Introduction
Glioblastoma (GBM) is the most common and aggressive
type of brain tumor affecting humans, with the survival of patients
being <2 years from the time of diagnosis (1-3).
The hallmarks of GBM include molecular and cellular heterogeneity,
uncontrolled proliferation, diffuse infiltration, necrosis,
angiogenesis, resistance to apoptosis and genomic instability
(4-7). Current treatment modalities involve
a combination of maximal tumor resection, radiotherapy and
concomitant chemotherapy; however, the median survival rate is 14.6
months (8). Toxicity in healthy
tissue is a common side-effect and tumor recurrence is almost
inevitable (9). There is
therefore an urgent need for the development of novel therapies
that act in a robust and GBM-selective manner.
One therapeutic candidate is tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL), a pro-apoptotic
cytokine. TRAIL holds much potential as an anticancer therapeutic
since it binds to death receptors (TRAIL-R1; DR4 and TRAIL-R2; DR5)
that are exclusively expressed on cancerous cells. The binding of
TRAIL induces the recruitment of Fas-associated death domain (FADD)
adaptor protein, which in turn recruits procaspase-8 to the
intracellular death domain of DR4/5 (10). Once cleaved, caspase-8 activates
subsequent executioner caspases-3/7, leading to the activation of
the extrinsic apoptotic cascade (11-13). Active caspase-8 can additionally
induce the intrinsic apoptotic pathway via the cleavage of BH3
interacting-domain death agonist (BID). The truncated form, tBID,
interacts with Bax and ultimately causes mitochondrial outer
membrane permeabilization and activation of the caspase cascade via
caspase-9 (14).
Although a number of established and patient-derived
GBM cell lines are sensitive to TRAIL, approximately half of these
display some degree of TRAIL resistance, thus limiting its
therapeutic potential (12,15-17). To increase the effectiveness of
TRAIL therapy, it is necessary to understand the mechanisms that
cause TRAIL resistance and develop therapeutic approaches that can
overcome resistance by sensitizing an otherwise TRAIL-resistant
population of cells. This has been tested using a combination
approach with either TRAIL-sensitizing agents, or biologics that
target different tumor-killing mechanisms to enhance death-inducing
signals (16,18-20). One such approach was used by
Horita et al, who treated GBMs with TRAIL in combination
with an epidermal growth factor receptor (EGFR)-targeted diphtheria
toxin (DT-EGF) (21). When TRAIL
and DT-EGF were used in combination, a synergy in GBM cell death
was observed compared to the cells treated with the monotherapies
alone. This effect was caused by DT-EGF depleting FLICE-inhibitory
protein (FLIP), an inhibitor of TRAIL-mediated apoptosis, thereby
potentiating TRAIL-induced apoptosis (21).
The authors have previously engineered
toxin-resistant somatic and human neural stem cells to continually
secrete 2 pseudomonas exotoxin (PE)-cytotoxins, IL13-PE and ENb-PE,
that target interleukin (IL)-13 receptor α2 (IL13Rα2) or EGFR,
respectively (22), expressed by
a number of GBMs (22-25). PE has a similar mechanism of
action as DT, resulting in the inhibition of protein synthesis and
the death of targeted cells (26-28). Previously, the authors
demonstrated efficacy in multiple GBM lines, and established an
association between GBM cell death and the expression of cognate
receptors (22). It would be
ideal to create a bifunctional stem cell (SC) that secretes
PE-cytotoxin and TRAIL; this combination may prove effective by
targeting multiple GBM subtypes and potentially sensitizing
TRAIL-resistant populations. Furthermore, SC delivery would
circumvent the short half-life, systemic toxicity and poor tissue
penetration attributed to standard chemotherapeutic administration
(9,29,30).
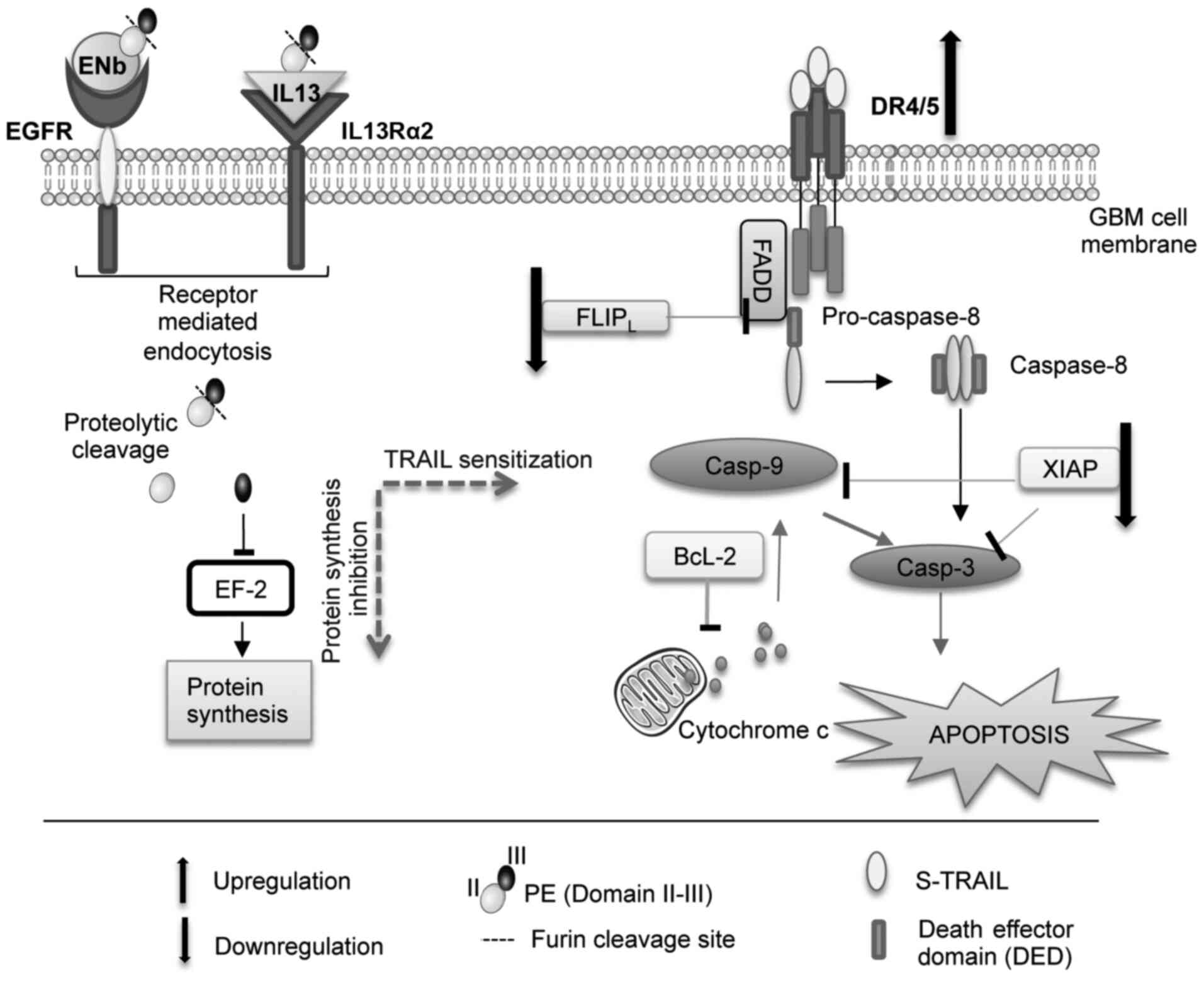
In the present study, it was demonstrated that the
exposure of TRAIL-resistant GBM lines to targeted PE-cytotoxins led
to the upregulation of DR4/5, and the depletion of the
anti-apoptotic proteins, FLIP and XIAP, thereby breaking TRAIL
resistance and sensitizing the cells to TRAIL. Furthermore,
combining pro-apoptotic TRAIL with the protein synthesis inhibitory
effects of PE-cytotoxins resulted in an enhanced GBM cell death by
potentiating both the extrinsic and intrinsic apoptotic
pathways.
Materials and methods
Preparation of lentiviral constructs
Based on the lentiviral transfer vectors,
pLV-CICS/IG and pLV-CICS, the following therapeutic and diagnostic
lentiviral vectors were engineered as previously described
(13,31): i) LV-TRAIL' ii) LV-ENb-PE' iii)
LV-IL13-PE; iv) LV-GFP/RLuc; v) LV-IL13Rα2-GFP/Rluc; vi)
LV-Fluc/mCherry; and vii) LV-GFP/Fluc. Recombinant IL13-PE was
constructed in the previously described Pico2 vector by replacing
Fluc with IL13-PE (32). IL13
was PCR-amplified using pORF5-hIL13 (Invitrogen; Thermo Fisher
Scientific, Inc.) as a template with primers encoding Nhe1
and PspX1. The PCR fragment was ligated into
Nhe1/PspX1-digested Pico2. To create IL13-PE, IL13 was
PCR-amplified as described above with primers encoding Nhe1
and EcoRV. PE, which lacks the receptor binding domain
sequence (domain Ia and Ib), was amplified by PCR with primers
encoding EcoRV and PspX1 using pJH8 (ATCC) as a
template. The two fragments were then ligated into
Nhe1/PspX1-digested Pico2. To construct the cytotoxic
variant of EGFR nanobodies, the cDNA encoding an 18-aa linker
sequence (lin) and PE sequence were amplified by PCR and
fused with the LV-ENb construct at the EcoRV and Xho1
sites. The EcorV-Linker-PE-Xho1 cDNA fragment were
amplified with EcoRVlinPE forward and Xho1-PE reverse
primers and directionally inserted in EcoRV-Xho1-digested
LV-ENb, resulting in LV-ENb-PE. To obtain a clinically-proven
lentiviral vector bearing IL13-PE-TRAIL, IL13-PE was first
PCR-amplified from the LV-(CMV)-IL13-PE vector and then
directionally sub-cloned into the previously described (31) LV-(EF)-Nb-TRAIL by replacing Nb
fragment with IL13-PE (since the C terminal of TRAIL contains
cytotoxic domain, we cloned IL13-PE in front of the N terminal of
TRAIL sequence). Lentiviral constructs were packaged as lentiviral
vectors in 293T/17 (ATCC) and in toxin-resistant 293 oligo cells
for cytotoxin constructs by using a helper virus-free packaging
system as previously described (31) and GBM cells were engineered to
express florescence and bioluminescence imaging agents as
previously described (32,33).
Cells and cell culture
The established GBM cell lines, LN229, U138 and
U251, were purchased from ATCC and grown in glioma growth medium;
DMEM supplemented with 10% (vol/vol) FBS and 1% (vol/vol)
penicillin/streptomycin. The highly invasive tumor-initiating
primary cell lines were grown in neural basal medium (Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 3 mM L-glutamine,
2 µg/ml heparin, 20 ng/ml epidermal growth factor (EGF) and
20 ng/ml basic fibroblast growth factor (bFGF).
Establishment of bio-imageable tumor
cells
GBM cells were transduced with either
LV-Fluc/mCherry or LV-GFP/Fluc at a multiplicity of infection (MOI)
of 2 and populations of transduced cells were visualized by
fluorescence microscopy for imageable reporter gene expression
(mCherry and GFP). To establish a pure cell line, transduced cells
with lentiviral vectors were sorted for high expressers of GFP and
mCherry using a FACSAria Ilu cell sorter (BD Biosciences). GBM
cells were transduced with pico2 vector were selected with
puromycin and sorted for high expressers of florescence proteins
using a FACSAria Ilu cell sorter (BD Biosciences).
Dot blot quantification
Serum-free conditioned medium from 293 oligo cells
engineered to express secretable forms of IL13-PE, ENb-PE and TRAIL
were collected 24 h following transfection and concentrated with
centrifugal filter units (Amicon, MilliporeSigma). GFP-conditioned
medium was collected and used for each control treatment. Purified
IL13 (Chemicon; Thermo Fisher Scientific, Inc.; 100 ng/µl)
was used as a positive control for the quantification of IL13-PE.
To determine the ENb-PE concentrations, IL13PE was used as a
positive control. Concentrated condition medium (20X) was loaded on
a nitrocellulose membrane (1 and 3 µl) and immunoblotted
using antibodies against IL13 (cat. no. ab106732, Abcam; antibody
dilution; 1:1,000) and PE (cat. no. P2318, Sigma-Aldrich; Merck
KGaA; antibody dilution; 1:20,000) for IL13-PE and ENb-PE,
respectively. The antibody incubations were performed at room
temperature for 1 h on a shaker. Following primary antibody
incubation, the membrane was also probed with HRP conjugated
secondary antibodies (1:5,000) goat anti-mouse (cat. no. ab6789,
Abcam) and goat anti-rabbit (cat. no. ab6702, Abcam) accordingly
for 30 min at room temperature. Band intensities and relative
concentration were quantified using NIH ImageJ software.
Cell viability assays
To examine the combined therapeutic effects of
PE-fused cytotoxins and S-TRAIL in vitro, a panel of GBM
lines was transduced with either LV-Rluc or LV-Fluc and seeded in
96-well plates (Matrical Bioscience). All patient derived GSC lines
(BT74, GBM4, GBM8, GBM18, GBM23 and GBM64) used in the present
study were previously established (34) and provided by Dr Wakimoto
(Department of Neurosurgery, Massachusetts General Hospital,
Harvard Medical School, Boston, MA, USA). A panel of cell lines
were firstly applied to a viability assay for TRAIL and PE
cytotoxin treatments. Both established TRAIL-resistant GBM lines
and patient-derived GSC lines (GBM23 and GBM64) expressing the
target receptors for PE cytotoxins were analyzed for TRAIL and PE
cooperation. The cells were pre-treated with conditioned media
containing 25 ng/ml of PE-cytotoxins and 500 ng/ml S-TRAIL (100
ng/ml S-TRAIL was added for TRAIL semi-sensitive lines) was
subsequently added after 24 h of toxin exposure. Cell viability was
measured with 15 µg/ml of D-luciferin (Biotium, Inc.) for
the Fluc signal or 1 µg/ml coelanterazine (Nanolight) for
the Rluc signal. For non-transduced cell lines, metabolic activity
was measured using ATP-dependent luminescent reagent
(CellTiter-Glo, Promega Corporation). For all in vitro
assays, photon emission was measured using a cryogenically cooled
high efficiency CCD camera system (Roper Scientific, Inc.).
BLI assay for apoptosis
To investigate the concentration dependency of
PE-fused cytotoxins in combination with S-TRAIL on GBM viability
and caspase activities, non-transduced GBM cells were treated as
described above and measured using CellTiterGlo and CaspaseGlo 3/7,
respectively (Promega Corporation) following manufacturer's
guidelines (Promega Corporation). All experiments were performed in
triplicate.
Western blot analysis
Receptor expression
Cell lysates from a panel of GBM cell lines were
prepared using Nonidet P-40 lysis buffer containing complete
protease inhibitor mixture (Roche Diagnostics) and protein amount
was quantified using a BCA Protein Assay (Bio-Rad Laboratories,
Inc.). The rotein amount in each sample was determined prior to the
SDS treatments using a DC Protein assay kit (Bio-Rad Laboratories,
Inc.) and a total of 25 µg protein samples were loaded in
each well of 4-15% polyacrylamide (gradient) gels (Bio-Rad
Laboratories, Inc.) and proteins were transferred onto
nitrocellulose membranes by wet blotting in ice for 90 min.
(Bio-Rad transfer system) The membranes were then blocked in 5%
bovine serum albumin (BSA) at room temperature for 90 min. Proteins
were examined by western blot analysis using either antibodies
against EGFR (cat. no. D38B1, Cell Signaling Technology, Inc.;
antibody dilution; 1:1,000) or IL13Rα2 (cat. no. AF146, R&D
Systems, Inc.; antibody dilution, 0.2 µg/ml). The membranes
were blocked in 5% BSA for 90 min. at room temperature and
incubated with proper primary antibodies overnight at 4°C.
Expression and secretion of
therapeutics
The cell lysates and serum-free conditioned medium
from 293.oligo-IL13-PE, or 293.oligo-ENb-PE or 293.oligo-GFP were
collected following transduction (16 h following transduction, the
infection medium was replaced with a serum-free medium and
following 24 h of incubation at 37°C 5% CO2 incubator,
the conditioned medium was collected) and examined by western blot
analysis using antibodies against IL-13 (cat. no. ab106732, Abcam;
antibody dilution, 1:1,000) or PE (cat. no. P2318, Sigma-Aldrich;
Merck KGaA; antibody dilution, 1:20,000). The membranes were
blocked in 5% BSA for 90 min and the antibody incubations were
performed overnight at 4°C. Following primary antibody incubation,
all membranes were also probed with HRP-conjugated secondary
antibodies (1:5,000) goat anti-mouse (ab6789, Abcam) and goat
anti-rabbit (ab6702, Abcam) accordingly for 1 h at room
temperature.
TRAIL sensitization upon targeted toxin
mono-treatments
LN229 and LN229-IL13Rα2 cells were treated with
ENb-PE and IL13-PE, respectively and cell lysates were analyzed
with antibodies against β-actin (cat. no. 4967, Cell Signaling
Technology, Inc.; antibody dilution, 1:2,000), DR4 (cat. no. 1139,
ProSci; antibody dilution, 1 µg/ml), DR5 (cat. no. 2019,
ProSci; antibody dilution, 1 µg/ml) and XIAP (cat. no.
14334, Cell Signaling Technology, Inc; antibody dilution, 1:20,00).
All membranes were blocked in 5% BSA for 90 min at room temperature
prior to antibody incubation. Following primary antibody
incubation, all membranes were also probed with HRP-conjugated
secondary antibodies (1:5,000) goat anti-mouse (ab6789, Abcam),
goat anti-rabbit (ab6702, Abcam) and donkey anti-goat (ab182021,
Abcam) accordingly for 1 h at room temperature.
Apoptosis on targeted toxin and TRAIL
combined treatments
Patient-derived GSC (glioma stem cell) lines (GBM23
and GBM64) and established GBM lines (U251, LN229 and
LN229-IL13Rα2) were pre-treated with PE-fused cytotoxins (25 ng/ml)
for 24 h, then treated with 500 ng/ml S-TRAIL (100 ng/ml for U251
cells). Following 24 h of S-TRAIL exposure, cell lysates were
analyzed using antibodies against β-actin (cat. no. 4967, Cell
Signaling Technology, Inc.) and apoptotic markers including cleaved
poly(ADP-ribose) polymerase (PARP) (cat. no. 5625, Cell Signaling
Technology, Inc.), caspase-8 (cat. no. 9746, Cell Signaling
Technology, Inc.) and caspase-9 (cat. no. 9508, Cell Signaling
Technology, Inc.). All membranes were blocked in 5% BSA for 90 min
at room temperature prior to antibody incubation. In this
experimental setting, primary antibody incubations were performed
at a 1:2,000 dilution for actin antibody and a 1:1,000 dilution for
all other antibodies. Following primary antibody incubation, all
membranes were also probed with HRP conjugated secondary antibodies
(1:5,000) goat anti-mouse (ab6789, Abcam) and goat anti-rabbit
(ab6702, Abcam) accordingly for 1 h at room temperature.
Data quantification
Protein bands on X-ray films were visualized with a
chemiluminescence substrate (Super Signal™ West Pico PLUS; Thermo
Fisher Scientific, Inc.) and the intensity of bands was quantified
using NIH ImageJ software.
Reverse transcription PCR
RNA samples were extracted by using RNeasy kit
(Qiagen, Inc.). Total RNA (0.5 µg) was reverse transcribed
to obtain cDNA using the Superscript VILO cDNA synthesis kit
(Invitrogen; Thermo Fisher Scientific, Inc.). A cDNA library was
obtained after 30 cycles of amplification (95°C, 30 sec.-60°C, 45
sec.-72°C, 45 sec-72°C, 10 min.) (Qiagen, Inc.) and Human IL13Rα2
chain was amplified using the primer pair (sense, 5′-ATG GCT TTC
GTT TGC TTG GCT AT-3′ and antisense, 5′-TCA TGT ATC ACA GAA AAA TTC
TGG-3′) yielding a product of 1,130 bp. A portion of PE was
amplified using the primer pair (sense, 5′-GAA CCC GAC GCA CGC GGC
CGG-3′ and antisense, 5′-CCG CTC GAG CTT CAG GTC CTC GCG CGG CG-3′)
to generate a 445 bp product. A human GAPDH primer pair (sense,
5′-GTC AGT GGT GGA CCT GAC CT-3′ and antisense, 5′-TGC TGT AGC CAA
ATT CGT TG-3′) was used yielding 245 bp PCR product as a positive
control.
Immunofluorescence staining
GBM cells were plated onto a coverslip containing
wells of 24 well plates 1×104 cells/well and treated for
24 h with 25 ng/ml IL13-PE; or ENb-PE; or GFP (control)
accordingly. The cells were then fixed with 4% PFA
(paraformaldehyde) for 10 min at room temperature and blocked with
1X phosphate-buffered saline (PBS) including 5% NGS (normal goat
serum) and 0.3% Triton-X at room temperature for 2 h. Primary
antibody against Ki-67 (M7240, Dako; Agilent Technologies, Inc.)
was added (1:100) to each well and incubated overnight at 4°C. The
following day, after washing with PBS, the cells were incubated
with Alexa-647 conjugated goat anti-mouse antibody (ab150115,
Abcam) and after washing with PBS, the cover slips were mounted
with Vectashield mounting medium with DAPI (H1200-10; Vector
Laboratories, Inc.) onto slides. They were then analyzed for Ki-67
staining.
Statistical analysis
A Student's t-test was used to compare data between
two groups and for multiple group comparisons, one-way ANOVA and
the Bonferroni post hoc test were applied. The differences were
considered statisticaly significant at P<0.05. Data are
expressed as the means ± SEM.
Results
Engineered toxin-resistant cells can
express secretable and functional forms of targeted toxins
293T cells were engineered for toxin resistance
using single-stranded oligonucleotides designed to encode mutant
elongation factor-2 (ssODN-mEF-2) as previously described by the
authors (22) (Fig. S1A). Cell viability assays
indicated toxin resistance (TR) in the 293T-ssODN (293 oligo) cells
compared with the controls post-treatment with various
concentrations of DT (Fig.
S1B). The expression and secretion of targeted toxins was
observed in the conditioned medium from toxin-resistant 293 oligo
cells transduced with LVs bearing either ENb-PE or IL13-PE and the
control (Fig. S1C-E).
Subsequently, to examine the functionality of ENb-PE or IL13-PE on
GBM cells, GBM lines expressing a luciferase reporter (destabilized
luciferase; dsluc) were established. Treatment of GBM cells with
IL13-PE and ENb-PE resulted in the blocking of protein synthesis
and a dynamic change in gene expression was detected according to
dsluc signal (Fig. S2A);
significant changes were also detected in cell proliferation and
viability (Fig. S2B-G).
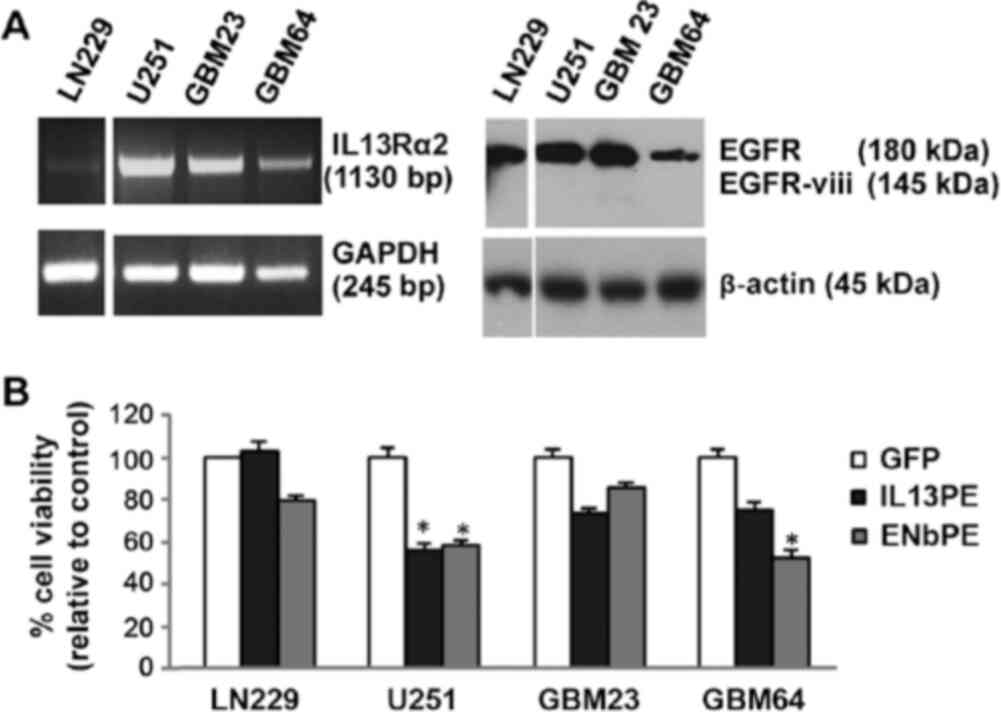
Subsequently, the EGFR and IL13Rα2 levels were assessed in a panel
of GBM lines, including patient-derived cells (Fig. 1A) and the efficacy of IL13-PE and
ENb-PE was evaluated on these cell lines. A direct association
between EGFR/IL13Rα2 expression and the response to ENb-PE/IL13-PE
was observed in all GBM lines tested (Fig. 1A and B). Taken together, these
results indicate that toxin-resistant 293oligo can be engineered to
express IL13-PE and ENb-PE, which have a targeted therapeutic
effect on GBM cells.
Targeted toxins upregulate DR4/5 and
induce a decrease in the expression of anti-apoptotic XIAP in
resistant GBM cells
GBM cells (LN229, GBM4, GBM18, GBM23, GBM64 and
BT74) exhibited varying degrees of TRAIL resistance (Fig. S3) and some exhibited significant
resistance to targeted toxins (Fig.
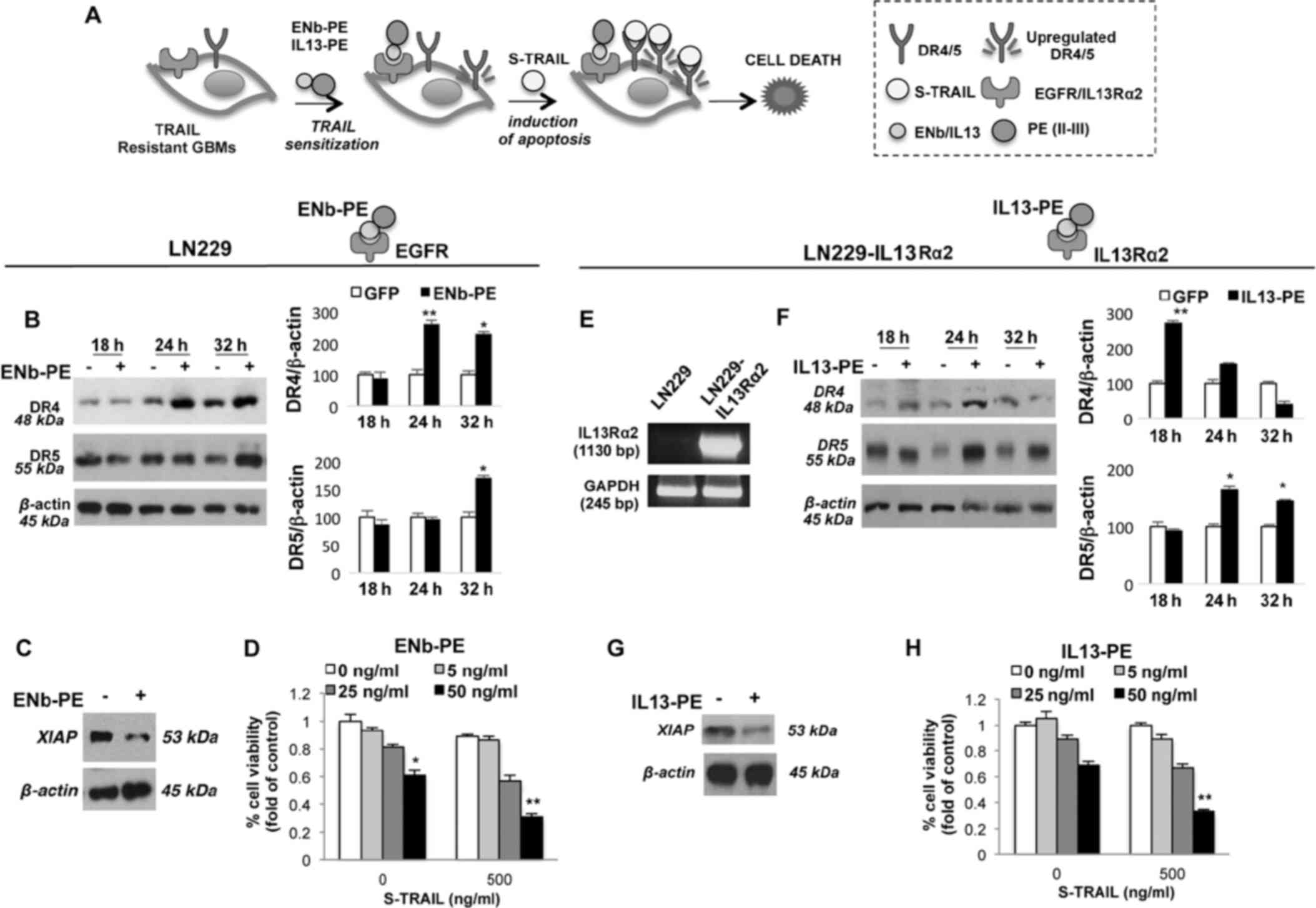
1B). To evaluate the possibility of sensitizing the GBM cells
the effects of targeted toxins to promote TRAIL-mediated,
TRAIL-resistant wild-type LN229 (wt) and LN229 cells engineered to
express IL13Rα2 (LN229-IL13Rα2) were treated with ENb-PE and
IL13-PE targeting their cognate receptors EGFR and IL13Rα2,
respectively (Fig. 2A). Western
blot analysis revealed an increase in the total DR4/5 levels
post-treatment with 25 ng/ml ENb-PE (Fig. 2B) and decreased anti-apoptotic
XIAP levels in the LN229 cells, respectively (Fig. 2C). To examine the in vitro
killing effect of the targeted toxin and TRAIL combination, a panel
of TRAIL-resistant GBM cells were pre-treated with ENb-PE (25 ng/ml
for 24 h) and these cells were then co-treated with 500 ng/ml TRAIL
for 24 h. ENb-PE stimulated the TRAIL killing of highly resistant
GBM lines with a significant decrease in cell viability (P<0.001
and P<0.05; Fig. 2D). For
assessing the combined therapeutic efficacy of IL13-PE and TRAIL, a
TRAIL-resistant line expressing IL13Rα2, LN229-IL13Rα2 was created
(Fig. 2E). Similar to ENb-PE,
western blot analysis revealed an increase in the total DR4/5
levels post-treatment with 25 ng/ml IL13-PE (Fig. 2F) and decreased anti-apoptotic
XIAP levels (Fig. 2G) in
LN229-IL13Rα2 cells treated with a combination of IL13-PE and
TRAIL. This combined treatment resulted in a significant decrease
in cell viability (P<0.001) in TRAIL-resistant LN229-IL13Rα2 GBM
lines (Fig. 2H). Subsequently,
the sensitizing effect of IL13-PE or ENb-PE treatment was assessed
in a TRAIL semi-sensitive GBM line, U251, in real-time by DR4/5
promoter-luciferase imaging. As was expected, DR4/5 receptor
expression levels were increased post-treatment with IL13-PE in the
U251 cells, which were previously engineered with the construct
bearing luciferase reporter under the DR4/5 promoter.
IL13-PE/ENb-PE and TRAIL combination was much more potent than any
mono-treatments on the semi-sensitive U251 cells. (Fig. S4A and C). These findings reveal
that treatment with IL13-PE and ENb-PE results in the upregulation
of DR4/5 and the downregulation of FLIP which then leads to the
sensitization of the cells to TRAIL-mediated apoptosis.
Co-operation of targeted toxins with
TRAIL kills GBM cells through both caspase-8- and
caspase-9-mediated apoptosis
TRAIL-mediated apoptosis is a rapid process
characterized by the activation of a cascade of intracellular
proteases, or caspases and the cleavage of numerous intracellular
proteins, resulting in cell death within 24-48 h post-TRAIL
treatment (12). In the present
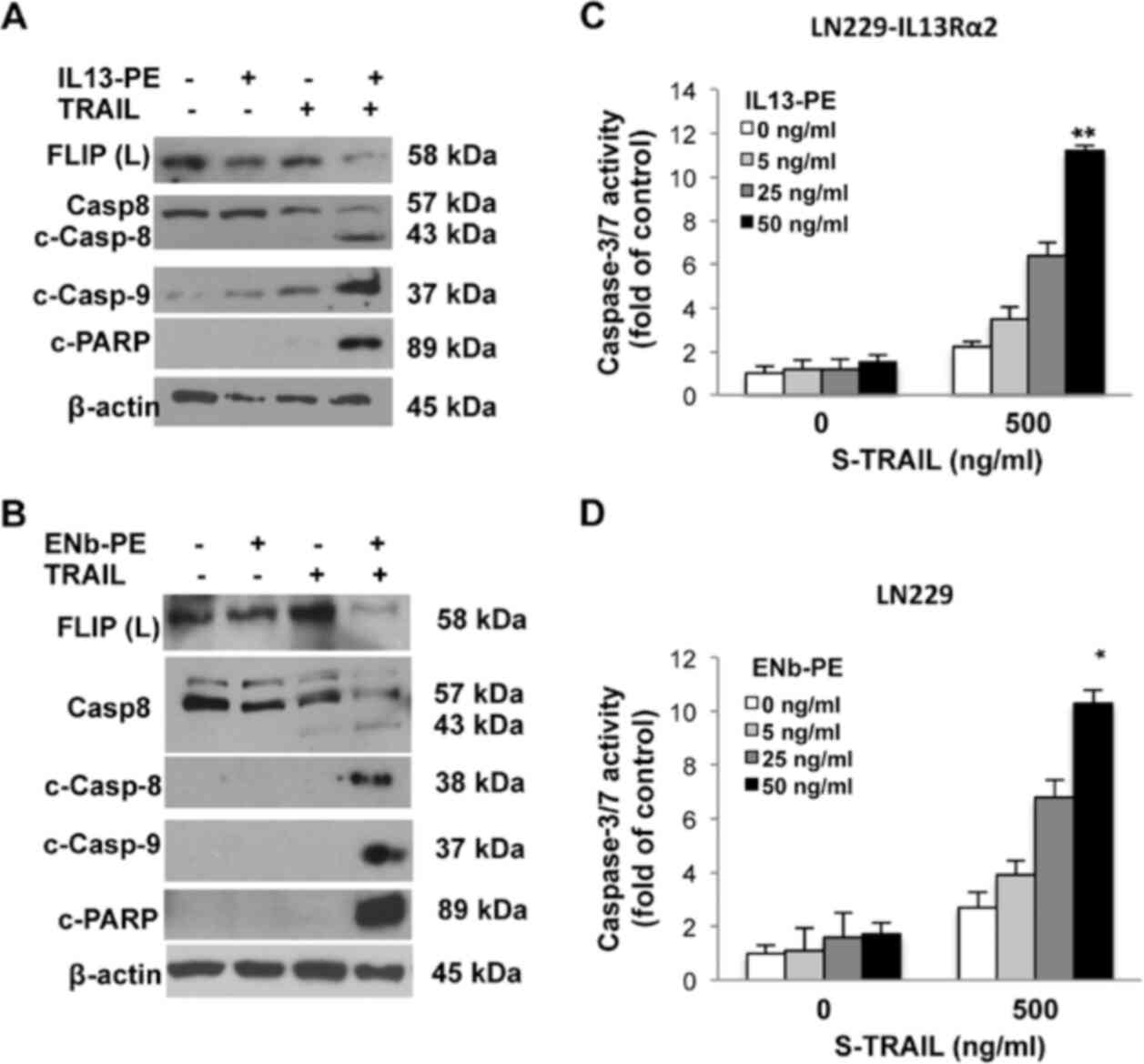
study, to examine the apoptosis of GBM cells exposed to a
combination of targeted toxins and TRAIL, since LN229 expresses
EGFR and lacks IL13Rα2 transcripts, the therapeutic efficacy of
ENb-PE was tested and the apoptosis of the LN229 cells was
analyzed. To examine the efficacy of IL13-PE, LN229-IL13Rα2 cells
were utilized, engineered as described above (Fig. 2E). The findings revealed that
both toxin fusion proteins targeted their cognate receptors and
sensitized the LN229 cells to TRAIL-mediated therapy. The
activation of caspase-8-mediated apoptosis was triggered in
TRAIL-resistant GBM cells when the cells were pre-treated with
targeted toxins, while TRAIL mono-treatment did not lead to the
cleavage of caspase-8 in any resistant lines. Similar results were
observed for caspase-9 and poly ADP ribose polymerase (PARP)
(Fig. 3A and B). A major
contributor of TRAIL resistance is a caspase-8 analogue, FLIP,
which is direct contact with DR4/5 and caspase-8 or -10 dynamics
(10-12). Thus, the present study evaluated
the interaction of FLIP and caspase-8 cleavage in GBM cells upon
treatment with the ENb-PE and TRAIL combinations. Western blot
analysis revealed that caspase-8 was cleaved when the FLIP
impediment was removed in GBM cells upon treatment with the
targeted toxins and TRAIL in combination, but not with treatment
with TRAIL or targeted toxins alone (Fig. 3A and B). In a more TRAIL
sensitive line, U251, caspase-9 cleavage was detected with the
TRAIL and IL13-PE/ENb-PE combination, while cleaved caspase-8 was
detected with both the TRAIL and TRAIL and IL13-PE/ENb-PE
combination treatments. Furthermore, the findings for PARP cleavage
were in accordance with those of caspase cleavage (Fig. S4B). In addition to the
biochemical analysis of apoptosis, relative caspase-3/7 activation
in GBM cells treated with increased concentrations of targeted
toxins and TRAIL, was quantified. The data revealed an association
between caspase-3/7 activation and GBM viability in a
concentration-dependent manner (Figs. 3C and D, and S4D). To sum up, these results revealed
that TRAIL combined with targeted toxins executed both caspase-8-
and caspase-9-dependent apoptotic machinery, and a final GBM cell
killing occurred in a synergistic manner, involving not only
intrinsic, but also extrinsic apoptotic pathways.
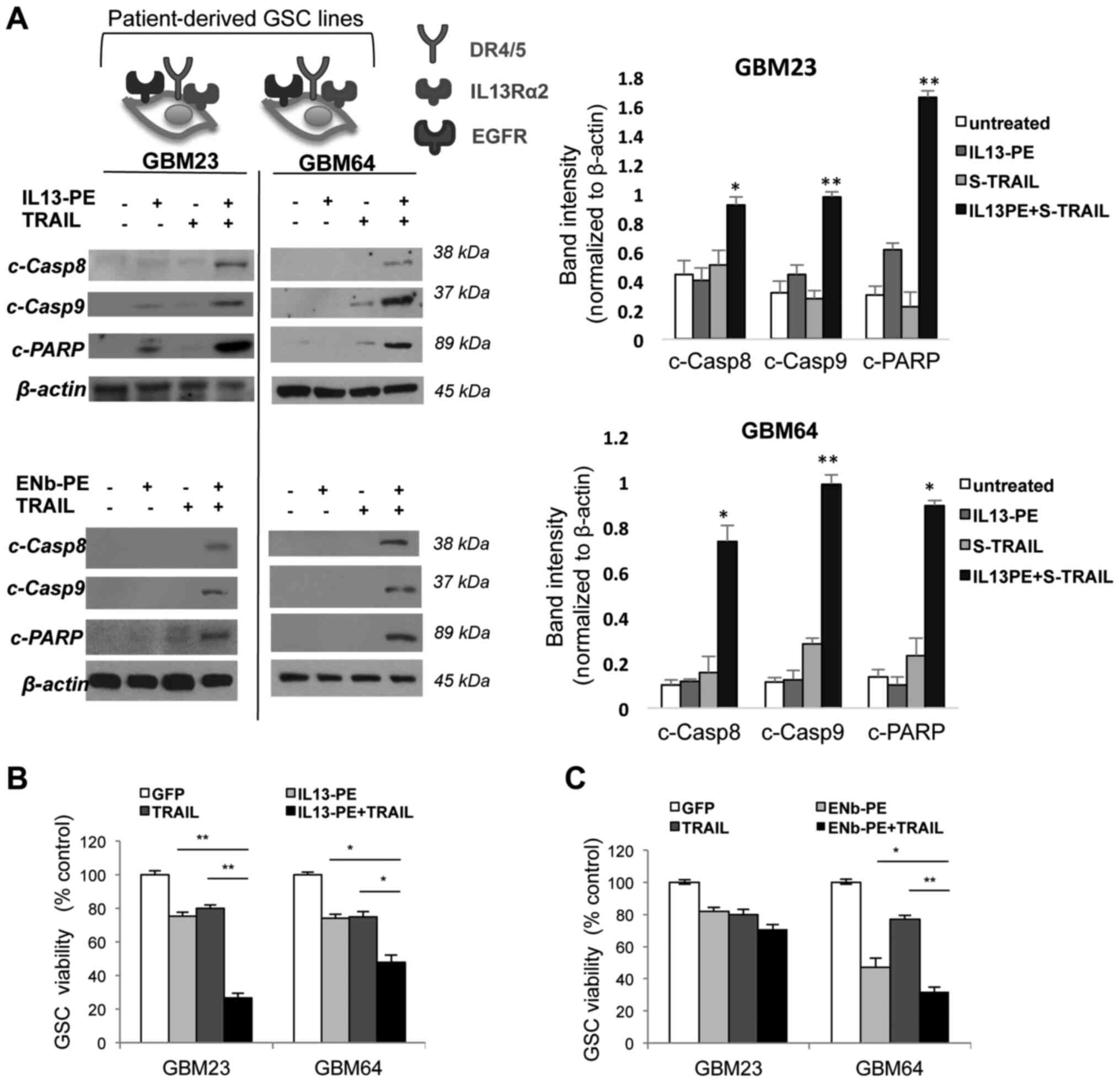
Combination of IL13-PE/ENb-PE and TRAIL
have therapeutic efficacy in patient-derived primary GBM lines
A number of studies have suggested that the majority
of the chemo- and radiotherapy-resistant cell population of GBM
arises from the CSC phenotype (35-38). In the present study, to
investigate the therapeutic response of PE cytotoxins and TRAIL
combination on CD133+ glioma stem cells, GSCs (also
known as glioma initiating cells), GSC lines that were previously
developed and characterized from human GBM tissue were used as a
xenograft model to examine several experimental therapeutics both
in vitro and in vivo. These CSC enriched GSCs can
grow in culture as spheres and when implanted intracerebrally into
immunodeficient mice, they can form highly invasive and angiogenic
tumors (34). The therapeutic
efficacy of the IL13-PE/ENb-PE and TRAIL combination in highly
TRAIL-resistant patient lines was associated with those established
lines expressing both 2 cognate receptors. Either IL13-PE (25
ng/ml) or ENb-PE (25 ng/ml) with TRAIL (500 ng/ml) post-treatment
induced apoptosis through the cleavage of caspase-8 and caspase-9
(Fig. 4A). Furthermore,
IL13-PE/ENb-PE and TRAIL co-treatment of the GCSs resulted in
significant cell death in resistant GSCs expressing both EGFR and
IL13Rα2 (Fig. 4B and C). These
findings revealed that TRAIL combination with targeted toxins
induced the mechanism-based killing of TRAIL-resistant GSC lines in
a similar manner with treated GBM cell lines (Fig. 5).
Discussion
The present study demonstrated a mechanism-based
rationale for combining targeted PE cytotoxins with TRAIL to
trigger tumor-killing in resistant GBMs. Targeted toxins result in
the upregulation of DR4/5 and the downregulation of FLIP and XIAP,
leading to the sensitization of resistant GBMs to TRAIL-mediated
apoptosis.
TRAIL is a novel pro-apoptotic cytokine targeting
cancer cells, whilst sparing healthy cells. A number of TRAIL-based
therapies have encountered difficulties in phase I/II clinical
trials with no marked success for use in phase III trials. On the
other hand, the authors, as well as other researchers have reported
that GBMs display various degrees of response to TRAIL monotherapy,
similar to a number of other cancer cells (16,17,39). There is therefore an urgent need
to overcome TRAIL resistance with combination therapy approaches.
To this end, a number of TRAIL sensitizers, as well as other
anticancer agents targeting different death-inducing mechanisms are
underway for the establishment of tumor regression and the
prevention of tumor reoccurrence. Barriers to these approaches
include the short half-life of therapeutic agents, blood-brain
barrier characteristics, normal tissue toxicity and limited
therapeutic effect caused by resistance mechanisms. In addition,
current TRAIL sensitizers are synthetic compounds and have similar
difficulties in therapeutic applications. In the present study,
secretable targeted toxins were developed that function as a TRAIL
sensitizers in GBMs. These findings have potential for development
as a new methodology for GBM therapy, combining three advantages:
i) Localized therapy ensures high concentration of the toxin at the
tumor site; ii) breaking TRAIL resistance with mechanism-oriented
sensitization; iii) maximizing residual tumor eradication using a
clinically relevant GBM resection model.
In tumor cells with a low or no response to TRAIL,
the cells can develop resistance to TRAIL via different mechanisms.
Previously, it was demonstrated that GBM cells have varying levels
of DR4/5 expression, which are the major determinants of
TRAIL-mediated apoptosis (16,39). Resistance to TRAIL can occur
either via the upregulation of the death receptors (DR4/5) or the
downregulation of anti-apoptotic proteins. cFLIP tends to bind the
death domain of DR4/5 through the FADD adaptor protein and competes
with procaspase-8, and therefore inhibits the activation of
downstream apoptotic cascades. Decreased cFLIP levels can enhance
signaling through caspase-8 and therefore the downstream caspases
(11-17). The depletion of some
anti-apoptotic proteins, such as XIAP, can trigger the intrinsic
apoptotic pathway, which leads to the cleavage of caspase-9. As the
present study detected both the upregulation of DR4/5 and the
downregulation of FLIP and XIAP, it was considered that both
mechanisms may play a role in the IL13-PE-mediated enhancement of
TRAIL-induced apoptosis. This indicates that toxin and TRAIL
treatments result in synergistic cell death involving the
activation of both the extrinsic and intrinsic apoptotic pathways.
Furthermore, a sensitizing agent that can upregulate DR4/5
expression potentially induces death signals through TRAIL-mediated
apoptosis. Additionally, a sensitizing agent may also influence
downstream effectors of the apoptotic cascade, such as FLIP, XIAP,
Bcl-2 family proteins, and others that can lead to increased cancer
cell death. In the present study, it was demonstrated that targeted
toxins (either IL13-PE or ENb-PE) can sensitize even highly
resistant cancer cells to TRAIL within 24 h and subsequently TRAIL
can kill these cells in <24 h. As shown in Fig. S2, the PE-mediated inhibitory
mechanism does not involve 100% translational inhibition within 24
h. This can be explained as follows: The PE toxin (domain III)
interaction with EF-2 makes EF-2 dysfunctional, while untargeted
EF-2 can still guide translation and be involved in maintaining
protein synthesis, resulting in upregulation of some proteins. As
such, DR4/5 is upregulated which then leads to more TRAIL binding
and enhanced death signaling. Such protein dynamics may also
explain why only approximately 50% of the protein synthesis
inhibition occurs upon toxin treatment. This also requires certain
time points to be chosen for TRAIL treatment post-PE treatment. The
present study observed the upregulation of DR4/5 at approximately
24 h and therefore optimized the TRAIL treatment time point at 24 h
post-PE treatment (Fig. 2B and
F).
As the present study detected both the upregulation
of DR4/5 and the downregulation of FLIP and XIAP, it was considered
that both mechanisms may play a role in the IL-13PE/ENb-PE-mediated
enhancement of TRAIL-induced apoptosis. This indicates that toxin
and TRAIL treatments result in synergistic cell death involving the
activation of both extrinsic and intrinsic apoptotic pathways. To
our knowledge, this is the first study linking targeted
toxin-mediated TRAIL sensitization with DR4/5 modulation. The
findings presented herein shed new light on the mechanisms through
which DR4/5 upregulation is regulated by targeted toxins. One
possibility is that unfolded proteins released during toxin
processing (protein synthesis inhibition at the translational
level) within the cell may cause ER stress and this may then result
in DR4/5 upregulation through reactive oxygen species (ROS)
activation. ROS are involved in the upstream modulation of TRAIL
signaling, which leads to an enhanced TRAIL sensitivity in various
cancer cells by inducing the expression of death receptors
(40). Alternatively, the
mechanism can be related to the modulation of transcription factors
which then drives DR4/5 gene expression. Further studies are
required to clarify the DR4/5 upregulation mechanism in
IL13-PE/ENb-PE-targeted toxin-treated cancer cells. Moreover, it
was demonstrated that GBM treatment with targeted toxins
downregulated FLIPL, resulting in the activation of
caspase-8-mediated apoptosis, since FLIP acts as a caspase-8/10
inhibitor and eventually an intracellular blocker of the apoptotic
TRAIL signal. Downstream of the apoptotic cascade, toxin-mediated
therapy decreases XIAP levels, which releases the blockage of
caspase-3 and caspase-9; hence, it enhances intracellular death
signals. As a proof of concept, wild-type TRAIL-resistant LN229
(LN229-wt) cells were engineered to overexpress IL13Rα2. The cells
were then treated with IL13-PE and TRAIL to evaluate TRAIL
sensitization upon cognate receptor expression. The results were in
accordance with the current findings on ENb-PE-treated LN229 cells.
ENb-PE-treated LN229 and IL13-PE pre-treatment of LN229-IL13Rα2
cells upregulated DR4/5, depleted FLIP and XIAP, and resulted in
significant cell death when combined with TRAIL. In line with this
finding, LN229-wt cells did not exhibit any significant decrease in
viability upon treatment with neither S-TRAIL alone nor IL13-PE and
S-TRAIL, since LN229-wt lacks the cognate IL13Rα2 receptor and is
highly resistant to TRAIL. These results reveal that toxin-mediated
TRAIL sensitization might occur in any resistant GBMs with cognate
receptor expression.
Moreover, TRAIL semi-sensitive GBMs undergo superior
cell death by the combination of TRAIL with targeted toxins.
According to the current findings, in GBMs, caspase-8-dependent
apoptosis occurred induced by TRAIL. Caspase-9-mediated apoptosis
also occurred due to the enhancement of death-inducing signals; the
cleaved form of caspase-9 was detected in only the toxin- and
TRAIL-treated groups suggesting that a subpopulation of cancer
cells (in terms of TRAIL response) in semi-sensitive GBMs exhibit
TRAIL resistance. In these cells, caspase-9-mediated apoptosis is
most likely induced in the presence of targeted toxins. Therefore,
in addition to the attenuation of anti-apoptotic proteins,
crosstalk between extrinsic and intrinsic apoptosis pathways may
exist explaining toxin and TRAIL co-operation. On the other hand,
targeted toxins can act as inhibitors of protein synthesis and lead
to cell death when used alone. The key point is that the PE toxins
alone lead to cell cycle arrest at 24 h and the killing effect is
measured in the following 48 h. For this reason, 24 h pre-treatment
with PE toxins is most likely sufficient to modulate protein levels
affected by synthesis inhibition, which then results in DR4/5
upregulation and depletion of anti-apoptotic proteins. To
summarize, in TRAIL-resistant GBMs, targeted toxins contribute to
cancer cell death via two dynamic mechanisms: i) TRAIL
sensitization in the first 24 h and subsequent TRAIL-mediated
apoptosis; ii) toxin-mediated killing via protein synthesis
blockage. To sum up, these two modes of toxin action can enable the
selective killing of GBMs in a synergistic manner using the
specific advantages of TRAIL and targeted PE toxin engagement.
Since GBM cells-similar to a number of other cancer
cells-exhibit some level of resistance to both TRAIL and PE
cytotoxins, the selective killing of primary GBMs with a
combination of targeted toxins and TRAIL is a favorable approach as
the combination induces cell death via different mechanisms. To
highlight the clinical potential of this strategy and considering
the heterogeneity of patient-derived GSCs to TRAIL therapy, the
present study analyzed toxin-directed TRAIL sensitization in a
panel of primary GBMs. The data demonstrated that both IL13-PE and
ENb-PE cytotoxins overcame TRAIL resistance and with TRAIL
involvement, greater cell death was achieved in primary GBM23 and
GBM64 cells expressing target receptors.
To the best of our knowledge, this is the first
report of TRAIL sensitization via the upregulation of DR4/5 by
targeted toxins. Since toxin and TRAIL co-operation leads to the
orchestrated killing of GBM cells, it will be of great interest to
utilize the same strategy for other malignancies, which are
difficult to treat with TRAIL monotherapy. Furthermore, the
biochemically-proven TRAIL sensitization effects of PE in
patient-derived GSC lines reveal the clinical importance of the
current findings and provides a novel strategy for GBM treatment
modalities.
Supplementary Data
Availability of data and materials
All the materials used and the data generated are
included in the present manuscript or are available from the
corresponding author upon reasonable request.
Authors' contributions
NK designed the study, and was responsible for the
provision of the study material, collection and assembly of data,
data analysis and interpretation, manuscript writing, revision and
the final approval of the manuscript. DS designed the study, and
was involved in the collection and assembly of data, data analysis
and interpretation, manuscript writing, and the final approval of
the manuscript. ERL was involved in the provision of the study
material, collection and assembly of data, data analysis and
interpretation, and the final approval of manuscript. KS was
involved in the conception and design of the study, and in the
provision of study material, data analysis and interpretation,
manuscript writing, revision and the final approval of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study used patient derived established
GSC (glioma stem cell) lines, the use of which was approved by the
Massachusetts General Hospital (MGH) Ethics Committee.
Patient consent for publication
Not applicable.
Competing interests
KS owns equity in and is a member of the Board of
Directors of AMASA Therapeutics, a company developing stem
cell-based therapies for cancer. The interests of KS were reviewed
and are managed by Brigham and Women's Hospital and Partners
HealthCare in accordance with their conflict of interest policies.
The authors declare no competing financial interests.
Acknowledgments
The authors would like to thank Dr Deepak Bhere
(Center for Stem Cell Therapeutics, Brigham and Woman's Hospital,
Harvard Stem Cell Institute, Cambridge, MA, USA) and Dr Clemens
Reinshagen (Center for Stem Cell Therapeutics, Brigham and Woman's
Hospital) for assisting with the western blot analysis and Dr
Hiroaki Wakimoto (Department of Neurosurgery, Massachusetts General
Hospital, Harvard Medical School, Boston, MA, USA) for the critical
reading of the manuscript.
Funding
The present study was supported by RO1 CA138922 (to KS), R01
CA201148 (to KS) and TUBITAK BIDEB#2211-A (to NK).
References
|
1
|
Su Z, Zang T, Liu ML, Wang LL, Niu W and
Zhang CL: Reprogramming the fate of human glioma cells to impede
brain tumor development. Cell Death Dis. 5:e14632014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martinez R, Rohde V and Schackert G:
Different molecular patterns in glioblastoma multiforme subtypes
upon recurrence. J Neurooncol. 96:321–329. 2010. View Article : Google Scholar :
|
|
5
|
Bastien JI, McNeill KA and Fine HA:
Molecular characterizations of glioblastoma, targeted therapy, and
clinical results to date. Cancer. 121:502–516. 2015. View Article : Google Scholar
|
|
6
|
Karsy M, Gelbman M, Shah P, Balumbu O, Moy
F and Arslan E: Established and emerging variants of glioblastoma
multiforme: Review of morphological and molecular features. Folia
Neuropathol. 50:301–321. 2012. View Article : Google Scholar
|
|
7
|
Cuddapah VA, Robel S, Watkins S and
Sontheimer H: A neurocentric perspective on glioma invasion. Nat
Rev Neurosci. 15:455–465. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carlsson SK, Brothers SP and Wahlestedt C:
Emerging treatment strategies for glioblastoma multiforme. EMBO Mol
Med. 6:1359–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fialho AM, Salunkhe P, Manna S, Mahali S
and Chakrabarty AM: Glioblastoma multiforme: Novel therapeutic
approaches. ISRN Neurol. 2012:6423452012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim K, Fisher MJ, Xu SQ and el-Deiry WS:
Molecular determinants of response to TRAIL in killing of normal
and cancer cells. Clin Cancer Res. 6:335–346. 2000.PubMed/NCBI
|
|
11
|
Suliman A, Lam A, Datta R and Srivastava
RK: Intracellular mechanisms of TRAIL: apoptosis through
mitochondrial-dependent and -independent pathways. Oncogene.
20:2122–2133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stuckey DW and Shah K: TRAIL on trial:
Preclinical advances in cancer therapy. Trends Mol Med. 19:685–694.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bagci-Onder T, Agarwal A, Flusberg D,
Wanningen S, Sorger P and Shah K: Real-time imaging of the dynamics
of death receptors and therapeutics that overcome TRAIL resistance
in tumors. Oncogene. 32:2818–2827. 2013. View Article : Google Scholar :
|
|
14
|
Deng Y, Lin Y and Wu X: TRAIL-induced
apoptosis requires Bax-dependent mitochondrial release of
Smac/DIABLO. Genes Dev. 16:33–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kauer TM, Figueiredo JL, Hingtgen S and
Shah K: Encapsulated therapeutic stem cells implanted in the tumor
resection cavity induce cell death in gliomas. Nat Neurosci.
15:197–204. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bagci-Onder T, Wakimoto H, Anderegg M,
Cameron C and Shah K: A dual PI3K/mTOR inhibitor, PI-103,
cooperates with stem cell-delivered TRAIL in experimental glioma
models. Cancer Res. 71:154–163. 2011. View Article : Google Scholar
|
|
17
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar
|
|
18
|
Du W, Uslar L, Sevala S and Shah K:
Targeting c-Met receptor overcomes TRAIL-resistance in brain
tumors. PLoS One. 9:e954902014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Finlay D, Richardson RD, Landberg LK,
Howes AL and Vuori K: Novel HTS strategy identifies
TRAIL-sensitizing compounds acting specifically through the
caspase-8 apoptotic axis. PLoS One. 5:e133752010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Qiu J, Wang Z, Huang N, Li X, Li
Q, Zhang Y, Zhao C, Luo C, Zhang N, et al: In vitro and in vivo
anti-tumor activities of anti-EGFR single-chain variable fragment
fused with recombinant gelonin toxin. J Cancer Res Clin Oncol.
138:1081–1090. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Horita H, Thorburn J, Frankel AE and
Thorburn A: EGFR-targeted diphtheria toxin stimulates TRAIL killing
of glioblastoma cells by depleting anti-apoptotic proteins. J
Neurooncol. 95:175–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stuckey DW, Hingtgen SD, Karakas N, Rich
BE and Shah K: Engineering toxin-resistant therapeutic stem cells
to treat brain tumors. Stem Cells. 33:589–600. 2015. View Article : Google Scholar :
|
|
23
|
Thaci B, Brown CE, Binello E, Werbaneth K,
Sampath P and Sengupta S: Significance of interleukin-13 receptor
alpha 2-targeted glioblastoma therapy. Neuro Oncol. 16:1304–1312.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heimberger AB, Suki D, Yang D, Shi W and
Aldape K: The natural history of EGFR and EGFRvIII in glioblastoma
patients. J Transl Med. 3:382005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Terabe M, Park JM and Berzofsky JA: Role
of IL-13 in regulation of anti-tumor immunity and tumor growth.
Cancer Immunol Immunother. 53:79–85. 2004. View Article : Google Scholar
|
|
26
|
Maletinska L, Blakely EA, Bjornstad KA,
Deen DF, Knoff LJ and Forte TM: Human glioblastoma cell lines:
Levels of low-density lipoprotein receptor and low-density
lipoprotein receptor-related protein. Cancer Res. 60:2300–2303.
2000.PubMed/NCBI
|
|
27
|
Wolf P and Elsasser-Beile U: Pseudomonas
exotoxin A: From virulence factor to anti-cancer agent. Int J Med
Microbiol. 299:161–176. 2009. View Article : Google Scholar
|
|
28
|
Weldon JE and Pastan I: A guide to taming
a toxin-recombinant immunotoxins constructed from Pseudomonas
exotoxin A for the treatment of cancer. FEBS J. 278:4683–4700.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kunwar S, Chang S, Westphal M, Vogelbaum
M, Sampson J, Barnett G, Shaffrey M, Ram Z, Piepmeier J, Prados M,
et al: Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel
wafers for recurrent glioblastoma. Neuro Oncol. 12:871–881. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Auffinger B, Thaci B, Nigam P, Rincon E,
Cheng Y and Lesniak MS: New therapeutic approaches for malignant
glioma: In search of the Rosetta stone. F1000 Med Rep. 4:182012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van de Water JA, Bagci-Onder T, Agarwal
AS, Wakimoto H, Roovers RC, Zhu Y, Kasmieh R, Bhere D, Van Bergen
en Henegouwen PM and Shah K: Therapeutic stem cells expressing
variants of EGFR-specific nanobodies have antitumor effects. Proc
Natl Acad Sci USA. 109:16642–16647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shah K, Hingtgen S, Kasmieh R, Figueiredo
JL, Garcia-Garcia E, Martinez-Serrano A, Breakefield X and
Weissleder R: Bimodal viral vectors and in vivo imaging reveal the
fate of human neural stem cells in experimental glioma model. J
Neurosci. 28:4406–4413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shah K, Tang Y, Breakefield X and
Weissleder R: Real-time imaging of TRAIL-induced apoptosis of
glioma tumors in vivo. Oncogene. 22:6865–6872. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wakimoto H, Kesari S, Farrell CJ, Curry WT
Jr, Zaupa C, Aghi M, Kuroda T, Stemmer-Rachamimov A, Shah K, Liu
TC, et al: Human glioblastoma-derived cancer stem cells:
Establishment of invasive glioma models and treatment with
oncolytic herpes simplex virus vectors. Cancer Res. 69:3472–3481.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gimple RC, Bhargava S, Dixit D and Rich
JN: Glioblastoma stem cells: Lessons from the tumor hierarchy in a
lethal cancer. Genes Dev. 33:591–609. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu G, Yuan X, Zeng Z, Tunici P, Ng H,
Abdulkadir IR, Lu L, Irvin D, Black KL and Yu JS: Analysis of gene
expression and chemoresistance of CD133+ cancer stem cells in
glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reinshagen C, Bhere D, Choi SH, Hutten S,
Nesterenko I, Wakimoto H, Le Roux E, Rizvi A, Du W, Minicucci C and
Shah K: CRISPR-enhanced engineering of therapy-sensitive cancer
cells for self-targeting of primary and metastatic tumors. Sci
Trans Med. 10:eaao32402018. View Article : Google Scholar
|
|
40
|
Dilshara MG, Jayasooriya RGPT, Molagoda
IMN, Jeong JW, Lee S, Park SR, Kim GY and Choi YH: Silibinin
sensitizes TRAIL-mediated apoptosis by upregulating DR5 through
ROS-induced endoplasmic reticulum stress-Ca(2+)-CaMKII-Sp1 pathway.
Oncotarget. 9:10324–10342. 2018. View Article : Google Scholar : PubMed/NCBI
|