Introduction
Endometriosis is a relatively common, enigmatic,
benign, estrogen-dependent gynecological illness, characterized by
the growth of endometrial tissue and the proliferation of
endometrial glands and stroma in ectopic sites, with most common
manifestations appearing in the pelvic cavity occurring in sites
other than the uterine cavity, most commonly in the pelvic cavity
(1). This condition is mainly
associated with pelvic pain, dysmenorrhea, dyspareunia and impaired
fertility (2). Previous gene
association studies, genome-wide association studies (GWAS) and
meta-analyses have identified various endometriosis-associated
loci, with the list of the novel ones still being enriched
(3,4).
Endometriosis markedly affects the health of women,
as well as the quality of their life. The gold standard for the
diagnosis of endometriosis involves laparoscopy and biopsy, that
is, a surgical visual inspection of the pelvic organs, while the
development of protocols concerning the treatment of this condition
aims for the preservation of patient fertility (5). Advances in modern technologies and
bioinformatics have greatly contributed to the generation of
large-scale biological data, thus leading biomedical sciences to
the -omics era. Currently, the search for novel biomarkers
for use in endometriosis continues, and the -omics
technologies have greatly contributed to this direction. The
-omics have revolutionized endometriosis research, and this
is proven by the vast number of related publications to date
(6). In a recent review,
multiple studies based on the high-throughput -omics
technologies were presented, in an attempt to gain insight into all
considerable advantages that they may confer to proper management
of endometriosis (7). The need
for non-invasive biomarkers is invaluable and urgent, considering
that the average delay between the first symptoms and the
laparoscopic diagnosis is estimated at approximately seven years
(7). The early diagnosis of
endometriosis in combination with proper genetic counseling may
facilitate couples to give birth to children at a younger age (of
the woman), at an earlier stage of endometriosis, which is
characterized by a decreased infertility. Furthermore, the use of
non-invasive biomarkers will lead to the elimination of unnecessary
laparoscopies (8). According to
the current literature, ~5% of adolescent girls aged between 15-19
experience severe dysmenorrhea not relieved by combined oral
contraceptives (COCs) and analgesics, a situation suggestive of
endometriosis. Furthermore, other common variable symptoms that may
present in young women with endometriosis include dyspareunia in
sexually active females, as well as gastrointestinal and urinary
tract disturbances (9). Of note,
endometriosis is reported to be a differential diagnosis for
chronic pelvic pain in adolescent and younger women. Although there
are silent (asymptomatic) cases of endometriosis, the majority of
symptoms are non-specific and may result in a delay in diagnosis
due to the overlapping clinical features with other gynecologic and
non-gynecologic conditions. Thus, the early and timely detection of
endometriosis with non-invasive procedures may prevent the delay in
diagnosis, which can interfere with the quality of life of patients
and may result in emotional distress. Moreover, the failure of
early recognition and sufficient management may exacerbate the
progression of the disease and the development of adhesions that
may affect fertility and the risk of the development and
maintenance of chronic pain (10).
Advanced techniques in modern genetics and the
increasing number of health studies related to genetic and genomic
data render precision medicine and consumer genetics a new reality
(11). The implementation of a
whole-genome (WGS) or whole-exome sequencing (WES) data set as a
principal test has provided beneficial information for a more
precise diagnosis, aiding and clarifying other conventional tests,
while decreasing the number of targeted genetic tests and
eventually the time required to perform a full genetic diagnosis
(12). The impact of
communicating genetic risks is increasingly important for the
prevention and treatment of a number of diseases and are rapidly
extended to the field of application and practice, as emerging
novel genomic pipelines permit more health experts to use
information concerning their patients' genetic profiles and gene
variants (11,13).
In recent decades, the rapid developments of new
technologies in the -omic sciences have produced vast
amounts of data. The processing and analysis of such large amounts
of data require the understanding of the type of data by inferring
structure or generalizations from the data and sophisticated
computational analyses towards drawing conclusions (14). The implementation of data mining
and semantic techniques in the field of bioinformatics has been
widely used for solving such issues, including problem definition,
data collection, data annotation, data preprocessing, modeling and
validation (12,15). The importance of applying such
efficient techniques will grow as researchers continue to generate
and integrate large quantities of genomics, proteomics,
transcriptomics, lipidomics, metabolomics, secretomics and other
-omics biological data. Examples of this type of specialized
analyses include GWAS, gene classification based on the literature
per disease, the clustering of gene expression data, single
nucleotide polymorphism (SNP) classification per disease,
regulatory networks of protein-protein interactions, and numerous
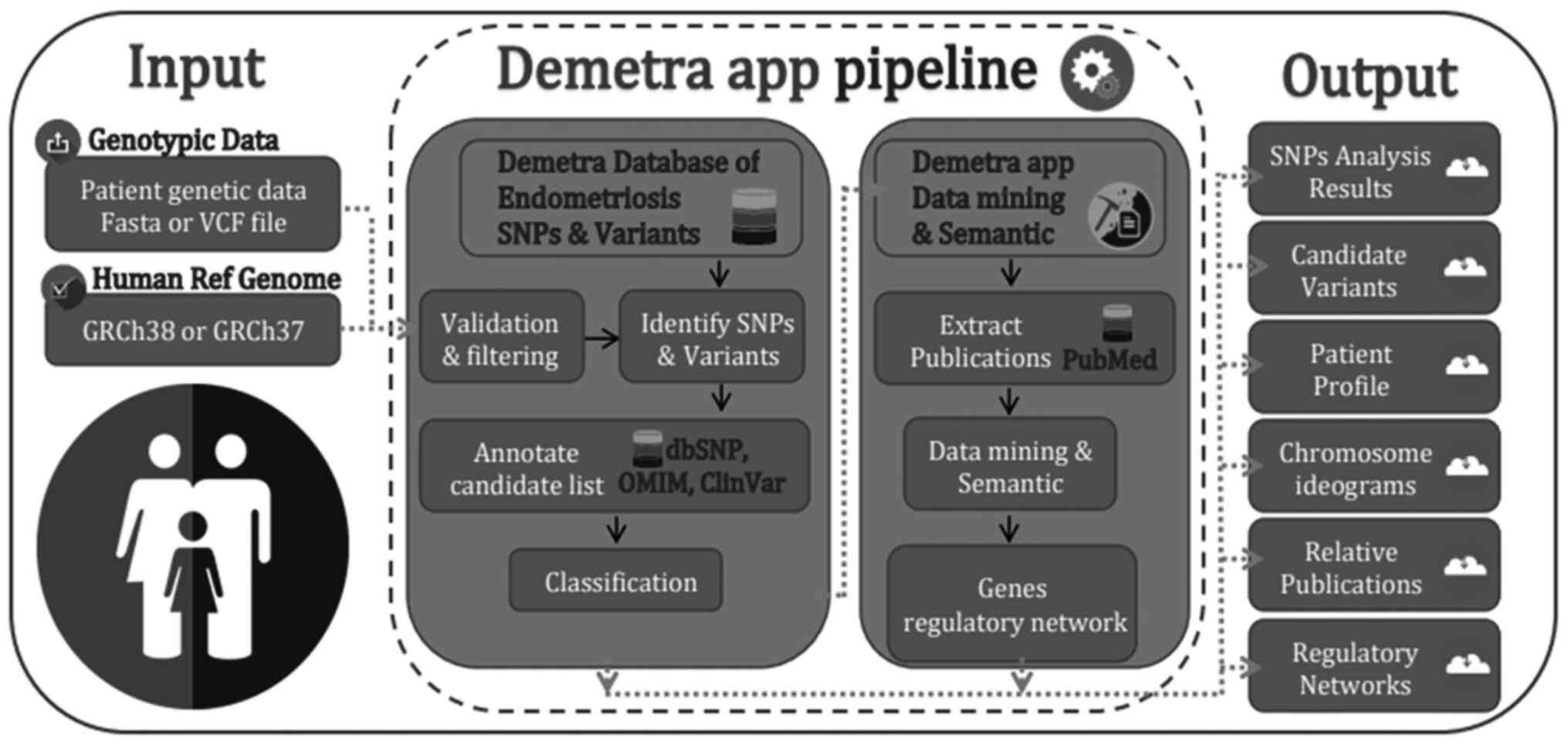
other applications (12,16,17). The Demetra Application (App)
webserver is an example that incorporates the application of
bioinformatics and data mining technologies to support the clinical
genomic diagnosis process of endometriosis (Fig. 1).
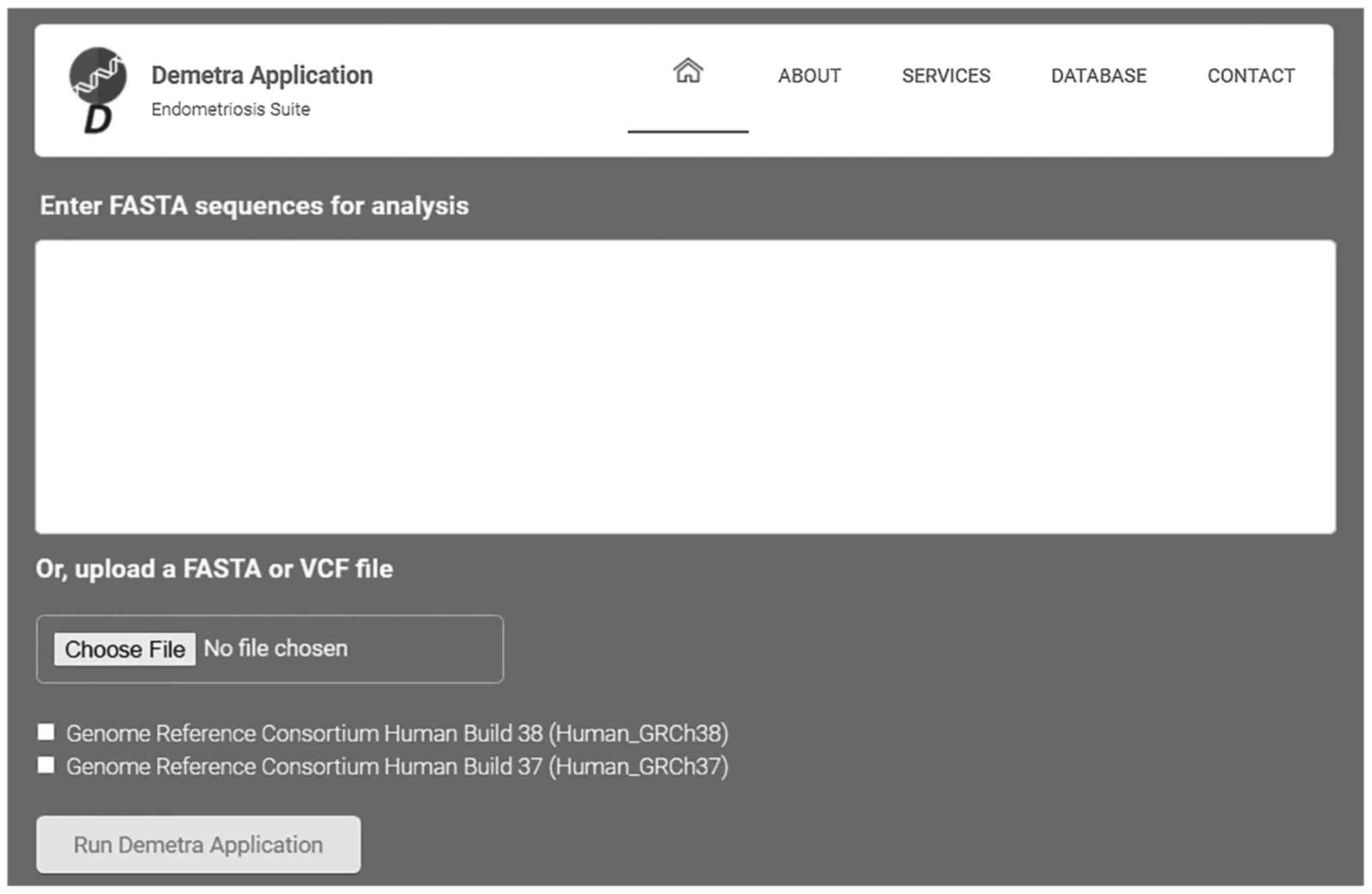
The present study demonstrates the Demetra App
toolkit, a webserver capable of facilitating the clinical genomic
diagnosis process of endometriosis. The user, by uploading the
patient's genetic data to the webserver, either as a FASTA or VCF
data file, automatically scans the nucleotide sequence against
thousands of relevant recorded SNPs. At the same time, the Demetra
App applies different filtering, processing and annotation
techniques, towards identifying and visualizing the most probable
dominant and relevant variants related to endometriosis. The
Demetra App toolkit identifies and classifies all the candidate
SNPs using an up-to-date curated database with SNPs and other
clinical information, and provides those gene variants and SNPs
with probably functional pathogenic effects in endometriosis,
guided by explanatory information and direct links to several
online databases such as the dbSNP and LitVar databases (18,19). Additionally, the Demetra App
extracts and exports other important information related to the
identified variants in the patient's profile, including chromosome
ideograms, statistics bars, a regulatory gene networks, and several
relevant publications from the PubMed database.
Data and methods
Demetra App Database (DAD) of SNPs and
variants for endometriosis
The DAD aimed to develop a resource with all genes,
SNPs and variants associated with endometriosis reported in the
online databases and the literature. The PubMed database depository
was initially mined in order to detect and extract entries related
to 'endometriosis'. The query was limited to human studies only.
The articles retrieved were curated using data mining techniques
aimed towards identifying those containing gene names by using a
dictionary from the gene database of the National Center for
Biotechnology Information (NCBI) (20). A search query was built using
regular expressions by combining each gene or variant with their
synonyms and the keyword 'endometriosis' (21). The extracted genes, SNPs and
variants referred in the article dataset were stored in DAD.
Furthermore, each relevant PubMed reference abstract was mined for
the provision of additional information, such as MeSH/MEDLINE
terms, polymorphisms/mutations described and other genes in the
reference studied for their role in endometriosis (21). Additional information was
extracted and added to the DAD from several available online
databases, including the Online Mendelian Inheritance in Man (OMIM)
database (22) and Endometriosis
Knowledgebase (3). All the
extracted SNPs and variants associated with endometriosis and
contained in the DAD were annotated using key terms and external
searches in the dbSNP, ClinVar and LitVar databases of the NCBI
(18,19,24), and representative FASTA files
were generated using the human reference genome, GRCh38, and the
human mitochondrial complete genome (NCBI: NC_012920.1). Preset
windows of ~201 bases (100 before and 100 after the change/deletion
or insertion of the polymorphism) were applied to the corresponding
genetic locus of each identified SNPs and representative FASTA
files were generated. Finally, the information contained in the DAD
was classified according the scoring function described below and
the final outcome was manually evaluated by medical experts in
endometriosis using the annotated information, results and the
sources of origin as follows:
where VNorFrePub represents the normalized frequency of the
identified SNPs from the PubMed dataset (Max = 1/Min = 0);
VNorFreLitVar represents the normalized frequency of the
identified SNPs based on the LitVar database output and
endometriosis connections (Scalar value, Max = 1/Min = 0); VClinVar
represents the Boolean Parameter (1 indicates that the SNP was
identified in the ClinVar database and has a connection with
endometriosis; 0 indicates that there is no profile in the ClinVar
database, or no connection to endometriosis); and
VMedExperts represents the Boolean Parameter (1 indicates
that the given SNP has been characterized as beiong associated with
the endometriosis by the medical experts team; and 0 indicates
equal to no connection.
VCF or FASTA file validation and
filtering
The uploaded file is validated for compliance with
the standardized formats including, FASTA format or VCF format
four, correspondingly (5). The
FASTA headers should contain the genetic data labels and key terms
and the genetic information sequence in a string of nucleotides
>250 characters. FASTA entries must begin with the symbol
'>', and a tab separated at the end, have each the suitable data
type, and have no duplicated header string names. Respectively, the
VCF header should include the format information and the defined
column names as they specified by the Global Alliance for Genomics
and Health (https://www.ga4gh.org/) (5). VCF file columns must be separated
with tabs, have no duplicated entries and each entry must contain
only the proper data type without gaps. In this initial version,
the file size that can be uploaded to the Demetra webserver must be
≤300 MB. In the next step of analysis in the Demetra webserver
pipeline, only SNPs and gene variants that have passed the quality
and filtering controls will be considered as an input structured
database.
Identification of SNPs and variants
The Demetra App webserver has two different SNP and
variant identification processes depending on the type of the
uploaded file (FASTA of VCF file). For each pipeline of the two
main processes, the webserver uses the DAD of SNPs and variants
associated with endometriosis to analyze and correlate the input
curated dataset. In the case of a FASTA file, the application
implements the process of the local alignments with the DAD. Input
entries identified with 100% identity in a range of a window of 200
bases within a given nucleotide sequence from DAD are reported and
marked to the system as a candidate mutation case endometriosis. In
the second case of the VCF file, all the endometriosis-related SNPs
and variants are identified based on the DAD's directory with the
reported positions of SNPs and variants on each chromosome.
Finally, all the identified cases in each case of the analysis are
collected in a separated list with all the annotated information
from the DAD.
Variant classification and interface
representation
The Demetra App classification procedure identifies
the most candidate and dominant deleterious SNPs and gene variants
in the list of exonic and non-coding polymorphisms. The graphic
representation interface enables the user to see the patient
endometriosis profile, which is presented through the three major
classes of polymorphisms according to the application scoring
function, namely 'Strong-associated SNPs', 'High-associated SNPs',
and 'Associated SNPs'. All the identified SNPs are classified in
these three major classes based on the annotated information
contained in the DAD. An additional list of all identified variants
with necessary information, such as 'snp_ name', 'chromosome',
'position', 'reference genome', 'change', 'gene_name',
'variant_type', 'disease', 'litvar' and 'class' is also provided to
the user. Moreover, for each identified variant, the application
provides an external link to the dbSNP and LitVar databases for
reference to additional information.
A more specialized representation with chart bars
and chromosome ideograms is presented based on the patient's
identified polymorphism profile. This enables the user to better
understand the general genetic profile for the patient, as well as
to draw beneficial conclusions about the association of each
chromosome in endometriosis development. With this more specialized
analysis, conclusions can be drawn on how genes may be involved in
endometriosis, not only as separate entities, but as part of
specific chromosomal regions or as a cluster in a network or in a
combination of both.
Data mining and semantics
The MEDLINE and PubMed databases were searched for
English-language publications that contain the key term
'endometriosis', with no date restrictions (21). The Matlab Bioinformatics toolbox
functions for data mining and semantics were used to extract gene
names from the abstracts of the selected publications using a
dictionary of the gene, allele and pseudogene names for Homo
sapiens (17,26). Furthermore, using the same
techniques, all the polymorphisms reported by at least two studies
from the dataset were extracted. A second-level analysis was
performed in order to estimate the internal links between genes
through selected publications. Internal links were created when
genes, alleles, pseudogenes, or transcription factors were
mentioned in the same publication. Finally, all the mining
knowledge was processed through semantic algorithms contained in
the Matlab 'Data Analysis for Computational Biology', towards
estimating correlations among genes and generating the regulator
network in a graph representation for endometriosis (26-28).
Demetra App web server security and
availability
The Demetra App web tool was used on a Secure XAMPP
HTTP Apache webserver hosted on the computing facility of the
School of Applied Biology and Biotechnology at the Agricultural
University of Athens (AUA). All DADs and third-party software
packages used are locally installed, and thus there are no
additional information transferred to other webservers. The user
genomic data uploaded in the webserver are used for the Demetra App
pipeline only, while the results are stored privately and securely
for a period of three months and subsequently deleted afterward.
The pipeline for identifying the most probable SNPs and gene
variants causing endometriosis described above is executed in the
webserver named, Demetra Application web tool, using Windows,
Apache, XAMPP, PHP, HTML, JavaScript, R and parallel computing
architecture, and is openly available online at http://geneticslab.aua.gr/.
Results
Demetra App
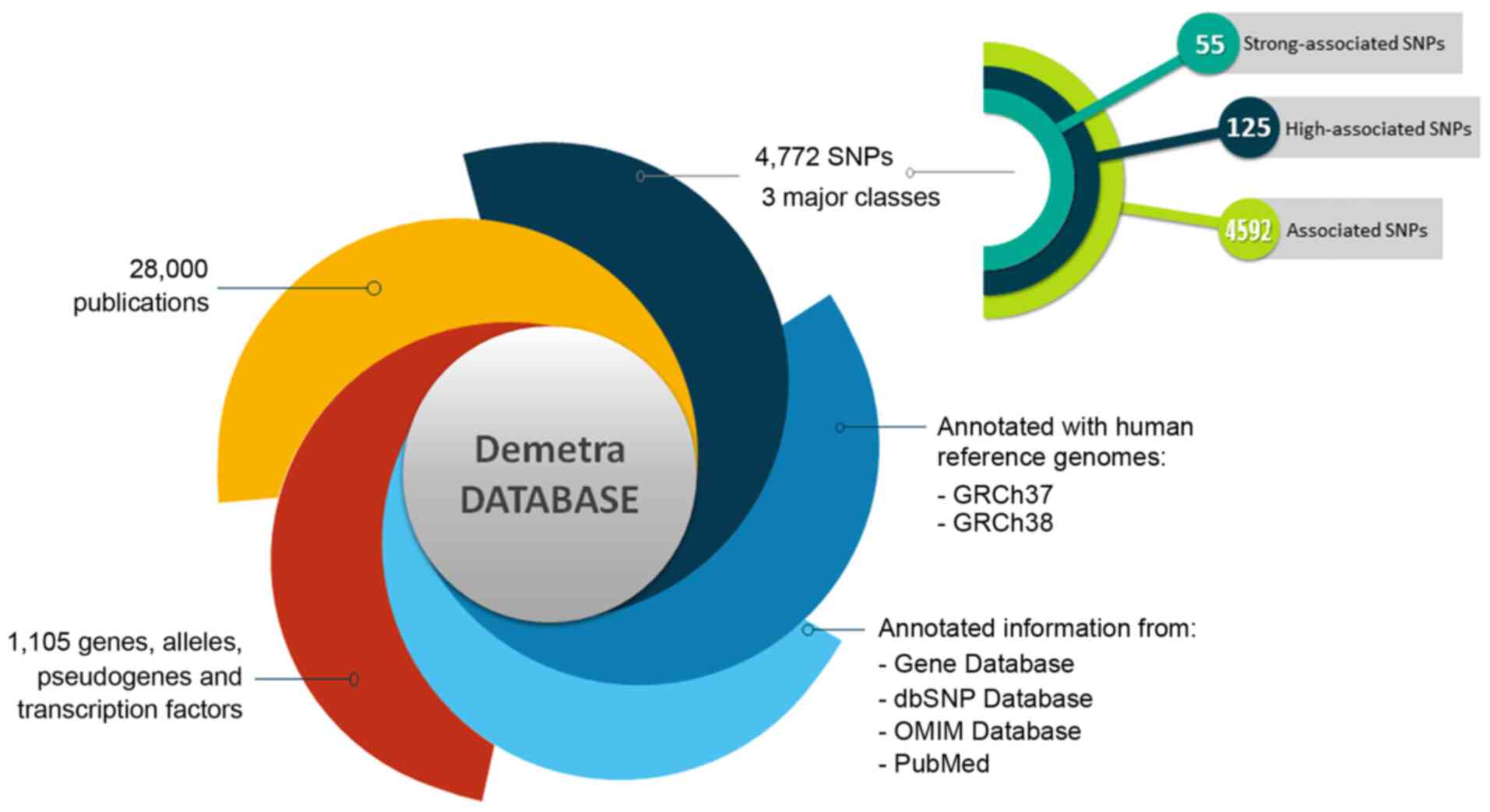
The Demetra App endometriosis database is an
integrated resource for genes, alleles, pseudogenes, transcription
factors and SNPs associated with endometriosis. The information and
the several fields of knowledge contained in the DAD were evaluated
and classified based on the novel pipeline and the specific scoring
function were descripted in the present study. The DAD currently
holds information on 1,105 genes, alleles, pseudogenes and
transcription factors, 4,772 SNPs and 28,000 related publications
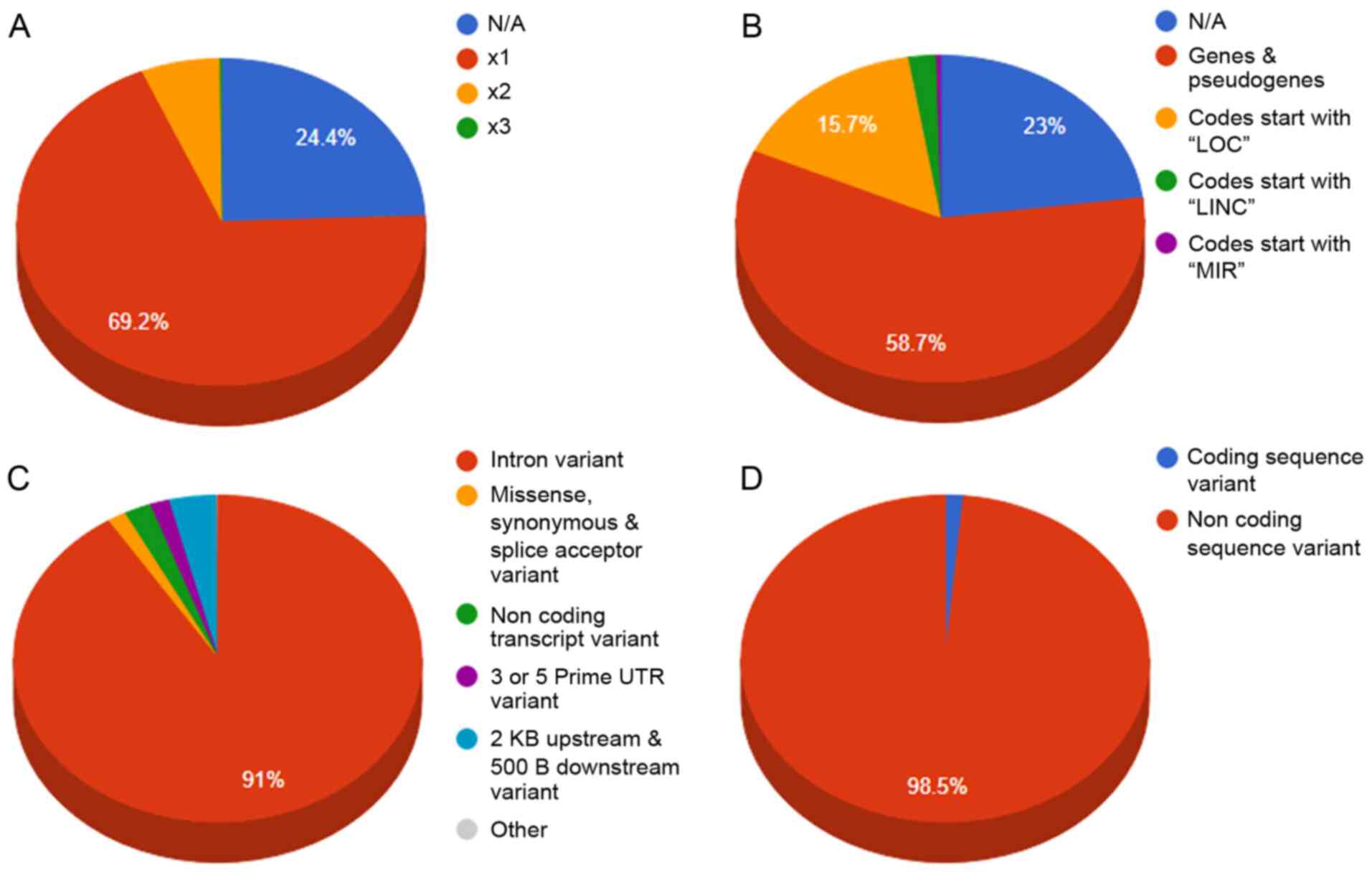
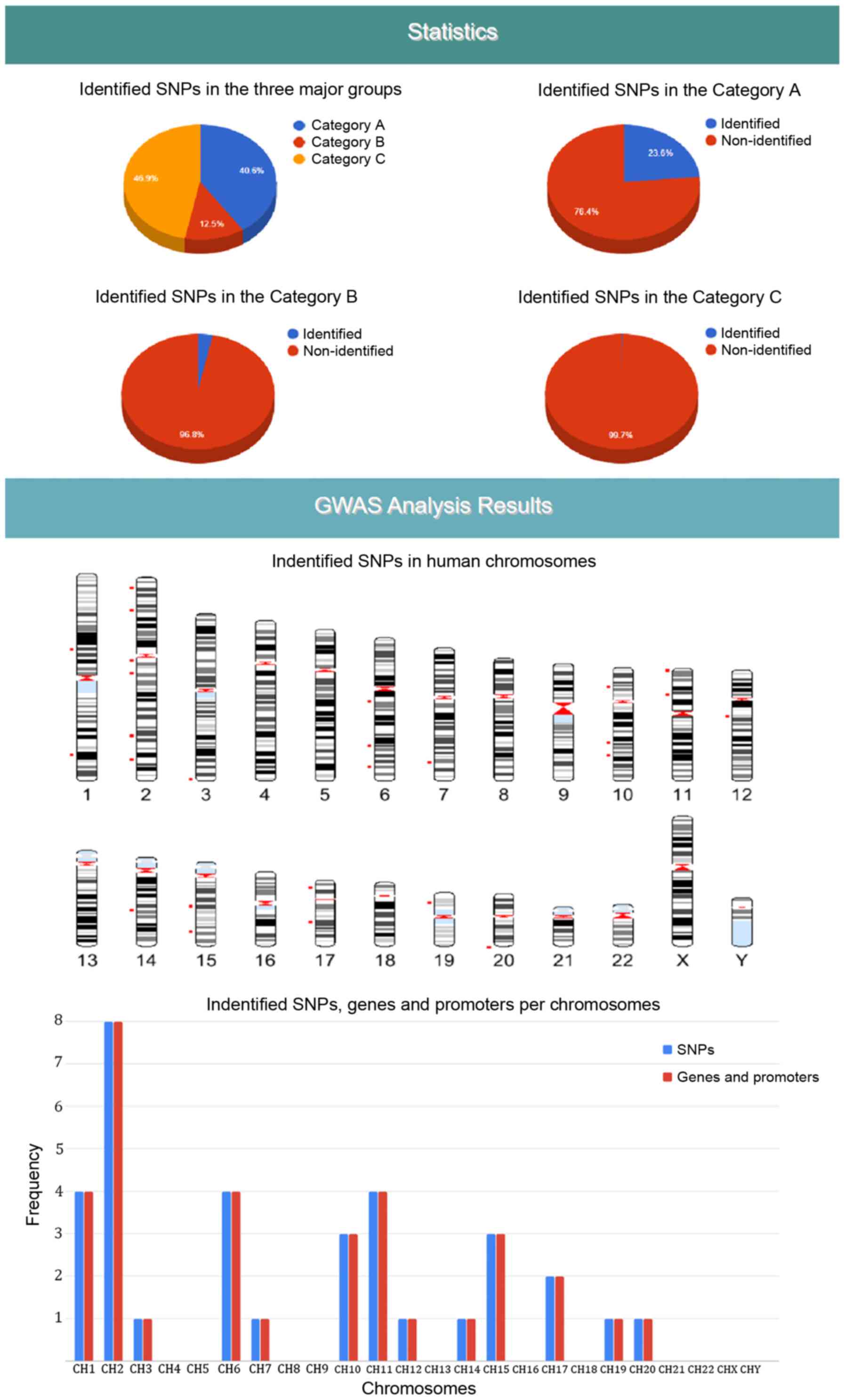
(Fig. 2). Moreover, 68 SNPs were
detected in the coding region sites of genes (Fig. 3).
All the SNPs associated with endometriosis were
manually curated and classified into three major classes, including
'strong-associated SNPs' with 55 members, 'high associated SNPs'
with 125 members and 'associated SNPs' with 4,592 members (Fig. 2). Moreover, each polymorphism is
described by a nucleotide sequence of ~200 bases using the Homo
sapiens reference genome, GRCh38. The database also includes
information from the Gene Database, dbSNP Database, LitVar
Database, ClinVar Database, OMIM Database and PubMed Database. The
information within the database is structured in several fields,
and the knowledge is organized in a specific manner in order to
serve the webserver application immediately and efficiently
(Fig. 3).
Data mining and semantic analysis for
endometriosis
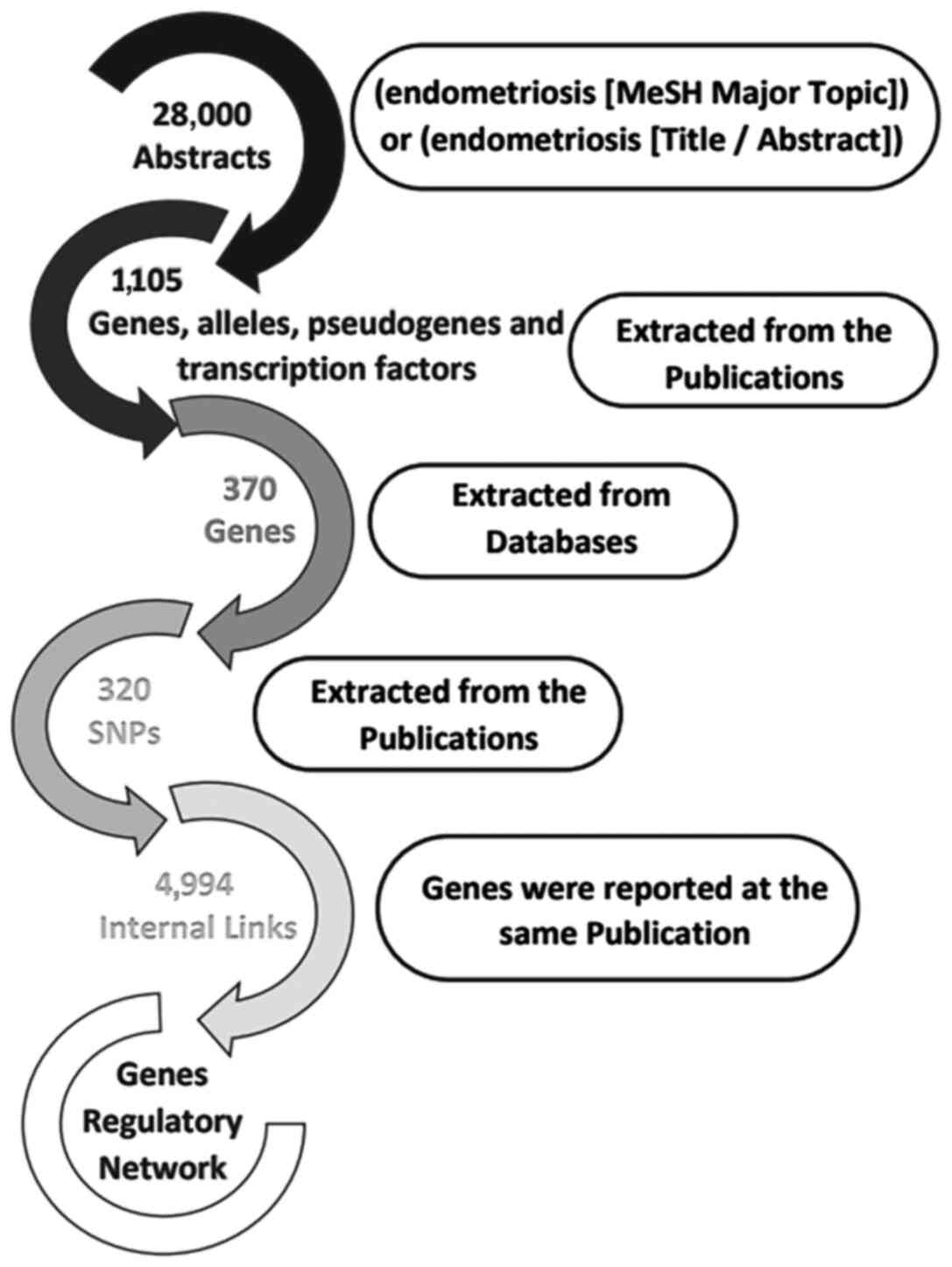
A systematic data mining and semantic analysis of
the most regularly reported genes and polymorphisms was performed
in order to identify those that may play a critical role in
endometriosis and may thus be of value in clinical genomics. For
the purpose of the present study, 28,000 publications were
analyzed, which contained the term 'endometriosis' in the title or
abstract of the MEDLINE file. In the first level of the analysis,
1,105 gene, allele, pseudogene and transcription factor names or
synonyms were identified, and 430 key terms were describing
endometriosis, which was present in >10 publications within the
dataset (Fig. 4). The 30 most
frequently identified key terms describing endometriosis are
presented in Table I. Moreover,
within the dataset, 320 different SNPs and 370 relative genes with
endometriosis were reported and imported from online databases.
Therefore, the analysis allowed the identification of polymorphisms
that could potentially be included in the DAD, alongside the other
SNPs that could definitely predispose to endometriosis. In the
second level of analysis, 4,994 internal links among genes,
alleles, pseudogenes and transcription factors were estimated
through publications, and the regulatory network was calculated in
a graph representation (Fig. 3).
The major goal of this step of the analysis was to provide an
exhaustive regulatory network in genes where are directly related
to endometriosis (Fig. S1).
 | Table IList of the 30 most frequently shown
key terms describing endometriosis within the dataset. |
Table I
List of the 30 most frequently shown
key terms describing endometriosis within the dataset.
| A/A | Key term | A/A | Key term |
|---|
| 1 | Laparoscopy | 16 | Genitalia |
| 2 | Infertility | 17 | Hysterectomy |
| 3 | Endometrium | 18 | Ovarian cancer |
| 4 | Endometrioma | 19 | Ovary |
| 5 | Family
planning | 20 | Fertility |
| 6 | Pelvic pain | 21 | Reproduction |
| 7 | Pregnancy | 22 | Ovarian
reserve |
| 8 | Contraception | 23 | Deep
endometriosis |
| 9 | Dysmenorrhea | 24 |
Endometriosis/complications |
| 10 | Uterus | 25 | Uterine
neoplasms |
| 11 | Adenomyosis | 26 | Apoptosis |
| 12 | Deep infiltrating
endometriosis | 27 | Hormones |
| 13 | Research
methodology | 28 | Endocrine
system |
| 14 | Urogenital
system | 29 | Endometrial
effects |
| 15 | Inflammation | 30 | Angiogenesis |
The extracted knowledge from the data mining and
semantic analysis for endometriosis is included in the Demetra App
in a seamless way, where for each patient profile, the pre-analyzed
information is used towards drawing the corresponding gene
regulatory network based on the identified genes from the SNPs
results. The Demetra App webserver contains all the pre-analyzed
data in an effort to calculate and draw the regulatory gene network
of each patient. The application generates a personalized
regulatory network graph based on patient profile using all the
identified SNPs related to genes, alleles, pseudogenes and
transcription factors from the previous steps of the described
pipeline. Thus, in addition to the detected polymorphisms, the
Demetra App is capable of returning a list of the genes directly
involved in several biological processes with the reference
identified genes. Furthermore, beyond the generated graph, all the
internal links are provided in a list along with genes and relative
publications.
Demetra App webserver
The Demetra App webserver assists the health expert
in confirming an endometriosis diagnosis for a patient using
genetic information. This effective and time-consuming otherwise
pipeline has been designed by geneticists able to benefit from
bioinformatics support and by medical experts in endometriosis
aiming to evaluate and classify all the determined variants and
genes related to endometriosis. Due to the large amount of data
required to be analyzed and the computational complexity of this
pipeline, advanced bioinformatics techniques and parallel
programming have been applied. It is estimated that using a
parallel programming webserver requires much less time (10-fold) to
analyze and extract the final results. Based on various tests
executed on the performance of this application, it was estimated
that this webserver has the ability to analyze a VCF file of 37,000
variants and create a personalized patient profile in <20 min.
The Demetra App has been designed to reduce complexity and minimize
probable errors, allowing health experts to inset only a patient's
genomic data from FASTA or VCF file towards estimating a clear and
concise output HTML file with the patient profile (Fig. 5).
The Demetra App is a state-of-the-art webserver,
designed for health experts in the scientific field of medicine and
clinical genomics who may not have advanced skills in computers to
filter, classify and annotate SNPs variants recognized in
sequencing studies, to be allowed to choose and summarize the SNPs
and gene variants that are associated with endometriosis. The
Demetra App output is an HTML file that describes the patient
profile through six major areas of results, including 'Server
output details', 'SNPs Analysis Results for Endometriosis',
'Statistic Charts', 'GWAS Analysis Results', 'Semantic and Data
mining of identified Genes' and 'Downloads' (Figs. 6-8). In the first results section, a
summary of the analyzed information is presented including, the
type of the data file analyzed, the number of the identified SNPs,
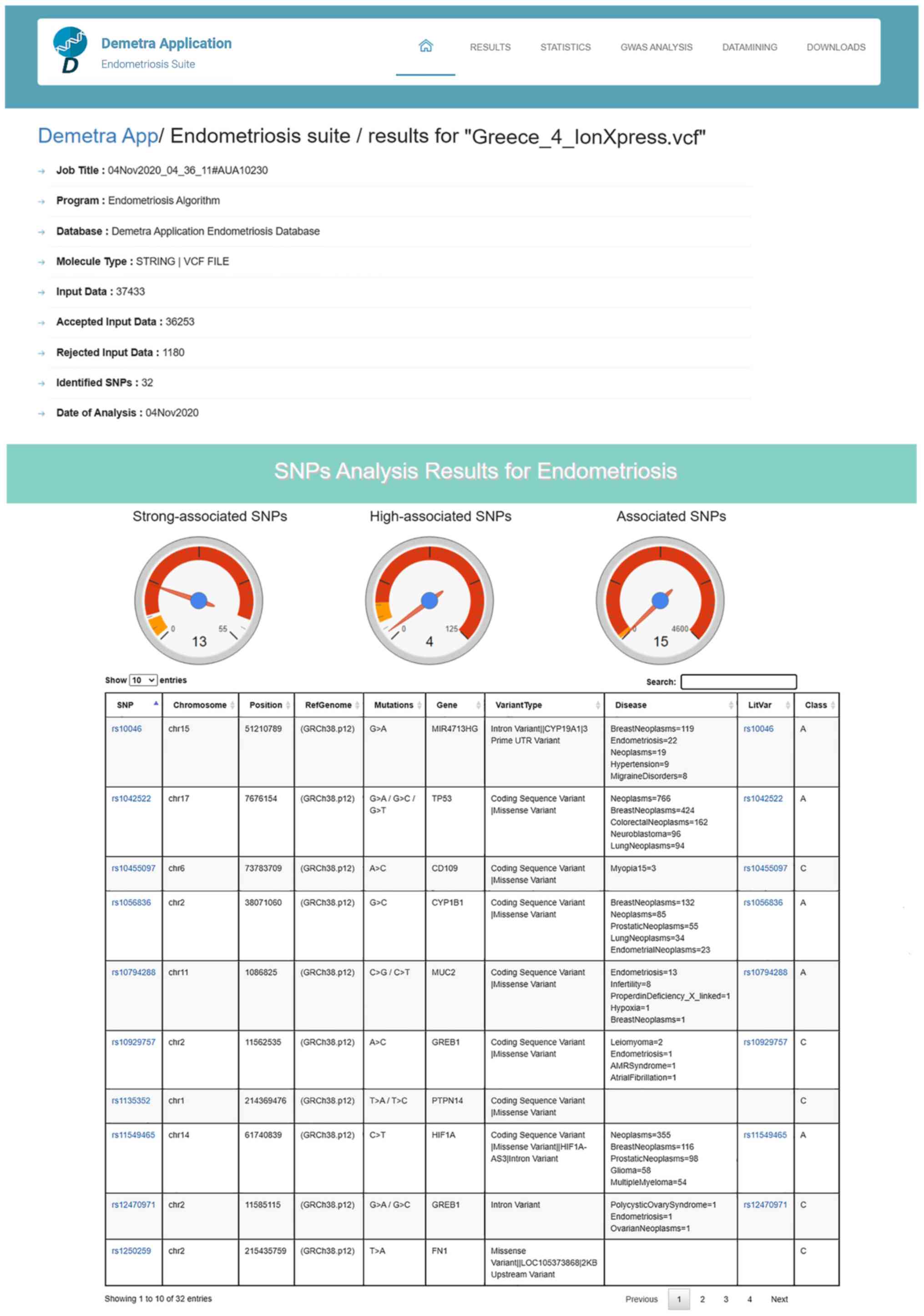
and the date the analysis was performed (Fig. 6). In the second section, the
results of the SNP classification are shown in three separated
charts and a list of all identified SNPs with extra information for
each SNP as extracted from the DAD (Fig. 6). The third results section is
concerned with various statistics charts regarding identified SNPs
and the overall SNPs contained in the DAD (Fig. 7). The fourth section provides
GWAS analysis results in a graphical representation of the
chromosome ideogram, where all the identified SNPs in each genetic
locus per chromosome have been marked. Moreover, a statistical
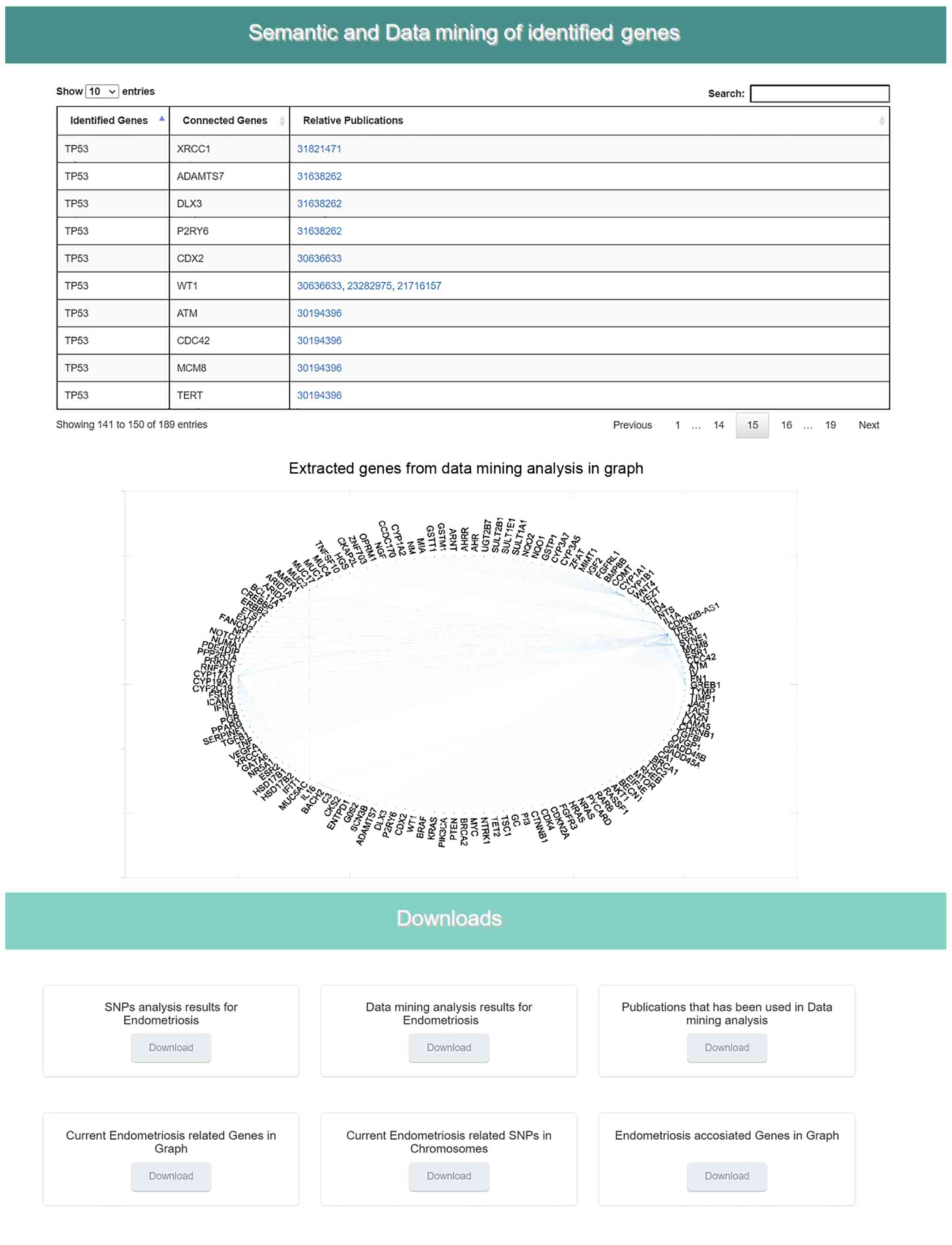
chart indicating the identified SNPs per chromosome (Fig. 7) is shown. In the sixth section,
the results from the data mining and semantic analysis are
presented (Fig. 8). A list of
all identified genes is provided with all the information mined
from the relative publications towards calculating and drawing the
regulatory network in a graph representation. The user can filter
the list in several ways and has the option to retrieve the
relevant publications that describe each internal link within the
network. Moreover, the beneficial knowledge of all connected genes
with the identified genes is provided to the users. In the last
results section, the user has the choice to download and save all
the generated results from the DAD webserver (Fig. 8).
Demetra App validation
Demetra App webserver validation was performed by a
retrospective study performed by Albertsen et al (29) on seven patients from a
three-generation family with endometriosis from the 'Venizeleio and
Pananio' General Hospital of Heraklion, Greece. The WES data of the
seven patients presented in the study by Albertsen et al
(29) in detail, were reanalyzed
using the Demetra App webserver. A list with all known genes that
were previously reported as 'endometriosis-associated' was properly
identified in the final output HTML profile per patient, and by
cross-comparison of the results, new findings have emerged. The
SNPs analysis performed identified the common pathogenic variants
that occurred within this family and were transmitted or imported
from generation to generation. Moreover, a list of
'high-associated' and 'strong-associated' polymorphisms that are
directly related to endometriosis were identified and classified in
each one of the seven patients (Table II). All tests were run with the
Demetra App using default parameters on the human reference genome
GRCh38 and the human mitochondrial complete genome (NCBI:
NC_012920.1). Furthermore, the Demetra App was also successfully
evaluated with different well-reported cases of SNPs located in
genes, which may play a critical role in the development of
endometriosis, as shown in Table
II.
| Table IIMajor SNP cases identified in the
seven patients with endometriosis. |
Table II
Major SNP cases identified in the
seven patients with endometriosis.
| SNP | Chromosome C | hange | Gene | Type | Class | Patients | Frequency |
|---|
| rs1056836 | chr2 | G>C | CYP1B1 | Coding sequence
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs13394619 | chr2 | G>A | GREB1 | Splice acceptor
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs2258447 | chr3 | T>A/T>C | MUC4 | Coding sequence
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs700518 | chr15 | T>C | CYP19A1 | Coding sequence
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs1042522 | chr17 | G>C/G>T | TP53 | Coding sequence
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs2427284 | chr20 | A>G/A>T | LAMA5 | Coding sequence
variant | A |
01|02|03|04|05|06|07 | 7 |
| rs10794288 | chr11 | C>G/C>T | MUC2 | Coding sequence
variant | A |
01|02|03|04|06|07 | 6 |
| rs743572 | chr10 | A>G/A>T | CYP17A1 | 5 Prime UTR
variant | A | 01|02|03|04|07 | 5 |
| rs10046 | chr15 | G>A | MIR4713HG | Intron variant | A | 01|02|03|04|06 | 5 |
| rs2304402 | chr2 | G>A | GREB1 | Coding sequence
variant | A | 01|02|03|04|07 | 5 |
| rs11549465 | chr14 | C>T | IF1A | Coding sequence
variant | A | 01|02|03|04 | 4 |
| rs1799930 | chr8 | G>A | NAT2 | Coding sequence
variant | A | 01|02|03 | 3 |
| rs4072111 | chr15 | C>T | IL16 | Coding sequence
variant | A | 04|05 | 2 |
| rs5498 | chr19 | A>G | ICAM1 | Coding sequence
variant | B |
01|02|03|04|05|06 | 6 |
| rs3783550 | chr2 | G>T | IL1A | Intron variant | B | 01|02|03|04|06 | 5 |
| rs7103978 | chr11 | A>G/A>T | MUC2 | Coding sequence
variant | B | 01|03|04|07 | 4 |
| rs113759408 | chr8 | G>A | CYP11B1 | Intron variant | B | 02|03 | 2 |
| rs280523 | chr19 | G>A/G>C | TYK2 | Coding sequence
variant | B | 01|07 | 2 |
| rs1801133 | chr1 | G>A | MTHFR | Coding sequence
variant | B | 03|07 | 2 |
| rs1802669 | chr10 | G>A/G>T | MLLT10 | Coding sequence
variant | B | 01|04 | 2 |
| rs605059 | chr17 |
G>A/G>C/G>T | HSD17B1 | Coding sequence
variant | B | 01 | 1 |
| rs500760 | chr11 | T>C | PGR | Coding sequence
variant | B | 06 | 1 |
| rs2304256 | chr19 | C>A | TYK2 | Coding sequence
variant | B | 06 | 1 |
| rs12720270 | chr19 | G>A | TYK2 | Intron variant | B | 06 | 1 |
| rs1135352 | chr1 | T>C | PTPN14 | Coding sequence
variant | C |
01|02|03|04|05|06|07 | 7 |
| rs3013451 | chr1 | G>A | PTPN14 | Intron variant | C |
01|02|03|04|05|06|07 | 7 |
| rs7550799 | chr1 | T>A/T>C | PTPN14 | Coding sequence
variant | C |
01|02|03|04|05|06|07 | 7 |
| rs2241820 | chr12 | C>A/C>T | HOXC9 | Coding sequence
variant | C |
01|02|03|04|05|06|07 | 7 |
| rs10929757 | chr2 | A>C | GREB1 | Coding sequence
variant | C |
01|02|03|04|05|06 | 6 |
| rs12470971 | chr2 | G>A | GREB1 | Intron variant | C |
01|02|03|04|05|06 | 6 |
| rs1250259 | chr2 | T>A | FN1 | Missense
variant | C |
01|02|03|04|05|06 | 6 |
| rs2278868 | chr17 | C>T | SKAP1 | Coding sequence
variant | C |
01|02|04|05|06|07 | 6 |
| rs7586970 | chr2 | T>C/T>G | FPI | Coding sequence
variant | C | 01|02|03|04|07 | 5 |
| rs6973420 | chr7 | A>G | CALD1 | Coding sequence
variant | C | 01|02|03|04|07 | 5 |
| rs2918308 | chr19 | A>C | NFILZ | 3 Prime UTR
variant | C | 02|03|04|05 | 4 |
| rs6169 | chr11 | C>T | FSHB | Coding sequence
variant | C | 01|02|03|04|07 | 5 |
| rs430600 | chr1 | T>A/T>C | PKN2 | Coding sequence
variant | C | 02|03|04|05|06 | 5 |
| rs6557210 | chr6 | G>A | SYNE1 | Intron variant | C | 01|02|04|05 | 4 |
| rs10455097 | chr6 | A>C | CD109 | Coding sequence
variant | C | 02|03|04|06 | 4 |
| rs2721939 | chr8 | C>T | TRPS1 | Intron variant | C | 01|05|07 | 3 |
| rs6904364 | chr6 | T>C | RMND1 | Intron variant | C | 05|06|07 | 3 |
| rs2293889 | chr8 | T>C/T>G | TRPS1 | Intron variant | C | 01|05 | 2 |
| rs1529868 | chr2 | C>T | GREB1 | Intron variant | C | 05|06 | 2 |
| rs17082236 | chr6 | C>A | SYNE1 | Coding sequence
variant | C | 01 | 1 |
Discussion
Demetra App services aid the diagnosis of
endometriosis using a patient's genetic profile through provided
information that will eventually help to identify a patient's
predisposition to endometriosis in the very early stages, even
without any symptoms. In the case where medical experts lack a
clear etiology for the patient's condition, Demetra App results can
provide useful information about the patient profile and a list of
the most critical polymorphisms present in the patient's genome and
their association with several biological pathways.
The quality of the data for variants identified in
the VCF file uploaded by the user many times may provide low
reliability and pause several limitations. To deal with such
issues, the Demetra App validates the VCF file and remove variants
that did not pass the quality control thresholds. On the other
hand, it can also enable the user to upload the raw sequences or
genotype data and provides a pre-processed analysis through which a
generated VCF file is passed into the main pipeline of the
webserver. Thus, the user has the option to analyze both VCF and
FASTA files without any restrictions.
DAD contains all the identified SNPs related to
endometriosis, classified into three major classes. The quality of
the information in the individual databases has possible
limitations, and clinical databases may include nonverified
annotations, as clinical research is being produced at ever faster
rates. In an effort to ensure the predictive performance and the
reliability of the system, so far, we opted for the manual update
of the SNP DAD following validation and classification of the
candidate SNPs by a team of medical experts.
In conclusion, endometriosis is an inherited
multifactorial illness that is usually detected at a fairly
advanced stage, preventing doctors from treating it well from an
early stage. The Demetra App was designed to support physician
diagnosis from the early stages by using the genomic data of the
patient. The comprehensible interface of the Demetra App was
designed to be used besides the clinical genomics scientists by
many other health experts. Its output presents the examined
patient's profile through which the user is provided with a
structured set of results in various categories, which are
generated based on the list of the most predictable candidate gene
variants related to endometriosis. The majority of the current
clinical genomics tools, web tools, and applications are
scientifically oriented for geneticists and bioinformaticians and
are not developed to be executed by medical doctors or other
scientists. In this sense, the Demetra App is an easy-to-use
integrated public web server for endometriosis, designed with the
aim of bringing personalized medicine and personal genomics tools
to the scientific community.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LP, DV, GNG, IM, MM, MIZ, DAS and EE substantially
contributed to the conception or design of the study, including the
acquisition, analysis, or interpretation of the data for the study.
LP, DV, GNG, IM, MM, MIZ, DAS and EE contributed towards drafting
the study or revising it critically for important intellectual
content and approved the version to be published. All authors
agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved. All authors
have read and approved the final manuscript. GNG and EE confirm the
authenticity of the datasets used. EE, DV and LP confirm the origin
of all data selected from public databases.
Ethics approval and consent to
participate
The test WES data used were from a previous study
(29), and thus no ethics
approval was required for the present study, as this was previously
obtained.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Hans M. Albertsen
of Juneau Biosciences (USA) for providing the test WES data from a
previous study (29) and for
providing critical view of the manuscript.
Funding
EE received funding by the project 'INSPIRED-The National
Research Infrastructures on Integrated Structural Biology, Drug
Screening Efforts and Drug Target Functional Characterization'
(Grant MIS 5002550) and by the project: 'OPENSCREENGR An
Open-Access Research Infrastructure of Chemical Biology and
Target-Based Screening Technologies for Human and Animal Health,
Agriculture and the Environment' (Grant MIS 5002691), which are
implemented under the Action 'Reinforcement of the Research and
Innovation Infrastructure', funded by the Operational Program
'Competitiveness, Entrepreneurship and Innovation' (NSRF 2014-2020)
and co-financed by Greece and the European Union (European Regional
Development Fund).
References
|
1
|
Halis G and Arici A: Endometriosis and
inflammation in infertility. Ann N Y Acad Sci. 1034:300–315. 2004.
View Article : Google Scholar
|
|
2
|
Zondervan KT, Becker CM, Koga K, Missmer
SA, Taylor RN and Viganò P: Endometriosis. Nat Rev Dis Primers.
4:92018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sapkota Y, Steinthorsdottir V, Morris AP,
Fassbender A, Rahmioglu N, De Vivo I, Buring JE, Zhang F, Edwards
TL, Jones S, et al: iPSYCH-SSI-Broad Group: Meta-analysis
identifies five novel loci associated with endometriosis
highlighting key genes involved in hormone metabolism. Nat Commun.
8:155392017. View Article : Google Scholar
|
|
4
|
Vassilopoulou L, Matalliotakis M, Zervou
MI, Matalliotaki C, Krithinakis K, Matalliotakis I, Spandidos DA
and Goulielmos GN: Defining the genetic profile of endometriosis.
Exp Ther Med. 17:3267–3281. 2019.PubMed/NCBI
|
|
5
|
Alborzi S, Hosseini-Nohadani A, Poordast T
and Shomali Z: Surgical outcomes of laparoscopic endometriosis
surgery: A 6 year experience. Curr Med Res Opin. 33:2229–2234.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Anastasiu CV, Moga MA, Elena Neculau A,
Bălan A, Scârneciu I, Dragomir RM, Dull AM and Chicea LM:
Biomarkers for the noninvasive diagnosis of endometriosis: state of
the art and future perspectives. Int J Mol Sci. 21:212020.
View Article : Google Scholar
|
|
7
|
Goulielmos GN, Matalliotakis M,
Matalliotaki C, Eliopoulos E, Matalliotakis I and Zervou MI:
Endometriosis research in the -omics era. Gene. 741:1445452020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Palmer SS and Barnhart KT: Biomarkers in
reproductive medicine: The promise, and can it be fulfilled? Fertil
Steril. 99:954–962. 2013. View Article : Google Scholar :
|
|
9
|
de Sanctis V, Matalliotakis M, Soliman AT,
Elsefdy H, Di Maio S and Fiscina B: A focus on the distinctions and
current evidence of endometriosis in adolescents. Best Pract Res
Clin Obstet Gynaecol. 51:138–150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Agarwal SK, Chapron C, Giudice LC, Laufer
MR, Leyland N, Missmer SA, Singh SS and Taylor HS: Clinical
diagnosis of endometriosis: a call to action. Am J Obstet Gynecol.
220:354.e1–354.e12. 2019. View Article : Google Scholar
|
|
11
|
Tam V, Patel N, Turcotte M, Bossé Y, Paré
G and Meyre D: Benefits and limitations of genome-wide association
studies. Nat Rev Genet. 20:467–484. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan R and Mittelman D: Consumer genomics
will change your life, whether you get tested or not. Genome Biol.
19:1202018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roberts J and Middleton A: Genetics in the
21st Century: Implications for patients, consumers and citizens.
F1000 Res. 6:20202017. View Article : Google Scholar
|
|
14
|
Perakakis N, Yazdani A, Karniadakis GE and
Mantzoros C: Omics, big data and machine learning as tools to
propel understanding of biological mechanisms and to discover novel
diagnostics and therapeutics. Metabolism. 87:A1–A9. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang J, Li Y, Liu Q, Li L, Feng A, Wang T,
Zheng S, Xu A and Lyu J: Brief introduction of medical database and
data mining technology in big data era. J Evid Based Med. 13:57–69.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu J, Kim S, Song M, Jeong M, Kim D, Kang
J, Rousseau JF, Li X, Xu W, Torvik VI, et al: Building a PubMed
knowledge graph. Sci Data. 7:2052020. View Article : Google Scholar
|
|
17
|
Liu JL and Zhao M: A PubMed-wide study of
endometriosis. Genomics. 108:151–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Allot A, Peng Y, Wei CH, Lee K, Phan L and
Lu Z: LitVar: A semantic search engine for linking genomic variant
data in PubMed and PMC. Nucleic Acids Res. 46(W1): W530–W536. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar :
|
|
20
|
Brown GR, Hem V, Katz KS, Ovetsky M,
Wallin C, Ermolaeva O, Tolstoy I, Tatusova T, Pruitt KD, Maglott
DR, et al: Gene: A gene-centered information resource at NCBI.
Nucleic Acids Res. 43(D1): D36–D42. 2015. View Article : Google Scholar :
|
|
21
|
Kim S, Yeganova L, Comeau DC, Wilbur WJ
and Lu Z: PubMed Phrases, an open set of coherent phrases for
searching biomedical literature. Sci Data. 5:1801042018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hamosh A, Scott AF, Amberger JS, Bocchini
CA and McKusick VA: Online Mendelian Inheritance in Man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids
Res. 33:D514–D517. 2005. View Article : Google Scholar :
|
|
23
|
Joseph S and Mahale SD: Endometriosis
Knowledgebase: a gene-based resource on endometriosis. Database
(Oxford). 2019. pp. baz0622019, View Article : Google Scholar
|
|
24
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46(D1): D1062–D1067. 2018. View Article : Google Scholar :
|
|
25
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al: 1000 Genomes Project Analysis Group: The variant call format
and VCFtools. Bioinformatics. 27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banchs RE: Text Mining With MATLAB.
Springer; New York, NY: 2013, View Article : Google Scholar
|
|
27
|
Xiao H, Yang L, Liu J, Jiao Y, Lu L and
Zhao H: Protein-protein interaction analysis to identify biomarker
networks for endometriosis. Exp Ther Med. 14:4647–4654.
2017.PubMed/NCBI
|
|
28
|
Jurca G, Addam O, Aksac A, Gao S, Özyer T,
Demetrick D and Alhajj R: Integrating text mining, data mining, and
network analysis for identifying genetic breast cancer trends. BMC
Res Notes. 9:2362016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Albertsen HM, Matalliotaki C,
Matalliotakis M, Zervou MI, Matalliotakis I, Spandidos DA, Chettier
R, Ward K and Goulielmos GN: Whole exome sequencing identifies
hemizygous deletions in the UGT2B28 and USP17L2 genes in a three
generation family with endometriosis. Mol Med Rep. 19:1716–1720.
2019.PubMed/NCBI
|