Introduction
IL-11 is a member of the IL-6 cytokine family and
has been reported to exert important effects on various liver
diseases (1). Lipid-laden
hepatocytes secrete IL-11, which acts via autocrine cis-signaling
and contributes to hepatocyte lipotoxicity in a reactive oxygen
species (ROS)-dependent manner (2). Furthermore, IL-11 receptor agonist
enhances the proliferation of hepatocytes and ameliorates oxidative
stress during acetaminophen (APAP)-induced liver injury. Among
acute liver injury models, IL-11 is markedly upregulated in
hepatocytes in response to APAP-induced liver injury (3) and liver ischemia (4). APAP is a commonly used drug for the
relief of pain and fever. Although it is considered to be safe at
therapeutic concentrations, its overdose can cause acute liver
damage (5). STAT3, a member of
the STAT family, can be activated by IL-6 family cytokines, and has
been found to exert anti-apoptotic and pro-proliferative effects on
APAP hepatotoxicity (6). These
functions mainly rely on the regulation of genes involved in cell
fate, such as the apoptosis regulators Bcl-2 and Bax (7,8).
Omega-3 polyunsaturated fatty acids (n-3 PUFAs),
which include eicosapentaenoic acid (EPA) and docosahexaenoic acid
(DHA), are important for human health. Several studies have
reported the effect of n-3 PUFAs on various liver diseases
(9-11). n-3 PUFAs attenuate systemic
inflammation by reducing the circulating level and gene expression
of IL-6 (9,12). Dietary n-3 PUFA intake is
associated with lower methylation at the IL-6 promoter, leading to
a decreased plasma IL-6 concentration (13). Reduced IL-6 production and
limited STAT3 phosphorylation have been observed in pancreatic
acinar cells treated with n-3 PUFAs during cerulein exposure
(14). Notably, previous studies
have reported that n-3 PUFAs inhibited the phosphorylation of STAT3
in various disease models, thereby influencing cell
differentiation, promoting cell death, inhibiting cell migration
and inducing autophagy (15-17). The complex of IL-6 and IL-6
receptor (IL-6R) binds to glycoprotein 130 (gp130), which dimerizes
and initiates intracellular STAT3 signaling (18). The presence of n-3 PUFAs may
reduce the surface expression of IL-6R and its association with
gp130 in lipid rafts, thereby leading to decreased downstream STAT3
activation (16). In addition,
n-3 PUFAs may reduce IL-6-induced gp130 dimerization and subsequent
STAT3 phosphorylation. Notably, n-3 PUFAs modulate STAT3 signaling
by enhancing the expression of Src homology region 2
domain-containing protein tyrosine phosphatase-1, which is a
well-known negative regulator of STAT3 signaling (19).
Fat-1 mice, which exhibit increased levels of
n-3 PUFAs in their organs and tissues compared with those of their
wild-type (WT) counterparts, have been reported to be a reliable
animal model to investigate n-3 PUFAs (20). The present study aimed to
investigate whether n-3 PUFAs modulate IL-11 expression and
downstream STAT3 signaling during APAP hepatotoxicity.
Materials and methods
Mice
WT C57BL/6J mice were obtained from the Laboratory
Animal Center of Southern Medical University (Guangzhou, China).
Fat-1 transgenic mice, which has been reported to carry the
gene that encodes the enzyme that coverts n-6 to n-3 PUFAs
endogenously (20), were
hybridized with WT C57BL/6J mice, and the fat-1 genotypes of
each animal were recognized as we previously described (21). A total of 79 WT C57BL/6J and 39
fat-1 mice (male, 8 weeks old, 20-25 g) were included in
this study. Mice included in the present study were housed under a
12 h light/dark cycle condition at a constant temperature (19-23°C)
and (55±10%) humidity, fed with commercial diet and had free access
to food and water. All animal experiments in this study were
approved by the Welfare and Ethical Committee for Experimental
Animal Care of Southern Medical University (approval no. L2018234).
All mice were euthanized with 5% isoflurane. Mice were sacrificed
at 0, 2, 6 and 24 h post-APAP injection. Before being sacrificed,
blood (50-100 µl) was obtained from the tail vein. After
standing for 4 h, blood was centrifugated at 1,400 × g for 10 min
at 4°C, and the supernatant was collected as serum.
APAP-induced liver injury model
APAP (cat. no. sc-203425; Santa Cruz Biotechnology,
Inc.) was intraperitoneally injected into 8-week-old male WT or
fat-1 mice at 400 mg/kg body weight (overdose) (22,23) or at 600 mg/kg body weight (lethal
dose) (24,25) as previously described. To
demonstrate the effect of exogenous n-3 PUFAs, WT mice were fed
with an n-3 PUFA-enriched diet for 3 weeks before APAP
administration. n-3 PUFA-enriched diets were modified to contain 60
g/kg oils from DHA ethyl ester-enriched fish oil (Ocean Nutrition
Canada) containing 540 g/kg DHA and 50 g/kg EPA. Commercial mouse
food was used for the normal diet (ND) control group.
Reagents
Antibodies (Abs) against phosphorylated (p)STAT3
(rabbit anti-mouse monoclonal; 1:2,000; cat. no. 9145), STAT3
(rabbit anti-mouse monoclonal; 1:1,000; cat. no. 12640), p-p44/42
MAPK (Erk1/2; rabbit anti-mouse monoclonal; 1:1,000; cat. no.
4376), p44/42 MAPK (Erk1/2) (rabbit anti-mouse monoclonal; 1:1,000;
cat. no. 4695) and GAPDH (rabbit anti-mouse monoclonal; 1:1,000;
cat. no. 5174) were obtained from Cell Signaling Technology, Inc.
Anti-IL-11 polyclonal antibody (rabbit anti-mouse; 1:1,000; cat.
no. A1902) for immunohistochemistry and immunoblotting was
purchased from ABclonal Biotech Co., Ltd. Anti-Fra-1 Ab (mouse
anti-mouse monoclonal; 1:100; cat. no. sc-271657) was obtained from
Santa Cruz Biotechnology, Inc. The antibody against Bcl-2 (mouse
anti-mouse monoclonal; 1:100; cat. no. YM3041) was from ImmunoWay
Biotechnology Company. Anti-Bax (rabbit anti-mouse polyclonal;
1:1,000; cat. no. DB123) anti-body was purchased from DB Biotech,
spol. s r.o.
Isolation of mouse hepatocytes and
hepatic mononuclear cells
Hepatocytes were isolated as previously described
(26). Briefly, livers were
perfused with calcium-free salt solution and type IV collagenase
through the portal vein in situ and then filtered with
polyamide mesh. After centrifugation at 50 × g for 1 min at 4°C,
the precipitants were collected as hepatocytes for reverse
transcription-quantitative PCR (RT-qPCR) and western blotting
assays, or for primary hepatocyte culture. The supernatants were
harvested for density gradient centrifugation using discontinuous
30/70% (v/v) Percoll gradients to obtain mononuclear cells for
RT-qPCR. For primary mouse hepatocyte (PMH) culture,
1×106 hepatocytes were seeded in 6-well dishes coated
with mouse tail collagen (cat. no. A1048301; Gibco; Thermo Fisher
Scientific, Inc.) in William's E medium (cat. no. A1217601; Gibco;
Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.). After incubation for 4 h, the culture was
replaced with serum-free RPMI-1640 medium (cat. no. 11875101;
Gibco; Thermo Fisher Scientific, Inc.). In some cases, cells were
treated with 10 µM Sc144 (cat. no. S7124; Selleck
Chemicals), 50 µM U0126 (cat. no. S1102; Selleck Chemicals)
or 10 µM C188-9 (cat. no. S8605; Selleck Chemicals) for 2 h
before APAP stimulation at 37°C.
Histology
Liver tissues were removed and collected in 1 ml 4%
paraformaldehyde for 24 h at room temperature. Sections were cut
into 5 µm. Samples were stained with hematoxylin and eosin
(H&E) at room temperature for 3 min to observe morphological
changes (Nikon Intensilight DS-Ri2; Nikon Corporation). The TUNEL
experiment was performed using a commercial kit (cat. no. C1091;
Beyotime Institute of Biotechnology) following the manufacturer's
protocols. Harris hematoxylin was used at room temperature for 3
min for nuclear staining and sections were mounted with neutral
resin. Three fields of view per section were observed (Nikon
Intensilight DS-Ri2). For immunohistochemical analysis, specimens
were embedded in paraffin and cut into 5-µm thick sections.
The sections were deparaffinized in xylene for 15 min at room
temperature and rehydrated with a graded series of alcohol,
following which a microwave antigen retrieval procedure was
performed using 10 mM sodium citrate buffer at 105°C for 10 min.
Upon blocking endogenous peroxidase activity, the samples were
treated with 5% BSA (cat. no. SRE0096; Sigma-Aldrich; Merck KGaA)
at room temperature for 1 h to block non-specific staining. The
sections were then incubated with an anti-IL-11 primary antibody
(1:50; cat. no. A1902; ABclonal Biotech Co., Ltd.) at 4°C
overnight, followed by incubation with the corresponding secondary
antibody (HRP-conjugated goat anti-rabbit; 1:200; cat. no. S0001;
Affinity Biosciences) at 37°C for 1 h. Peroxidase activity was
detected (Nikon Intensilight DS-Ri2) using DAB solution (Beyotime
Institute of Biotechnology).
Serum alanine aminotransferase (ALT)
assay and cytokine assessment
The serum was collected at the indicated time points
after APAP injection. Serum ALT activity was measured with a
commercial kit (cat. no. C009-2; Nanjing Jiancheng Bioengineering
Institute), based on the manufacturer's instructions. Different
cytokine levels in the serum and cell culture supernatants were
detected using commercial ELISA kits purchased from eBioscience
(Thermo Fisher Scientific, Inc.), including IL-6 (cat. no.
BMS603-2), IL-11 (cat. no. EMIL11), IL-13 (cat. no. BMS6015) and
IL-22 (cat. no. BMS6022).
Flow cytometry
The Annexin V/PI apoptosis kit was obtained from
Hangzhou Multi Sciences (Lianke) Biotech Co., Ltd., cells were
stained for 5 min in the dark at room temperature before
monitoring. Annexin V+/PI+ was identified as
late apoptosis and Annexin V+/PI− was
identified as early apoptosis. Cells were stained with JC-1 dye
(Nanjing KeyGen Biotech Co., Ltd.) for 30 min at room temperature
in the dark to evaluate mitochondrial membrane potential. The cells
were analyzed using the FACS LSRFortessa™ flow cytometer (BD
Biosciences) with BD FACSDiva™ software (version 8.0.1; BD
Biosciences).
Western blotting
Protein samples were obtained from hepatocytes
isolated from APAP-treated mice or primary hepatocytes lysed with
RIPA buffer (50 mM Tris, 150 mM NaCl and 1% Nonidet P-40, pH 7.4).
Protein concentration was measured using a BCA assay kit (cat. no.
23225; Thermo Fisher Scientific, Inc.). Protein samples (20
µg/lane) were separated on 10% SDS-polyacrylamide gels and
then transferred onto polyvinylidene fluoride membranes
(MilliporeSigma; Merck KGaA). The membranes were blocked with 5%
BSA for 1 h at room temperature, and subsequently incubated with
the indicated primary antibodies at 4°C overnight. Primary
antibodies, including phosphorylated (p)STAT3 (rabbit anti-mouse
monoclonal; 1:2,000; cat. no. 9145), STAT3 (rabbit anti-mouse
monoclonal; 1:1,000; cat. no. 12640), p-p44/42 MAPK (Erk1/2; rabbit
anti-mouse monoclonal; 1:1,000; cat. no. 4376), p44/42 MAPK
(Erk1/2) (rabbit anti-mouse monoclonal; 1:1,000; cat. no. 4695) and
GAPDH (rabbit anti-mouse monoclonal; 1:1,000; cat. no. 5174) were
obtained from Cell Signaling Technology, Inc. Anti-IL-11 polyclonal
antibody (rabbit anti-mouse; 1:1,000; cat. no. A1902) was purchased
from ABclonal Biotech Co., Ltd. Anti-Fra-1 (mouse anti-mouse
monoclonal; 1:100; cat. no. sc-271657) was obtained from Santa Cruz
Biotechnology, Inc. The anti-body against Bcl-2 (mouse anti-mouse
monoclonal; 1:100; cat. no. YM3041) was from ImmunoWay
Biotechnology Company. Anti-Bax (rabbit anti-mouse polyclonal;
1:1,000; cat. no. DB123) antibody was purchased from DB Biotech,
spol. s r.o. Subsequently, the membranes were incubated with the
corresponding HRP-conjugated secondary antibody (goat anti-rabbit;
1:10,000; cat. no. S0001; Affinity Biosciences) at room temperature
for 1 h. The results were visualized with ECL substrate (cat. no.
1705062; Bio-Rad Laboratories, Inc.). Each western blot analysis
was performed three times.
RT-qPCR analysis
Isolated hepatocytes were homogenized in 1 ml
TRIzol® (Thermo Fisher Scientific, Inc.), and total RNA
was extracted based on the manufacturer's instruction. Then, 1,000
ng RNA was synthesized into cDNA using TransScript® Fly
First-Stand cDNA Synthesis SuperMix (TransGen Biotech Co., Ltd.)
with RNA removal reagent at 50°C for 10 min and 85°C for 5 sec.
SYBR Green Real-Time PCR Master Mix (cat. no. A46112; Applied
Biosystems; Thermo Fisher Scientific, Inc.) was applied for qPCR on
an ABI Prism 7500 Sequence Detection System (Applied Biosystems;
Thermo Fisher Scientific, Inc.), according to the following
thermocycling conditions: Initial denaturation at 94°C for 30 sec,
followed by 40 cycles of denaturation at 94°C for 5 sec, and
extension at 60°C for 30 sec. The expression levels of the target
genes were normalized to GAPDH gene expression and calculated using
the 2−∆∆Cq method (27). The primer sequences used in this
experiment are shown in Table
I.
 | Table IPrimers for the target genes. |
Table I
Primers for the target genes.
| Gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| Bcl-2 |
GGACTTGAAGTGCCATTGGT | AGCCCCTCTGTGACA
GCTTA |
| Bax |
GGATGCGTCCACCAAGAAGC |
GGAGGAAGTCCAGTGTCCAGCC |
| IL-11 |
GTTTACAGCTCTTGATGTCTC |
GAGTCTTTAACAACAGCAGG |
| IL-13 |
CCTGGCTCTTGCTTGCCTT |
GGTCTTGTGTGATGTTGCTCA |
| Il-22 |
TGACGACCAGAACATCCAGA |
AATCGCCTTGATCTCTCCAC |
| IL-6 |
TACCACTTCACAAGTCGGAGGC |
CTGCAAGTGCATCATCGTTGTTC |
| Fra-1 |
TCATCTGGAGAGGTGGGTCC |
CTGCGGTTCTGACTCACTCG |
| GAPDH |
CGTCCCGTAGACAAAATGGT |
TTGATGGCAACAATCTCCAC |
Statistical analysis
All experiments were independently repeated in
triplicate. Statistical analysis was performed using SPSS 12.0
(SPSS, Inc.). The data are presented as the mean ± SD. An unpaired
Student's t-test was performed between WT and fat-1 mice or
ND control and n-3 PUFA-enriched diet mice at each time point.
Comparison of the survival curves was analyzed with the
Kaplan-Meier method and log-rank test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Endogenous n-3 PUFAs exacerbate
APAP-induced liver injury
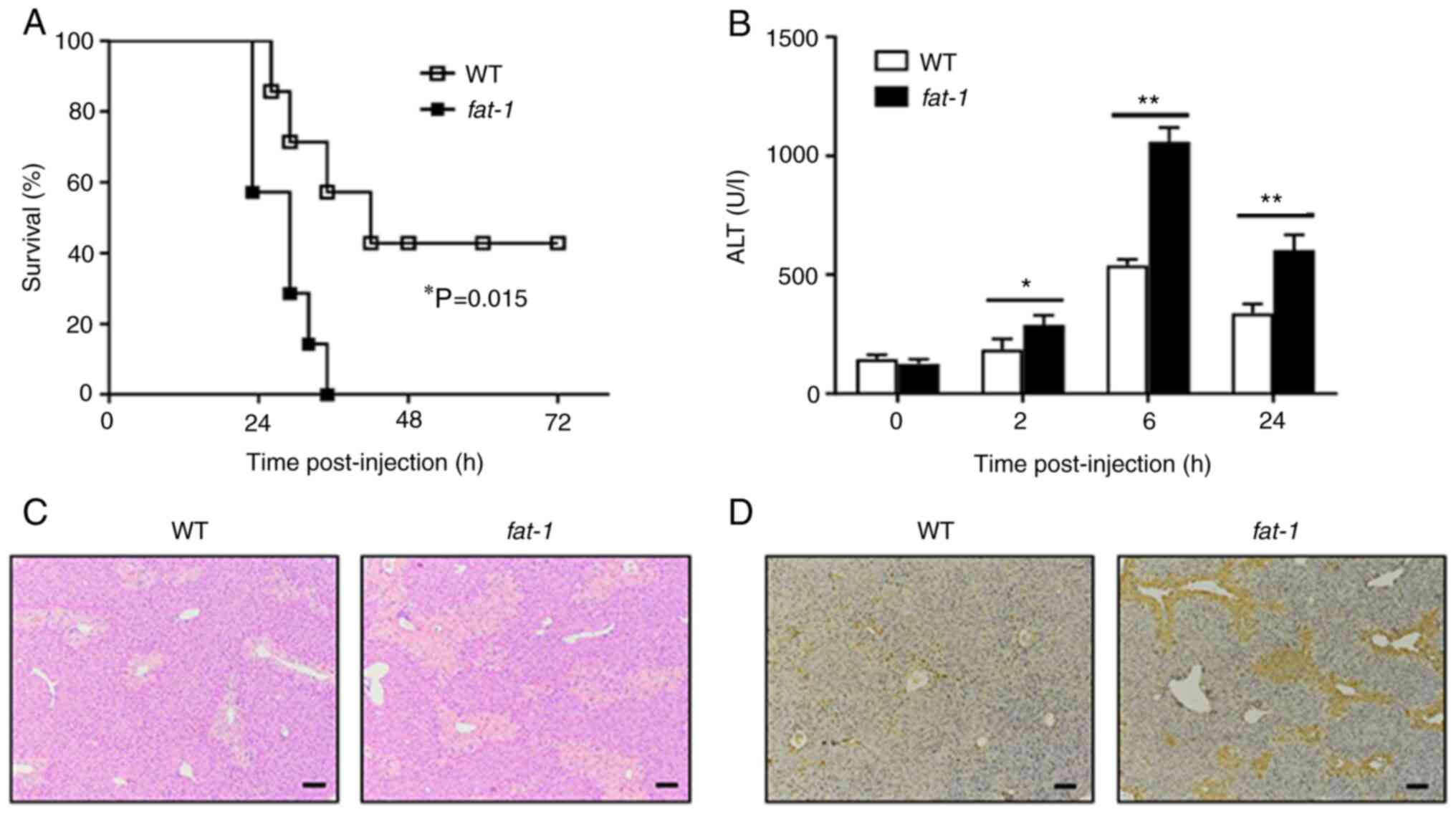
To confirm the role of n-3 PUFAs on APAP-induced
liver injury, WT or fat-1 transgenic mice were challenged
with a lethal dose of APAP. Animal mortality was markedly increased
in fat-1 transgenic mice compared with that of WT mice
(Fig. 1A). To further evaluate
the effect of n-3 PUFAs on APAP-induced liver injury, WT or
fat-1 transgenic mice were intraperitoneally injected with
400 mg/kg APAP, followed by examination of serum ALT activity and
liver pathological changes. Serum samples collected from
fat-1 transgenic mice showed markedly elevated levels of ALT
activity compared with that of serum samples from WT mice following
APAP administration (Fig. 1B).
Liver H&E staining revealed severe hepatic damage in
APAP-treated fat-1 transgenic mice compared with that of the
WT controls (Fig. 1C). In
addition, TUNEL-positive cells were markedly more abundant in
livers from fat-1 transgenic mice than in livers obtained
from APAP-treated WT mice (Fig.
1D). Taken together, these results indicated a potential role
of endogenous n-3 PUFAs in aggravating the pathogenesis of
APAP-induced liver damage.
n-3 PUFAs promote APAP-induced hepatocyte
apoptosis by inhibiting STAT3 phosphorylation
As hepatic STAT3 signaling is activated by the IL-6
cytokine family and exerts a protective effect against APAP
overdose (6), the present study
investigated whether the effect of n-3 PUFAs on APAP hepatotoxicity
involves STAT3 signaling. The results revealed attenuated protein
levels of pSTAT3 in hepatocytes derived from APAP-treated
fat-1 transgenic mice compared with those derived from WT
controls (Fig. 2A). Similarly,
decreased Bcl-2 and enhanced Bax expression were found in
hepatocytes obtained from fat-1 mice at both the
transcriptional and translational levels compared with those
observed in hepatocytes from WT controls (Fig. 2B and C). Considering that the Bcl
family plays an essential role in mitochondrial homeostasis
(7,8), JC-1 dye was used to detect the
mitochondrial membrane potential by flow cytometry. The results
showed an increased JC-1 monomer ratio in hepatocytes from
fat-1 transgenic mice compared with that found in
hepatocytes from WT mice upon APAP injection, which indicated more
severe mitochondrial injury in fat-1 transgenic mice
(Fig. 2D). To further validate
these results, primary hepatocytes from WT or fat-1 mice
were isolated and treated with APAP. As expected, attenuated
phosphorylation of STAT3 was observed in the hepatocytes obtained
from fat-1 mice (Fig.
2E). Furthermore, APAP treatment induced a higher apoptosis
ratio in hepatocytes from fat-1 transgenic mice than that
observed in hepatocytes from the WT controls. Of note, pretreatment
with SC144, a gp130 inhibitor, abrogated the difference in cell
apoptosis between hepatocytes from WT and fat-1 mice during
APAP exposure (Fig. 2F).
Consistently, the protein levels of Bcl-2 and Bax were comparable
between APAP-treated WT and fat-1 mice in the presence of
SC144 pretreatment (Fig. 2G).
Similarly, C188-9, a STAT3-specific inhibitor, also abrogated the
effect of n-3 PUFAs on Bcl-2 and Bax expression (Fig. S1). Collectively, these data
suggested that STAT3 inactivation was responsible for the effect of
n-3 PUFAs on APAP-induced liver injury.
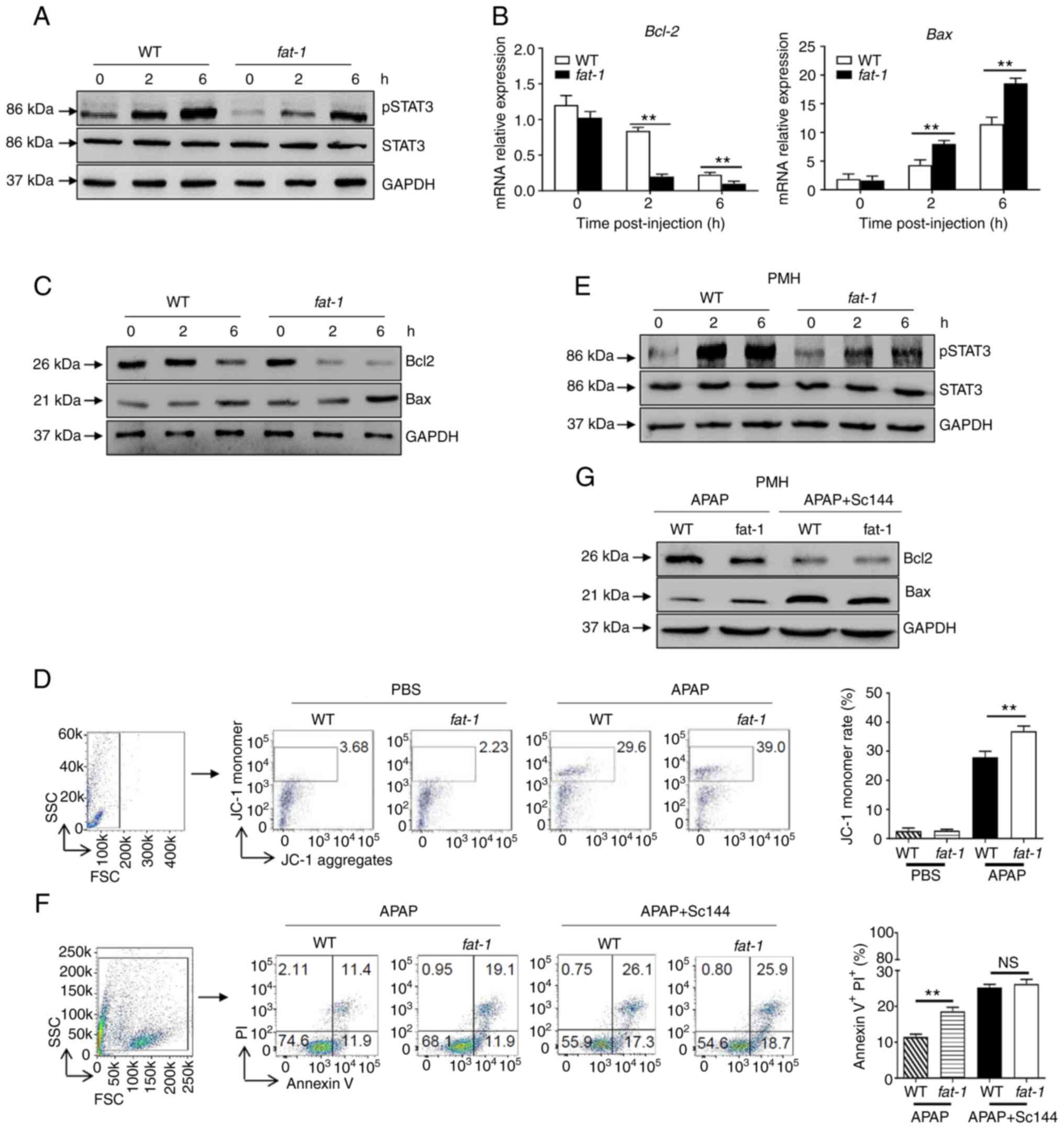 | Figure 2N-3 PUFAs promote hepatocytes
apoptosis through dephosphorylation of STAT3 signaling. (A-D)
Hepatocytes were isolated from WT and fat-1 transgenic mice
(n=5) at the indicated time after APAP (400 mg/kg) injection. (A)
Protein levels of pSTAT3, STAT3 and GAPDH were determined by
western blotting analysis. (B) The mRNA levels of Bcl-2 and Bax
were measured by reverse transcription-quantitative PCR and
expressed as a ratio to GAPDH. (C) The hepatic protein levels of
Bcl-2, Bax and GAPDH were evaluated at different time points by
western blotting analysis. (D) Mitochondrial membrane potential
(ΔΨm) was detected in mouse hepatocytes by flow cytometry 6 h
post-APAP injection. (E-G) Primary hepatocytes were isolated from
WT and fat-1 mice (n=5) and were stimulated with 20 mM APAP
in vitro. In some cases, cells were pretreated with 10
µM Sc144 for 2 h before APAP stimulation. (E) The levels of
pSTAT3, STAT3 and GAPDH were detected by western blotting analysis.
(F) Cellular apoptosis was measured by Annexin V-PI staining 24 h
post-APAP treatment. (G) The protein levels of Bcl-2, Bax and GAPDH
expression were determined by western blotting analysis 6 h
post-APAP treatment. **P<0.01. NS, not significant;
N-3 PUFAs, omega-3 polyunsaturated fatty acids; APAP,
acetaminophen; WT, wild-type; p, phosphorylated. |
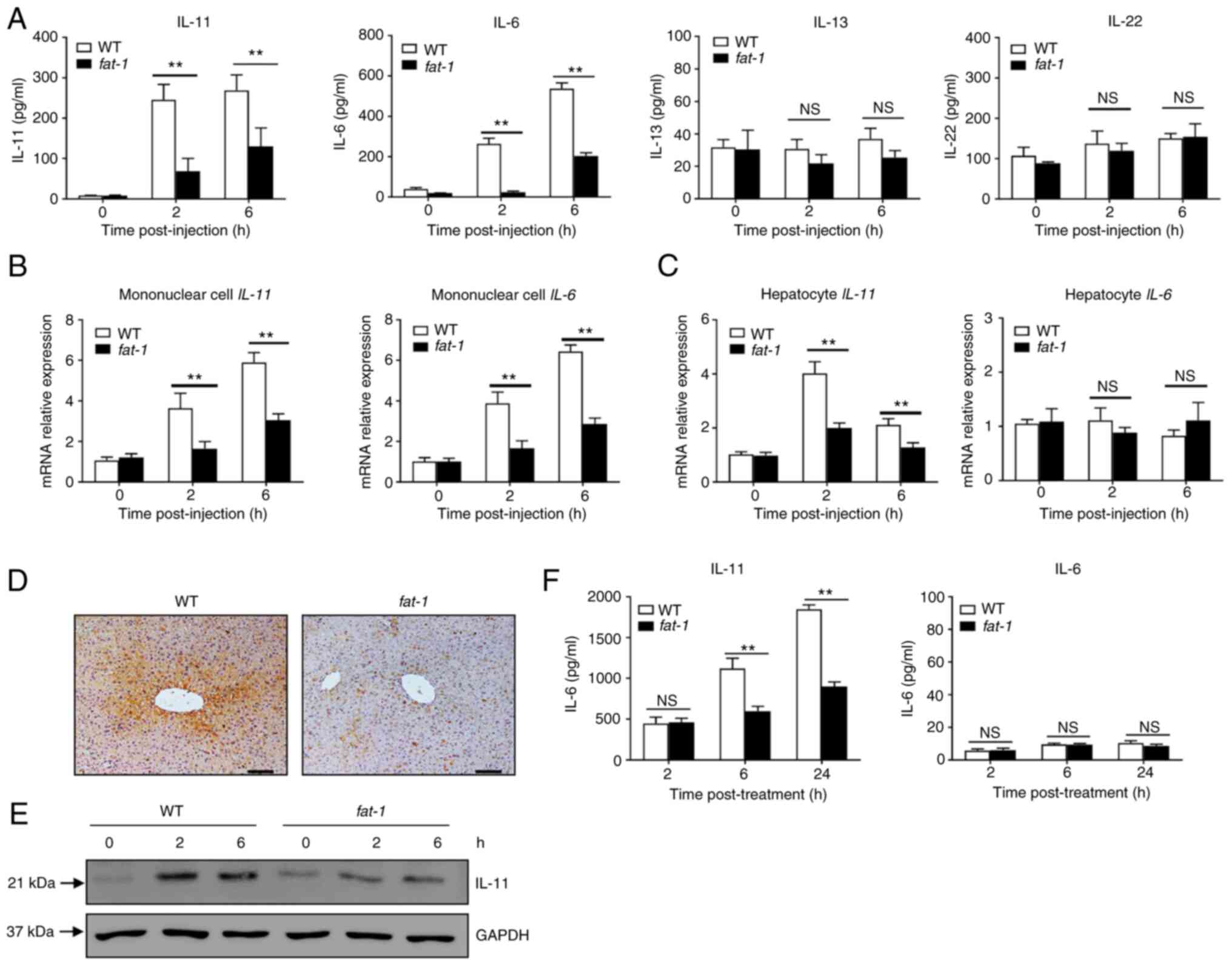
n-3 PUFAs inhibit the production of IL-11
in hepatocytes
Several STAT3-activating cytokines, including IL-6,
IL-11, IL-13 and IL-22, are able to directly target hepatocytes
(6,7). Therefore, the present study first
assessed the levels of cytokines in the serum of APAP-treated mice.
Compared with those of WT mice, APAP-challenged fat-1
transgenic mice showed significantly lower levels of IL-6 and
IL-11, but not of IL-13 or IL-22 (Fig. 3A). Next, the mRNA expression of
cytokines in hepatocytes and mononuclear cells was evaluated. The
results showed that the levels of IL-11 and IL-6 were lower in
hepatic mononuclear cells from APAP-treated mice compared with
those observed in their WT counterparts (Fig. 3B). Notably, hepatocytes from
APAP-treated fat-1 transgenic mice produced less IL-11 than
those from WT controls (Fig.
3C), while there was no significant difference in the mRNA
level of IL-6 (Fig. 3C).
Similarly, immunohistochemical staining showed that the hepatic
level of IL-11 was lower in fat-1 transgenic mice than in WT
mice upon APAP administration (Fig.
3D). In addition, decreased IL-11 expression was found in
hepatocytes obtained from fat-1 mice compared with that
found in hepatocytes from WT mice (Fig. 3E). Furthermore, ELISA showed
decreased IL-11 production in primary hepatocytes from fat-1
transgenic mice compared with that observed in their WT
counterparts (Fig. 3F). Taken
together, these results suggested that endogenous n-3 PUFAs limited
the expression of IL-11 in hepatocytes during APAP-induced liver
damage.
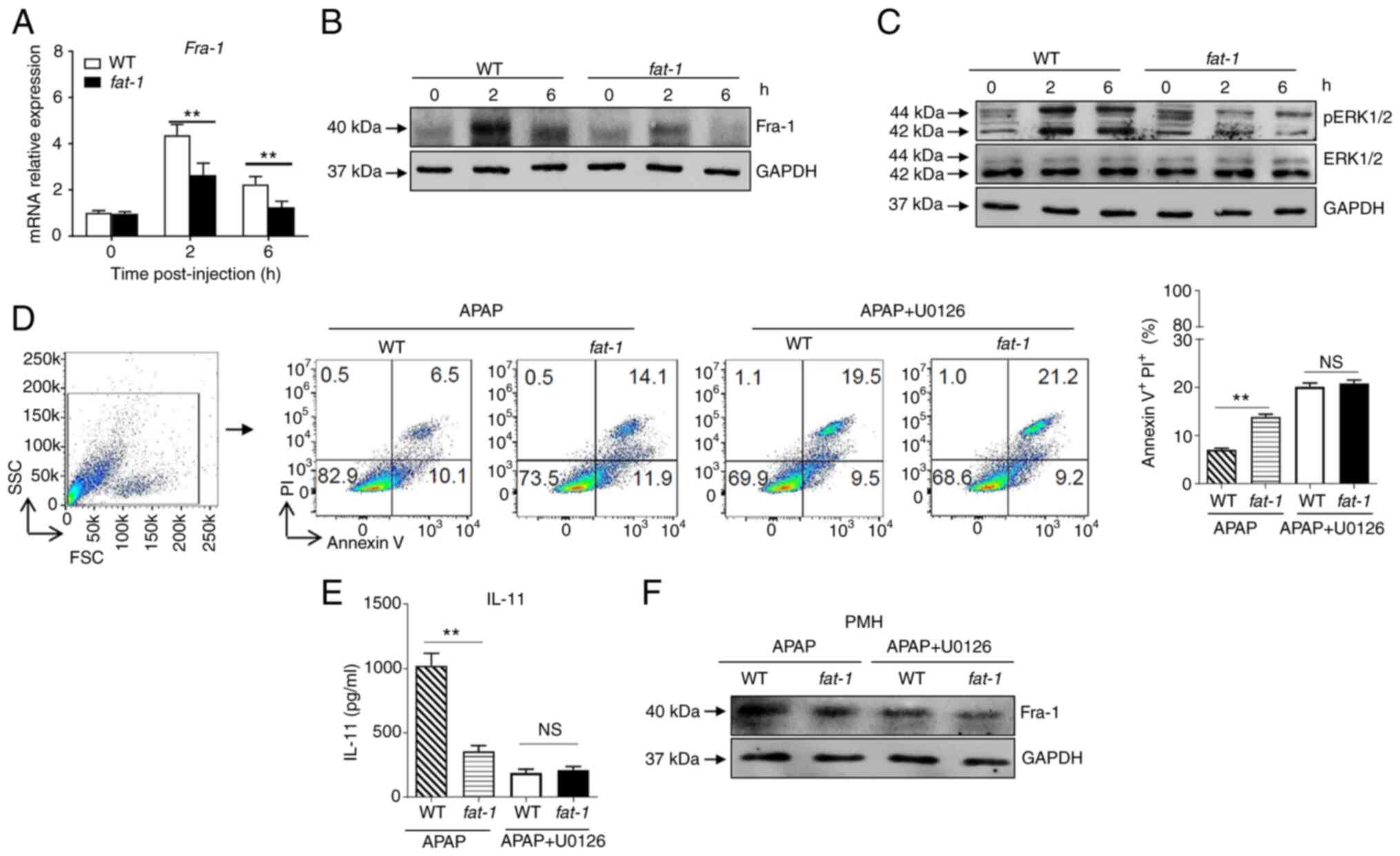
n-3 PUFAs inhibit the production of IL-11
via ERK1/2-dependent decreased Fra-1 levels in hepatocytes
It has been reported that Fra-1, which is recruited
to the IL-11 promoter and enhances IL-11 gene transcription, is
essential for the production of IL-11 (3). The present results revealed
attenuated expression of Fra-1 at the mRNA and protein level in
hepatocytes isolated from APAP-treated fat-1 transgenic mice
compared with that observed in WT controls (Fig. 4A and B). Considering that Fra-1
expression is mainly dependent on ERK1/2 activity, ERK1/2
activation in hepatocytes from APAP-injected mice was assessed. The
results showed that APAP induced a lower level of phosphorylation
of ERK1/2 in hepatocytes from APAP-treated fat-1 transgenic
mice than that observed in hepatocytes derived from their WT
counterparts (Fig. 4C). To
identify whether ERK1/2 signaling is involved in the effect of n-3
PUFAs on APAP hepatotoxicity, hepatocytes from WT and fat-1
transgenic mice were treated with U0126, a widely used ERK1/2
inhibitor, to block ERK1/2 activation prior to incubation with
APAP. After ERK1/2 inhibition, the level of cell apoptosis was
comparable between WT and fat-1 transgenic hepatocytes with
U0126 pretreatment (Fig. 4D).
Notably, the production of IL-11 (Fig. 4E) and the expression of Fra-1
(Fig. 4F) were similar between
hepatocytes from WT mice and those from fat-1 transgenic
mice upon APAP stimulation when ERK1/2 signaling was blocked. Taken
together, these data suggested that n-3 PUFAs inhibited ERK1/2
phosphorylation following APAP stimulation, which resulted in
limited expression of Fra-1 and consequently reduced IL-11
production.
Exogenous DHA exacerbates APAP-induced
liver damage
To further demonstrate the effect of n-3 PUFAs on
IL-11 production in APAP-induced liver damage, WT mice were fed
daily with a n-3 PUFA-enriched diet for 3 weeks prior to APAP
administration. As expected, it was found that dietary n-3 PUFAs
efficiently enhanced the severity of APAP-induced liver damage, as
demonstrated by hepatic pathology (Fig. 5A) and serum ALT levels (Fig. 5B). In addition, a markedly
reduced IL-11 level was determined in the serum (Fig. 5C) and liver tissue (Fig. 5D) of mice with a n-3
PUFA-enriched diet compared with that of mice in the ND group.
Furthermore, exogenous n-3 PUFAs suppressed ERK1/2 phosphorylation
and Fra-1 expression in hepatocytes stimulated with APAP, as
evaluated by immunoblotting (Fig.
5E). Notably, exogenous n-3 PUFAs reduced STAT3 phosphorylation
and Bcl-2 expression while enhancing Bax expression during the APAP
challenge (Fig. 5F).
Collectively, these findings indicated that exogenous n-3 PUFAs
also exacerbated APAP-induced liver damage and share the same
underlying mechanism as endogenous n-3 PUFAs.
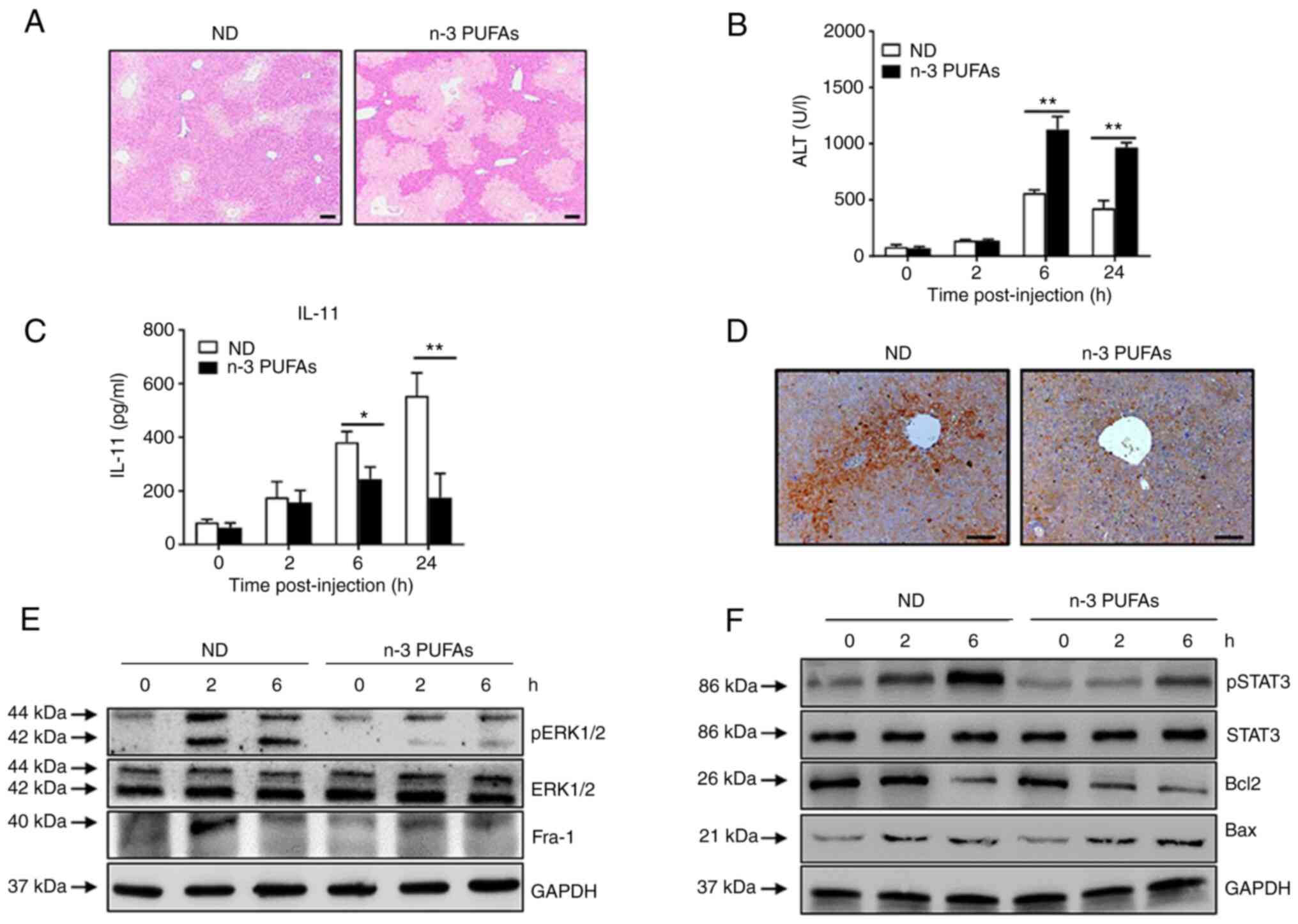 | Figure 5Exogenous DHA aggravate APAP-induced
liver damage through dephosphorylated ERK-mediated decreased IL-11
production. (A-D) An overdose of APAP (400 mg/kg) was
intraperitoneally injected into WT mice fed with ND or n-3
PUFA-enriched diet (n=5). (A) Histological analysis of mouse livers
was performed by hematoxylin & eosin staining. Scale bar, 100
µm. (B) Serum ALT activities at different time points were
measured. (C) The serum level of IL-11 was determined at the
indicated time points post-APAP injection. (D) The protein level of
IL-11 in the liver tissues was detected by immunohistochemical
staining 6 h post-APAP injection. Scale bar, 50 µm. (E and
F) Hepatocytes were isolated from WT mice fed with ND or n-3
PUFA-enriched diet (n=5) at the indicated time post-400 mg/kg APAP
administration. (E) The levels of pERK, ERK, Fra-1 and GAPDH were
determined by western blotting analysis. (F) The protein levels of
pSTAT3, STAT3, Bcl-2, BAX and GAPDH were detected by western
blotting analysis. *P<0.05, **P<0.01.
N-3 PUFAs, omega-3 polyunsaturated fatty acids; APAP,
acetaminophen; WT, wild-type; ALP, alanine aminotransferase; Fra-1,
Fos-like-1; p, phosphorylated; ND, normal diet. |
Discussion
n-3 PUFAs have been reported to exert an inhibitory
effect on IL-6 production (9,12,13), while their effect on IL-11
expression remains unknown. The present study found that n-3 PUFAs
exacerbated APAP-induced liver damage. The data revealed that n-3
PUFAs inhibited IL-11 production in hepatocytes and STAT3
phosphorylation. In addition, it was observed that limited
phosphorylation of ERK1/2 and reduced Fra-1 expression were
associated with the effect of n-3 PUFAs on IL-11 production.
STAT3 signaling plays an essential role in acute
liver injury (6,28). It has been reported that myeloid
STAT3 activation inhibits T cell-mediated hepatitis through
suppression of type 1 T helper cytokines (28). Furthermore, mice lacking IL-6 or
gp130-STAT3 signaling in hepatocytes are more susceptible to
supplemented ethionine or lipopolysaccharide-induced liver injury
(29,30). The present study found limited
STAT3 phosphorylation and Bcl-2 expression levels alongside
increased Bax expression in fat-1 mice compared with the
findings in their WT counterparts. In addition, n-3 PUFAs did not
affect APAP-induced liver injury when STAT3 signaling was inhibited
by SC144, indicating that the effect of n-3 PUFAs on APAP
hepatotoxicity depends on the regulation of the STAT3 signaling
pathway. Notably, SC144 exerts an inhibitory effect on STAT3
phosphorylation through deglycosylation of gp130, the shared
β-receptor of IL-6 and IL-11 signaling transduction, which
indicates that the effect of n-3 PUFAs on STAT3 phosphorylation may
rely on STAT3-inducing cytokines.
IL-11 belongs to the IL-6 family and shares the same
gp130 homodimer receptor complex with IL-6. Specificity is gained
through an individual IL-6/IL-11 α-receptor (31). Previous studies have demonstrated
that IL-6 and IL-11 exert the opposite effect on immune regulation.
It has been reported that IL-6 promotes type 2 T helper (Th2) cell
response in an asthma model, while IL-11 inhibits Th2 type
inflammation (32). IL-6 and
IL-11 exhibit similar biological outcomes in the induction of the
acute-phase response (33). In a
previous study, both IL-11 and IL-6 increased the level of
phosphorylated STAT3 in hepatocytes, however, only IL-6, but not
IL-11, could induce STAT3 phosphorylation in Kupffer cells, which
indicates that hepatocytes may be the target of IL-11 (3). In the present study, decreased
levels of IL-6 and IL-11 were observed in the sera of APAP-injected
fat-1 mice compared with those found in their WT
counterparts, but only IL-11 could be detected, and its expression
was found to be attenuated in hepatocytes obtained from
fat-1 mice, which indicated that the inhibitory effect of
n-3 PUFAs on IL-11 acts via an autocrine loop in hepatocytes during
APAP hepatotoxicity.
IL-11 is a pleiotropic cytokine with biological
functions in different cell types (2). It is mainly produced by hepatocytes
in response to ROS during acute liver injury in mice (3). In a previous study, livers of mice
receiving IL-11 exhibited damage, with elevated markers of
fibrosis, hepatocyte cell death and inflammation (34). Notably, inhibiting IL-11
signaling by neutralizing antibodies could reduce hepatocyte death,
inflammation and hyperglycemia in mouse models of diet-induced
steatohepatitis (34). A
previous study reported the pathological function of IL-11 in
hepatocytes during the early stages of metabolic liver disease
(2). MAPK/ERK activation is
crucial for the oxidative stress-induced production of IL-11
(3). Treatment with ERK
inhibitors can efficiently suppress TNF-α-induced IL-11 induction
in murine embryonic fibroblasts (3). Fra-1, a member of the activator
protein 1 family, was found to bind to the IL-11 promoter before
stimulation, but recruitment of Fra-1 was further enhanced after
oxidative stimulation (3,35).
Gillies et al (36) found
that Fra-1 is expressed in proportion to the amplitude and duration
of ERK1/2 activity. A previous study reported that the expression
of IL-11 in hepatocytes is regulated by ERK1/2-mediated Fra-1
expression (37). n-3 PUFAs
appear to act via receptors or sensors, thereby controlling
cellular signaling processes that influence gene expression
patterns (38). Several studies
have reported that the anticancer properties of n-3 PUFAs involve
the altered phosphorylation of ERK1/2 (39,40). It has been reported that n-3
PUFAs induce apoptosis in human breast cancer cells through
inhibition of ERK1/2 activation (40). n-3 PUFAs are able to suppress
VEGF expression in colon cancer cells by limiting ERK1/2
phosphorylation and hypoxia-inducible factor 1α overexpression
(41). In addition, the
anti-inflammatory effect of n-3 PUFAs on the endothelium depends on
the dephosphorylation of ERK1/2 (42). The present study demonstrated
that n-3 PUFAs inhibited ERK1/2 phosphorylation and Fra-1
expression in hepatocytes in response to the APAP treatment.
Notably, inhibition of ERK1/2 activation attenuated n-3
PUFA-induced hepatic IL-11 production and subsequent liver injury
in APAP-treated mice, suggesting that n-3 PUFAs affect IL-11
production and APAP hepatotoxicity via regulation of the ERK1/2
signaling pathway. However, the specific underlying mechanism by
which n-3 PUFAs regulate ERK1/2 signaling needs further
investigation. Fluorescence labeled n-3 PUFAs could be used to
evaluate whether there is a physical interaction between n-3 PUFAs
and ERK1/2 protein in vitro, while IL-11 receptor antagonist
or IL-11/IL-11 receptor knockout by CRISPR/CRISPR-associated
protein 9 could be used to further identify whether the effect of
n-3 PUFAs on APAP-induced liver injury depends on the production of
IL-11.
In summary, the present results revealed that n-3
PUFAs inhibit IL-11 production and downstream STAT3 phosphorylation
in hepatocytes, thereby aggravating APAP-induced liver injury. The
present study demonstrated that the ERK1/2-Fra-1 axis is important
for the regulatory function of n-3 PUFAs on IL-11 expression.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YL, ZC and DZ conceived and designed the present
study. ZC and DZ provided administrative support. JZ and DZ were
responsible for the provision of study materials and patients. YL,
JL and YC were responsible for the collection and assembly of data.
YL, ZL, JZ, XL, ZC and DZ analyzed and interpreted the data. YL, ZC
and DZ confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The authors are accountable for all aspects of the
work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved. All animal protocols in this study were approved by
the Welfare and Ethical Committee for Experimental Animal Care of
Southern Medical University (approval no. L2018234; Guangzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported in part by the National Natural Science
Foundation of China (grant nos. 82071781, 81873872 and 81771771),
and the Innovation Team of Chronic Kidney Disease with Integrated
Traditional Chinese and Western Medicine (grant no.
2019KCXTD014).
References
|
1
|
Cook SA and Schafer S: Hiding in plain
sight: Interleukin-11 emerges as a master regulator of fibrosis,
tissue integrity, and stromal inflammation. Annu Rev Med.
71:263–276. 2020. View Article : Google Scholar
|
|
2
|
Dong J, Adami E, Chothani SP, Viswanathan
S, Ng B, Lim WW, Sing BK, Zhou J, Ko NSJ, Shekeran SG, et al:
Autocrine IL11 cis-signaling in hepatocytes is an initiating nexus
between lipotoxicity and non-alcoholic steatohepatitis. BioRxiv.
2020.
|
|
3
|
Nishina T, Komazawa-Sakon S, Yanaka S,
Piao X, Zheng DM, Piao JH, Kojima Y, Yamashina S, Sano E, Putoczki
T, et al: Interleukin-11 links oxidative stress and compensatory
proliferation. Sci Signal. 5:ra52012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu M, Lu B, Cao Q, Wu Z, Xu Z, Li W, Yao
X and Liu F: IL-11 attenuates liver ischemia/reperfusion injury
(IRI) through STAT3 signaling pathway in mice. PLoS One.
10:e01262962015. View Article : Google Scholar
|
|
5
|
Bernal W, Auzinger G, Dhawan A and Wendon
J: Acute liver failure. Lancet. 376:190–201. 2010. View Article : Google Scholar
|
|
6
|
Mühl H: STAT3, a key parameter of
cytokine-driven tissue protection during sterile inflammation-the
case of experimental acetaminophen (paracetamol)-induced liver
damage. Front Immunol. 7:1632016. View Article : Google Scholar
|
|
7
|
Harrison DA: The Jak/STAT pathway. Cold
Spring Harb Perspect Biol. 4:a0112052012. View Article : Google Scholar
|
|
8
|
Nielsen M, Kaestel CG, Eriksen KW,
Woetmann A, Stokkedal T, Kaltoft K, Geisler C, Röpke C and Odum N:
Inhibition of constitutively activated Stat3 correlates with
altered Bcl-2/Bax expression and induction of apoptosis in mycosis
fungoides tumor cells. Leukemia. 13:735–738. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmöcker C, Weylandt KH, Kahlke L, Wang
J, Lobeck H, Tiegs G, Berg T and Kang JX: Omega-3 fatty acids
alleviate chemically induced acute hepatitis by suppression of
cytokines. Hepatology. 45:864–869. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Tang Y, Wang S, Zhou J, Zhou J, Lu
X, Bai X, Wang XY, Chen Z and Zuo D: Endogenous n-3 polyunsaturated
fatty acids attenuate T cell-mediated hepatitis via autophagy
activation. Front Immunol. 7:3502016. View Article : Google Scholar
|
|
11
|
Yang J, Fernández-Galilea M,
Martínez-Fernández L, González-Muniesa P, Pérez-Chávez A, Martínez
JA and Moreno-Aliaga MJ: Oxidative stress and non-alcoholic fatty
liver disease: Effects of omega-3 fatty acid supplementation.
Nutrients. 11:8722019. View Article : Google Scholar :
|
|
12
|
Kelley DS, Siegel D, Fedor DM, Adkins Y
and Mackey BE: DHA supplementation decreases serum C-reactive
protein and other markers of inflammation in hypertriglyceridemic
men. J Nutr. 139:495–501. 2009. View Article : Google Scholar
|
|
13
|
Ma Y, Smith CE, Lai CQ, Irvin MR, Parnell
LD, Lee YC, Pham LD, Aslibekyan S, Claas SA, Tsai MY, et al: The
effects of omega-3 polyunsaturated fatty acids and genetic variants
on methylation levels of the interleukin-6 gene promoter. Mol Nutr
Food Res. 60:410–419. 2016. View Article : Google Scholar :
|
|
14
|
Song EA, Lim JW and Kim H: Docosahexaenoic
acid inhibits IL-6 expression via PPARγ-mediated expression of
catalase in cerulein-stimulated pancreatic acinar cells. Int J
Biochem Cell Biol. 88:60–68. 2017. View Article : Google Scholar
|
|
15
|
D'Eliseo D, Di Renzo L, Santoni A and
Velotti F: Docosahexaenoic acid (DHA) promotes immunogenic
apoptosis in human multiple myeloma cells, induces autophagy and
inhibits STAT3 in both tumor and dendritic cells. Genes Cancer.
8:426–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Allen MJ, Fan YY, Monk JM, Hou TY,
Barhoumi R, McMurray DN and Chapkin RS: n-3 PUFAs reduce T-helper
17 cell differentiation by decreasing responsiveness to
interleukin-6 in isolated mouse splenic CD4+ T cells. J Nutr.
144:1306–1313. 2014. View Article : Google Scholar :
|
|
17
|
Tasaki S, Horiguchi A, Asano T, Ito K,
Asano T and Asakura H: Docosahexaenoic acid inhibits the
phosphorylation of STAT3 and the growth and invasion of renal
cancer cells. Exp Ther Med. 14:1146–1152. 2017. View Article : Google Scholar :
|
|
18
|
O'Shea JJ, Schwartz DM, Villarino AV,
Gadina M, McInnes IB and Laurence A: The JAK-STAT pathway: Impact
on human disease and therapeutic intervention. Annu Rev Med.
66:311–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiong A, Yu W, Liu Y, Sanders BG and Kline
K: Elimination of ALDH+ breast tumor initiating cells by
docosahexanoic acid and/or gamma tocotrienol through SHP-1
inhibition of Stat3 signaling. Mol Carcinog. 55:420–430. 2016.
View Article : Google Scholar
|
|
20
|
Kang JX, Wang J, Wu L and Kang ZB:
Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature.
427:5042004. View Article : Google Scholar
|
|
21
|
Liu Y, Chen Y, Xie X, Yin A, Yin Y, Liu Y,
Dong L, Zhu Z, Zhou J, Zeng Q, et al: Gender difference on the
effect of omega-3 polyunsaturated fatty acids on
acetaminophen-induced acute liver failure. Oxid Med Cell Longev.
2020:80968472020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Henderson MW, Sparkenbaugh EM, Wang S,
Ilich A, Noubouossie DF, Mailer RK, Renné T, Flick MJ, Luyendyk JP,
Chen ZL, et al: Plasmin-mediated cleavage of high molecular weight
kininogen contributes to acetaminophen-induced acute liver failure.
Blood. Apr 7–2021.Epub ahead of print. View Article : Google Scholar
|
|
23
|
Saha B and Nandi D: Farnesyltransferase
inhibitors reduce Ras activation and ameliorate
acetaminophen-induced liver injury in mice. Hepatology.
50:1547–1557. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang C, Feng J, Du J, Zhuo Z, Yang S,
Zhang W, Wang W, Zhang S, Iwakura Y, Meng G, et al:
Macrophage-derived IL-1α promotes sterile inflammation in a mouse
model of acetaminophen hepatotoxicity. Cell Mol Immunol.
15:973–982. 2018. View Article : Google Scholar
|
|
25
|
Torres S, Baulies A, Insausti-Urkia N,
Alarcón-Vila C, Fucho R, Solsona-Vilarrasa E, Núñez S, Robles D,
Ribas V, Wakefield L, et al: Endoplasmic reticulum stress-induced
upregulation of STARD1 promotes acetaminophen-induced acute liver
failure. Gastroenterology. 157:552–568. 2019. View Article : Google Scholar
|
|
26
|
Osawa Y, Uchinami H, Bielawski J, Schwabe
RF, Hannun YA and Brenner DA: Roles for C16-ceramide and
sphingosine 1-phosphate in regulating hepatocyte apoptosis in
response to tumor necrosis factor-alpha. J Biol Chem.
280:27879–27887. 2005. View Article : Google Scholar
|
|
27
|
Pang Y, Liu Z, Han H, Wang B, Li W, Mao C
and Liu S: Peptide SMIM30 promotes HCC development by inducing
SRC/YES1 membrane anchoring and MAPK pathway activation. J Hepatol.
73:1155–1169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lafdil F, Wang H, Park O, Zhang W,
Moritoki Y, Yin S, Fu XY, Gershwin ME, Lian ZX and Gao B: Myeloid
STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1
cytokine and interleukin-17 production. Gastroenterology.
137:2125–2135. e1–e2. 2009. View Article : Google Scholar
|
|
29
|
Kroy DC, Beraza N, Tschaharganeh DF,
Sander LE, Erschfeld S, Giebeler A, Liedtke C, Wasmuth HE,
Trautwein C and Streetz KL: Lack of interleukin-6/glycoprotein
130/signal transducers and activators of transcription-3 signaling
in hepatocytes predisposes to liver steatosis and injury in mice.
Hepatology. 51:463–473. 2010. View Article : Google Scholar
|
|
30
|
Streetz KL, Wüstefeld T, Klein C, Kallen
KJ, Tronche F, Betz UA, Schütz G, Manns MP, Müller W and Trautwein
C: Lack of gp130 expression in hepatocytes promotes liver injury.
Gastroenterology. 125:532–543. 2003. View Article : Google Scholar
|
|
31
|
Garbers C and Scheller J: Interleukin-6
and interleukin-11: Same same but different. Biol Chem.
394:1145–1161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Homer RJ, Hong L, Cohn L, Lee CG,
Jung S and Elias JA: IL-11 selectively inhibits
aeroallergen-induced pulmonary eosinophilia and Th2 cytokine
production. J Immunol. 165:2222–2231. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benigni F, Fantuzzi G, Sacco S, Sironi M,
Pozzi P, Dinarello CA, Sipe JD, Poli V, Cappelletti M, Paonessa G,
et al: Six different cytokines that share GP130 as a receptor
subunit, induce serum amyloid A and potentiate the induction of
interleukin-6 and the activation of the
hypothalamus-pituitary-adrenal axis by interleukin-1. Blood.
87:1851–1854. 1996. View Article : Google Scholar
|
|
34
|
Widjaja AA, Singh BK, Adami E, Viswanathan
S, Dong JR, D'Agostino GA, Ng B, Lim WW, Tan J, Paleja BS, et al:
Inhibiting interleukin 11 signaling reduces hepatocyte death and
liver fibrosis, inflammation, and steatosis in mouse models of
nonalcoholic steatohepatitis. Gastroenterology. 157:777–792.e14.
2019. View Article : Google Scholar
|
|
35
|
Shin SY, Choi C, Lee HG, Lim Y and Lee YH:
Transcriptional regulation of the interleukin-11 gene by oncogenic
Ras. Carcinogenesis. 33:2467–2476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gillies TE, Pargett M, Minguet M, Davies
AE and Albeck JG: Linear integration of ERK activity predominates
over persistence detection in Fra-1 regulation. Cell Syst.
5:549–563.e5. 2017. View Article : Google Scholar :
|
|
37
|
Nishina T, Deguchi Y, Miura R, Yamazaki S,
Shinkai Y, Kojima Y, Okumura K, Kumagai Y and Nakano H: Critical
contribution of nuclear factor erythroid 2-related factor 2 (NRF2)
to electrophile-induced interleukin-11 production. J Biol Chem.
292:205–216. 2017. View Article : Google Scholar
|
|
38
|
Calder PC: Mechanisms of action of (n-3)
fatty acids. J Nutr. 142:592S–599S. 2012. View Article : Google Scholar
|
|
39
|
Serini S and Calviello G: Modulation of
Ras/ERK and phosphoinositide signaling by long-chain n-3 PUFA in
breast cancer and their potential complementary role in combination
with targeted drugs. Nutrients. 9:1852017. View Article : Google Scholar
|
|
40
|
Sun H, Hu Y, Gu Z, Owens RT, Chen YQ and
Edwards IJ: Omega-3 fatty acids induce apoptosis in human breast
cancer cells and mouse mammary tissue through syndecan-1 inhibition
of the MEK-Erk pathway. Carcinogenesis. 32:1518–1524. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Calviello G, Di Nicuolo F, Gragnoli S,
Piccioni E, Serini S, Maggiano N, Tringali G, Navarra P, Ranelletti
FO and Palozza P: n-3 PUFAs reduce VEGF expression in human colon
cancer cells modulating the COX-2/PGE2 induced ERK-1 and -2 and
HIF-1alpha induction pathway. Carcinogenesis. 25:2303–2310. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu KL, Yang YC, Yao HT, Chia TW, Lu CY,
Li CC, Tsai HJ, Lii CK and Chen HW: Docosahexaenoic acid inhibits
inflammation via free fatty acid receptor FFA4, disruption of TAB2
interaction with TAK1/TAB1 and downregulation of ERK-dependent
Egr-1 expression in EA.hy926 cells. Mol Nutr Food Res. 60:430–443.
2016. View Article : Google Scholar
|