1. Introduction
Spastin, encoded by the SPG4 gene, was identified
from mutations in patients with hereditary spastic paraplegia
(HSP). HSPs are characterized by progressive lower limb spasticity
and weakness. Spastin mutation is the chief cause of HSP and is
inherited as an autosomal dominant trait, accounting for 60% of
autosomal dominant cases and 15% of sporadic cases (1,2).
HSP pathogenesis is associated with the degradation of the distal
ends of corticospinal axons, which are the major central nervous
system pathways connecting the motor cerebral cortex to the spinal
cord (3,4). However, the underlying molecular
mechanisms of spastin in neural development and HSP have remained
to be fully clarified. Recently, numerous spastin-related studies
have been performed to identify its cellular roles and elucidate
how it affects axonal behaviors, thereby aiding in the development
of therapeutic strategies against HSPs.
Spastin isoforms add complexity to the understanding
of the pathology and etiology of HSP-SPG4. Spastin has two major
isoforms, M1 and M87, attributed to different initiation codons. An
additional two spastin isoforms are caused by alternative mRNA
splicing of exon 4 (4,5). Physiologically, M1 is mainly
distributed in the adult spinal cord, where the corticospinal axons
degenerate in patients with HSP. However, M87 is more abundant and
ubiquitously expressed than M1 (6,7).
Genetic analyses have revealed that both M1 and M87 are involved in
HSP pathology (8-10). With the exclusive hairloop
domain, M1 spastin particularly localizes to the endoplasmic
reticulum (ER) membrane and lipid droplets (LDs), thus regulating
serious cellular activity involving the ER or LDs (11). To date, numerous studies have
revealed the cellular function of M1, including ER shaping
(12), LD metabolism (11,13), membrane remodeling (14,15), endosomal fission (16-18) and fast axonal transport (19,20). However, how M87 works and
coordinates with M1 remains largely elusive. Recent studies further
highlight the important roles of spastin during neurogenesis
(21), axonal development
(22-24), synapse formation and spine
maturation (25-27). Furthermore, an innovative
severing model has been proposed, which provides novel insight
regarding microtubule (MT)-related activities, thus reasonably
elucidating the mechanisms of the roles of spastin in physiological
and pathological conditions (28,29). The present review focuses on
recent biological advances and provides insight into the cellular
mechanism of spastin and its role in neural development and
disease.
2. Structure and functions of spastin
Domain architecture and structure of
spastin
Spastin is a highly conserved protein whose complex
functions are attributed to its multiple modular domain structure
and different isoforms. Full-length spastin consists of four
functional domains: The hydrophobic domain (HD) situated between
residues 1-87, the MT-interacting and trafficking domain (MIT)
spinning (residues 116-194), the MT-binding domain (MTBD) situated
in residues 270-328 and the adenosine triphosphatases associated
with diverse cellular activities (AAA) domain spinning (residues
342-599). The HD has a high affinity for the membrane structure and
determines the subcellular distribution of full-length spastin. MIT
is essential for spastin's interaction with endosomal sorting
complexes required for transport (ESCRT)-III proteins responsible
for cytokinesis completion, endosomal trafficking and other related
cellular processes. Similarly, MTBD mediates spastin's interaction
with tubulin, which is a prerequisite for severing. By contrast,
the AAA domain mediates hexamer formation and catalyzes the
severing of MTs (3,30,31).
M1 and M87 are localized in different tissues and
subcellular locations. M1 is largely distributed in the cytosol due
to the strong nuclear export signal (NES) sequence of 50-87, while
M87 is distributed in the whole cell. Spastin also has a nuclear
export signal sequence situated in residues 195-204 that spans the
MIT and exon 4 region and a nuclear localization signal (NLS)
sequence (32,33). Fig. 1 presents the spastin domain
architecture and highlights the NES and NLS locations. Although
spastin is found in the nucleus, its function in the nucleus
remains elusive. A recent study postulated that extracellular
histone 2B is able to recruit spastin at the midbody during the
late stages of abscission (34).
By using proteomics analysis, our group also identified the
interaction between spastin and numerous histone isoforms (not
published). Since histones are vital for the morphology of
chromosomes, it may be inferred that spastin is involved in the
consolidation of chromosomes or in the regulation of protein
transcription. However, this assumption remains to be
experimentally validated.
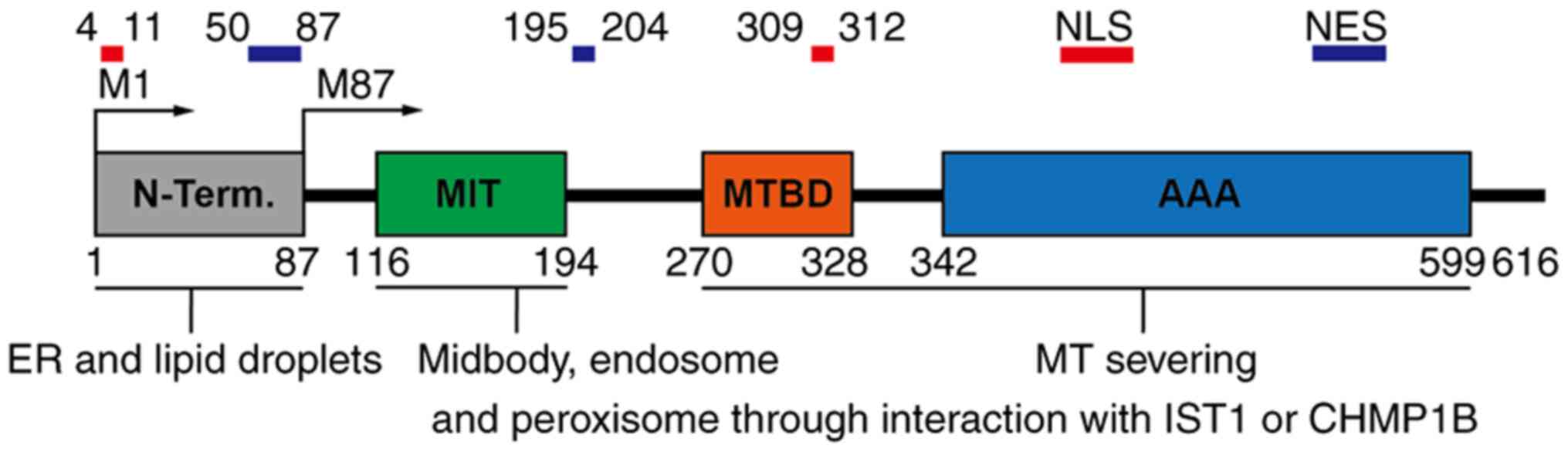 | Figure 1Spastin domain architecture. The
domains recruit spastin to specific subcellular localizations to
mediate MT severing. The N-terminal domain is responsible for
wedging into the ER and lipid droplet membrane. The ESCRT-III
proteins IST1 and CHMP1B interact with the MIT domain and recruit
spastin to ESCRT-III regions, such as midbody, endosome and
peroxisome. MTBD is required for MT binding, while the AAA domain
is necessary for MT severing. Two nuclear localization signals
(amino acids 4-11 and 309-312) and two nuclear export signals
(amino acids 50-87 and 195-204) are responsible for protein
shuttling between the nucleus and cytoplasm. MT, microtubule; ER,
endoplasmic reticulum; MIT domain, MT interacting and trafficking
domain; ESCRT, endosomal sorting complexes required for transport;
NES, nuclear export signal; NLS, nuclear localization signal; MTBD,
MT-binding domain; AAA, adenosine triphosphatases associated with
diverse cellular activities; IST1, increased sodium tolerance 1;
CHMP1B, charged multivesicular body protein 1B. |
Severing model of spastin
Studies that determined that spastin's structure
binds with peptides have revealed its severing mechanism. Spastin
forms hexamers through its AAA region in the presence of ATP. The
hexamer structure has a central spiral ring structure that is
positively charged. The loop subsequently engages with the
negatively charged C-termini of tubulin, thereby mechanically
extracting tubulin from the lattice using the energy provided by
ATP hydrolysis (35-37). Mechanistically, spastin forms
open spirals, as the subunits are mutated in an ATP-binding manner.
The hexamer moves up to form a closed structure when the spastin
structure with adenosine diphosphate-beryllium fluoride represents
a 'broken spiral' in which the sixth AAA is in the apo state
(36).
Questions to be addressed are whether MT severase is
able to depolymerize or amplify MTs, and how severase controls MT
length and mass. Recent studies provide answers to these questions
and postulate that spastin acts as an amplifier of total MT length
and mass (29,38). However, spastin's potential to
induce MT regrowth or catastrophe (i.e. the conversion of a growing
microtubule into a shrinking one depends on the MT concentration
(29,39,40). To date, two major theories for MT
rescue after severing action by spastin have been postulated.
According to the first theory, spastin grips the C-terminus of
tubulin in an adenosine triphosphate (ATP) hydrolysis manner,
followed by refilling the damaged lattice with guanosine
triphosphate-tubulin. The MTs severed by spastin are then
stabilized and act as seeds to mediate MT regrowth (38). In the second theory, spastin
concentrates at the newly generated plus end of MTs to block
depolymerizing activity, similar to other MT-associated proteins
(MAPs), such as calmodulin regulated spectrin associated protein
family member 3. The key evidence for this theory is that the
stimulation of increasing the MT length and mass is independent of
ATP hydrolysis (29). In
general, both theories indicate that spastin is a dual-function
enzyme that promotes MT regrowth or catastrophe and provide novel
insight regarding its severing activity during physiological
conditions.
Over the past decades, spastin was thought to cut
long MTs into short MTs to regulate MT dynamics (3). A question may be put forward
regarding how MTs behave after severing and how severed MTs
contribute to cellular functions. The observations discussed above
provide an intuitionistic model and form a foundation to understand
MT behavior after severing and provide comprehensive insight into
numerous molecular and cellular mechanisms of spastin. The cellular
functions of spastin introduced below are based on this model.
Cellular functions of spastin based on
its domain interactions
Spastin is an MT-severing enzyme containing MTBD and
AAA domains with adequate severing activity. In addition, the HD
and MIT domains interact with various proteins and recruit spastin
to specific subcellular localizations to sever the spatial MTs.
Understanding the domain-based interactions may provide insight
into spastin-severing activities at the cellular level. The
cellular functions of spastin based on its domain interactions are
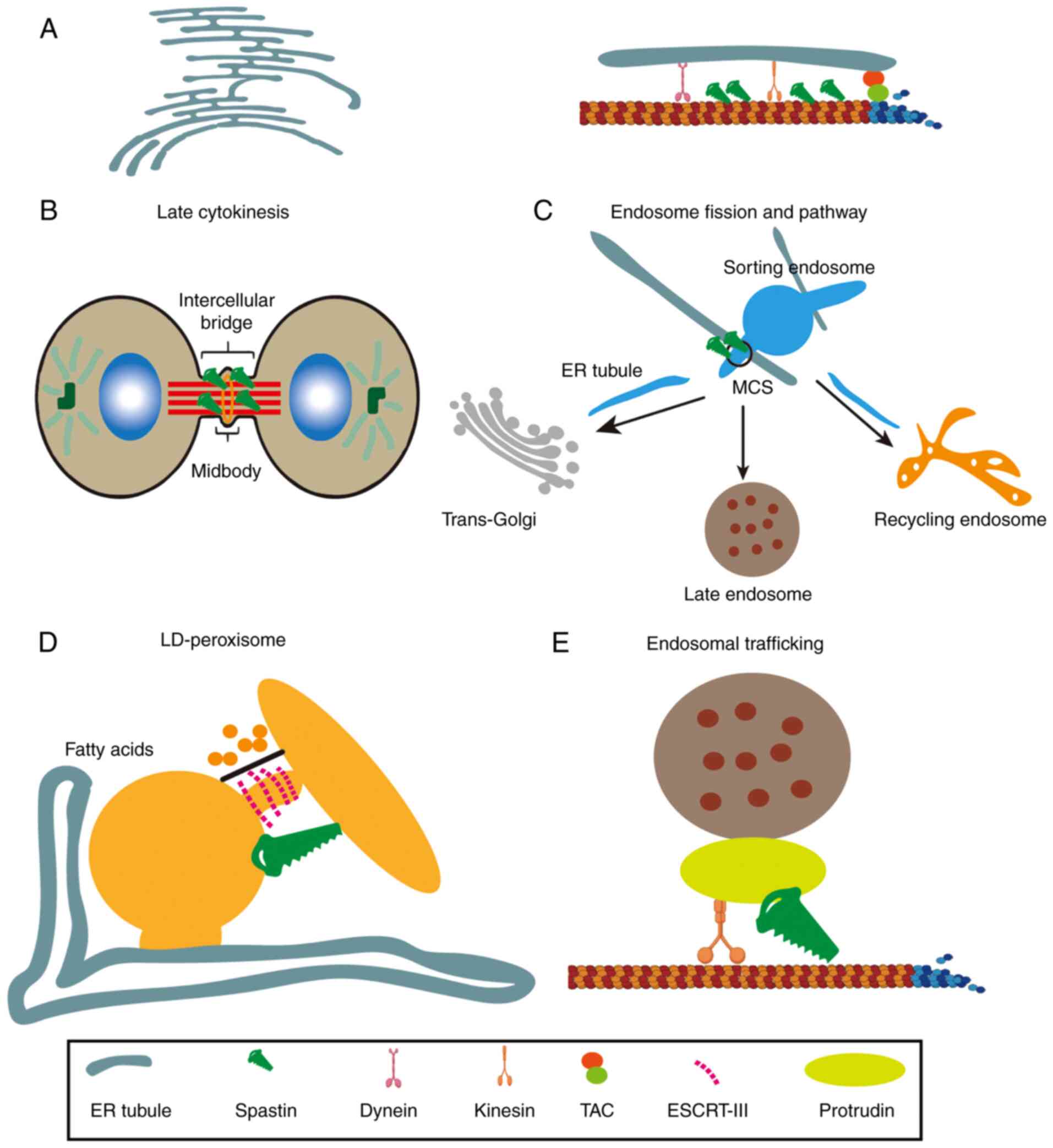
described below and illustrated in Fig. 2.
HD domain wedged into the membrane
structure ER shaping and Ca2+ handling
M1 spastin has an HD region that forms a hairpin
loop that embeds into the membrane lipid bilayer, causing spastin
to be wedged into the membrane. Three other HSP-related proteins,
receptor expression enhancing proteins (REEPs), alastins and
reticulons, also have a similar hairpin loop and are also recruited
in the ER. Thus, these proteins shape the tubular ER (12,41).
Spastin shapes the ER network and affects
Ca2+ handling (42).
However, the mechanisms of spastin in ER shaping remain to be
explored, although numerous HSP-related proteins involved in ER
shaping have been largely investigated. The ER sheets in
Drosophila melanogaster neurons are larger and have much
fewer tubules when the dominant-negative variant of the MT-severing
protein spastin is used to induce MT stabilization. Furthermore,
the store-operated calcium entry (SOCE) process is inhibited,
affecting the flies' flight ability. However, promoting MT dynamics
by applying the MT-destabilizing drug vinblastine restores flies'
ER morphology, SOCE and flight ability (42). These results suggest that the MT
dynamics provided by spastin have a decisive role in forming ER
tubules. An important question is whether the severing activity
mediated by spastin affects ER morphology. Currently, there is no
direct evidence of how spastin affects MTs surrounding the ER. As
discussed above, MT dynamics have an important role in ER
morphogenesis. There are two conditions under which MTs direct the
fate of ER morphology: i) Kinesin-MT-mediated transport drives ER
tubules; and ii) the ER tubules move along the growing MT by
docking onto the plus-end through a tip attachment complex
(43-45). Combined with the innovative
hypothesis of the severing model, it is reasonable that spastin
promotes ER tubule formation by increasing MT fragments with more
MT-plus ends (38). Furthermore,
the increase in the total length of the MT potentially leads to
more kinesin loading, resulting in more efficient transport.
Another potential mechanism of spastin-mediated ER
shaping is through interactions with protrudin. Protrudin is also
an ER-resident protein and regulates the sheet-to-tubule balance
(46,47). Kinesin-related protein 5 (KIF5)
belongs to the kinesin motor family, which drives long-distance
cargos along the MT 'tracts' in the anterograde direction using the
energy generated by ATP hydrolysis (48,49). Protrudin interacts with KIF5
(16,49), thus influencing ER morphology in
an MT-dependent mode. Furthermore, protrudin extracts lipids from
the ER by interacting with PDZ domain-containing protein 8 (PDZD8),
thus proving that these proteins coordinate with each other to
influence ER morphology (50,51). M1 spastin is able to interact
with protrudin, thereby regulating ER morphology, probably by
modulating the MT dynamics necessary for protrudin-mediated kinesin
motor activities (47,52).
Overall, spastin contributes to ER tubule formation
and inhibits the SOCE process. Although the clear attribution of
the ER sheet-tubule balance to cellular functions remains largely
elusive, it is now known that Ca2+ signaling is
affected. Furthermore, casein kinase 2 (CK2) is also activated upon
spastin deficits (19). Whether
other signaling pathways are activated or inhibited when the ER
sheet-tubule balance is affected by spastin remains to be explored.
Given that the ER is distributed throughout neurons, how spastin
affects ER shaping, the calcium pathway and other signaling
pathways in neural development and disease is an important issue in
understanding the physiological and pathological process, which
still requires further investigation. Answers to these questions
will further elucidate the pathogenesis of HSP.
Spastin and LD formation
LDs are highly dynamic and complex organelles
responsible for the assembly and storage of neutral lipids
(53). They are specialized
compartments of the tubular ER, from which they are generated in a
stepwise process (54). Lipids
from LDs are transported to the cell membranes and regulate
intracellular signals (55,56). Various studies postulated that
spastin anchors to LDs through its hydrophobic domain (57-86). Spastin depletion or
overexpression of the dominant-negative variant impairs the
formation of LDs in the nerves and skeletal muscles of fruit flies,
while overexpression of spastin promotes the formation of LDs
(11,57). These results suggest that M1
spastin is associated with the formation of LDs. Although there is
no direct evidence indicating how spastin affects the formation of
LDs, it may be hypothesized that the newly formed LDs may be ER
shaping products, as LDs are specialized compartments of the
tubular ER. However, how LDs function remains largely elusive.
Whether LDs transported to the cell membrane or intracellular
signals are changed still requires further research. In addition,
it has not been demonstrated that dysfunction of LDs is involved in
HSP pathogenesis. Whether LD abnormalities are a common defect or
rather a readout of different dysfunctional pathways remains
elusive. Therefore, LD function should be investigated in patients
with HSP to further the understanding of HSP and offer novel
therapeutic opportunities.
Interaction between the MIT domain and
ESCRT-III
The MIT domain is adequate for binding two ESCRT-III
proteins: increased sodium tolerance-1 (IST1) and charged
multivesicular body protein 1B (CHMP1B) (17,58,59). This interaction allows the
recruitment of spastin to specific subcellular localizations, thus
regulating spatial MT dynamics. The ESCRT-III machinery is vital
for membrane scission in numerous membrane remodeling processes,
including viral budding from the cell surface, budding of endosomes
to form vesicles of late endosomes and cytokinetic abscission
(14,60,61). In addition, the ESCRT-III
proteins recruit spastin to regulate cytokinetic abscission,
endosomal trafficking and fatty acid (FA) metabolism.
Cytokinetic abscission
The role of spastin during cytokinesis has been
extensively elucidated and reviewed in various studies (30,58,62). In the present review, the process
is briefly discussed. During the late stages of the anaphase, the
nuclear envelope, which is frequently intersected by spindle MTs,
is progressively reassembled around the reforming daughter nuclei.
In this process, spastin recruitment by IST1 helps to remove the
remaining spindle MTs for nuclear envelope sealing. Failure of this
process leads to the accumulation of DNA damage.
Similarly, the daughter cells remain connected by an
inter-cellular bridge with the midbody ring overlapping with MTs at
its center during the late stages of abscission. CHMP1B thus
recruits spastin to remove the MTs in the intercellular bridge.
Depletion of spastin results in abscission failure, causing the
daughter cells to remain connected by elongated MT-filled
bridges.
Endosomal trafficking
Endosomes are sorting organelles that mediate the
internalization of extracellular transmembrane molecules and
ligands. Endosomes are categorized into early, recycling and late
endosomes based on their internalization stage. Internalized
molecules may be recycled into the cell membrane using recycling
endosomes or degraded using late endosomes (63,64). Endosomal trafficking regulates
plasma membrane receptor concentrations, which are critical
extracellular responses. The ESCRT machinery recruits spastin into
endosomes to regulate endosome fission and trafficking pathways
(65,66).
Existing evidence suggests that spastin regulates
endosome trafficking in two ways. i) Spastin may promote the
tubulation and fission of endosomes, thereby inhibiting their
pathway to the lysosome (17,18,67). This process is accomplished by
the actin-dependent and MT-dependent pulling force and membrane
scission by dynamin (68,69).
In this process, the ESCRT-III protein increased sodium tolerance 1
recruits spastin into the endosome through the interaction of its
MIT domain. Previously, severing activities mediated by spastin
were thought to generate more MTs (38), thus ensuring that MTs were loaded
with more dynein to enhance the driving force, thereby resulting in
an enhancement of endosome tabulation and fission. However, this
point of view lacks experimental evidence. Of note, a recent study
provided direct evidence that the ER and endosome membrane contact
sites (MCSs) are vital for endosome tubulation and fission
(18). Overall, a better
explanation for the tubulation and fission of endosomes may be the
result of spastin-mediated ER reorganization. The changed ER
network reconstructs the MCSs and contributes to promoting endosome
tabulation and fission, while how spastin affects the MCSs between
the ER and endosomes remains to be elucidated. ii) Spastin is able
to regulate endosome trafficking by antagonizing protrudin, thereby
regulating endosome trafficking in a generalized manner (16). Protrudin is an ER-resident
protein that interacts with Rab7 and phosphatidylinositol
3-phosphate. These interactions facilitate the transfer of KIF5 to
the late endosomal motor adaptor FYVE and coiled-coil domain
containing 1, thus promoting endosomal transport to the plasma
membrane (70,71). Although the interaction of
spastin with KIF5 remains to be proven, studies have indicated that
it is able to interact with protrudin (52). Regardless of whether it is direct
or indirect, this interaction may recruit spastin to KIF5,
resulting in MT 'railway breakage', which blocks the late endosome
toward the cell periphery.
To date, the process of endosomal behaviors mediated
by spastin has remained largely elusive. As described above,
spastin promotes endosome tabulation and fission to inhibit the
lysosome pathway. Spastin regulates endosome trafficking in a
generalized manner. Of note, the destination of the endosome cargo
remains to be determined. Taking BMP receptors as an example, it is
unlikely that the BMP receptor retransports to the cell periphery,
as its level on the membrane increases upon deficits of spastin.
Furthermore, spastin inhibits the lysosomal pathway, and it remains
to be determined how cargo is metabolized in the cell. In addition,
the specific cargos transported through spastin-mediated endosomal
trafficking remain to be identified. Answering these questions may
lead to the elucidation of the specific mechanisms of endosomal
trafficking mediated by spastin and pave the road for the
development of therapies for HSP. With the rapid progression of
biological analysis techniques, it may be promising to identify
endosome components and trace these cargos, thus elucidating the
endosomal trafficking mediated by spastin and revealing the
molecular and cellular pathogenic mechanisms leading to HSP.
LD-peroxisome tethering
Membrane contact sites between LDs and peroxisomes
have been observed for decades. However, the molecular underpinning
that connects these organelles remains largely elusive (72,73). Peroxisomes harvest FAs from LDs
for energy production and maintain cellular homeostasis by
recycling lipids and protecting cells from oxidative stress and
damage (74,75). It has been postulated that
spastin tethers to peroxisomes through the interaction between the
MIT domain and the peroxisome resident protein ATP-binding cassette
sub-family D member 1 and mediates FA flux from LDs to peroxisomes
(13). Furthermore,
overexpression of M1 spastin reduces peroxidized lipid accumulation
in cells exposed to oxidative stress. On the other hand, the
dominant-negative variant mutant spastin (K388R) is not able to
promote LD peroxisome tethering and fails to lower peroxidized
lipid levels in LDs (13,76).
In addition, HSP patient-derived cells with mutations in spastin
also indicated impaired peroxisome movement and distribution. These
observations have established an intuitionistic model to describe
the mechanism of cellular organelle crosstalk between LDs and
peroxisomes and the process of FA trafficking and metabolism.
However, there is still a lack of evidence that FA impairment is
involved in patients with HSP, which requires further validation.
Overall, this mechanism strongly suggests that LD peroxisome
tethering has an important role in the neuropathology of HSP and
may provide an opportunity of targeting FA metabolism for HSP
therapy.
3. Regulation of spastin's stability and
activity
Spastin's stability and activity are
spatiotemporally regulated at different transcriptional levels and
with posttranslational modifications and coordination of other
MAPs. Understanding the precise mechanisms involved in regulating
spastin protein levels and activity may provide novel therapeutic
targets for HSP based on haploinsufficiency (77,78). To date, no relevant spastin
regulators have been identified in patients with HSP. However,
numerous factors have been reported to regulate its stability and
activity in in vitro cell models. For instance, numerous
transcription factors (NRF1 and SOX11) and microRNAs (miRs),
including miR-30, -33a, -96 and -182, are able to regulate the
expression levels of spastin (79-81). At the posttranscriptional level,
spastin may be phosphorylated at S268 and recruited to the midbody
(62); SUMOylation of spastin at
K427 may alter the severing ability of spastin (26); ubiquitination of spastin at K554
mediates degradation (82). In
addition, the consolidation of protein on MT (Tau, collapsin
response mediator protein 5) also regulates the severing activity
of spastin (83,84). The Dpy-30 histone
methyltransferase complex regulatory subunit gene, which modifies
in hereditary spastic paraplegia, has an epistatic interaction with
spastin (85). Understanding
spastin's regulatory mechanisms may help identify novel therapeutic
strategies against HSP. Table I
highlights the specific regulators and their impact on spastin.
Further putative regulators of spastin should be identified through
proteomic and genomic approaches to understand the precise
regulatory mechanisms of spastin in cellular activities.
 | Table ISpastin's stability and activity
regulators. |
Table I
Spastin's stability and activity
regulators.
| Regulators | Function | (Refs.) |
|---|
| Transcription
factors | | |
| NRF1, SOX11 | Positive regulation
of spastin expression | (79) |
| Posttranscriptional
regulators | | |
| miR96, miR-182,
miR-30, miR-33a | Negative regulation
of spastin expression | (79-81) |
| Posttranscriptional
modifications | | |
| Phosphorylation at
S268 | Midbody
localization to regulate abscission | (62) |
| SUMOylation at
K427 | Regulation of
severing activity | (26) |
| Ubiquitination at
K554 | Protein
degradation | (82) |
| MAPs | | |
| Tau | Consolidation of
this protein on MTs regulates the severing activity of spastin | (83) |
| CRMP5 | Consolidation of
this protein on MTs regulates the severing activity of spastin | (23,84) |
| Epistatic
interaction | | |
| DYP30 | Epistatic
interaction with spastin during disease onset | (85) |
4. Role of spastin in neural
development
To date, no breakthrough has been made in the
treatment of HSP-SPG4. However, a deeper understanding of the role
of spastin in neural development contributes to more comprehensive
insight into its etiology, thereby providing novel ideas for
effective HSP-SPG4 treatment. Developing neurons require dynamic
MTs to remodel their shapes and mediate kinesin and dynein-based
cargo transport, thus promoting and maintaining axonal and
dendritic processes (86,87).
The MT dynamics mediated by spastin in different subcellular
locations highlight its vital role in neural development. The role
of spastin in neural development has been gradually reported in
recent years. In the chapter below, recent advances in
spastin-mediated neurogenesis, axon elongation and branching,
synapse formation and maturation, axon transportation and axon
maintenance are discussed and existing problems that should be
explored are highlighted. The roles of spastin during neural
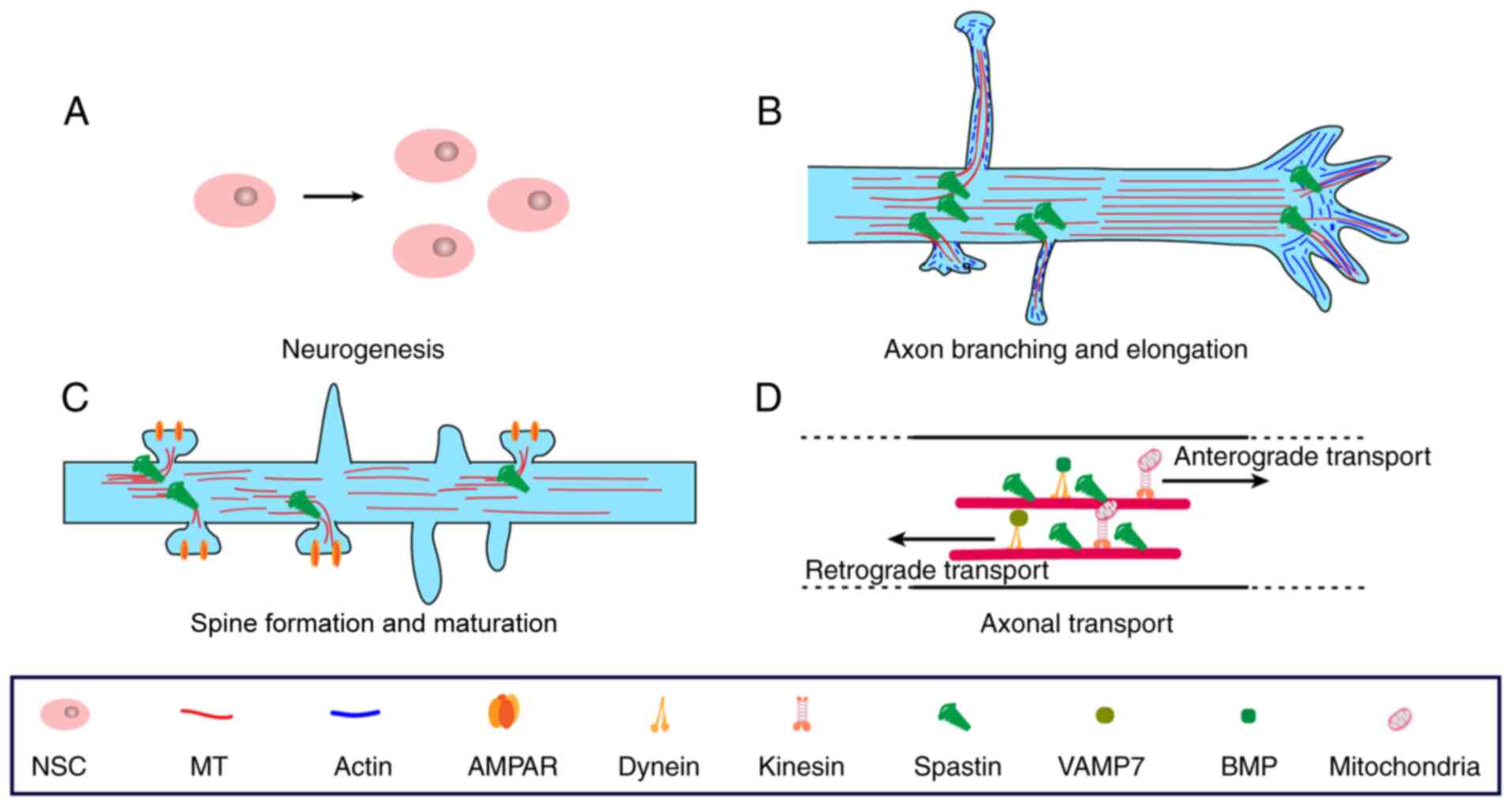
development are illustrated in Fig.
3.
Neurogenesis
Neurogenesis is active during the embryonic period,
as it is responsible for producing new neurons. In this period,
neural stem and progenitor cells go through symmetric proliferative
division until a sufficient population of neural stem cells (NSCs)
is produced and subsequently develops into neurons (88,89). It has been reported that both M1
and M87 spastin is enriched in proliferative NSCs and embryonic
brain tissues. Spastin depletion using short hairpin RNA led to a
decrease in NSC proliferation and neuronal lineage differentiation.
Of note, this phenomenon was partly reversed using the
MT-destabilizing drug nocodazole, indicating that NSC proliferation
requires the dynamic MTs provided by spastin (90). This observation suggests that
spastin participates in neurogenesis by promoting MT dynamics. As
the Sonic hedgehog (Shh) signaling pathway is responsible for
regulating and coordinating cellular growth and development in the
embryo, this study also revealed that the Shh signaling pathway was
inhibited by spastin depletion (83). However, the inhibition mechanism
of the Shh signaling pathway by spastin depletion should be further
evaluated.
Axonal elongation and branching
Growth cones at the end of an axon mediate its
extension and turning after axon differentiation. They sense the
extracellular environment and regulate axonal development and
turning. They also have a vital role in intercellular signaling
transmission (91,92). In general, axon development
undergoes three stages: Protrusion, engorgement and consolidation.
In the growth cone, the MTs are highly dynamic in contrast to the
axonal draft. Highly dynamic MTs contribute to the high sensitivity
of the cones to guidance cues. Furthermore, filopodia and
lamellipodia consolidation also requires the mechanical force
provided by dynamic MTs (93,94).
MTs form highly stable and polarized bundles in
axons, providing structural support and 'railways' to guide
MT-dependent transport. During axon branching, the cytoskeleton
must be remodeled, which requires actin filament accumulation to
form axonal filopodia. Soon after that, the MTs then penetrate the
actin-rich filopodia, allowing the maturation and continuous
extension of the branches (95,96). This phenomenon proves that MT
dynamics are a vital factor during axon branching and
formation.
As described above, M1 spastin is distributed in the
cytosol and mainly at the membrane sites, such as the ER and LD.
However, M87 spastin is distributed in the whole cell and was
reported to be enriched in the growth cone (97), and part of the axonal branch
points are likely to generate new branches (24,98). There is abundant evidence
suggesting that spastin contributes to axonal branching and
elongation. Furthermore, the new MT severing theory suggests that
spastin and other severing enzymes may act as amplifiers to
generate more MT seeds during regrowth (29,38). Cumulative evidence has indicated
that neurons are sensitive to severing activities. Appropriate
severing by spastin and katanin, which are highly expressed in
neurons during development, promotes axonal elongation (77,99-101). Of note, overexpression of
spastin significantly promotes the formation of axonal branches,
whereas katanin does not. The accumulation of spastin in the branch
points strongly suggests that branch formation requires the
accumulation of actin filaments and MTs (100). Thus, it may be inferred that
spastin potentially affects the actin cytoskeleton by interacting
with related actin-associated proteins through MIT and MTBD
domains. However, the actual mechanism involved in spastin-mediated
remodeling of the actin cytoskeleton should be further
evaluated.
Dendritic spine formation and
maturation
The formation and maturation of dendritic spines
contribute to the connections between neighboring neurons, which is
vital for responding to novel stimuli throughout neural development
and adulthood (102,103). In the last decade, research on
spastin has primarily focused on axonal behavioral mechanisms. It
is worth noting that M87 spastin is distributed in the entire cell
compartment, including dendrites and dendritic spines (26,100). How M87 spastin affects dendrite
behaviors remains largely elusive.
A decade ago, spastin was reported to regulate MT
dynamics during the elimination of neuromuscular junctions in fly
knockout models (27). Recent
studies further indicated that spastin is closely related to
learning and memory. Furthermore, it participates in the formation
and maturation of dendritic spines and regulates the transport of
the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
(AMPAR) subunit GluA1 (26) and
GluA2 (25). Lopes et al
(25) established a
spastin-knockout mouse model and determined that the formation and
maturation of dendritic spines were inhibited upon spastin
depletion and that KIF5-mediated GluA2 transport was also blocked
upon spastin depletion. AMPAR trafficking may induce long-term
potential or long-term depression, thereby regulating the maturity
of dendritic spines (104,105). As such, spastin potentially
regulates dendritic spine morphology by altering MT-dependent
transport. However, another study suggested that overexpression of
spastin leads to an increase in the proportion of immature
dendritic spines and leads to a decrease in GluA1 membrane
expression (26). That study
also indicated that GluA1 trafficking is closely related to
spastin-mediated endosomal trafficking. The different effects of
spastin on GluA1 and GluA2 trafficking may be attributed to the
different trafficking pathways. GluA1 membrane trafficking is
primarily dependent on the endocytosis or exocytosis of endosomes,
while GluA2 binds directly to glutamate receptor interacting
protein (GRIP) and is then subjected to KIF5-mediated transport
(106). Of note, GRIP is able
to bind the GluA2 subunit but not to GluA1 or GluA4 (107). Furthermore, neural activities
are sensitive to the dosage of spastin levels, and it still
requires to be elucidated how spastin works during synapse
formation under physiological conditions. Collectively, these
studies postulate that spastin-mediated cargo delivery is closely
related to the formation and maturation of dendritic spines by
regulating AMPAR trafficking. Furthermore, studies have also
confirmed that KIF5 is also able to transport GABA receptors toward
the dendritic spine (108).
However, whether spastin is able to regulate this process requires
further exploration.
Although the actin cytoskeleton is the primary
regulator of spine morphology, MTs may also penetrate the spine
transiently. Invasive MTs are able to transport relevant cargo
proteins, such as p140Cap, to promote the polymerization of actin
filaments (109). It may be
assumed that spastin contributes to actin polymerization by
interacting with actin-binding proteins, as it acts as an amplifier
to generate more MT seeds during regrowth. However, this assumption
should be verified.
Axonal transport and maintenance
Axon transport is essential for regulating axon
composition, neural development, maintenance and survival (110,111). The process is mediated by motor
molecules (kinesin or dynein) to deliver different cargos, such as
organelles, signal molecules and RNAs, along the MTs in an
ATP-dependent manner. For instance, the kinesin motor mediates the
anterograde transport responsible for delivering RNAs, proteins and
organelles toward the growth cones and synapses. Similarly,
dynein-mediated retrograde transport delivers nutrient factors and
proteins that require degradation by the cell body, thus responding
to the extracellular environment (112-114). This process is highly dependent
on the MTs. As such, spastin is potentially involved, as it alters
the MT dynamics and arrays.
Numerous studies have reported spastin involvement
in MT-mediated cargo transportation. Of note, spastin mutations
identified in patients with HSP lead to axonal swellings and axonal
transport abnormalities (19,20,115). To date, spastin has also been
reported to regulate the transportation of mitochondria (20,116), peroxisomes (117,118), amyloid precursor protein
(20), vesicle-associated
membrane protein 7 (VAMP7) (119) and BMPII receptors (120). In particular, M1 spastin is
crucial during fast axonal transport using isolated squid axoplasm
but not M87 (6,19). Consistent with these
observations, M1 spastin has a pivotal role in VAMP7 transport in
cortical neurons but not M87 (121). However, Connell et al
(16) postulated that both M1
and M87 antagonize KIF5-mediated BMPII transport to the cell
membrane. These studies confirm that both M1 and M87 spastin are
involved in axonal transport despite the differences in each cargo
transportation mechanism and their transport direction.
M1 and M87 are different in the process of axon
transportation. M1 has an additional hairpin loop domain that
determines its location in the ER and LDs (32). It was also reported that M1
spastin is more efficient in promoting endosomal tubule fission.
(17). Thereafter, M1 spastin
regulates cargo transportation in ER-dependent pathways, while M87
primarily directs cargo transport by altering MT dynamics.
Furthermore, M1 spastin is able to potentially recruit M87 from the
cytosolic pool, as spastin requires hexamerization to sever MTs
(28). Therefore, it may be
hypothesized that M1 is more efficient than M87 in axonal transport
via ER-dependent pathways.
As described previously, M1 spastin contributes to
tubular ER formation by regulating ER-surrounded MT dynamics. The
ER is distributed throughout the whole cell and is conducive to the
physical continuity of axons (122). The ER and other organelle
membrane contact sites allow communication with each other, thereby
regulating intracellular membrane trafficking (123,124). For instance, Allison et
al (18) revealed that
spastin promotes endosomal fission and regulates its trafficking
using ER-endosome MCSs. Furthermore, ER-resident proteins, such as
protrudin and PDZD8, potentially contribute to endosome trafficking
(50) after ER morphology is
altered by M1 spastin. ER morphology alteration is closely
associated with calcium flux (125,126). The process is required to
maintain axonal transport (126). Of note, spastin also
potentially influences the calcium signaling pathway in axons, thus
indirectly contributing to normal axonal transport. As such, the M1
spastin-mediated mechanism involved in the regulation of axonal
transport may be ER-dependent, which requires further experimental
confirmation.
5. Role of spastin in HSP
HSP is characterized by progressive weakness and
paraplegia of the lower limbs. Its major pathological feature is
corticospinal tract degeneration, which leads to axonal swellings,
resulting in neurological disorders. A breakthrough in
understanding this disease is that spastin was discovered to be an
MT severing enzyme that acts as an MT amplifier to generate more
MTs (90), thus providing a
reasonable etiological basis for understanding spastin-induced HSP.
To date, the major cause of HSP-SPG4 is postulated to be
insufficient MT cutting caused by haploinsufficiency of spastin
(127). However,
haploinsufficiency alone cannot explain numerous phenomena observed
in HSP-SPG4 models (128,129). Furthermore, the
gain-of-function scenario is also postulated to be one of the
onsets of HSP (130,131). Differences between spastin
isoforms also add complexity to the understanding of the disease.
The recent series of innovative studies on spastin provides a
theoretical basis for understanding its pathogenic mechanisms. The
present review primarily focuses on discussing the pathogenic
mechanism of spastin combined with existing observations of spastin
in cellular activities to elucidate how spastin mutations lead to
HSP occurrence.
The haploinsufficiency scenario posits that the
pathogenesis of HSP-SPG4 is a consequence of decreased functional
spastin protein due to the presence of the mutant protein, which
cannot function normally (132). However, gain-of-function is
another different mechanism. It is proposed that mutant spastin
aggregates and elicits toxicity. One of the most obvious features
is that a small number of spastin protein mutations may lead to
negative effects. This phenomenon is supported by evidence
indicating that spastin mutants activate CK2. CK2 phosphorylates
both kinesin-1 and dynein, which mediate cargo release from the
motor (19). A recent review
postulated that gain-of-function may be the primary cause of
HSP-SPG4 pathogenesis, while haploinsufficiency makes axons more
vulnerable to a second insult (31).
With the increase in the current knowledge of
spastin, the molecular and cellular mechanisms have been revealed
by intense studies. These mechanisms contribute to a comprehensive
understanding of this disease. It may be suggested that
haploinsufficiency and gain-of-function are not controversial but
complementary to each other, causing HSP. Furthermore, numerous
gain-of-function observations may be explained from the current
literature and require further exploration. A more reasonable
explanation combined with recent discoveries of the molecular and
cellular mechanisms of spastin may thus be proposed.
First, the gain-of-function scenario is supported by
evidence that M1 mutants cause hyperactivation of CK2, which is an
effect not shared with the M87 mutant (19). However, there is no direct
evidence of how M1 spastin activates CK2. As described above, M1
spastin mutants lead to ER shaping abnormalities. Furthermore, CK2
activation in numerous cell types, including neurons, is closely
associated with ER stress (133-135). Therefore, it is conjectured
that the insufficient ER surrounding MT severing mediated by the
mutant spastin potentially leads to indirect CK2 activation. The
gain-of-function phenomenon is also supported by MT destabilization
caused by the mutant spastin. Second, the gain-of-function scenario
is also supported by the fact that mutant spastin may result in MT
destabilization. Regarding this phenomenon, MT destabilization,
referring to a decreased proportion of acetylated and detyrosinated
tubulin, was detected by western blot analysis (130). Combined with the severing model
described above, spastin deficits may lead to a loss of MTs, as
spastin acts as an MT amplifier under physiological conditions.
Finally, the gain-of-function scenario is further supported by
knockout and knockdown models. Spastin knockout and knockdown mice
have only a mild phenotype of axonal swellings. For instance,
SPASTC448Y mice exhibit locomotor phenotypes far more
reminiscent of HSP than any other mouse despite corticospinal
degeneration (130). From our
perspective, this is not controversial, as the knockout and
knockdown mice would have compensatory mechanisms and other related
proteins, such as REEP1 and alastin, to rescue the phenotype caused
by insufficient spastin dosage. The ER sites in
SPASTC448Y mice may be occupied by mutant spastin, thus
inhibiting other rescue effects through the dominant-negative
mechanism. However, whether the expression of other proteins is
altered in spastin knockout or knockdown mice should be further
examined.
Another question that should be addressed is whether
M1 or M87 mutations cause the occurrence of HSP. Axonal transport
impairment is documented as the main HSP feature associated with
spastin mutations. Regarding whether M1 or M87 induce the
impairment of axonal transport, Solowska et al (6) demonstrated that axonal transport is
affected by M1 spastin but not M87 by using squid axoplasm with
different HSP-related mutant spastin. Furthermore, M1 spastin is
highly expressed in the corticospinal tract, which is consistent
with HSP pathology (6,19). Cognizant of this, M1 mutation
appears to be more relevant to HSP. In addition, M1 mutation was
markedly more toxic than mutant M87 to the effects of motor neurons
in transgenic flies and neurite outgrowth in primary cortical
neurons (131). However,
studies postulate that the vast majority of spastin disease
mutations affect both protein isoforms. Connell et al
(16) and Allison et al
(18) provided a reasonable
explanation for the relationship between M1 and M87 isoforms based
on the coordination of spastin-mediated endosome trafficking. They
hypothesized that M1 localizes and nucleates in the ER and
subsequently binds with M87 by recruiting it from the cytosolic
pool to form functional hexamers. This observation was further
supported by the evidence that M1 is more efficient than M87 in
endosome tubule fission. Collectively, both spastin isoforms are
involved in HSP pathogenesis, with M1 mutations possibly being more
efficient than M87 mutations.
Understanding the etiology of HSP is vital in
designing appropriate therapeutic strategies for patients with HSP.
At present, HSP-SPG4 is mostly thought to be mediated by
haploinsufficiency and insufficient MT severing. Null spastin
homologs have been used to identify drugs that are able to reverse
the HSP phenotype (128). For
instance, the application of phenazine, methylene blue and
N-acetyl-cysteine improves locomotor activity. These compounds have
been approved by the Food and Drug Administration and hold great
promise for translation to therapy (1). In addition, vinblastine and
nocodazole are able to abrogate axonal swellings associated with
spastin-induced impaired axonal transport in cortical neurons of
knockout mice. However, application of these drugs would cause side
effects (136). Furthermore,
overexpression of M1 or M87 spastin isoforms restores neurite
length, branching and the number of primary neurites and reduces
swelling in neuronal cells (10). However, this strategy, combined
with other approaches, should be empirically designed and tested in
animal models prior to roll-out. As such, targeting drugs for
HSP-SPG4 remains an area of focus for future extensive studies.
6. Concluding remarks
HSP is a disease caused by different gene mutations
with diverse phenotypes. However, the onset of HSP is usually
caused by a single gene mutation. Therefore, it is promising to
develop precise drugs for targeted treatment of single genes.
Spastin is the most common mutated gene of HSP, accounting for 60%
of autosomal dominant inheritance and 15% of sporadic cases. Taking
HSP-SPG4 as a starting point, research on spastin in the past two
decades has revealed numerous underlying molecular mechanisms in
cell function, aiding in understanding the potential mechanism of
axon degeneration. However, the cellular functions of spastin
remain to be fully clarified and gene therapy targeting spastin for
HSP is still full of challenges.
The key discovery to understand spastin's function
is that it is an MT-severing enzyme that is able to cut long MTs
into short fragments. Spastin is a protein with multiple modular
domains. By interacting with other proteins or membrane structures,
spastin is able to bring severing activity to various cellular
subregions and participate in different cellular activities, such
as endosomal transport, ER shaping, MT-based transport and lipid
metabolism. However, their molecular mechanisms have yet to be
fully elucidated. In addition, certain innovative cellular
mechanisms mediated by spastin, such as LD formation and
peroxisome-mediated FA metabolism in patients with HSP, remain to
be explored. High-resolution image visualization technology is
required to observe the ultramicrocellular structure of axon
degeneration in patients or models of HSP.
Although devising drugs targeting spastin appears to
be more promising, there are still numerous obstacles that hinder
the development of precision therapeutic drugs. First, the etiology
of HSP-SPG4 remains elusive. Specifically, it remains to be
clarified whether haploinsufficiency or gain-of-function cause the
degeneration of axons. If the gain-of-function mechanism truly
exists, the toxic mutant spastin is imperative to be removed or the
toxic effects caused by mutant spastin should be inhibited. For
instance, using CK2 inhibitor to activate the axonal transport
could reduce the axonal swellings in HSP patients. Furthermore, it
remains to be determined whether M1 and M87 mutations mediate the
occurrence of HSP independently or coordinatively and how M1 and
M87 coordinate to function. Both isoforms are able to restore
neurite length and branching and reduce axon swellings in induced
pluripotent stem cells from a patient with a spastin nonsense
mutation, and it remains to be determined which isoform or whether
both of them are vital for recovery. It should also be clarified
what isoform should be used as a therapeutic target. In addition,
the use of MT depolymerization drugs such as vinblastine improves
the phenotype of HSP-SPG4, but MT depolymerization drugs are not
targeted. It remains to be elucidated whether they change the MT
dynamics in other cellular regions and cause other side effects.
Finally, excessive spastin destroys the cytoskeleton and is toxic
to neurons. It is thus required to determine how to administer the
accurate dosage of spastin according to physiological conditions to
reach the axon degeneration site.
Fast progress in understanding the molecular
mechanisms of spastin holds promise to unveil the most basic
cellular biological mechanism of HSP-SPG4 in the near future.
Resolving these questions will help provide more precise and
innovative treatments for patients with HSP.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
QLL performed the literature search and wrote the
manuscript. GWZ contributed to the molecular mechanisms. ZSJ and
HSL provided supervision and revised the manuscript. All authors
read and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
HSP
|
hereditary spastic paraplegia
|
|
ER
|
endoplasmic reticulum
|
|
LD
|
lipid droplet
|
|
HD
|
hydrophobic domain
|
|
MT
|
microtubule
|
|
MIT
|
MT-interacting and trafficking
domain
|
|
MTBD
|
MT-binding domain
|
|
AAA
|
ATPases associated with diverse
cellular activities
|
|
ESCRT
|
endosomal sorting complexes required
for transport
|
|
NES
|
nuclear export signal
|
|
NLS
|
nuclear localization signal
|
|
ATP
|
adenosine triphosphate
|
|
PDZD8
|
PDZ domain-containing protein 8
|
|
SOCE
|
store-operated calcium entry
|
|
MAP
|
MT-associated protein
|
|
KIF5
|
kinesin-related protein 5
|
|
MCS
|
membrane contact site
|
|
NSC
|
neural stem cell
|
|
GRIP
|
glutamate receptors interacting
protein
|
|
AMPAR
|
α-amino-3-hydroxy-5-methy
l-4-isoxazolepropionic acid receptor
|
|
CK2
|
casein kinase 2
|
References
|
1
|
Shribman S, Reid E, Crosby AH, Houlden H
and Warner TT: Hereditary spastic paraplegia: From diagnosis to
emerging therapeutic approaches. Lancet Neurol. 18:1136–1146. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schüle R, Wiethoff S, Martus P, Karle KN,
Otto S, Klebe S, Klimpe S, Gallenmüller C, Kurzwelly D, Henkel D,
et al: Hereditary spastic paraplegia: Clinicogenetic lessons from
608 patients. Ann Neurol. 79:646–658. 2016. View Article : Google Scholar
|
|
3
|
Solowska JM and Baas PW: Hereditary
spastic paraplegia SPG4: What is known and not known about the
disease. Brain. 138:2471–2484. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fink JK: Hereditary spastic paraplegia:
Clinico-pathologic features and emerging molecular mechanisms. Acta
Neuropathol. 126:307–328. 2013. View Article : Google Scholar
|
|
5
|
Salinas S, Carazo-Salas RE, Proukakis C,
Schiavo G and Warner TT: Spastin and microtubules: Functions in
health and disease. J Neurosci Res. 85:2778–2782. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Solowska JM, Morfini G, Falnikar A, Himes
BT, Brady ST, Huang D and Baas PW: Quantitative and functional
analyses of spastin in the nervous system: Implications for
hereditary spastic paraplegia. J Neurosci. 28:2147–2157. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deluca GC, Ebers GC and Esiri MM: The
extent of axonal loss in the long tracts in hereditary spastic
paraplegia. Neuropathol Appl Neurobiol. 30:576–584. 2004.
View Article : Google Scholar
|
|
8
|
Lumb JH, Connell JW, Allison R and Reid E:
The AAA ATPase spastin links microtubule severing to membrane
modelling. Biochim Biophys Acta. 1823:192–197. 2012. View Article : Google Scholar
|
|
9
|
Schickel J, Pamminger T, Ehrsam A, Münch
S, Huang X, Klopstock T, Kurlemann G, Hemmerich P, Dubiel W, Deufel
T and Beetz C: Isoform-specific increase of spastin stability by
N-terminal missense variants including intragenic modifiers of SPG4
hereditary spastic paraplegia. Eur J Neurol. 14:1322–1328. 2007.
View Article : Google Scholar
|
|
10
|
Havlicek S, Kohl Z, Mishra HK, Prots I,
Eberhardt E, Denguir N, Wend H, Plötz S, Boyer L, Marchetto MC, et
al: Gene dosage-dependent rescue of HSP neurite defects in SPG4
patients' neurons. Hum Mol Genet. 23:2527–2541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arribat Y, Grepper D, Lagarrigue S, Qi T,
Cohen S and Amati F: Spastin mutations impair coordination between
lipid droplet dispersion and reticulum. PLoS Genet.
16:e10086652020. View Article : Google Scholar :
|
|
12
|
Park SH, Zhu PP, Parker RL and Blackstone
C: Hereditary spastic paraplegia proteins REEP1, spastin, and
atlastin-1 coordinate microtubule interactions with the tubular ER
network. J Clin Invest. 120:1097–1110. 2010. View Article : Google Scholar
|
|
13
|
Chang CL, Weigel AV, Ioannou MS, Pasolli
HA, Xu CS, Peale DR, Shtengel G, Freeman M, Hess HF, Blackstone C,
et al: Spastin tethers lipid droplets to peroxisomes and directs
fatty acid trafficking through ESCRT-III. J Cell Biol.
218:2583–2599. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vietri M, Radulovic M and Stenmark H: The
many functions of ESCRTs. Nat Rev Mol Cell Biol. 21:25–42. 2020.
View Article : Google Scholar
|
|
15
|
Guo EZ and Xu Z: Distinct mechanisms of
recognizing endosomal sorting complex required for transport III
(ESCRT-III) protein IST1 by different microtubule interacting and
trafficking (MIT) domains. J Biol Chem. 290:8396–8408. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Connell JW, Allison RJ, Rodger CE, Pearson
G, Zlamalova E and Reid E: ESCRT-III-associated proteins and
spastin inhibit protrudin-dependent polarised membrane traffic.
Cell Mol Life Sci. 77:2641–2658. 2020. View Article : Google Scholar
|
|
17
|
Allison R, Lumb JH, Fassier C, Connell JW,
Ten Martin D, Seaman MN, Hazan J and Reid E: An ESCRT-spastin
interaction promotes fission of recycling tubules from the
endosome. J Cell Biol. 202:527–543. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Allison R, Edgar JR, Pearson G, Rizo T,
Newton T, Günther S, Berner F, Hague J, Connell JW, Winkler J, et
al: Defects in ER-endosome contacts impact lysosome function in
hereditary spastic paraplegia. J Cell Biol. 216:1337–1355. 2017.
View Article : Google Scholar :
|
|
19
|
Leo L, Weissmann C, Burns M, Kang M, Song
Y, Qiang L, Brady ST, Baas PW and Morfini G: Mutant spastin
proteins promote deficits in axonal transport through an
isoform-specific mechanism involving casein kinase 2 activation.
Hum Mol Genet. 26:2321–2334. 2017. View Article : Google Scholar
|
|
20
|
Kasher PR, De Vos KJ, Wharton SB, Manser
C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller
CC, et al: Direct evidence for axonal transport defects in a novel
mouse model of mutant spastin-induced hereditary spastic paraplegia
(HSP) and human HSP patients. J Neurochem. 110:34–44. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeong B, Kim TH, Kim DS, Shin WH, Lee JR,
Kim NS and Lee DY: Spastin contributes to neural development
through the regulation of microtubule dynamics in the primary cilia
of neural stem cells. Neuroscience. 411:76–85. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goyal U, Renvoisé B, Chang J and
Blackstone C: Spastin-interacting protein NA14/SSNA1 functions in
cytokinesis and axon development. PLoS One. 9:e1124282014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji Z, Zhang G, Chen L, Li J, Yang Y, Cha
C, Zhang J, Lin H and Guo G: Spastin interacts with CRMP5 to
promote neurite outgrowth by controlling the microtubule dynamics.
Dev Neurobiol. 78:1191–1205. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wood JD, Landers JA, Bingley M, McDermott
CJ, Thomas-McArthur V, Gleadall LJ, Shaw PJ and Cunliffe VT: The
microtubule-severing protein Spastin is essential for axon
outgrowth in the zebrafish embryo. Hum Mol Genet. 15:2763–2771.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lopes AT, Hausrat TJ, Heisler FF, Gromova
KV, Lombino FL, Fischer T, Ruschkies L, Breiden P, Thies E,
Hermans-Borgmeyer I, et al: Spastin depletion increases tubulin
polyglutamylation and impairs kinesin-mediated neuronal transport,
leading to working and associative memory deficits. PLoS Biol.
18:e30008202020. View Article : Google Scholar :
|
|
26
|
Ji ZS, Liu QL, Zhang JF, Yang YH, Li J,
Zhang GW, Tan MH, Lin HS and Guo GQ: SUMOylation of spastin
promotes the internalization of GluA1 and regulates dendritic spine
morphology by targeting microtubule dynamics. Neurobiol Dis.
146:1051332020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sherwood NT, Sun Q, Xue M, Zhang B and
Zinn K: Drosophila spastin regulates synaptic microtubule networks
and is required for normal motor function. PLoS Biol. 2:e4292004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roll-Mecak A and Vale RD: Structural basis
of microtubule severing by the hereditary spastic paraplegia
protein spastin. Nature. 451:363–367. 2008. View Article : Google Scholar
|
|
29
|
Kuo YW, Trottier O, Mahamdeh M and Howard
J: Spastin is a dual-function enzyme that severs microtubules and
promotes their regrowth to increase the number and mass of
microtubules. Proc Natl Acad Sci USA. 116:5533–5541. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Connell JW, Lindon C, Luzio JP and Reid E:
Spastin couples microtubule severing to membrane traffic in
completion of cytokinesis and secretion. Traffic. 10:42–56. 2009.
View Article : Google Scholar
|
|
31
|
Qiang L, Piermarini E and Baas PW: New
hypothesis for the etiology of SPAST-based hereditary spastic
paraplegia. Cytoskeleton (Hoboken). 76:289–297. 2019. View Article : Google Scholar
|
|
32
|
Sakoe K, Shioda N and Matsuura T: A newly
identified NES sequence present in spastin regulates its
subcellular localization and microtubule severing activity. Biochim
Biophys Acta Mol Cell Res. 1868:1188622021. View Article : Google Scholar
|
|
33
|
Beetz C, Brodhun M, Moutzouris K,
Kiehntopf M, Berndt A, Lehnert D, Deufel T, Bastmeyer M and
Schickel J: Identification of nuclear localisation sequences in
spastin (SPG4) using a novel Tetra-GFP reporter system. Biochem
Biophys Res Commun. 318:1079–1084. 2004. View Article : Google Scholar
|
|
34
|
Monteonofrio L, Valente D, Rinaldo C and
Soddu S: Extrachromosomal Histone H2B contributes to the formation
of the abscission site for cell division. Cells. 8:13912019.
View Article : Google Scholar
|
|
35
|
Sandate CR, Szyk A, Zehr EA, Lander GC and
Roll-Mecak A: An allosteric network in spastin couples multiple
activities required for microtubule severing. Nat Struct Mol Biol.
26:671–678. 2019. View Article : Google Scholar :
|
|
36
|
Han H, Schubert HL, McCullough J, Monroe
N, Purdy MD, Yeager M, Sundquist WI and Hill CP: Structure of
spastin bound to a glutamate-rich peptide implies a hand-over-hand
mechanism of substrate translocation. J Biol Chem. 295:435–443.
2020. View Article : Google Scholar :
|
|
37
|
White SR, Evans KJ, Lary J, Cole JL and
Lauring B: Recognition of C-terminal amino acids in tubulin by pore
loops in Spastin is important for microtubule severing. J Cell
Biol. 176:995–1005. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vemu A, Szczesna E, Zehr EA, Spector JO,
Grigorieff N, Deaconescu AM and Roll-Mecak A: Severing enzymes
amplify microtubule arrays through lattice GTP-tubulin
incorporation. Science. 361:eaau15042018. View Article : Google Scholar
|
|
39
|
Kuo YW, Trottier O and Howard J: Predicted
effects of severing enzymes on the length distribution and total
mass of microtubules. Biophys J. 117:2066–2078. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saltini M and Mulder BM: Critical
threshold for microtubule amplification through templated severing.
Phys Rev E. 101:0524052020. View Article : Google Scholar
|
|
41
|
Rao K, Stone MC, Weiner AT, Gheres KW,
Zhou C, Deitcher DL, Levitan ES and Rolls MM: Spastin, atlastin,
and ER relocalization are involved in axon but not dendrite
regeneration. Mol Biol Cell. 27:3245–3256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vajente N, Norante R, Redolfi N, Daga A,
Pizzo P and Pendin D: Microtubules stabilization by mutant spastin
affects ER morphology and Ca2+ handling. Front Physiol.
10:15442019. View Article : Google Scholar
|
|
43
|
Pendin D, McNew JA and Daga A: Balancing
ER dynamics: Shaping, bending, severing, and mending membranes.
Curr Opin Cell Biol. 23:435–442. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Farías GG, Fréal A, Tortosa E, Stucchi R,
Pan X, Portegies S, Will L, Altelaar M and Hoogenraad CC:
Feedback-driven mechanisms between microtubules and the endoplasmic
reticulum instruct neuronal polarity. Neuron. 102:184–201.e8. 2019.
View Article : Google Scholar
|
|
45
|
Liu X, Guo X, Niu L, Li X, Sun F, Hu J,
Wang X and Shen K: Atlastin-1 regulates morphology and function of
endoplasmic reticulum in dendrites. Nat Commun. 10:5682019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hashimoto Y, Shirane M, Matsuzaki F, Saita
S, Ohnishi T and Nakayama KI: Protrudin regulates endoplasmic
reticulum morphology and function associated with the pathogenesis
of hereditary spastic paraplegia. J Biol Chem. 289:12946–12961.
2014. View Article : Google Scholar :
|
|
47
|
Chang J, Lee S and Blackstone C: Protrudin
binds atlastins and endoplasmic reticulum-shaping proteins and
regulates network formation. Proc Natl Acad Sci USA.
110:14954–14959. 2013. View Article : Google Scholar
|
|
48
|
Iworima DG, Pasqualotto BA and Rintoul GL:
Kif5 regulates mitochondrial movement, morphology, function and
neuronal survival. Mol Cell Neurosci. 72:22–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Matsuzaki F, Shirane M, Matsumoto M and
Nakayama KI: Protrudin serves as an adaptor molecule that connects
KIF5 and its cargoes in vesicular transport during process
formation. Mol Biol Cell. 22:4602–4620. 2011. View Article : Google Scholar :
|
|
50
|
Shirane M, Wada M, Morita K, Hayashi N,
Kunimatsu R, Matsumoto Y, Matsuzaki F, Nakatsumi H, Ohta K, Tamura
Y and Nakayama KI: Protrudin and PDZD8 contribute to neuronal
integrity by promoting lipid extraction required for endosome
maturation. Nat Commun. 11:45762020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shirane M: Lipid transfer-dependent
endosome maturation mediated by protrudin and PDZD8 in neurons.
Front Cell Dev Biol. 8:6156002020. View Article : Google Scholar
|
|
52
|
Zhang C, Li D, Ma Y, Yan J, Yang B, Li P,
Yu A, Lu C and Ma X: Role of spastin and protrudin in neurite
outgrowth. J Cell Biochem. 113:2296–2307. 2012. View Article : Google Scholar
|
|
53
|
Olzmann JA and Carvalho P: Dynamics and
functions of lipid droplets. Nat Rev Mol Cell Biol. 20:137–155.
2019. View Article : Google Scholar :
|
|
54
|
Walther TC, Chung J and Farese RV Jr:
Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 33:491–510. 2017.
View Article : Google Scholar
|
|
55
|
Welte MA and Gould AP: Lipid droplet
functions beyond energy storage. Biochim Biophys Acta Mol Cell Biol
Lipids. 1862:1260–1272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Velázquez AP, Tatsuta T, Ghillebert R,
Drescher I and Graef M: Lipid droplet-mediated ER homeostasis
regulates autophagy and cell survival during starvation. J Cell
Biol. 212:621–631. 2016. View Article : Google Scholar
|
|
57
|
Papadopoulos C, Orso G, Mancuso G, Herholz
M, Gumeni S, Tadepalle N, Jüngst C, Tzschichholz A, Schauss A,
Höning S, et al: Spastin binds to lipid droplets and affects lipid
metabolism. PLoS Genet. 11:e10051492015. View Article : Google Scholar :
|
|
58
|
Vietri M, Schink KO, Campsteijn C, Wegner
CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C and
Stenmark H: Spastin and ESCRT-III coordinate mitotic spindle
disassembly and nuclear envelope sealing. Nature. 522:231–235.
2015. View Article : Google Scholar
|
|
59
|
Reid E, Connell J, Edwards TL, Duley S,
Brown SE and Sanderson CM: The hereditary spastic paraplegia
protein spastin interacts with the ESCRT-III complex-associated
endosomal protein CHMP1B. Hum Mol Genet. 14:19–38. 2005. View Article : Google Scholar
|
|
60
|
Christ L, Raiborg C, Wenzel EM, Campsteijn
C and Stenmark H: Cellular functions and molecular mechanisms of
the ESCRT membrane-scission machinery. Trends Biochem Sci.
42:42–56. 2017. View Article : Google Scholar
|
|
61
|
Henne WM, Buchkovich NJ and Emr SD: The
ESCRT pathway. Dev Cell. 21:77–91. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pisciottani A, Biancolillo L, Ferrara M,
Valente D, Sardina F, Monteonofrio L, Camerini S, Crescenzi M,
Soddu S and Rinaldo C: HIPK2 phosphorylates the
microtubule-severing enzyme spastin at S268 for abscission. Cells.
8:6842019. View Article : Google Scholar :
|
|
63
|
Scott CC, Vacca F and Gruenberg J:
Endosome maturation, transport and functions. Semin Cell Dev Biol.
31:2–10. 2014. View Article : Google Scholar
|
|
64
|
Tu Y, Zhao L, Billadeau DD and Jia D:
Endosome-to-TGN trafficking: Organelle-vesicle and
organelle-organelle interactions. Front Cell Dev Biol. 8:1632020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang J, Fedoseienko A, Chen B, Burstein E,
Jia D and Billadeau DD: Endosomal receptor trafficking: Retromer
and beyond. Traffic. 19:578–590. 2018. View Article : Google Scholar :
|
|
66
|
Vagnozzi AN and Praticò D: Endosomal
sorting and trafficking, the retromer complex and
neurodegeneration. Mol Psychiatry. 24:857–868. 2019. View Article : Google Scholar :
|
|
67
|
Allison R, Edgar JR and Reid E: Spastin
MIT Domain Disease-Associated mutations disrupt lysosomal function.
Front Neurosci. 13:11792019. View Article : Google Scholar :
|
|
68
|
Skjeldal FM, Strunze S, Bergeland T,
Walseng E, Gregers TF and Bakke O: The fusion of early endosomes
induces molecular-motor-driven tubule formation and fission. J Cell
Sci. 125:1910–1919. 2012.
|
|
69
|
Hoyer MJ, Chitwood PJ, Ebmeier CC,
Striepen JF, Qi RZ, Old WM and Voeltz GK: A novel class of ER
membrane proteins regulates ER-associated endosome fission. Cell.
175:254–265.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Raiborg C, Wenzel EM, Pedersen NM, Olsvik
H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, et
al: Repeated ER-endosome contacts promote endosome translocation
and neurite outgrowth. Nature. 520:234–238. 2015. View Article : Google Scholar
|
|
71
|
Elbaz-Alon Y, Guo Y, Segev N, Harel M,
Quinnell DE, Geiger T, Avinoam O, Li D and Nunnari J: PDZD8
interacts with Protrudin and Rab7 at ER-late endosome membrane
contact sites associated with mitochondria. Nat Commun.
11:36452020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Joshi AS, Nebenfuehr B, Choudhary V,
Satpute-Krishnan P, Levine TP, Golden A and Prinz WA: Lipid droplet
and peroxisome biogenesis occur at the same ER subdomains. Nat
Commun. 9:29402018. View Article : Google Scholar :
|
|
73
|
Joshi AS and Cohen S: Lipid droplet and
peroxisome biogenesis: Do they go hand-in-hand? Front Cell Dev
Biol. 7:922019. View Article : Google Scholar
|
|
74
|
Walker CL, Pomatto LCD, Tripathi DN and
Davies KJA: Redox regulation of homeostasis and proteostasis in
peroxisomes. Physiol Rev. 98:89–115. 2018. View Article : Google Scholar
|
|
75
|
Islinger M, Voelkl A, Fahimi HD and
Schrader M: The peroxisome: An update on mysteries 2.0. Histochem
Cell Biol. 150:443–471. 2018. View Article : Google Scholar :
|
|
76
|
Henne WM: Spastin joins LDs and
peroxisomes in the interorganelle contact ballet. J Cell Biol.
218:2439–2441. 2019. View Article : Google Scholar :
|
|
77
|
Riano E, Martignoni M, Mancuso G, Cartelli
D, Crippa F, Toldo I, Siciliano G, Di Bella D, Taroni F, Bassi MT,
et al: Pleiotropic effects of spastin on neurite growth depending
on expression levels. J Neurochem. 108:1277–1288. 2009. View Article : Google Scholar
|
|
78
|
Denton KR, Lei L, Grenier J, Rodionov V,
Blackstone C and Li XJ: Loss of spastin function results in
disease-specific axonal defects in human pluripotent stem
cell-based models of hereditary spastic paraplegia. Stem Cells.
32:414–423. 2014. View Article : Google Scholar
|
|
79
|
Henson BJ, Zhu W, Hardaway K, Wetzel JL,
Stefan M, Albers KM and Nicholls RD: Transcriptional and
post-transcriptional regulation of SPAST, the gene most frequently
mutated in hereditary spastic paraplegia. PLoS One. 7:e365052012.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jiang T, Cai Z, Ji Z, Zou J, Liang Z,
Zhang G, Liang Y, Lin H and Tan M: The lncRNA MALAT1/miR-30/Spastin
axis regulates hippocampal neurite outgrowth. Front Cell Neurosci.
14:5557472020. View Article : Google Scholar
|
|
81
|
Nakazeki F, Tsuge I, Horie T, Imamura K,
Tsukita K, Hotta A, Baba O, Kuwabara Y, Nishino T, Nakao T, et al:
MiR-33a is a therapeutic target in SPG4-related hereditary spastic
paraplegia human neurons. Clin Sci (Lond). 133:583–595. 2019.
View Article : Google Scholar
|
|
82
|
Sardina F, Pisciottani A, Ferrara M,
Valente D, Casella M, Crescenzi M, Peschiaroli A, Casali C, Soddu
S, Grierson AJ and Rinaldo C: Spastin recovery in hereditary
spastic paraplegia by preventing neddylation-dependent degradation.
Life Sci Alliance. 3:e2020007992020. View Article : Google Scholar :
|
|
83
|
Tan R, Lam AJ, Tan T, Han J, Nowakowski
DW, Vershinin M, Simó S, Ori-McKenney KM and McKenney RJ:
Microtubules gate tau condensation to spatially regulate
microtubule functions. Nat Cell Biol. 21:1078–1085. 2019.
View Article : Google Scholar :
|
|
84
|
Jin Z, Shou HF, Liu JW, Jiang SS, Shen Y,
Cheng WY and Gao LL: Spastin interacts with CRMP5 to promote
spindle organization in mouse oocytes by severing microtubules.
Zygote. 1–12. 2021. View Article : Google Scholar
|
|
85
|
Newton T, Allison R, Edgar JR, Lumb JH,
Rodger CE, Manna PT, Rizo T, Kohl Z, Nygren AOH, Arning L, et al:
Mechanistic basis of an epistatic interaction reducing age at onset
in hereditary spastic paraplegia. Brain. 141:1286–1299. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kapitein LC and Hoogenraad CC: Building
the neuronal microtubule cytoskeleton. Neuron. 87:492–506. 2015.
View Article : Google Scholar
|
|
87
|
Kelliher MT, Saunders HA and Wildonger J:
Microtubule control of functional architecture in neurons. Curr
Opin Neurobiol. 57:39–45. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bond AM, Ming GL and Song H: Adult
mammalian neural stem cells and neurogenesis: Five decades later.
Cell Stem Cell. 17:385–395. 2015. View Article : Google Scholar :
|
|
89
|
Katsimpardi L and Lledo PM: Regulation of
neurogenesis in the adult and aging brain. Curr Opin Neurobiol.
53:131–138. 2018. View Article : Google Scholar
|
|
90
|
McNally FJ and Roll-Mecak A:
Microtubule-severing enzymes: From cellular functions to molecular
mechanism. J Cell Biol. 217:4057–4069. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kahn OI and Baas PW: Microtubules and
growth cones: Motors drive the turn. Trends Neurosci. 39:433–440.
2016. View Article : Google Scholar :
|
|
92
|
Dent EW and Gertler FB: Cytoskeletal
dynamics and transport in growth cone motility and axon guidance.
Neuron. 40:209–227. 2003. View Article : Google Scholar
|
|
93
|
Lowery LA and Van Vactor D: The trip of
the tip: Understanding the growth cone machinery. Nat Rev Mol Cell
Biol. 10:332–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Dent EW, Gupton SL and Gertler FB: The
growth cone cytoskeleton in axon outgrowth and guidance. Cold
Spring Harb Perspect Biol. 3:a0018002011. View Article : Google Scholar
|
|
95
|
Rao AN and Baas PW: Polarity sorting of
microtubules in the axon. Trends Neurosci. 41:77–88. 2018.
View Article : Google Scholar
|
|
96
|
Tas RP, Chazeau A, Cloin BMC, Lambers MLA,
Hoogenraad CC and Kapitein LC: Differentiation between oppositely
oriented microtubules controls polarized neuronal transport.
Neuron. 96:1264–1271.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Claudiani P, Riano E, Errico A, Andolfi G
and Rugarli EI: Spastin subcellular localization is regulated
through usage of different translation start sites and active
export from the nucleus. Exp Cell Res. 309:358–369. 2005.
View Article : Google Scholar
|
|
98
|
Butler R, Wood JD, Landers JA and Cunliffe
VT: Genetic and chemical modulation of spastin-dependent axon
outgrowth in zebrafish embryos indicates a role for impaired
microtubule dynamics in hereditary spastic paraplegia. Dis Model
Mech. 3:743–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Karabay A, Yu W, Solowska JM, Baird DH and
Baas PW: Axonal growth is sensitive to the levels of katanin, a
protein that severs microtubules. J Neurosci. 24:5778–5788. 2004.
View Article : Google Scholar
|
|
100
|
Yu W, Qiang L, Solowska JM, Karabay A,
Korulu S and Baas PW: The microtubule-severing proteins spastin and
katanin participate differently in the formation of axonal
branches. Mol Biol Cell. 19:1485–1498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Conde C and Cáceres A: Microtubule
assembly, organization and dynamics in axons and dendrites. Nat Rev
Neurosci. 10:319–332. 2009. View Article : Google Scholar
|
|
102
|
Herms J and Dorostkar MM: Dendritic spine
pathology in neurodegenerative diseases. Annu Rev Pathol.
11:221–250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Stein IS and Zito K: Dendritic spine
elimination: Molecular mechanisms and implications. Neuroscientist.
25:27–47. 2019. View Article : Google Scholar
|
|
104
|
Park M: AMPA receptor trafficking for
postsynaptic potentiation. Front Cell Neurosci. 12:3612018.
|
|
105
|
Chater TE and Goda Y: The role of AMPA
receptors in postsynaptic mechanisms of synaptic plasticity. Front
Cell Neurosci. 8:4012014. View Article : Google Scholar
|
|
106
|
Hanley JG: AMPA receptor trafficking
pathways and links to dendritic spine morphogenesis. Cell Adh Migr.
2:276–282. 2008. View Article : Google Scholar
|
|
107
|
Dong H, O'Brien RJ, Fung ET, Lanahan AA,
Worley PF and Huganir RL: GRIP: A synaptic PDZ domain-containing
protein that interacts with AMPA receptors. Nature. 386:279–284.
1997. View Article : Google Scholar
|
|
108
|
Nakajima K, Yin X, Takei Y, Seog DH, Homma
N and Hirokawa N: Molecular motor KIF5A is essential for GABA(A)
receptor transport, and KIF5A deletion causes epilepsy. Neuron.
76:945–961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jaworski J, Kapitein LC, Gouveia SM,
Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di
Stefano P, Demmers J, et al: Dynamic microtubules regulate
dendritic spine morphology and synaptic plasticity. Neuron.
61:85–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Okabe S and Hirokawa N: Axonal transport.
Curr Opin Cell Biol. 1:91–97. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Millecamps S and Julien JP: Axonal
transport deficits and neurodegenerative diseases. Nat Rev
Neurosci. 14:161–176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Gibbs KL, Greensmith L and Schiavo G:
Regulation of axonal transport by protein kinases. Trends Biochem
Sci. 40:597–610. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Guedes-Dias P and Holzbaur ELF: Axonal
transport: Driving synaptic function. Science. 366:eaaw99972019.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cyr JL and Brady ST: Molecular motors in
axonal transport. Cellular and molecular biology of kinesin Mol
Neurobiol. 6:137–155. 1992.
|
|
115
|
Fuerst JC, Henkel AW, Stroebel A, Welzel
O, Groemer TW, Kornhuber J and Bönsch D: Distinct intracellular
vesicle transport mechanisms are selectively modified by spastin
and spastin mutations. J Cell Physiol. 226:362–368. 2011.
View Article : Google Scholar
|
|
116
|
McDermott CJ, Grierson AJ, Wood JD,
Bingley M, Wharton SB, Bushby KM and Shaw PJ: Hereditary spastic
paraparesis: Disrupted intracellular transport associated with
spastin mutation. Ann Neurol. 54:748–759. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wali G, Sutharsan R, Fan Y, Stewart R,
Tello Velasquez J, Sue CM, Crane DI and Mackay-Sim A: Mechanism of
impaired microtubule-dependent peroxisome trafficking and oxidative
stress in SPAST-mutated cells from patients with Hereditary Spastic
Paraplegia. Sci Rep. 6:270042016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wali G, Liyanage E, Blair NF, Sutharsan R,
Park JS, Mackay-Sim A and Sue CM: Oxidative stress-induced axon
fragmentation is a consequence of reduced axonal transport in
hereditary spastic paraplegia SPAST patient neurons. Front
Neurosci. 14:4012020. View Article : Google Scholar :
|
|
119
|
Plaud C, Joshi V, Marinello M, Pastré D,
Galli T, Curmi PA and Burgo A: Spastin regulates VAMP7-containing
vesicles trafficking in cortical neurons. Biochim Biophys Acta Mol
Basis Dis. 1863:1666–1677. 2017. View Article : Google Scholar
|
|
120
|
Jardin N, Giudicelli F, Ten Martín D,
Vitrac A, De Gois S, Allison R, Houart C, Reid E, Hazan J and
Fassier C: BMP- and neuropilin 1-mediated motor axon navigation
relies on spastin alternative translation. Development.
145:dev1627012018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Plaud C, Joshi V, Kajevu N, Poüs C, Curmi
PA and Burgo A: Functional differences of short and long isoforms
of spastin harboring missense mutation. Dis Model Mech.
11:dmm0337042018. View Article : Google Scholar
|
|
122
|
Öztürk Z, O'Kane CJ and Pérez-Moreno JJ:
Axonal endoplasmic reticulum dynamics and its roles in
neurodegeneration. Front Neurosci. 14:482020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Henne WM, Liou J and Emr SD: Molecular
mechanisms of inter-organelle ER-PM contact sites. Curr Opin Cell
Biol. 35:123–130. 2015. View Article : Google Scholar
|
|
124
|
Phillips MJ and Voeltz GK: Structure and
function of ER membrane contact sites with other organelles. Nat
Rev Mol Cell Biol. 17:69–82. 2016. View Article : Google Scholar
|
|
125
|
Chung WY, Jha A, Ahuja M and Muallem S:
Ca2+ influx at the ER/PM junctions. Cell Calcium.
63:29–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Friel D: Interplay between ER
Ca2+ uptake and release fluxes in neurons and its impact
on [Ca2+] dynamics. Biol Res. 37:665–674. 2004.
View Article : Google Scholar
|
|
127
|
Rehbach K, Kesavan J, Hauser S,
Ritzenhofen S, Jungverdorben J, Schüle R, Schöls L, Peitz M and
Brüstle O: Multiparametric rapid screening of neuronal process
pathology for drug target identification in HSP patient-specific
neurons. Sci Rep. 9:96152019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Julien C, Lissouba A, Madabattula S,
Fardghassemi Y, Rosenfelt C, Androschuk A, Strautman J, Wong C,
Bysice A, O'sullivan J, et al: Conserved pharmacological rescue of
hereditary spastic paraplegia-related phenotypes across model
organisms. Hum Mol Genet. 25:1088–1099. 2016. View Article : Google Scholar :
|
|
129
|
Connell JW, Allison R and Reid E:
Quantitative gait analysis using a motorized treadmill system
sensitively detects motor abnormalities in mice expressing ATPase
defective spastin. PLoS One. 11:e01524132016. View Article : Google Scholar :
|
|
130
|
Qiang L, Piermarini E, Muralidharan H, Yu
W, Leo L, Hennessy LE, Fernandes S, Connors T, Yates PL, Swift M,
et al: Hereditary spastic paraplegia: Gain-of-function mechanisms
revealed by new transgenic mouse. Hum Mol Genet. 28:1136–1152.
2019. View Article : Google Scholar
|
|
131
|
Solowska JM, D'Rozario M, Jean DC,
Davidson MW, Marenda DR and Baas PW: Pathogenic mutation of spastin
has gain-of-function effects on microtubule dynamics. J Neurosci.
34:1856–1867. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yip AG, Dürr A, Marchuk DA, Ashley-Koch A,
Hentati A, Rubinsztein DC and Reid E: Meta-analysis of age at onset
in spastin-associated hereditary spastic paraplegia provides no
evidence for a correlation with mutational class. J Med Genet.
40:e1062003. View Article : Google Scholar
|
|
133
|
Wu F, Qiu J, Fan Y, Zhang Q, Cheng B, Wu Y
and Bai B: Apelin-13 attenuates ER stress-mediated neuronal
apoptosis by activating Gαi/Gαq-CK2 signaling
in ischemic stroke. Exp Neurol. 302:136–144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Manni S, Brancalion A, Tubi LQ, Colpo A,
Pavan L, Cabrelle A, Ave E, Zaffino F, Di Maira G, Ruzzene M, et
al: Protein kinase CK2 protects multiple myeloma cells from ER
stress-induced apoptosis and from the cytotoxic effect of HSP90
inhibition through regulation of the unfolded protein response.
Clin Cancer Res. 18:1888–1900. 2012. View Article : Google Scholar
|
|
135
|
Hessenauer A, Schneider CC, Götz C and
Montenarh M: CK2 inhibition induces apoptosis via the ER stress
response. Cell Signal. 23:145–151. 2011. View Article : Google Scholar
|
|
136
|
Fassier C, Tarrade A, Peris L, Courageot
S, Mailly P, Dalard C, Delga S, Roblot N, Lefèvre J, Job D, et al:
Microtubule-targeting drugs rescue axonal swellings in cortical
neurons from spastin knockout mice. Dis Model Mech. 6:72–83.
2013.
|