Introduction
Epilepsy is a common and widespread disease of the
central nervous system (CNS) (1); ~5 million individuals are diagnosed
annually (2,3), >65 million are affected by
epilepsy and epileptic comorbidities worldwide (4), and ~15-30% of patients with
epilepsy experience epilepsy-related comorbidities (5). Neuronal injury and
neuroinflammation caused by long-term recurrent seizures are
characteristics of epilepsy that further result in behavioral
disorders (6,7). Anti-epileptic drug (AED) therapy
remains the preferred treatment for epilepsy, and although 30 types
of AEDs with diverse molecular targets are available, numerous
challenges are associated with their use (8), among which AED resistance and side
effects are the primary issues (9,10). In addition, ~30% of patients with
epilepsy fail to respond to AEDs, and long-term use affects
cognitive function and causes psychological impairments (8,11). Therefore, research into novel
AEDs with greater effectiveness and fewer side effects is urgently
required.
Previous studies have suggested that
neuroinflammatory processes play a key role in epileptogenesis.
Seizures cause neuroinflammation, which in turn aggravates epilepsy
(12,13). The NLRP3 inflammasome results
from the assembly of NLR family pyrin domain containing 3 (NLRP3),
apoptosis-associated speck-like protein (ASC) and caspase-1
(14), and promotes the
secretion of inflammatory cytokines, influencing the
pathophysiology of epilepsy (14). Additionally, in animal and
clinical epilepsy-related research, increased levels of the NLRP3
inflammasome and neuroinflammatory cytokines have been detected in
hippocampal tissues (15,16),
suggesting their close association with epileptogenesis. Moreover,
numerous studies have reported that inhibiting NLRP3 activation and
inflammatory cytokine secretion decreases to occurrence of
epileptic seizures and ameliorates cognitive dysfunction (17,18). These findings suggest that
inhibition of NLRP3 inflammasome activity and the associated
cascade reactions may be a promising approach for adjuvant
treatment of epilepsy.
Initially, glucagon like peptide-1 (GLP-1) was
widely regarded as a peripheral incretin hormone. Natural GLP-1 has
a short half-life and is quickly degraded by proteases, which
promotes GLP-1 analogue generation (19). Originally, GLP-1 analogues were
used as second-line hypoglycemic drugs, and further research has
demonstrated that GLP-1 exerts anti-inflammatory and
neuroprotective effects, as well as the ability to ameliorate
cognitive dysfunction (20).
Numerous studies have also demonstrated that GLP-1 analogues reduce
tissue apoptosis by suppressing NLRP3 inflammasome activation and
downstream inflammatory cytokine secretion (21,22). In the brain, preproglucagon
neurons synthesize GLP-1; subsequently, GLP-1 is distributed in the
nucleus of the solitary tract, where it functions as a
neuropeptide. GLP-1 receptors are widely expressed in various
structures, such as the brainstem, cerebral cortex and hippocampus
(23,24). Moreover, an increasing number of
studies have suggested that GLP-1 signaling may serve as a
potential drug target for the management of various neurological
disorders, including epilepsy and its associated comorbidities
(20,25). Semaglutide entered the market in
2017 as a novel once-weekly GLP-1 analogue for the treatment of
type II diabetes (26). In
addition, semaglutide has been shown to reduce apoptosis and
ameliorate cognitive impairment in cerebrovascular disease
(27), Alzheimer's disease (AD)
(28) and Parkinson's disease
(PD) (29). Moreover, a number
of reports have indicated that patients with AD and AD model mice
are markedly susceptible to unprovoked seizures (30), and AD-related pathological
changes have been detected in epileptic foci (31). However, limited research has been
conducted on the potential role of semaglutide in
epileptogenesis.
Therefore, the aim of the present study was to
investigate whether semaglutide affects seizure severity and
epilepsy-related cognitive impairment. The results obtained herein,
including those of mouse and cellular models, may support further
investigation into semaglutide use in patients with epilepsy and
associated comorbidities.
Materials and methods
Cell lines and culture
BV2 cells (Guangzhou Cellcook Biotech Co., Ltd.)
were maintained in DMEM supplemented with 10% fetal bovine serum
(FBS) (both Gibco; Thermo Fisher Scientific, Inc.) and 1%;
penicillin/streptomycin (Beijing Solarbio Science & Technology
Co., Ltd.) and cultured at 37°C with 5% CO2.
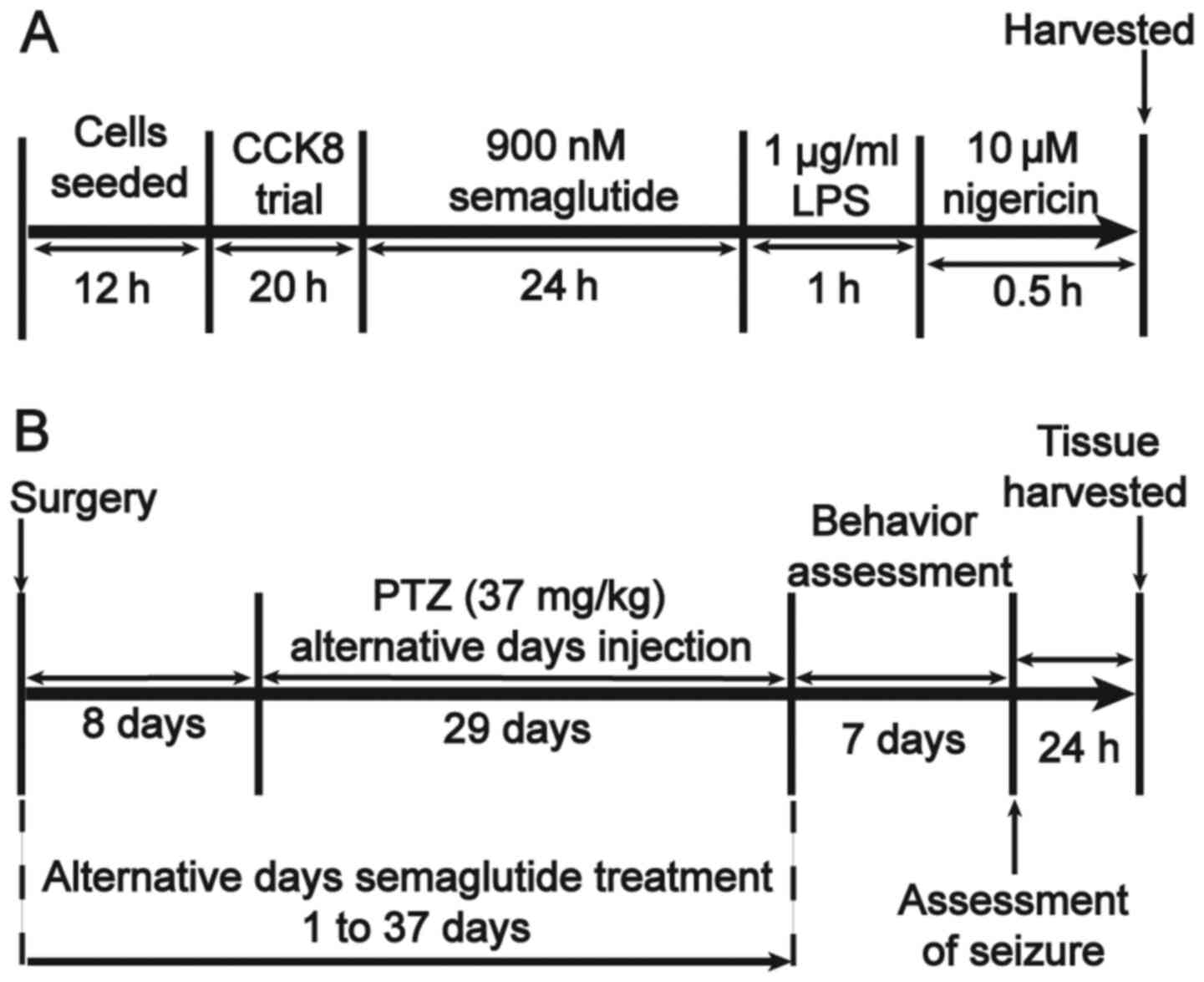
The in vitro study protocols are outlined in
Fig. 1A. BV2 cells were assigned
to four groups as follows: i) The control group, treated with 1%
DMSO; ii) the semaglutide group, treated with 900 nM semaglutide
(purity, ≥98%; cat. no. HY-114118; MedChemExpress); iii) the
lipopolysaccharide (LPS; cat. no. L2630; Sigma-Aldrich; Merck KGaA)
+ nigericin (cat. no. HY-100381; MedChemExpress) group, treated
with 1 µg/ml LPS (32)
and 10 µM nigericin (33); and iv) the semaglutide + LPS +
nigericin group, treated with 900 nM semaglutide followed by 1
µg/ml LPS and 10 µM nigericin.
Cellular proliferation
Cellular proliferation was evaluated using the Cell
Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Inc.). BV2
cells were seeded into 96-well plates (1×104 cells/well)
for 12 h, and then treated with different doses (300, 600, 900 or
1,000 nM) of semaglutide for 20 h at 37°C. A blank well with
culture medium was used to detect background, and five replicate
wells were established per group. After 20 h, 10% CCK-8 reagent
(100 µl) was added to each well, and the cells were cultured
at 37°C in 5% CO2 for 1 h. The absorbance value was
recorded at 450 nm using a microplate reader (BioTek Instruments,
Inc.), and the optical density (OD) value was recorded for
evaluation.
ELISA
The supernatants of each group of BV2 cells were
harvested, and the levels of IL-1β (cat. no. JL18442), IL-18 (cat.
no. JL20253), IL-6 (cat. no. JL20268) and TNF-α (cat. no. JL10484)
were evaluated using the associated ELISA kits (Jianglai Industrial
Limited By Share, Ltd.) according to the manufacturer's
instructions. A microplate reader was used to record the absorbance
and OD values at 450 nm.
Lactate dehydrogenase (LDH) assay
Each group of BV2 cell supernatants was collected,
and the LDH concentration was determined using an LDH assay kit
(Nanjing KeyGen Biotech Co., Ltd.) according to the manufacturer's
instructions. The absorbance at 450 nm was determined using a
microplate reader, and the OD value was recorded for further
evaluation.
Western blotting (WB)
Total protein was extracted from BV2 cells and
hippocampal tissues using a Whole Protein Extraction kit, and the
protein concentrations were determined using a BCA Protein Assay
kit (both Nanjing KeyGen Biotech Co., Ltd.). Equal amounts of
protein (50-60 µg per lane) were separated by 10% SDS-PAGE
and then transferred to 0.22-mm PVDF membranes (MilliporeSigma). To
block non-specific binding, 4% bovine serum albumin (BSA) was
applied at room temperature (RT) for 1.5 h, after which the
membrane was incubated with the following primary antibodies for 17
h at 4°C: Anti-NLRP3 (cat. no. bs-10021R), anti-ASC (cat. no.
bs-6741R), anti-IL-1β, (cat. no. bs-0812R) and anti-caspase-1p20
(cat. no. bs-10442R) (all 1:500); anti-IL-6, (cat. no. bs-6309R),
anti-IL-18 (cat. no. bs-4988R), anti-TNF-α (cat. no. bs-10802R),
anti-active caspase-3 (cat. no. bsm-33199M), anti-Bax (cat. no.
bs-0127R), anti-Bcl-2 (cat. no. bsm-52304R) and anti-β-actin (cat.
no. bs-0061R) (all 1:1,000) (BIOSS); and anti-β-actin (cat. no.
66009-1-Ig; 1:5,000; ProteinTech Group, Inc.). Next, the membranes
were washed with Tris-buffered saline (TBS) containing 5‰ Tween-20,
and then incubated with goat anti-mouse IgG 800RD (cat. no.
926-32210; 1:5,000) or goat anti-rabbit IgG 680RD (cat. no.
925-68071; 1:5,000) (both BD Biosciences) at RT for 1.5 h. Finally,
Odyssey CLX software (9141-00; BD Biosciences) was used for
imaging, and protein signals were quantified using ImageJ 8.0
software (National Institutes of Health). All experiments were
conducted ≥3 times.
Animals and treatment
C57BL/6J mice (male, 10±2 weeks old; weight, 25±3 g)
were obtained from Beijing, China Fukang Biotechnology Co., Ltd.
All protocols were approved by the Ethics Committee of Ningxia
Medical University [approval no. SCXK(Ning)2019-203]. All mice were
raised in the independent ventilation cage (n=5 per cage) at 25±1°C
with 50-60% relative humidity, free access to food and water, and
12-h light/dark cycles. All mice were anesthetized with 60 mg/kg
sodium pentobarbital and decapitated for hippocampal tissue
removal. Briefly, electrode implantation surgery was performed on
the first day, and semaglutide was administered every other day for
37 days. At 8 days post-surgery, the mice received
pentylenetetrazole (PTZ) every other day for 29 days. From the 38th
day, a behavioral test was conducted for 7 days. Brain tissues were
collected on the 45th day (Fig.
1B). The animals were randomized into five groups (20 per
group), all of which were treated by intraperitoneal (i.p.)
injection. In the control group (1% DMSO, i.p.), mice received 20
injections in total - 19 injections every other day for 37 days,
and a final injection on the 45th day. For the semaglutide group
[25 nM/kg semaglutide i.p. (29)], the administration schedule was
the same as that for the control group. In the PTZ group [37 mg/kg
PTZ, i.p. (1)], the mice
received a total of 16 injections - one injection every other day
from the 9th to the 37th day for a total of 15 injections, and a
final injection on the 45th day. For the low-dose semaglutide group
[37 mg/kg PTZ + 10 nM/kg semaglutide, i.p. (27)], the semaglutide administration
schedule was the same as that for the control group, and the PTZ
administration schedule was the same as that for the PTZ group, 30
min after each injection of semaglutide. For the high-dose
semaglutide group (37 mg/kg PTZ + 25 nM/kg semaglutide, i.p.), the
administration schedule was the same as that for the low-dose
semaglutide group.
Electrocorticography (ECoG)
Intracranial electrodes were implanted 8 days before
PTZ injection. Briefly, after the mice were anaesthetized with 60
mg/kg sodium pentobarbital, a stereotactic device was used to
implant electrodes into the cerebral cortex on both sides of the
bregma. The specific method for implanting the intracranial
electrodes was the same as that used in our previous study
(25). On the 45th day, seizure
severity was assessed, and ECoG was recorded using an information
integrated biological signal acquisition and processing system
(BL-420 N; Techman).
Kindling model protocol
Mice were administered PTZ (37 mg/kg) every other
day for 29 days. Seizure severity was evaluated using the modified
Racine scale as follows: Stage one, mouse and facial twitching;
stage two, nodding and clonus; stage three, mild unilateral or
bilateral limb twitch; stage four, bilateral forelimb vibration
accompanied by standing; stage five, generalized convulsions
accompanied by falling; stage six, tonic convulsions with hindlimb
extension; and stage seven, death. Three consecutive 4- or
higher-stage convulsions indicated successful kindling (34).
Behavioral assessment
Modified novel object recognition
(NOR)
The NOR assessment evaluates the ability of the
mouse to recognize a novel object under a specific condition, using
the Smart 3.0 behavior video recording and analysis system
(Panlab). The modified NOR test consists of the habituation phase,
training phase and test phase. Each mouse was tested and evaluated
in a sound-attenuated room. During the habituation phase, each
mouse was adapted for 3 min before training. During the training
phase with similar objects, a single mouse was placed in a closed
square-shaped plane (arena, 55×55×55 cm) containing two plastic
objects of the same shape (square, 7×7×7 cm) for 6 min. The test
phase followed the training phase, and was conducted with different
objects; the mouse was returned to the arena, one object was
replaced with a novel plastic object (cylindrical shape; radius,
3.5 cm; and height, 7 cm) and the mouse was placed into the square
for testing for 6 min at 20 min intervals. The arena and objects
were cleaned between the two tests, completely eliminating
olfactory cues. The discriminant index (DI) was defined as the time
spent by the mouse exploring the new object as a percentage of the
memory retention period. The total time was calculated as the time
that the mouse spent exploring both objects. A DI of 50% is equal
to the random chance percentage, and a higher DI indicates that the
mouse prefers a certain object, indicating that it can remember
similar objects and locations.
Shuttle box active avoidance test
These trials were performed in a two-way shuttle box
(MED-APAP-D1R; MED Associates, Inc.) consisting of two compartments
accessible to each other by a guillotine door. Each experimental
session included 30 trials and was repeated on five consecutive
days. Each experiment included conditioned stimulation
(simultaneous presentation of light and sound, 5 sec) and
unconditioned stimulation (electric shock, 0.3 mA, 50 Hz, 10 sec)
delivered via stainless steel rods on the floor of the apparatus.
When the mouse walked into the other side of the shuttle box, the
stimulus was immediately turned off. The number of total active
avoidance responses (AARs) was recorded with Med-PC (version 5.1;
MED Associates, Inc.) software for evaluation. Mice were tested on
the 5th day.
Passive avoidance test
These trials were performed in a one-way shuttle box
(MED-APAP-D1R; MED Associates, Inc.) with two equal-sized
compartments (darkened/lit apparatus) separated by a guillotine
door. The memory test was performed over 2 days. On the first day,
a mouse was released into the lit cage and moved freely around for
5 min for habituation to the compartment. Then, the mouse was
placed in the lit cage again, and the door opened; when the mouse
entered the dark cage, the door was closed, and the grid floor
delivered a mild electrical shock (0.3 mA for 3 sec). After 120
sec, the animal was removed from the dark compartment and placed in
the lit compartment again until it remained in the lit compartment
for 120 sec, at which point the experiment was terminated. The
number of electric shocks was recorded; 24 h later, passive
avoidance learning was evaluated using the shuttle box, the latency
time was recorded, and 120 sec was set as the maximum latency time
for each mouse.
Morris water maze (MWM)
assessment
The MWM (WMT-100S; Techman Software) was performed
in a pool with a diameter of 80 cm and a height of 50 cm, filled
with 23±2°C water, and a white circular platform with a diameter of
5 cm that allows the mouse to escape the water. The experimental
method was as previously described (1). Briefly, the pool was divided into
four quadrants, and the circular platform was placed in the first
quadrant (target quadrant). Several distal cues were set around the
pool and were maintained in the same positions throughout all the
tests. The MWM assessment consisted of three parts: i) A visible
circular platform trial; ii) a navigation trail; and iii) a spatial
probe trail. The visual platform trial was first performed to
assess the vision and motor function of each mouse, and the
platform was placed 1.5 cm under the surface of the water. The mice
were tested for 5 days, the four quadrants were tested each day,
with a test interval of 15 min. On the sixth day, the spatial probe
test began. Mice were placed in the third quadrant and allowed to
swim for 60 sec. The swimming tracking data were recorded.
Reverse transcription-quantitative (RT-q)
PCR
Total RNA was extracted from the hippocampal tissues
using an RNA simple Total RNA kit (Tiangen Biotech Co., Ltd.), and
the FastKing gDNA Dispelling RT SuperMix kit (Tiangen Biotech Co.,
Ltd.) was used to synthesize cDNA. RT-qPCR was performed using the
FastFire qPCR PreMix (SYBR-Green; Tiangen Biotech Co., Ltd.)
according the manufacturer's protocol. The following primers were
used for qPCR: NLRP3 forward, 5′-GGAGGAA GAAGAAGAGAGGAGAGGAG-3′ and
reverse, 5′-CTTGAGA AGAGACCACGGCAGAAG-3′; ASC forward, 5′-ACAATGA
CTGTGCTTAGAGACA-3 and reverse, 5′-CACAGCTCCAGA CTCTTCTTTA-3′;
caspase-1 p20 forward, 5′-TGAATACAAC CACTCGTACACGTCTTG-3′ and
reverse, 5′-CCAGATCCT CCAGCAGCAACTTC-3′; and GAPDH (mouse
endogenous reference gene primers; cat. no. B661304). All primers
were purchased from Shenggong Bioengineering Co., Ltd. The reaction
conditions were in accordance with the manufacturer's instructions:
Denaturation at 95° for 15 min, followed by 40 cycles of
denaturation at 95° for 10 sec, annealing at 61.5° for 20 sec and
extension at 72° for 30 sec; iQ™5 software (Bio-Rad Laboratories,
Inc.) was used to conduct PCR amplification. The RNA expression
levels were evaluated using the 2−ΔΔCq method (35). PRISM 8 software (GraphPad
Software, Inc.) was used to evaluate the relative RNA level. All
experiments were carried out >3 times.
Nissl staining
After the mice were anaesthetized, they were
intracardially perfused with 4% paraformaldehyde. Then, the brain
tissue was collected and soaked in fixative for 12 h at RT.
Specimens were dehydrated and immersed in paraffin, and were then
sliced to a thickness of 5 µm at RT for later use. The
paraffin sections were sequentially incubated in xylene solution A
and xylene solution B for 19 min, followed by incubation in
anhydrous ethanol solution A, anhydrous ethanol solution B, and 75%
alcohol for 6 min at RT. The sections were then washed with flowing
water.
Mouse tissue sections were placed in staining
solution for 6 min and slightly differentiated with 1% glacial
acetic acid at RT. The reaction was terminated by washing with tap
water. The degree of differentiation was controlled by evaluation
under a fluorescent microscope. For transparent mounting, the
slices were incubated in fresh xylene for 6 min and mounted with
neutral gum at RT. Sections were imaged with Pannoramic DESK,
p-MIDI, p250 (3D HISTECHE, Ltd.), and analyzed using ImageJ 8.0
software.
Immunofluorescence staining
The method used to prepare the tissue sections was
the same as that used for Nissl staining. Subsequently,
ethylenediaminetetraacetic acid (pH 8.0) was used for antigen
retrieval at RT, after which the sections were washed with PBS (pH
7.4). Subsequently, the sections were incubated in an
autofluorescence quencher agent reagent (cat. no. G1221; Wuhan
Servicebio Technology Co., Ltd.) for 5 min at RT, rinsed with
running water for 10 min, and then incubated with 5% BSA for 30 min
at RT to block nonspecific reactions. Next, the sections were
incubated with anti-active caspase-3, anti-Bax, anti-Bcl-2,
anti-NLRP3, anti-ASC, anti-caspase-1 p20, anti-NeuN (cat. no.
bs-10394R; BIOSS) and anti-Iba-1 [ab283319, Abcam (all 1:500)]
antibodies for 15 h at 4°C. Then, the sections were washed 3 times
with PBS and subsequently incubated with the associated secondary
antibodies (all 1:1,000) for 90 min at RT. Finally, the sections
were covered with a sealer containing DAPI, imaged using Pannoramic
DESK, p-MIDI, p250 and analyzed using ImageJ 8.0 software.
Statistical analysis
PRISM 8 software was used for statistical analysis.
The results are expressed as the mean ± standard deviation. All
experiments were performed at least three times. Differences
between two factors was compared using unpaired Student's t-test,
and multivariate differences were compared by one-way or repeated
measures two-way ANOVA followed by Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Semaglutide attenuates LPS- and
nigericin-induced inflammatory responses by blocking the NLRP3
inflammasome in vitro
Seizures promote brain inflammation and activate
microglia; activated microglia in turn exacerbate inflammation,
which further aggravates epilepsy (36). Therefore, in the present study,
the anti-inflammatory effect of semaglutide was evaluated using BV2
cells. The most appropriate concentration of semaglutide for the
treatment of BV2 cells was determined using the CCK-8. The findings
suggested that semaglutide did not exhibit cytotoxicity and
minimally impacted cell viability at either low or high doses.
Therefore, 900 nM was selected as the optimal semaglutide
concentration to assess cell viability for subsequent
experimentation (Fig. 2A;
P<0.001).
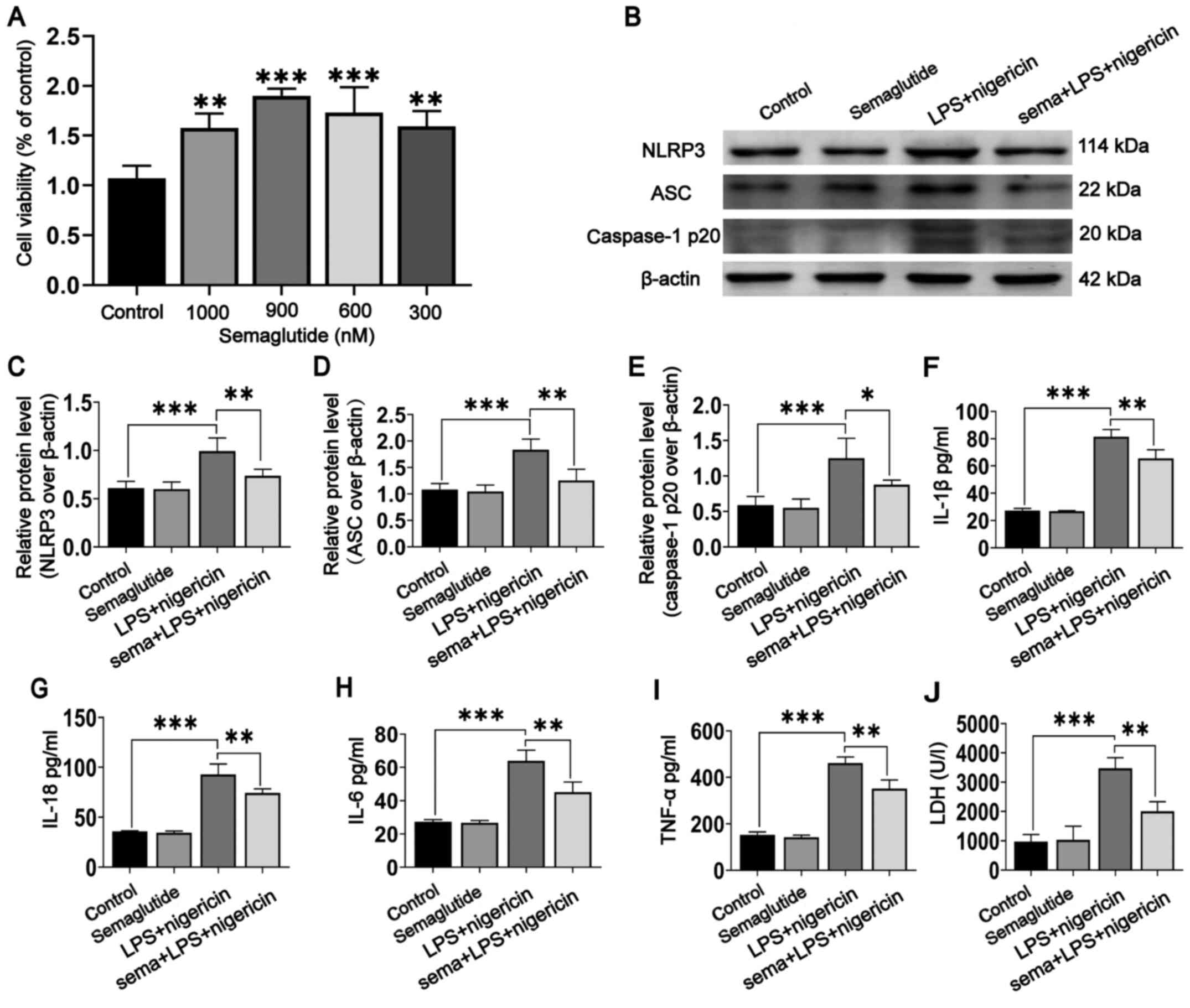 | Figure 2Effect of semaglutide treatment on
BV2 cells mediated by LPS and nigericin. (A) Proliferation of BV2
cells treated with semaglutide at different concentrations (300,
600, 900 and 1,000 nM) was assessed using Cell Counting Kit-8
analysis followed by one-way ANOVA. (B) Representative WB images in
different BV2 cell groups. (C-E) Statistical results of WB band
intensities (Student's t-test). (F-I) Statistical results of IL-1β,
IL-18, IL-6 and TNF-α secretion, as assessed by ELISA (Student's
t-test). (J) Semaglutide inhibited LPS- and nigericin-mediated LDH
release in BV2 cells (Student's t-test). Data are presented as the
means ± SD. *P<0.05, **P<0.01 and
***P<0.001. N>3. LPS, lipopolysaccharide; WB,
western blotting/blot; LDH, lactate dehydrogenase; NLRP3, NLR
family pyrin domain containing 3; ASC, apoptosis-associated
speck-like protein. |
WB analysis was used to evaluate the expression of
the NLRP3 inflammasome in BV2 cells (Fig. 2B). NLRP3 inflammasome expression
was not significantly different between the semaglutide group and
the controls (Fig. 2C-E), but
was increased in the LPS + nigericin group (P<0.001). Indeed,
semaglutide pretreatment followed by LPS + nigericin significantly
reduced NLRP3 inflammasome activation (P<0.05). In addition, the
ELISA results showed that the levels of IL-1β, IL-18, IL-6 and
TNF-α were significantly higher in the LPS + nigericin group than
in the control groups (Fig.
2F-I; P<0.001). Semaglutide pretreatment significantly
suppressed the secretion of inflammatory cytokines mediated by LPS
+ nigericin (P<0.01), but semaglutide alone did not affect these
levels. These data revealed that semaglutide attenuated LPS +
nigericin-induced NLRP3 inflammasome activation in BV2 cells.
Semaglutide inhibits LPS- and
nigericin-mediated LDH release in BV2 cells
Pyroptosis is a type of programmed necrosis that is
involved in inflammatory cell death. NLRP3 inflammasome activation
produces active caspase-1 and promotes the release of the
inflammatory factor IL-1β from cells, which is an important
mechanism leading to pyroptosis. LDH release is often measured to
assess the level of pyroptosis after inflammasome activation
(37). Semaglutide was confirmed
to inhibit the release of caspase-1 p20 and IL-1β after LPS- and
nigericin-mediated NLRP3 inflammasome activation. Therefore, the
potential effects of semaglutide on BV2 cell pyroptosis were
further investigated. Analysis of cell supernatants showed that
stimulation with LPS and nigericin promoted the release of LDH
(Fig. 2J; P<0.001) and that
semaglutide inhibited LDH release mediated by LPS + nigericin
(P<0.01), indicating that semaglutide can inhibit LPS +
nigericin-mediated pyroptosis.
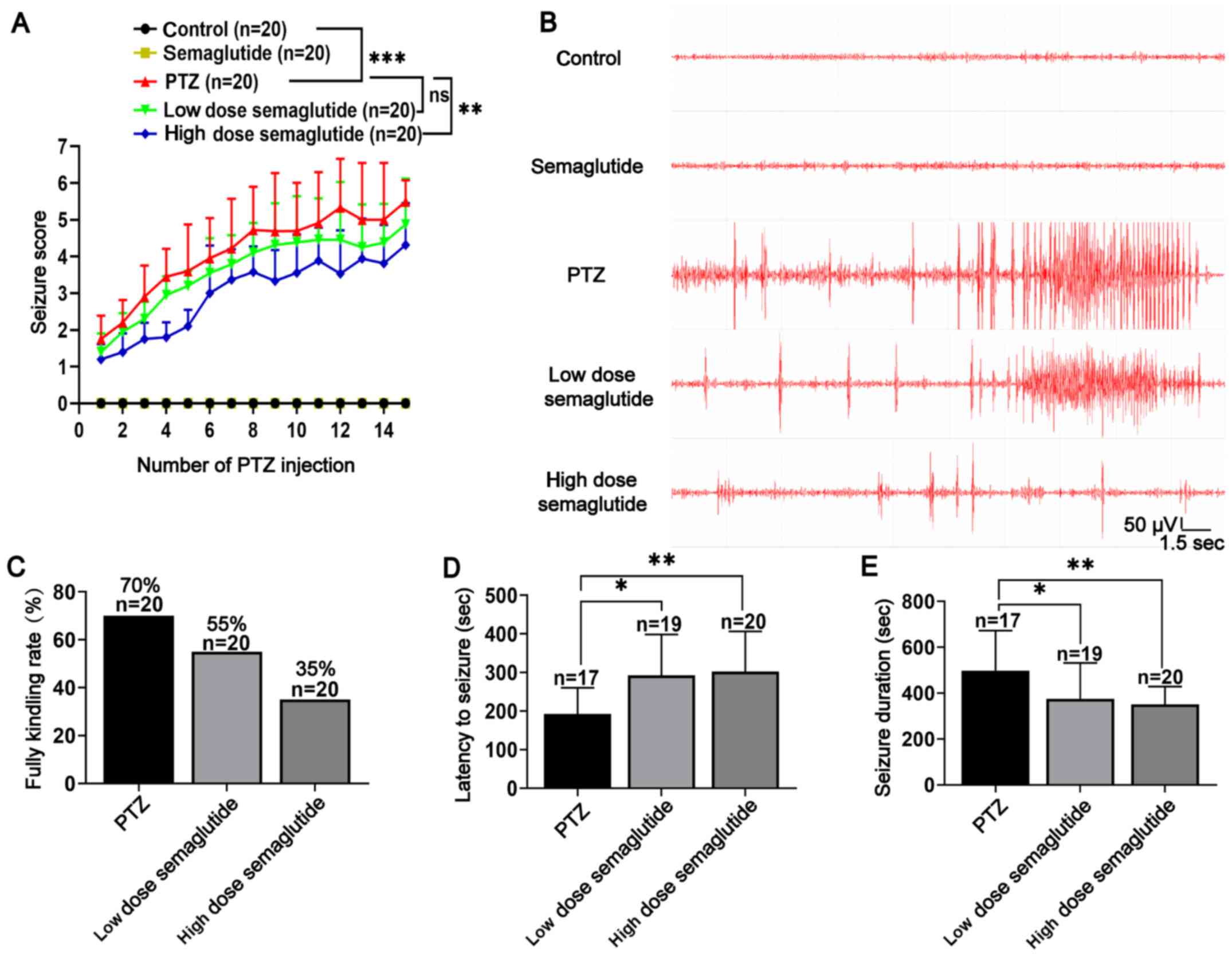
Semaglutide reduces seizure severity in
PTZ-kindled mice
Semaglutide exerted anti-inflammatory and
anti-pyroptotic effects on LPS- and nigericin-activated BV2 cells.
Therefore, the potential effects of semaglutide on PTZ-kindled mice
were also investigated. The modified Racine score and rate of
complete kindling were recorded every other day for 29 days (15
injections). After the last (16th) injection on the 45th day, the
latency to seizure, seizure duration and ECoG were recorded.
Pretreatment with semaglutide (10 and 25 nM/kg, i.p.) decreased the
seizure score (Fig. 3A;
P>0.05 and P<0.01, respectively) and reduced the rate of
complete kindling during 15 injections of PTZ (55 and 35%,
respectively) compared with the PTZ group (70%) (Fig. 3A and C). Both doses of
semaglutide (10 and 25 nM/kg; i.p.) partially increased the latency
to generalized seizures (Fig.
3D; P<0.05 and P<0.01, respectively) and shortened the
seizure duration (Fig. 3E;
P<0.05 and P<0.01, respectively). Furthermore, the ECoG
results showed a lower frequency and amplitude of spike-wave
discharges in the low- and high-dose semaglutide groups than in the
PTZ group (Fig. 3B).
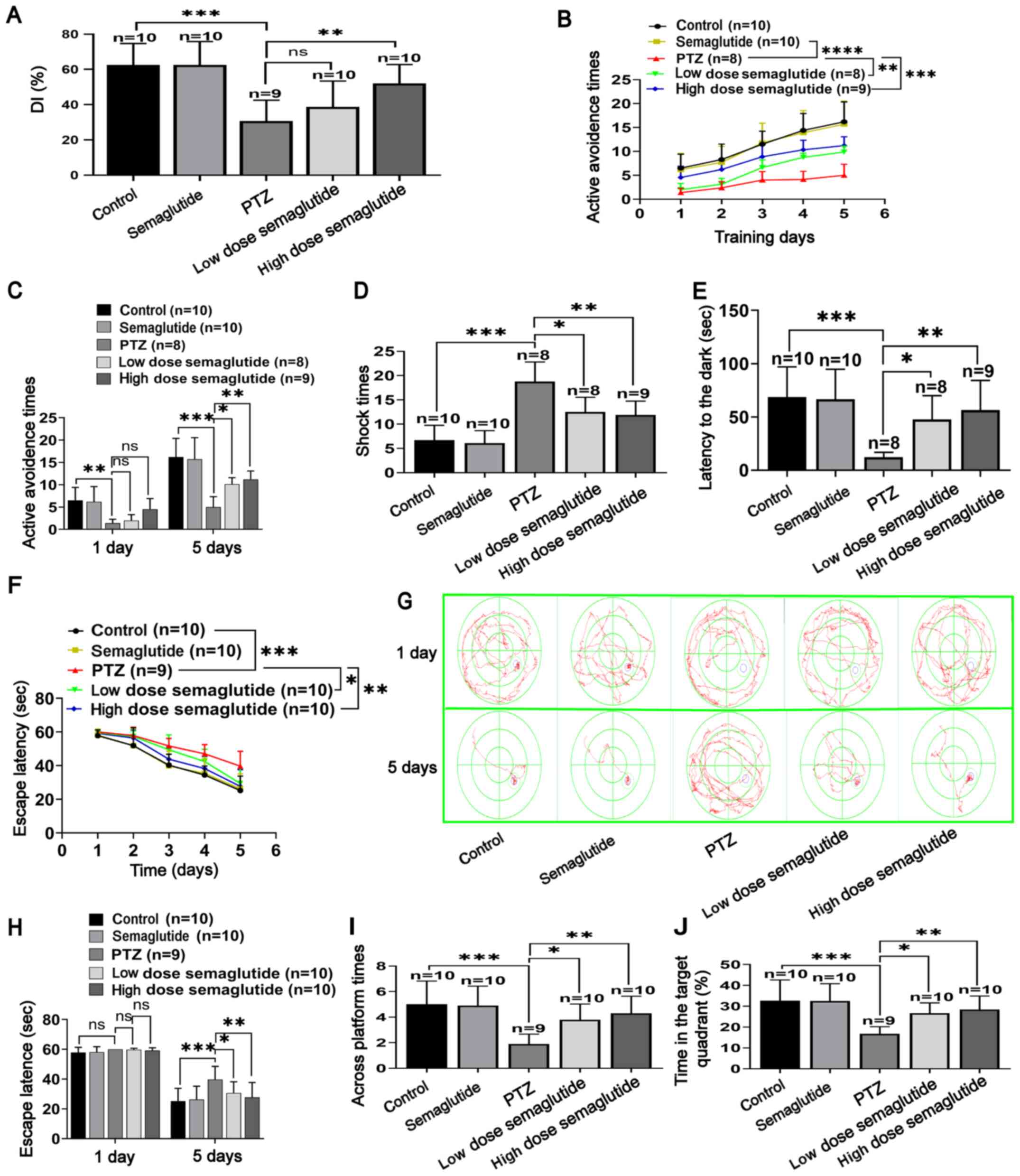
Semaglutide affects learning and memory
in PTZ-kindled mice
NOR test
No significant differences were observed between the
two similar objects during the test and training phases. The
results of the NOR test revealed no significant differences in the
DI between the semaglutide-treated and control mice (Fig. 4A). Conversely, the PTZ mice had a
lower DI than the controls (P<0.001). In addition, mice treated
with either dose of semaglutide (10 or 25 nM/kg) spent more time in
the area of the new object and had a higher DI than the PTZ mice
(P>0.05 and P<0.01, respectively).
Shuttle box active avoidance test
The number of AARs increased gradually over the 5
days of training in all groups. However, the AAR time was lower in
the PTZ mice than in the control mice (Fig. 4B; P<0.0001). In both
semaglutide groups (low- and high-dose), the number of AARs was
increased compared with that in the PTZ mice (P<0.01 and
P<0.001, respectively). The number of AARs in the semaglutide
mice and the controls was not significantly different. Moreover, on
the first day, the number of AARs was not significantly different
in any group. However, on the fifth day, the success rate on the
active avoidance test was increased in both semaglutide groups
(low- and high-dose) with respect to the PTZ mice (Fig. 4C; P<0.05 and P<0.01,
respectively).
Passive avoidance test
Before receiving a shock, the latency time to enter
the dark cage was not significantly different between any of the
groups. In addition, in the training test, the number of training
sessions did not differ between the semaglutide-treated mice and
controls (Fig. 4D). However, the
number of training sessions was significantly higher in the
PTZ-treated mice compared with the controls (P<0.001). In both
semaglutide groups (low- and high-dose), the number of training
sessions was significantly decreased with respect to that in the
PTZ mice (P<0.05 and P<0.01, respectively). The test trial
was carried out 1 day later in the training trial. The delayed
period to enter the dark cage did not differ between the
semaglutide and control mice (Fig.
4E). Notably, the delay to enter the dark cage was shorter in
the PTZ mice than in the controls (P<0.001) and longer in mice
in both semaglutide groups (low- and high-dose) than in the PTZ
mice (P<0.05 and P<0.01, respectively).
MWM test
In the visible platform trial, the escape latency
did not significantly differ between the groups. In the positioning
cruise test, the escape latency gradually decreased in all groups
(Fig. 4F-G). For the semaglutide
mice, the escape latency did not differ with respect to the
controls. However, in the PTZ mice, the escape latency was
significantly longer with respect to the control mice (P<0.001).
The escape latency was partially reduced by both concentrations of
semaglutide (low- and high-dose) with respect to the PTZ alone
(P<0.05 and P<0.01, respectively). On the first training day,
there was no significant difference in escape latency between any
of the groups (Fig. 4H). On the
last training day (day 5), the escape latency was longer in the PTZ
mice than in the controls (P<0.001). In both semaglutide groups
(low- and high-dose), the escape latency was partially reduced
compared with that in the PTZ mice (P<0.05 and P<0.01,
respectively).
In the spatial probe trial, the time spent in the
target quadrant and the number of crossed times were not
significantly different between the semaglutide mice and the
controls (Fig. 4I-J). However,
the number of crossings in the PTZ group was significantly lower
with respect to the controls over a 60-sec period (P<0.001).
Both doses of semaglutide (10 and 25 nM/kg) partially increased the
number of crossings compared with the PTZ-alone group (P<0.05
and P<0.01, respectively). Collectively, these data suggested
that semaglutide minimized PTZ- induced cognitive impairment in
mice.
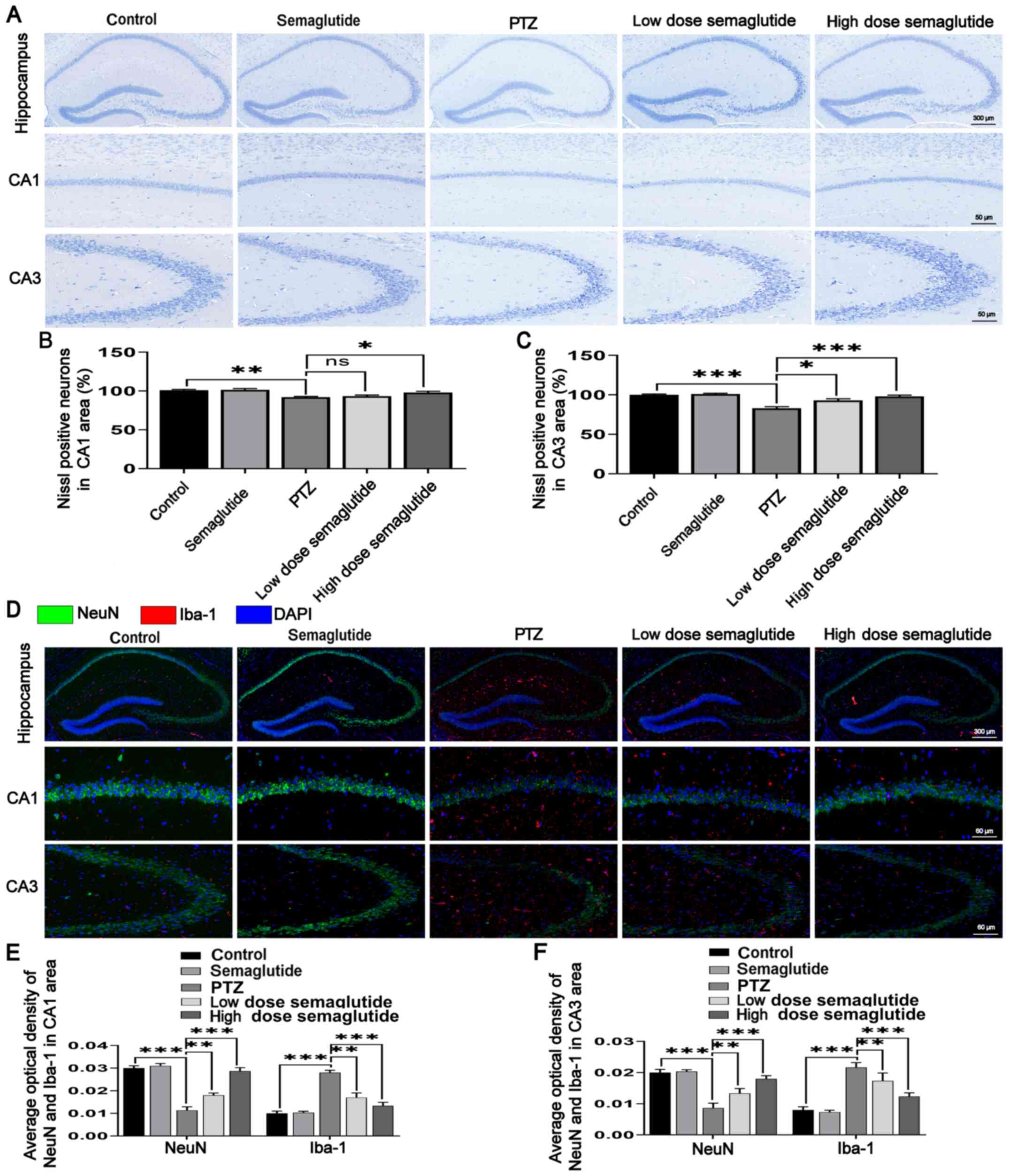
Semaglutide decreases hippocampal neuron
apoptosis in PTZ kindled-mice
Cognitive dysfunctions are closely related to
hippocampal structure (20);
therefore, the effect of semaglutide on hippocampal neuronal
apoptosis was further evaluated.
First, Nissl staining was used for histological
examination. The number of hippocampal neurons was similar between
the semaglutide mice and the controls, and there were no obvious
abnormalities in cell morphology or structure in the semaglutide
mice (Fig. 5A-C). However, the
number of the CA1 and CA3 hippocampal neurons were reduced in the
PTZ mice (P<0.01 and P<0.001, respectively), and evaluation
of neuronal morphology and structure revealed nuclear pyknosis and
fragmentation relative to the controls (Fig. 5A). The number of the CA1
hippocampal neurons was increased in both semaglutide groups (low-
and high-dose), and reduced nuclear pyknosis and fragmentation were
observed relative to those in the PTZ mice (Fig. 5A and B; P>0.05 and P<0.05).
The number of CA3 hippocampal neurons was increased and nuclear
pyknosis and fragmentation were decreased in these mice relative to
the PTZ mice (Fig. 5A and C;
P<0.05 and P<0.001).
NeuN is located in the nucleus and cytoplasm; DAPI
is located in the nucleus. NeuN has a larger fluorescence range,
and the overall color after merging is green. Double
immunofluorescence staining for NeuN and Iba-1 showed no
significant differences in the semaglutide mice with respect to the
controls (Fig. 5D-F). However,
the fluorescence intensity of NeuN was decreased, but the
fluorescence intensity of Iba-1 was higher in the PTZ mice with
respect to the controls (P<0.001). By contrast, the fluorescence
intensity of NeuN was higher and the fluorescence intensity of
Iba-1 was lower in mice in both semaglutide groups (low- and
high-dose) with respect to the PTZ mice (P<0.01 and P<0.001,
respectively).
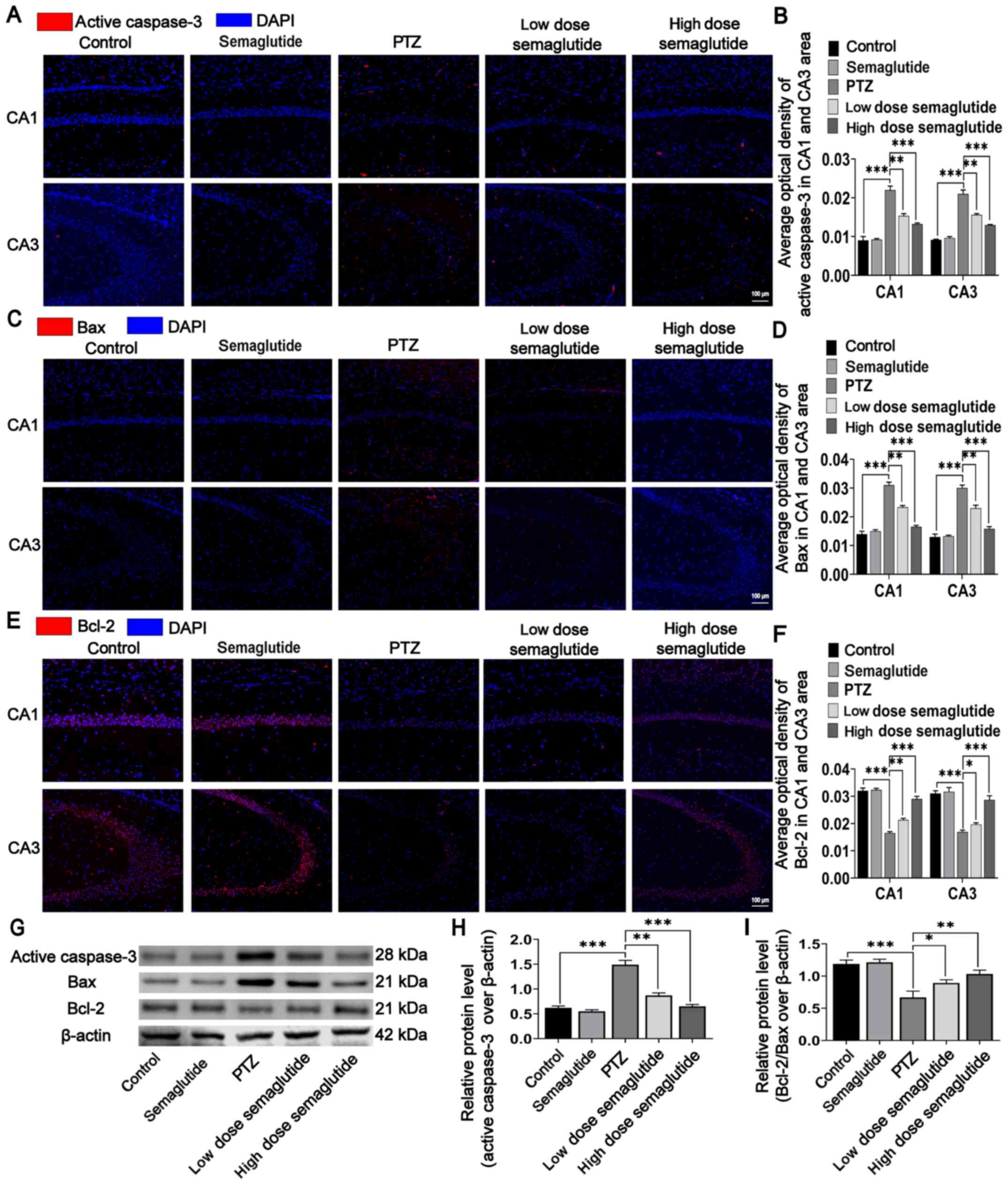
In addition, active caspase-3, Bax, and Bcl-2 were
detected in hippocampal tissues using immunofluorescence staining
and WB analysis. There were no significant differences in the
immunofluorescence intensities of active caspase-3, Bax and Bcl-2
between the semaglutide mice and the control mice (Fig. 6A-F). The immunofluorescence
intensities of active caspase-3 and Bax were significantly higher
(Fig. 6A-D; P<0.001), while
the immunofluorescence intensities of Bcl-2 was lower (Fig. 6E and F; P<0.001) in the PTZ
mice than in the controls. However, the effects of PTZ were
partially blocked by both semaglutide doses (low- and high-dose):
the fluorescence intensities of active caspase-3 and Bax were lower
and that of Bcl-2 was higher in the CA1 region in both semaglutide
mouse groups than in PTZ mice (P<0.01 and P<0.001). Moreover,
both semaglutide doses (low- and high-dose) reduced the
fluorescence intensities of active caspase-3 and Bax (P<0.01 and
P<0.001, respectively) and increased the fluorescence intensity
of Bcl-2 (P<0.05 and P<0.001, respectively) with respect to
the PTZ mice in the CA3 region.
Finally, the WB results were similar to the
immunofluorescence results. With respect to the corresponding
values in the controls, the protein level of active caspase-3 was
increased and the ratio of Bcl-2/Bax was decreased in the PTZ mice
(Fig. 6G-I; P<0.001).
However, the expression of active caspase-3 was partially decreased
in both semaglutide groups (low- and high-dose) with respect to the
PTZ mice (Fig. 6H; P<0.01 and
P<0.001, respectively). The ratio of Bcl-2/Bax was also
decreased in both semaglutide groups (low- and high-dose) with
respect to the PTZ mice (Fig.
6I; P<0.05 and P<0.01, respectively). Injection of
semaglutide alone did not significantly affect the levels of
apoptosis-related proteins.
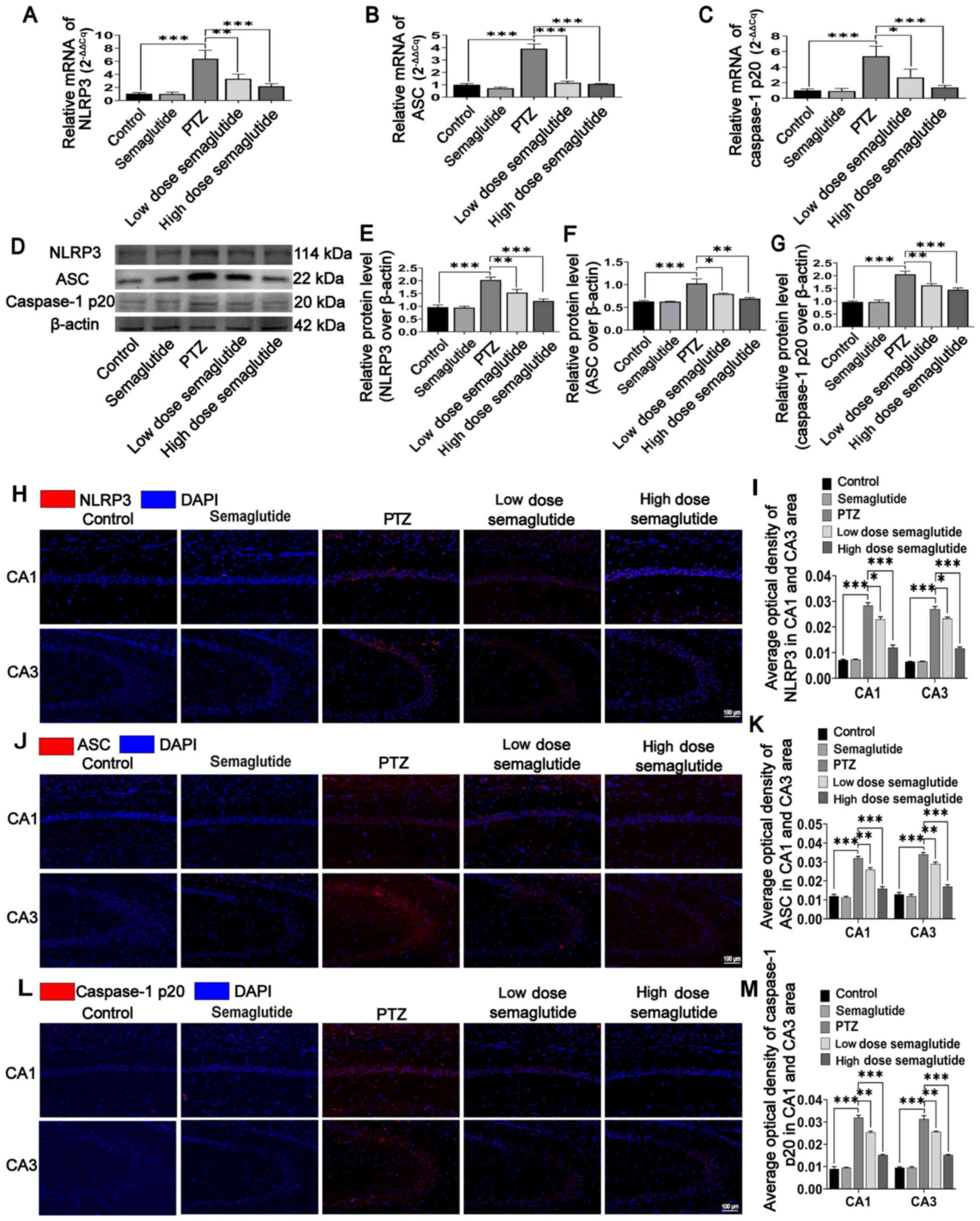
Semaglutide blocks NLRP3 inflammasome
activation in PTZ kindled-mice
It was confirmed that semaglutide attenuated the
LPS- and nigericin-mediated inflammatory response by blocking the
effects of NLRP3 inflammasome activation in vitro.
Semaglutide attenuated seizure severity, ameliorated cognitive
impairment and decreased hippocampal neuronal apoptosis in
PTZ-kindled mice. Therefore, the potential for semaglutide to block
NLRP3 inflammasome activation was then investigated in vivo.
The mRNA levels in the hippo-campus were assessed by RT-qPCR, and
the protein levels were evaluated by WB analysis and
immunofluorescence staining. In the PTZ mice, the mRNA level of the
NLRP3 inflammasome in the hippocampus was significantly higher with
respect to those in the controls (Fig. 7A-C; P<0.001), while the mRNA
level of the NLRP3, ASC and caspase-1p20 were decreased by low-dose
(P<0.01, P<0.001 and P<0.05, respectively) and high-dose
(all P<0.001) semaglutide-treated mice compared with PTZ mice
(Fig. 7A-C). However,
semaglutide alone had no significant influence with respect to the
controls.
The protein expression of the NLRP3 inflammasome was
similar to its RNA level. NLRP3 inflammasome expression was higher
in the PTZ mice with respect to the control mice (Fig. 7D-G; P<0.001). The protein
levels of NLRP3, ASC and caspase-1 p20 were partially inhibited in
low-dose (P<0.01, P<0.05 and P<0.01, respectively) and
high-dose semaglutide mice (P<0.001, P<0.01 and P<0.001,
respectively) with respect to the PTZ mice (Fig. 7E-G). However, semaglutide alone
failed to influence the protein expression of the NLRP3
inflammasome with respect to that in the controls.
The immunofluorescence results were similar to those
of WB and RT-qPCR. The immunofluorescence intensity of NLRP3
successively decreased from the PTZ mice (Fig. 7H-I; P<0.001) to the low-dose
semaglutide mice (P<0.05) to the high-dose semaglutide mice
(P<0.001) to the control and semaglutide mice (P>0.05). The
immunofluorescence intensities of ASC and caspase-1 p20
successively decreased from the PTZ mice (Fig. 7J-M; P<0.001) to the low-dose
semaglutide mice (P<0.01) to the high-dose semaglutide mice
(P<0.001) to the control and semaglutide mice (P>0.05). These
results showed that semaglutide alleviated hippocampal neuronal
insult and cognitive impairment in mice, possibly via suppression
of NLRP3 inflammasome activation.
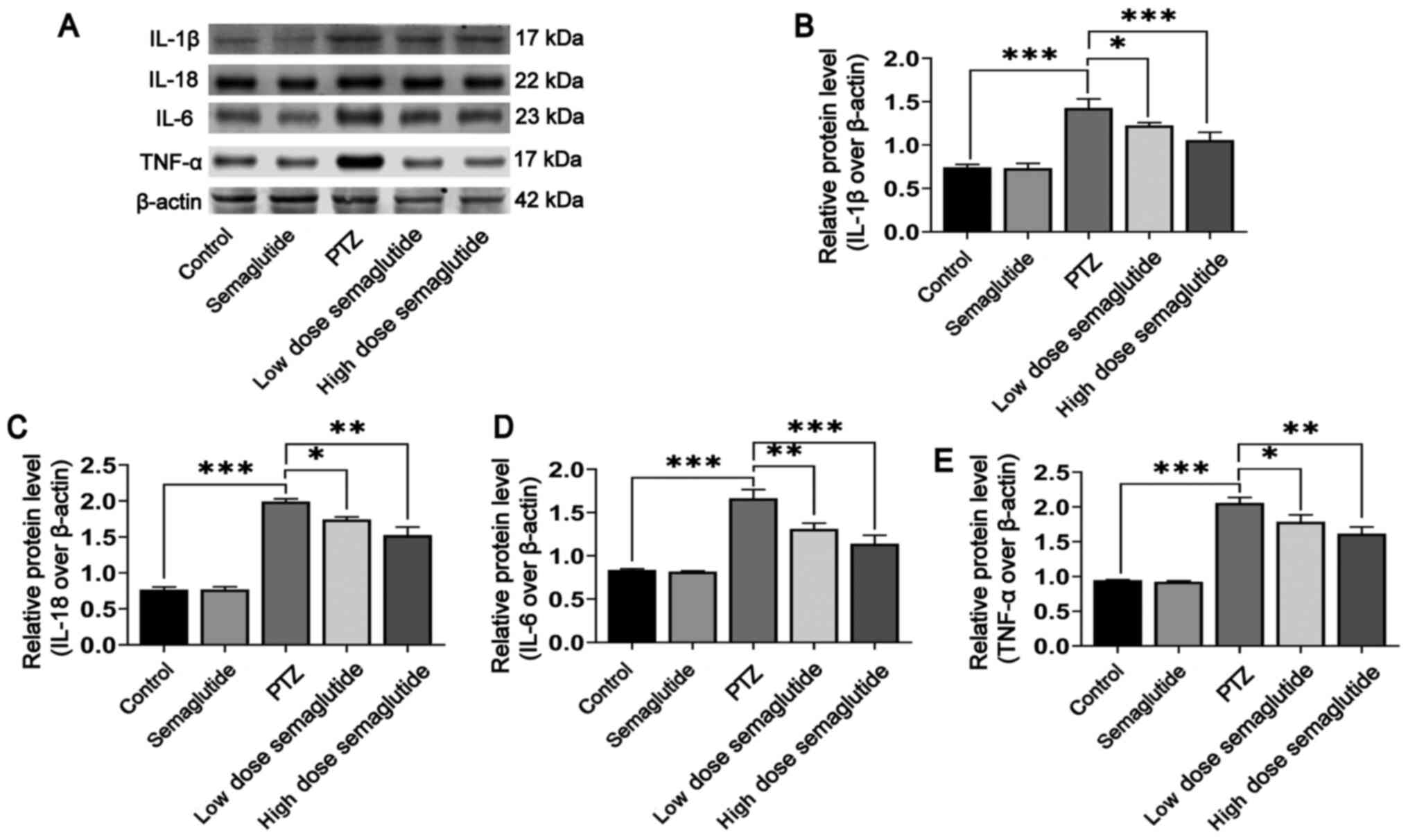
Semaglutide decreases inflammatory
cytokine secretion in PTZ-kindled mice
In various respects, inflammasome activation
contributes to the release of inflammatory cytokines, which play a
pivotal role in epileptogenesis (14,15,38). The present study demonstrated
that semaglutide blocked NLRP3 inflammasome activation.
Subsequently, WB was used to detect the protein levels of
inflammatory cytokines in the hippocampus of PTZ-kindled mice.
Inflammatory cytokine release were increased in the PTZ mice with
respect to the controls (Fig.
8A-E; P<0.001). Nonetheless, the expression levels of the
inflammatory cytokines IL-1β, IL-18, IL-6 and TNF-α were partially
downregulated in the low-dose (Fig.
8B, P<0.05; Fig. 8C,
P<0.05; Fig. 8D, P<0.01;
Fig. 8E, P<0.05,
respectively) and high-dose semaglutide mice with respect to the
PTZ mice (P<0.001, P<0.01, P<0.001 and P<0.01,
respectively), though expression levels between the semaglutide
alone mice and the controls were not significantly different. These
results indicated that semaglutide also blocked the secretion of
inflammatory cytokines in PTZ-kindled mice.
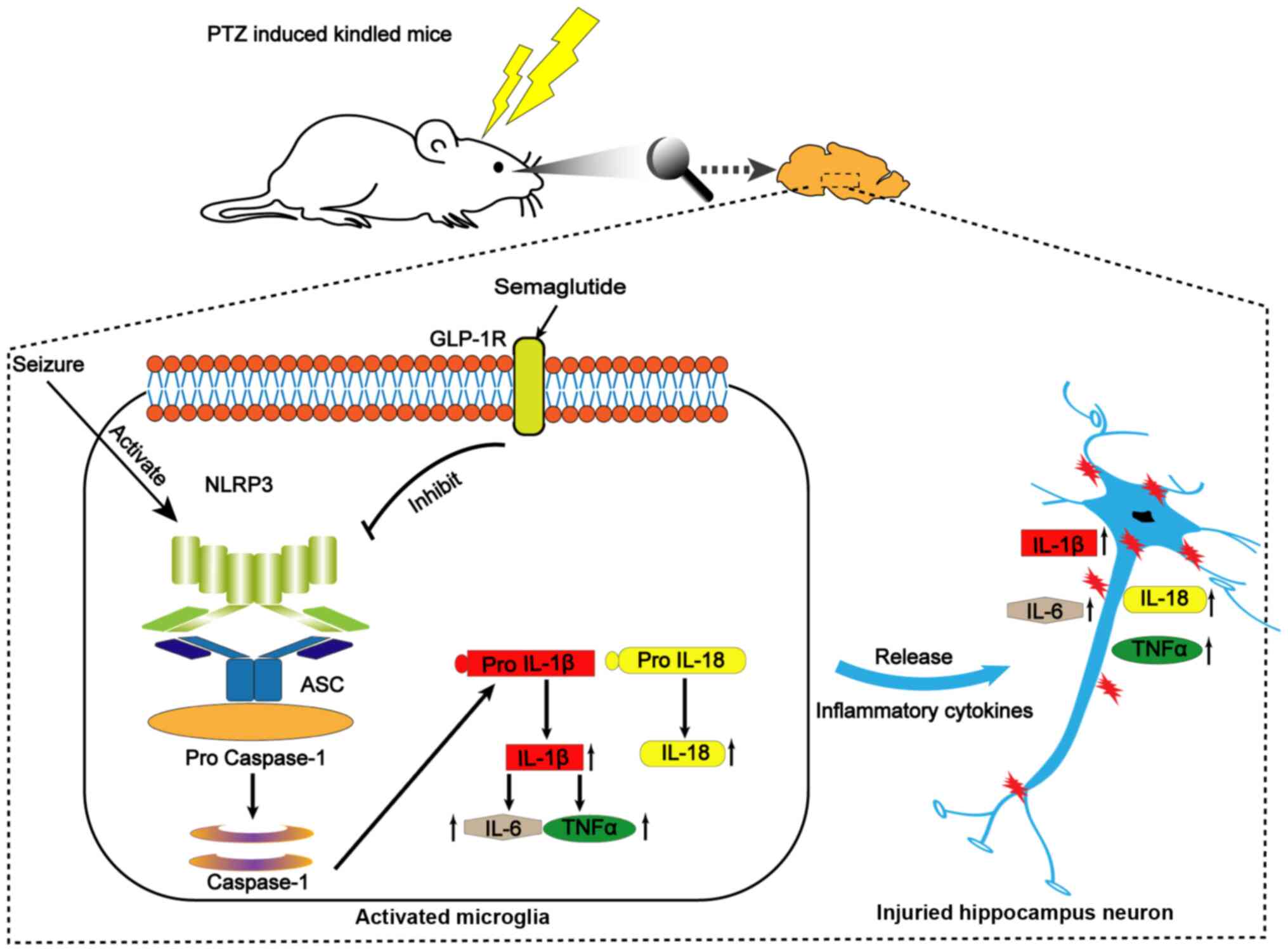
Discussion
In the present study, semaglutide was found to
alleviate the LPS-and nigericin-mediated inflammatory response and
LDH release by inhibiting NLRP3 inflammasome activation in
vitro. Semaglutide was also shown to decrease kindling rate and
seizure severity in vivo, and to alleviate hippocampal
neuronal injury and cognitive dysfunction in PTZ-kindled mice.
Furthermore, the results showed that semaglutide blocked NLRP3
inflammasome activation and decreased inflammatory cytokine release
in the hippocampal tissues of PTZ-kindled mice. These findings
indicated that semaglutide reduced seizure severity, exerted
neuroprotective effects and ameliorated cognitive dysfunction,
possibly by inhibiting NLRP3 inflammasome activation and decreasing
inflammatory cytokine secretion (Fig. 9).
These findings were in line with previous studies
indicating that GLP-1 analogues reduce tissue apoptosis or
pyroptosis by inhibiting NLRP3 activation (21,39). Based on these in vitro
findings, a chronic epilepsy model was established using
PTZ-kindled mice. In vivo, preventive administration of
either 10 or 25 nM/kg semaglutide every other day decreased seizure
severity in PTZ-kindled mice. This result was similar to the
results of daily injection of liraglutide and sitagliptin in
previous studies (25,40).
Epilepsy is frequently accompanied by cognitive
dysfunction (41). Indeed;
learning impairment and cognitive dysfunction have been observed in
patients with epilepsy and various animal models, and even if the
seizures were controlled, the cognitive dysfunction was not
markedly improved (11,41). Therefore, after finding that
semaglutide decreased seizure severity in PTZ-kindled mice,
behavioral tests were used in the current study to assess whether
semaglutide ameliorated cognitive dysfunction as a result of PTZ
kindling. In the NOR, shuttle box and MWM tests, both 10 and 25
nM/kg semaglutide ameliorated cognitive dysfunction in PTZ-kindled
mice. This result was consistent with previous findings in cerebral
ischemia (27), AD (28) and PD (29) models. The results showed that
semaglutide ameliorated cognitive dysfunction in CNS diseases,
including epilepsy. However, in the NOR test, the DI of the
low-dose semaglutide mice showed an increasing trend with respect
to that in the PTZ mice (P>0.05), but the DI in the high-dose
semaglutide mice was significantly higher with respect to that in
the PTZ mice (P<0.01). These results may be caused by the
difference in the semaglutide dose; the 25 nM/kg dose may more
effectively ameliorate cognitive dysfunction than the 10 nM/kg dose
in PTZ-kindled mice.
Numerous lines of evidence indicate that the
hippocampal CA1 and CA3 areas are more vulnerable to neuronal
injury in patients with epilepsy. Neuronal injury further promoted
learning and memory impairment (20), and hippocampal CA3 neurons are
more sensitive to epilepsy-related excitotoxicity than CA1 neurons
(42). Seizures aggravate injury
to hippocampal neurons and cause cognitive dysfunction. Multiple
GLP-1 analogues have been shown to exert neuroprotective and nerve
regenerative functions under pathological conditions in previous
epilepsy studies (25,36,40). Our previous research showed that
semaglutide reduced seizure severity and ameliorated cognitive
dysfunction; thus, in the present study, whether semaglutide
improved hippocampal neuronal apoptosis was further investigated.
The results suggested that both semaglutide doses ameliorated
neuronal injury in the hippocampal tissue, decreased the level of
the apoptosis-related protein active caspase-3 and increased the
Bcl-2/Bax ratio in hippocampal neurons in PTZ-kindled mice. These
results suggested that the ameliorative effect of semaglutide on
cognitive dysfunction may be associated with reduced hippocampal
neuronal apoptosis and enhanced nerve function recovery in
PTZ-kindled mice. These effects were similar to those of
semaglutide in other central nervous system diseases (28,29).
Accumulating evidence has demonstrated that
neuroinflammation plays a pivotal role in the pathological
mechanism of epilepsy (12,13). Activation of the NLRP3
inflammasome induces the secretion of IL-1β and IL-18 (38), and IL-1β further induces the
release of IL-6 and TNF-α (43).
Both inflammasomes and neuroinflammation affect the occurrence and
development of epilepsy, and anti-inflammasome and
anti-neuroinflammation therapies may be novel treatments for
epilepsy (16). In the present
study, semaglutide attenuated NLRP3 inflammasome activation and
inflammatory cytokine secretion in LPS- and nigericin-induced
inflammatory BV2 cells. Therefore, semaglutide may also possess
anti-inflammatory effects in vivo. In PTZ-kindled mice, both
doses of semaglutide (10 and 25 nM/kg) decreased NLRP3 inflammasome
activation and lowered inflammatory cytokine secretion in the
present study. These results indicated that semaglutide reduced
neuronal apoptosis, ameliorated cognitive dysfunction and
attenuated seizure severity, possibly via an inhibitory effect on
the NLRP3 inflammasome and inflammatory cytokines.
However, the precise mechanisms by which semaglutide
affects NLRP3 inflammasome signaling are unclear. Previous studies
have shown that GLP-1 agonists inhibit NLRP3 inflammasome
activation through sirtuin1 signaling to alleviate cardiovascular
impairment (44,45). Another study found that GLP-1
agonists alleviated inflammation and attenuated peripheral nerve
injury via the p38 MAPK/NF-κB pathway (46). A previous study also indicated
that ibuprofen has antiepileptic and neuroprotective effects
through the cyclooxygenase-2/NLRP3 pathway in PTZ-kindled mice
(18). However, further research
is necessary to clarify the precise mechanisms.
In conclusion, through in vitro
experimentation, the present study revealed that semaglutide
blocked NLRP3 inflammasome activation, inhibited inflammatory
cytokine secretion, and reduced LDH release. In an in vivo
experiment, semaglutide was found to decrease seizure severity,
reduce neuronal damage and ameliorate cognitive impairment,
potentially via inhibition of NLRP3 inflammasome activation and
related cascade reactions. These results offer novel insights into
the treatment of epilepsy and indicate that semaglutide may be a
promising adjuvant therapeutic.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TS and FW designed the study. LW, JD and CZ
conducted the experiments. LW, JD and CZ wrote the paper. BG, WY,
WH, XL, YW and WL analyzed the data. LW and TS confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The animal study was reviewed and approved by
institutional Animal Care and Use Committee of Ningxia Medical
University [IACUC Animal Use Certificate No. SCXK (Ning)
2019-203].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors are grateful to the Ningxia Key
Laboratory of Cerebrocranial Disease of Ningxia Medical University
for their kind help on technical expertise.
References
|
1
|
Rong S, Wan D, Fan Y, Liu S, Sun K, Huo J,
Zhang P, Li X, Xie X, Wang F, et al: Amentoflavone affects
epileptogenesis and exerts neuroprotective effects by inhibiting
NLRP3 inflammasome. Front Pharmacol. 10:8562019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Juvale IIA and Che Has AT: Possible
interplay between the theories of pharmacoresistant epilepsy. Eur J
Neurosci. 53:1998–2026. 2021. View Article : Google Scholar
|
|
3
|
Xiao D, Lv J, Zheng Z, Liu Y, Zhang Y, Luo
C, Qi L, Qin B and Liu C: Mechanisms of microRNA 142 in
mitochondrial autophagy and hippocampal damage in a rat model of
epilepsy. Int J Mol Med. 47:982021. View Article : Google Scholar
|
|
4
|
González-H G, Contreras-García IJ,
Sánchez-Huerta K, Queiroz CM, Gallardo Gudiño LR,
Mendoza-Torreblanca JG and Zamudio SR: Levetiracetam reduced the
basal excitability of the dentate gyrus without restoring impaired
synaptic plasticity in rats with temporal lobe epilepsy. Brain Sci.
10:E6342020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guiard BP and Di Giovanni G: Central
serotonin-2A (5-HT2A) receptor dysfunction in depression and
epilepsy: The missing link? Front Pharmacol. 6:462015. View Article : Google Scholar :
|
|
6
|
Fisher RS, van Emde Boas W, Blume W, Elger
C, Genton P, Lee P and Engel J Jr: Epileptic seizures and epilepsy:
Definitions proposed by the International League Against Epilepsy
(ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia.
46:470–472. 2005. View Article : Google Scholar
|
|
7
|
Baroli G, Sanchez JR, Agostinelli E,
Mariottini P and Cervelli M: Polyamines: The possible missing link
between mental disorders and epilepsy (Review). Int J Mol Med.
45:3–9. 2020.
|
|
8
|
Perucca E, Brodie MJ, Kwan P and Tomson T:
30 years of second-generation antiseizure medications: Impact and
future perspectives. Lancet Neurol. 19:544–556. 2020. View Article : Google Scholar
|
|
9
|
Wang Y and Chen Z: An update for epilepsy
research and antiepileptic drug development: Toward precise circuit
therapy. Pharmacol Ther. 201:77–93. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Löscher W, Potschka H, Sisodiya SM and
Vezzani A: Drug resistance in epilepsy: Clinical impact, potential
mechanisms, and new innovative treatment options. Pharmacol Rev.
72:606–638. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
West PJ, Saunders GW, Remigio GJ, Wilcox
KS and White HS: Antiseizure drugs differentially modulate θ-burst
induced long-term potentiation in C57BL/6 mice. Epilepsia.
55:214–223. 2014. View Article : Google Scholar :
|
|
12
|
Vezzani A, Fujinami RS, White HS, Preux
PM, Blümcke I, Sander JW and Löscher W: Infections, inflammation
and epilepsy. Acta Neuropathol. 131:211–234. 2016. View Article : Google Scholar :
|
|
13
|
Espinosa-Garcia C, Zeleke H and Rojas A:
Impact of stress on epilepsy: Focus on neuroinflammation-A mini
review. Int J Mol Sci. 22:40612021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shao BZ, Xu ZQ, Han BZ, Su DF and Liu C:
NLRP3 inflammasome and its inhibitors: A review. Front Pharmacol.
6:2622015. View Article : Google Scholar
|
|
15
|
Wu C, Zhang G, Chen L, Kim S, Yu J, Hu G,
Chen J, Huang Y, Zheng G and Huang S: The role of NLRP3 and IL-1β
in refractory epilepsy brain injury. Front Neurol. 10:14182020.
View Article : Google Scholar
|
|
16
|
de Brito Toscano EC, Vieira ÉL, Dias BB,
Caliari MV, Gonçalves AP, Giannetti AV, Siqueira JM, Suemoto CK,
Leite RE, Nitrini R, et al: NLRP3 and NLRP1 inflammasomes are
up-regulated in patients with mesial temporal lobe epilepsy and may
contribute to overexpression of caspase-1 and IL-β in sclerotic
hippocampi. Brain Res. 1752:1472302021. View Article : Google Scholar
|
|
17
|
Kegler A, Caprara AL, Pascotini ET, Arend
J, Gabbi P, Duarte MM, Furian AF, Oliveira MS, Royes LF and Fighera
MR: Apoptotic markers are increased in epilepsy patients: A
relation with manganese superoxide dismutase Ala16Val polymorphism
and deizure type through IL-1β and IL-6 pathways. BioMed Res Int.
2020:62504292020. View Article : Google Scholar
|
|
18
|
Liu R, Wu S, Guo C, Hu Z, Peng J, Guo K,
Zhang X and Li J: Ibuprofen exerts antiepileptic and
neuroprotective effects in the rat model of
pentylenetetrazol-induced epilepsy via the COX-2/NLRP3/IL-18
pathway. Neurochem Res. 45:2516–2526. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tran KL, Park YI, Pandya S, Muliyil NJ,
Jensen BD, Huynh K and Nguyen QT: Overview of glucagon-like
peptide-1 receptor agonists for the treatment of patients with type
2 diabetes. Am Health Drug Benefits. 10:178–188. 2017.
|
|
20
|
Koshal P and Jamwal S: An insight review.
Neuropharmacology. 136:271–279. 2018. View Article : Google Scholar
|
|
21
|
Zhu W, Feng PP, He K, Li SW and Gong JP:
Liraglutide protects non-alcoholic fatty liver disease via
inhibiting NLRP3 inflammasome activation in a mouse model induced
by high-fat diet. Biochem Biophys Res Commun. 505:523–529. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu X, Hao M, Liu Y, Ma X, Lin W, Xu Q,
Zhou H, Shao N and Kuang H: Liraglutide ameliorates non-alcoholic
steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis
activation via mitophagy. Eur J Pharmacol. 864:1727152019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cork SC, Richards JE, Holt MK, Gribble FM,
Reimann F and Trapp S: Distribution and characterisation of
Glucagon-like peptide-1 receptor expressing cells in the mouse
brain. Mol Metab. 4:718–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Graham DL, Durai HH, Trammell TS, Noble
BL, Mortlock DP, Galli A and Stanwood GD: A novel mouse model of
glucagon-like peptide-1 receptor expression: A look at the brain. J
Comp Neurol. 528:2445–2470. 2020. View Article : Google Scholar :
|
|
25
|
Liu S, Jin Z, Zhang Y, Rong S, He W, Sun
K, Wan D, Huo J, Xiao L, Li X, et al: The glucagon-like peptide-1
analogue liraglutide reduces seizures susceptibility, cognition
dysfunction and neuronal apoptosis in a mouse model of dravet
syndrome. Front Pharmacol. 11:1362020. View Article : Google Scholar
|
|
26
|
Dhillon S: Semaglutide: First global
approval. Drugs. 78:275–284. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang X, Feng P, Zhang X, Li D, Wang R, Ji
C, Li G and Hölscher C: The diabetes drug semaglutide reduces
infarct size, inflammation, and apoptosis, and normalizes
neurogenesis in a rat model of stroke. Neuropharmacology.
158:1077482019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang YF, Zhang D, Hu WM, Liu DX and Li L:
Semaglutide-mediated protection against Aβ correlated with
enhancement of autophagy and inhibition of apotosis. J Clin
Neurosci. 81:234–239. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang L, Zhang L, Li L and Hölscher C:
Semaglutide is neuro-protective and reduces α-synuclein levels in
the chronic MPTP mouse model of Parkinson's disease. J Parkinsons
Dis. 9:157–171. 2019. View Article : Google Scholar
|
|
30
|
Minkeviciene R, Rheims S, Dobszay MB,
Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany
T, Pitkänen A, et al: Amyloid beta-induced neuronal
hyperexcitability triggers progressive epilepsy. J Neurosci.
29:3453–3462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan XX, Cai Y, Shelton J, Deng SH, Luo XG,
Oddo S, Laferla FM, Cai H, Rose GM and Patrylo PR: Chronic temporal
lobe epilepsy is associated with enhanced Alzheimer-like
neuropathology in 3xTg-AD mice. PLoS One. 7:e487822012. View Article : Google Scholar
|
|
32
|
Nam HY, Nam JH, Yoon G, Lee JY, Nam Y,
Kang HJ, Cho HJ, Kim J and Hoe HS: Ibrutinib suppresses LPS-induced
neuroinflammatory responses in BV2 microglial cells and wild-type
mice. J Neuroinflammation. 15:2712018. View Article : Google Scholar :
|
|
33
|
Zeng QZ, Yang F, Li CG, Xu LH, He XH, Mai
FY, Zeng CY, Zhang CC, Zha QB and Ouyang DY: Paclitaxel Enhances
the Innate Immunity by Promoting NLRP3 Inflammasome Activation in
Macrophages. Front Immunol. 10:722019. View Article : Google Scholar :
|
|
34
|
Koshal P and Kumar P: Neurochemical
modulation involved in the beneficial effect of liraglutide, GLP-1
agonist on PTZ kindling epilepsy-induced comorbidities in mice. Mol
Cell Biochem. 415:77–87. 2016. View Article : Google Scholar
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
36
|
Zheng Z, Liang P, Hou B, Lu X, Ma Q, Yu X,
Han S, Peng B, Chen T, Liu W, et al: The effect of dipeptidyl
peptidase IV on disease-associated microglia phenotypic
transformation in epilepsy. J Neuroinflammation. 18:1122021.
View Article : Google Scholar :
|
|
37
|
Vezzani A, Balosso S and Ravizza T:
Neuroinflammatory pathways as treatment targets and biomarkers in
epilepsy. Nat Rev Neurol. 15:459–472. 2019. View Article : Google Scholar
|
|
38
|
Swanson KV, Deng M and Ting JP: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Birnbaum Y, Tran D, Bajaj M and Ye Y:
DPP-4 inhibition by linagliptin prevents cardiac dysfunction and
inflammation by targeting the Nlrp3/ASC inflammasome. Basic Res
Cardiol. 114:352019. View Article : Google Scholar
|
|
40
|
Safar MM, Shahin NN, Mohamed AF and
Abdelkader NF: Suppression of BACE1 and amyloidogenic/RAGE axis by
sitagliptin ameliorates PTZ kindling-induced cognitive deficits in
rats. Chem Biol Interact. 328:1091442020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu L, Chen L, Xu P, Lu D, Dai S, Zhong L,
Han Y, Zhang M, Xiao B, Chang L, et al: Genetic and molecular basis
of epilepsy-related cognitive dysfunction. Epilepsy Behav.
104:1068482020. View Article : Google Scholar
|
|
42
|
Lai MC, Lin KM, Yeh PS, Wu SN and Huang
CW: The novel effect of immunomodulator-glatiramer acetate on
epileptogenesis and epileptic seizures. Cell Physiol Biochem.
50:150–168. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Alomar SY, Gentili A, Zaibi MS, Kępczyńska
MA and Trayhurn P: IL-1β (interleukin-1β) stimulates the production
and release of multiple cytokines and chemokines by human
preadipocytes. Arch Physiol Biochem. 122:117–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen A, Chen Z, Xia Y, Lu D, Yang X, Sun
A, Zou Y, Qian J and Ge J: Liraglutide attenuates NLRP3
inflammasome-dependent pyroptosis via regulating SIRT1/NOX4/ROS
pathway in H9c2 cells. Biochem Biophys Res Commun. 499:267–272.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Luo X, Hu Y, He S, Ye Q, Lv Z, Liu J and
Chen X: Dulaglutide inhibits high glucose-induced endothelial
dysfunction and NLRP3 inflammasome activation. Arch Biochem
Biophys. 671:203–209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ma J, Shi M, Zhang X, Liu X, Chen J, Zhang
R, Wang X and Zhang H: GLP 1R agonists ameliorate peripheral nerve
dysfunction and inflammation via p38 MAPK/NF-κB signaling pathways
in streptozotocin induced diabetic rats. Int J Mol Med.
41:2977–2985. 2018.PubMed/NCBI
|