Type 2 diabetes mellitus (T2DM) and non-alcoholic
fatty liver disease (NAFLD), two common metabolic syndromes, are
emerging as global epidemics, whose incidence are rising annually
(1,2). T2DM is predominantly characterized
by an assembly of hyperglycemia, hyperinsulinemia, insulin
resistance and insulin deficiency (3). According to the Diabetes Atlas 9th
edition published by the International Diabetes Federation (IDF),
463 million adults aged 20-79 years are suffering from diabetes
mellitus worldwide, with the prevalence of diabetes mellitus in
that age group being ~9.3%, and the total number of diabetic
patients predicted to rise to 700 million (10.9%) by 2045 (1). In total, >90% of the diabetic
patients belong to T2DM, as estimated by IDF. NAFLD, currently the
most common chronic liver disease, covers a wide disease spectrum,
ranging from simple steatosis to non-alcoholic steatohepatitis
(NASH), hepatic fibrosis, cirrhosis and hepatocellular carcinoma
(HCC), which finally causes liver-associated mortality (4). A meta-analysis on NAFLD
epidemiology reported a global prevalence of 25.24% in 2016
(2). In China, the prevalence
has risen from 25.4% in 2008-2010 to 32.3% in 2015-2018 (5).
T2DM and NAFLD are closely associated. According to
clinical data, the overall incidence of NAFLD is 55.5% among
patients with T2DM (6), and
NAFLD is an independent risk factor for T2DM, indicating a strong
bi-directional relationship between T2DM and NAFLD (7,8).
T2DM is a risk factor for progression from simple steatosis to NASH
and advanced fibrosis. T2DM is associated with a high morbidity of
NASH (9,10). Patients with simple steatosis
often have a benign prognosis, whereas NASH can progress to
cirrhosis, with patients eventually developing HCC (11,12). The prevalence of HCC is ~5-fold
higher when the disease progresses from simple steatosis to NASH,
leading to a markedly higher mortality rate (13,14). In addition, the presence of NAFLD
in patients with T2DM is highly associated with the incidence of
macro- and micro-vascular diabetic complications (15). It is harder to control blood
glucose levels in patients with T2DM with NAFLD, compared with
patients with only T2DM (16).
Both T2DM and NAFLD can be caused by metabolic disorders, and share
familiar or even the same risk factors and pathological mechanisms.
Although studies have shown that the existing pathogenic mechanisms
of T2DM and NAFLD include insulin resistance, hyperglycemia,
hepatic lipid accumulation and inflammation (17-19), due to the multifaceted and
intricate correlations between these two diseases, the underlying
molecular mechanisms require further exploration.
Both T2DM and NAFLD can be largely influenced by
dietary structure. The intake of Western diet contributes to the
onset and development of T2DM and NAFLD (20-22). A Western diet is mainly
characterized by high amounts of saturated fatty acids (such as
palmitic acid), simple carbohydrates (corn syrup and fructose), low
levels of polyunsaturated fatty acids (PUFAs; n-6 and n-3 PUFAs),
and insufficient intake of protein and dietary fibers (22,23). In addition, there is a high
intake of n-6 PUFAs (particularly linoleic acid) and a low intake
of n-3 PUFAs [such as α-linolenic acid (ALA)] in this dietary
pattern among patients with T2DM and NAFLD, which cause a high
ratio of n-6/n-3 PUFAs (24).
The intake of Western diets results in an increased level of
n-6-PUFA-derived arachidonic acid (AA) and subsequent eicosanoid
production [particularly 2-series prostaglandins (PGs)], and there
is a decreased level of those derivatives from n-3 PUFAs in
patients with T2DM and NAFLD (25-27). Decreased n-3 PUFAs, which are
partly caused by an impaired ALA desaturation in the liver, can
repress fatty acid oxidation and contribute to pro-lipogenic
outcome by downregulating peroxisome proliferator-activated
receptor-α (PPAR-α); they can also promote lipogenic and glycogenic
capacity by upregulating sterol regulatory element-binding protein
1c (SREBP-1c) (24).
Furthermore, the downregulation of PPAR-α by n-3 PUFAs depletion
activates the nuclear factor-κB (NF-κB) and activating protein 1 in
the liver, leading to a pro-inflammatory effect in patients with
NAFLD (24). On the other hand,
the increased n-6 PUFAs and its derivatives can influence the
inflammatory state and disturb glucose and lipid metabolism
(28-32). Linoleic acid can alter fatty acid
transportation, mitochondrial function, inflammatory responses and
oxidative stress by increasing PG release and activating PPAR-γ,
interleukin-8 (IL-8) and the NF-κB signaling pathway (30,32). The J2-series PGs can promote
adipocyte differentiation by directly activating PPAR-γ (31). Therefore, n-6 PUFAs and n-3 PUFAs
exert various vital metabolic effects, and the levels of n-6 and
n-3 PUFAs, which can be mediated by similar dietary patterns of
T2DM and NAFLD (particularly a Western diet) are important for the
pathological development of these two diseases.
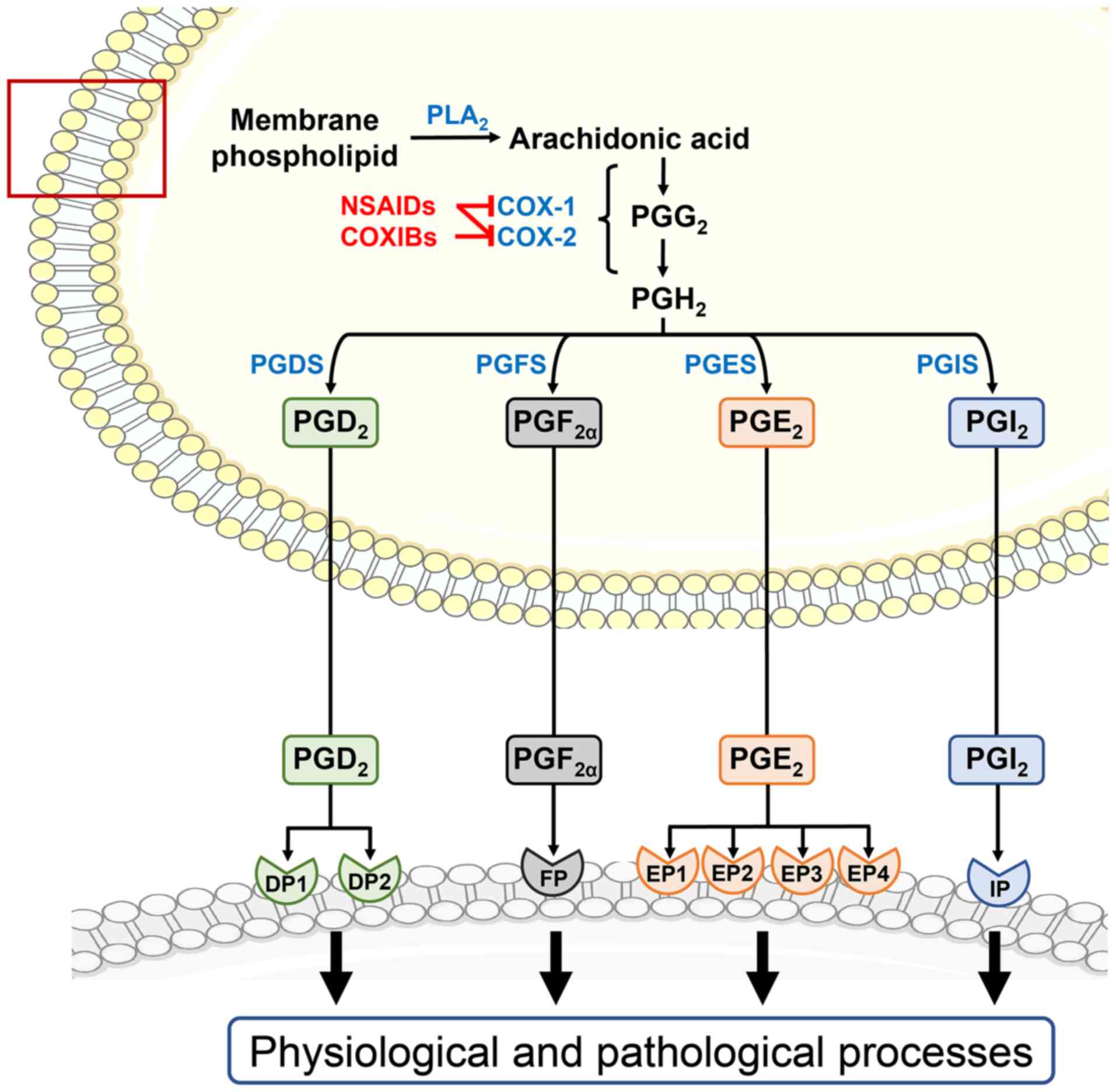
As important derivatives of n-6 PUFAs, 2-series PGs
are widely distributed in almost every tissue. In the PG synthesis
pathway, four principal bioactive 2-series PGs are generated,
including PGD2, PGE2, PGF2α and
PGI2 (33). Clinical
and experimental evidence has indicated that 2-series PGs are
involved in the initiation and progression of numerous diseases,
including diabetes mellitus (34), hypertension (35), obesity (36), fatty liver disease (37), vascular diseases (38), carcinoma (39), inflammatory bowel disease
(40), rheumatoid arthritis
(41), asthma and allergic
diseases (42) and Alzheimer's
disease (43). Studies have
revealed that 2-series PGs play complex and interlinked roles in
mediating metabolic homeostasis and systemic chronic inflammation
(34,44-48). Moreover, 2-series PGs have
bidirectional effects on insulin secretion and pancreatic β-cell
proliferation during hyperglycemia (34). As PPAR-γ modulators, 2-series PGs
regulate adipogenesis and lipolysis in lipid metabolism, leading to
excessive fat deposit (44-46). In addition, 2-series PGs are
involved in immune response by affecting various cytokines and
immune cells, such as macrophages and monocytes, under insulin
resistance, hyperlipidemic and diabetic status (44,47,48). Of note, the nonsteroidal
anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2-selective
inhibitors (COXIBs) can interfere with 2-series PG synthesis by
inhibiting cyclooxygenases (COXs), and have been widely used in
anti-inflammation, analgesia, antiplatelet aggregation and
anti-tumorigenesis treatment (49-51). However, the potential application
of NSAIDs and COXIBs in the treatment of T2DM and NAFLD requires
further investigation.
To the best of our knowledge, 2-series PGs play an
important role in the development of T2DM and NAFLD. However, few
studies have focused on the therapeutic effect of targeting the
2-series PG pathway in these two metabolic syndromes. Herein, the
way in which 2-series PGs exert multifunctional effects on the
highly intertwined pathogenesis of T2DM and NAFLD, including
insulin resistance, hyperglycemia, hepatic lipid accumulation and
inflammation, were systematically reviewed, and it was revealed
that targeting the 2-series PG pathway may be an important
therapeutic strategy in T2DM and NAFLD treatment.
PGs belong to eicosanoids and have 1-, 2- and
3-series homologues. Each series of PGs is biosynthesized from
different PUFAs, including dihomo-γ-linolenic acid (DGLA), AA and
eicosapentaenoic acid (EPA) (52). DGLA is catalyzed by COXs to
produce 1-series PGs (such as PGE1, PGG1 and
PGD1), and can also be converted to AA by the enzyme
Δ5 desaturase (53).
AA is the precursor of multiple important bioactive lipid
mediators, including the 2-series PGs (such as PGE2,
PGD2, PGF2α and PGI2) lipoxins,
leukotrienes, resolvins, protectins and maresins (54).
To the best of our knowledge, the 1-series
metabolites may be less closely associated with the correlation
between T2DM and NAFLD, since a limited number of studies have been
conducted. Furthermore, the 3-series PGs (such as PGF3α
and PGE3) produced by EPA generally have a lower
biological activity than their 1- and 2-series homologues (55). Therefore, the present review
focused on the 2-series PGs that are the principal PGs derived from
AA with a biological significance in T2DM and NAFLD.
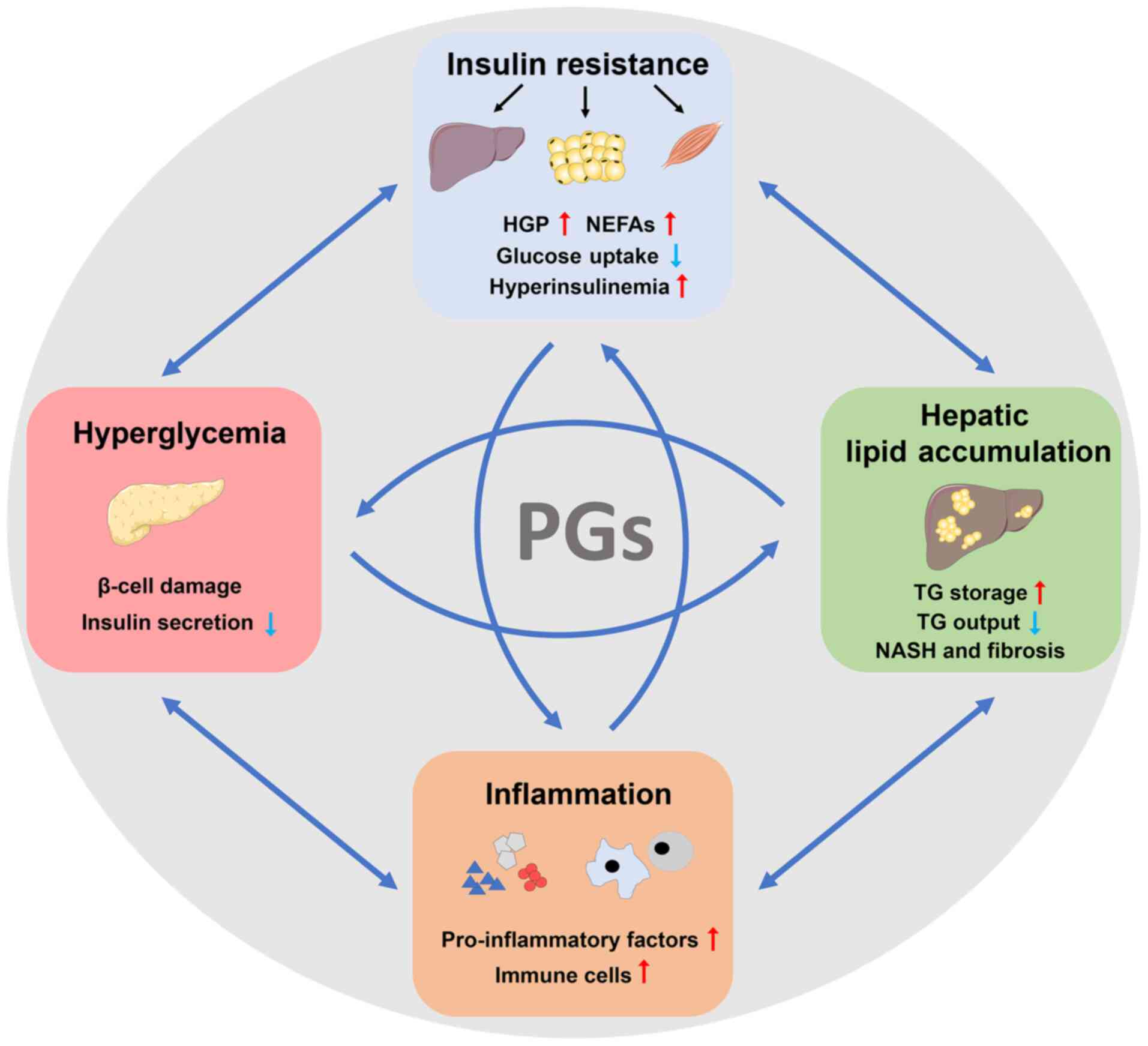
The synthesized 2-series PGs affect various
physiological and pathological processes, particularly the
important pathogenesis of T2DM and NAFLD, which includes insulin
resistance, hyperglycemia, hepatic lipid accumulation and
inflammation (Fig. 2) (17-19). In the next section, the role of
PGs in these pathogenic mechanisms will be further reviewed
(Fig. 3).
Insulin resistance is regarded as a key pathogenic
mechanism that accounts for the interplay between T2DM and NAFLD.
Insulin resistance is characterized by an impaired insulin
sensitivity of liver and peripheral tissues, including skeletal
muscle and adipose tissue. It is a crucial contributor to the other
related pathogeneses, which includes hyperinsulinemia,
hyperglycemia, dyslipidemia, ectopic lipid accumulation (such as in
the liver) and inflammation (Fig.
2) (61). More specifically,
first, insulin resistance contributes to hyperglycemia. Hepatic
insulin resistance markedly increases hepatic glucose production,
while peripheral insulin resistance enhances circulating
non-esterified fatty acids (NEFAs) and decreases glucose uptake,
together leading to elevated glycemia (62-65). Secondly, hepatic de novo
lipogenesis, a primary initiation mechanism of liver fat formation,
is facilitated by compensatory hyperinsulinemia and increased
substrates (such as glucose and NEFAs) under insulin-resistant
status in liver (64). Thirdly,
insulin resistance is of great significance in the
steatosis-to-NASH progression, as it is closely linked to
aggravated inflammation, apoptosis and fibrogenesis in the liver
(66). As for peripheral insulin
resistance, adipose insulin resistance also triggers chronic
low-grade inflammation by the release of adipokines and cytokines,
which in turn maintains or even exacerbates the development of T2DM
and NAFLD (67,68).
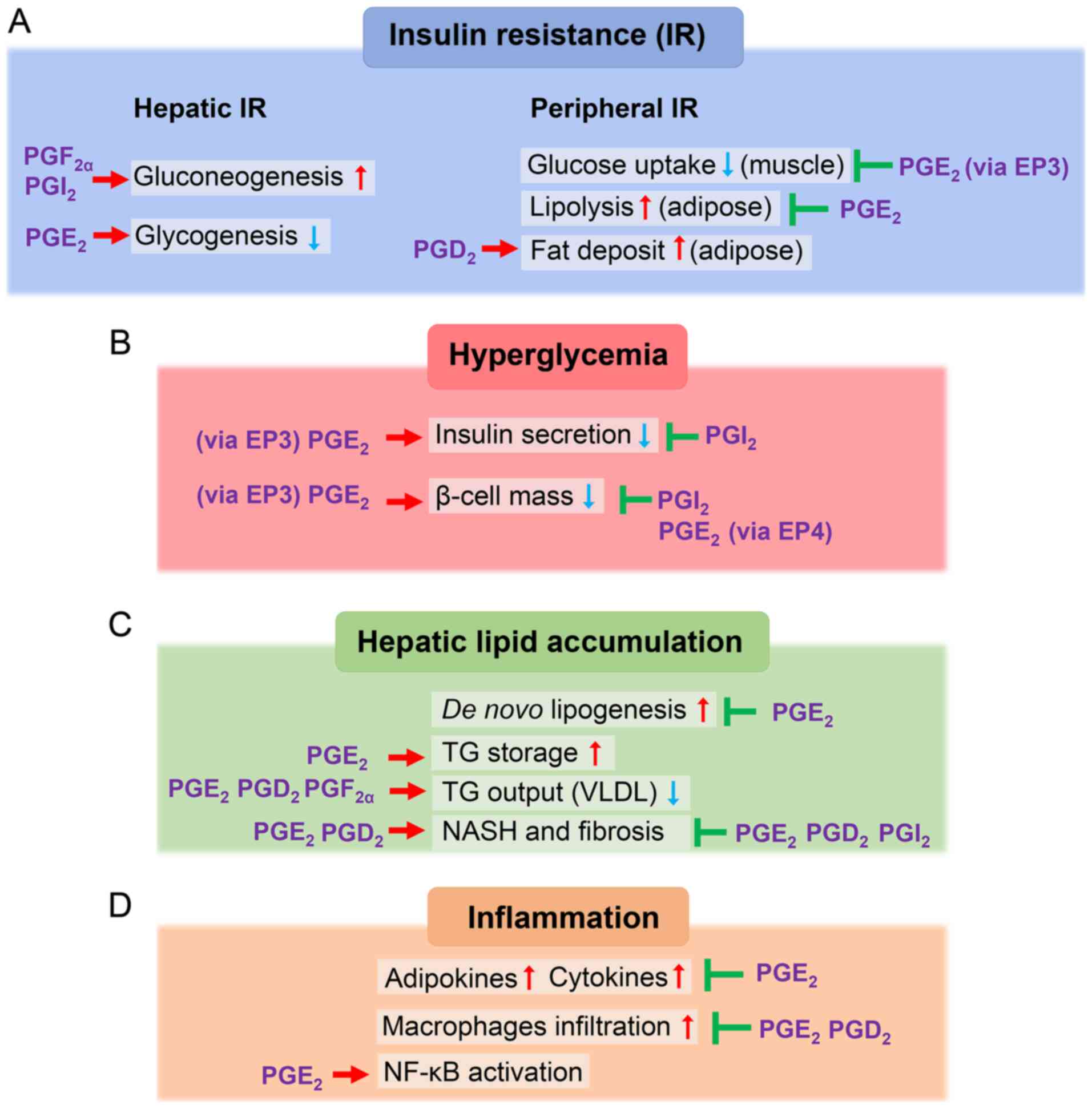
Accumulating evidence has revealed the important
role of 2-series PGs in the development of insulin resistance
(Fig. 3A) (37). Herein, the role of 2-series PGs
in both hepatic and peripheral insulin resistance was
discussed.
Hepatic insulin resistance is the key
pathophysiological event during the development of T2DM and NAFLD,
which is characterized by suppressed glycogenesis, increased
gluconeogenesis and glycogenolysis, and augmented de novo
lipogenesis (62-64). Insulin signaling has a different
effect on hepatic glucose and lipid metabolism. Under insulin
resistance, glucose metabolism becomes resistant to insulin action,
while lipid metabolism remains sensitive to insulin or even
enhanced by hyperinsulinemia (69). In combination, these metabolic
alterations enhance hepatic glucose production, finally leading to
hyperglycemia and liver lipid accumulation.
PGs have a dual effect on mediating hepatic insulin
signaling; however, their impact remains inconclusive. These
metabolites can be generated in hepatocytes, such as parenchymal
hepatocytes (70) and Kupffer
cells (71), acting as negative
mediators for insulin signaling. Previous experimental research has
shown that the use of COX-2 inhibitors in an obese rat model
resulted in decreased PGE metabolites and improved systemic insulin
sensitivity by increasing glucose uptake, repressing hepatic
glucose production and decreasing hepatic triglyceride (TG)
contents (37). Furthermore,
PGE2 can disrupt hepatic insulin signaling, which most
likely resembles the IL-6-induced interference on insulin signaling
(72). Via EP3 receptor,
PGE2 activates extracellular signal-regulated kinase 1/2
(ERK1/2) and subsequently promotes serine phosphorylation of
insulin receptor substrate (IRS) 1. This finally prevents glycogen
synthesis in cultured hepatocytes by interfering with
insulin-dependent serine/threonine kinase (Akt) activation
(72). Another study revealed
that PGE2-induced oncostatin M (OSM) production in liver
Kupffer cells attenuated insulin-dependent IRS/PI3K/Akt signaling,
leading to a repressed glucokinase expression and increased TG
accumulation in hepatocytes (71). The intrinsic mechanism is that
increased OSM promotes phosphorylation of signal transducer and
activator of transcription 3 (STAT3) to induce transcription of
cytokine signaling 3 (SOCS3) (71). Consistent with in vitro
results, this mechanism is also responsible for the development of
hepatic insulin resistance, steatosis and elevated plasma glucose
level in murine NAFLD models. It is recommended that the
PGE2-dependent feed-forward loop for NAFLD development
is most likely due to the suppression of fatty acid and TG
consuming pathways (fatty acid oxidation and TG export),
independently of the inhibition of insulin-induced fatty acid
synthesis (71).
The negative effects of PGs on insulin signaling are
closely associated with hepatic glucose homeostasis (particularly
gluconeogenesis). Gluconeogenic action is considerably increased
under insulin resistance (73).
A previous study revealed that the suppression of the hepatic
PGF2α-FP axis improved insulin resistance and glucose
homeostasis in ob/ob mice partially via decreased hepatic
gluconeogenesis (74). Under
fasting conditions, PGF2α activates FP receptors in
hepatocytes to upregulate gene expression levels of gluconeogenic
rate-limiting enzymes, phosphoenolpyruvate carboxykinase (PCK1),
and glucose-6-phosphatase (G6Pase) (74). The precise underlying mechanism
is that FP receptor coupling with G protein Gq facilitates
Ca2+ release and subsequently activates
Ca2+/calmodulin-dependent protein kinase II γ, which
accelerates p38-dependent forkhead box protein O1 (FOXO1) nuclear
translocation (74). Another
study revealed that treatment with high doses of acetylsalicylic
acid suppressed hepatic gluconeogenesis through the inhibition of
the COX-2/PGI2/IP axis for further improvement of
diabetes (75). Hepatic
gluconeogenesis was revealed to be inhibited by the downregulation
of PGI2 or disruption of IP receptor in a mouse model of
T2DM through the activation of the Gαs/protein kinase A
(PKA)/cAMP-response element binding protein pathway and inhibition
of Gβγ/PI3K-γ/protein kinase C
(PKC)-ζ/tribbles homolog 3/Akt/FOXO1 pathway, which is
involved in insulin signaling, both of which subsequently repressed
the expression of G6Pase and PCK1 in hepatocytes (75). These results demonstrated that
PGs can promote gluconeogenesis under insulin resistance.
Of note, PGs can also exert a protective effect on
hepatic insulin signaling through the regulation of COX-2 under the
stress of lipid overload, although COX-2 is widely recognized as a
pro-inflammatory mediator. Hepatic COX-2 overexpression in mice fed
with high-fat diet (HFD) caused a threefold increase of
PGE2 and elicited preservation against hepatic insulin
resistance. COX-2-dependent PG synthesis has been revealed to
mediate insulin signaling by increasing the Akt and AMP-activated
protein kinase phosphorylation level and decreasing the protein
tyrosine phosphatase-1β expression level in fatty livers or
hepatocytes exposed to fatty acids (76).
Insulin action in adipocytes and muscles is closely
correlated with glucose and lipid metabolism in T2DM and NAFLD. The
adipocyte insulin resistance can decrease intracellular TG storage
and induce lipolysis, which decreases fat content and increases the
release of NEFAs. Elevated circulating NEFAs can further lead to a
redistribution of fat depot from adipose tissue into the liver and
muscles, namely ectopic fat accumulation (65). Furthermore, adipose insulin
resistance facilitates the release of adipokines (such as
adiponectin, leptin and resistin) and cytokines [such as tumor
necrosis factor α (TNF-α), IL-6 and IL-1β], leading to chronic
low-grade inflammation in T2DM and NAFLD (67,68). On the other hand, insulin
resistance primarily impairs glucose uptake in muscle tissue, which
results in hyperglycemia and subsequently increases glucose
delivery to the liver for further hepatic lipogenesis (77).
PGs mostly exert preventive effects against adipose
insulin resistance and mediate adipogenesis in adipocytes. PGs may
improve insulin sensitivity by altering inflammatory status,
alleviating hepatic steatosis and overweight under obese status
(78). In both subcutaneous and
epididymal adipose tissues, the increased COX-2 activity enhances
various PGs levels, including PGE2, which further
improves the inflammatory profile including increased levels of
TNF-α, IL-33 and IL-4. This subsequently contributes to increased
insulin sensitivity in adipocytes and downregulates mRNA levels of
PPAR-γ and CCAAT/enhancer-binding protein α (37,78). In addition, particularly under
HFD treatment, mice with selective COX-2 overexpression in
adipocytes resulted in a mass reduction of inguinal white adipose
tissue (WAT) and decreased hepatic steatosis when compared with the
littermate control (78). This
finding suggested that COX-2-derived PGs may be benign mediators of
type 2 immunity cues in subcutaneous WAT under deranged
metabolism.
In addition to their impacts on adipose insulin
resistance, PGs are also correlated with muscle insulin resistance.
During the development of T2DM and NAFLD, increased delivery of
NEFAs can accelerate intramyocellular lipid accumulation, which
causes muscle insulin resistance. In addition, insulin-stimulated
glucose transport is impaired in insulin-resistant muscles, which
can happen prior to the occurrence of overt T2DM (90-92). PGs have been implicated in the
translation of insulin-dependent glucose uptake into skeletal
muscle (93) and, meanwhile,
PGE2 enhances insulin sensitivity to increase muscle
glycolysis (94). In addition,
COX-2-induced PGE2 production alleviates the fatty
acid-induced inflammatory process in skeletal muscle cells
(95). Furthermore,
intramuscular fat accumulation was observed in global deletion of
EP3 receptors in diabetic mice with diet-induced obesity (79). These results suggested that the
PGE2 signaling pathway may improve muscle insulin
resistance. However, the intrinsic mechanism remains poorly
understood.
In the aforementioned pathogenic mechanisms,
2-series PGs are most likely to aggravate hepatic insulin
resistance but prevent peripheral insulin resistance. The
aggravated hepatic insulin resistance eventually initiates or
exacerbates hyperglycemia, hepatic lipid accumulation and
inflammation, which in turn can be affected by these metabolic
stresses during the progressive disease course. Moreover, the
improved peripheral insulin resistance ameliorates blood
biochemical indexes and inflammatory status, but leads to excess
fat storage in muscle or adipose tissue. To date, the understanding
of how 2-series PGs affect the insulin signaling pathway and the
underlying molecular mechanism is lacking. Further investigations
of key molecular targets will shed light onto the translational
application in the treatment of T2DM and NAFLD.
Hyperglycemia is a hallmark of dysregulated glucose
metabolism that contributes to the initiation and progression of
T2DM and NAFLD (Fig. 2). When
insulin resistance occurs, chronic hyperglycemia can induce insulin
release by pancreatic β-cells, thus contributing to
hyperinsulinemia (96). Under
insulin resistance and hyperinsulinemia, elevated glycemia and
circulating NEFAs can cause the deleterious impairment of various
organs and tissues, processes that are referred to as glucotoxicity
and lipotoxicity, respectively (97,98). In the pancreas, glucotoxicity and
lipotoxicity can account for β-cell failure and subsequent insulin
secretion deficiency (97-99). In addition, hepatic
gluconeogenesis can be facilitated by insulin resistance, most
likely contributing to hyperglycemia by increasing the hepatic
glucose output. These mechanisms collectively contribute to
hyperglycemia during the development of T2DM and NAFLD.
A previous study has demonstrated the close
correlation between 2-series PG action and the development of
hyperglycemia (Fig. 3B). First,
COX-1 and COX-2 participate in the control of glycemia (100). In a 2-week clinical trial of
high-dose aspirin treatment among nine patients with T2DM, aspirin
treatment was revealed to reduce fasting plasma glucose and
improves insulin sensitivity in cases with diabetes (101). In addition, the increased
formation of PGs and PG metabolites has been observed in T2DM,
including PGE2, PGI2 in islet or blood, and
15-keto-dihydro-PGF2α, 8-iso-PGF2α in urine
(102-106). However, interference with the
PGE2/EP3 signaling pathway through the blockade of the
EP3 receptor in mice has been reported to predispose to systemic
insulin resistance; in addition, insulin secretion also increases,
finally contributing to hyperglycemia (79,107). These observations imply the
multifunctional involvement of 2-series PGs in the development of
hyperglycemia.
β-cell failure includes β-cell dysfunction and
β-cell mass deficiency, which remain the two major causes of
hyperglycemic pathogenesis. β-cell dysfunction and apoptosis reduce
insulin secretion and deplete β-cell mass, respectively (108). Due to their involvement in
inflammation and oxidative stress signaling pathways, PGs mainly
act as an initial and deteriorative pathological element for β-cell
failure, leading to hyperglycemia.
Glucose-stimulated insulin secretion (GSIS)
commonly occurs when β-cells are constantly exposed to high glucose
stimulation (109). To a
certain extent, PGs act as a potential negative modulator of GSIS.
A number of studies have demonstrated that PGE2
attenuates GSIS. PGE2 is the predominant E-series PG in
islets formed by COX-2, the dominant form of COX in the pancreas
(110-112). COX-2 expression is
significantly upregulated in pancreatic islets under hyperglycemic
conditions (113,114). COX-2-dependent PGE2
generation is augmented by group X secretory phospholipase
A2 and eventually suppresses GSIS in vitro and
in vivo (115).
PGE2 equally inhibits both two phases of GSIS in HIT
cells, which is associated with reduced cAMP accumulation mediated
by pertussis toxin-sensitive G protein (Gi) (116). Among PGE receptors, EP3
receptor is the most abundant PGE receptor type in islets (112), which is overexpressed in islets
from patients with T2DM (117,118). Previous research has indicated
that PGE2 coupling with EP3 receptor is highly
associated with a reduction of insulin secretion in terms of β-cell
dysfunction. Meng et al (118) revealed that the
PGE2-stimulated gene expression of PG EP3 receptor
subtype led to intracellular cAMP reduction, accompanied by a
downregulated phosphorylation level of Akt and forkhead box 'Other'
(Foxo) in HIT-T15 cells (118).
Kimple et al (103)
confirmed that this active PGE2/EP3 receptor pathway in
islets depended on coupling to G-proteins of the Gi subfamily in
vivo. In addition, EP3 receptor agonists can antagonize
glucagon-like peptide-1 (GLP-1) signaling, leading to reduced cAMP
production and attenuated GSIS (103). Hence, since GLP-1 treatment is
not effective in all patients with T2DM (119), as a non-competitive antagonist
of GLP-1 receptor, EP3 receptor may be a potent target for
improving the GLP-1 effect in anti-diabetic therapeutics (103,120). Another observation revealed
that PGE2 presents an impotent influence on GSIS
suppression in rat islets exposed to epinephrine-induced glucose
overload (121). Under
hyperglycemic states, crosstalk between PGs and other inflammatory
factors has a profound effect on glycemic control. Systemic
inflammatory responses are upregulated in T2DM individuals
(122), characterized by
elevated levels of lipid molecules, including PGs and cytokines
such as TNF-α (123), IL-1β
(124), IL-6 (125) and IL-8 (126), in correspondence with the
decline of their natural antibodies (127). In PG signaling, COX-2 is
involved in IL-1β-induced auto-stimulation in islets (111). The COX-2 expression and
activity are upregulated by IL-1β-induced NF-κB activation,
resulting in a negative effect on GSIS caused by increased
PGE2 via EP3 receptor (112). Recently, the
IL-1β/COX-2/PGE2 pathway loop has been revealed as the
underlying mechanism for the onset and progression of diabetes,
which leads to β-cell inflammatory impairments by downregulating
the expression of β-cell functional genes pancreatic and duodenal
homeobox 1, NK6 homeobox 1 and MAF bZIP transcription factor A
(124). As previously
mentioned, PGE2 can impact GSIS through different
receptors and also by interacting with inflammatory reaction in
hyperglycemia.
Deficient β-cell mass is recognized as another
essential event that results in elevated glycemia in T2DM
progression (128,129). Apart from delaying the GSIS
process, COX-2/PGE2 signaling also plays a role in the
regulation of β-cell proliferation and apoptosis. In a model of
transgenic mice overexpressing COX-2 and microsomal prostaglandin E
synthase 1 (mPGES-1), increased PGE2 appeared to be
associated with a significant reduction in the number of β-cells
and further caused severe hyperglycemia (130). A different study concluded that
the blockade of EP3 receptor and activation of EP4 receptor
enhanced human β-cell proliferation and survival ex vivo,
suggesting a reciprocal effect of different EP receptors on the
mediation of β-cell failure in T2DM (117). Furthermore, an EP3 receptor
antagonist improved β-cell proliferation partly by enhancing
phospholipase C-γ1 activity in young mouse islets rather than in
old ones, while the EP4 receptor was activated to exert the same
protective effect in human β-cells only with combination of EP3
inhibition. In terms of promotion of β-cell survival, forkhead box
protein M1, a critical β-cell proliferation factor, is upregulated
by EP3 antagonist and EP4 agonist in islets from obese T2DM
individuals (117). However,
EP4 has further been revealed to be involved in PKA signaling
activation through a GS-coupled mechanism in the
survival of mouse β-cells, which is proposed to facilitate the
phosphorylation of eukaryotic initiation factor 4E and PKC-ε in a
putative downstream mechanism (117). In addition, α-subunit of the
heterotrimeric Gz protein (Gαz), a member of
the Gαi family, may couple to EP3 in pancreatic β-cells
(131). The global deletion of
Gαz can block the PGE2/EP3 pathway, which
subsequently results in a robust increase in β-cell mass and
augments GSIS by cAMP upregulation in mice with both insulin
resistance and glucose intolerance (120). In addition, EP3 receptor
knockout in HFD-fed mice was revealed to promote β-cell
proliferation (79), which is
consistent with the aforementioned Gαz-null data.
Notably, in human islets from patients with T2DM and MIN6 β-cells,
palmitate can upregulate the expression levels of COX-2 and EP3
receptor, which initiates β-cell apoptosis through the
COX-2/PGE2/EP3 pathway (132). These results demonstrated that
the PGE2 pathway can inhibit proliferation and induce
apoptosis in β-cells exposed to glucotoxicity and lipotoxicity.
Elevated circulating lipid contents (such as
cholesterol, TG and NEFAs) are a characteristic of T2DM and NAFLD
(Fig. 2) (133-135). Lipotoxicity alone or in
combination with glucotoxicity is highly associated with the
impairment of insulin sensitivity in various organs. Previous
clinical studies have revealed that lipid infusion contributes to
hepatic insulin resistance (136). In addition, the accumulation of
hepatic lipid contents, particularly TGs, is an initiation of liver
steatosis. Liver steatosis is the first hit in the progression of
NAFLD, whose onset is due to insulin resistance (19,137). The direct contributors to
excess lipid storage in the liver include increased circulating
NEFAs, accelerated de novo lipogenesis, overloaded dietary
fat and inadequate lipid oxidation (64,65,134,138). In turn, hepatic steatosis
induces subacute intrahepatic inflammation through the NF-κB
pathway as a pathogenic mechanism for exacerbated hepatic and
systematic insulin resistance both in NAFLD and T2DM (19,139).
PGs markedly contribute to the dysregulation of the
lipid metabolism in hepatic lipid accumulation (Fig. 3C). PGE2 acts
synergistically with insulin in the pathogenesis of hepatic
steatosis, but their roles remain discordant and controversial.
PGE2 decreases the activity of lipogenic enzymes in
primary hepatocytes in vitro through sustained ERK1/2
activation, thereby attenuating insulin-dependent phosphorylation
of Akt kinase. This finally abrogates insulin signaling and further
alleviates SREBP-1c pathway in hepatic de novo lipogenesis
(140). Furthermore, short-term
blockade of PGE2 signaling by EP3 antagonist in mice
with diet-induced obesity caused a significant reduction of TG
content in skeletal muscle and slightly increased hepatic TGs
(107). As a result, it can be
hypothesized that PGE2 elicits preservation against
hepatic steatosis. However, other observations vary from this
hypothesis. A previous study has indicated that extracellular
PGD2, PGE2 and PGF2α diminish the
secretion of very low-density lipoprotein (VLDL)-apolipoprotein B
(apoB) to promote steatosis in primary hepatocytes (141). The reduction of VLDL-apoB is
correlated with decreased TG transportation and impaired cellular
TG recycling, which finally results in a reduced TG output. In
addition, only PGE2 can completely antagonize the
IL-6-induced secretion of VLDL-apoB in hepatocytes (141). Furthermore, PGE2
acts synergistically with insulin and enhances the incorporation of
glucose into TGs in hepatocytes. PGE2 and insulin
synergistically inhibit lipolysis, mitochondrial β-oxidation and
VLDL synthesis, which are mediated by PGE2-dependent
suppression of adipose TG lipase, carnitine palmitoyltransferase-1
and apoB-mediated lipidation, respectively (140). Moreover, apoB and microsomal
transfer protein are downregulated by PPAR-γ-coactivator-1α and
PCK1 in insulin-dependent and PGE2-dependent manners. In
combination, these events contribute to a reduced TG breakdown and
increased fat droplets in hepatocytes (140). In terms of NAFLD development
in vivo, under HFD feeding, increased COX-2 activity and
PGE2 concentration also results in hepatic steatosis in
mice mostly through NF-κB activation and lipid peroxidation
enhancement. An aggravation of insulin resistance also appears with
increased levels of serum alanine aminotransferase and total
hepatic fatty acids (142).
Another putative mechanism of hepatic steatosis formation involves
CD36-mediated PG levels in the liver. Although the expression level
of CD36 was 5-fold higher in hepatic steatosis liver than in normal
liver, the global deletion of CD36 in ob/ob mice aggravated
hepatic lipid accumulation by significantly suppressing the outputs
of VLDL, apoB and TGs by increasing hepatic PGD2,
PGE2 and PGF2α (143). Based on these experiments, PGs
including PGD2, PGE2 and PGF2α may
accelerate the initiation and progression of hepatic steatosis.
Since the mediation of PGs on hepatic lipid
accumulation is ambiguous, the precise mechanism requires further
study. Considering the findings of the aforementioned studies, it
can be hypothesized that various PGs (PGD2,
PGE2 and PGF2α) promote hepatic lipid
accumulation, mostly through facilitating TG storage and inhibiting
TG output by repressing lipolysis, fatty acid oxidation and VLDL
synthesis. In addition, under insulin resistance, PGs can increase
de novo lipogenesis and promote the development of hepatic
steatosis. As hepatic lipid accumulation is the initial step of
NAFLD as well as a risk factor for T2DM, the inhibition of the
2-series PG pathway may be a potential option for treating
NAFLD.
Systemic chronic inflammation is a health-damaging
phenotype that plays a central role in multiple metabolic
syndromes, including T2DM and NAFLD (Fig. 2) (145,146). 2-Series PGs have
multifunctional effects on the promotion and resolution of
inflammation following the occurrence of insulin resistance, β-cell
failure and hepatic steatosis (Fig.
3D) (37,71,78,79,82,112,142). PGs, TNF-α, IL-1β and IL-6 have
been recognized as the major inflammatory mediators in T2DM and
NAFLD (102,145-148). There are multifaceted
interactions between PGs and other inflammatory molecules or cells
in the pathogenic mechanisms of T2DM and NAFLD. As aforementioned,
COX-2-derived PGs disrupt insulin signaling by activating the
STAT3/SOCS3 signaling pathway in the liver or interacting with
TNF-α and ILs in WAT (37,71,78), whereas, in obesity,
PGD2 and PGE2 mediate macrophage polarization
and infiltration with downregulated TNF-α, MCP-1 and IL-6 in WAT,
leading to an improvement in peripheral insulin resistance
(79,82). In turn, when peripheral insulin
resistance occurs, adipokines and cytokines are released from
dysfunctional adipose tissues and subsequently induce inflammation,
which is associated with β-cell failure and hepatic steatosis.
COX-2-derived PGE2 contributes to β-cell dysfunction in
GSIS via IL-1β-induced NF-κB activation (112), and hepatic lipid accumulation
via NF-κB activation (142).
NF-κB-mediated inflammation is important in the pathogenesis of
T2DM and NAFLD. PGs may be involved in inflammatory processes
mostly through NF-κB pathway activation with or without coaction
with other inflammatory factors. The inhibitor κB kinase β
(IKK-β)/NF-κB pathway plays a critical role in chronic hepatic
inflammation, leading to insulin resistance and steatohepatitis, in
which TNF-α and IL-1β are also involved (19,139).
Hepatic inflammation is a landmark in the
development of NASH, which is also triggered by progressed insulin
resistance and other injurious stimuli, such as glucotoxicity and
lipotoxicity (149-152). Progressively, NASH-related
hepatic fibrosis and cirrhosis become long-term manifestations of
NAFLD. Hepatic fibrosis is characterized by high-density
extracellular matrix protein deposition (153). Both NASH and liver fibrosis can
be exacerbated in NAFLD with comorbidity of T2DM (154,155). T2DM-promoted NASH is attributed
to peripheral insulin resistance, intrahepatic lipotoxicity and M1
macrophage recruitment in adipose tissue (156-158). The activated M1 macrophages
secrete pro-inflammatory cytokines, including MCP-1, TNF-α and
IL-1β, which induce systemic inflammation. These cytokines are
further delivered to the liver and cause steatohepatitis (159). Therefore, insulin resistance,
hyperglycemia, hyperinsulinemia and hyperlipidemia are key factors
for pro-inflammatory status in hepatic inflammation, in which PGs
are involved as inflammatory mediators. In addition, the
upregulation of transforming growth factor-β (TGF-β) and connective
tissue growth factor in T2DM can lead to NAFLD-related fibrosis
progression (160,161).
PGs are correlated with the progression from
hepatic steatosis to NASH. The upregulated expression of COX-2 and
mPGES-1, the key enzymes of PGE2 synthesis, is closely
associated with NASH activity score in human liver from patients
with NASH (162). Lipidomics
profiling was performed in a clinical cohort that attempted to
describe the hepatic inflammatory characteristic of NASH. As a
result, the plasma PGE2 level was revealed to be
elevated only in patients with NASH, while the level of
13,14-dihydro-15-keto-PGD2, a metabolite degraded from
PGD2, was found to be remarkably higher in the NASH
group, compared with the simple steatosis or control groups
(163). As a consequence, it is
reasonable to suggest that PGs, particularly PGE2, may
aggravate the course of NAFLD.
However, there are some discrepancies in the impact
of PGs and COX-2 activity on the development of NASH under dietary
nutritious stress. An in vivo study revealed that
hepatocyte-specific COX-2 transgenic mice (hCOX-2-Tg) with an
increased level of PGE2 improved intrahepatic steatosis,
ballooning and inflammation (164). This was partially achieved by
decreasing the plasma levels of pro-inflammatory cytokines (such as
IL-1β, IL-6, TNF-α and MCP-1), and inhibiting macrophage
recruitment and infiltration in steatohepatitis liver induced by a
methionine- and choline-deficient diet (MCDD) (164). In addition, there are
ameliorations of augmented oxidative stress and apoptosis in liver
samples with NASH (164).
Similarly, under a NASH diet, hepatic PGE2 production
derived from mPGES-1 is increased to potently inhibit
monocyte-derived macrophage infiltration, which is associated with
PGE2-induced suppression of TNF-α-triggered responses in
hepatocytes. These responses consist of pro-inflammatory cytokine
IL-1β production and hepatocyte apoptosis (162). These results suggest a combined
action of PGs and other inflammatory factors in NASH development.
Furthermore, the blockade of L-PGDS in PGD2 signaling
rapidly accelerates non-alcoholic simple steatosis to severe
steatohepatitis in nutrition overload or normal conditions
(165). This progression to
NASH is also accompanied by enhanced lipogenic gene expression
(such as SREBP-1c and liver X receptor α) and deranged metabolic
features, including progressed insulin resistance and increased
fasting glucose, insulin and lipid levels in the blood (165). With regards to the
PGI2/IP pathway, under MCDD conditions,
IP-receptor-knockout (IP-KO) mice had accelerated progression to
steatohepatitis, with greater iron deposition due to marked
oxidative stress. PGI2-IP signaling prevents the
development of NASH in anti-inflammatory response, as evidenced by
the suppressed expression of MCP-1 and TNF-α in
lipopolysaccharide-stimulated Kupffer cells in vitro.
Consistently, the Kupffer cell-induced expression levels of MCP-1
and TNF-α were progressively increased in IP-KO mice, and the
oxidative stress-induced hepatic iron deposition was reduced in the
MCDD-induced steatohepatitis liver, suggesting that PGI2
signaling inhibits inflammation and influences the antioxidant
reaction in NASH (166). Thus,
PG appears to play a protective role against hepatic
steatohepatitis, most likely under disturbed metabolism in NAFLD
and T2DM progression.
The key mechanisms of hepatic fibrosis include a
disbalance between fibrogenesis and fibrinolysis and the activation
of hepatic stellate cells (HSCs) and Kupffer cells in response to
various stimuli (167,168). Numerous studies have revealed
that PGs facilitate the development of hepatic steatosis,
steatohepatitis and fibrosis (169-171). In a prospective cohort research
of 361 patients with NAFLD, daily aspirin use induced less severity
of histologic characteristics of NAFLD and significantly decreased
the risk of fibrosis initiation and progression in a
duration-dependent manner, when compared with the non-regular use
of aspirin (172). It was
further suggested that the antifibrotic effect of long-term aspirin
treatment is attributed to its involvement in inhibiting NF-κB and
IKK-β signaling (173).
Furthermore, plasma bioactive lipids, such as PGE2 and
PGI2, have been regarded as useful markers for prognosis
in liver cirrhosis (174). In
accordance with clinical evidence, the upregulation of COX-2 was
positively correlated with fibrosis formation in liver from a
carbon tetrachloride (CCl4)-induced fibrotic mouse model
(175). Conversely, it was
revealed that COX-2-derived PGE2 could suppress
fibrogenesis and NASH progression (176,177). In hCOX-2 Tg mice with
diet-induced NASH, PGE2 attenuated
CCl4-induced liver fibrosis by decreasing the activation
and proliferation of HSCs and increasing apoptosis by suppressing
microRNA (miR)-23a and miR-28a expression (164,178). In addition, COX-2-derived
PGE2 was revealed to suppress collagen synthesis through
the downregulation of collagen type I α1, α smooth muscle actin and
collagen binding protein-1 in HSCs under TGF-β1-induced conditions
(178,179). These results demonstrated that
the COX-2/PGE2 pathway prevents the development of liver
fibrosis through growth-suppressive and pro-apoptotic effects on
HSCs.
To sum up, during the progression of T2DM and
NAFLD, PGs may primarily act by interacting with other inflammatory
factors, as well as mediating the NF-κB signaling pathway, which
plays an important role in the chronic inflammation caused by
glucotoxicity and lipotoxicity in a variety of organs. PGs can
serve as pro-inflammatory mediators in the impairment of insulin
sensitivity, glycemia and hepatic lipid metabolism. However,
PGD2, PGE2 and PGI2 also exert
anti-inflammatory effects and improve peripheral insulin
resistance, NASH and related fibrosis. Due to the complex action of
PGs in the inflammatory process, the use of COX inhibitors in T2DM
and NAFLD treatment should be given more consideration, and further
explorations are highly warranted.
The comorbidity of T2DM and NAFLD is well
recognized and has become an area of increased investigation over
past decades. Nowadays, considerable evidence has highlighted the
roles of 2-series PGs in the pathogenesis of T2DM and NAFLD.
2-Series PGs are important lipid molecules that are widely
distributed in various organs. These exert multifunctional effects
on the four highly intertwined pathogeneses of T2DM and NAFLD,
including insulin resistance, hyperglycemia, hepatic lipid
accumulation and chronic inflammation. PGs potently mediate insulin
resistance, which subsequently induces pathological alterations
including hyperinsulinemia, hyperglycemia, dyslipidemia and ectopic
lipid accumulation. In addition, PGs can directly impact
hyperglycemia by decreasing insulin secretion, pancreatic β-cell
proliferation and increasing gluconeogenesis. In addition, PGs
contribute to hepatic lipid accumulation by enhancing hepatic
lipogenesis and decreasing TG output. Moreover, PGs distinctly
establish a close interaction with inflammatory processes in the
progression of T2DM and NAFLD.
Most 2-series PGs exert negative effects on the
progression of T2DM and NAFLD. Therefore, the application of COX
inhibitors such as aspirin and celecoxib beyond their conventional
use on vascular diseases, rheumatoid arthritis and pain is emerging
as a promising option for T2DM and NAFLD treatment. However,
certain aspects of the application of PG pathways should be
considered. First, some PGs are beneficial to the prevention of
T2DM and NAFLD development to a certain extent, suggesting that the
clinical use of COX inhibitors requires careful consideration and
highlighting the potential therapeutic use of PGs and their
derivates in the prevention and control of T2DM and NAFLD.
Secondly, since the existing NSAIDs and COXIBs are associated with
several side effects, it is meaningful to perform molecular
modification of these drugs and develop new treatment strategies,
to aim to accurately modulate the PG pathway in related organs such
as the pancreas, liver and adipose tissues. Overall, due to the
important role of 2-series PGs in T2DM and NAFLD, additional
studies associated with the molecular mechanisms of PGs in the
pathogenesis of T2DM and NAFLD are highly warranted. These studies
will provide new and more precise therapeutic strategies based on
targeting PG pathways in the treatment of these two diseases.
Not applicable.
JG, WW and XZ conceived the study. WW and XZ wrote
and prepared the original manuscript. JG and WW contributed to the
review of the manuscript. JG and WW were responsible for the
funding acquisition. All authors read the final manuscript and
agree to be accountable for the content of the work.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The authors would like to thank Professor Lexun
Wang from the Institute of Chinese Medicine Sciences, Guangdong
Pharmaceutical University, Guangzhou, China for editing and
reviewing our manuscript.
This work was supported by the National Key R&D plan
'Research on modernization of traditional Chinese medicine' (grant
no. 2018YFC1704200), the Major Basic And Applied Basic Research
Projects of Guangdong Province of China (grant no. 2019B030302005),
the Basic and Applied Basic Research fund of Guangdong Province
(grant no. 2021A1515012553), the Innovative Strong School Project
of Guangdong Pharmaceutical University (grant no. 2018KQNCX130),
the Basic and Applied Basic Research Fund of Guangdong Province
(grant no. 2019A1515110123), and the Medical Science and Technology
Research Fund of Guangdong Province (grant no. A2019531).
|
1
|
International Diabetes Federation(IDF):
IDF diabetes atlas. 9th edition. IDF; Brussels: 2019
|
|
2
|
Younossi ZM, Koenig AB, Abdelatif D, Fazel
Y, Henry L and Wymer M: Global epidemiology of nonalcoholic fatty
liver disease-meta-analytic assessment of prevalence, incidence,
and outcomes. Hepatology. 64:73–84. 2016. View Article : Google Scholar
|
|
3
|
Chatterjee S, Khunti K and Davies MJ: Type
2 diabetes. Lancet. 389:2239–2251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Younossi Z, Anstee QM, Marietti M, Hardy
T, Henry L, Eslam M, George J and Bugianesi E: Global burden of
NAFLD and NASH: Trends, predictions, risk factors and prevention.
Nat Rev Gastroenterol Hepatol. 15:11–20. 2018. View Article : Google Scholar
|
|
5
|
Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX,
Zhang P, She ZG, Zhu L, Cai J and Li H: Unexpected rapid increase
in the burden of NAFLD in China from 2008 to 2018: A systematic
review and meta-analysis. Hepatology. 70:1119–1133. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Younossi ZM, Golabi P, de Avila L, Paik
JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A and Nader F: The
global epidemiology of NAFLD and NASH in patients with type 2
diabetes: A systematic review and meta-analysis. J Hepatol.
71:793–801. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sung KC, Jeong WS, Wild SH and Byrne CD:
Combined influence of insulin resistance, overweight/obesity, and
fatty liver as risk factors for type 2 diabetes. Diabetes Care.
35:717–722. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wild SH, Morling JR, McAllister DA,
Kerssens J, Fischbacher C, Parkes J, Roderick PJ, Sattar N and
Byrne CD; Scottish and Southampton Diabetes and Liver Disease
Group: Scottish Diabetes Research Network Epidemiology Group: Type
2 diabetes and risk of hospital admission or death for chronic
liver diseases. J Hepatol. 64:1358–1364. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leite NC, Villela-Nogueira CA, Pannain VL,
Bottino AC, Rezende GF, Cardoso CR and Salles GF: Histopathological
stages of nonalcoholic fatty liver disease in type 2 diabetes:
Prevalences and correlated factors. Liver Int. 31:700–706. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prashanth M, Ganesh HK, Vima MV, John M,
Bandgar T, Joshi SR, Shah SR, Rathi PM, Joshi AS, Thakkar H, et al:
Prevalence of nonalcoholic fatty liver disease in patients with
type 2 diabetes mellitus. J Assoc Physicians India. 57:205–210.
2009.PubMed/NCBI
|
|
11
|
Dam-Larsen S, Franzmann M, Andersen IB,
Christoffersen P, Jensen LB, Sørensen TI, Becker U and Bendtsen F:
Long term prognosis of fatty liver: Risk of chronic liver disease
and death. Gut. 53:750–755. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ratziu V, Bonyhay L, Di Martino V,
Charlotte F, Cavallaro L, Sayegh-Tainturier MH, Giral P, Grimaldi
A, Opolon P and Poynard T: Survival, liver failure, and
hepatocellular carcinoma in obesity-related cryptogenic cirrhosis.
Hepatology. 35:1485–1493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ekstedt M, Franzén LE, Mathiesen UL,
Thorelius L, Holmqvist M, Bodemar G and Kechagias S: Long-term
follow-up of patients with NAFLD and elevated liver enzymes.
Hepatology. 44:865–873. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rafiq N, Bai C, Fang Y, Srishord M,
McCullough A, Gramlich T and Younossi ZM: Long-term follow-up of
patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol.
7:234–238. 2009. View Article : Google Scholar
|
|
15
|
Targher G, Bertolini L, Padovani R,
Rodella S, Tessari R, Zenari L, Day C and Arcaro G: Prevalence of
nonalcoholic fatty liver disease and its association with
cardiovascular disease among type 2 diabetic patients. Diabetes
Care. 30:1212–1218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ryysy L, Häkkinen AM, Goto T, Vehkavaara
S, Westerbacka J, Halavaara J and Yki-Järvinen H: Hepatic fat
content and insulin action on free fatty acids and glucose
metabolism rather than insulin absorption are associated with
insulin requirements during insulin therapy in type 2 diabetic
patients. Diabetes. 49:749–758. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lomonaco R, Bril F, Portillo-Sanchez P,
Ortiz-Lopez C, Orsak B, Biernacki D, Lo M, Suman A, Weber MH and
Cusi K: Metabolic impact of nonalcoholic steatohepatitis in obese
patients with type 2 diabetes. Diabetes Care. 39:632–638. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marchesini G, Brizi M, Bianchi G,
Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S,
Forlani G and Melchionda N: Nonalcoholic fatty liver disease: A
feature of the metabolic syndrome. Diabetes. 50:1844–1850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai D, Yuan M, Frantz DF, Melendez PA,
Hansen L, Lee J and Shoelson SE: Local and systemic insulin
resistance resulting from hepatic activation of IKK-beta and
NF-kappaB. Nat Med. 11:183–190. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun G, Jackson CV, Zimmerman K, Zhang LK,
Finnearty CM, Sandusky GE, Zhang G, Peterson RG and Wang YJ: The
FATZO mouse, a next generation model of type 2 diabetes, develops
NAFLD and NASH when fed a Western diet supplemented with fructose.
BMC Gastroenterol. 19:412019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garcia-Jaramillo M, Spooner MH, Löhr CV,
Wong CP, Zhang W and Jump DB: Lipidomic and transcriptomic analysis
of western diet-induced nonalcoholic steatohepatitis (NASH) in
female Ldlr-/-mice. PLoS One. 14:e02143872019. View Article : Google Scholar
|
|
22
|
Verboven M, Deluyker D, Ferferieva V,
Lambrichts I, Hansen D, Eijnde BO and Bito V: Western diet given to
healthy rats mimics the human phenotype of diabetic cardiomyopathy.
J Nutr Biochem. 61:140–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hernandez-Rodas MC, Valenzuela R and
Videla LA: Relevant aspects of nutritional and dietary
interventions in non-alcoholic fatty liver disease. Int J Mol Sci.
16:25168–25198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Valenzuela R and Videla LA: The importance
of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in
development of non-alcoholic fatty liver associated with obesity.
Food Funct. 2:644–648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taha AY, Cheon Y, Faurot KF, Macintosh B,
Majchrzak-Hong SF, Mann JD, Hibbeln JR, Ringel A and Ramsden CE:
Dietary omega-6 fatty acid lowering increases bioavailability of
omega-3 polyunsaturated fatty acids in human plasma lipid pools.
Prostaglandins Leukot Essent Fatty Acids. 90:151–157. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wood KE, Lau A, Mantzioris E, Gibson RA,
Ramsden CE and Muhlhausler BS: A low omega-6 polyunsaturated fatty
acid (n-6 PUFA) diet increases omega-3 (n-3) long chain PUFA status
in plasma phospholipids in humans. Prostaglandins Leukot Essent
Fatty Acids. 90:133–138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xia F, He C, Ren M, Xu FG and Wan JB:
Quantitative profiling of eicosanoids derived from n-6 and n-3
polyunsaturated fatty acids by twin derivatization strategy
combined with LC-MS/MS in patients with type 2 diabetes mellitus.
Anal Chim Acta. 1120:24–35. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li N, Yue H, Jia M, Liu W, Qiu B, Hou H,
Huang F and Xu T: Effect of low-ratio n-6/n-3 PUFA on blood
glucose: A meta-analysis. Food Funct. 10:4557–4565. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu HQ, Qiu Y, Mu Y, Zhang XJ, Liu L, Hou
XH, Zhang L, Xu XN, Ji AL, Cao R, et al: A high ratio of dietary
n-3/n-6 polyunsaturated fatty acids improves obesity-linked
inflammation and insulin resistance through suppressing activation
of TLR4 in SD rats. Nutr Res. 33:849–858. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shrestha N, Cuffe JSM, Holland OJ, Perkins
AV, McAinch AJ and Hryciw DH: Linoleic acid increases prostaglandin
E2 release and reduces mitochondrial respiration and cell viability
in human trophoblast-like cells. Cell Physiol Biochem. 52:94–108.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kliewer SA, Lenhard JM, Willson TM, Patel
I, Morris DC and Lehmann JM: A prostaglandin J2 metabolite binds
peroxisome proliferator-activated receptor gamma and promotes
adipocyte differentiation. Cell. 83:813–819. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hennig B, Toborek M, Joshi-Barve S, Barger
SW, Barve S, Mattson MP and McClain CJ: Linoleic acid activates
nuclear transcription factor-kappa B (NF-kappa B) and induces
NF-kappa B-dependent transcription in cultured endothelial cells.
Am J Clin Nutr. 63:322–328. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Seo MJ and Oh DK: Prostaglandin synthases:
Molecular characterization and involvement in prostaglandin
biosynthesis. Prog Lipid Res. 66:50–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schaid MD, Zhu Y, Richardson NE,
Patibandla C, Ong IM, Fenske RJ, Neuman JC, Guthery E, Reuter A,
Sandhu HK, et al: Systemic metabolic alterations correlate with
islet-level prostaglandin E2 production and signaling mechanisms
that predict β-cell dysfunction in a mouse model of type 2
diabetes. Metabolites. 11:582021. View Article : Google Scholar
|
|
35
|
Nasrallah R, Robertson SJ, Karsh J and
Hébert RL: Celecoxib modifies glomerular basement membrane,
mesangium and podocytes in OVE26 mice, but ibuprofen is more
detrimental. Clin Sci (Lond). 124:685–694. 2013. View Article : Google Scholar
|
|
36
|
Chan PC, Hsiao FC, Chang HM, Wabitsch M
and Hsieh PS: Importance of adipocyte cyclooxygenase-2 and
prostaglandin E2-prostaglandin E receptor 3 signaling in the
development of obesity-induced adipose tissue inflammation and
insulin resistance. FASEB J. 30:2282–2297. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen
CH and Shih KC: COX-2-mediated inflammation in fat is crucial for
obesity-linked insulin resistance and fatty liver. Obesity (Silver
Spring). 17:1150–1157. 2009. View Article : Google Scholar
|
|
38
|
Szerafin T, Erdei N, Fulop T, Pasztor ET,
Edes I, Koller A and Bagi Z: Increased cyclooxygenase-2 expression
and prostaglandin-mediated dilation in coronary arterioles of
patients with diabetes mellitus. Circ Res. 99:e12–e17. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang T, Cai H, Zheng W, Michel A, Pawlita
M, Milne G, Xiang YB, Gao YT, Li HL, Rothman N, et al: A
prospective study of urinary prostaglandin E2 metabolite,
helicobacter pylori antibodies, and gastric cancer risk. Clin
Infect Dis. 64:1380–1386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Na YR, Jung D, Stakenborg M, Jang H, Gu
GJ, Jeong MR, Suh SY, Kim HJ, Kwon YH, Sung TS, et al:
Prostaglandin E2 receptor PTGER4-expressing macrophages promote
intestinal epithelial barrier regeneration upon inflammation. Gut.
Feb 7–2021.Epub ahead of print. View Article : Google Scholar
|
|
41
|
McCoy JM, Wicks JR and Audoly LP: The role
of prostaglandin E2 receptors in the pathogenesis of rheumatoid
arthritis. J Clin Invest. 110:651–658. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fajt ML, Gelhaus SL, Freeman B, Uvalle CE,
Trudeau JB, Holguin F and Wenzel SE: Prostaglandin D2
pathway upregulation: Relation to asthma severity, control, and TH2
inflammation. J Allergy Clin Immunol. 131:1504–1512. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoshino T, Nakaya T, Homan T, Tanaka K,
Sugimoto Y, Araki W, Narita M, Narumiya S, Suzuki T and Mizushima
T: Involvement of prostaglandin E2 in production of amyloid-beta
peptides both in vitro and in vivo. J Biol Chem. 282:32676–32688.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feingold KR, Doerrler W, Dinarello CA,
Fiers W and Grunfeld C: Stimulation of lipolysis in cultured fat
cells by tumor necrosis factor, interleukin-1, and the interferons
is blocked by inhibition of prostaglandin synthesis. Endocrinology.
130:10–16. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yokota T, Meka CS, Medina KL, Igarashi H,
Comp PC, Takahashi M, Nishida M, Oritani K, Miyagawa J, Funahashi
T, et al: Paracrine regulation of fat cell formation in bone marrow
cultures via adiponectin and prostaglandins. J Clin Invest.
109:1303–1310. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Forman BM, Tontonoz P, Chen J, Brun RP,
Spiegelman BM and Evans RM: 15-Deoxy-delta 12, 14-prostaglandin J2
is a ligand for the adipocyte determination factor PPAR gamma.
Cell. 83:803–812. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Litherland SA, Xie XT, Hutson AD,
Wasserfall C, Whittaker DS, She JX, Hofig A, Dennis MA, Fuller K,
Cook R, et al: Aberrant prostaglandin synthase 2 expression defines
an antigen-presenting cell defect for insulin-dependent diabetes
mellitus. J Clin Invest. 104:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yasui M, Tamura Y, Minami M, Higuchi S,
Fujikawa R, Ikedo T, Nagata M, Arai H, Murayama T and Yokode M: The
prostaglandin E2 receptor EP4 regulates obesity-related
inflammation and insulin sensitivity. PLoS One. 10:e01363042015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Edelman MJ, Wang X, Hodgson L, Cheney RT,
Baggstrom MQ, Thomas SP, Gajra A, Bertino E, Reckamp KL, Molina J,
et al: Phase III randomized, placebo-controlled, double-blind trial
of celecoxib in addition to standard chemotherapy for advanced
non-small-cell lung cancer with cyclooxygenase-2 overexpression:
CALGB 30801 (Alliance). J Clin Oncol. 35:2184–2192. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pathan SA, Mitra B, Straney LD, Afzal MS,
Anjum S, Shukla D, Morley K, Al Hilli SA, Al Rumaihi K, Thomas SH
and Cameron PA: Delivering safe and effective analgesia for
management of renal colic in the emergency department: A
double-blind, multigroup, randomised controlled trial. Lancet.
387:1999–2007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bath PM, Woodhouse LJ, Appleton JP,
Beridze M, Christensen H, Dineen RA, Duley L, England TJ, Flaherty
K, Havard D, et al: Antiplatelet therapy with aspirin, clopidogrel,
and dipyridamole versus clopidogrel alone or aspirin and
dipyridamole in patients with acute cerebral ischaemia (TARDIS): A
randomised, open-label, phase 3 superiority trial. Lancet.
391:850–859. 2018. View Article : Google Scholar :
|
|
52
|
Norambuena F, Mackenzie S, Bell JG, Callol
A, Estévez A and Duncan N: Prostaglandin (F and E, 2- and 3-series)
production and cyclooxygenase (COX-2) gene expression of wild and
cultured broodstock of Senegalese sole (Solea senegalensis). Gen
Comp Endocrinol. 177:256–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang X, Lin H and Gu Y: Multiple roles of
dihomo-γ-linolenic acid against proliferation diseases. Lipids
Health Dis. 11:252012. View Article : Google Scholar
|
|
54
|
Sonnweber T, Pizzini A, Nairz M, Weiss G
and Tancevski I: Arachidonic acid metabolites in cardiovascular and
metabolic diseases. Int J Mol Sci. 19:32852018. View Article : Google Scholar :
|
|
55
|
Cheng Z, Abayasekara DR and Wathes DC: The
effect of supplementation with n-6 polyunsaturated fatty acids on
1-, 2- and 3-series prostaglandin F production by ovine uterine
epithelial cells. Biochim Biophys Acta. 1736:128–135. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Smith WL, Urade Y and Jakobsson PJ:
Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis.
Chem Rev. 111:5821–5865. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Seibert K, Zhang Y, Leahy K, Hauser S,
Masferrer J, Perkins W, Lee L and Isakson P: Pharmacological and
biochemical demonstration of the role of cyclooxygenase 2 in
inflammation and pain. Proc Natl Acad Sci USA. 91:12013–12017.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kirkby NS, Zaiss AK, Urquhart P, Jiao J,
Austin PJ, Al-Yamani M, Lundberg MH, MacKenzie LS, Warner TD,
Nicolaou A, et al: LC-MS/MS confirms that COX-1 drives vascular
prostacyclin whilst gene expression pattern reveals non-vascular
sites of COX-2 expression. PLoS One. 8:e695242013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz
E, Jiao J, Berglund LM, Verdu EF, Ahmetaj-Shala B, Wallace JL,
Herschman HR, et al: Systematic study of constitutive
cyclooxygenase-2 expression: Role of NF-κB and NFAT transcriptional
pathways. Proc Natl Acad Sci A. 113:434–439. 2016. View Article : Google Scholar
|
|
60
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: Structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tilg H, Moschen AR and Roden M: NAFLD and
diabetes mellitus. Nat Rev Gastroenterol Hepatol. 14:32–42. 2017.
View Article : Google Scholar
|
|
62
|
Oakes ND, Cooney GJ, Camilleri S, Chisholm
DJ and Kraegen EW: Mechanisms of liver and muscle insulin
resistance induced by chronic high-fat feeding. Diabetes.
46:1768–1774. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Krssak M, Brehm A, Bernroider E, Anderwald
C, Nowotny P, Dalla Man C, Cobelli C, Cline GW, Shulman GI,
Waldhäusl W and Roden M: Alterations in postprandial hepatic
glycogen metabolism in type 2 diabetes. Diabetes. 53:3048–3056.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Smith GI, Shankaran M, Yoshino M,
Schweitzer GG, Chondronikola M, Beals JW, Okunade AL, Patterson BW,
Nyangau E, Field T, et al: Insulin resistance drives hepatic de
novo lipogenesis in nonalcoholic fatty liver disease. J Clin
Invest. 130:1453–1460. 2020. View Article : Google Scholar :
|
|
65
|
McQuaid SE, Hodson L, Neville MJ, Dennis
AL, Cheeseman J, Humphreys SM, Ruge T, Gilbert M, Fielding BA,
Frayn KN and Karpe F: Downregulation of adipose tissue fatty acid
trafficking in obesity: A driver for ectopic fat deposition?
Diabetes. 60:47–55. 2011. View Article : Google Scholar :
|
|
66
|
Garcia-Monzón C, Lo Iacono O, Mayoral R,
González-Rodriguez A, Miquilena-Colina ME, Lozano-Rodriguez T,
Garcia-Pozo L, Vargas Castrillón J, Casado M, Boscá L, et al:
Hepatic insulin resistance is associated with increased apoptosis
and fibrogenesis in nonalcoholic steatohepatitis and chronic
hepatitis C. J Hepatol. 54:142–152. 2011. View Article : Google Scholar
|
|
67
|
Weisberg SP, McCann D, Desai M, Rosenbaum
M, Leibel RL and Ferrante AW Jr: Obesity is associated with
macrophage accumulation in adipose tissue. J Clin Invest.
112:1796–1808. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xu H, Barnes GT, Yang Q, Tan G, Yang D,
Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA and Chen H:
Chronic inflammation in fat plays a crucial role in the development
of obesity-related insulin resistance. J Clin Invest.
112:1821–1830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cook JR, Langlet F, Kido Y and Accili D:
Pathogenesis of selective insulin resistance in isolated
hepatocytes. J Biol Chem. 290:13972–13980. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H,
Erdélyi K, Hao E, Holovac E, Haskó G, Cravatt BF, Nomura DK and
Pacher P: Monoacylglycerol lipase controls endocannabinoid and
eicosanoid signaling and hepatic injury in mice. Gastroenterology.
144:808–817.e15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Henkel J, Gärtner D, Dorn C, Hellerbrand
C, Schanze N, Elz SR and Püschel GP: Oncostatin M produced in
Kupffer cells in response to PGE2: Possible contributor to hepatic
insulin resistance and steatosis. Lab Invest. 91:1107–1117. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Henkel J, Neuschäfer-Rube F,
Pathe-Neuschäfer-Rube A and Püschel GP: Aggravation by
prostaglandin E2 of interleukin-6-dependent insulin resistance in
hepatocytes. Hepatology. 50:781–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bock G, Chittilapilly E, Basu R, Toffolo
G, Cobelli C, Chandramouli V, Landau BR and Rizza RA: Contribution
of hepatic and extrahepatic insulin resistance to the pathogenesis
of impaired fasting glucose: Role of increased rates of
gluconeogenesis. Diabetes. 56:1703–1711. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang Y, Yan S, Xiao B, Zuo S, Zhang Q,
Chen G, Yu Y, Chen D, Liu Q, Liu Y, et al: Prostaglandin
F2α facilitates hepatic glucose production through
CaMKIIγ/p38/FOXO1 signaling pathway in fasting and obesity.
Diabetes. 67:1748–1760. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yan S, Zhang Q, Zhong X, Tang J, Wang Y,
Yu J, Zhou Y, Zhang J, Guo F, Liu Y, et al: I prostanoid
receptor-mediated inflammatory pathway promotes hepatic
gluconeogenesis through activation of PKA and inhibition of AKT.
Diabetes. 63:2911–2923. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Francés DE, Motiño O, Agrá N,
González-Rodríguez Á, Fernández-Álvarez A, Cucarella C, Mayoral R,
Castro-Sánchez L, García-Casarrubios E, Boscá L, et al: Hepatic
cyclooxygenase-2 expression protects against diet-induced
steatosis, obesity, and insulin resistance. Diabetes. 64:1522–1531.
2015. View Article : Google Scholar
|
|
77
|
Petersen KF, Dufour S, Savage DB, Bilz S,
Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et
al: The role of skeletal muscle insulin resistance in the
pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA.
104:12587–12594. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Banhos Danneskiold-Samsøe N, Sonne SB,
Larsen JM, Hansen AN, Fjære E, Isidor MS, Petersen S, Henningsen J,
Severi I, Sartini L, et al: Overexpression of cyclooxygenase-2 in
adipocytes reduces fat accumulation in inguinal white adipose
tissue and hepatic steatosis in high-fat fed mice. Sci Rep.
9:89792019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ceddia RP, Lee D, Maulis MF, Carboneau BA,
Threadgill DW, Poffenberger G, Milne G, Boyd KL, Powers AC,
McGuinness OP, et al: The PGE2 EP3 receptor regulates dietinduced
adiposity in male mice. Endocrinology. 157:220–232. 2016.
View Article : Google Scholar
|
|
80
|
Garcia-Alonso V, Titos E, Alcaraz-Quiles
J, Rius B, Lopategi A, López-Vicario C, Jakobsson PJ, Delgado S,
Lozano J and Clària J: Prostaglandin E2 exerts multiple regulatory
actions on human obese adipose tissue remodeling, inflammation,
adaptive thermogenesis and lipolysis. PLoS One. 11:e01537512016.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fujitani Y, Aritake K, Kanaoka Y, Goto T,
Takahashi N, Fujimori K and Kawada T: Pronounced adipogenesis and
increased insulin sensitivity caused by overproduction of
prostaglandin D2 in vivo. FEBS J. 277:1410–1419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Virtue S, Masoodi M, de Weijer BA, van
Eijk M, Mok CY, Eiden M, Dale M, Pirraco A, Serlie MJ, Griffin JL
and Vidal-Puig A: Prostaglandin profiling reveals a role for
haematopoietic prostaglandin D synthase in adipose tissue
macrophage polarisation in mice and humans. Int J Obes (Lond).
39:1151–1160. 2015. View Article : Google Scholar
|
|
83
|
Hernandez-Carretero A, Weber N, La Frano
MR, Ying W, Lantero Rodriguez J, Sears DD, Wallenius V, Borgeson E,
Newman JW and Osborn O: Obesity-induced changes in lipid mediators
persist after weight loss. Int J Obes (Lond). 42:728–736. 2018.
View Article : Google Scholar
|
|
84
|
Fujimori K, Aritake K, Oishi Y, Nagata N,
Maehara T, Lazarus M and Urade Y: L-PGDS-produced PGD2 in
premature, but not in mature, adipocytes increases obesity and
insulin resistance. Sci Rep. 9:19312019. View Article : Google Scholar :
|
|
85
|
Fujimori K, Maruyama T, Kamauchi S and
Urade Y: Activation of adipogenesis by lipocalin-type prostaglandin
D synthase-generated Δ¹2-PGJ2 acting through
PPARγ-dependent and independent pathways. Gene. 505:46–52. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wakai E, Aritake K, Urade Y and Fujimori
K: Prostaglandin D2 enhances lipid accumulation through suppression
of lipolysis via DP2 (CRTH2) receptors in adipocytes. Biochem
Biophys Res Commun. 490:393–399. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Groop LC, Bonadonna RC, DelPrato S,
Ratheiser K, Zyck K, Ferrannini E and DeFronzo RA: Glucose and free
fatty acid metabolism in non-insulin-dependent diabetes mellitus.
Evidence for multiple sites of insulin resistance. J Clin Invest.
84:205–213. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Skurk T, Alberti-Huber C, Herder C and
Hauner H: Relationship between adipocyte size and adipokine
expression and secretion. J Clin Endocrinol Metab. 92:1023–1033.
2007. View Article : Google Scholar
|
|
89
|
Lê KA, Mahurkar S, Alderete TL, Hasson RE,
Adam TC, Kim JS, Beale E, Xie C, Greenberg AS, Allayee H and Goran
MI: Subcutaneous adipose tissue macrophage infiltration is
associated with hepatic and visceral fat deposition,
hyperinsulinemia, and stimulation of NF-κB stress pathway.
Diabetes. 60:2802–2809. 2011. View Article : Google Scholar
|
|
90
|
Utriainen T, Takala T, Luotolahti M,
Rönnemaa T, Laine H, Ruotsalainen U, Haaparanta M, Nuutila P and
Yki-Järvinen H: Insulin resistance characterizes glucose uptake in
skeletal muscle but not in the heart in NIDDM. Diabetologia.
41:555–559. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pratipanawatr W, Pratipanawatr T, Cusi K,
Berria R, Adams JM, Jenkinson CP, Maezono K, DeFronzo RA and
Mandarino LJ: Skeletal muscle insulin resistance in normoglycemic
subjects with a strong family history of type 2 diabetes is
associated with decreased insulin-stimulated insulin receptor
substrate-1 tyrosine phosphorylation. Diabetes. 50:2572–2578. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Weyer C, Bogardus C, Mott DM and Pratley
RE: The natural history of insulin secretory dysfunction and
insulin resistance in the pathogenesis of type 2 diabetes mellitus.
J Clin Invest. 104:787–794. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Dietze G, Wicklmayr M, Böttger I and Mayer
L: Insulin action on glucose uptake into skeletal muscle:
Inhibition of endogenous biosynthesis of prostaglandins. FEBS Lett.
92:294–298. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Leighton B, Budohoski L, Lozeman FJ,
Challiss RA and Newsholme EA: The effect of prostaglandins E1, E2
and F2 alpha and indomethacin on the sensitivity of glycolysis and
glycogen synthesis to insulin in stripped soleus muscles of the
rat. Biochem J. 227:337–340. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Coll T, Palomer X, Blanco-Vaca F,
Escolà-Gil JC, Sánchez RM, Laguna JC and Vázquez-Carrera M:
Cyclooxygenase 2 inhibition exacerbates palmitate-induced
inflammation and insulin resistance in skeletal muscle cells.
Endocrinology. 151:537–548. 2010. View Article : Google Scholar
|
|
96
|
Smith GI, Polidori DC, Yoshino M, Kearney
ML, Patterson BW, Mittendorfer B and Klein S: Influence of
adiposity, insulin resistance, and intrahepatic triglyceride
content on insulin kinetics. J Clin Invest. 130:3305–3314. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Solomon TP, Knudsen SH, Karstoft K,
Winding K, Holst JJ and Pedersen BK: Examining the effects of
hyperglycemia on pancreatic endocrine function in humans: Evidence
for in vivo glucotoxicity. J Clin Endocrinol Metab. 97:4682–4691.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hughan KS, Bonadonna RC, Lee S,
Michaliszyn SF and Arslanian SA: β-Cell lipotoxicity after an
overnight intravenous lipid challenge and free fatty acid elevation
in African American versus American white overweight/obese
adolescents. J Clin Endocrinol Metab. 98:2062–2069. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Goodpaster BH, Thaete FL and Kelley DE:
Thigh adipose tissue distribution is associated with insulin
resistance in obesity and in type 2 diabetes mellitus. Am J Clin
Nutr. 71:885–892. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Solini A, Rossi C, Duranti E, Taddei S,
Natali A and Virdis A: Saxagliptin prevents vascular remodeling and
oxidative stress in db/db mice. Role of endothelial nitric oxide
synthase uncoupling and cyclooxygenase. Vascul Pharmacol. 76:62–71.
2016. View Article : Google Scholar
|
|
101
|
Hundal RS, Petersen KF, Mayerson AB,
Randhawa PS, Inzucchi S, Shoelson SE and Shulman GI: Mechanism by
which high-dose aspirin improves glucose metabolism in type 2
diabetes. J Clin Invest. 109:1321–1326. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Helmersson J, Vessby B, Larsson A and Basu
S: Association of type 2 diabetes with cyclooxygenase-mediated
inflammation and oxidative stress in an elderly population.
Circulation. 109:1729–1734. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kimple ME, Keller MP, Rabaglia MR, Pasker
RL, Neuman JC, Truchan NA, Brar HK and Attie AD: Prostaglandin E2
receptor, EP3, is induced in diabetic islets and negatively
regulates glucose- and hormone-stimulated insulin secretion.
Diabetes. 62:1904–1912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Batchu SN, Majumder S, Bowskill BB, White
KE, Advani SL, Brijmohan AS, Liu Y, Thai K, Azizi PM, Lee WL and
Advani A: Prostaglandin I2 receptor agonism preserves β-cell
function and attenuates albuminuria through nephrin-dependent
mechanisms. Diabetes. 65:1398–1409. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Arablou T, Aryaeian N, Valizadeh M,
Sharifi F, Hosseini A and Djalali M: The effect of ginger
consumption on glycemic status, lipid profile and some inflammatory
markers in patients with type 2 diabetes mellitus. Int J Food Sci
Nutr. 65:515–520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhu CF, Li GZ, Peng HB, Zhang F, Chen Y
and Li Y: Treatment with marine collagen peptides modulates glucose
and lipid metabolism in Chinese patients with type 2 diabetes
mellitus. Appl Physiol Nutr Metab. 35:797–804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ceddia RP, Downey JD, Morrison RD, Kraemer
MP, Davis SE, Wu J, Lindsley CW, Yin H, Daniels JS and Breyer RM:
The effect of the EP3 antagonist DG-041 on male mice with
diet-induced obesity. Prostaglandins Other Lipid Mediat.
144:1063532019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Weir GC, Gaglia J and Bonner-Weir S:
Inadequate β-cell mass is essential for the pathogenesis of type 2
diabetes. Lancet Diabetes Endocrinol. 8:249–256. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Grodsky GM: A threshold distribution
hypothesis for packet storage of insulin and its mathematical
modeling. J Clin Invest. 51:2047–2059. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Persaud SJ, Muller D, Belin VD,
Kitsou-Mylona I, Asare-Anane H, Papadimitriou A, Burns CJ, Huang
GC, Amiel SA and Jones PM: The role of arachidonic acid and its
metabolites in insulin secretion from human islets of langerhans.
Diabetes. 56:197–203. 2007. View Article : Google Scholar
|
|
111
|
Tran PO, Gleason CE, Poitout V and
Robertson RP: Prostaglandin E(2) mediates inhibition of insulin
secretion by interleukin-1beta. J Biol Chem. 274:31245–31248. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Tran PO, Gleason CE and Robertson RP:
Inhibition of interleukin-1beta-induced COX-2 and EP3 gene
expression by sodium salicylate enhances pancreatic islet beta-cell
function. Diabetes. 51:1772–1778. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Shanmugam N, Todorov IT, Nair I, Omori K,
Reddy MA and Natarajan R: Increased expression of cyclooxygenase-2
in human pancreatic islets treated with high glucose or ligands of
the advanced glycation endproduct-specific receptor (AGER), and in
islets from diabetic mice. Diabetologia. 49:100–107. 2006.
View Article : Google Scholar
|
|
114
|
Persaud SJ, Burns CJ, Belin VD and Jones
PM: Glucose-induced regulation of COX-2 expression in human islets
of langerhans. Diabetes. 53(Suppl 1): S190–S192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Shridas P, Zahoor L, Forrest KJ, Layne JD
and Webb NR: Group X secretory phospholipase A2 regulates insulin
secretion through a cyclooxygenase-2-dependent mechanism. J Biol
Chem. 289:27410–27417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Seaquist ER, Walseth TF, Nelson DM and
Robertson RP: Pertussis toxin-sensitive G protein mediation of PGE2
inhibition of cAMP metabolism and phasic glucose-induced insulin
secretion in HIT cells. Diabetes. 38:1439–1445. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Carboneau BA, Allan JA, Townsend SE,
Kimple ME, Breyer RM and Gannon M: Opposing effects of
prostaglandin E2 receptors EP3 and EP4 on mouse and human β-cell
survival and proliferation. Mol Metab. 6:548–559. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Meng ZX, Sun JX, Ling JJ, Lv JH, Zhu DY,
Chen Q, Sun YJ and Han X: Prostaglandin E2 regulates Foxo activity
via the Akt pathway: Implications for pancreatic islet beta cell
dysfunction. Diabetologia. 49:2959–2968. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Anderson SL, Trujillo JM, McDermott M and
Saseen JJ: Determining predictors of response to exenatide in type
2 diabetes. J Am Pharm Assoc (2003). 52. pp. 466–471. 2012,
View Article : Google Scholar
|
|
120
|
Kimple ME, Moss JB, Brar HK, Rosa TC,
Truchan NA, Pasker RL, Newgard CB and Casey PJ: Deletion of GαZ
protein protects against diet-induced glucose intolerance via
expansion of β-cell mass. J Biol Chem. 287:20344–20355. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zawalich WS, Zawalich KC and Yamazaki H:
Divergent effects of epinephrine and prostaglandin E2 on
glucose-induced insulin secretion from perifused rat islets.
Metabolism. 56:12–18. 2007. View Article : Google Scholar
|
|
122
|
Igoillo-Esteve M, Marselli L, Cunha DA,
Ladrière L, Ortis F, Grieco FA, Dotta F, Weir GC, Marchetti P,
Eizirik DL and Cnop M: Palmitate induces a pro-inflammatory
response in human pancreatic islets that mimics CCL2 expression by
beta cells in type 2 diabetes. Diabetologia. 53:1395–1405. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Gokulakrishnan K, Mohanavalli KT,
Monickaraj F, Mohan V and Balasubramanyam M: Subclinical
inflammation/oxidation as revealed by altered gene expression
profiles in subjects with impaired glucose tolerance and Type 2
diabetes patients. Mol Cell Biochem. 324:173–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wang G, Liang R, Liu T, Wang L, Zou J, Liu
N, Liu Y, Cai X, Liu Y, Ding X, et al: Opposing effects of
IL-1β/COX-2/PGE2 pathway loop on islets in type 2 diabetes
mellitus. Endocr J. 66:691–699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Pradhan AD, Manson JE, Rifai N, Buring JE
and Ridker PM: C-reactive protein, interleukin 6, and risk of
developing type 2 diabetes mellitus. JAMA. 286:327–334. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Abou-Shousha S, Abd El-Megeed MH and
Sultan HK: Interleukin-8, ferritin and soluble transferrin
receptors in type II diabetes mellitus. Egypt J Immunol. 13:19–25.
2006.
|
|
127
|
Cai W, Qiu C, Zhang H, Chen X, Zhang X,
Meng Q and Wei J: Detection of circulating natural antibodies to
inflammatory cytokines in type-2 diabetes and clinical
significance. J Inflamm (Lond). 14:242017. View Article : Google Scholar
|
|
128
|
Rahier J, Guiot Y, Goebbels RM, Sempoux C
and Henquin JC: Pancreatic beta-cell mass in European subjects with
type 2 diabetes. Diabetes Obes Metab. 10(Suppl 4): S32–S42. 2008.
View Article : Google Scholar
|
|
129
|
Butler AE, Janson J, Bonner-Weir S, Ritzel
R, Rizza RA and Butler PC: Beta-cell deficit and increased
beta-cell apoptosis in humans with type 2 diabetes. Diabetes.
52:102–110. 2003. View Article : Google Scholar
|
|
130
|
Oshima H, Taketo MM and Oshima M:
Destruction of pancreatic beta-cells by transgenic induction of
prostaglandin E2 in the islets. J Biol Chem. 281:29330–29336. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kimple ME, Nixon AB, Kelly P, Bailey CL,
Young KH, Fields TA and Casey PJ: A role for G(z) in pancreatic
islet beta-cell biology. J Biol Chem. 280:31708–31713. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Amior L, Srivastava R, Nano R, Bertuzzi F
and Melloul D: The role of Cox-2 and prostaglandin E2 receptor EP3
in pancreatic β-cell death. FASEB J. 33:4975–4986. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lewis B, Mancini M, Mattock M, Chait A and
Fraser TR: Plasma triglyceride and fatty acid metabolism in
diabetes mellitus. Eur J Clin Invest. 2:445–453. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Diraison F, Moulin P and Beylot M:
Contribution of hepatic de novo lipogenesis and reesterification of
plasma non esterified fatty acids to plasma triglyceride synthesis
during non-alcoholic fatty liver disease. Diabetes Metab.
29:478–485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Tirosh A, Shai I, Bitzur R, Kochba I,
Tekes-Manova D, Israeli E, Shochat T and Rudich A: Changes in
triglyceride levels over time and risk of type 2 diabetes in young
men. Diabetes Care. 31:2032–2037. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Kashyap S, Belfort R, Gastaldelli A,
Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L,
DeFronzo R and Cusi K: A sustained increase in plasma free fatty
acids impairs insulin secretion in nondiabetic subjects genetically
predisposed to develop type 2 diabetes. Diabetes. 52:2461–2474.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sakurai M, Takamura T, Ota T, Ando H,
Akahori H, Kaji K, Sasaki M, Nakanuma Y, Miura K and Kaneko S:
Liver steatosis, but not fibrosis, is associated with insulin
resistance in nonalcoholic fatty liver disease. J Gastroenterol.
42:312–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Svegliati-Baroni G, Saccomanno S,
Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, Faraci G,
Pacetti D, Vivarelli M, Nicolini D, et al: Glucagon-like peptide-1
receptor activation stimulates hepatic lipid oxidation and restores
hepatic signalling alteration induced by a high-fat diet in
nonalcoholic steatohepatitis. Liver Int. 31:1285–1297. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Arkan MC, Hevener AL, Greten FR, Maeda S,
Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J and Karin M:
IKK-beta links inflammation to obesity-induced insulin resistance.
Nat Med. 11:191–198. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
140
|
Henkel J, Frede K, Schanze N, Vogel H,
Schürmann A, Spruss A, Bergheim I and Püschel GP: Stimulation of
fat accumulation in hepatocytes by PGE2-dependent
repression of hepatic lipolysis, β-oxidation and VLDL-synthesis.
Lab Invest. 92:1597–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Pérez S, Aspichueta P, Ochoa B and Chico
Y: The 2-series prostaglandins suppress VLDL secretion in an
inflammatory condition-dependent manner in primary rat hepatocytes.
Biochim Biophys Acta. 1761:160–171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Chung MY, Mah E, Masterjohn C, Noh SK,
Park HJ, Clark RM, Park YK, Lee JY and Bruno RS: Green tea lowers
hepatic COX-2 and prostaglandin E2 in rats with dietary fat-induced
nonalcoholic steatohepatitis. J Med Food. 18:648–655. 2015.
View Article : Google Scholar
|
|
143
|
Nassir F, Adewole OL, Brunt EM and Abumrad
NA: CD36 deletion reduces VLDL secretion, modulates liver
prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J
Lipid Res. 54:2988–2997. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Sato N, Kaneko M, Tamura M and Kurumatani
H: The prostacyclin analog beraprost sodium ameliorates
characteristics of metabolic syndrome in obese Zucker (fatty) rats.
Diabetes. 59:1092–1100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Duncan BB, Schmidt MI, Pankow JS,
Ballantyne CM, Couper D, Vigo A, Hoogeveen R, Folsom AR and Heiss
G; Atherosclerosis Risk in Communities Study: Low-grade systemic
inflammation and the development of type 2 diabetes: The
atherosclerosis risk in communities study. Diabetes. 52:1799–1805.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Haukeland JW, Damås JK, Konopski Z, Løberg
EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K and
Aukrust P: Systemic inflammation in nonalcoholic fatty liver
disease is characterized by elevated levels of CCL2. J Hepatol.
44:1167–1174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kamari Y, Shaish A, Vax E, Shemesh S,
Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E,
et al: Lack of interleukin-1α or interleukin-1β inhibits
transformation of steatosis to steatohepatitis and liver fibrosis
in hypercholesterolemic mice. J Hepatol. 55:1086–1094. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Masters SL, Dunne A, Subramanian SL, Hull
RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen
Z, et al: Activation of the NLRP3 inflammasome by islet amyloid
polypeptide provides a mechanism for enhanced IL-1β in type 2
diabetes. Nat Immunol. 11:897–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Chitturi S, Abeygunasekera S, Farrell GC,
Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D,
Liddle C, et al: NASH and insulin resistance: Insulin
hypersecretion and specific association with the insulin resistance
syndrome. Hepatology. 35:373–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Sanyal AJ, Campbell-Sargent C, Mirshahi F,
Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML and Clore
JN: Nonalcoholic steatohepatitis: Association of insulin resistance
and mitochondrial abnormalities. Gastroenterology. 120:1183–1192.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Yang ZH, Miyahara H, Takeo J and Katayama
M: Diet high in fat and sucrose induces rapid onset of
obesity-related metabolic syndrome partly through rapid response of
genes involved in lipogenesis, insulin signalling and inflammation
in mice. Diabetol Metab Syndr. 4:322012. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ren LP, Chan SM, Zeng XY, Laybutt DR,
Iseli TJ, Sun RQ, Kraegen EW, Cooney GJ, Turner N and Ye JM:
Differing endoplasmic reticulum stress response to excess
lipogenesis versus lipid oversupply in relation to hepatic
steatosis and insulin resistance. PLoS One. 7:e308162012.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Brunt EM, Kleiner DE, Wilson LA, Unalp A,
Behling CE and Lavine JE: Portal chronic inflammation in
nonalcoholic fatty liver disease (NAFLD): A histologic marker of
advanced NAFLD-clinicopathologic correlations from the nonalcoholic
steatohepatitis clinical research network. Hepatology. 49:809–820.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Angulo P, Keach JC, Batts KP and Lindor
KD: Independent predictors of liver fibrosis in patients with
nonalcoholic steatohepatitis. Hepatology. 30:1356–1362. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Hossain N, Afendy A, Stepanova M, Nader F,
Srishord M, Rafiq N, Goodman Z and Younossi Z: Independent
predictors of fibrosis in patients with nonalcoholic fatty liver
disease. Clin Gastroenterol Hepatol. 7:1224–1229. 1229.e1–e2. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zeyda M, Farmer D, Todoric J, Aszmann O,
Speiser M, Györi G, Zlabinger GJ and Stulnig TM: Human adipose
tissue macrophages are of an anti-inflammatory phenotype but
capable of excessive pro-inflammatory mediator production. Int J
Obes (Lond). 31:1420–1428. 2007. View Article : Google Scholar
|
|
157
|
Itani SI, Ruderman NB, Schmieder F and
Boden G: Lipid-induced insulin resistance in human muscle is
associated with changes in diacylglycerol, protein kinase C, and
IkappaB-alpha. Diabetes. 51:2005–2011. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hotamisligil GS, Arner P, Caro JF,
Atkinson RL and Spiegelman BM: Increased adipose tissue expression
of tumor necrosis factor-alpha in human obesity and insulin
resistance. J Clin Invest. 95:2409–2415. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
du Plessis J, van Pelt J, Korf H, Mathieu
C, van der Schueren B, Lannoo M, Oyen T, Topal B, Fetter G, Nayler
S, et al: Association of adipose tissue inflammation with
histologic severity of nonalcoholic fatty liver disease.
Gastroenterology. 149:635–648.e14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Paradis V, Perlemuter G, Bonvoust F,
Dargere D, Parfait B, Vidaud M, Conti M, Huet S, Ba N, Buffet C and
Bedossa P: High glucose and hyperinsulinemia stimulate connective
tissue growth factor expression: A potential mechanism involved in
progression to fibrosis in nonalcoholic steatohepatitis.
Hepatology. 34:738–744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ota T, Takamura T, Kurita S, Matsuzawa N,
Kita Y, Uno M, Akahori H, Misu H, Sakurai M, Zen Y, et al: Insulin
resistance accelerates a dietary rat model of nonalcoholic
steatohepatitis. Gastroenterology. 132:282–293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Henkel J, Coleman CD, Schraplau A, Jöhrens
K, Weiss TS, Jonas W, Schürmann A and Püschel GP: Augmented liver
inflammation in a microsomal prostaglandin E synthase 1
(mPGES-1)-deficient diet-induced mouse NASH model. Sci Rep.
8:161272018. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Loomba R, Quehenberger O, Armando A and
Dennis EA: Polyunsaturated fatty acid metabolites as novel
lipidomic biomarkers for noninvasive diagnosis of nonalcoholic
steatohepatitis. J Lipid Res. 56:185–192. 2015. View Article : Google Scholar :
|
|
164
|
Motiño O, Agra N, Brea Contreras R,
Dominguez-Moreno M, Garcia-Monzón C, Vargas-Castrillón J, Carnovale
CE, Boscá L, Casado M, Mayoral R, et al: Cyclooxygenase-2
expression in hepatocytes attenuates non-alcoholic steatohepatitis
and liver fibrosis in mice. Biochim Biophys Acta. 1862:1710–1723.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Kumar S, Srivastava A, Palaia T, Hall C,
Lee J, Stevenson M, Zhao CL and Ragolia L: Lipocalin-type
prostaglandin D2 synthase deletion induces dyslipidemia and
non-alcoholic fatty liver disease. Prostaglandins Other Lipid
Mediat. 149:1064292020. View Article : Google Scholar
|
|
166
|
Kumei S, Yuhki KI, Kojima F, Kashiwagi H,
Imamichi Y, Okumura T, Narumiya S and Ushikubi F: Prostaglandin
I2 suppresses the development of diet-induced
nonalcoholic steatohepatitis in mice. FASEB J. 32:2354–2365. 2018.
View Article : Google Scholar
|
|
167
|
Meng F, Wang K, Aoyama T, Grivennikov SI,
Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al:
Interleukin-17 signaling in inflammatory, Kupffer cells, and
hepatic stellate cells exacerbates liver fibrosis in mice.
Gastroenterology. 143:765–776.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Ikejima K, Takei Y, Honda H, Hirose M,
Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T
and Sato N: Leptin receptor-mediated signaling regulates hepatic
fibrogenesis and remodeling of extracellular matrix in the rat.
Gastroenterology. 122:1399–1410. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Kim SM, Park KC, Kim HG and Han SJ: Effect
of selective cyclooxygenase-2 inhibitor meloxicam on liver fibrosis
in rats with ligated common bile ducts. Hepatol Res. 38:800–809.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yu J, Ip E, Dela Peña A, Hou JY, Sesha J,
Pera N, Hall P, Kirsch R, Leclercq I and Farrell GC: COX-2
induction in mice with experimental nutritional steatohepatitis:
Role as pro-inflammatory mediator. Hepatology. 43:826–836. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Paik YH, Kim JK, Lee JI, Kang SH, Kim DY,
An SH, Lee SJ, Lee DK, Han KH, Chon CY, et al: Celecoxib induces
hepatic stellate cell apoptosis through inhibition of Akt
activation and suppresses hepatic fibrosis in rats. Gut.
58:1517–1527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Simon TG, Henson J, Osganian S, Masia R,
Chan AT, Chung RT and Corey KE: Daily aspirin use associated with
reduced risk for fibrosis progression in patients with nonalcoholic
fatty liver disease. Clin Gastroenterol Hepatol. 17:2776–2784.e4.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Vilar-Gomez E and Chalasani N: Daily
aspirin use reduces risk of fibrosis progression in patients with
nonalcoholic fatty liver disease, providing new uses for an old
drug. Clin Gastroenterol Hepatol. 17:2651–2653. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sui G, Cheng G, Yuan J, Hou X, Kong X and
Niu H: Interleukin (IL)-13, Prostaglandin E2 (PGE2), and
Prostacyclin 2 (PGI2) Activate Hepatic Stellate Cells via Protein
kinase C (PKC) pathway in hepatic fibrosis. Med Sci Monit.
24:2134–2141. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hanson A, Wilhelmsen D and DiStefano JK:
The role of long non-coding RNAs (lncRNAs) in the development and
progression of fibrosis associated with nonalcoholic fatty liver
disease (NAFLD). Noncoding RNA. 4:182018.
|
|
176
|
Kamada Y, Mori K, Matsumoto H, Kiso S,
Yoshida Y, Shinzaki S, Hiramatsu N, Ishii M, Moriwaki K, Kawada N,
et al: N-Acetylglucosaminyltransferase V regulates TGF-β response
in hepatic stellate cells and the progression of steatohepatitis.
Glycobiology. 22:778–787. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Hui AY, Leung WK, Chan HL, Chan FK, Go MY,
Chan KK, Tang BD, Chu ES and Sung JJ: Effect of celecoxib on
experimental liver fibrosis in rat. Liver Int. 26:125–136. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Brea R, Motiño O, Francés D, García-Monzón
C, Vargas J, Fernández-Velasco M, Boscá L, Casado M, Martín-Sanz P
and Agra N: PGE2 induces apoptosis of hepatic stellate
cells and attenuates liver fibrosis in mice by downregulating
miR-3a-5p and miR-28a-5p. Biochim Biophys Acta Mol Basis Dis.
1864:325–337. 2018. View Article : Google Scholar
|
|
179
|
Hui AY, Dannenberg AJ, Sung JJ,
Subbaramaiah K, Du B, Olinga P and Friedman SL: Prostaglandin E2
inhibits transforming growth factor beta 1-mediated induction of
collagen alpha 1(I) in hepatic stellate cells. J Hepatol.
41:251–258. 2004. View Article : Google Scholar : PubMed/NCBI
|