Introduction
Traumatic brain injury (TBI) is a type of acute
brain injury caused by the mechanical energy of external force
acting on the head (1). TBI
poses an enormous financial and societal burden with high morbidity
and mortality (2). Following the
damage caused at the time of brain injury (primary injury), various
events that occur within minutes to days after the injury can lead
to secondary injury (3). The
long-term outcomes of patients with TBI can be ameliorated by
prompt and effective treatment after trauma (4). Thus, further elucidation of the
complicated cellular and molecular events contributing to secondary
injury is crucial. Neuroinflammation is reported to be a critical
factor inducing secondary brain injury, and suppression of
inflammation is a favorable approach to alleviate secondary brain
injury and improve prognosis in TBI (5). The inflammatory cascade is
activated by the release of pro-inflammatory and anti-inflammatory
cytokines (6). Cytokines, such
as interleukin (IL)-17, are notably upregulated after TBI, which is
concerned with the pathogenesis of TBI (7). An imbalance in Th17/Treg cells is
considered a pivotal factor in the progression of the inflammatory
response (8,9). Therefore, maintaining the Th17/Treg
balance may be an effective approach for managing TBI.
Propofol, a short-acting intravenous anesthetic, has
been extensively utilized in the management of craniocerebral
injury for induction, maintenance, and sedation during surgery
(10). Propofol also reduces the
generation of pro-inflammatory cytokines (11) and has been reported to exert a
neuroprotective effect on diverse neuronal injuries, such as
ischemic stroke and TBI (12).
However, the detailed mechanism underlying propofol's protective
effect on TBI has not been elucidated.
Emerging evidence has shown that propofol plays an
anti-inflammatory and neuroprotective role by regulating microRNAs
(miRs) and their downstream targets (12,13). miRs are endogenous non-coding
RNAs that regulate protein-coding genes post-transcriptionally
(14). Dysregulation of miRs has
been reported to be involved in the pathological process of TBI
(15). For example, Henry et
a. revealed that the suppression of miR-155 can reduce
neuroinflammation and promote functional restoration after TBI in
mice (16). In view of this, a
TBI model in rats was established to explore the mechanism by which
propofol protects against brain injury by modulating miR and its
downstream targets. The findings may provide promising therapeutic
targets for patients with TBI to attenuate brain injury and improve
prognosis.
Materials and methods
Ethics statement
The study was approved by the Ethics committee of
the Guangdong Provincial People's Hospital. All experimental
procedures were performed in accordance with the Ethical Guidelines
for the Study of Experimental Pain in Conscious Animals.
Establishment of TBI model in rats and
grouping (17)
Male Sprague Dawley (SD) rats (aged 12-16 weeks,
weighing 360-400 g), purchased from the Animal Research Center of
Wuhan University [SCXK (Hubei) 2019-0004, Wuhan, Hubei, China] were
kept at the experimental site for at least one week prior to
surgery. SD rats were raised in separate cages in an animal room at
23-25°C and 50-60% humidity under a 12-h light/dark cycle, and
given normal clean feed. The rats had free access to food and
water. The padding was replaced every other day to keep the cage
clean. The rats were anesthetized via intraperitoneal injection of
1% pentobarbital (30 mg/kg). The rats were then fixed, shaven, and
disinfected, and the scalp was cut to expose the skull. A
5-mm-diameter hole (2.0 mm from the posterior dura and 2.0 mm to
the right of the sagittal suture) was drilled in the rats' skulls,
and the dura was exposed. Moderate TBI was caused in the rats by a
40-g weight-drop from a height of 25 cm. The rats in the
sham-operation group were operated on without a weight-drop. After
the weight-drop, the scalps of the rats were sutured, and the rats
recovered from anesthesia. All the operations were performed using
aseptic techniques. TBI was confirmed by the observation of limb
convulsions, transient apnea, and unconsciousness in rats.
The rats with TBI were assigned to five groups (n=18
in each group) as follows: Sham group, TBI group, TBI + propofol
group (TBI+P), TBI + fat emulsion group (TBI+FB), TBI + propofol +
antigomiR-145-3p group (TBI+P+ant-145), and TBI + propofol +
antigomiR NC group (TBI+P+ant-NC). In the TBI+P group, the rats
were intravenously injected with propofol over 5 min (12.5 mg/kg) 1
h after TBI and then infused intravenously at 40 mg/kg/h over 2 h.
Clinical doses of propofol were used based on preliminary
experiments and pharmacological calculations. Fat emulsion, which
was injected intravenously over 5 min 1 h after TBI and then
continuously infused for 2 h at the same dosage and infusion rate
as that of propofol in the TBI+P group, was used as the solvent
control for propofol. Then, 1 h after establishing TBI, 0.5 nmol of
ant-NC (Invitrogen, Inc.) was administered into the brains of rats
by intracerebroventricular injection and repeated every 1 h for
three total injections.
Blood (1.5 ml) was collected from the rats' orbits
24 h after establishing TBI and then anticoagulated for
preservation [0.6 ml blood was anticoagulated with ethylene diamine
tetraacetic acid (EDTA) for the detection of Treg cells using flow
cytometry; 0.9 ml blood was anticoagulated with heparin sodium for
the detection of Th17 cells using flow cytometry and the detection
of IL-17 content using enzyme-linked immunosorbent assay (ELISA)].
After neurological severity scoring, the rats were euthanized by
intraperitoneal injection of ≥100 mg/kg pentobarbital sodium, and
their brains were immediately removed. From each group, six brain
tissues were used to detect protein and gene expression in tissue
homogenates, six brain tissues were embedded in paraffin sections
for histopathological observation, and six brain tissues were used
for brain water content testing.
Neurological severity assessment
The neurological functioning of the rats was studied
using the modified neurological severity score (18). It comprised a combination of
motor, sensory, and reflex tests. The score ranged from 0 to 18,
according to the severity of injury. The higher score indicated the
most severe injury (0, no injury; 1-6, mild injury; 7-12, average
moderate injury; 13-18, severe injury; 18, maximum injury).
Neurological function was assessed one day after TBI by an observer
who was blinded to the treatment groups. Scoring criteria are shown
in Table SI.
Brain water content
The injured brain tissues of rats with TBI were
collected and weighed on an electronic balance to obtain the wet
weight. The brain tissues were then dried in an oven at 85°C for 24
h and the dry weight was obtained. Brain water content (%) was
calculated as; (wet weight-dry weight)/wet weight ×100.
Histological staining
Brain tissues were fixed, dehydrated, and embedded
in paraffin: The brain tissues were fixed with 4% paraformaldehyde
at room temperature for 24 h, then dehydrated with gradient
ethanol, soaked in xylene for 30 min, embedded in paraffin, and
sliced into tissue sections (5 μ.m). Next, the paraffin
sections were deparaffinized and hydrated. The sections were then
stained with hematoxylin and eosin (H&E) for 5 min and
differentiated in ethanol with 0.6% hydrochloric acid for 30 sec,
followed by counterstaining with acidified eosin alcohol (pH 4.2)
for 3 min. Subsequently, the tissues were dehydrated and cleared.
The pathological areas of the brain were observed under an optical
microscope (Olympus Optical Co., Ltd.).
Terminal deoxynucleotidyl transferase (TdT)-mediated
dUTP nick end-labeling (TUNEL) staining was performed on the
sections of the rats' brain tissues according to the manufacturer's
instructions (Thermo Fisher). The sections were fixed in ethanol
and acetic acid (2:1), washed with phosphate-buffered saline (PBS),
and then incubated with protease K (100 μ.g/ml) at room
temperature for 15-30 min. Subsequently, the sections were washed
with PBS, cultured in the presence of 3% hydrogen peroxide, and
permeabilized with 0.5% Triton X-100. After rewashing, the sections
were incubated with the TUNEL reaction mixture at 37°C for 1 h.
Finally, the sections were developed with 0.03% 2,4-diaminobutyric
acid (DAB), counterstained with hematoxylin, and observed under a
fuorescence microscope (Olympus IX71; Olympus Corporation) after
sealing.
The brain tissue sections were incubated with the
primary antibodies (rabbit anti-rat IL-17A polyclonal antibody,
1:500, ab214588, Abcam; rabbit TNF-α polyclonal antibody, 1:50,
PA1-40281, Invitrogen-Thermo Fisher Scientific; rabbit IL-1β
polyclonal antibody, 1:400, BS-6319R, Invitrogen) at 4°C overnight
and then incubated with the secondary antibody (goat anti-rabbit
IgG H&L (horseradish peroxidase), 1:2,000, ab205718; Abcam) at
37°C for 2 h. After washing with PBS, the sections were developed
with DAB as the substrate and H2O2 as the
catalyst. The sections were observed and analyzed under a
microscope after sealing.
Flow cytometry
Treg cells in the peripheral blood were detected
according to the manufacturer's instructions (19,20). Peripheral venous blood and
cluster for differentiation of antibodies, including CD4 (1:250,
ab59474; Abcam), CD25 (1:250, ab210330; Abcam), and forkhead box P3
(Foxp3; 1:250, ab210232; Abcam) were added to the detection tube,
with immunoglobulin G (IgG) (1:250, ab205718; Abcam) acting as the
isotype control. The tube was then placed in the dark at 4°C,
followed by detection using a flow cytometer, and subsequently,
Th17 cells in the peripheral blood were detected. Peripheral blood
mononuclear cells (PBMCs) were also isolated. Subsequently, 1 ml
cell suspension, 50 ng phorbol 12-myristate 13-acetate (PMA), 1
μ.g ionomycin, and 0.7 μ.l monensin were added into
each well of a 6-well plate and then cultured at 37°C for 6 h.
After 5 min of centrifugation at room temperature and 200 × g, the
cells were transferred to a flow detection tube, resuspended in
PBS, incubated with CD3 (1:1,000, ab16669; Abcam) and CD8
antibodies (1:1,000, ab101500; Abcam), and supplemented with a
fixed membrane-penetrating agent. After washing and resuspending,
the cells were incubated with antibody IL-17A (1:250, ab193955;
Abcam) with IgG acting as the isotype control, and then the cells
were placed in the dark, followed by detection using a flow
cytometer.
The cells were collected and resuspended at
1×106 cells/ml. Annexin V-FITC and propidium iodide (PI)
(5 μ.l) were added into each tube, which were then incubated
in the dark at room temperature for 10 min. Thereafter, the stained
cells were analyzed by flow cytometry.
ELISA
The inflammatory factor levels of IL-17, tumor
necrosis factor-α (TNF-α), and IL-1β in the brain tissues or cells
after the different treatments were measured based on the
enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems)
instructions.
Reverse transcriptase-quantitative
polymerase chain reaction
Total RNA was extracted from brain tissue using a
TRIzol® reagent (Invitrogen). The purity of the
extracted high-quality RNA was measured by ultraviolet analysis and
formaldehyde deformation electrophoresis. Reverse
transcriptase-quantitative polymerase chain reaction (RT-qPCR) was
performed using an RT-qPCR kit (Thermo Fisher Scientific). Primers
(Table I) were designed and
synthesized by Sangon Biotech Co., Ltd. (Shanghai).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as an
internal reference. Amplification and dissolution curves were
confirmed after the reaction. The relative expression of genes was
calculated by the 2−ΔΔCq method (21).
 | Table IPrimer sequences for RT-qPCR. |
Table I
Primer sequences for RT-qPCR.
| Gene name | Primer
sequence |
|---|
| U. | F:
5′-CGCTTCGGCAGCACATATAC-3′ |
| R:
5′-AATATGGAACGCTTCACGA-3′ |
|
miR-145-3. | F:
5′-GGATTCCTGGAAATACTGT-3′ |
| R:
5′-AGAACAGTATTTCCAGGAATC-3′ |
|
miR-28-.p | F:
5′-CACTAGATTGTGAGCTCCT-3′ |
| R:
5′-TCCAGGAGCTCACAATCTAG-3′ |
|
miR-532-5. | F:
5′-CATGCCTTGAGTGTAGGACC-3′ |
| R:
5′-CGGTCCTACACTCAAGGCATG-3′ |
|
miR-29b-3. | F:
5′-TAGCACCATTTGAAATCAGTG-3′ |
| R:
5′-AACACTGATTTCAAATGGTGC-3′ |
| GAPD. | F:
5′-GGGAGCCAAAAGGGTCAT-3′ |
| R:
5′-GAGTCCTTCCACGATACCAA-3′ |
| NFATC. | F:
5′-ATGCAGAGAGAGGCTGCGTTCAG-3′ |
| R:
5′-TCATAATATGTTTTGTATCCAGC-3′ |
Western blot analysis
The brain tissues were added with pre-cooled
Tris-HCl buffer (pH 7.4) at the ratio of 1:3, and made into brain
tissue homogenate with a glass homogenizer in an ice bath, and then
the cells or tissue homogenates were lysed in cold RIPA containing
a protease inhibitor cocktail [50 mM Tris (pH 7.4), 150 mM NaCl, 1%
NP-40, 0.1% SDS, 1 mM PMSF, 1 mM NaF, 1 mM
Na3VO4, 1% protease inhibitor cocktail from
Sigma, which was added right before use], for 30 min. The lysate
was then centrifuged at 4°C for 20 min at 16,000 × g to collect the
supernatant. The protein concentration was determined using the
Pierce bicinchoninic acid assay kit (Beyotime Biotechnology Co.,
Ltd.). Equal amounts of protein (20 μ.l/well) were separated
by 7.5% SDS-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes. The membranes were blocked
with 5% skimmed milk for 1 h and incubated overnight at 4°C with
primary antibodies as follows: Recombinant nuclear factor of
activated T cells c2 (NFATc2) (1:1,000, ab2722; Abcam), p65
(1:1,000, ab16502; Abcam), p-p65 (1:2,000, ab86299; Abcam), IκBα
(1:1,000, ab32518; Abcam), and p-IκBα (1:10,000, ab133462; Abcam).
The membranes were then incubated with secondary antibody IgG
(1:2,000, ab205718; Abcam). The gray value of the target band was
analyzed using ImageJ software (Rawak Software, Inc.) with β-actin
as the internal reference.
Bioinformatics analysis
The target binding bite between miR-145-3p and
NFATc2 was analyzed and predicted through Targetscan (http://www.targetscan.org/) (22).
Dual-luciferase reporter gene assay
The NFATc2 fragment containing the miR-145-3p
binding site was cloned into the pmirGLO vector (Promega).
Subsequently, pmirGLO-N FATc2-wild-type (N FATc2-WT) and
pmirGLO-NFATc2-mutant-type (NFATc2-MUT) plasmids were constructed.
The constructed vectors were transfected into 293T cells, and then
the cells were transfected with miR-145-3p mimic or miR-NC.
Luciferase activity was detected 48 h after transfection using the
dual-luciferase reporter gene assay system (Promega), and the
relative activity was calculated as the ratio of firefly luciferase
activity to that of Renill. luciferase activity.
Statistical analysis
Data analysis was performed using SPSS 21.0 (IBM
Corp.) and GraphPad Prism 8.0 (GraphPad Software Inc.). The
Shapiro-Wilk test confirmed that the data were normally
distributed. Data are expressed as mean ± standard deviation. The
unpaired t-test was performed to compare analyses between the two
groups. One-way analysis of variance (ANOVA) or two-way ANOVA was
performed for comparisons among multiple groups, and Tukey's
multiple comparison test was used as the post hoc test after ANOVA.
The P-value was obtained from the two-tailed test and P<0.05
indicated statistical significance.
Results
Propofol reduced brain injury in TBI
rats, inhibited IL-17 expression, and maintained Th17/Treg
balance
The neurological score of the rats in the TBI+P
group was significantly lower than that in the TBI group (Fig. 1A). The brain water content of
rats after TBI was increased markedly, and propofol decreased the
brain water content (Fig. 1B).
The results of H&E and TUNEL staining showed that propofol
notably relieved brain injury and reduced the neuronal apoptosis
rate (Fig. 1C). IL-17 expression
in rat peripheral blood was detected using ELISA, and the ratio of
Th17/Treg cells was measured using flow cytometry. Compared with
the TBI group, the TBI+P group had significantly reduced IL-17
expression (Fig. 1D), Th17/Treg
cell ratio (Fig. 1E) and
inflammation in the rats' brains (Fig. 1F) (all P<0.05).
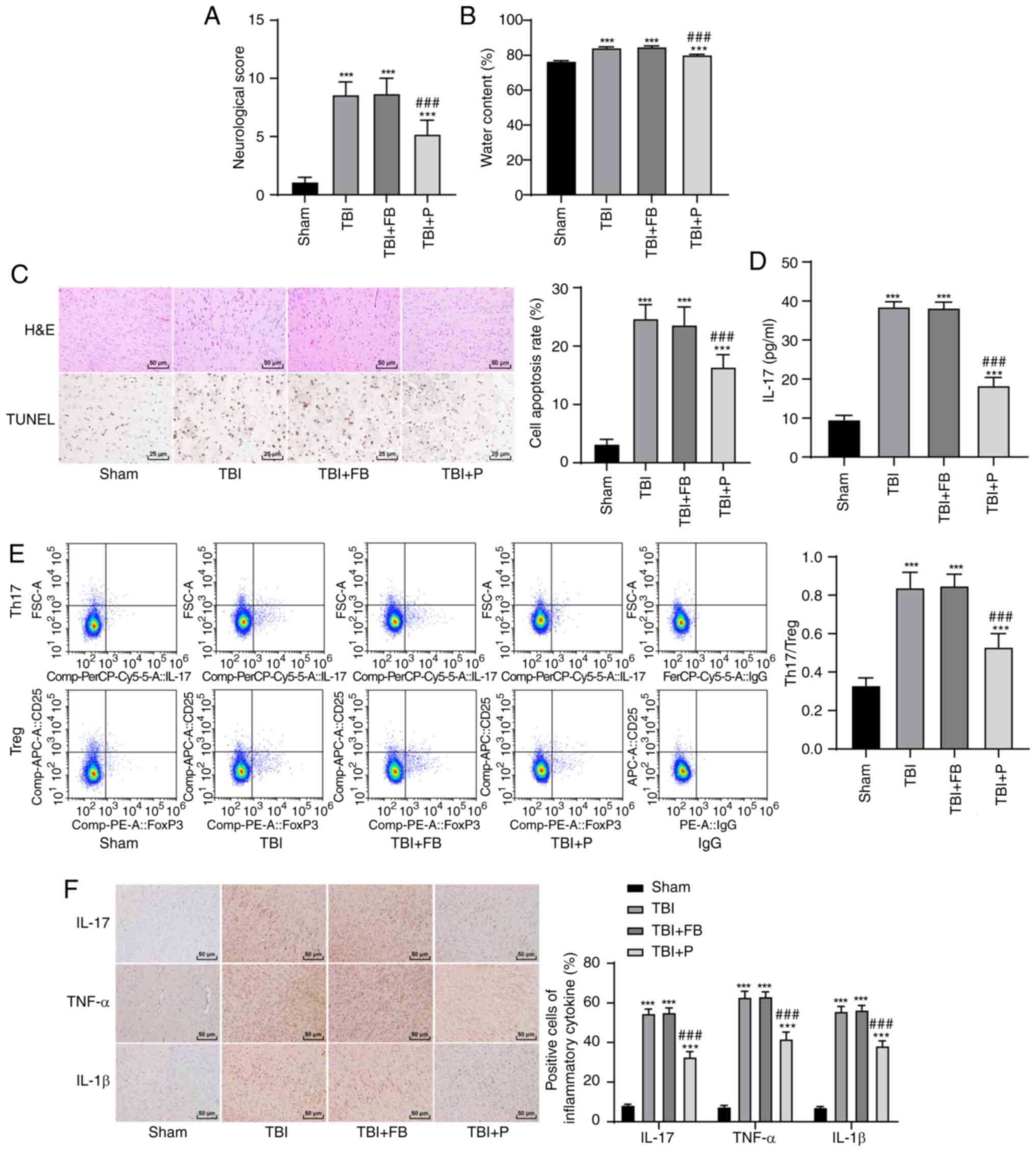 | Figure 1Propofol reduced brain injury in rats
with TBI, inhibited IL-17 expression, and maintained Th17/Treg
balance. (A) Neurological score, n=18; (B) brain water content,
n=6; (C) brain tissue sections of rats with TBI were stained with
H&E and TUNEL, and the apoptotic rate was expressed using the
TUNEL-positive cell rate, n=6; (D) IL-17 expression in the rats'
peripheral blood was detected using ELISA, n=18; (E) the ratio of
Th17/Treg cells in the rats' peripheral blood was detected by flow
cytometry, n=18; (F) the cells that were positive for inflammatory
factors in the rats' brains were detected using
immunohistochemistry, n=6. Each experiment was repeated three
times. Data were analyzed using one-way ANOVA, followed by Tukey's
multiple comparisons test. ***Compared with the sham
group, P<0.001; ###Compared with the TBI group,
P<0.001. |
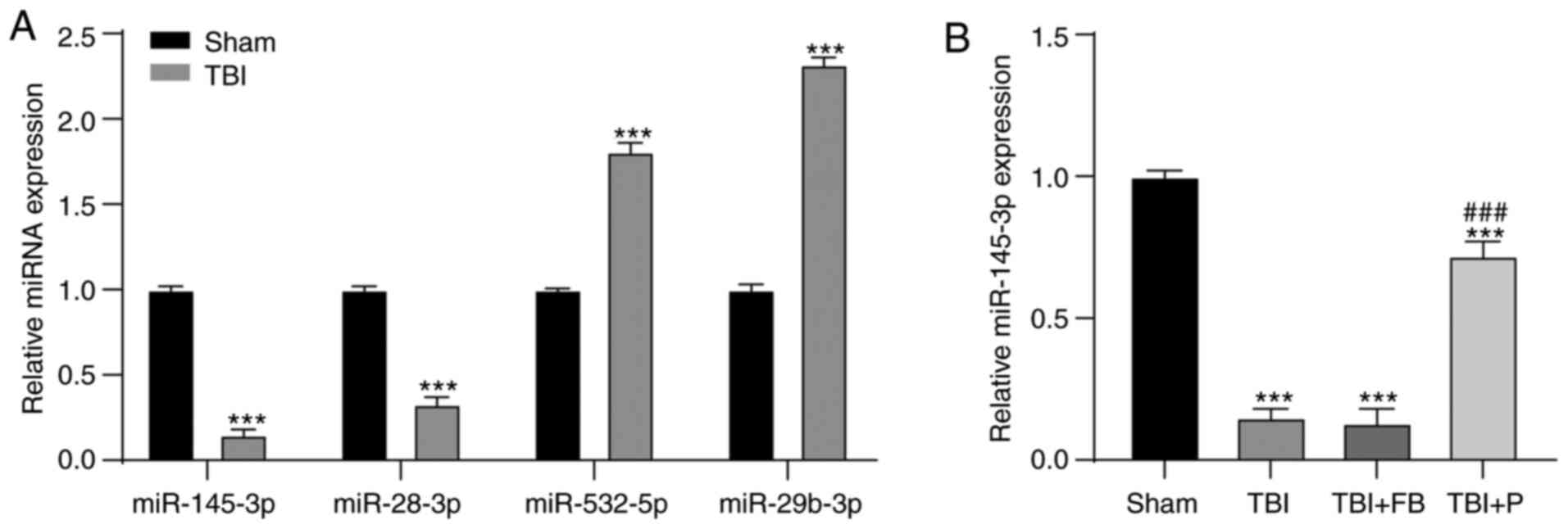
Propofol upregulated miR-145-3p
expression in the brain of rats with TBI
It has been reported that miR is differentially
expressed in the plasma of rats with TBI, and miR-145-3p expression
is decreased after TBI (23). We
used RT-qPCR to verify some differentially expressed miRs mentioned
in the literature and found that the expression of miR-145-3p was
significantly reduced after TBI (Fig. 2A), which has been reported to
affect the response of Th17 cells (24). Our study demonstrated that
propofol upregulated miR-145-3p expression in the brain of rats
with TBI (Fig. 2B) (all
P<0.05). Therefore, we focused on miR-145-3p.
Inhibition of miR-145-3p reversed the
effect of propofol on brain injury
To verify that the effect of propofol on rats with
TBI was realized through the upregulation of miR-145-3p, we
injected ant-145 into the lateral ventricles of the rats (Fig. 3A). Subsequently, we found that
the inhibition of miR-145-3p reversed the ameliorative effect of
propofol on brain injury. The neurological score, brain water
content, neuronal apoptosis rate, IL-17 expression in peripheral
blood, Th17/Treg cell ratio, and inflammatory factor-positive cell
rates were significantly higher in the TBI+P+ant-145 group than in
the TBI+P group (Fig. 3B-G) (all
P<0.05). This indicated that the inhibition of miR-145-3p
reversed the effect of propofol on brain injury.
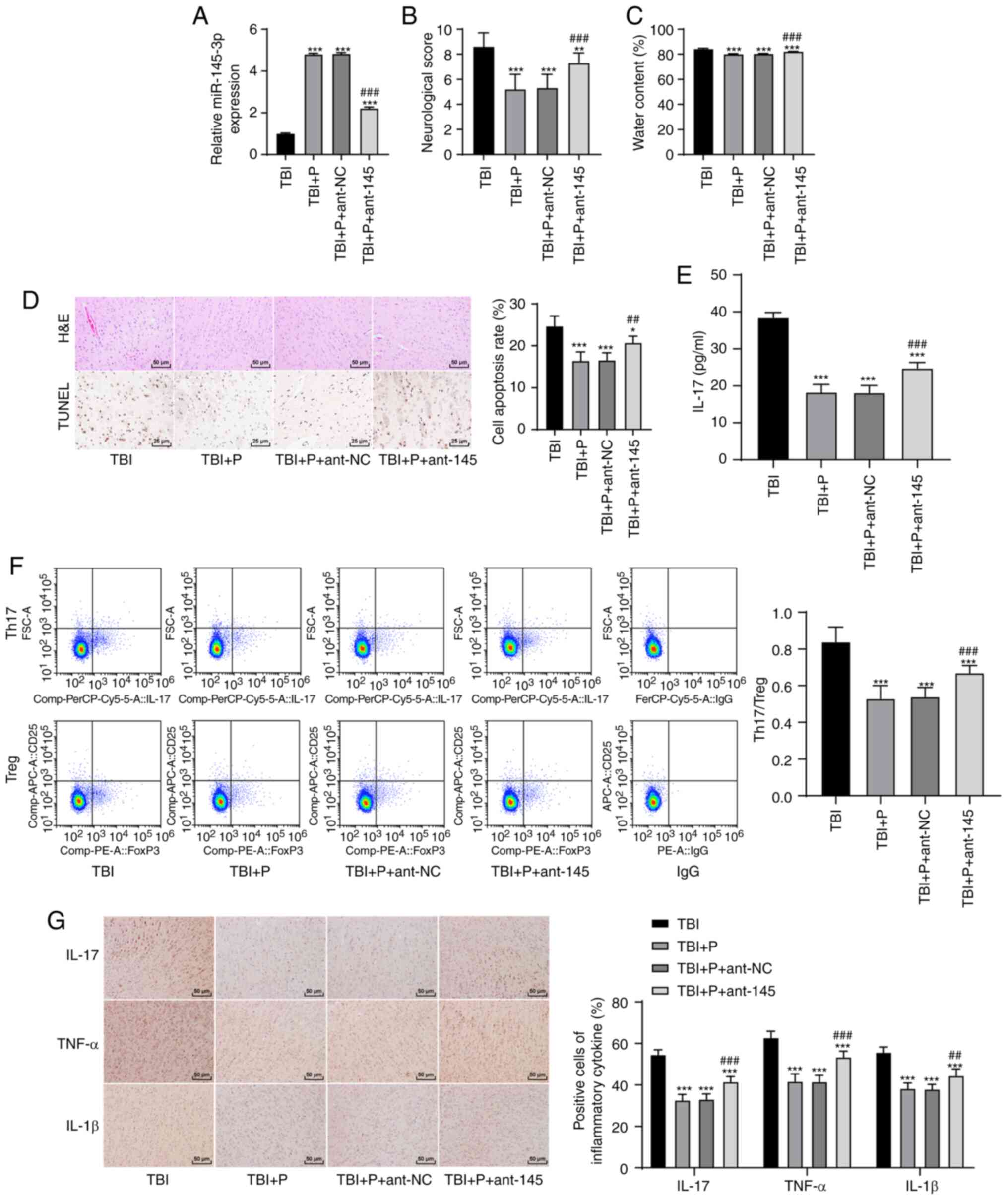 | Figure 3Inhibition of miR-145-3p reversed the
effect of propofol on brain injury. (A) Expression of miR-145-3p in
brain tissues of rats after injection of antagomiR into lateral
ventricle was detected using RT-qPCR; (B) neurological score, n=18;
(C) brain water content, n=6; (D) brain tissue sections of rats
with TBI were stained with H&E and TUNEL, and the apoptotic
rate was expressed using the TUNEL-positive cell rate, n=6; (E)
IL-17 expression in the rats' peripheral blood was detected using
ELISA, n=18; (F) the ratio of Th17/Treg cells in the rats'
peripheral blood was detected by flow cytometry, n=18; (G) the
cells that were positive for inflammatory factors in the rats'
brains were detected using immunohistochemistry, n=6. Each
experiment was repeated three times. Data were analyzed using
one-way ANOVA, followed by Tukey's multiple comparisons test.
*P<0.05, **P<0.01,
***P<0.001, compared with the TBI group;
##P<0.01, ###P<0.001, compared with the
TBI+P group. |
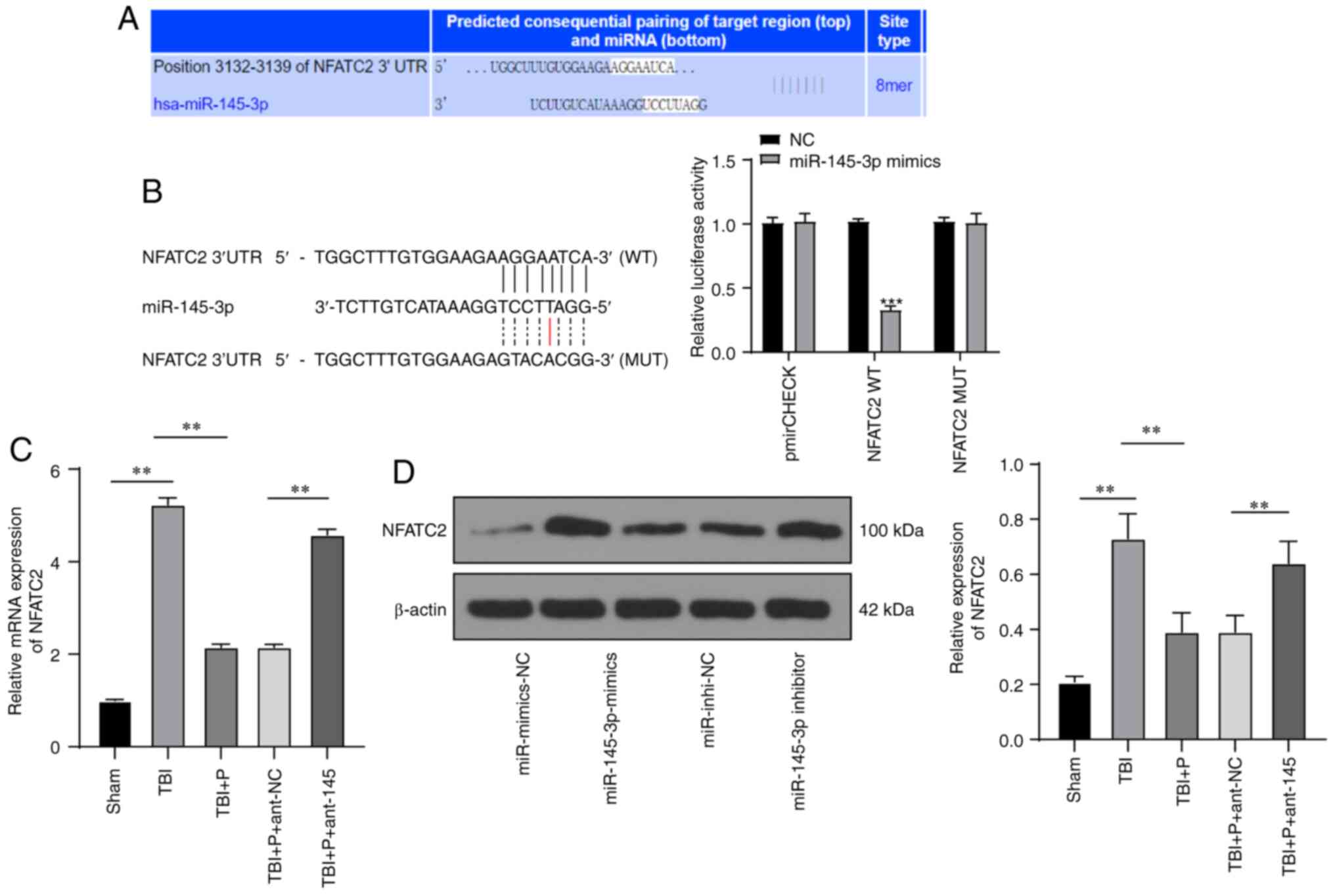
miR-145-3p inhibited NFATc2
expression
Through analysis and prediction using the
bioinformatics website (http://www.targetscan.org/), we found that miR-145-3p
targeted NFATc2 (Fig. 4A)
(22), and the dual-luciferase
reporter gene assay confirmed this targeted relationship (Fig. 4B). NFATc2 expression in the brain
tissues of rats in different treatment groups was detected using
RT-qPCR and western blot analysis. The results demonstrated that
compared with those in the sham group, the rat brain tissues in the
TBI group showed increased NFATc2 mRNA expression and protein
level; the upregulation of miR-145-3p induced by propofol reduced
the NFATc2 mRNA expression and protein level, while the inhibition
of miR-145-3p partially reversed NFATc2 mRNA expression and protein
level (Fig. 4C and D) (all
P<0.05). These results suggested that miR-145-3p negatively
regulated the expression of NFATc2.
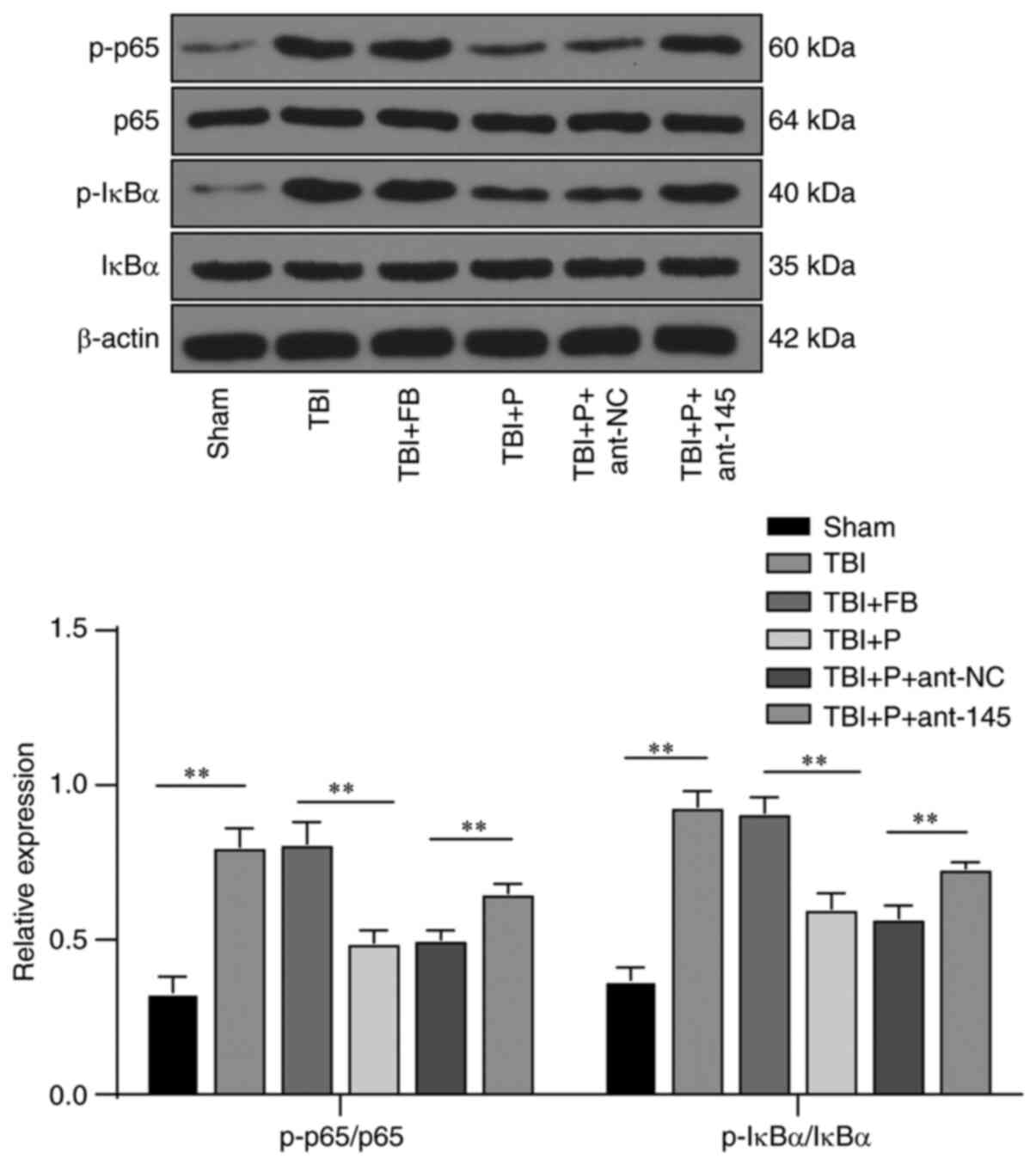
Propofol inhibited brain inflammation in
rats with TBI via the miR-145-3p/NFATC2/NF-κB pathway
The NF-κB pathway is a classic inflammation pathway.
We observed the inhibitory effect of propofol on inflammation in
rats with TBI and found that propofol exerted therapeutic effects
by upregulating miR-145-3p expression. We speculated that the
downstream effects of miR-145-3p may be associated with the NF-κB
pathway. Phosphorylation levels of NF-κB pathway-related proteins
(p-p65 and p-IκBα) were detected and found to be significantly
increased in rats with TBI compared with sham rats. Propofol
inhibited the phosphorylation levels of these proteins, while
inhibition of miR-145-3p notably increased their phosphorylation
levels (Fig. 5) (all P<0.01).
It was suggested that propofol inhibited brain inflammation in rats
with TBI via the miR-145-3p/NFATC2/NF-κB pathway.
Discussion
After the primary mechanical insult, TBI causes
secondary injury through the activation of pro-inflammatory
factors, loss of neuronal and cerebral edema, resulting in
neurological dysfunction (25).
Inflammation has been demonstrated to play a vital role in the
progression of secondary brain injury (26). Propofol is a clinical anesthetic
commonly used to treat patients with TBI (27). Although propofol has been
reported to inhibit the inflammatory response (28), research on the underlying
mechanism of propofol in TBI has been less encouraging. In this
study, we demonstrated that propofol inhibited brain inflammation
in rats with TBI via the miR-145-3p/NFATc2/NF-κB axis, thereby
alleviating brain injury in rats with TBI.
First, the TBI model was established in rats. The
neuroinflammatory response after TBI represents a critical
secondary injury factor that can drive sustained neuronal damage
(29). IL-17 is an inflammatory
mediator that influences the pathogenesis of brain injury (30,31). We found that the neurological
score, brain edema, and neuronal apoptosis rate of the
propofol-treated rats with TBI were significantly decreased,
indicating that propofol could relieve brain injury in rats with
TBI. In addition, Th17 cells contribute to autoimmunity and
inflammation, while Treg cells maintain immune homeostasis
(32). An imbalance of Th17/Treg
cells is therefore also associated with autoimmune and inflammatory
diseases (33). To the best of
our knowledge, this is the first study to demonstrate that propofol
inhibits IL-17 expression in rats with TBI and maintains the
balance of Th17/Treg cells. It is worth noting that the expression
of some miRs in the cerebral cortex and hippocampus of rats with
TBI was abnormal, which could accelerate or suppress secondary
brain injury (34). Previous
findings demonstrated that propofol attenuates hypoxia-induced
brain injury by upregulating miR-137 expression (35). miR-145-3p is widely acknowledged
to be a tumor suppressor (36);
however, relatively little is known about the role of miR-145-3p in
TBI. In this study, we found that miR-145-3p expression was notably
decreased in rats with TBI and increased after propofol treatment.
Many miRs have been found to be differentially expressed in rats
with TBI, including miR-145-3p (23). In this study, the inhibition of
miR-145-3p reversed the effect of propofol on brain injury, which
confirmed that the upregulation of miR-145-3p is a vital part of
propofol's protective effect on rats with TBI. Overexpression of
miR-145 has been demonstrated to ameliorate astrocyte injury in
cerebral ischemic stroke by targeting aquaporin 4 (37). Wang et a. have also shown
that lncRNA MALAT1 silencing protects against cerebral
ischemia-reperfusion injury by upregulating miR-145 expression
(38).
The miR-145-3p target gene was then explored. NFAT
is a carcinogenic transcription factor that is induced under
inflammatory conditions (39).
NFATc2, a member of the NFAT family, promotes the generation of
inflammatory cytokines (40). Of
the various miR-149-5p target genes proposed by the bioinformatics
website used, we focused on NFATc2. Gerlach et a. revealed
the key role of NFATc2 in the initiation of inflammation-related
colorectal tumors (41). In this
study, the targeting relationship between miR-145-3p and NFATc2 was
verified using a dual-luciferase reporter gene assay. The
upregulation of miR-145-3p reduced NFATc2 mRNA and protein levels,
suggesting that miR-145-3p reduces the expression of NFATc2.
Intriguingly, Furman showed that blocking NFAT signaling was
conducive to normalizing hippocampal synaptic function in rats with
TBI (42). NFATc has also been
reported to be involved in calcineurin-mediated ischemic brain
injury (43). Subsequently, we
investigated the signaling pathways modulated by miR-145-3p/NFATc2.
NF-κB is a central mediator in inflammatory progression, and the
activation of NF-κB has been deemed a contributor to disease
(44,45). Accumulated evidence has shown
that suppression of the NF-κB signaling pathway could alleviate
early brain injury (46,47). Consistent with those findings,
our data demonstrated that the phosphorylation levels of NF-κB
pathway-related proteins in rats with TBI were significantly
higher, and propofol could inhibit the phosphorylation levels of
these proteins.
In summary, our study demonstrated that propofol
could maintain Th17/Treg cell balance and attenuate inflammation in
rats with TBI by upregulating miR-145-3p expression and
inactivating the NF-κB pathway. Our results provide essential
evidence of the protective mechanism of propofol in brain injury.
Another limitation of this study lies in that the mechanism of
propofol protecting TBI rats from brain injury through the
miR-145-3p/NFATc2 axis lacks of further cell experimental
verification. In future, primary microglia isolated and cultured
from SD rats may be used to carry out further cell experiment in
vitr., and prospective trials are to be conducted to refine our
clinical guidance.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CC and DZ completed the experimental design and were
the major contributors in writing the manuscript. KS, HL, JH and LX
participated in the design and operation of the experiments. GL,
YG, and JH were responsible for literature research and data
collection. JC, GL and LN performed the statistical analysis. YC,
YG and DY completed the data interpretation and revised the
manuscript. WY, PW, and YS ensured the manuscript preparation and
the integrity of project management and confirmed the authenticity
of all original data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was performed with the approval of the
Medical Research of Guangdong Provincial People's Hospital
(Guangdong Academy of Medical Sciences), GDREC2012116H (R1),
January 15, 2016. All procedures were strictly performed according
to the code of ethics. Great efforts were made to minimize pain in
experimental animals.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was supported by the Science and Technology Planning
Project of Guangdong Province, China (2012B031800161), the Medical
Scientific Research Foundation of Guangdong Province, China
(A2016573), the Natural Science Foundation of Guangdong Province,
China (2018A0303130236), the Natural Science Foundation of
Guangdong Province, China (2018A0303130297); the Science and
Technology Program of Guangzhou, China (201904010080), the Project
of Administration of Traditional Chinese Medicine of Guangdong
Province, China (20191006), the Foundation for Basic and Applied
Basic Research of Guangdong Province, China (2019A1515110063), and
the Medical Scientific Research Foundation of Guangdong Province,
China (A2020038).
References
|
1
|
Gardner AJ and Zafonte R:
Neuroepidemiology of traumatic brain injury. Handb Clin Neurol.
138:207–223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Georgiou AP and Manara AR: Role of
therapeutic hypothermia in improving outcome after traumatic brain
injury: A systematic review. Br J Anaesth. 110:357–367. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ziebell JM and Morganti-Kossmann MC:
Involvement of pro- and anti-inflammatory cytokines and chemokines
in the pathophysiology of traumatic brain injury.
Neurotherapeutics. 7:22–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boer C, Franschman G and Loer SA:
Prehospital management of severe traumatic brain injury: Concepts
and ongoing controversies. Curr Opin Anaesthesiol. 25:556–562.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Si L, Wang H and Wang L: Suppression of
miR-193a alleviates neuroinflammation and improves neurological
function recovery after traumatic brain injury (TBI) in mice.
Biochem Biophys Res Commun. 523:527–534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Helmy A, Carpenter KL, Menon DK, Pickard
JD and Hutchinson PJ: The cytokine response to human traumatic
brain injury: Temporal profiles and evidence for cerebral
parenchymal production. J Cereb Blood Flow Metab. 31:658–670. 2011.
View Article : Google Scholar :
|
|
7
|
Li T, Zhang YM, Han D, Hua R, Guo BN, Hu
SQ, Yan XL and Xu T: Involvement of IL-17 in secondary brain injury
after a traumatic brain injury in rats. Neuromolecular Med.
19:541–554. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo T, Ji WJ, Yuan F, Guo ZZ, Li YX, Dong
Y, Ma YQ, Zhou X and Li YM: Th17/Treg imbalance induced by dietary
salt variation indicates inflammation of target organs in humans.
Sci Rep. 6:267672016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen Y, Tang XY, Yang YC, Ke X, Kou W, Pan
CK and Hong SL: Impaired balance of Th17/Treg in patients with
nasal polyposis. Scand J Immunol. 74:176–185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fulton B and Sorkin EM: Propofol. An
overview of its pharmacology and a review of its clinical efficacy
in intensive care sedation. Drugs. 50:636–657. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marik PE: Propofol: An immunomodulating
agent. Pharmacotherapy. 25:28S–33S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng X, Huang H, Liu J, Li M, Liu M and
Luo T: Propofol attenuates inflammatory response in LPS-activated
microglia by regulating the miR-155/SOCS1 pathway. Inflammation.
41:11–19. 2018. View Article : Google Scholar
|
|
13
|
Yu S, Xin W, Jiang Q and Li A: Propofol
exerts neuroprotective functions by down-regulating microRNA-19a in
glutamic acid-induced PC12 cells. Biofactors. 46:934–942. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shioya M, Obayashi S, Tabunoki H, Arima K,
Saito Y, Ishida T and Satoh J: Aberrant microRNA expression in the
brains of neurodegenerative diseases: miR-29a decreased in
Alzheimer disease brains targets neurone navigator 3. Neuropathol
Appl Neurobiol. 36:320–330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Di Pietro V, Yakoub KM, Scarpa U, Di
Pietro C and Belli A: MicroRNA signature of traumatic brain injury:
From the biomarker discovery to the point-of-care. Front Neurol.
9:4292018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Henry RJ, Doran SJ, Barrett JP, Meadows
VE, Sabirzhanov B, Stoica BA, Loane DJ and Faden AI: Inhibition of
miR-155 limits neuroinflammation and improves functional recovery
after experimental traumatic brain injury in mice.
Neurotherapeutics. 16:216–230. 2019. View Article : Google Scholar :
|
|
17
|
Li Z, Wang Y, Zeng G, Zheng X, Wang W,
Ling Y, Tang H and Zhang J: Increased miR-155 and heme oxygenase-1
expression is involved in the protective effects of formononetin in
traumatic brain injury in rats. Am J Transl Res. 9:5653–5661.
2017.
|
|
18
|
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M
and Chopp M: Therapeutic benefit of intravenous administration of
bone marrow stromal cells after cerebral ischemia in rats. Stroke.
32:1005–1011. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miyara M, Yoshioka Y, Kitoh A, Shima T,
Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al:
Functional delineation and differentiation dynamics of human
CD4+ T cells expressing the FoxP3 transcription factor.
Immunity. 30:899–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kustrimovic N, Comi C, Magistrelli L,
Rasini E, Legnaro M, Bombelli R, Aleksic I, Blandini F, Minafra B,
Riboldazzi G, et al: Parkinson's disease patients have a complex
phenotypic and functional Th1 bias: Cross-sectional studies of
CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated
patients. J Neuroinflammation. 15:2052018. View Article : Google Scholar
|
|
21
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar :
|
|
23
|
Wang P, Ma H, Zhang Y, Zeng R, Yu J, Liu
R, Jin X and Zhao Y: Plasma exosome-derived MicroRNAs as novel
biomarkers of traumatic brain injury in rats. Int J Med Sci.
17:437–448. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Zheng S, Xin N, Dou C, Fu L, Zhang
X, Chen J, Zhang Y, Geng D, Xiao C, et al: Identification of novel
MicroRNA signatures linked to experimental autoimmune myasthenia
gravis pathogenesis: Down-regulated miR-145 promotes pathogenetic
Th17 cell response. J Neuroimmune Pharmacol. 8:1287–1302. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu F, Chen MR, Liu J, Zou Y, Wang TY, Zuo
YX and Wang TH: Propofol administration improves neurological
function associated with inhibition of pro-inflammatory cytokines
in adult rats after traumatic brain injury. Neuropeptides. 58:1–6.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Goodman JC, Van M, Gopinath SP and
Robertson CS: Pro-inflammatory and pro-apoptotic elements of the
neuroinflammatory response are activated in traumatic brain injury.
Acta Neurochir Suppl. 102:437–439. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu Y, Jian MY, Wang YZ and Han RQ:
Propofol ameliorates calpain-induced collapsin response mediator
protein-2 proteolysis in traumatic brain injury in rats. Chin Med J
(Engl). 128:919–927. 2015. View Article : Google Scholar
|
|
28
|
Shi SS, Zhang HB, Wang CH, Yang WZ, Liang
RS, Chen Y and Tu XK: Propofol attenuates early brain injury after
subarachnoid hemorrhage in rats. J Mol Neurosci. 57:538–545. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Corrigan F, Mander KA, Leonard AV and Vink
R: Neurogenic inflammation after traumatic brain injury and its
potentiation of classical inflammation. J Neuroinflammation.
13:2642016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuwabara T, Ishikawa F, Kondo M and
Kakiuchi T: The role of IL-17 and related cytokines in inflammatory
autoimmune diseases. Mediators Inflamm. 2017:39080612017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang DD, Zhao YF, Wang GY, Sun B, Kong QF,
Zhao K, Zhang Y, Wang JH, Liu YM, Mu LL, et al: IL-17 potentiates
neuronal injury induced by oxygen-glucose deprivation and affects
neuronal IL-17 receptor expression. J Neuroimmunol. 212:17–25.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee GR: The balance of Th17 versus Treg
cells in autoimmunity. Int J Mol Sci. 19:7302018. View Article : Google Scholar :
|
|
33
|
Guo H, He Z, Li M, Wang T and Zhang L:
Imbalance of peripheral blood Th17 and Treg responses in children
with refractory Mycoplasma pneumoniae pneumonia. J Infect
Chemother. 22:162–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pan YB, Sun ZL and Feng DF: The role of
MicroRNA in traumatic brain injury. Neuroscience. 367:189–199.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chang J, Yan X and Zeng Y: Propofol
weakens hypoxia-aroused apoptosis and autophagy via elevating
microRNA-137 in neurocytes. Exp Mol Pathol. 112:1043272020.
View Article : Google Scholar
|
|
36
|
Mataki H, Seki N, Mizuno K, Kamikawaji K,
Kumamoto T, Koshizuka K, Goto Y and Inoue H: Dual-strand
tumor-suppressor microRNA-145 (miR-145-5p and miR-145-3p)
coordinately targeted MTDH in lung squamous cell carcinoma.
Oncotarget. 7:72084–72098. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng L, Cheng W, Wang X, Yang Z, Zhou X
and Pan C: Overexpression of MicroRNA-145 ameliorates astrocyte
injury by targeting aquaporin 4 in cerebral ischemic stroke. Biomed
Res Int. 2017:95309512017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang H, Zheng X, Jin J, Zheng L, Guan T,
Huo Y, Xie S, Wu Y and Chen W: LncRNA MALAT1 silencing protects
against cerebral ischemia-reperfusion injury through miR-145 to
regulate AQP4. J Biomed Sci. 27:402020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baumgart S, Chen NM, Zhang JS, Billadeau
DD, Gaisina IN, Kozikowski AP, Singh SK, Fink D, Ströbel P, Klindt
C, et al: GSK-3β Governs Inflammation-Induced NFATc2 Signaling Hubs
to Promote Pancreatic Cancer Progression. Mol Cancer Ther.
15:491–502. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qi H, Yang Z, Dai C, Wang R, Ke X, Zhang
S, Xiang X, Chen K, Li C, Luo J, et al: STAT3 activates
MSK1-mediated histone H3 phosphorylation to promote NFAT signaling
in gastric carcinogenesis. Oncogenesis. 9:152020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gerlach K, Daniel C, Lehr HA, Nikolaev A,
Gerlach T, Atreya R, Rose-John S, Neurath MF and Weigmann B:
Transcription factor NFATc2 controls the emergence of colon cancer
associated with IL-6-dependent colitis. Cancer Res. 72:4340–4350.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Furman JL, Sompol P, Kraner SD, Pleiss MM,
Putman EJ, Dunkerson J, Mohmmad Abdul H, Roberts KN, Scheff SW and
Norris CM: Blockade of astrocytic Calcineurin/NFAT signaling helps
to normalize hippocampal synaptic function and plasticity in a rat
model of traumatic brain injury. J Neurosci. 36:1502–1515. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang YJ, Mei HS, Wang C, Wang YL and
Zhang YJ: Involvement of nuclear factor of activated T-cells
(NFATc) in calcineurin-mediated ischemic brain damage in vivo. Yao
Xue Xue Bao. 40:299–305. 2005.PubMed/NCBI
|
|
44
|
Choi MC, Jo J, Park J, Kang HK and Park Y:
NF-κB signaling pathways in osteoarthritic cartilage destruction.
Cells. 8:7342019. View Article : Google Scholar
|
|
45
|
DiDonato JA, Mercurio F and Karin M: NF-κB
and the link between inflammation and cancer. Immunol Rev.
246:379–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qu J, Zhao H, Li Q, Pan P, Ma K, Liu X,
Feng H and Chen Y: MST1 suppression reduces early brain injury by
inhibiting the NF-κB/MMP-9 pathway after subarachnoid hemorrhage in
mice. Behav Neurol. 2018:64709572018. View Article : Google Scholar
|
|
47
|
Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang
S, Deng X, Xie Z and Zheng S: Isoliquiritigenin alleviates early
brain injury after experimental intracerebral hemorrhage via
suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome
activation by promoting Nrf2 antioxidant pathway. J
Neuroinflammation. 14:1192017. View Article : Google Scholar
|