Introduction
Asthma is the most common chronic childhood disease
(1). Neutrophilic asthma (NA) is
a subtype of asthma, which occurs in 15-25% of cases and responds
poorly to glucocorticoid treatment (2). NA is characterized by
neutrophil-mediated inflammation of the airway, and can result in
reduced bronchodilator reversibility and fixed airflow obstruction,
which are also the features of chronic obstructive pulmonary
disease (3,4). Understanding the immunopathology of
NA may result in the discovery of targeted treatments for patients
(3).
MicroRNAs (miRNAs/miRs) are small non-coding RNAs
that regulate the post-transcriptional expression of multiple genes
by complementary binding to the 3′ untranslated region (3′UTR) of
the target gene (5). miR-29a-3p
has been demonstrated to participate in a variety of diseases and
different pathological processes, including cell proliferation,
apoptosis, fibrosis and immunomodulation (6,7).
Epithelial-mesenchymal transition (EMT) is a process in which
epithelial cells lose the epithelial phenotype and undergo the
transition to typical mesenchymal characteristics. Recent evidence
has suggested that EMT is involved in airway remodeling and the
development of asthma (8,9).
More importantly, miR-29a-3p has been implicated in the inhibition
of gene expression in EMT and metastasis (6). However, the effect of miR-29a-3p on
EMT in NA remains unknown.
Thus, the present study aimed to investigate the
role of miR-29a-3p in NA. To study the role of miR-29a-3p in NA, a
mouse model of NA was established and these animals were compared
to normal controls. Both groups of mice were subjected to lung
function tests and histopathological analysis to confirm the
induction of the NA model. Human bronchial epithelial cells (16HBE)
were grown in culture, and in vitro experiments were
performed. The expression levels of miR-29a-3p, secreted protein
acidic rich in cysteine (SPARC) and EMT-related markers were
measured and luciferase reporter assays were performed to identify
the direct regulatory relationship between miR-29a-3p and
SPARC.
Materials and methods
Animals
Female C57BL/6 mice (n=12; certificate no. SCXK
2020-0003) aged 6-8 weeks and weighing 18-20 g were purchased from
Guangxi Medical University Animal Center (license no. SYXK
2020-0004). Mice were housed under specific pathogen-free
conditions with a relatively stable temperature (20-24°C) and
humidity (55±10%) at 12-h light/dark cycles. All animal experiments
were approved by The Ethics Committee of The First Affiliated
Hospital of Guangxi Medical University [approval no. 2019
(KY-E-035); Nanning, China].
Animal groups
Mice were randomly divided into the normal control
group (NC) and a NA group (NA) (n=6 in each group). The mouse model
with NA was established using previously outlined protocols
(10,11). Mice were sensitized by airway
delivery of 100 µg ovalbumin (OVA; Grade II & V;
Sigma-Aldrich; Merck KGaA) and 1 µg lipopolysaccharide (LPS;
Sigma-Aldrich; Merck KGaA) in a total volume of 50 µl PBS on
days 1, 7 and 14. The mixture was instilled along the posterior
oropharyngeal wall and inhaled into the airway, followed by a
challenge with 1% OVA aerosol for 1 h from day 21 for 7 consecutive
days. The mice in the NC group received the equivalent amount of
PBS treatment instead of OVA + LPS for sensitization and
challenge.
Lung function measurements
Mice were anesthetized with 1% pentobarbital sodium
(50 mg/kg body weight) by intraperitoneal injection. Measurements
of dynamic resistance were assessed using whole-body
plethysmography (Buxco® FinePointe Noninvasive Airway
Mechanics; Data Sciences International) and induced with
methacholine (Sigma-Aldrich; Merck KGaA) at doses of 12.5, 25 and
50 mg/ml 24 h after the final OVA challenge. Each mouse was exposed
to aerosolized PBS (baseline) for 3 min followed by the
administration of increasing concentrations of methacholine
solutions. Airway resistance [enhanced pause (Penh)] values were
then evaluated for 5 min. The results were expressed as the
percentage of the baseline Penh value for each concentration of
methacholine used (12).
Cell classification of bronchoalveolar
fluid (BALF)
Mice were sacrificed 24 h after the final
aerosolization. Cervical dislocation was used for euthanasia and
death was confirmed by the onset of rigor mortis, according to The
National Institutes of Health Guide for the Care and Use of
Laboratory Animals. The lungs were subjected to bronchoalveolar
lavage twice 24 h after the final aerosolization with 0.5 ml PBS
(recovery rate ≥80%) and the total volume of BALF was 0.8 ml. Total
and differential cell counts in BALF were determined by Diff-Quick
staining (Beijing Solarbio Science & Technology Co., Ltd.) for
1 min at room temperature, according to previously outlined
protocols (11).
Histopathological analysis
Lungs were fixed in 4% paraformaldehyde solution for
24 h at room temperature, and then subjected to gradient alcohol
dehydration and paraffin-embedding. Then, the lungs were sliced
into 5-7-µm thick sections and subsequently stained with
hematoxylin and eosin (H&E) at room temperature for 2-3 min. An
optical microscope (Olympus Corporation) was used to evaluate the
general inflammation using previously outlined protocols (11).
Cell culture and transfection
Human bronchial epithelial cells (16HBE) were
purchased from FuHeng Biology (cat. no. FH1013) and maintained in
Keratinocyte medium (cat. no. 2101; ScienCell Research
Laboratories, Inc.) in a humidified atmosphere of 5% CO2
at 37°C. Cells were authenticated using the STR genotype test and
passed the mycoplasma testing. Cell passage four was used for this
study. For lentiviral transfection,
pLV-h-SPARC-CMV-MCS-3FLAG-EF1-ZsGreen-PURO [Lentivirus (LV)-SPARC]
and pLV-CMV-MCS-3FLAG-EF1-Zs-Green-T2A-PURO (LV-control; both
purchased from Sangon Biotech Co., Ltd.) were transfected into 293T
cells (National Collection of Authenticated Cell Cultures; National
Science & Technology Infrastructure), 2nd generation system was
used [ratio of the lentiviral plasmid (10 µg):packaging
vector (10 µg):envelope (5 µg)]. For virus
collection, the supernatant was collected 48 and 72 h after
transfection of the packaged cells, centrifuged at 2,000 × g for 10
min to remove cell debris, and ultracentrifuged at 82,700 × g for
120 min, then the pellet was resuspend in the culture medium to
determine the titer. miR-29a-3p mimic and miR-29a-3p inhibitor were
provided by Sangon Biotech Co., Ltd. 16HBE cells (1×106
cells/ml) at 60-80% confluence were treated with LV-SPARC [at 50
multiplicity of infection (MOI)] for 24 h, and then a miR-29a-3p
mimic or miR-29a-3p inhibitor (100 nM) mixed with
Lipofectamine® 6000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) was transfected into cells at room
temperature for 5 min, and incubated for 24 h. The negative control
group (NeC; untreated cells) was set. miRNAs were labeled with FAM,
and transfection efficacy was determined by the expression of
immunofluorescence using fluorescence microscopy (Olympus
Corporation). The cells were lysed for reverse
transcription-quantitative (RT-q)PCR and western blotting. The
sequences were as follows: miR-29a-3p mimic, 5′-UAG CAC CAU CUG AAA
UCG GUU A-3′; miR-29a-3p inhibitor, 5′-UAA CCG AUU UCA GAU GGU GCU
A-3′; miR-29a-3p mimic control, 5′-UUU GUA CUA CAC AAA AGU ACU
G-3′; and miR-29a-3p inhibitor control, 5′-CAG UAC UUU UGU GUA GUA
CAA A-3′.
Luciferase reporter gene assay
The target gene of miR-29a-3p was predicted using
the online bioinformatics software, TargetScan3.1 (http://www.targetscan.org/vert_71/). Cells were
co-transfected with miR-29a-3p mimic or miR mimic control, and
psiCHECK-2-SPARC-3′-UTR-wild-type (WT) plasmid or a
psiCHECK-2-SPARC-3′-UTR-mutant (MUT) plasmid (Promega Corporation)
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Luciferase activity was measured 48 h after
transfection using Dual-Luciferase system (Promega Corporation) in
comparison with Renilla luciferase activity.
RT-qPCR
The lung samples and cultured cells were subjected
to RNA extraction using TRIzol (cat. no. 15596-026; Invitrogen;
Thermo Fisher Scientific, Inc.). cDNA for RNA and miRNA were
synthesized using the PrimeScript™ RT Reagent kit with gDNA Eraser
(cat. no. RR047A; Takara Bio, Inc.) and Mir-X™ miRNA First-Strand
Synthesis kit (cat. no. 638313; Takara Bio, Inc.) according to the
manufacturer's protocol, respectively. qPCR was performed using an
ABI 7500 Real-Time PCR instrument (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with TB Green® Premix Ex Taq™
II (Tli RNase H Plus) (cat. no. RR820A; Takara Bio, Inc.) using the
following primers: U6 forward, 5′-GCT TCG GCA GCA CAT ATA CTA AAA
T-3′ and reverse, 5′-CGC TTC ACG AAT TTG CGT GTC AT-3′; miR-29a-3p,
5′-CGC TAG CAC CAT CTG AAA TCG GTT A-3′; glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) forward, 5′-CCT CTG CGC CCT TGA GCT AGG A-3′
and reverse, 5′-CAC AAG AAG ATG CGG CCG TCT C-3′; SPARC forward,
5′-GCT CCC ATT GGC GAG TTT G-3′ and reverse, 5′-GAT GTA GTC CAG GTG
GAG CTT GTG-3′; E-cadherin forward, 5′-CAC CGA TGG TGA GGG TAC ACA
G-3′ and reverse, 5′-GGC TTC AGG AAT ACA TGG ACA AAG A-3′; Vimentin
forward, 5′-AAA GCG TGG CTG CCA AGA A-3′ and reverse, 5′-ACC TGT
CTC CGG TAC TCG TTT GA-3′; U6 (human) forward, 5′-CTC GCT TCG GCA
GCA CA-3′ and reverse, 5′-AAC GCT TCA CGA ATT TGC GT-3′; miR-29a-3p
(human) forward, 5′-TAG CAC CAT CTG AAA TCG GTT A-3′ and reverse,
5′-TGG TGT CGT GGA GTC G-3′; GAPDH (human) forward, 5′-GCA CCG TCA
AGG CTG AGA AC-3′ and reverse, 5′-TGG TGA AGA CGC CAG TGG A-3′;
SPARC (human) forward, 5′-ACA TAA GCC CAG TTC ATC ACC A-3′ and
reverse, 5′-ACA ACC GAT TCA CCA ACT CCA-3′; E-cadherin (human)
forward, 5′-GGA TTG CAA ATT CCT GCC ATT C-3′ and reverse,
5′-AACGTTGTCCCGGGTGTCA-3′; and vimentin (human) forward, 5′-GGA AGG
CGA GGA GAG CAG GAT T-3′ and reverse, 5′-TTC AAG GTC ATC GTG ATG
CTG AGA AG-3′. qPCR thermocycling conditions were as follows:
Denaturation at 95°C for 30 sec, annealing at 95°C for 5 sec, and
extension at 60°C for 34 sec for 40 cycles. The miRNA and mRNA
levels were normalized to U6 or GAPDH, respectively. The
fold-change for each gene was calculated using the
2−ΔΔCq method (13).
Western blotting
The lung tissues and cells were lysed in a mixture
of RIPA lysis buffer (Beyotime Institute of Biotechnology) and
protease inhibitors (Thermo Fisher Scientific, Inc.). The protein
concentration of samples was measured using a BCA protein assay kit
(Applygen Technologies, Inc.). Equal amounts of total protein (20
µl) were loaded onto 10% sodium dodecyl
sulfate-polyacrylamide gels and subjected to electrophoresis, and
then separated proteins were electro-transferred onto PVDF
membranes (Thermo Fisher Scientific, Inc.). Membranes were blocked
with 5% skimmed milk or 5% BSA (cat. no. A8020; Beijing Solarbio
Science & Technology Co., Ltd.) for 1 h and incubated at 4°C
overnight with primary antibodies against the following proteins:
SPARC (cat. no. 8725; 1:1,000; Cell Signaling Technology, Inc.),
E-cadherin (cat. no. 3195; 1:1,000; Cell Signaling Technology,
Inc.), vimentin (cat. no. 5741; 1:1,000; Cell Signaling Technology,
Inc.), ERK (cat. no. 4695; 1:1,000; Cell Signaling Technology,
Inc.), p-ERK (cat. no. 4370; 1:2,000; Cell Signaling Technology,
Inc.) and GADPH (cat. no. 21612; 1:5,000; Signalway Antibody LLC).
Following which, membranes were incubated at room temperature for 1
h with a secondary antibody (anti-rabbit IgG; cat. no. L3012;
1:5,000; Signalway Antibody LLC). An ECL kit (cat. no. BL520A;
Biosharp Life Sciences) was used to view the protein bands, and
ImageJ software version 1.8.0 (National Institutes of Health) was
applied for the analysis of the relative intensities of protein
bands.
Statistical analysis
Data were analyzed using GraphPad Prism 6 software
(GraphPad Software, Inc.) and expressed as the mean ± SD of three
independent experimental repeats. The statistical significance was
determined by an unpaired Student's t-test or one-way ANOVA with
Tukey's post hoc test for multiple comparisons. For non-normally
distributed data, the significance was determined using a
Kruskal-Wallis test with a post hoc Dunn's multiple comparison
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Establishment of the NA mouse model
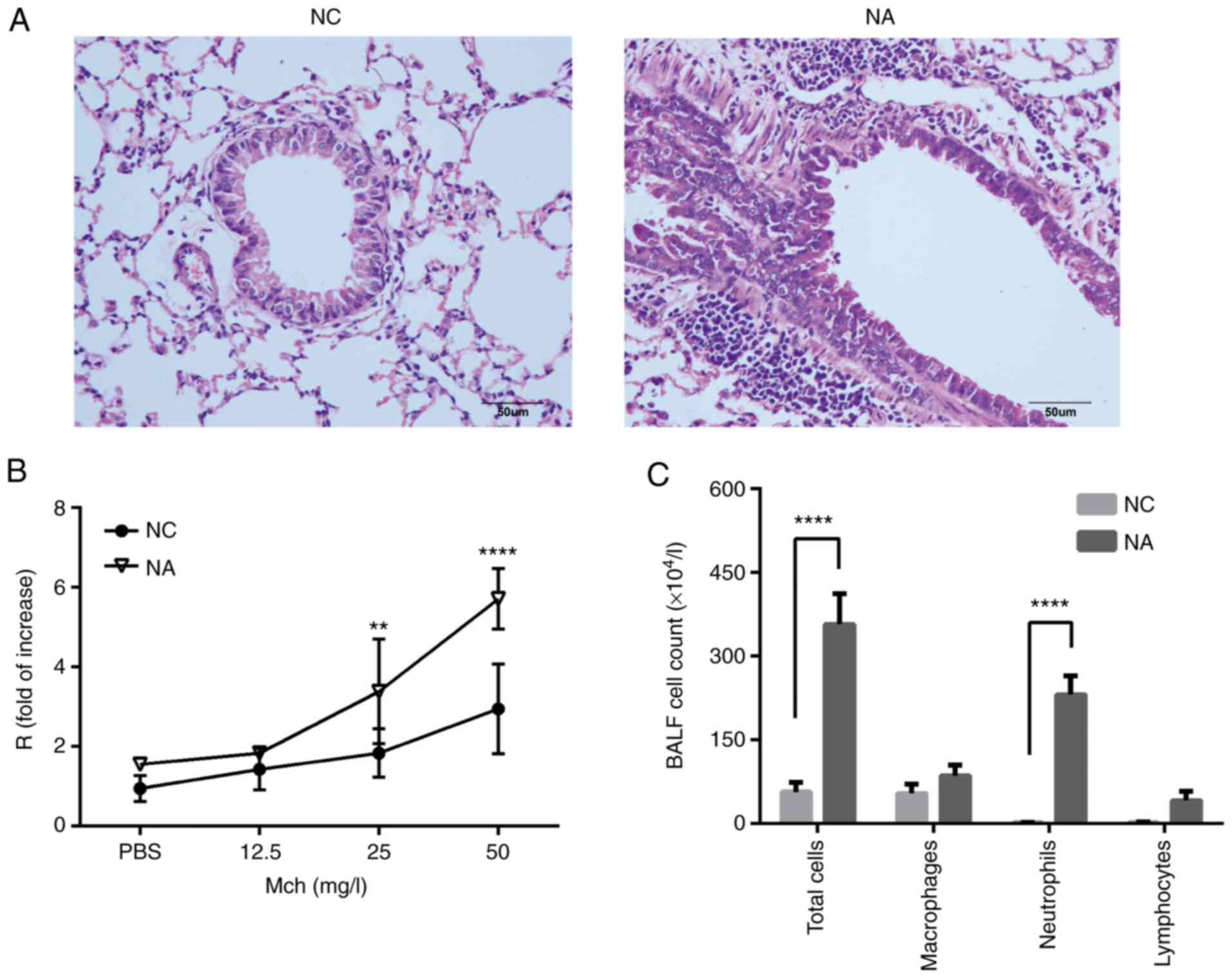
The mouse model was established and presented with
the following features of NA: Presence of bronchial
hyperresponsiveness, the accumulation of inflammatory cells in the
lung, particularly increased neutrophils, as well as a high number
of neutrophils in the BALF. As shown in the H&E staining
images, NA mice exhibited a thick basement membrane and increased
inflammatory cell infiltration around the bronchus (Fig. 1A). Airway resistance was elevated
in the NA group when compared with the NC group after methacholine
challenges at doses of 25 and 50 mg/ml (Fig. 1B). Compared with the NC mice, the
total number of cells and neutrophils were significantly increased,
while no eosinophilic cells were observed in the BALF of NA mice
(Fig. 1C).
miR-29a-3p and SPARC are involved in
NA
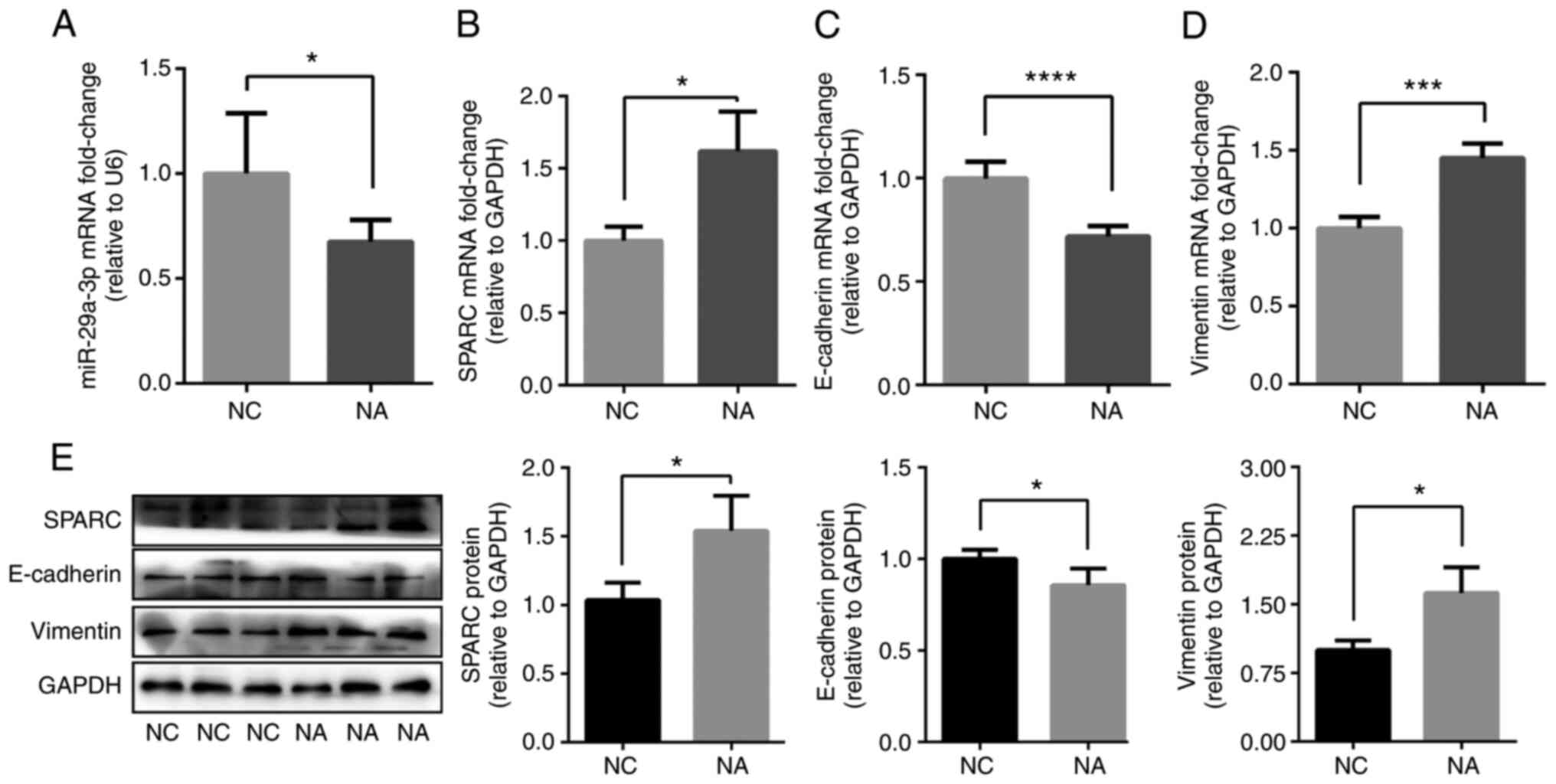
miR-29a-3p and SPARC expression levels were
determined via RT-qPCR. Compared with the NC group, miR-29a-3p
expression was decreased (Fig.
2A), while SPARC expression was increased in NA mice (Fig. 2B and E). The levels of
EMT-related markers in the lung of NA mice were then determined.
The results showed that the mRNA and protein expression levels of
the epithelial marker, E-cadherin, were decreased in NA mice
compared with the NC group (Fig. 2C
and E). On the contrary, the mRNA and protein expression levels
of the mesenchymal marker, vimentin, were increased in NA mice
compared with the NC group (Fig. 2D
and E). The data revealed that the NA mouse model exhibited an
EMT phenotype with decreased levels of miR-29a-3p and increased
SPARC.
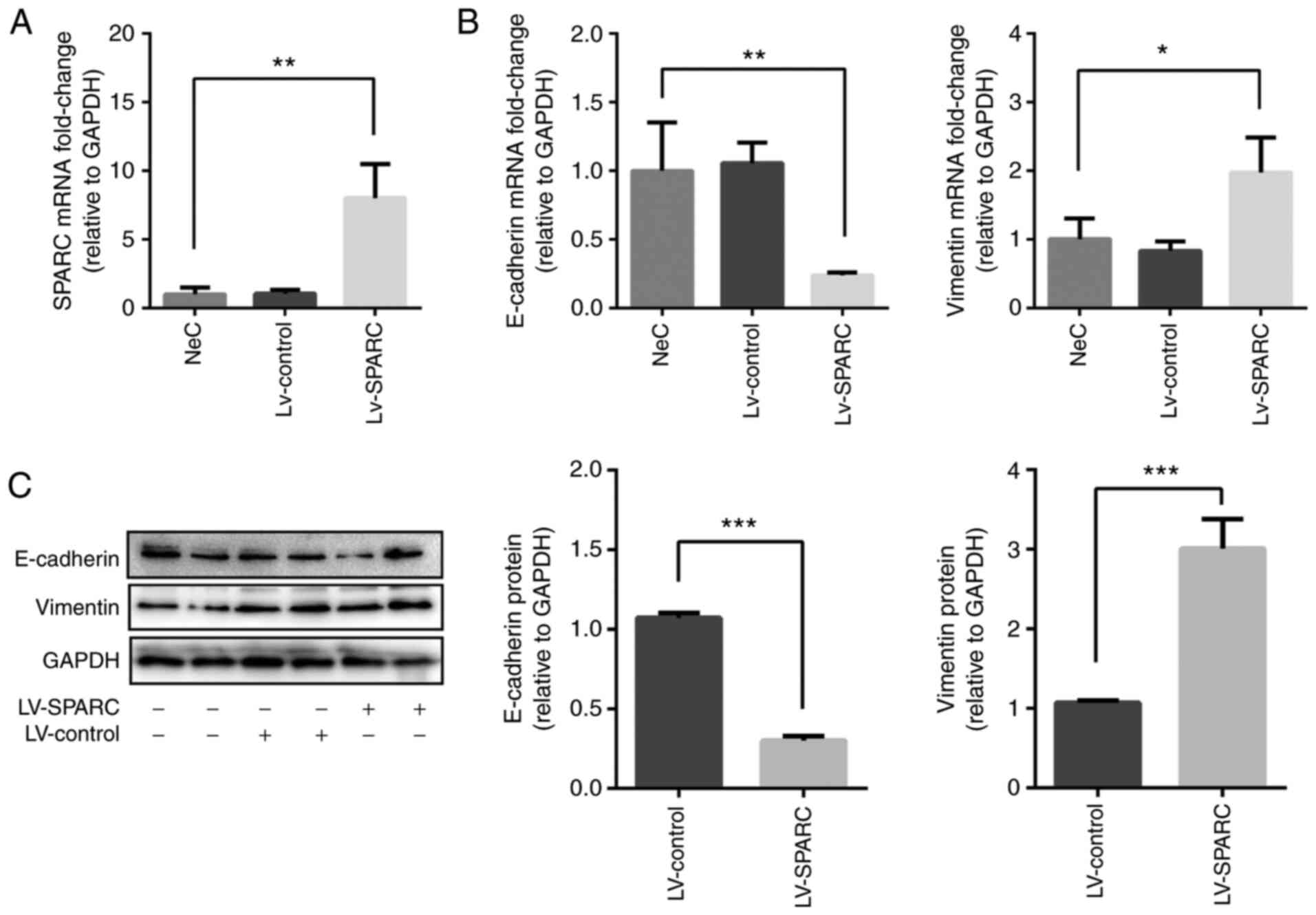
SPARC regulates EMT in 16HBE cells
Next, the function of SPARC during EMT was
investigated in 16HBE cells. SPARC was overexpressed in 16HBE cells
following LV-SPARC transfection (Fig. 3A). The mRNA expression of
E-cadherin was significantly reduced in the LV-SPARC group compared
with the NeC group, whereas mRNA expression of vimentin was
significantly increased by LV-SPARC administration (Fig. 3B). E-cadherin protein expression
was reduced, while vimentin expression was elevated in
SPARC-treated 16HBE cells compared with the LV-control group, as
analyzed via western blotting (Fig.
3C), suggesting that SPARC participated in the EMT of 16HBE
cells.
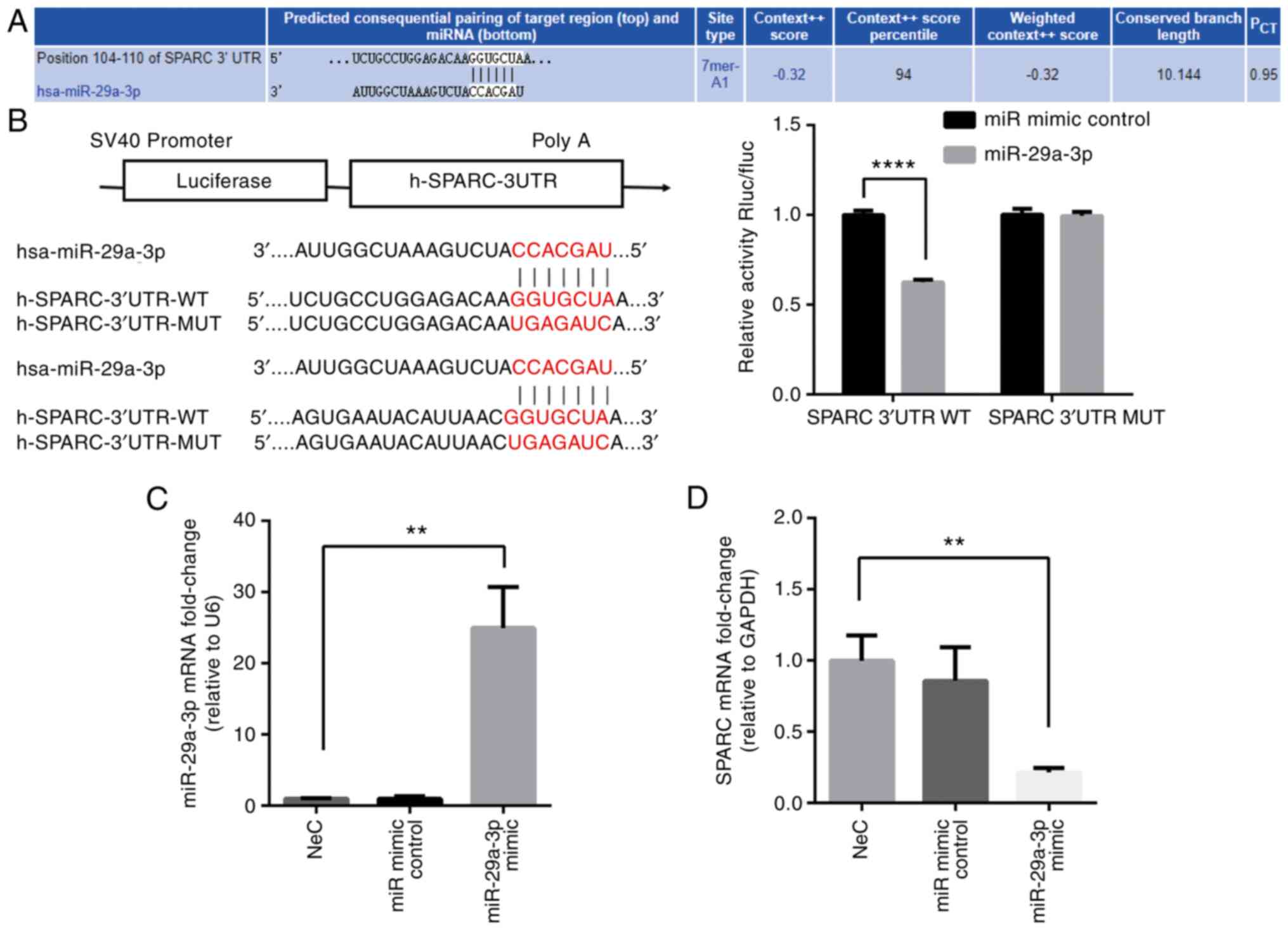
miR-29a-3p regulates SPARC expression in
vitro
miRNAs are hypothesized to be involved in various
diseases where they inhibit gene expression through binding to the
3′UTR of the target gene (6). In
the present study, the target gene of miR-29a-3p was predicted
using the online bioinformatics software, TargetScan. The results
showed that SPARC was a target gene for miR-29a-3p and the
predicted binding site is shown in Fig. 4A. Dual-luciferase reporter gene
assays demonstrated the direct regulation of miR-29a-3p by SPARC
(Fig. 4B). Furthermore, the
expression of SPARC in cultured 16HBE cells treated with a
miR-29a-3p mimic was determined. miR-29a-3p expression following
transfection is shown in Fig.
4C. It was found that SPARC mRNA expression was reduced after
transfection of 16HBE cells with a miR-29a-3p mimic compared with
the NeC group (Fig. 4D). These
data supported the concept that reducing the expression of
miR-29a-3p can regulate the mRNA synthesis of SPARC.
Effect of miR-29a-3p on EMT in 16HBE
cells
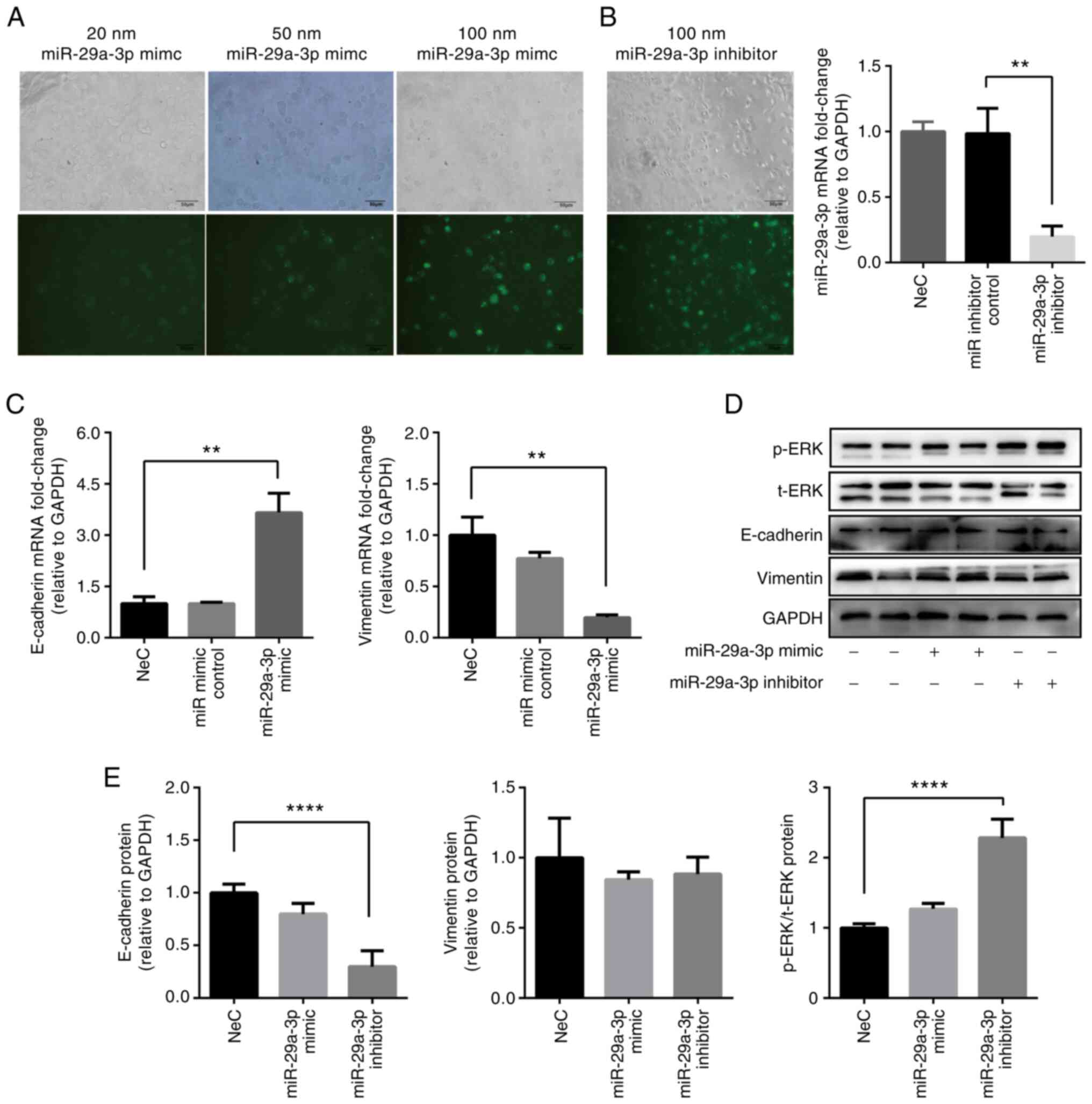
Transfection efficacy was higher in 16HBE cells
administrated with miR-29a-3p mimic at 100 nM, rather than at 20 or
50 nM, as determined using fluorescence microscopy (Fig. 5A). Transfection of 16HBE cells
with a miR-29a-3p inhibitor was shown to successfully induce the
knockdown of miR-29a-3p compared with the miR inhibitor control
group (Fig. 5B). E-cadherin
expression was increased, while vimentin expression was decreased
in 16HBE cells transfected with the miR-29a-3p mimic at 100 nM
compared with the NeC group, as measured via RT-qPCR (Fig. 5C). E-cadherin expression was
decreased, while p-ERK was increased in the miR-29a-3p inhibitor
group compared with the NeC group, which was verified via western
blotting (Fig. 5D and E).
Vimentin protein was not affected by administration of the
miR-29a-3p inhibitor. These data indicated that miR-29a-3p
regulated the mRNA expression of EMT-related markers, while the
effect on protein expression was not significant. We speculated
that a post-translational modification may be involved in the
protein expression process. p-ERK expression increased after
inhibitor administration, implying that ERK may participate in the
process of EMT formation.
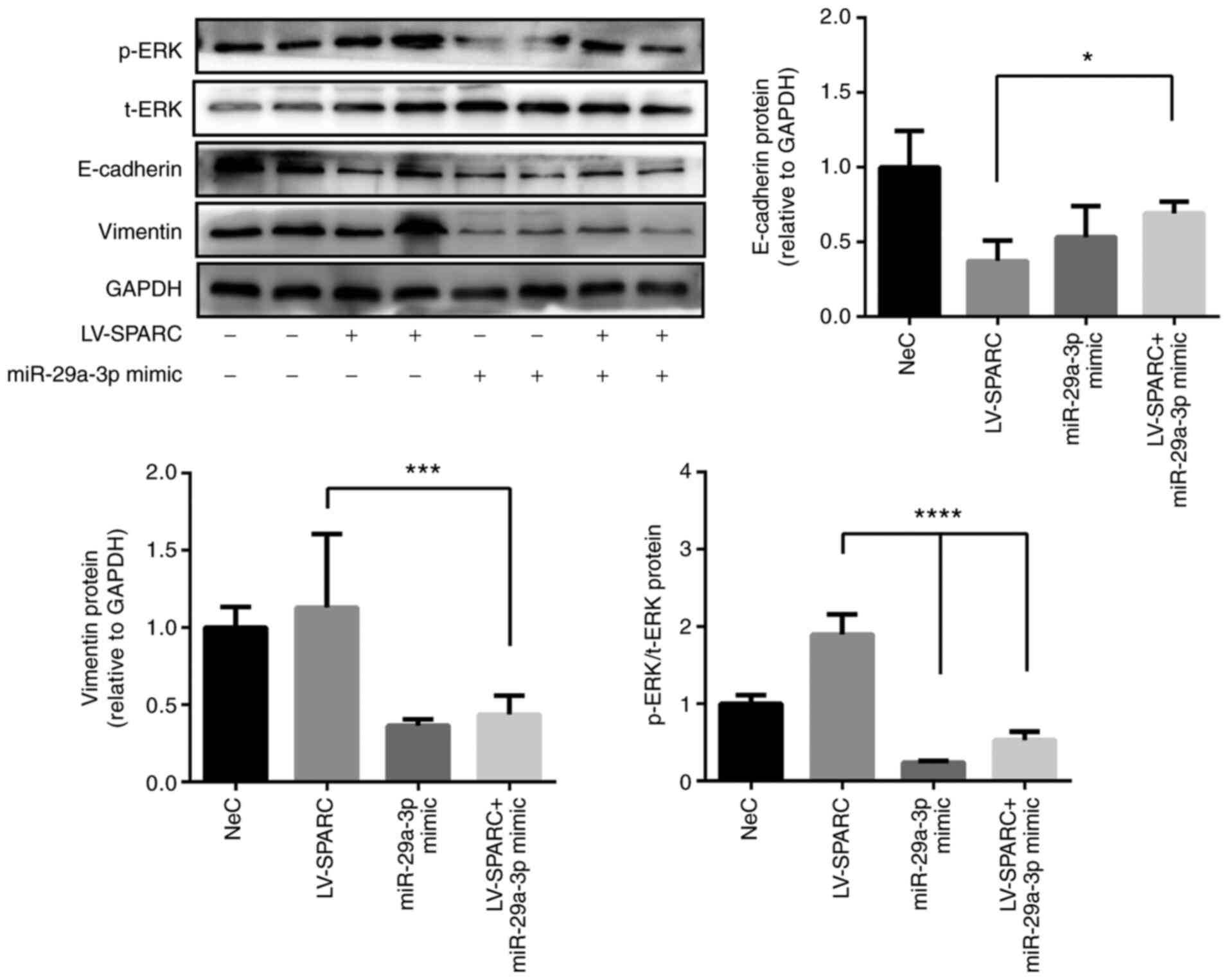
miR-29a-3p reduces SPARC-induced EMT in
16HBE cells
Transfection with the miR-29a-3p mimic increased
E-cadherin expression, and decreased vimentin expression in 16HBE
cells pretreated with LV-SPARC compared with transfection with
LV-SPARC alone, as determined via western blotting (Fig. 6). This suggested that miR-29a-3p
administration reversed the SPARC-induced EMT observed in 16HBE
cells. p-ERK expression was upregulated by transfection with
LV-SPARC alone, but this was reversed by administration of the
miR-29a-3p mimic (Fig. 6). These
results implied that ERK may be involved in the EMT process and
that it is regulated by miR-29a-3p.
Discussion
It is commonly known that patients with NA may
exhibit irreversible airflow obstruction and poor response to
treatment (3,4). Dysregulated extracellular matrix
(ECM) remodeling has also been reported to result in EMT, which is
considered to be involved in airway remodeling, which is the main
cause of fixed airflow limitations that occur during asthma attacks
(8). A previous study
demonstrated that the loss of miR-29a-3p is crucial for ECM protein
deposit and pulmonary fibrosis (14). However, findings concerning the
underlying function of miR-29a-3p in asthma are still limited. The
present study reported that miR-29a-3p was significantly decreased
in the lung of the mouse model with NA and may mediate EMT via
SPARC/ERK signaling pathway.
miR-29a-3p has been reported in a multitude of
diseases, ranging from intrauterine inflammation in extremely
preterm infants (15) and airway
epithelial remodeling in chronic obstructive pulmonary disease
(16) to proliferation and
differentiation in retinal progenitors (17). Studies have reported that
miR-29a-3p could be a therapeutic tool for certain diseases, such
as pulmonary fibrosis (14) and
Alzheimer's disease (18). The
existing data revealed that miR-29a-3p inhibited Th17 cell
differentiation and activation in a disease model with inflammatory
bowel syndrome (19). Our
previous study showed that NA was characterized by airway
neutrophil inflammation and Th17 cell dominance (11).
In the present study, it was that miR-29a-3p was
downregulated in NA when compared with the control mice. miR-29a-3p
is well-characterized by its ability to regulate ECM proteins,
including collagen, elastin and fibrillin, which play important
roles in airway remodeling (20). For instance, downregulation of
miR-29a-3p was found to result in the enhanced expression of the
collagen proteins in pulmonary fibrosis (21). Moreover, it could attenuate
TGF-β1-induced fibrosis in primary human endometrial stromal cells
(22) and mediate the remodeling
process of airway epithelial cells (16). Therefore, we suggest that
miR-29a-3p has a possible role in airway remodeling during NA. EMT
is one of the mechanisms of airway remodeling in asthma (23). In the present study, it was found
that the NA mouse model exhibited the EMT phenotype with decreased
miR-29a-3p levels, suggesting it may be involved in airway
remodeling of NA. It was demonstrated that miR-29a-3p regulated EMT
in cultured 16HBE cells, which was similar to the regulation of
miR-29a-3p in fibroblast accumulation in the kidneys (24). However, the present study also
demonstrated that while E-cadherin and vimentin mRNA expression
changed significantly, protein expression did not show the same
obvious alternations. We speculated that this phenomenon may be
related to post-translational modification. This deduction needs
further research and discussion, which is a limitation of this
study.
miRNAs regulate gene expression by binding to the
3′UTR of the target genes (25).
An increasing number of studies have demonstrated that miR-29a-3p
exhibits negative regulation of mRNAs encoding ECM proteins that
play essential roles in matrix deposition and EMT (21,22). In the present study, the online
bioinformatics software, TargetScan, was used to predict the target
genes for miR-29a-3p and SPARC. SPARC is a molecule that regulates
cell proliferation, differentiation, ECM deposition and EMT, as
well as participating in airway remodeling of chronic airways
disease (26). Studies have
shown that SPARC-null mice exhibited reduced collagen deposition
and tissue fibrosis (27,28).
SPARC inhibition may, therefore, represent a potential therapeutic
approach in fibrotic diseases (29). The present data showed that SPARC
was upregulated in the lung of the mouse model with NA, implying
that it may participate in airway remodeling. SPARC was reported to
participate in the EMT of cancer cells (30,31). In the current study, it was found
that SPARC overexpression increased vimentin and decreased
E-cadherin expression in vitro, suggesting that this
molecule may contribute to EMT of 16HBE cells.
The direct link between miR-29a-3p and SPARC was
verified using dual-luciferase reporter gene assays. Moreover, it
was found that miR-29a-3p administration reversed the SPARC-induced
EMT formation in 16HEB cells, supporting the concept that this
miRNA inhibited EMT by suppressing SPARC synthesis directly. The
data implied that the regulation of miR-29a-3p may participate in
EMT of a mouse model with NA. The regulation of miR-29a-3p by SPARC
was not verified in vivo, due to restricted access to the
animal center due to COVID-19, and this is a limitation of the
current study.
The potential signaling pathways involved in the
miR-29a-3p-mediated effects on the EMT of 16HBE cells were also
studied in the present study. The function of SPARC has been
reported to be mediated by the phosphorylation of c-Jun N-terminal
kinase and p38-MAPK signaling pathways on limbal epithelial stem
cells (32). SPARC is
upregulated during the EMT process in lung cancer cells and
overexpression of SPARC can induce the increased expression of
p-Akt and p-ERK (31).
Consistent with the existing studies, the present study found that
SPARC overexpression led to the elevated expression of p-ERK and
miR-29a-3p inhibited this increase. This suggested that ERK may
participate in the SPARC-induced EMT process mediated by miR-29a-3p
and SPARC observed in NA. Presenting the ratio of p-protein/total
protein may improve the evidence for the activation of this
signaling pathway. The band for total protein expression of ERK was
not present because of the experimental design and the restricted
laboratory access due to COVID-19, which was a limitation of this
study.
In conclusion, the current study found that
miR-29a-3p expression was decreased, while SPARC was elevated in a
mouse model of NA. SPARC was observed to induce EMT in cultured
16HBE cells in vitro and this was directly targeted by
miR-29a-3p, and may be mediated by p-ERK. The data suggested that
miR-29a-3p may participate in the airway remodeling of NA.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ conceived the present study, performed the
experiments, analyzed the data, and wrote the manuscript. JX and HS
conducted the animal studies. QW analyzed and interpreted the data.
GN designed and supervised the study, critically revised the
manuscript for intellectual content, and was primarily responsible
for the writing of the manuscript. XZ and JX confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of The First Affiliated Hospital of Guangxi Medical
University [approval no. 2019 (KY-E-035); Nanning, China].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Dev Sooranna of
Imperial College London (London, UK) for editing the
manuscript.
References
|
1
|
Szefler SJ, Fitzgerald DA, Adachi Y, Doull
IJ, Fischer GB, Fletcher M, Hong J, García-Marcos L, Pedersen S,
Østrem A, et al: A worldwide charter for all children with asthma.
Pediatr Pulmonol. 55:1282–1292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seys SF, Lokwani R, Simpson JL and Bullens
DMA: New insights in neutrophilic asthma. Curr Opin Pulm Med.
25:113–120. 2019. View Article : Google Scholar
|
|
3
|
Gibson PG and Foster PS: Neutrophilic
asthma: Welcome back! Eur Respir J. 54:19018462019.
|
|
4
|
Postma DS and Rabe KF: The asthma-COPD
overlap syndrome. N Engl J Med. 373:1241–1249. 2015. View Article : Google Scholar
|
|
5
|
Jonas S and Izaurralde E: Towards a
molecular understanding of microRNA-mediated gene silencing. Nat
Rev Genet. 16:421–433. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kwon JJ, Factora TD, Dey S and Kota J: A
systematic review of miR-29 in cancer. Mol Ther Oncolytics.
12:173–194. 2018. View Article : Google Scholar
|
|
7
|
Gao Y, Qiao H, Lu Z and Hou Y: miR-29
promotes the proliferation of cultured rat neural stem/progenitor
cells via the PTEN/AKT signaling pathway. Mol Med Rep.
20:2111–2118. 2019.PubMed/NCBI
|
|
8
|
Sohal SS, Ward C and Walters EH:
Importance of epithelial mesenchymal transition (EMT) in COPD and
asthma. Thorax. 69:7682014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haddad A, Gaudet M, Plesa M, Allakhverdi
Z, Mogas AK, Audusseau S, Baglole CJ, Eidelman DH, Olivenstein R,
Ludwig MS and Hamid Q: Neutrophils from severe asthmatic patients
induce epithelial to mesenchymal transition in healthy bronchial
epithelial cells. Respir Res. 20:2342019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilson RH, Whitehead GS, Nakano H, Free
ME, Kolls JK and Cook DN: Allergic sensitization through the airway
primes Th17-dependent neutrophilia and airway hyperresponsiveness.
Am J Respir Crit Care Med. 180:720–730. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Zhang M, Jiang M and Nong G:
Effect of IL-7 on Th17 cell responses in a mouse model of
neutrophilic asthma. Mol Med Rep. 22:1205–1112. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hou C, Kong J, Liang Y, Huang H, Wen H,
Zheng X, Wu L and Chen Y: HMGB1 contributes to allergen-induced
airway remodeling in a murine model of chronic asthma by modulating
airway inflammation and activating lung fibroblasts. Cell Mol
Immunol. 12:409–423. 2015. View Article : Google Scholar :
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Xiao J, Meng XM, Huang XR, Chung AC, Feng
YL, Hui DS, Yu CM, Sung JJ and Lan HY: miR-29 inhibits
bleomycin-induced pulmonary fibrosis in mice. Mol Ther.
20:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pavlek LR, Vudatala S, Bartlet CW,
Buhimschi IA, Buhimschi CS and Rogers LK: miR-29b is associated
with perinatal inflammation in extremely preterm infants. Pediatr
Res. 89:889–893. 2021. View Article : Google Scholar
|
|
16
|
Chi Y, Di Q, Han G, Li M and Sun B:
Mir-29b mediates the regulation of Nrf2 on airway epithelial
remodeling and Th1/Th2 differentiation in COPD rats. Saudi J Biol
Sci. 26:1915–1921. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Shen B, Zhang D, Wang Y, Tang Z,
Ni N, Jin X, Luo M, Sun H and Gu P: miR-29a regulates the
proliferation and differentiation of retinal progenitors by
targeting Rbm8a. Oncotarget. 8:31993–32008. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pereira PA, Tomás JF, Queiroz JA,
Figueiras AR and Sousa F: Recombinant pre-miR-29b for Alzheimer s
disease therapeutics. Sci Rep. 6:199462016. View Article : Google Scholar
|
|
19
|
Fukata T, Mizushima T, Nishimura J,
Okuzaki D, Wu X, Hirose H, Yokoyama Y, Kubota Y, Nagata K,
Tsujimura N, et al: The supercarbonate apatite-MicroRNA complex
inhibits dextran sodium sulfate-induced colitis. Mol Ther Nucleic
Acids. 12:658–671. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He Y, Huang C, Lin X and Li J: MicroRNA-29
family, a crucial therapeutic target for fibrosis diseases.
Biochimie. 95:1355–1359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cushing L, Kuang P and Lü J: The role of
miR-29 in pulmonary fibrosis. Biochem Cell Biol. 93:109–118. 2015.
View Article : Google Scholar
|
|
22
|
Li J, Cen B, Chen S and He Y: MicroRNA-29b
inhibits TGF-β1-induced fibrosis via regulation of the TGF-β1/Smad
pathway in primary human endometrial stromal cells. Mol Med Rep.
13:4229–4237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pain M, Bermudez O, Lacoste P, Royer PJ,
Botturi K, Tissot A, Brouard S, Eickelberg O and Magnan A: Tissue
remodelling in chronic bronchial diseases: From the epithelial to
mesenchymal phenotype. Eur Respir Rev. 23:118–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Srivastava SP, Hedayat AF, Kanasaki K and
Goodwin JE: microRNA crosstalk influences
epithelial-to-mesenchymal, endothelial-to-mesenchymal, and
macrophage-to-mesenchymal transitions in the kidney. Front
Pharmacol. 10:9042019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wong SL and Sukkar MB: The SPARC protein:
An overview of its role in lung cancer and pulmonary fibrosis and
its potential role in chronic airways disease. Br J Pharmacol.
174:3–14. 2017. View Article : Google Scholar :
|
|
27
|
Trombetta-Esilva J and Bradshaw AD: The
function of SPARC as a mediator of fibrosis. Open Rheumatol J.
6:146–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bradshaw AD: The role of secreted protein
acidic and rich in cysteine (SPARC) in cardiac repair and fibrosis:
Does expression of SPARC by macrophages influence outcomes? J Mol
Cell Cardiol. 93:156–161. 2016. View Article : Google Scholar
|
|
29
|
Wang JC, Lai S, Guo X, Zhang X, de
Crombrugghe B, Sonnylal S, Arnett FC and Zhou X: Attenuation of
fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res Ther.
12:R602010. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang F, Zhang Y, Da J, Jia Z, Wu H and Gu
K: Downregulation of SPARC expression decreases cell migration and
invasion involving epithelial-mesenchymal transition through the
p-FAK/p-ERK pathway in esophageal squamous cell carcinoma. J
Cancer. 11:414–420. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun W, Feng J, Yi Q, Xu X, Chen Y and Tang
L: SPARC acts as a mediator of TGF-β1 in promoting
epithelial-to-mesenchymal transition in A549 and H1299 lung cancer
cells. Biofactors. 44:453–464. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu J, Wang LY, Li CY, Wu JY, Zhang YT,
Pang KP, Wei Y, Du LQ, Liu M and Wu XY: SPARC promotes self-renewal
of limbal epithelial stem cells and ocular surface restoration
through JNK and p38-MAPK signaling pathways. Stem Cells.
38:134–145. 2020. View Article : Google Scholar
|