Introduction
Glioblastoma is one of the most malignant types of
brain cancer, characterized by high recurrence and mortality rates,
and a low cure rate (1,2). Due to the invasive growth of
glioma, tumor tissue cannot be completely removed with surgery, and
it can easily relapse in situ, making it difficult to cure.
The prognosis of patients with glioblastoma is unsatisfactory, with
an annual survival rate <30% and a 5-year survival rate <5%
in the United States between 2000 and 2014 (3,4).
The resistance of glioma chemotherapeutic drugs is the leading
cause of treatment failure (5).
Traditional chemotherapy agents, such as carmustine, cisplatin and
teniposide, are commonly used; however, they are only ~20%
efficient, with the efficiency of the newest drug, temozolomide, at
only 35% (6). Therefore,
developing new strategies and drugs to effectively inhibit glioma
growth has become one of the main research goals of scientists
worldwide.
A number of studies have demonstrated that
extracellular adenosine signaling is associated with glioma cell
proliferation, migration, invasion and mortality in animal models
(7). Although the concentration
of adenosine in the extracellular fluid of glioma tissue is in the
micromolar range, it is sufficient to activate all known adenosine
receptor subtypes (A1, A2A, A2B and A3) (8). Among them, A2A and A2B receptors
are mainly distributed on epithelial and a variety of immune cells,
and the A1 and A3 adenosine receptors are distributed on the tumor
cell membrane and serve a key role in tumor cell proliferation and
invasion (9). Under hypoxic and
normal oxygen environments, the same adenosine receptor may exert
different biological effects (10). Due to the regulatory effects of
adenosine receptors and their downstream signaling pathways,
chemical compounds or drugs that affect these receptor may inhibit
the proliferation and angiogenesis of glioma cells.
In our previous studies, a number of saponins (e.g.
saponin 6 of Anemone taipaiensis) with antiglioma activity
were isolated and identified from various medicinal plants, and the
feasibility of saponin treatment on malignant glioma was determined
by interstitial chemotherapy (11-13). Rhizoma Paridis (RP; the rhizome
of Paris) has a wide range of medicinal activities,
including anticancer, and has been used for the treatment of lung
cancer, brain tumors and malignant lymphomas (14). In previous studies, saponins from
RP showed good effects in inhibiting tumor effects, including
inducing apoptosis and cell cycle arrest, inhibiting angiogenesis
and increasing immunity (15,16). Paris saponin H (PSH), a
steroid saponin component of RP, shares similar characteristics
with Polyphyllin VII, Polyphyllin D and Paris Saponin II
(17). Polyphyllin VII and
Polyphyllin D have been demonstrated to exert their antiglioma
effects in vitro by inducing apoptosis (18,19). Paris Saponin II serves an
anti-angiogenetic role through reducing the levels of VEGF and
blocking the activity of VEGFR2 (20). Total steroidal saponins extracted
from the RP have been demonstrated to activate adenosine receptors
in rat platelets, and several saponins (including
N6-cyclohexyladenosine and 8-cyclopentyl1,
3-dipropyl-2,3-(N)-xanthine) exert inhibitory effects on A1 and A3
adenosine receptors (21,22).
However, the mechanism of PSH against glioma remains
unclear. Based on the aforementioned previous experiments, we
hypothesized that PSH may bind to the A1 and A3 adenosine receptors
on glioma cell membranes, inhibit their downstream signaling
pathways, promote tumor cell apoptosis and inhibit glioma growth.
On the other hand, the inhibition of the A3 receptor activation may
reduce hypoxia-inducible factor-1α (HIF-1α) synthesis, which may in
turn inhibit glioma angiogenesis and cell proliferation.
Materials and methods
Chemicals and reagents
PSH (purity, 98%) was obtained from the Department
of Natural Medicine, School of Pharmacy, Fourth Military Medical
University. Cell Counting Kit-8 (CCK-8) was purchased from Shanghai
7Sea PharmTech Co. Ltd. Human astrocytoma U251 cells were purchased
from the Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences. RIPA buffer, CHAPS buffer, caspase-3 (cat. no.
C1116) and caspase-9 (cat. no. C1158) activity assay kits were
purchased from Beyotime Institute for Biotechnology. VEGF ELISA kit
(cat. no. PDVE00) was purchased from R&D Systems, Inc.
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum
(FBS) were purchased from Gibco; Thermo Fisher Scientific, Inc. The
Annexin V-FITC/PI staining kit was purchased from Nanjing Jiancheng
Bioengineering Institute. The primary antibodies against HIF-1α
(cat. no. 36169), VEGF (cat. no. 2463), MAP kinase kinase 1/2
(MEK1/2) (cat. no. 8727), phosphorylated (P-)MEK1/2 (cat. no.
9154), P38 mitogen-activated protein kinase (MAPK; cat. no. 8690),
P-P38 MAPK (cat. no. 4511), Akt (cat. no. 4691), P-Akt (cat. no.
4060), P44/42 MAPK (cat. no. 4695), P-P44/42 MAPK (cat. no. 4370),
P27 (cat. no. 3686) and CDK4 (cat. no. 12790) were purchased from
Cell Signaling Technology, Inc. The peroxidase-conjugated
anti-rabbit (cat. no. BA1056) and anti-mouse (cat. no. BA1050)
secondary antibodies were purchased from Wuhan Boster Biological
Technology, Ltd. LY294002 (cat. no. 19-142), U0126 (cat. no.
19-147), 2-Chloro-N6-cyclopentyladenosine (CCPA; cat. no.
37739-05-2), 2-chloro-N6-(3-iodobenzyl)
adenosine-50-N-methylcarboxamide (CI-IB-MECA; cat. no.
163042-96-4), 9-chloro-2-(2-furanyl)-5-(phenylacetyl)
amino-(1,2,4)
triazolo (1,5-c) quinazoline (MRS1220; cat. no. 183721-15-5) and
other reagents for buffer preparations were purchased from
Sigma-Aldrich; Merck KGaA.
Cell culture
U251 cells were cultured in DMEM supplemented with
10% FBS, 100 U/ml streptomycin and 100 µg/ml penicillin at
37°C with 5% CO2. The culture medium was changed every
other day, and the cells were subcultured when 90% confluence was
reached. The cells were inoculated on suitable culture plates
specific to each assay and treated with 25, 50 or 100 µg/ml
PSH for 72 h at 37°C. The cells in the control group were cultured
in normal medium for the same amount of time. Treatment with
LY294002 (20 µM), U0126 (20 µM), CCPA (20 nM),
CI-IB-MECA (15 µM) and MRS1220 (1 µM) were performed
simultaneously with PSH treatment.
Cell viability
U251 cells were seeded into 96-well plates at a
density of 5×105 cells/well and cultured for 24 h,
followed by continuous culture with PSH for 72 h at 37°C. Cell
viability was examined by CCK-8 assay. Briefly, after the
treatments, 10 µl CCK-8 solution was added into each well
and incubated at 37°C for 4 h, and the absorbance was measured at
455 nm. The cells in the control group were cultured in normal
medium. Data are presented as the fold-change relative to the
control group.
Apoptotic rate measurement
The PSH-induced apoptosis rate of U251 cells was
determined using Annexin V-FITC/PI staining. U251 cells were seeded
into 6-well plates at a density of 1×106 cells/well and
cultured for 24 h at 37°C. Following the aforementioned treatments,
the cells were washed with PBS, trypsinized with 0.125 g/l trypsin,
centrifuged at 800 × g for 5 min at room temperature and collected.
The cells were resuspended in 100 µl 1X binding buffer, and
5 µl Annexin V-FITC and 5 µl PI were added to each
well and incubated for 15 min at 37°C in the dark. Subsequently,
400 µl binding buffer was added, and apoptosis was analyzed
on a FACSCalibur flow cytometer with BD Accuri™ C6 software (both
BD Biosciences). A total of 1×104 cells were collected
per well, and early and late apoptotic cells were recorded.
Cell cycle analysis
The inhibitory effects of PSH on cell proliferation
were measured by flow cytometry. Following the aforementioned
treatments, U251 cells were collected and fixed with 70% ethanol at
4°C for 2 h. Following centrifugation (800 × g for 5 min at 4°C),
the cells were resuspended in RNase A solution and 0.02 mg/ml PI
(Sigma-Aldrich; Merck KGaA), and incubated for 30 min at 4°C. The
fluorescence of 1×104 cells was recorded by a
FACSCalibur C6 flow cytometer (BD Biosciences) and analyzed by
ModFit software (Verity Software House).
Invasion assay
To determine the effects of PSH on cell invasiveness
in vitro, Matrigel Basement Membrane Matrix (Corning,
Inc.)-coated Transwell inserts (Costar; Corning, Inc.) were used.
The Transwell inserts were coated with 0.8 mg/ml Matrigel at 37°C
for 2 h, and cells were seeded at a density of 1×106 and
treated with various concentrations of PSH. Following incubation
for 72 h at 37°C, the non-invasive cells were removed by a cotton
swab, and the cells that passed through the membrane into the lower
wells were fixed with 5% glutaraldehyde at 37°C for 30 min and
stained with 0.1% crystal violet at 37°C for 30 min. Images in five
random fields per sample were captured using a light microscope,
and the cells were counted by ImageJ software (version 1.42,
National Institutes of Health). The results were expressed as the
percentage of total cells in the lower and upper wells.
Wound healing assay
To investigate the effects of PSH on cell migration,
a wound healing assay was performed. Cells were seeded into 6-well
plates at 1×106 cells/ml, and cultured until a confluent
monolayer was formed. The cell surface was wounded by a
200-µl pipette tip. PBS was used to remove the cells from
the wound area, and the cells were treated with different doses of
PSH in DMEM containing 1% FBS; the serum concentration was based on
previous studies (23,24). Images of the wound areas were
captured by an inverted microscope (×200 magnification) at the
start of the experiment (0 h) and following treatment for 72 h. For
each experiment, five replicates were performed, and the experiment
was repeated three times. The gap width was calculated using ImageJ
software (version 1.42), and the width was expressed in arbitrary
units. The wound healing rate was calculated as follows: Wound
healing (%)=(1-B/A) × 100%, where A is the gap width at 0 h, and B
is the gap width at 72 h.
Caspase activation analysis
To determine the caspase activity, 5×105
cells were seeded in 6-well plates and treated as aforementioned.
Caspase-3 and 9 activation was measured using specific kits, and
the direction and absorption values were recorded at 405 nm using a
microtiter plate reader (Bio-Rad Laboratories, Inc.).
20S proteasome enzymatic activity
analysis
To avoid affecting the enzymatic activity of the
proteasome, 0.5% CHAPS buffer was used to prepare the total
cellular protein lysate. The proteasome activity was determined in
the total protein lysates using a Proteasome 20S Activity Assay kit
(cat. no. ab112154; Abcam) according to the manufacturer's
instructions. The fluorescence intensity was measured at the
excitation/emission wavelengths of 490/525 nm using a VICTOR X2
fluorescent microplate reader (PerkinElmer, Inc.).
Immunoblotting
Following the aforementioned treatments, the cells
were harvested and homogenized in RIPA buffer at 4°C for 30 min.
The protein concentration was determined by the BCA assay (Beyotime
Institute of Biotechnology). Protein (30 µg) from each
sample was resolved by 10% SDS-PAGE, followed by transfer to a PVDF
membrane (Bio-Rad Laboratories, Inc.). Following blocking with 5%
non-fat dry milk at 37°C for 30 min, the membranes were probed with
anti-HIF-α (1:1,000), anti-VEGF (1:1,000), anti-P27 (1:1,000),
anti-CDK4 (1:1,000), anti-cleaved caspase-9 (1:1,000), anti-cleaved
caspase 3 (1:1,000), anti-Akt (1:1,000), anti-P-Akt (1:1,000),
anti-P38 (1:1,000), anti-P-P38 (1:1,000), anti-p44/42 MAPK
(1:1,000), anti-P-p44/42 MAPK (1:1,000), anti-ARA1 (1:200),
anti-ARA3 (1:200) and anti-GAPDH (1:1,000) primary antibodies at
4°C overnight. Subsequently, the membranes were washed with PBS +
1% Tween-20 three times and incubated with horseradish
peroxidase-conjugated anti-rabbit or mouse IgG (1:5,000) at 37°C
for 1 h. The signals were visualized using an ECL Plus western blot
detection system. The grayscale values of the bands were calculated
using Image J software (version 1.42) and presented as the
fold-change relative to the control group.
Statistical analysis
All experiments were repeated three times, and the
data are presented as the mean ± SD. The differences between two
groups were evaluated by either one-way analysis of variance
followed by the Dunnett's or Bonferroni post hoc test using using
GraphPad Prism 7.0 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
PSH inhibits U251 cell viability
To evaluate the inhibitory effects of PSH on U251
cells, the cells were treated with a range concentrations of PSH
for various durations, and the cell viability was measured by CCK-8
assay. As presented in Fig. 1A,
PSH inhibited the viability of U251 cells by ~60% at a
concentration of 100 µg/ml. Therefore, in subsequent
experiments, 25, 50 and 100 µg/ml PSH was used to study the
concentration-dependent effects.
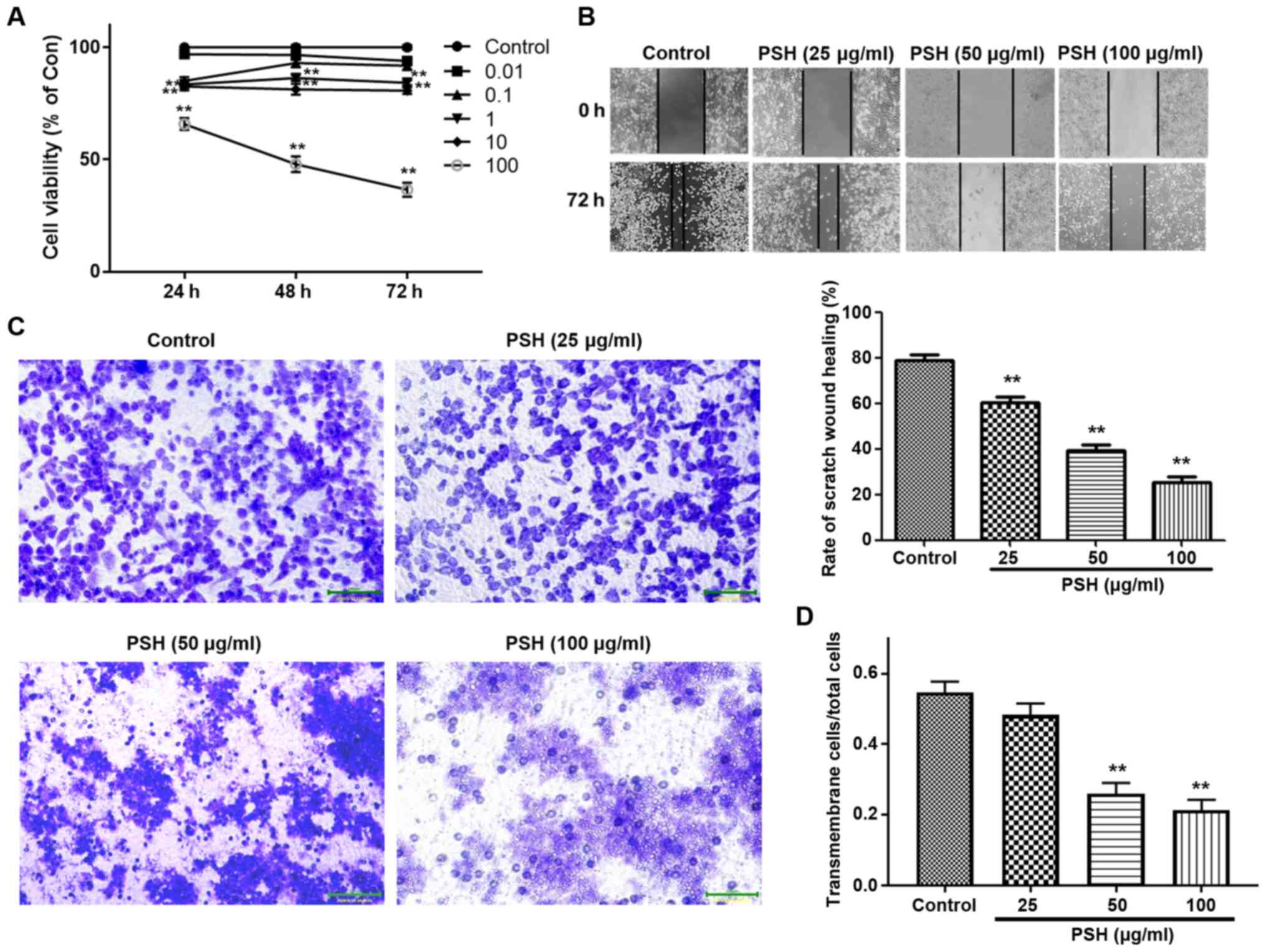 | Figure 1Effects of PSH on U251 cell
viability, migration and invasion. (A) Cells were treated with
0.01, 0.1, 1, 10 and 100 µg/ml PSH for 24, 48 and 72 h, and
the cell viability was measured by Cell Counting Kit-8 assay. (B)
Effects of PSH on cell migration. After confluence was reached,
cells were treated with 25, 50 and 100 µg/ml PSH, and the
wound healing assay was performed. (C) Effects of PSH on U251 cell
invasion. (D) Quantitative data of the cell invasion assay results.
**P<0.01 vs. control. PSH, Paris saponin H;
Con, control. |
To evaluate the inhibitory effects of PSH on U251
migration and invasion, wound healing and Transwell assays were
carried out. As demonstrated in Fig.
1B, PSH significantly inhibited cell migration. In the invasion
assay, a large number of control cells passed through the filter
into the lower wells; 25 µg/ml PSH exhibited an inhibitory
effect, but there was no significant difference in the invasive
cell number compared with that in the control group (Fig. 1C and D). PSH at 50 and 100
µg/ml significantly decreased the number of cells in the
lower wells, suggesting that PSH exerted an inhibitory effect on
U251 cell invasion.
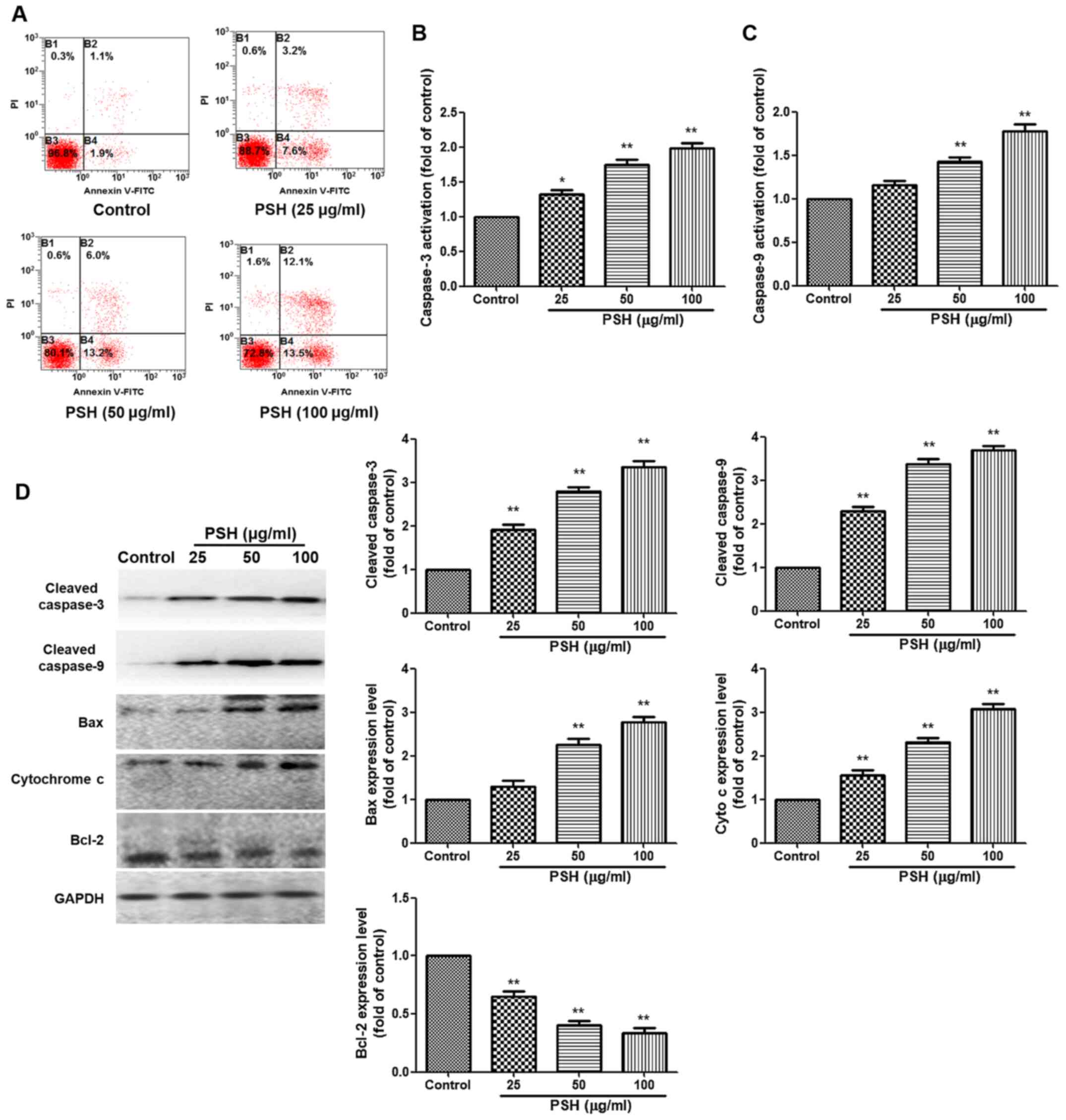
PSH induces apoptosis in U251 cells
To evaluate the effects of PSH on apoptosis, Annexin
V-FITC/PI staining was used. Treatment with PSH increased the
apoptotic rate of U251 cells compared with that in the control
group. The caspase 3 and caspase 9 activation analysis demonstrated
that 50 and 100 µg/ml PSH significantly increased the
activation of caspase 3 and caspase 9 compared with that in the
control group (Fig. 2B and C).
Western blotting results also revealed that compared with those in
the control group, PSH treatment significantly increased the
protein expression levels of cleaved caspase 3, cleaved caspase 9,
Bcl2-associated X protein and cytochrome c. Furthermore, the
expression levels of the anti-apoptotic protein Bcl-2 were reduced
in all PSH-treated groups compared with those in the control group.
These results suggested that PSH induced apoptosis in U251
cells.
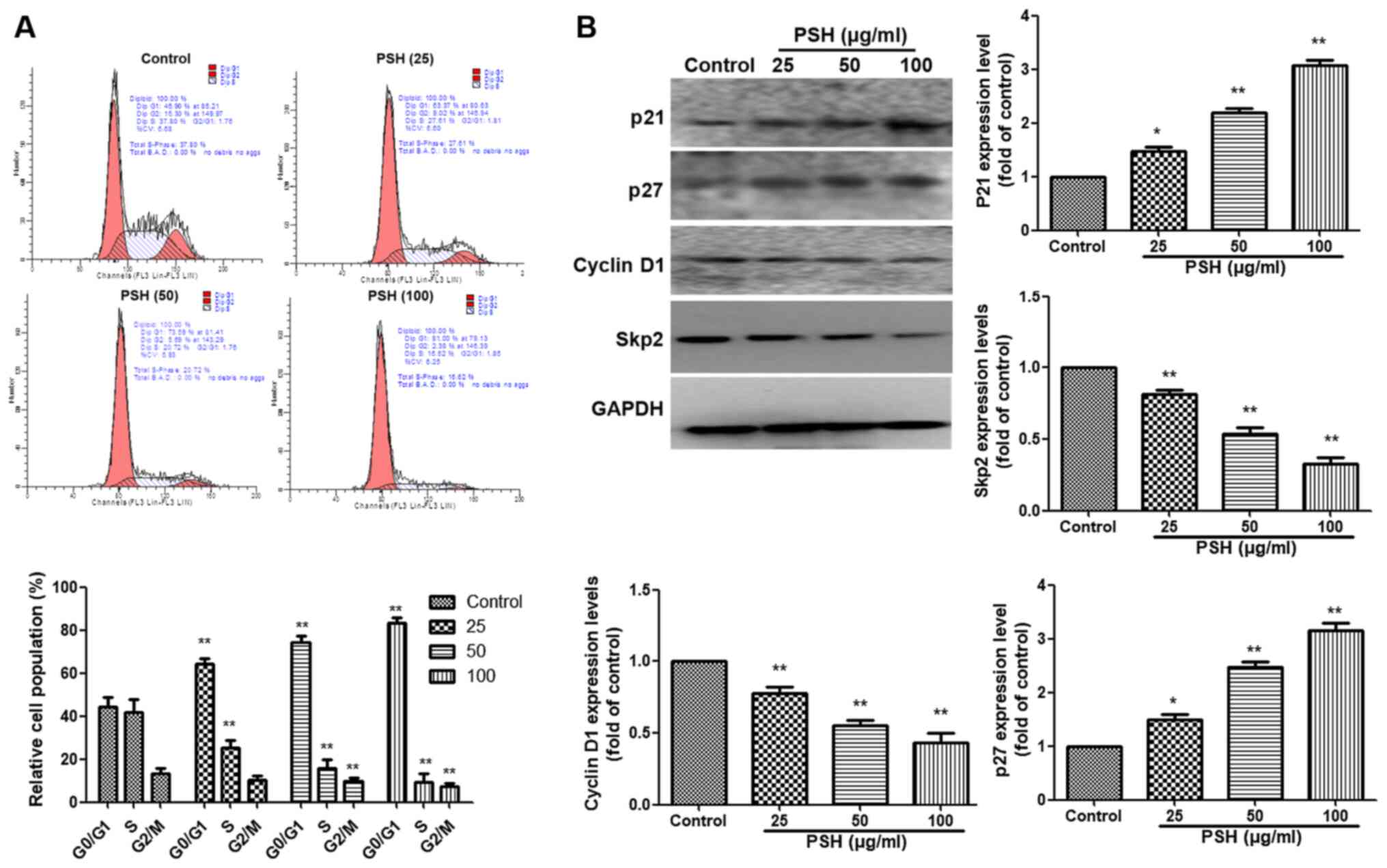
PSH induces cell cycle arrest at the G1
phase in U251 cells
To further evaluate whether the inhibitory effects
of PSH on cell proliferation were associated with the induction of
cell cycle arrest, PI staining was used and analyzed by flow
cytometry. As demonstrated in Fig.
3A, PSH induced cell accumulation at the G1 phase, and a
reduction at the S and G2 phases compared with the control group.
The western blotting results also revealed that compared with those
in the control group, treatment with 25, 50 or 100 µg/ml PSH
increased the protein expression levels of P21 and P27, and
inhibited those of cyclin D1 and S-phase kinase associated protein
2 (Skp2) (P<0.01). These results suggested that PSH induced cell
cycle arrest at the G1 phase in U251 cells.
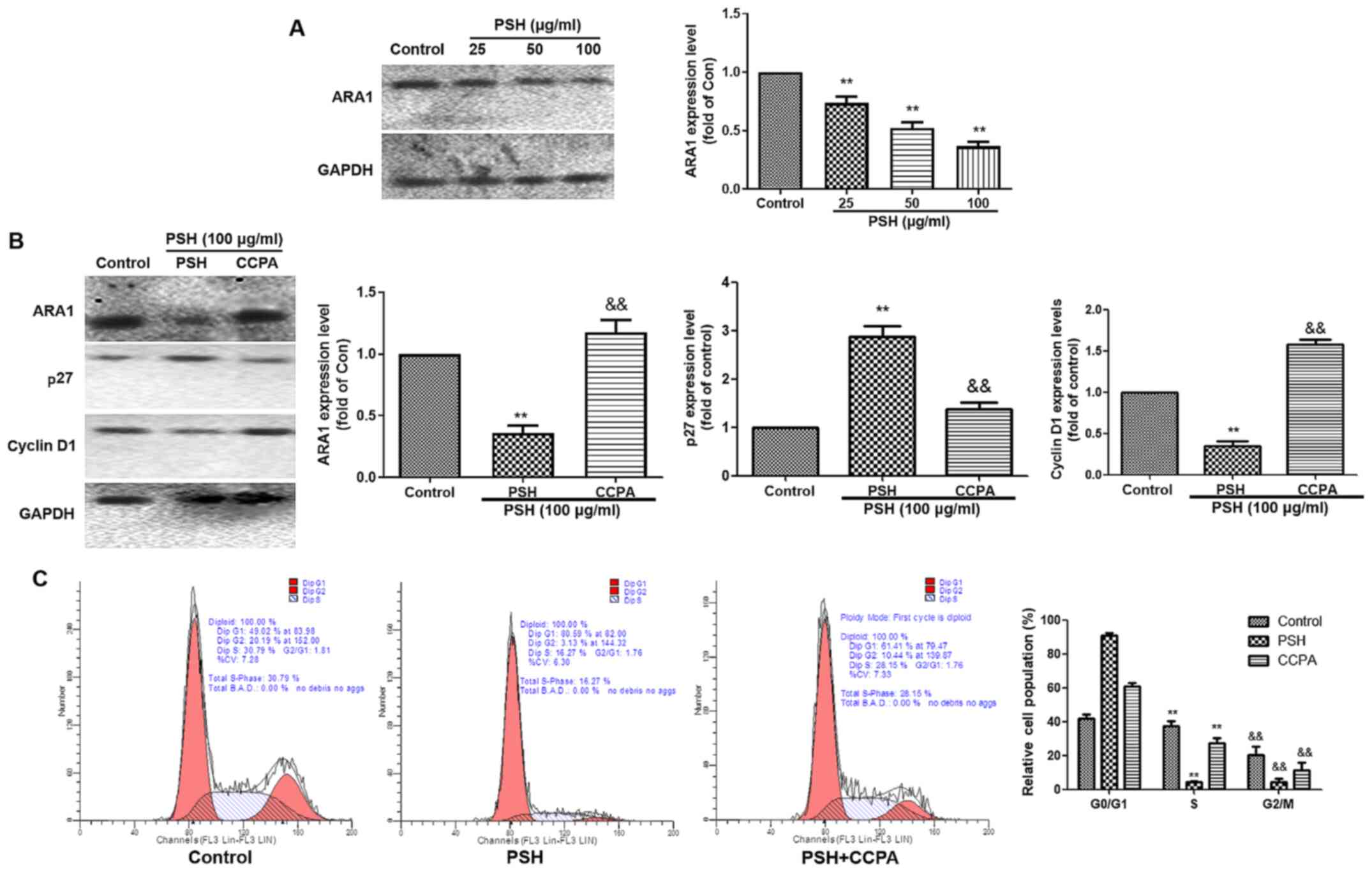
PSH induces cell cycle arrest via
ARA1
To investigate whether the PSH-induced cell cycle
arrest in U251 cells was ARA1-dependent, the expression of ARA1 was
determined by western blotting (Fig.
4A). Following 48-h treatment with 25, 50 or 100 µg/ml
PSH, the protein expression levels of ARA1 were significantly
decreased compared with those in the control group. To clarify the
role of ARA1, the ARA1 agonist CCPA was used. As demonstrated in
Fig. 4B, the effects of PSH on
the expression levels of ARA1, P27 and cyclin D1 were reversed by
CCPA. The PSH-induced cell cycle arrest was also abolished by CCPA
(Fig. 4C). These results
suggested that the PSH-induced cell cycle arrest was partly
dependent on ARA1.
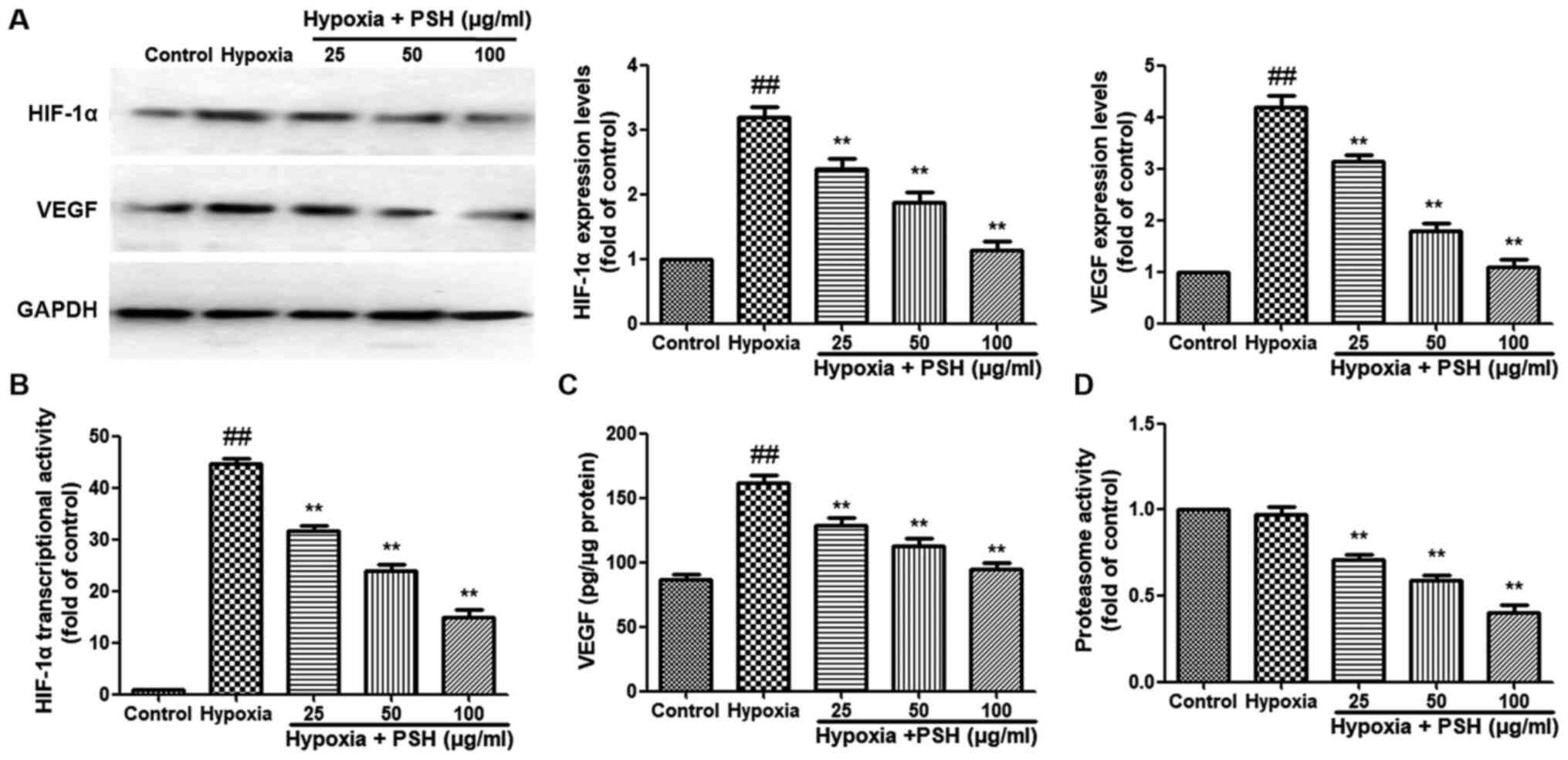
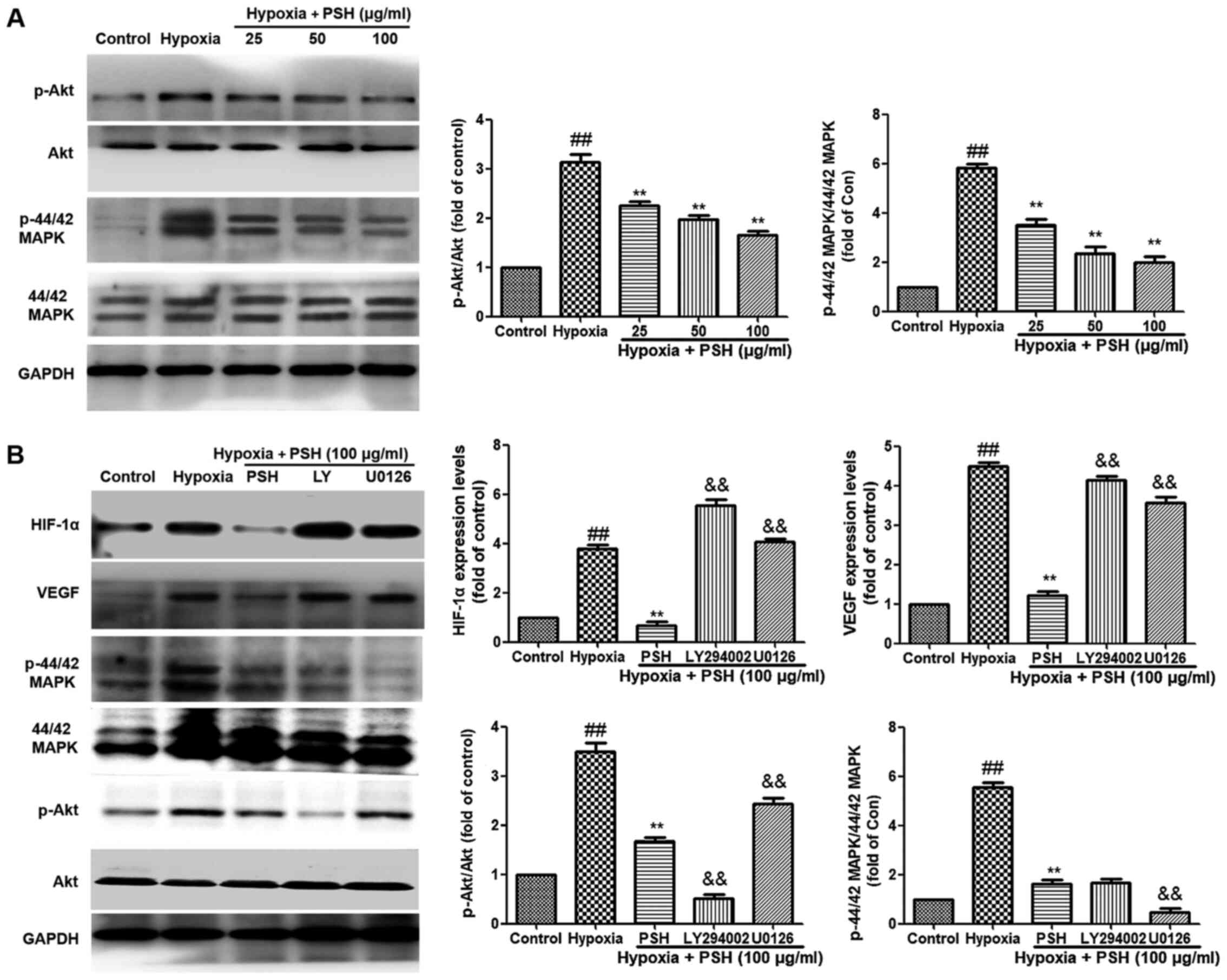
PSH attenuates HIF-1α and VEGF expression
in U251 cells under hypoxia
The effects of PSH on the expression of HIF-1α and
VEGF were analyzed in U251 cells under hypoxic conditions. The
expression levels of HIF-1α and VEGF were significantly increased
following exposure to hypoxia for 48 h compared with those under
normoxic conditions. However, treatment with 25, 50 or 100
µg/ml PSH significantly reversed the effects of hypoxia on
HIF-1α and VEGF expression levels (Fig. 5A). Next, the effects of PSH on
the HIF-1α transcriptional activity were examined; as demonstrated
in Fig. 5B, hypoxia induced a
significant increase in HIF-1α transcriptional activity compared
with that under normoxia. Treatment with 25, 50 or 100 µg/ml
PSH resulted in a significant reduction of HIF-1α transcriptional
activity compared with that in the hypoxia group The production of
VEGF was also analyzed. Hypoxia significantly induced VEGF
secretion compared with that under normoxic conditions, and PSH
inhibited this effect (Fig. 5C).
Subsequently, 20S proteasome activity in U251 cells was measured
following PSH treatment. The results demonstrated that treatment
with 25, 50 or 100 µg/ml PSH resulted in a significant
reduction in U251 cell proteasome activity, which compared with
that in the hypoxia group (Fig.
5D).
PSH inhibits the PI3K/Akt and MAPK
signaling pathways in U251 cells under hypoxia
To verify the inhibitory effects of PSH on the
expression of HIF-1α and VEGF, as well as the potential roles of
the PI3K/Akt and MAPK signaling pathways in the underlying
mechanism, the effects of PSH on these pathways were determined. As
demonstrated in Fig. 6A, the
phosphorylation levels of Akt and 44/42 MAPK in U251 cells were
upregulated by hypoxia compared with those in the control group,
but inhibited by 25, 50 or 100 µg/ml PSH compared with those
in the hypoxia group. Furthermore, the specific PI3K/Akt inhibitor
LY294002 and the P44/42 MAPK inhibitor U0126 were used to clarify
these effects. As presented in Fig.
6B, compared with those in the PSH treatment group, LY294002
and U0126 inhibited the phosphorylation levels of PI3K/Akt and
P44/42 MAPK, respectively. In addition, LY294002 and U0126
abolished the inhibitory effects of PSH on the protein expression
levels of HIF-1α and VEGF in U251 cells. These results indicated
that the PI3K/Akt and MAPK signaling pathways were involved in the
effects of PSH on HIF-1α and VEGF expression.
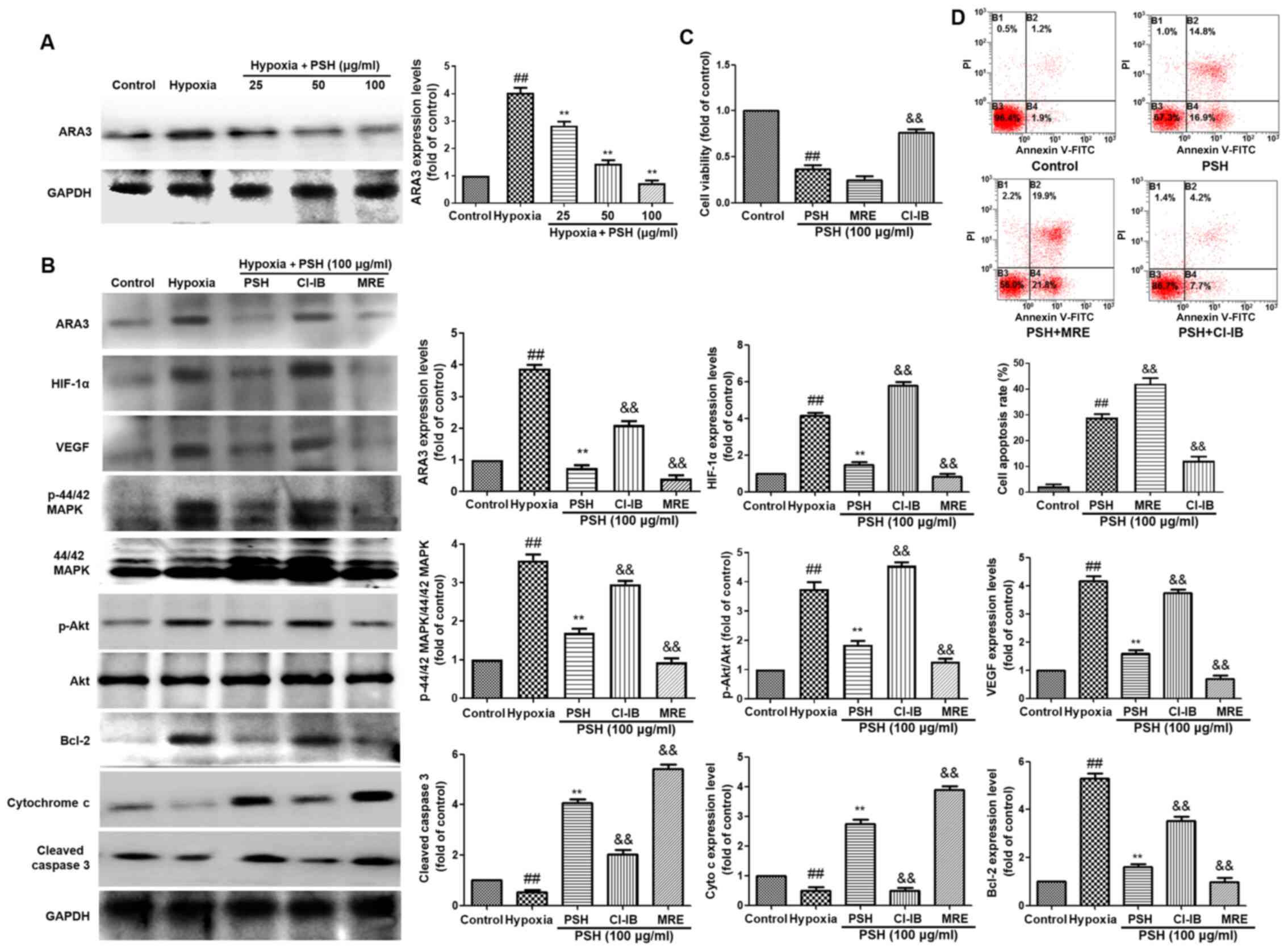
PSH decreases HIF-1α expression via ARA3
inactivation
To determine whether ARA3 was involved in
PSH-induced cytotoxicity, the protein expression levels of ARA3
were analyzed using western blotting. As demonstrated in Fig. 7A, treatment with 25, 50 or 100
µg/ml PSH decreased ARA3 expression levels compared with
those in the hypoxia group. To confirm the effects of ARA3 on
PSH-induced cytotoxicity, the selective ARA3 agonist CI-IB-MECA (15
µM) and the ARA3 antagonist MRS1220 (1 µM) were used
in the subsequent experiments. The inhibitory effects of PSH on the
protein expression levels of ARA3, HIF-1α, VEGF, P-Akt, P-44/42
MAPK, cleaved-caspase-3 and cytochrome c were abolished by
CI-IB-MECA and enhanced by MRS1220 treatment (Fig. 7B). The effects of PSH on cell
viability (Fig. 7C) and
apoptosis (Fig. 7D) were also
reversed by treatment with CI-IB-MECA and enhanced by MRS1220 in
U251 cells. These results suggested that the effects of PSH on U251
cells were exerted via the inhibition of ARA3 activity.
Discussion
Glioblastoma is one of the most common and malignant
brain tumors (25,26). Common clinical treatments for
glioblastoma include surgery, chemotherapy and radiotherapy;
however, their therapeutic effects are poor, and the median
survival time of patients with glioblastoma is only 6-14 months
(3,27). Glioblastomas are resistant to the
currently used chemotherapeutic strategies, thus limiting their
efficacy (28). Traditional
Chinese medicine has been used for thousands of years, and its
anticancer effects have attracted increasing attention among
pharmacists and scientists (29). Saponins from herbs such as RP
possess anticancer activities (30); however, the molecular mechanisms
underlying their effects are largely unknown.
Previous studies have demonstrated that the crude
extracts from RP and saponins exert inhibitory effects on tumor
cell proliferation and progression (31,32). Results from a structure-activity
relationship analysis have revealed that the variety at the C-3
position of the aglycone contributes to the antitumor effects of
saponins from RP (19). Previous
studies have suggested that polyphyllin D exerts inhibitory effects
on tumor cell proliferation and angiogenesis (33-35). By screening the antitumor
activity of RP saponins on U87 and U251 glioblastoma cells, it was
observed that PSH exhibited the strongest antiglioma effects (data
not shown). However, the antitumor activity of PSH and its possible
mechanism have remained largely unknown to date. Therefore, the
present study verified the anticancer effect of PSH and elucidated
the underlying mechanisms.
In the present study, the results of the CCK-8
viability assay demonstrated that PSH inhibited cell viability, and
100 µg/ml PSH reduced U251 cell viability to 60% of that in
untreated cells. The effects of PSH on cell migration and invasion
were examined by wound healing and Transwell invasion assays,
respectively, and the results demonstrated that PSH inhibited U251
cell migration and invasion. These results suggested that PSH
exhibited anticancer effects in the U251 glioma cell line. The
anticancer effects of PSH may be exerted through several
mechanisms, such as cell cycle progression interference and
apoptosis promotion. When U251 cells were exposed to PSH in the
present study, the apoptotic rates were measured by Annexin V/PI
assay, and the results demonstrated that the numbers of apoptotic
cells were increased in the PSH treatment groups compared with
those in the control group. ELISA results also revealed that the
caspase-3 and caspase-9 levels were significantly increased in U251
cells following PSH treatment compared with those in the untreated
cells, suggesting that PSH may induce caspase-dependent apoptosis
in U251 cells. Further western blotting results demonstrated that
PSH induced the protein expression of apoptosis-related proteins
cleaved caspase-3, cleaved caspase-9, Bax and cytochrome
c.
Cell cycle checkpoints prevent cells from genome
damage by undergoing DNA replication or mitosis (36). Interfering with the cell cycle
progression is a potentially effective treatment strategy for
inhibiting cancer cell proliferation (37). Cell cycle progression is promoted
by cyclins D1, D2 and D3, which serve important roles in the
modulation of the transition from the G1 to the S phase during the
cell cycle, and CDKs, and inhibited by CDK inhibitors, such as p21
and p27 (38,39). The results of the present study
demonstrated that PSH induced cell cycle arrest at the G1 phase,
increased p21 and p27 protein expression levels, and decreased
those of cyclin D1 and Skp2 compared with those in the untreated
cells. These results suggested that DNA synthesis in U251 cells was
inhibited by PSH.
A previous study has reported that, in the
peritumoral area of C6 glioma cell-injected animals, the levels of
ARA1 were significantly upregulated compared with those in normal
tissues (40). Although the
regulatory mechanism of the ARA1 increase is unknown, the levels of
ARA1 around tumors represent a useful diagnostic and prognostic
marker for glioblastoma progression (41). To determine whether ARA1 was
involved in the PSH-induced cell cycle arrest, the expression
levels of ARA1 were analyzed in the present study. The results
demonstrated that compared with those in the control group, PSH
significantly inhibited the expression levels of ARA1. The ARA1
agonist CCPA was used to verify these results; CCPA treatment
abolished the effects of PSH on the cell cycle and the related
protein expression. These results suggested that PSH induced U251
cell cycle progression potentially by inhibiting ARA1.
Previous studies have reported that hypoxia is
prevalent in solid tumor and hypoxic microenvironments,
contributing to radiotherapy resistance (42,43). HIF-1, a transcriptional
activator, serves an important role in adapting to low oxygen and
nutrition conditions, and promotes tumor progression by regulating
the expression of a number of genes, such as AKT and MAPK, involved
in angiogenesis, apoptosis, invasion and cell metabolism (44,45). HIF-1 contains two subunits,
HIF-1α and HIF-1β. HIF-1α is activated by hypoxia and translocates
into the nucleus, where it forms a heterodimer with HIF-1β,
inducing the expression of a range of transcription factors,
including VEGF (46). In the
present study, hypoxia upregulated the expression levels of HIF-1α
and VEGF in U251 cells compared with those under normoxia. PSH
significantly reversed these effects. The 20S proteasome activity
was also decreased by PSH treatment compared with that in the
hypoxia group. These results suggested that PSH inhibited U251 cell
proliferation by regulating HIF-1α and VEGF expression.
HIF-1α is activated by several oncogenic pathways,
including phosphatase and tensin homolog (PTEN), as well as growth
factors, such as hepatocyte growth factor (HGF) and stromal-derived
factor-1a (SDF-1a) (47,48). PTEN negatively regulates Akt
expression, which can increase HIF-1α protein synthesis and induce
VEGF production under hypoxia (49). P44/42 (also termed ERK1/2), which
is a member of the MAPK signaling pathway, activates HIF-1α by
promoting the formation of the HIF-p300/CBP complex and modulating
its transactivation activity (50). P44/42 MAPK also increases the
HIF-1 transcriptional activity by directly phosphorylating HIF-1α
(51). P44/42 MAPK induces HIF-1
activation by inhibiting the chromosomal maintenance 1-dependent
nuclear export of HIF-1α (52).
To determine through which pathway PSH was involved in controlling
HIF-1α, the phosphorylation levels of PI3K/Akt and P44/42 MAPK were
analyzed in the present study. The results demonstrated that PSH
inhibited the phosphorylation levels of PI3K/Akt and P44/42 MAPK.
The specific PI3K/Akt inhibitor LY294002 and P44/42 MAPK inhibitor
U0126 were further used to confirm these results; LY294002 and
U0126 abolished the inhibitory effects of PSH on the protein
expression levels of HIF-1α and VEGF. These results suggested that
PSH-mediated inhibition of HIF-1α and VEGF expression was possibly
dependent on the PI3K/Akt and p44/42 MAPK pathways.
Under normal conditions, ARA3 inhibits protein
kinase A (PKA) and Akt, and activates glycogen synthase kinase 3β,
which inhibits tumor cell proliferation (53). However, under hypoxic conditions,
ARA3 is activated and upregulates the expression levels of HIF-1α
and VEGF, which induces angiogenesis (54). Hypoxia-induced chemoresistance of
glioblastoma cells depends on the activation of ARA3, which
subsequently induces Akt activation and the inactivation of the
pro-apoptotic protein Bad, leading to cell survival (55). In U87 cells, the activation of
ARA3 also induces the upregulation of matrix metalloproteinase-9,
which increases tumor cell migration (56). In the present study, the results
demonstrated that hypoxia induced, whereas PSH inhibited the
expression of ARA3. CI-IB-MECA and MRS1220 were used to confirm the
role of ARA3 in PSH treatment; the results revealed that the
inhibitory effects of PSH on ARA3, HIF-1α, VEGF, P-Akt, P-44/42
MAPK, cleaved-caspase-3 and cytochrome c expression as well
as cell viability were abolished by CI-IB-MECA and enhanced by
MRS1220. These results suggested that the effects of PSH were
exerted through the inhibition of the ARA3 activity.
Taken together, the results of the present study
demonstrated that PSH inhibited the viability and induced cell
cycle arrest and apoptosis in U251 cells. The effects of PSH may
involve the modulation of adenosine receptor interaction via the
inhibition of the P44/42 MAPK and Akt pathways.
Funding
This study was funded by the National Natural
Science Foundation of China (grant no. 81703761).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LB and HT designed the study. LB, YLiu1, QY, XZ,
YLiu2 and HL performed the study. LB, YLu and JL analyzed the data.
LB and HT confirmed the authenticity of all the raw data. LB wrote
the manuscript. HT revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Ordys BB, Launay S, Deighton RF, McCulloch
J and Whittle IR: The role of mitochondria in glioma
pathophysiology. Mol Neurobiol. 42:64–75. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Batash R, Asna N, Schaffer P, Francis N
and Schaffer M: Glioblastoma multiforme, diagnosis and treatment;
recent literature review. Curr Med Chem. 24:3002–3009. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Almeida Sassi F, Lunardi Brunetto A,
Schwartsmann G, Roesler R and Abujamra AL: Glioma revisited: From
neurogenesis and cancer stem cells to the epigenetic regulation of
the niche. J Oncol. 2012:5378612012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang L, Lin C, Wang L, Guo H and Wang X:
Hypoxia and hypoxia-inducible factors in glioblastoma multiforme
progression and therapeutic implications. Exp Cell Res.
318:2417–2426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shahar T, Nossek E, Steinberg DM, Rozovski
U, Blumenthal DT, Bokstein F, Sitt R, Freedman S, Corn BW, Kanner
AA and Ram Z: The impact of enrollment in clinical trials on
survival of patients with glioblastoma. J Clin Neurosci.
19:1530–1534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patil SA, Hosni-Ahmed A, Jones TS, Patil
R, Pfeffer LM and Miller DD: Novel approaches to glioma drug design
and drug screening. Expert Opin Drug Discov. 8:1135–1151. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Torres Á, Erices JI, Sanchez F, Ehrenfeld
P, Turchi L, Virolle T, Uribe D, Niechi I, Spichiger C, Rocha JD,
et al: Extracellular adenosine promotes cell migration/invasion of
glioblastoma stem-like cells through A3 adenosine
receptor activation under hypoxia. Cancer Lett. 446:112–122. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Melani A, De Micheli E, Pinna G, Alfieri
A, Corte LD and Pedata F: Adenosine extracellular levels in human
brain gliomas: An intraoperative microdialysis study. Neurosci
Lett. 346:93–96. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fredholm BB, IJzerman AP, Jacobson KA,
Klotz KN and Linden J: International union of pharmacology. XXV.
Nomenclature and classification of adenosine receptors. Pharmacol
Rev. 53:527–552. 2001.PubMed/NCBI
|
|
10
|
Castillo CA, León D, Ruiz MA, Albasanz JL
and Martín M: Modulation of adenosine A1 and A2A receptors in C6
glioma cells during hypoxia: Involvement of endogenous adenosine. J
Neurochem. 105:2315–2329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang XY, Chen XL, Tang HF, Gao H, Tian XR
and Zhang PH: Cytotoxic triterpenoid saponins from the rhizomes of
Anemone taipaiensis. Planta Med. 77:1550–1554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang XY, Gao H, Zhang W, Li Y, Cheng G,
Sun XL and Tang HF: Bioactive oleanane-type saponins from the
rhizomes of Anemone taipaiensis. Bioorg Med Chem Lett.
23:5714–5720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji C, Cheng G, Tang H, Zhang Y, Hu Y,
Zheng M and Fei Z: Saponin 6 of Anemone taipaiensis inhibits
proliferation and induces apoptosis of U87 MG cells. Xi Bao Yu Fen
Zi Mian Yi Xue Za Zhi. 31:484–486. 2015.In Chinese. PubMed/NCBI
|
|
14
|
Zhang W, Zhang D, Ma X, Liu Z, Li F and Wu
D: Paris saponin VII suppressed the growth of human cervical cancer
Hela cells. Eur J Med Res. 19:412014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang T, Liu H, Liu XT, Xu DR, Chen XQ and
Wang Q: Qualitative and quantitative analysis of steroidal saponins
in crude extracts from Paris polyphylla var. yunnanensis and P.
polyphylla var. chinensis by high performance liquid chromatography
coupled with mass spectrometry. J Pharm Biomed Anal. 51:114–124.
2010. View Article : Google Scholar
|
|
16
|
Sun J, Liu BR, Hu WJ, Yu LX and Qian XP:
In vitro anticancer activity of aqueous extracts and ethanol
extracts of fifteen traditional Chinese medicines on human
digestive tumor cell lines. Phytother Res. 21:1102–1104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Negi JS, Bisht VK, Bhandari AK, Bhatt VP,
Singh P and Singh N: Paris polyphylla: Chemical and biological
prospectives. Anticancer Agents Med Chem. 14:833–839. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pang D, Li C, Yang C, Zou Y, Feng B, Li L,
Liu W, Geng Y, Luo Q, Chen Z and Huang C: Polyphyllin VII promotes
apoptosis and autophagic cell death via ROS-inhibited AKT activity,
and sensitizes glioma cells to temozolomide. Oxid Med Cell Longev.
2019:18056352019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu Q, Li Q, Lu P and Chen Q: Polyphyllin D
induces apoptosis in U87 human glioma cells through the c-Jun
NH2-terminal kinase pathway. J Med Food. 17:1036–1042. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao X, Yang M, Xiao J, Zou J, Huang Q,
Yang K, Zhang B, Yang F, Liu S, Wang H and Bai P: Paris saponin II
suppresses the growth of human ovarian cancer xenografts via
modulating VEGF-mediated angiogenesis and tumor cell migration.
Cancer Chemother Pharmacol. 73:807–818. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cong Y, Liu X, Kang L, Yu Z, Zhao Z, Li J,
Ma B and Cong Y: Pennogenin tetraglycoside stimulates
secretion-dependent activation of rat platelets: Evidence for
critical roles of adenosine diphosphate receptor signal pathways.
Thromb Res. 129:e209–e216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cohen FR, Lazareno S and Birdsall NJ: The
effects of saponin on the binding and functional properties of the
human adenosine A1 receptor. Br J Pharmacol. 117:1521–1529. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie X, Zhu H, Zhang J, Wang M, Zhu L, Guo
Z, Shen W and Wang D: Solamargine inhibits the migration and
invasion of HepG2 cells by blocking epithelial-to-mesenchymal
transition. Oncol Lett. 14:447–452. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
D'Agostino A, Stellavato A, Busico T, Papa
A, Tirino V, Papaccio G, La Gatta A, De Rosa M and Schiraldi C: In
vitro analysis of the effects on wound healing of high- and
low-molecular weight chains of hyaluronan and their hybrid
H-HA/L-HA complexes. BMC Cell Biol. 16:192015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crocetti E, Trama A, Stiller C, Caldarella
A, Soffietti R, Jaal J, Weber DC, Ricardi U, Slowinski J and
Brandes A; RARECARE working group: Epidemiology of glial and
non-glial brain tumours in Europe. Eur J Cancer. 48:1532–1542.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ricard D, Idbaih A, Ducray F, Lahutte M,
Hoang-Xuan K and Delattre JY: Primary brain tumours in adults.
Lancet. 379:1984–1996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Trembath DG, Lal A, Kroll DJ, Oberlies NH
and Riggins GJ: A novel small molecule that selectively inhibits
glioblastoma cells expressing EGFRvIII. Mol Cancer. 6:302007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frosina G: DNA repair and resistance of
gliomas to chemotherapy and radiotherapy. Mol Cancer Res.
7:989–999. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma L, Wen S, Zhan Y, He Y, Liu X and Jiang
J: Anticancer effects of the Chinese medicine matrine on murine
hepatocellular carcinoma cells. Planta Med. 74:245–251. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang MY, Zhang LL, Ding J and Lu JJ:
Anticancer drug discovery from Chinese medicinal herbs. Chin Med.
13:352018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu GX, Wang TT, Hu WJ, Qian XP, Yu LX and
Liu BR: Anticancer effect of Rhizoma Paridis on primary cancer
cells isolated from malignant pleural effusion and ascites. Pract
Geriatr. 22:1012008.
|
|
32
|
Li X, Wang JH and Xiao YX: Effect of
paridis extract on proliferation of human colon cancer SW480 cells
and mechanism of the effect. Chin J Biol. 23:619–622+632. 2010.
|
|
33
|
Chan JY, Koon JC, Liu X, Detmar M, Yu B,
Kong SK and Fung KP: Polyphyllin D, a steroidal saponin from Paris
polyphylla, inhibits endothelial cell functions in vitro and
angiogenesis in zebrafish embryos in vivo. J Ethnopharmacol.
137:64–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiao X, Bai P, Bui Nguyen TM, Xiao J, Liu
S, Yang G, Hu L, Chen X, Zhang X, Liu J and Wang H: The antitumoral
effect of Paris saponin I associated with the induction of
apoptosis through the mitochondrial pathway. Mol Cancer Ther.
8:1179–1188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Man SL, Wang YL, Li YY, Gao WY, Huang XX
and Ma CY: Phytochemistry, pharmacology, toxicology, and
structure-cytotoxicity relationship of paridis rhizome saponin.
Chin Herbal Med. 5:33–46. 2013.
|
|
36
|
Shackelford RE, Kaufmann WK and Paules RS:
Cell cycle control, checkpoint mechanisms, and genotoxic stress.
Environ Health Perspect. 107(Suppl 1): S5–S24. 1999.
|
|
37
|
Mork C, Faller D and Spanjaard R: A
mechanistic approach to anticancer therapy: Targeting the cell
cycle with histone deacetylase inhibitors. Curr Pharm Des.
11:1091–1104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Evron E, Umbricht CB, Korz D, Raman V,
Loeb DM, Niranjan B, Buluwela L, Weitzman SA, Marks J and Sukumar
S: Loss of cyclin D2 expression in the majority of breast cancers
is associated with promoter hypermethylation. Cancer Res.
61:2782–2787. 2001.PubMed/NCBI
|
|
39
|
Harper JW, Elledge SJ, Keyomarsi K,
Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley
L, Swindell E, et al: Inhibition of cyclin-dependent kinases by
p21. Mol Biol Cell. 6:387–400. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dehnhardt M, Palm C, Vieten A, Bauer A and
Pietrzyk U: Quantifying the A1AR distribution in peritumoural zones
around experimental F98 and C6 rat brain tumours. J Neurooncol.
85:49–63. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stone TW, Ceruti S and Abbracchio MP:
Adenosine receptors and neurological disease: Neuroprotection and
neurodegeneration. Handb Exp Pharmacol. 45:535–587. 2009.
View Article : Google Scholar
|
|
42
|
Thienpont B, Steinbacher J, Zhao H, D'Anna
F, Kuchnio A, Ploumakis A, Ghesquière B, Van Dyck L, Boeckx B,
Schoonjans L, et al: Tumour hypoxia causes DNA hypermethylation by
reducing TET activity. Nature. 537:63–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Badowska-Kozakiewicz AM, Sobol M and
Patera J: Expression of multidrug resistance protein P-glycoprotein
in correlation with markers of hypoxia (HIF-1α, EPO, EPO-R) in
invasive breast cancer with metastasis to lymph nodes. Arch Med
Sci. 13:1303–1314. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Semenza GL: Regulation of cancer cell
metabolism by hypoxia-inducible factor 1. Semin Cancer Biol.
19:12–16. 2009. View Article : Google Scholar
|
|
45
|
Veschini L, Belloni D, Foglieni C, Cangi
MG, Ferrarini M, Caligaris-Cappio F and Ferrero E:
Hypoxia-inducible transcription factor-1 alpha determines
sensitivity of endothelial cells to the proteosome inhibitor
bortezomib. Blood. 109:2565–2570. 2007. View Article : Google Scholar
|
|
46
|
Boddy JL, Fox SB, Han C, Campo L, Turley
H, Kanga S, Malone PR and Harris AL: The androgen receptor is
significantly associated with vascular endothelial growth factor
and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a,
and the prolyl hydroxylases in human prostate cancer. Clin Cancer
Res. 11:7658–7663. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ceradini DJ, Kulkarni AR, Callaghan MJ,
Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP
and Gurtner GC: Progenitor cell trafficking is regulated by hypoxic
gradients through HIF-1 induction of SDF-1. Nat Med. 10:858–864.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kitajima Y, Ide T, Ohtsuka T and Miyazaki
K: Induction of hepatocyte growth factor activator gene expression
under hypoxia activates the hepatocyte growth factor/c-Met system
via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci.
99:1341–1347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sang N, Stiehl DP, Bohensky J, Leshchinsky
I, Srinivas V and Caro J: MAPK signaling up-regulates the activity
of hypoxia-inducible factors by its effects on p300. J Biol Chem.
278:14013–14019. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Richard DE, Berra E, Gothié E, Roux D and
Pouysségur J: p42/p44 mitogen-activated protein kinases
phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and
enhance the transcriptional activity of HIF-1. J Biol Chem.
274:32631–32637. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mylonis I, Chachami G, Samiotaki M,
Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S and
Simos G: Identification of MAPK phosphorylation sites and their
role in the localization and activity of hypoxia-inducible
factor-1alpha. J Biol Chem. 281:33095–33106. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fishman P, Bar-Yehuda S, Madi L and Cohn
I: A3 adenosine receptor as a target for cancer therapy. Anticancer
Drugs. 13:437–443. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Merighi S, Benini A, Mirandola P, Gessi S,
Varani K, Leung E, Maclennan S and Borea PA: Adenosine modulates
vascular endothelial growth factor expression via hypoxia-inducible
factor-1 in human glioblastoma cells. Biochem Pharmacol. 72:19–31.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Merighi S, Benini A, Mirandola P, Gessi S,
Varani K, Leung E, Maclennan S, Baraldi PG and Borea PA: Hypoxia
inhibits paclitaxel-induced apoptosis through adenosine-mediated
phosphorylation of bad in glioblastoma cells. Mol Pharmacol.
72:162–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gessi S, Sacchetto V, Fogli E, Merighi S,
Varani K, Baraldi PG, Tabrizi MA, Leung E, Maclennan S and Borea
PA: Modulation of metalloproteinase-9 in U87MG glioblastoma cells
by A3 adenosine receptors. Biochem Pharmacol. 79:1483–1495. 2010.
View Article : Google Scholar : PubMed/NCBI
|