Introduction
Skeletal muscle atrophy is a debilitating response
to chronic infection and other systemic diseases, including
acquired immunodeficiency syndrome, tuberculosis, cancer, chronic
obstructive pulmonary disease and chronic kidney disease (1). Skeletal muscle atrophy is
characterized by muscle wasting and partial or complete loss of
muscle function, which becomes more pronounced as the disease
progresses (1-3). Several categories of drugs,
including statins, antiviral therapies and immunosuppressants
(e.g., glucocorticoids), cause muscle atrophy (4,5).
Skeletal muscle atrophy severely affects quality of life, and
current therapies display limited effects (6). Therefore, investigating the
pathogenic mechanism underlying muscle atrophy and identifying
effective therapeutic targets is of significant importance.
Jagoe et al (7,8)
confirmed that under atrophic conditions, protein degradation rates
in muscles were increased primarily via activation of the atrophy
program. Muscle-specific expression of atrogin1 and muscle
RING-finger protein-1 (MURF1) is dramatically induced during
atrophy (9,10). Additionally, the levels of
several proteins are often markedly decreased in atrophying
muscles, including myoblast determination protein 1 (MyoD), myosin
heavy chain (MyHC) and mTOR, which have been reported to serve
critical roles in muscle metabolism and are used as biomarkers
(11-13).
Forkhead box O (FoxO) genes belong to the forkhead
box gene family of transcription factors that contain the forkhead
domain (14). In mammals, four
FoxO genes have been identified: FoxO1, FoxO3a, FoxO4 and FoxO6
(15-17). Previous studies have demonstrated
that FoxO genes are associated with cellular processes, including
metabolism, the cell cycle, apoptosis and cellular homeostasis, and
mediate cell responses to oxidative stress and antitumor drug
treatment (14-18). The roles of other members of the
FoxO family, including FoxO1, FoxO3 and FoxO4, in the regulation of
skeletal muscle mass have been demonstrated in several studies
(19,20). FoxO1 or FoxO3a overexpression in
muscle is sufficient to induce skeletal muscle atrophy in
vivo (21-24). Moreover, FoxO4 is necessary for
TNF-induced atrogin1 expression (25).
FoxO6 is the most recently identified FoxO-encoding
gene (17). In mammals, FoxO6
was initially observed in the central nervous system and was then
reported to be ubiquitously expressed in various tissues, with
higher expression levels in tissues undergoing oxidative stress
(17). In the hippocampus, FoxO6
is negatively regulated by insulin/insulin like growth factor 1
signaling via the PI3K/AKT signaling pathway (26,27). Additionally, in muscles
undergoing oxidative stress, FoxO6 can form a regulatory loop with
peroxisome proliferator-activated receptor γ coactivator 1-α
(PGC-1α) to establish the level of oxidative metabolism (28). In 2018, Sun et al
(29) reported that FoxO6
controls the growth of the craniofacial complex via the Hippo
signaling pathway. However, the effects of FoxO6 on skeletal muscle
atrophy are not completely understood.
The present study investigated the role of FoxO6 in
maintaining the proliferation and differentiation of skeletal
muscle, as well as the underlying mechanism, in vitro and in
vivo.
Materials and methods
Reagents and antibodies
The primary antibodies targeted against mTOR (cat.
no. 2983) and β-actin (cat. no. 4970) were purchased from Cell
Signaling Technology, Inc. The primary antibodies targeted against
FoxO6 (cat. no. 19122-1-AP) and MURF1 (cat. no. 55456-1-AP) were
purchased from ProteinTech Group, Inc. The primary antibodies
targeted against atrogin1 (cat. no. ab168372), MyoD (cat. no.
ab203383), FoxO3a (cat. no. ab70315), FoxO1 (cat. no. ab52857) and
MyHC (cat. no. ab11083) were purchased from Abcam. Anti-mouse (cat.
no. 7076) and anti-rabbit (cat. no. 7074) IgG HRP-conjugated
antibodies were purchased from Cell Signaling Technology, Inc.
TNF-α (cat. no. AF-315-01A) was acquired from PeproTech EC Ltd.
Phalloidin (cat. no. P5282) was purchased from Sigma-Aldrich (Merck
KGaA). Goat anti-mouse IgG (H+L) Cross-Adsorbed secondary antibody,
Alexa Fluor (488; cat. no. A11001) were purchased from Thermo
Fisher Scientific, Inc. Cell Counting Kit-8 (cat. no. CK04) and
DAPI (cat. no. D212) were purchased from Dojindo Molecular
Technologies, Inc. Horse serum (cat. no. BL209A; Beijing Lanjieke
Technology Co., Ltd.). Control small interfering RNA [si;
si-negative control (NC)] and FoxO6-specific siRNAs were purchased
from Beijing Viewsolid Biotech Co., Ltd. GenMute siRNA transfection
reagent (cat. no. SL100568) was from SignaGen Laboratories.
Cell line and culture
The murine myoblast cell line
C2C12 (The Cell Bank of Type Culture
Collection of Chinese Academy of Sciences) was used within the
first 10 passages. Cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Biowest), 100
U/ml penicillin and 100 uU/ml streptomycin in a humidified
atmosphere with 5% CO2 at 37°C. To obtain
C2C12 myotubes, C2C12
cells were differentiated in DMEM/2% horse serum for 10 days in a
humidified atmosphere with 5% CO2 at 37°C, changing the
medium every 2 days. The mouse hepatocyte cell line AML12 was
purchased from The Cell Bank of Type Culture Collection of Chinese
Academy of Sciences and cultured in DMEM supplemented with 10% FBS,
40 ng/ml dexamethasone (cat. no. BS134A; Biosharp Life Sciences),
and 1% insulin, transferrin and selenium (Invitrogen; Thermo Fisher
Scientific, Inc.) in a humidified atmosphere with 5% CO2
at 37°C. For the muscle atrophy model in vitro,
C2C12 myotubes were treated with TNF-α (50
ng/ml) for 6 h with 5% CO2 at 37°C.
Cell proliferation assay
Relative cell proliferation of si-FoxO6-treated
C2C12 cells was determined using the CCK-8
kit. Cells were seeded (3×103/well) into 96-well plates
and transfection with si-NC (50 nM) or si-FoxO6-3 (50 nM) was
performed for 24, 48, 72, 96 or 120 h at room temperature using
GenMute siRNA transfection reagent according to the manufacturer's
protocol. Following removal of the culture medium, 100 µl
medium and 10 µl CCK-8 reagent were added to each well and
incubated for 3 h at 37°C in the dark. Absorbance was measured at a
wavelength of 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.). The assay was performed in triplicate.
Western blotting
Mouse muscle tissues were ground in liquid nitrogen.
Total protein was extracted from tissues and cells using M-PER
Mammalian Protein Extraction Reagent (cat. no. 78503; Pierce;
Thermo Fisher Scientific, Inc.). For mouse muscle tissues, 1% SDS
lysis reagent (cat. P0013G; Beyotime Institute of Biotechnology)
was also used for protein extraction. Protein concentrations of
cell and tissue lysates were quantified using a Qubit Protein Assay
kit (cat. no. 2157145; Invitrogen; Thermo Fisher Scientific, Inc.)
and a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Equal amounts of
protein (30 µg) were separated via 8 or 10% SDS-PAGE and
transferred to PVDF membranes (EMD Millipore). Following blocking
5% skimmed milk at 37°C for 2 h, the membranes were incubated
overnight at 4°C with the following primary antibodies: Anti-MyoD
(1:800), anti-β-actin (1:1,000), anti-MyHC (1:1,000), anti-mTOR
(1:2,000), anti-FoxO6 (1:1,000), anti-FoxO3a (1:2,000), anti-FoxO1
(1:1,000), anti-MURF1 (1:1,000) and anti-atrogin1 (1:2,000). After
washing in TBST (1% Tween-20), the membranes were incubated with
secondary antibodies (1:2,000) at 37°C for 2 h. Protein bands were
visualized using enhanced chemiluminescence reagents (Amersham;
Cytiva). β-actin was used as the loading control.
FoxO6 knockdown
C2C12 cells were plated at a
density of 1.5×105/well and grown overnight.
Subsequently, cells were transfected with 50 nM si-FOXO6-1,
si-FOXO6-2, si-FOXO6-3 or si-NC using GenMute transfection reagent
(cat. no. SL100568) at room temperature according to the
manufacturer's protocol. At 48 h post-transfection, cells were
lysed using TRIzol® reagent for RNA extraction
(Invitrogen; Thermo Fisher Scientific, Inc.) and M-PER Mammalian
Protein Extraction Reagent (cat. no. 78503; Pierce; Thermo Fisher
Scientific, Inc.) for protein extraction. Subsequently,
transfection efficiency was assessed via reverse
transcription-quantitative PCR (RT-qPCR) and western blotting.
siRNA oligomers were designed and synthesized by Beijing Viewsolid
Biotech Co., Ltd (China), as follows: si-FoxO6-1 forward, 5′-GGU
CGG ACC CUU GCG GAA ATT dTd T-3′ and reverse, 3′-dTd TUU UCC GCA
AGG GUC CGA CCT T-5′; si-FoxO6-2 forward, 5′-GGC ACU GGC AAG AGU
UCA UTT dTd T-3′ and reverse, 3′-dTd TAU GAA CUC UUG CCA GUG CCT
T-5′; and si-FoxO6-3 forward, 5′CCA UCA UCC UCA ACG ACU UTT dTd
T-3′ and reverse, 3′-dTd TAA GUC GUU GAG GAU GAU GGT T-5′. The
scrambled NC siRNA (si-NC) was purchased from Beijing Viewsolid
Biotech Co., Ltd.
RT-qPCR
Total RNA from cells and tissues was extracted using
TRIzol. RT-qPCR was performed according to the manufacturer's
protocol. Briefly, total RNA was reverse transcribed into cDNA
using PrimeScript™ RT Master Mix (Perfect Real Time; cat. no.
RR036A; Takara Bio, Inc.). Subsequently, qPCR was performed using
SYBR Premix Ex Taq II (cat. no. RR820A; Takara Bio, Inc.) and a CFX
Connect instrument (Bio-Rad Laboratories, Inc.). The following
primers were used for qPCR: Mouse FoxO6 forward, 5′-CAG CAA CCC TCT
TCG TTC ACA-3′ and reverse, 5′-CAG GAC TGG TTA AGA TGG GAG ACT-3′
(101 bp); and mouse β-actin forward, 5′-AGA TTA CTG CTC TGG CTC CTA
GC-3′ and reverse, 5′-ACT CAT CGT ACT CCT GCT TGC T-3′ (147 bp).
The following thermocycling conditions were used for qPCR: 95°C for
30 sec; 40 cycles of 5 sec at 95°C and 30 sec at 56°C; and melting
curve analysis. mRNA expression levels were quantified using the
2−ΔΔCq method (30)
and normalized to the internal reference gene β-actin using CFX
Connect instrument software (CFX Maestro 2.0; Bio-Rad Laboratories,
Inc.). RT-qPCR was performed in duplicate.
Myotube diameter assay
At 48 h post-transfection,
C2C12 cells were treated with differentiation
medium for 10 days. The differentiation medium was changed every 2
days. Subsequently, myotubes were fixed in 4% polyformaldehyde at
room temperature for 30 min, then stained with 50 µg/ml
fluorescent phalloidin conjugate solution in PBS for 40 min at room
temperature. Stained myotubes were observed using a Ti2 inverted
fluorescence microscope (Nikon Corporation; magnification, ×20) and
analyzed using NIS-Elements BR Analysis software (version 5.20.02;
Nikon Instruments, Inc.).
Immunofluorescence staining
C2C12 myotubes cultured in
6-well plates (1.5×105/well) were fixed with 4%
paraformaldehyde at room temperature for 20 min, permeabilised with
0.5% Triton X-100 for 20 min at 37°C and blocked in PBS buffer
containing 5% BSA (cat. no. A8020; Beijing Solarbio Science &
Technology Co., Ltd.) at 4°C overnight. Cells were incubated with
anti-MyHC (1:4,000) overnight at 4°C. Subsequently, cells were
incubated with a goat anti-mouse fluorescein-conjugated Alexa Fluor
488 antibody (1:1,000) for 1.5 h at room temperature. Cell nuclei
were stained with DAPI for 10 min. Stained cells were visualized
using a Ti2 inverted fluorescence microscope (Nikon Corporation;
magnification, ×20) and analyzed using NIS-Elements BR Analysis
software (version 5.20.02; Nikon Instruments, Inc.).
Animals
A total of 16 6-week-old healthy male C57BL/6J mice
(weight, 16-18 g) and five 8-week-old (weight, 20-22 g) healthy
male C57BL/6J mice were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. All animals were subjected
to equivalent feeding conditions. Mice were housed at a constant
temperature (22-24°C) with humidity (50-60%), 12-h light/dark
cycles and ad libitum access to water and food. In the
present study, for the measurement of FoxO6 mRNA and protein
expression levels, the 8-week-old healthy male C57BL/6J mice were
used to obtain liver, heart, lung, colon and skeletal muscle
tissues, respectively. The 16 6-week-old healthy male C57BL/6J mice
were used for the AAV9-siRNA knockdown experiment. According to the
siRNA knockdown efficiency in vitro, adeno-associated virus
9 (AAV9)-shFoxO6 (based on the si-FOXO6-3 sequence) was prepared.
Control AAV9 (shRNA-scramble) and AAV9-shFoxO6 particles were
packaged, purified and titrated by ViGene Biosciences, Inc. For
preliminary experiments, six six-week-old healthy male C57BL/6J
mice were used to verify the efficiency of AAV9-shFoxO6
transfection into skeletal muscles (n=3 per group; control AAV9=3
and AAV9-shFoxO6=3). For subsequent experiments, 50 µl AAV9
(1×1011 genome copies) carrying control AAV9 (n=5) or
AAV9-shFoxO6 (n=5) was injected into the skeletal muscles. After 4
weeks, except for slightly reduced activity of AAV9-shFoxO6 mice,
no obvious abnormalities, such as diet or drinking water, were
observed. Subsequently, AAV9-shFoxO6 mice and control AAV9 mice
were euthanatized by the intraperitoneal injection of 150 mg/kg
sodium pentobarbital. Skeletal muscles were collected and stored at
-80°C until further analysis.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 6; GraphPad Software, Inc.). Data are
presented as the mean ± SEM from three independent experiments.
SPSS (version 21.0; IBM Corp.) software was used to assess the
normal distribution and homogeneity of variances of the data using
the Shapiro-Wilk test and the Levene test. Comparisons between two
groups were analyzed using the unpaired Student's t-test.
Comparisons between multiple groups were analyzed using one-way
ANOVA followed by Dunnett's post-hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
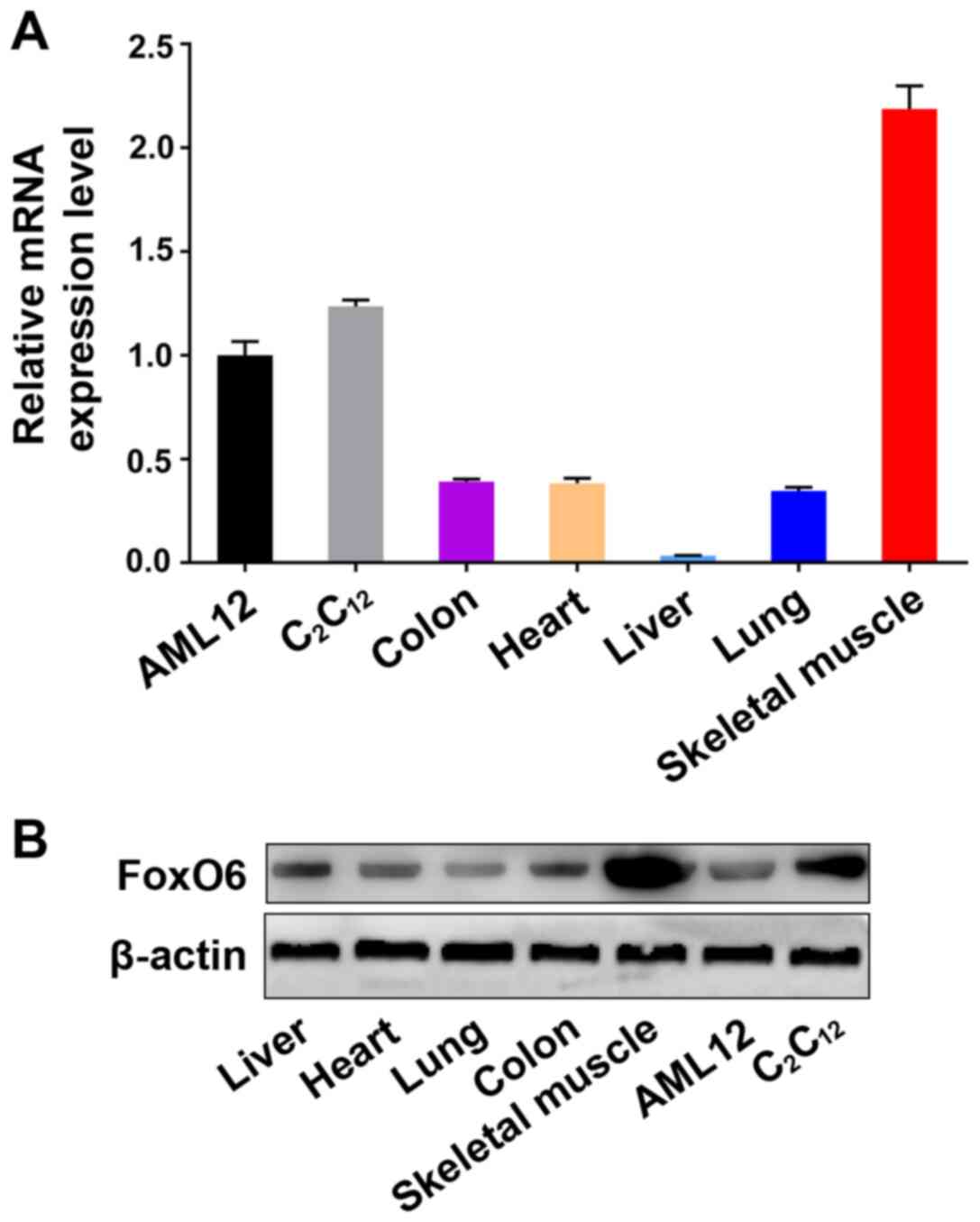
FoxO6 is highly expressed in skeletal
muscle cells
Among the FoxO family members, FoxO1, FoxO3a, and
FoxO4 are expressed in almost all tissues (14-16). The present study measured FoxO6
mRNA and protein expression levels in the major tissues of five
8-week-old (weight, 20-22 g) healthy male C57BL/6J mice and in cell
lines by performing RT-qPCR and western blotting, respectively. The
investigated samples included the following: Mouse liver, mouse
heart, mouse lung, mouse colon and mouse skeletal muscle obtained
from healthy mice, C2C12 cells and AML12
cells. FoxO6 was expressed in the cell lines and tissues,
especially in mouse skeletal muscle tissues and the
C2C12 myoblast cell line (Fig. 1), which suggested that FoxO6
might be essential for maintaining the structure and function of
muscle.
Efficient FoxO6 knockdown using
siRNA
To determine whether FoxO6 served a key role in
muscle metabolism, three siRNA oligomer screening assays in
C2C12 myoblast cells were designed as
follows: si-FoxO6-1 forward, 5′-GGU CGG ACC CUU GCG GAA ATT dTd
T-3′ and reverse, 3′-dTd TUU UCC GCA AGG GUC CGA CCT T-5′;
si-FoxO6-2 forward, 5′-GGC ACU GGC AAG AGU UCA UTT dTd T-3′ and
reverse, 3′-dTd TAU GAA CUC UUG CCA GUG CCT T-5′; and si-FoxO6-3
forward, 5′CCA UCA UCC UCA ACG ACU UTT dTd T-3′ and reverse, 3′-dTd
TAA GUC GUU GAG GAU GAU GGT T-5′. To identify the most efficient
si-FoxO6 oligomer, in the initial screen,
C2C12 cells were transfected with one of the
three si-FoxO6 oligomers or the si-NC oligomer for 48 h.
Subsequently, cells were lysed, and then RNA and protein expression
levels were determined via RT-qPCR and western blotting,
respectively. According to the RT-qPCR results, the oligomer that
most significantly knocked down FoxO6 expression levels compared
with the si-NC group was si-FoxO6-3, and similar results were
obtained by western blotting (Fig.
S1). Therefore, si-FoxO6-3 was used for subsequent
experiments.
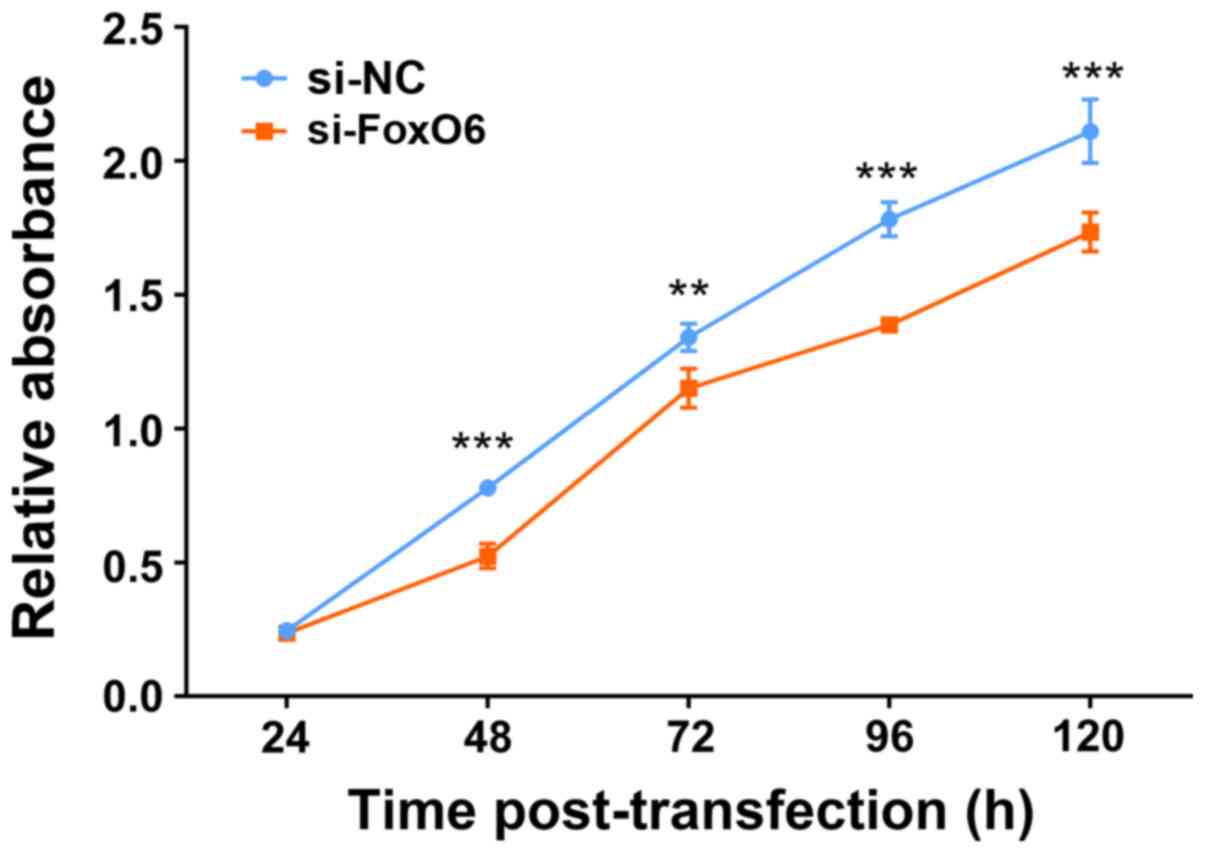
C2C12 myoblast cell
proliferation is decreased by FoxO6 knockdown
The aforementioned results suggested that high FoxO6
expression in mouse skeletal muscle and C2C12
myoblast cells might be related to the structure and function of
muscle. To determine whether FoxO6 was necessary for myoblast cell
proliferation, si-FoxO6 oligomer was transfected into
C2C12 myoblast cells to assess the effect of
FoxO6 knockdown on cell proliferation by performing the CCK-8
assay. The results demonstrated that compared with the si-NC group,
cell proliferation was significantly inhibited by FoxO6 knockdown
at 48, 72, 96 and 120 h post-transfection (Fig. 2).
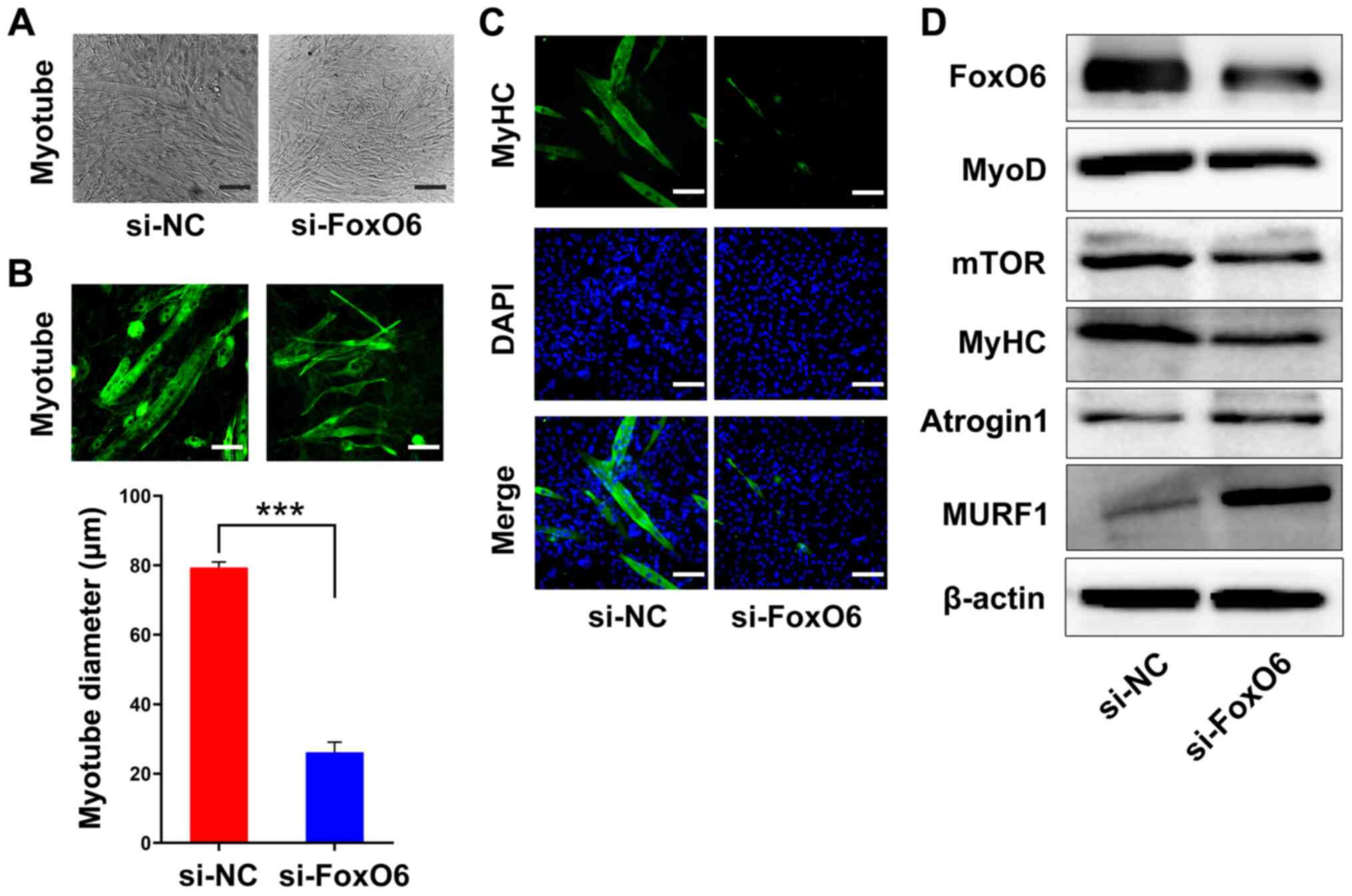
FoxO6 knockdown inhibits
C2C12 myotube differentiation
The results demonstrated that FoxO6 was highly
expressed in muscle at the RNA and protein levels, and FoxO6
knockdown significantly inhibited C2C12 cell
proliferation compared with the si-NC group. Moreover, the results
indicated that FoxO6 knockdown inhibited myotube differentiation,
as morphologically demonstrated by the significantly decreased
diameter of differentiated C2C12 myotubes in
the si-FoxO6 group compared with the si-NC group (Fig. 3A and B). In addition, the
immunofluorescence assay results indicated that FoxO6 knockdown
notably decreased the number of myotubes expressing MyHC compared
with the si-NC group (Figs. 3C
and S2).
Furthermore, to investigate the possible mechanism
underlying FoxO6-maintained myotube activity, the protein
expression levels of several critical biomarkers involved in muscle
metabolism, including MyoD, MyHC, mTOR, atrogin1 and MURF1, were
determined via western blotting. In differentiated
C2C12 myotubes, FoxO6 was knocked down using
siRNA for 48 h. Compared with the si-NC group, FoxO6 knockdown
markedly downregulated FoxO6, mTOR, MyHC and MyoD expression
levels, but notably upregulated atrogin1 and MURF1 expression
levels (Fig. 3C). Collectively,
the results suggested that FoxO6 was required for myotube
differentiation and maintenance in C2C12
cells.
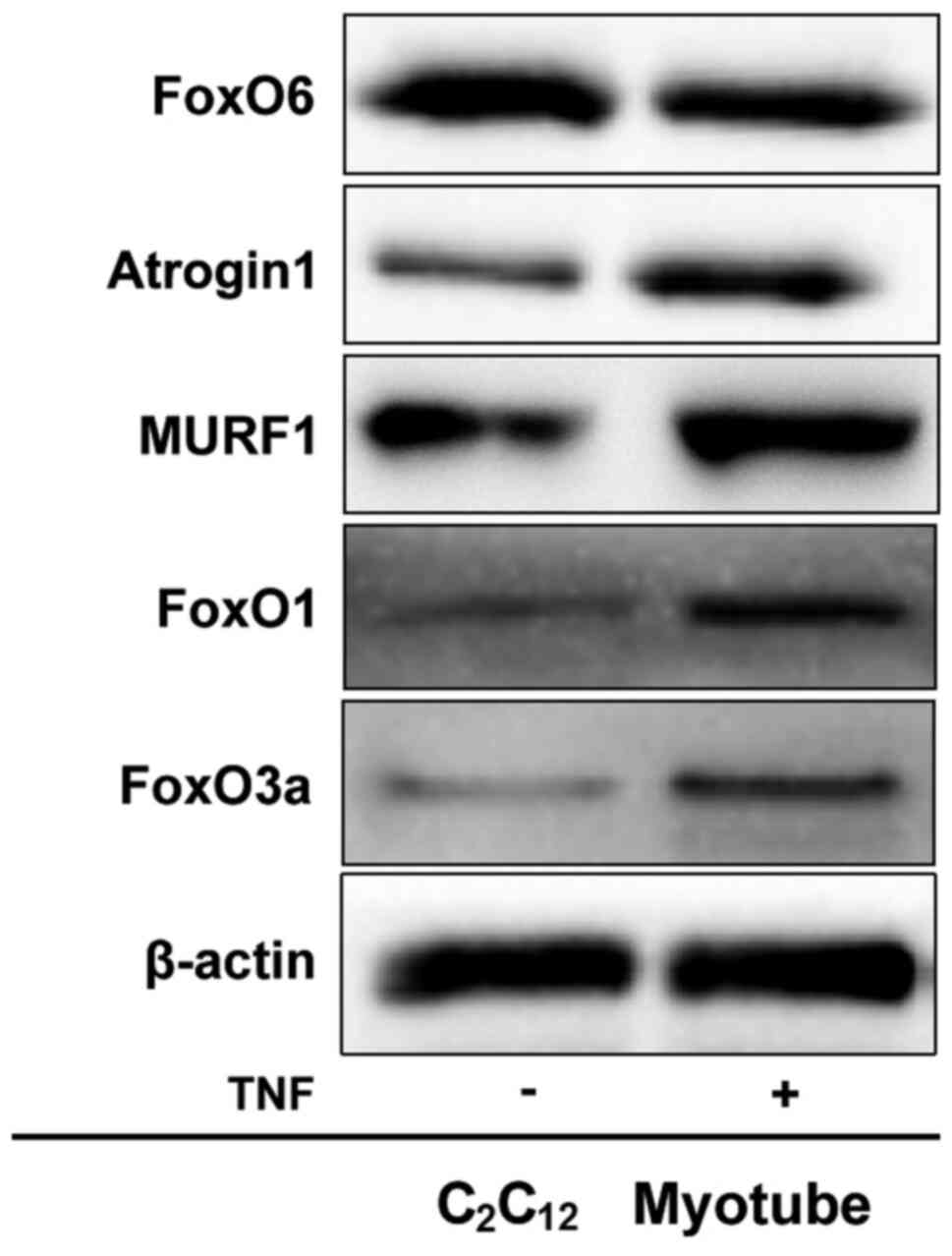
FoxO6 is downregulated in TNF-α-induced
C2C12 myotube atrophy
To further verify the aforementioned results, an
in vitro model of TNF-α-induced myotube atrophy, which has
been proven valid by Eley and Chen (31,32), was utilized. Atrogin1 and MURF1
expression levels were markedly upregulated in atrophied
C2C12 myotubes compared with the control
group (Fig. 4). Furthermore,
FoxO6 expression levels were notably downregulated, but FoxO1 and
FoxO3a expression levels were markedly upregulated in atrophied
C2C12 myotubes compared with the control
group. In other words, the results indicated that, unlike FoxO1 or
FoxO3a, FoxO6 was required for the maintenance of
C2C12 myotubes. The aforementioned results
indicated that FoxO6 regulated C2C12 myotubes
in an in vitro model of TNF-α-induced myotube atrophy.
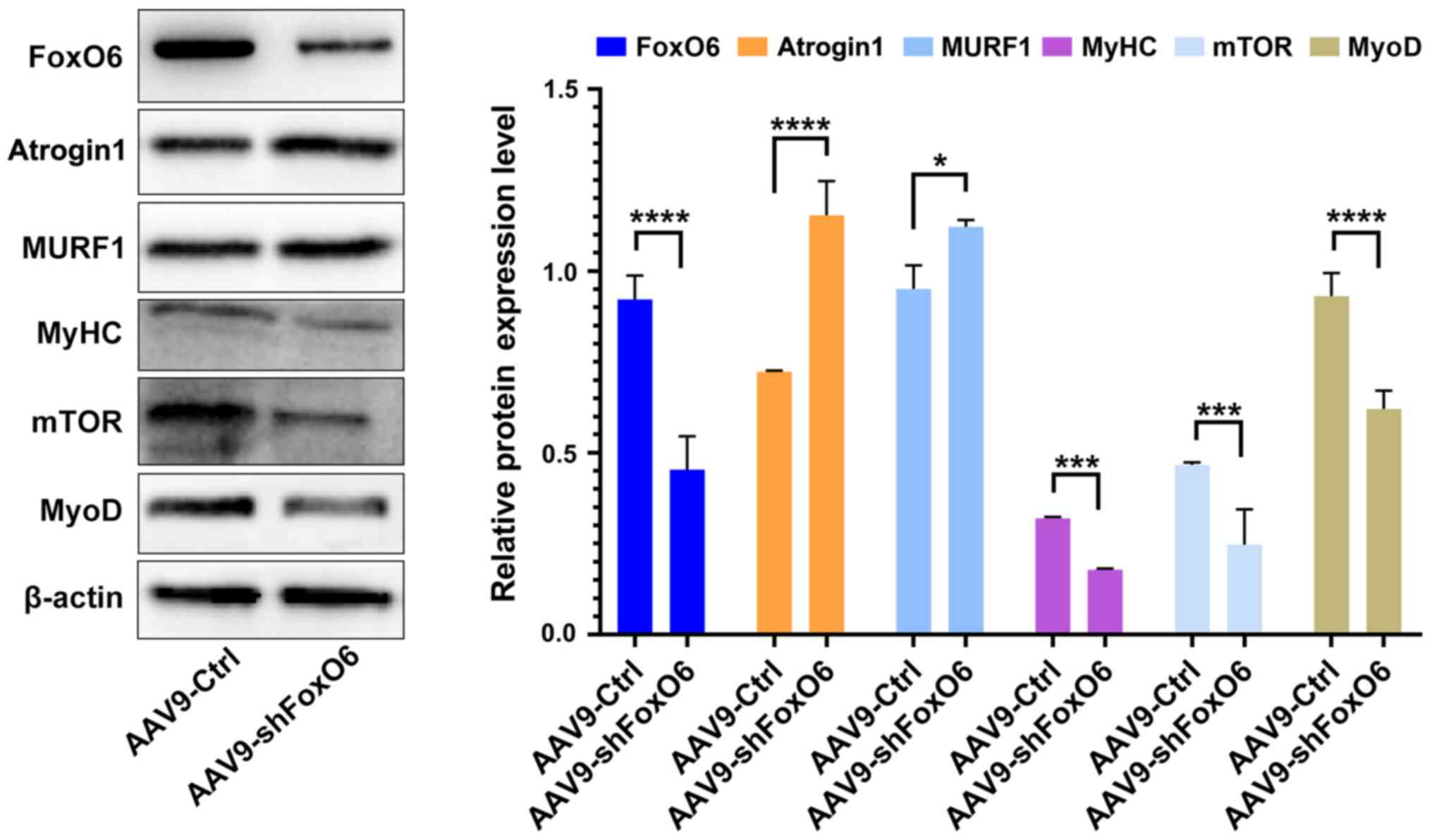
FoxO6 knockdown causes myofiber atrophy
in mice
The aforementioned results indicated that FoxO6
served a key role in maintaining C2C12
myotubes in vitro. Compared with the si-NC group, FoxO6
knockdown induced the downregulation of major muscle proteins,
including MyoD, MyHC and mTOR, and the upregulation of ubiquitin
ligase (atrogin1) and MURF1. Therefore, whether AAV9-shFoxO6
resulted in skeletal muscle fiber atrophy in vivo was
investigated. According to the knockdown efficiency of siFoxO6-3
in vitro, AAV9-shFoxO6 (based on the siFoxO6-3 sequence) was
prepared. AAV9-control (control group; n=5) or AAV9-shFoxO6
(knockdown group; n=5) was injected into the skeletal muscles of
each mouse. At 4 weeks post-injection, FoxO6 RNA expression levels
were significantly decreased by 65% in the AAV9-Ctrl group compared
with the AAV9-shFoxO6 group (Fig.
S3). Subsequently, the protein expression levels of atrogin1
and MURF1 in myofibers of AAV9-control or AAV9-shFoxO6 mice were
measured. Atrogin1 and MURF1 expression levels were significantly
increased in AAV9-shFoxO6 mice compared with AAV9-control mice
(Fig. 5). Moreover, the
expression levels of MyoD, MyHC and mTOR were significantly
decreased in AAV9-shFoxO6 mice compared with AAV9-control mice.
Thus, the results suggested that FoxO6 knockdown induced myofiber
atrophy. Collectively, the results of the present study indicated
that FoxO6 prevented skeletal muscle atrophy and was required for
muscle activity.
Discussion
Skeletal muscle atrophy is one of the major
characteristics of patients with chronic disease, and the
associated muscle loss becomes more pronounced as the disease
progresses (1-3,20). Several categories of drugs,
including statins, antiviral therapies and immunosuppressants, also
cause muscle atrophy (4,5). Although significant progress has
been made towards understanding skeletal muscle loss, the relevant
molecules and underlying cellular mechanisms are not completely
understood.
Previous studies have demonstrated that the majority
of members of the FoxO family are associated with the regulation of
skeletal muscle metabolism (19,20). In muscle, FoxO1 or FoxO3a could
induce skeletal muscle atrophy in vivo (21,22,24). FoxO4 was also involved in the
physiological regulation of mammalian skeletal muscle hypertrophy
and atrophy (20,33). In C2C12
myotubes, Moylan et al (25) reported that the TNF-induced
atrogin1 expression was dependent on FoxO4 expression, but not on
FoxO1/3 signaling. Chung et al (28) also demonstrated that FoxO6 could
form a regulatory loop with PGC-1α to establish the level oxidative
metabolism in muscles undergoing oxidative stress. However, as the
most recently discovered FoxO family member, the functions and
exact mechanism underlying FoxO6 in skeletal muscle metabolism are
not completely understood.
The present study investigated the role of FoxO6
in vitro with si-FoxO6 and in vivo with AAV9-shFoxO6.
FoxO6 expression levels in several samples, including samples of
mouse liver, heart, lung, colon and skeletal muscle, as well as the
C2C12 and AML12 cell lines, were assessed.
FoxO6 expression was particularly high in mouse skeletal muscles
and C2C12 myoblast cells. To assess the
hypothesis that FoxO6 might serve as an important regulator in the
maintenance of skeletal muscle function, the effect of FoxO6
knockdown on C2C12 cell proliferation was
investigated. Compared with the si-NC group, FoxO6 knockdown
significantly inhibited C2C12 cell
proliferation. Furthermore, C2C12 myotubes
were used to verify whether si-FoxO6 resulted in myotube atrophy.
The morphological characteristics were examined, and a significant
decrease in myotube diameter was observed following FoxO6 knockdown
compared with the si-NC group. Moreover, as the major component of
skeletal muscle (13,34,35), MyHC expression was analyzed via
immunofluorescence staining. The results demonstrated that FoxO6
knockdown notably downregulated the expression level of MyHC in
myotubes compared with the si-NC group. Collectively, the results
of the present study suggested that FoxO6 might be required for
C2C12 myotube differentiation and
maintenance.
Skeletal muscle mass is essential for motility,
whole body metabolism and viability. Patients suffering from muscle
atrophy often become weaker and unable to perform normal
activities, and in severe cases, muscle atrophy can result in
death. The primary characteristics of patients suffering from
muscle atrophy are extreme thinness and wasting. Under poor
conditions, muscle-related proteins are often severely degraded
(1-3,6).
Consistent with the important role of FoxO1 or FoxO3a during muscle
atrophy, the aforementioned FoxO6 knockdown-mediated effects were
noted in atrophic C2C12 myotubes.
Interestingly, the results of the present study suggested that,
unlike FoxO1 or FoxO3a, FoxO6 protected C2C12
myotubes against atrophy.
Increasing evidence has demonstrated that the loss
of muscle proteins in skeletal muscle leads to severe myopathy,
which in turn leads to a series of muscular disorders associated
with muscle protein dysfunction (11-13,36). The expression of muscle proteins
is significantly reduced when skeletal muscle atrophy occurs
(34,35). Therefore, to investigate the
possible mechanism underlying FoxO6-induced maintenance of myotube
activity, the present study assessed several critical biomarkers in
muscle, including MyoD, MyHC and mTOR, via western blotting. Under
several conditions, including fasting, a variety of diseases (e.g.,
cancer, diabetes mellitus and Cushing's Syndrome) and in specific
muscles upon denervation or disuse, muscle atrophy participates in
a common mechanism that upregulates atrogin1 and MURF1 (1,7,9,10). In the present study, compared
with the si-NC group, FoxO6 knockdown notably downregulated mTOR,
MyHC and MyoD expression levels, and markedly upregulated atrogin1
and MURF1 expression levels in C2C12
myotubes. Additionally, an in vitro model of TNF-α-induced
myotube atrophy was utilized. Similar results were obtained in the
in vitro model. For example, FoxO6 expression levels were
notably downregulated, and atrogin1 and MURF1 expression levels
were markedly upregulated in atrophied C2C12
myotubes compared with the control group. The results also
demonstrated that FoxO1 and FoxO3a expression levels were notably
upregulated in atrophied C2C12 myotubes
compared with the control group. In other words, the results
indicated that FoxO6 maintained C2C12
myotubes and protected against atrophy, which was different from
the known functions of FoxO1 or FoxO3a.
Consistent with the in vitro results, similar
results were also obtained in vivo. Atrogin1 and MURF1
expression levels were significantly increased in skeletal muscles
isolated from AAV9-shFoxO6 mice compared with AAV9-control mice.
Moreover, MyoD, MyHC and mTOR expression levels were notably
decreased in AAV9-shFoxO6 mice compared with AAV9-control mice. In
conclusion, the results of the present study indicated that FoxO6
was required for maintaining C2C12 myotubes
and protecting against atrophy in vitro and in vivo;
thus, the present study highlighted the protective effects of FoxO6
in muscle protein metabolism.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZH and CJ contributed to the conceptualization of
the study and acquisition of funding. LZ and CJ were involved in
designing and performing the experiments. YZ, MZ and SW provided
materials, and participated in performing the experiments and
analyzing the data. LZ drafted the manuscript. ZH and CJ
contributed to revising the manuscript. SW and TL performed the
animal experiments and generated the figures. All authors are
responsible for all aspects of the study in ensuring that questions
relevant to the accuracy or integrity of any part of the study are
appropriately investigated and resolved. All authors read and
approved the final version of manuscript.
Ethics approval and consent to
participate
All animal procedures were performed in accordance
with the Guiding Opinions on the Good Treatment of Laboratory
Animals (Sichuan Provincial Laboratory Animal Public Service
Center) guidelines and were approved by the Institutional Animal
Care and Treatment Committee of Sichuan Province (approval no.
2018328A), which was affiliated to Sichuan Academy of Medical
Sciences & Sichuan Provincial People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mr Guanyu Zhou
(Chengdu University of Traditional Chinese Medicine) for assisting
with the data analysis, and Mr Qiang Wang (Chengdu University of
Traditional Chinese Medicine) and Ms Wanping Xia (Chengdu
University of Traditional Chinese Medicine) for their valuable
assistance in the animal experiments.
Funding
The present study was supported by the Sichuan Science and
Technology Program (grant nos. 2020YFS0422, 2016JY0207, 2019YJ0150,
2019YFS0263, 2017FZ0091 and 2018RZ0091).
References
|
1
|
Sandri M: Autophagy in skeletal muscle.
FEBS Lett. 584:1411–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lecker SH, Goldberg AL and Mitch WE:
Protein degradation by the ubiquitin-proteasome pathway in normal
and disease states. J Am Soc Nephrol. 17:1807–1819. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lecker SH, Jagoe RT, Gilbert A, Gomes M,
Baracos V, Bailey J, Price SR, Mitch WE and Goldberg AL: Multiple
types of skeletal muscle atrophy involve a common program of
changes in gene expression. FASEB J. 18:39–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fappi A, de Carvalho Neves J, Sanches LN,
Silva PV, Sikusawa GY, Brandão TPC, Chadi G and Zanoteli E:
Skeletal muscle response to deflazacort, dexamethasone and
methylprednisolone. Cells. 8:4062019. View Article : Google Scholar :
|
|
5
|
Gupta A and Gupta Y:
Glucocorticoid-Induced myopathy: Pathophysiology, diagnosis, and
treatment. Indian J Endocrinol Metab. 17:913–916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cohen S, Nathan JA and Goldberg AL: Muscle
wasting in disease: Molecular mechanisms and promising therapies.
Nat Rev Drug Discov. 14:58–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jagoe RT and Goldberg AL: What do we
really know about the ubiquitin-proteasome pathway in muscle
atrophy? Curr Opin Clin Nutr Metab. 4:183–190. 2001. View Article : Google Scholar
|
|
8
|
Jagoe RT, Lecker SH, Gomes M and Goldberg
AL: Patterns of gene expression in atrophying skeletal muscles: The
response to food deprivation. FASEB J. 16:1697–1712. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gomes MD, Lecker SH, Jagoe RT, Navon A and
Goldberg AL: Atrogin-1, a muscle-specific F-box protein highly
expressed during muscle atrophy. Proc Natl Acad Sci USA.
98:14440–14445. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bodine SC, Latres E, Baumhueter S, Lai VK,
Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K,
et al: Identification of ubiquitin ligases required for skeletal
muscle atrophy. Science. 294:1704–1708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Megeney LA, Kablar B, Garrett K, Anderson
JE and Rudnicki MA: MyoD is required for myogenic stem cell
function in adult skeletal muscle. Genes Dev. 10:1173–1183. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bodine SC, Stitt TN, Gonzalez M, Kline WO,
Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC,
Glass DJ and Yancopoulos GD: Akt/mTOR pathway is a crucial
regulator of skeletal muscle hypertrophy and can prevent muscle
atrophy in vivo. Nat Cell Biol. 3:1014–1019. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Agbulut O, Noirez P, Beaumont F and
Butler-Browne G: Myosin heavy chain isoforms in postnatal muscle
development of mice. Biol Cell. 95:399–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hannenhalli S and Kaestner KH: The
evolution of fox genes and their role in development and disease.
Nat Rev Genet. 10:233–240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Link W: Introduction to FOXO biology.
Methods Mol Biol. 1890:1–9. 2019. View Article : Google Scholar
|
|
16
|
Monsalve M and Olmos Y: The complex
biology of FOXO. Curr Drug Targets. 12:1322–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jacobs FM, van der Heide LP, Wijchers PJ,
Burbach JPH, Hoekman MFM and Smidt MP: FoxO6, a novel member of the
foxo class of transcription factors with distinct shuttling
dynamics. J Biol Chem. 278:35959–35967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu W, Li Y and Luo B: Current perspective
on the regulation of FOXO4 and its role in disease progression.
Cell Mol Life Sci. 77:651–663. 2020. View Article : Google Scholar
|
|
19
|
Reed SA, Sandesara PB, Senf SM and Judge
AR: Inhibition of FoxO transcriptional activity prevents muscle
fiber atrophy during cachexia and induces hypertrophy. FASEB J.
26:987–1000. 2012. View Article : Google Scholar :
|
|
20
|
Sandri M, Sandri C, Gilbert A, Skurk C,
Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH and Goldberg
AL: Foxo transcription factors induce the atrophy-related ubiquitin
ligase atrogin-1 and cause skeletal muscle atrophy. Cell.
117:399–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kamei Y, Miura S, Suzuki M, Kai Y,
Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H,
et al: Skeletal muscle FOXO1 (FKHR) transgenic mice have less
skeletal muscle mass, down-regulated type I (slow twitch/red
muscle) fiber genes, and impaired glycemic control. J Biol Chem.
279:41114–41123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu J, Li RS, Workeneh B, Dong Y, Wang X
and Zhaoyong Hu: Transcription factor FoxO1, the dominant mediator
of muscle wasting in chronic kidney disease, is inhibited by
microRNA-486. Kidney Int. 82:401–411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao J, Brault JJ, Schild A, Cao P, Sandri
M, Schiaffino S, Lecker SH and Goldberg AL: FoxO3 coordinately
activates protein degradation by the autophagic/lysosomal and
proteasomal pathways in atrophying muscle cells. Cell Metabol.
6:472–483. 2007. View Article : Google Scholar
|
|
24
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Piccolo PD, Burden SJ, Lisi RD, Sandri C, Zhao JH, et
al: Foxo3 controls autophagy in skeletal muscle in vivo. Cell
Metabol. 6:458–471. 2007. View Article : Google Scholar
|
|
25
|
Moylan JS, Smith JD, Chambers MA,
McLoughlin TJ and Reid MB: TNF induction of atrogin-1/MAFbx mRNA
depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am J
Physiol Cell Physiol. 295:C986–C993. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoekman MF, Jacobs FM, Smidt MP and
Burbach JP: Spatial and temporal expression of FoxO transcription
factors in the developing and adult murine brain. Gene Expr
Patterns. 6:134–140. 2006. View Article : Google Scholar
|
|
27
|
Salih DA, Rashid AJ, Colas D, de la
Torre-Ubieta L, Zhu RP, Morgan AA, Santo EE, Ucar D, Devarajan K,
Cole CJ, et al: FoxO6 regulates memory consolidation and synaptic
function. Genes Dev. 26:2780–2801. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chung SY, Huang WC, Su CW, Lee KW, Chi HS,
Lin CT, Chen ST, Huang KM, Tsai MS, Yu HP and Chen SL: FoxO6 and
PGC-1α form a regulatory loop in myogenic cells. Biosci Rep.
33:e000452013. View Article : Google Scholar
|
|
29
|
Sun Z, da Fontoura CSG, Moreno M, Holton
NE, Sweat M, Sweat Y, Lee MK, Arbon J, Bidlack FB, Thedens DR, et
al: FoxO6 regulates hippo signaling and growth of the craniofacial
complex. PLoS Genet. 14:e10076752018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-(-Delta Delta C(T))method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Eley HL, Russell ST and Tisdale MJ:
Mechanism of attenuation of muscle protein degradation induced by
tumor necrosis factor-alpha and angiotensin II by
beta-hydroxy-beta-methylbutyrate. Am J Physiol Endocrinol Metab.
295:E1417–E1426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Wu Y, Yang T, Wei M, Wang Y, Deng
X, Shen C, Li W, Zhang H, Xu W, et al: Salidroside alleviates
cachexia symptoms in mouse models of cancer cachexia via activating
mTOR signalling. J Cachexia Sarcopenia Muscle. 7:225–232. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mandai S, Mori T, Nomura N, Furusho T,
Arai Y, Kikuchi H, Sasaki E, Sohara E, Rai T, Uchida S, et al: WNK1
regulates skeletal muscle cell hypertrophy by modulating the
nuclear localization and transcriptional activity of FOXO4. Sci
Rep. 14:91012018. View Article : Google Scholar
|
|
34
|
Derde S, Hermans G, Derese I, Güiza F,
Hedström Y, Wouters PJ, Bruyninckx F, D'Hoore A, Larsson L, Van den
Berghe G and Vanhorebeek I: Muscle atrophy and preferential loss of
myosin in prolonged critically ill patients. Crit Care Med.
40:79–89. 2012. View Article : Google Scholar
|
|
35
|
Banduseela V, Ochala J, Lamberg K, Kalimo
H and Larsson L: Muscle paralysis and myosin loss in a patient with
cancer cachexia. Acta Myol. 26:136–144. 2007.
|
|
36
|
Risson V, Mazelin L, Roceri M, Sanchez H,
Moncollin V, Corneloup C, Richard-Bulteau H, Vignaud A, Baas D,
Defour A, et al: Muscle inactivation of mTOR causes metabolic and
dystrophin defects leading to severe myopathy. J Cell Biol.
187:859–874. 2009. View Article : Google Scholar : PubMed/NCBI
|