
|
245
|
de Gramont A, Figer A, Seymour M, Homerin
M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer
G, et al: Leucovorin and fluorouracil with or without oxaliplatin
as first-line treatment in advanced colorectal cancer. J Clin
Oncol. 18:2938–2947. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Douillard JY, Cunningham D, Roth AD,
Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J,
Alakl M, et al: Irinotecan combined with fluorouracil compared with
fluorouracil alone as first-line treatment for metastatic
colorectal cancer: A multicentre randomised trial. Lancet.
355:1041–1047. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Tournigand C, André T, Achille E, Lledo G,
Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, et
al: FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced
colorectal cancer: A randomized GERCOR study. J Clin Oncol.
22:229–237. 2004. View Article : Google Scholar
|
|
248
|
Colucci G, Gebbia V, Paoletti G, Giuliani
F, Caruso M, Gebbia N, Cartenì G, Agostara B, Pezzella G, Manzione
L, et al: Phase III randomized trial of FOLFIRI versus FOLFOX4 in
the treatment of advanced colorectal cancer: A multicenter study of
the gruppo oncologico Dell'Italia meridionale. J Clin Oncol.
23:4866–4875. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Cassidy J, Clarke S, Díaz-Rubio E,
Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS,
Rivera F, et al: Randomized phase III study of capecitabine plus
oxaliplatin compared with fluorouracil/folinic acid plus
oxaliplatin as first-line therapy for metastatic colorectal cancer.
J Clin Oncol. 26:2006–2012. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Skof E, Rebersek M, Hlebanja Z and Ocvirk
J: Capecitabine plus irinotecan (XELIRI regimen) compared to
5-FU/LV plus Irinotecan (FOLFIRI regimen) as neoadjuvant treatment
for patients with unresectable liver-only metastases of metastatic
colorectal cancer: A randomised prospective phase II trial. BMC
Cancer. 9:1202009. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Obrand DI and Gordon PH: Incidence and
patterns of recurrence following curative resection for colorectal
carcinoma. Dis Colon Rectum. 40:15–24. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Brodsky FM: Monoclonal antibodies as magic
bullets. Pharm Res. 5:1–9. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Lee YT, Tan YJ and Oon CE: Molecular
targeted therapy: Treating cancer with specificity. Eur J
Pharmacol. 834:188–196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Martinelli E, Ciardiello D, Martini G,
Troiani T, Cardone C, Vitiello PP, Normanno N, Rachiglio AM,
Maiello E, Latiano T, et al: Implementing anti-epidermal growth
factor receptor (EGFR) therapy in metastatic colorectal cancer:
Challenges and future perspectives. Ann Oncol. 31:30–40. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Oh DY and Bang YJ: HER2-targeted
therapies-a role beyond breast cancer. Nat Rev Clin Oncol.
17:33–48. 2020. View Article : Google Scholar
|
|
256
|
Ferguson FM and Gray NS: Kinase
inhibitors: The road ahead. Nat Rev Drug Discov. 17:353–377. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Tariman JD: Changes in cancer treatment:
Mabs, Mibs, Mids, Nabs, and Nibs. Nurs Clin North Am. 52:65–81.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
André T, Blons H, Mabro M, Chibaudel B,
Bachet JB, Tournigand C, Bennamoun M, Artru P, Nguyen S, Ebenezer
C, et al: Panitumumab combined with irinotecan for patients with
KRAS wild-type metastatic colorectal cancer refractory to standard
chemotherapy: A GERCOR efficacy, tolerance, and translational
molecular study. Ann Oncol. 24:412–419. 2013. View Article : Google Scholar
|
|
259
|
Papadatos-Pastos D, Rabbie R, Ross P and
Sarker D: The role of the PI3K pathway in colorectal cancer. Crit
Rev Oncol Hematol. 94:18–30. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
260
|
Katz LH, Li Y, Chen JS, Muñoz NM, Majumdar
A, Chen J and Mishra L: Targeting TGF-β signaling in cancer. Expert
Opin Ther Targets. 17:743–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Jin D, Fang Y, Li Z, Chen Z and Xiang J:
Epithelial-mesenchymal transition-associated microRNAs in
colorectal cancer and drug-targeted therapies (Review). Oncol Rep.
33:515–525. 2015. View Article : Google Scholar
|
|
262
|
Qiao L and Wong BC: Role of Notch
signaling in colorectal cancer. Epithelial-mesenchymal
transition-associated microRNAs in colorectal cancer and
drug-targeted therapies (Review). Carcinogenesis. 30:1979–1986.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Katoh M and Katoh M: Precision medicine
for human cancers with Notch signaling dysregulation (Review). Int
J Mol Med. 45:279–297. 2020.PubMed/NCBI
|
|
264
|
Fragoulis GE, Mclnnes IB and Siebert S:
JAK-inhibitors New players in the field of immune-mediated diseases
beyond rheumatoid arthritis. Rheumatology (Oxford). 58(Suppl 1):
pp. i42–i54. 2019, View Article : Google Scholar
|
|
265
|
Wang SW, Hu J, Guo QH, Zhao Y, Cheng JJ,
Zhang DS, Fei Q, Li J and Sun YM: AZD1480, a JAK inhibitor,
inhibits cell growth and survival of colorectal cancer via
modulating the JAK2/STAT3 signaling pathway. Oncol Rep.
32:1991–1998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
266
|
Gulino A, Ferretti E and De Smaele E:
Hedgehog signalling in colon cancer and stem cells. EMBO Mol Med.
1:300–302. 2009. View Article : Google Scholar
|
|
267
|
Wu JY, Xu XF, Xu L, Niu PQ, Wang F, Hu GY,
Wang XP and Guo CY: Cyclopamine blocked the growth of colorectal
cancer SW116 cells by modulating some target genes of Gli1 in
vitro. Hepatogastroenterology. 58:1511–1518. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Varnat F, Duquet A, Malerba M, Zbinden M,
Mas C, Gervaz P and Ruiz i Altaba A: Human colon cancer epithelial
cells harbour active HEDGEHOG-GLI signalling that is essential for
tumour growth, recurrence, metastasis and stem cell survival and
expansion. EMBO Mol Med. 1:338–351. 2009. View Article : Google Scholar
|
|
269
|
Gonzalez-Donquiles C, Alonso-Molero J,
Fernandez-Villa T, Vilorio-Marqués L, Molina AJ and Martín V: The
NRF2 transcription factor plays a dual role in colorectal cancer: A
systematic review. PLoS One. 12:e01775492017. View Article : Google Scholar : PubMed/NCBI
|
|
270
|
Wu WK, Wang XJ, Cheng AS, Luo MX, Ng SS,
To KF, Chan FK, Cho CH, Sung JJ and Yu J: Dysregulation and
crosstalk of cellular signaling pathways in colon carcinogenesis.
Crit Rev Oncol Hematol. 86:251–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
271
|
Hurwitz H, Fehrenbacher L, Novotny W,
Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S,
Holmgren E, et al: Bevacizumab plus irinotecan, fluorouracil, and
leucovorin for metastatic colorectal cancer. N Engl J Med.
350:2335–2342. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
272
|
Passardi A, Nanni O, Tassinari D, Turci D,
Cavanna L, Fontana A, Ruscelli S, Mucciarini C, Lorusso V,
Ragazzini A, et al: Effectiveness of bevacizumab added to standard
chemotherapy in metastatic colorectal cancer: Final results for
first-line treatment from the ITACa randomized clinical trial. Ann
Oncol. 26:1201–1207. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
273
|
Tang PA, Cohen SJ, Kollmannsberger C,
Bjarnason G, Virik K, MacKenzie MJ, Lourenco L, Wang L, Chen A and
Moore MJ: Phase II clinical and pharmacokinetic study of
aflibercept in patients with previously treated metastatic
colorectal cancer. Clin Cancer Res. 18:6023–6031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Van Cutsem E, Sobrero AF, Siena S, Falcone
A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, et
al: Phase III CORRECT trial of regorafenib in metastatic colorectal
cancer (mCRC). J Clin Oncol. 30(15 Suppl): S35022012. View Article : Google Scholar
|
|
275
|
Tabernero J, Yoshino T, Cohn AL,
Obermannova R, Bodoky G, Garcia-Carbonero R, Ciuleanu TE, Portnoy
DC, Van Cutsem E, Grothey A, et al: Ramucirumab versus placebo in
combination with second-line FOLFIRI in patients with metastatic
colorectal carcinoma that progressed during or after first-line
therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine
(RAISE): A randomised, double-blind, multicentre, phase 3 study.
Lancet Oncol. 16:499–508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
276
|
Bendell JC, Ervin TJ, Gallinson D, Singh
J, Wallace JA, Saleh MN, Vallone M, Phan SC and Hack SP: Treatment
rationale and study design for a randomized, double-blind,
placebo-controlled phase II study evaluating onartuzumab (MetMAb)
in combination with bevacizumab plus mFOLFOX-6 in patients with
previously untreated metastatic colorectal cancer. Clin Colorectal
Cancer. 12:218–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Shao Z, Pan H, Tu S, Zhang J, Yan S and
Shao A: HGF/c-Met axis: The advanced development in digestive
system cancer. Front Cell Dev Biol. 8:8012020. View Article : Google Scholar : PubMed/NCBI
|
|
278
|
Ganesh K, Stadler ZK, Cercek A, Mendelsohn
RB, Shia J, Segal NH and Diaz LA Jr: Immunotherapy in colorectal
cancer: Rationale, challenges and potential. Nat Rev Gastroenterol
Hepatol. 16:361–375. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
279
|
Passardi A, Canale M, Valgiusti M and
Ulivi P: Immune checkpoints as a target for colorectal cancer
treatment. Int J Mol Sci. 18:13242017. View Article : Google Scholar :
|
|
280
|
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
281
|
Giannakis M, Mu XJ, Shukla SA, Qian ZR,
Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et
al: Genomic correlates of immune-cell infiltrates in colorectal
carcinoma. Cell Rep. 15:857–865. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Llosa NJ, Cruise M, Tam A, Wicks EC,
Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS,
et al: The vigorous immune microenvironment of microsatellite
instable colon cancer is balanced by multiple counter-inhibitory
checkpoints. Cancer Discov. 5:43–51. 2015. View Article : Google Scholar :
|
|
283
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
284
|
Marcus L, Lemery SJ, Keegan P and Pazdur
R: FDA approval summary: Pembrolizumab for the treatment of
microsatellite instability-high solid tumors. Clin Can Res.
25:3753–3758. 2019. View Article : Google Scholar
|
|
285
|
Andre T, Shiu KK, Kim TW, Jensen BV,
Jensen LH, Punt CJA, Smith DM, Garcia-Carbonero R, Benavides M,
Gibbs P, et al: Pembrolizumab vs chemotherapy for microsatellite
instability-high/mismatch repair deficient metastatic colorectal
cancer: The phase 3 KEYNOTE-177 study. J Clin Oncol. 38(18 Suppl):
LBA42020. View Article : Google Scholar
|
|
286
|
U.S. Food and Drug (FDA): FDA grants
nivolumab accelerated approval for MSI-H or dMMR colorectal cancer
FDA. Silver Spring, MD: 2017, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accel-erated-approval-msi-h-or-dmmr-colorectal-cancer.
Accessed January 8, 2017.
|
|
287
|
Sun J, Zheng Y, Mamun M, Li X, Chen X and
Gao Y: Research progress of PD-1/PD-L1 immunotherapy in
gastrointestinal tumors. Biomed Pharmacother. 129:1105042020.
View Article : Google Scholar : PubMed/NCBI
|