Introduction
Although multiple risk factors have been linked to
the development of lung cancer, cigarette smoke remains the leading
cause of the disease (1-3). Tobacco contains multiple
carcinogens; when these are inhaled into the lungs, bronchial
epithelial (BE) cells are damaged which may initiate
carcinogenesis. Carcinogens identified in tobacco exhibit a myriad
of effects, ranging from DNA damage to metastasis promotion
(4,5). Cigarette smoke can cause damage to
BE cells in a multitude of ways; much of this damage is usually
corrected by cell self-repair mechanisms. However, this damage may
build up and lead to the development of lung cancer.
Smoking-induced human bronchial epithelial (HBE) cell carcinoma
develops across multiple stages, including: DNA damage, BE
hyperplasia, cell dysplasia, early carcinogenesis and finally
invasive carcinoma development (6). In this multi-step carcinogenesis
process, the formation of DNA adducts is considered to be a key
cancer initiator (7). Therefore,
the use of natural or synthetic agents in the prevention of
smoking-induced, BE cell, DNA adduct formation, may play a role in
the prevention of lung cancer. In lung cancer, cigarette smoke can
also promote disease progression by altering the regulation of
multiple genes including: TSPO, AP-2α and EGF (8-11). Cigarette smoke has also been
demonstrated to be involved in carcinogenesis through the
modulation of key signaling pathways (12-14). Cigarette smoke also causes DNA
methylation and site-specific histone modification (12-14). Cigarette smoke is involved in
multiple processes that promote carcinogenesis; this renders
combating its effects complicated (4). Due to this, a broad-spectrum
anticancer agent may be the best approach for addressing cigarette
smoke-mediated BE cell damage and subsequent carcinogenesis. The
polyphenol epigallocatechin-3-gallate (EGCG), found in green tea,
may be one such preventative, due to its array of anticancer
properties.
EGCG is the most abundant and pharmacologically
active polyphenol found in green tea (15). It is widely used due to its
health benefits, including its chemoprevention and anticancer
activity (16,17). Whilst there is some dispute as to
whether drinking green tea reduces the risk of lung cancer,
experimental evidence has indicated that the polyphenols found in
green tea may be protective against carcinogen-induced lung cancer
(18-22). EGCG inhibits cell proliferation,
migration, promotes apoptosis and inhibits the self-renewal
capability of lung cancer stem-like cells; all of this may help it
combat lung cancer (23-28). EGCG interacts with several
signaling pathways to exude anticancer effects; these include: the
JNK, PI3K/Akt, nuclear factor-κB (NF-κB), Wnt/β-catenin (29-32), ERK and ERK1/2/NEAT1 pathways
(33,34). The multi-pronged approach by
which EGCG acts as a chemopreventive agent, (through the modulation
of multiple signaling pathways and strong antioxidant effect) may
be used to prevent BE cell lung carcinogenesis. However, the
expression of various genes and signaling pathway activation are
regulated by microRNA (miRNA) and epigenetic modifications.
Molecular changes in lung cancer, such as the dysregulation of
miRNA expression, have been linked to tobacco smoke (35). It is considered that EGCG affects
the expression of various long non-coding RNAs and miRNAs in the
cells, therefore affecting cell function (36). Moreover, EGCG treatment can
modulate miRNAs that play a significant role in specific signaling
pathways (37,38). However, cigarettes contain
thousands of chemical substances (39), including the carcinogen
benzopyrene (40); there are
several other carcinogens found in cigarette smoke, including:
tobacco-specific nitrosamines (TSNA) (41), polycyclic aromatic hydrocarbons
(PAH) (39,40), aromatic amines, benzene, dioxin,
catechol and carcinogenic quinones and hydrazine. The pathogenesis
of smoking-related cancer formation is very complex; as such,
elucidating protective mechanisms is equally complicated. Whether
EGCG can prevent smoking-related lung cancer formation by reducing
smoking-induced DNA damage in BE cells, as well as regulating the
expression of miRNA in BE cells, remains to be investigated.
Materials and methods
Cell culture
HBE cells were purchased from the Xiangya Hospital
Cell Bank (Central South University, Changsha, China). The base
growth medium was Dulbecco's modified Eagle's medium (DMEM;
HyClone; Cytiva). This was supplemented with fetal bovine
serum(FBS; 10%; Sigma-Aldrich; Merck KGaA) as well as
penicillin/streptomycin (100 U/ml penicillin/100 µg/ml
streptomycin). HBE cells had been stored in liquid nitrogen, before
being thawed rapidly in a 37°C water bath. The cells were then
dispensed into a 75 cm2 culture flask; the flask was
then stored in an incubator at 5% CO2. The growth medium
was changed the following day.
Cigarette smoke extract (CSE) treatment
and EGCG treatment
The original CSE solution was prepared using an
aqueous medium; when smoke from burnt cigarettes passed through a
sealed jar containing water, the toxic compounds contained in the
smoke were collected in the solvent. During the experiment, the
same cigarette brand was used and the original CSE solution was
prepared on the day of the experiment. HBE cells were cultured at
37°C for 24 h with DMEM. Following this, the base medium was
replaced with growth medium containing the final CSE. Flasks
received growth media with CSE concentrations of 2.5, 5, 7.5, 10,
12.5 or 15%. The original CSE solution was prepared with DMEM on
the day of the experiment, with the final concentration determined
by spectrophotometry. At 24, 48, and 72 h post-CSE treatment, an
immunofluorescence assay was performed on the cells to detect
benzopyrene-DNA adducts. The optimal time-point and CSE
concentration for benzopyrene-DNA adduct formation was recorded.
These parameters were then used for subsequent experiments. With
reference to the literature and previous experiments (26,27), the concentrations used for EGCG
treatment were 0, 5, 10, 20 and 40 µM. A gene microarray
technique was used to detect the differential miRNA expression
profiles of the treated HBE cells.
Cell Counting Kit-8 (CCK-8) assay
A CCK-8 (MedChemExpress) assay was used to detect
the survival rate of HBE cells. Cells were plated in 96-well plates
at a density of 8×104 cells/ml before CSE and EGCG
treatments were performed. These cells were then cultured at 37°C
in an incubator at 5% CO2 for 24, 48 and 72 h. The
treated medium was replaced with 100 µl of the CCK-8
solution (CCK-8: PBS, 1:9). The plates were then wrapped in tin
foil before being incubated at 37°C for a further 2 h. The
absorbance was then measured at 450 nm on an enzyme-labeling
instrument. Each experiment was repeated 3 times.
Immunofluorescence cytochemistry
HBE cells were cultured on cell slides in 24-well
plates. Following the treatment regimens, an immunofluorescence
assay was performed. A specific BPDE-DNA adduct mouse monoclonal
antibody (1:100; cat. no. sc-52625; Santa Cruz Biotechnology, Inc.)
was used to detect DNA lesions in the HBE cells. The slide was
incubated in a wet box at 4°C overnight. The cells were stained
with DAPI (10 µg/ml; cat. no. D1306; Thermo Fisher
Scientific, Inc.) at room temperature for 10 min. The slides were
then sealed with an anti-fluorescence quenching agent (containing
90% glycerin; Thermo Fisher Scientific, Inc.) before images were
captured under a fluorescence microscope (1:4 and 1:400).
Gene microarray assay and analysis
The treated HBE cells were washed with PBS. Cells
were lysed with TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA was extracted using a Mini miRNA
extraction kit (cat. no. 217004; Qiagen China Co., Ltd.). RNA
integrity was assessed using RNA denaturation electrophoresis
(Bioanalyzer 2100; Agilent). miRNA microarray hybridization
experiments and data collection were performed by the Wuhan Google
Biotechnology Co., Ltd. A miRNA microarray hybridization chip (cat.
no. G4474A; Agilent Technologies, Inc.) was scanned using an
Agilent Microarray Scanner. The featured extraction software
version 10.7.1.1 (Agilent Scan Control software) was used to
collect and analyze data. The fold change of differentially
screened miRNA was at least two-fold compared with the control
group. Functional changes to miRNA expression levels following EGCG
treatment were determined by comparative analysis. Furthermore,
bioinformatics technology was used to predict the target genes and
signaling pathways affected by EGCG treatment. The miRNA target
gene prediction softwares, miRDB (http://mirdb.org/miRDB), Diana microT v3.0 (http://diana.cslab.ece.ntua.gr/microT)
and TargetScan 3.0 (http://www.targetscan.org/), were used to predict the
target genes of the selected miRNA. Targeted signaling pathways
were predicted by the signaling pathway analysis software
'Ingenuity Pathway Analysis' (http://www.ingenuity.com). The most important miRNA
was further validated in tissue samples by fluorescence in
situ hybridization. The experiment was carried out according to
the instructions of a FAM-Labeled Probe Detection kit (Wuhan
Servicebio Technology Co., Ltd.). The sequence of the gene-specific
oligonucleotide probe was 5′-FAM-ACA GGC ACC CCA CTC CAC AGA-FAM-3′
for miRNA-7114-5p. The sections were dewaxed with xylene for 15
min. For RNA retrieval, the sections were placed into boiling water
for 15 min and cooled naturally. Subsequently, the sections were
digested with protease K (20 µg/ml; Wuhan Servicebio
Technology Co., Ltd.) at 37°C for 30 min. Following incubation with
pre-hybridization solution at 37°C for 1 h, the sections were
treated with probe miRNA-7214-5p hybridization solution (8
ng/µl) and placed in an incubator at 37°C overnight. Tissue
sections were stained with DAPI (10 µg/ml), incubated in
darkness for 8 min, and sealed with anti-fluorescence quenching
agent. The images were collected under a fluorescence
microscope.
Immunohistochemical analysis of patient
tissues
Non-small cell lung cancer specimens and adjacent
tissues, atypical hyperplasia, and chronic inflammatory tissues
were collected. Tissue samples were collected from 30 patients,
including 19 males and 11 females, aged 37 to 68 years. This
research protocol was approved by the Medical Ethics Committee of
Xiangya Hospital of Central South University (approval no.
201703133), and paraffin-embedded tissue samples were selected from
the specimen bank of the Department of Pathology of Xiangya
Hospital. These samples were preserved and paraffin-embossed after
surgical treatment at the Department of Thoracic Surgery of Xiangya
Hospital from May 2018 to October 2019. All patients provided
written informed consent before surgery. Retrospective
investigation of clinical data revealed that all patients with
non-small cell lung cancer had a lung mass or nodule which was
found by enhanced CT examination, and were confirmed as non-small
cell lung cancer by postoperative pathological diagnosis.
Para-carcinoma tissue was defined as lung tissue removed from the
margin of lung cancer tissue of more than 20 mm. The patients with
atypical hyperplasia or pulmonary inflammation were those who had a
lung mass or pulmonary nodule suspected diagnosis such as lung
cancer (possibly lung cancer) by enhanced CT scan before operation
and confirmed atypical hyperplasia or pulmonary inflammation by
pathological diagnosis after surgical resection. Continuous
4-µm thick tissue sections were sliced. Immunohistochemistry
was used to detect protein expression in different lung tissues.
For immunohistochemical protein detection, xylene was used to dewax
the paraffin sections. For antigen retrieval, the sections were
placed into a sodium citrate buffer (pH 6.0) and heated in a
microwave for 2 min before being placed in a water bath (98°C), for
a further 20 min. The expression levels of NF-κB (1:200),
transforming growth factor (TGF)-β (1:200) and CYP1A1 (1:200)
proteins were detected by their respective specific
rabbit-antibodies: product no. ADI-KAS-TF110-D (Enzo Life Sciences,
Inc.); and cat. nos. MBS462142 and MBS127670; MyBioSource, Inc.).
The slides were incubated in a wet box at 4°C overnight. Tissue
known to express the target protein was used as a positive control,
while tissue known to have an absence of the protein was used as a
negative control. DAB staining (at room temperature for 15 min) was
used to assess the results. A scoring system from 0-3 was devised;
unstained cells scored 0, mild yellow staining scored 1, strong
yellow staining scored 2 and brown staining scored 3. The
percentage of cells positively stained was also scored; <5% was
0 points, 5-25% was 1 point, 26-50% was 2 points, and >50% was 3
points. These scores were then multiplied to provide an overall
grading; a score of 0-1 was negative (−), 2-3 was weakly positive
(+), 4-6 was positive (++) and a score of 6-9 was strongly positive
(+++).
Animals and treatments
Female Sprague Dawley (SD) rats, aged 6-8 weeks and
weighing 180-200 g, were purchased from the Department of
Experimental Animals (Central South University, Changsha, China). A
total of 60 rats were randomly divided into three groups. Animals
were caged in groups of five. The rats were kept at room
temperature (24±2°C), 50% humidity and in a 12-h light/dark cycle
with adequate food and water supply. The control and cigarette
smoke (CS) treatment groups were administered normal drinking
water, while the EGCG group received water supplemented with EGCG
(0.3%) (13,42). The EGCG solution was prepared and
changed daily. Following 2 weeks of drinking the solution, the rats
inhaled CS. All rats in the CS +/− EGCG treatment groups were
exposed to 90 min of CS a day, 5 days a week (43-45). The rats were treated in a
transparent glass cage. A total of 10 cigarettes were lit at a
time. In each round of exposure, the rats were treated with passive
smoke for 15 min, the sealed cover of the cage was then opened and
the rats were allowed to breathe 'clean' air for 15 min. During the
next round of exposure, rats were passively exposed to smoke for 90
min, with close attention paid to the activity of the rats during
this period. Following this exposure, smoking treatment was
immediately discontinued if there was evidence of breathing
difficulties (rapid breathing, salivation, and cyanosis). On the 4,
8, 12 and 16th weekend following the introduction of CS, 5 rats
from each treatment group were sacrificed. Rats received an
intraperitoneal injection of a chloral hydrate solution (300
mg/kg), before being sacrificed under anesthesia. In brief, after
weighing the rats with an electronic balance, the required 10%
chloral hydrate solution was calculated. The anesthetic solution
was extracted with a syringe and injected into the abdominal cavity
of the rats. The rats quickly lost consciousness after the
injection, and none of the rats exhibited signs of peritonitis,
pain or discomfort. After the breathing of the rat became slow and
weak and it did not respond to stimulation, the rat was sacrificed
by exsanguination from the carotid artery under the conditions of
unconsciousness and painlessness. All of the animals were treated
humanely and in compliance with the Animal Welfare Act of America.
The experiment was approved (approval no. 201703133) by the Ethics
Department of Xiangya Hospital, Central South University (Changsha,
China).
Histopathological examination
Tissue from each lung of the treated rats was
obtained, completely immersed in 4% paraformaldehyde at room
temperature, fixed for 24 h. This was then transferred to a 0.2%
sodium azide solution at room temperature, and tissue continued to
be fixed for 24 h before being embedded in paraffin. Each lobe was
sectioned in 5 consecutive slices at a thickness of 5 µm.
Each section was stained with 0.5% hematoxylin for 10 min and 0.5%
eosin for 3 min and analysis was performed independently by two
pathologists.
Immunohistochemical analysis of rat
tissues
Prepared rat lung sections were dewaxed in xylene
and then alcohol. These sections were then placed in a 0.1 mol/l
citric acid buffer solution (pH=6) and microwaved for 20 min to
retrieve antigens (microwave oven PM100). The tissue was blocked
using 10% goat serum (Beijing Zhongshan Jinqiao Biotechnology,
Inc.) at room temperature for 1 h in a wet box. Staining for
benzopyrene-DNA adducts in the rat lung samples was performed using
an anti-BPDE-adduct mouse monoclonal antibody (1:100) for 1 h at
room temperature. The sections were then rinsed and incubated with
a secondary goat anti-mouse antibody (1:200; LS-C56298; LifeSpan
BioSciences, Inc.) for 60 min at room temperature. Positive and
negative samples, where benzopyrene-DNA adducts were present or
absent, were used as controls. After the experiment was completed,
the glass slides were sealed with an anti-fluorescence quenching
agent. The benzopyrene-DNA adducts were observed and images were
captured using a fluorescence microscope, before the average
optical density was assessed by ImageJ software v1.8.0 (National
Institutes of Health).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA of rat lung tissue was extracted according
to the Direct-zol™ RNA MiniPrep Quick Protocol (Zymo Research
Corp). RNA was reverse transcribed to cDNA using the High Capacity
cDNA Reverse Transcription Kit (cat. no. 4368813; Applied
Biosystems; Thermo Fisher Scientific, Inc.). A total of
50-µl reaction volumes, containing 500 ng of sample RNA,
were prepared for cDNA amplification. Using a Bio-Rad MyCycler™,
the reaction was carried out at 25°C for 10 min, 37°C for 120 min
and held at 4°C upon completion. The GAPDH gene was amplified as a
positive control, an RT negative sample was produced using the same
reaction mixture without reverse transcriptase. The cDNA products
were diluted at 1:10 for use in qPCR. qPCR was performed using an
ABI Quantstudio™ 3 Real-Time PCR System, with SYBR Green Master Mix
(Bio-Rad Laboratories, Inc.). Primers were synthesized by Shanghai
Biotechnology Co., Ltd. and were as follows: NF-κB forward, 5′-GGC
TTC TAT GAG GCT GAA CTC TGC-3′ and reverse, 5′-CTT GCT CCA GGT CTC
GCT TCT TC-3′; CYP1A1 forward, 5′-CAG GAC AGG AGG CTG GAC GAG-3′
and reverse, 5′-ACC AGG TAC ATG AGG CTC CAA GAG-3′; GAPDH forward,
5′-GAC ATG CCG CCT GGA GAA AC-3′ and reverse, 5′-AGC CCA GGA TGC
CCT TTA GT-3′. The q-PCR reactions were carried out at 94°C initial
denaturation for 2 min; 40 of cycles at 94°C denaturation for 15
sec, 60°C annealing, elongation and fluorescence was read for 1
min; and 60°C final extension for 4 min. All data were analyzed
using the 2−ΔΔCq method (46).
Western blot analysis
Rat lung tissue was collected to determine the level
of NF-κB and CYP1A1 protein expression. Lung tissue was washed
twice with cold TBS and crushed in a 1.5-ml homogenate tube, before
being placed on ice and suspended in RIPA buffer (cat. no. G2002;
Wuhan Servicebio Technology Co., Ltd.) with protein inhibitor. The
mixture was placed in an ice-bath for 30 min, centrifuged 13,000 ×
g at 4°C for 15 min, before the supernatant was removed in a clean
tube and stored at -80°C. To perform the western blot analysis, 16
µl of sample containing 150 µg of protein was loaded
along with 1.6 µl of 10X SDS buffer and 4 µl of SDS
buffer per well of a 10% SDS-PAGE gel, then electrophoresis was
performed at 80 V for 100 min. Electrophoresis was stopped and the
protein on the gel was transferred to a nitrocellulose membrane at
60 V for 120 min. The membrane was then blocked with 5% fat-free
milk for 1 h at room temperature. The membrane was then incubated
with mouse specific NF-κB (1:1,000; cat. no. GB11142; Wuhan
Servicebio Technology Co., Ltd.) and CYP1A1 (1:1,000; cat. no.
sc-101828; Santa Cruz Biotechnology, Inc.) antibodies overnight at
4°C. Next, the membrane was washed three times with TBS-T (0.1%
Tween-20) buffer for 10 min. The membrane was then incubated with a
goat anti-rat secondary antibody (1:3,000; GB23302; Wuhan
Servicebio Technology Co., Ltd.) for 2 h at room temperature,
before the washing step was repeated. Finally, the blot was
developed using a prepared A (luminol) and B (hydrogen peroxide),
1:1 reagent mix and incubated at room temperature for 5 min, before
images were captured using a ChemiDoc™ XRS+ system with Image Lab™
Software V6.0 (Bio-Rad Laboratories, Inc.).
Statistical analysis
The data are presented as the mean ± SD of three
repeats. SPSS 24.0 (IBM Corp.) statistical software was used to
analyze the data. A paired Student's t-test was used for comparison
between two groups. One-way analysis of variance (ANOVA) was used
for comparison between multiple groups. The pairwise comparisons
among the multiple groups were conducted using Tukey's post hoc
test. All statistics were bilateral. The test level was set as
α=0.05, and P<0.05 was considered to indicate a statistically
significant difference. GraphPad Prism v. 6 (GraphPad Software,
Inc.) was used to create all the graphs.
Results
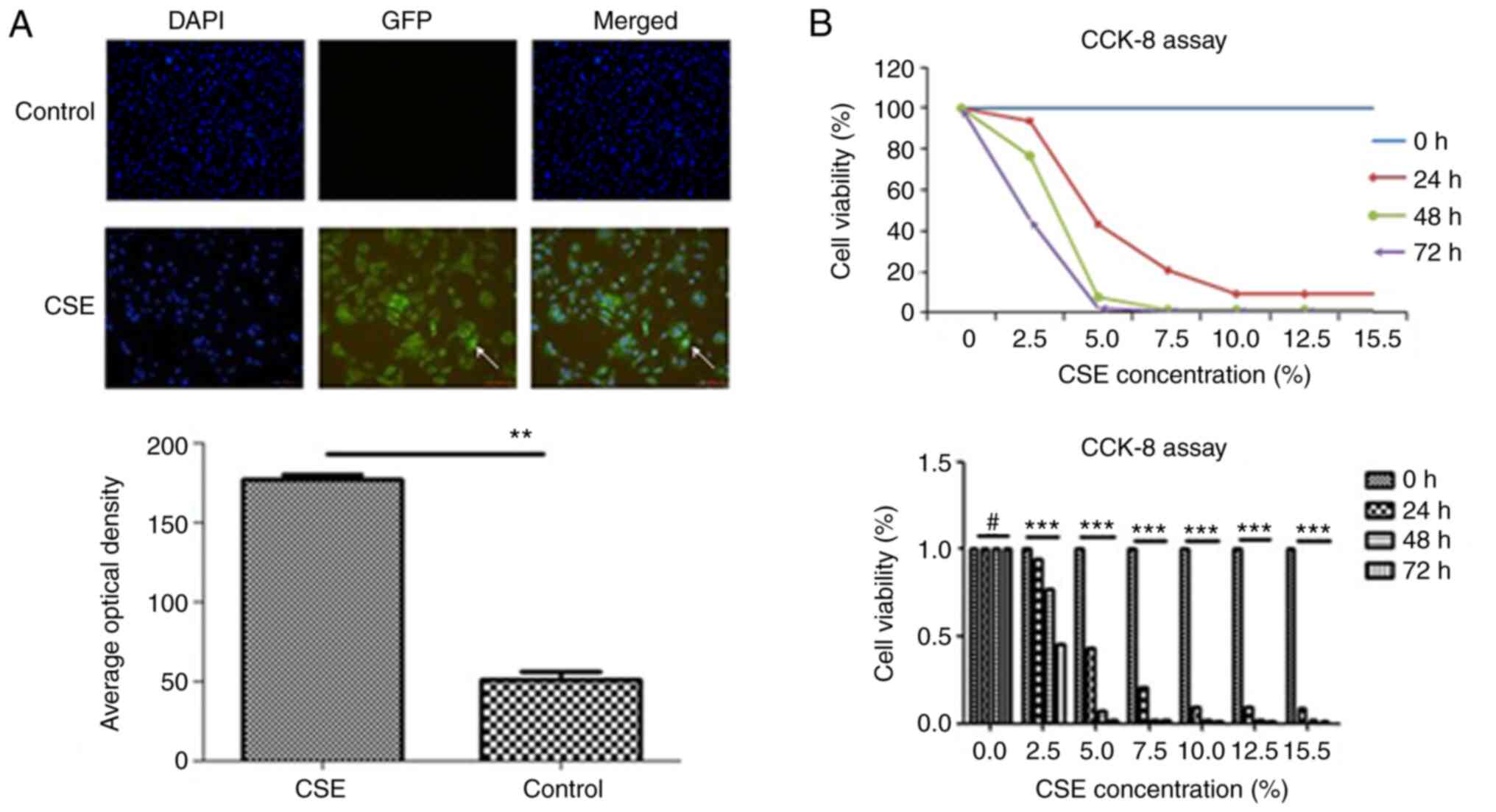
HBE cell damage by CSE
After CSE treatment, two types of damage occurred in
the HBE cells. Acute toxicity was associated with a decreased
survival of HBE cells. When the CSE concentration reached 5%, the
survival rate of HBE cells was <50%. The effect of CSE on HBE
cell survival was revealed to be time and concentration-dependent
(Fig. 1). CSE-induced DNA
damage, was observed in the surviving HBE cells. In addition,
benzopyrene-DNA adducts were detected in HBE cells after 24 h of 5%
CSE treatment (Fig. 1).
Effect of EGCG on HBE cell damage induced
by CSE
HBE cells were treated with different concentrations
of EGCG (0, 5, 10, 20 and 40 µM). The cell survival rate
between each treatment group and the control was not significantly
different (P>0.05). No apparent cytotoxicity was observed at any
concentration of EGCG (Fig. 2).
Compared with the CSE only treatment group, the HBE cells treated
with 5% CSE+EGCG revealed a significantly higher survival rate.
This indicated that EGCG protected HBE cells against CSE-mediated
acute injury. The immunofluorescence assay revealed that HBE cells
treated with CSE+EGCG formed less benzopyrene-DNA adducts than the
CSE group. EGCG significantly reduced the formation of
benzopyrene-DNA adducts induced by CSE exposure (Fig. 2).
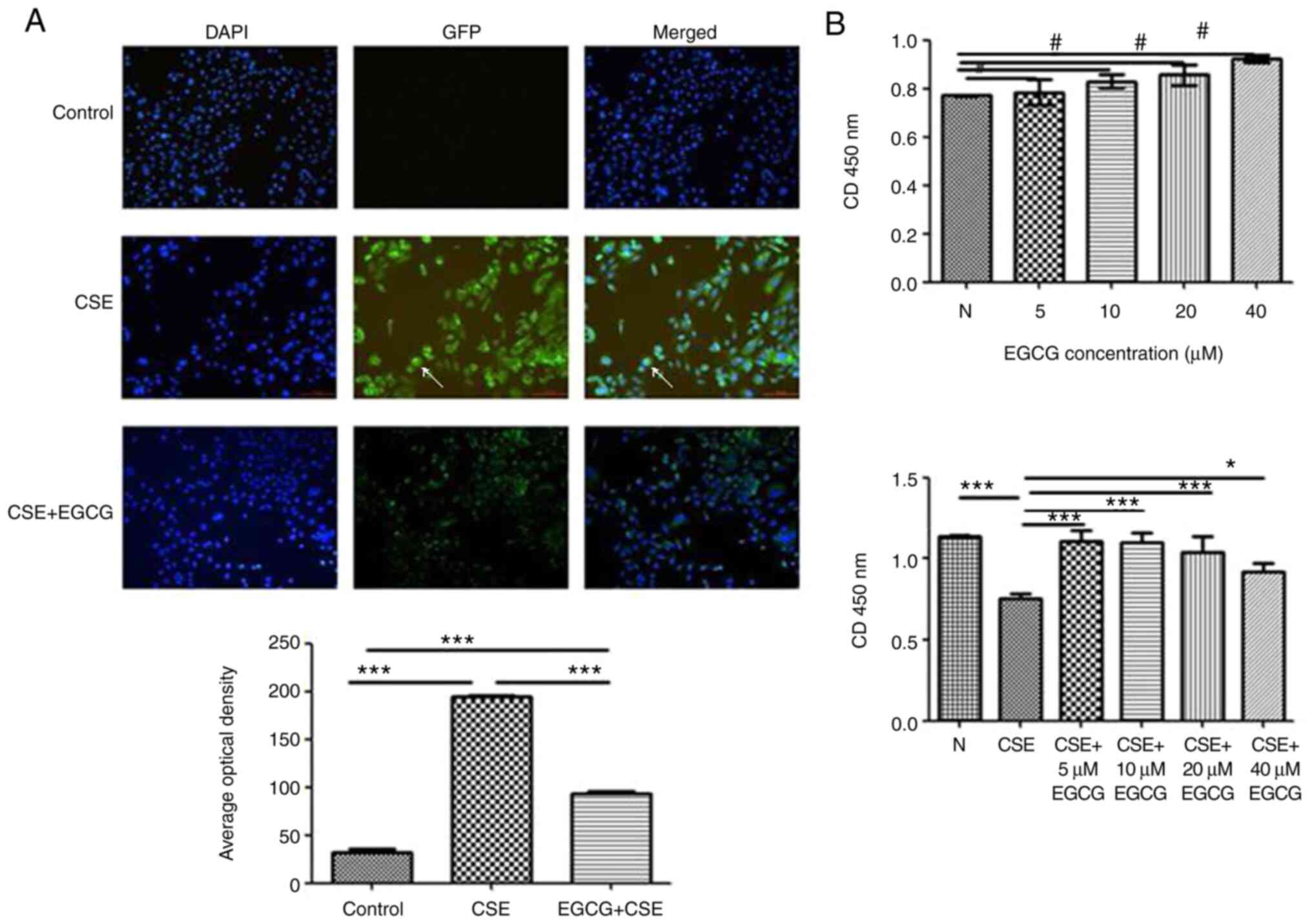 | Figure 2Effect of EGCG on HBE cell damage
induced by CSE. (A) In panel GFP and panel Merged (DAPI Merged with
GFP), immunofluorescence revealed a benzopyrene-DNA adduct in
cells. In the control cells, there was no evidence of
benzopyrene-DNA adducts. After CSE treatment, benzopyrene-DNA
adducts were detected in the nucleus of HBE cells. Cells treated
with CSE+EGCG had significantly less benzopyrene-DNA adducts than
cells treated with CSE alone. (B) HBE cells were treated with
different concentrations of EGCG (0, 5, 10, 20 and 40 µM).
The cell survival rate between each treatment group and the control
was not significantly different. No apparent cytotoxicity was
observed at any concentration of EGCG. Compared with the CSE only
treatment group, the HBE cells treated with 5% CSE+EGCG revealed a
significantly higher survival rate. #P>0.05,
*P<0.05 and ***P<0.001. EGCG,
epigallocatechin-3-gallate; HBE, human bronchial epithelial; CSE,
cigarette smoke extract; GFP, green fluorescent protein. |
miRNA differential expression
HBE cell miRNA expression profiles were detected by
microarray assay. The miRNA expression profiles of HBE cells
treated with CSE were significantly different when compared with
the expression profiles of untreated HBE cells. The expression of
multiple miRNAs was upregulated (Table I), while the expression of
several other miRNAs was downregulated (Table II). When HBE cells were treated
with CSE and EGCG, the miRNA expression profile was significantly
different compared with treatment with CSE only. EGCG treatment
inhibited the CSE-mediated upregulation of several miRNAs (Table III). Moreover, new miRNAs were
upregulated following EGCG treatment (Table IV).
 | Table IUpregulated miRNA expression in human
bronchial epithelial cells following cigarette smoke extract
treatment. |
Table I
Upregulated miRNA expression in human
bronchial epithelial cells following cigarette smoke extract
treatment.
| Systematic
name |
active_sequence |
miRBase_accession_No | Fold change |
|---|
| hsa-miR-6131 |
CACTCCCATCTGACC | MIMAT0024615 | 7.372937839 |
|
hsa-miR-5581-5p |
TCTCCATTTCTCCTGGA | MIMAT0022275 | 7.227041457 |
|
hsa-miR-4716-3p |
TCTCCATGTTTCCTTCC | MIMAT0019827 | 6.90081197 |
| hsa-miR-3198 |
TCTCCATTCCCCAGG | MIMAT0015083 | 6.316905823 |
| hsa-miR-1305 |
TCTCTCCCATTAGAGTTGA | MIMAT0005893 | 5.62008685 |
|
hsa-miR-6717-5p |
TCTCTACATCCCCACATC | MIMAT0025846 | 5.504413227 |
|
hsa-miR-6767-5p |
TCTCCATGTGTCCCTG | MIMAT0027434 | 5.357791135 |
|
hsa-miR-4713-3p |
TTCTCCCACTGTCTGG | MIMAT0019821 | 4.86793705 |
|
hsa-miR-6734-5p |
TCTCCACCTCATTCTCC | MIMAT0027369 | 4.702717565 |
|
hsa-miR-6740-5p |
TCTCCTCTCTCCATCCC | MIMAT0027381 | 4.505579757 |
|
hsa-miR-6879-5p |
CTCTCCCACCTTCCC | MIMAT0027658 | 4.294047462 |
|
hsa-miR-6780b-5p |
TCTTCCCTGCCAAGC | MIMAT0027572 | 3.974158682 |
|
hsa-miR-6875-5p |
TCTCCTGTCCTGGGT | MIMAT0027650 | 3.699151613 |
| hsa-miR-6124 |
TCCTCCCCCTTCCTT | MIMAT0024597 | 3.644386611 |
| hsa-miR-6127 | CCTCCCACCCACTC | MIMAT0024610 | 3.329050106 |
| hsa-miR-4442 |
CCTCCCTCTTGTCCG | MIMAT0018960 | 3.290982071 |
| hsa-miR-575 |
GCTCCTGTCCAACTGGCT | MIMAT0003240 | 3.112871166 |
| hsa-miR-4788 |
GCCTCCCTTAGCTGG | MIMAT0019958 | 2.802568783 |
|
hsa-miR-6763-5p | CTCCCCAGCCACTC | MIMAT0027426 | 2.635624935 |
| hsa-miR-5739 |
GCTCCCCATTCTCTCT | MIMAT0023116 | 2.48844758 |
| hsa-miR-6165 | CTCCCCTCACCTCC | MIMAT0024782 | 2.431995318 |
| hsa-miR-7641 |
GCTTAGCTTCCGAGATC | MIMAT0029782 | 2.360662434 |
| hsa-miR-4499 |
TCCCTCCTCTCAGTCT | MIMAT0019035 | 2.276882434 |
|
hsa-miR-4653-3p |
TCTCCAAGCAACCCTT | MIMAT0019719 | 2.249377984 |
| hsa-miR-1275 | GACAGCCTCTCCCC | MIMAT0005929 | 2.194037944 |
|
hsa-miR-7114-5p | ACAGGCACCCCACT | MIMAT0028125 | 2.118378937 |
|
hsa-miR-6826-5p |
AGGTCCCACCTCTTTC | MIMAT0027552 | 2.115317566 |
|
hsa-miR-6749-5p | GCTCCCCCAACCC | MIMAT0027398 | 2.052827758 |
| hsa-miR-197-5p | CCTCCCACTGCCC | MIMAT0022691 | 2.011052693 |
| Table IIDownregulated miRNA expression in
human bronchial epithelial cells following cigarette smoke extract
treatment. |
Table II
Downregulated miRNA expression in
human bronchial epithelial cells following cigarette smoke extract
treatment.
| Systematic
name |
active_sequence |
miRBase_accession_No | Fold change |
|---|
| hsa-miR-185-5p |
TCAGGAACTGCCTTTCT | MIMAT0000455 | 0.494826228 |
|
hsa-miR-200b-5p |
TCCAATGCTGCCCAG | MIMAT0004571 | 0.494208987 |
|
hsa-miR-1304-3p | GGGGTTCGAGGCT | MIMAT0022720 | 0.491609208 |
| hsa-miR-4291 |
AGCTGTTCCTGCTGAA | MIMAT0016922 | 0.486833224 |
|
hsa-miR-3162-3p | TGGGGAGTGGAGGG | MIMAT0019213 | 0.486455419 |
| hsa-miR-10b-5p |
CACAAATTCGGTTCTACAGGG | MIMAT0000254 | 0.480239071 |
|
hsa-miR-6737-3p | CTGGGTAGGGGTGA | MIMAT0027376 | 0.475216109 |
| hsa-miR-8485 |
ATACGTGTGTGTGTGTG | MIMAT0033692 | 0.469232286 |
|
hsa-miR-4649-3p | TGGGGAGAGGCAGG | MIMAT0019712 | 0.46559046 |
| hsa-miR-6132 | TGCAATCCCCAGCC | MIMAT0024616 | 0.447294549 |
| hsa-miR-149-5p |
GGGAGTGAAGACACGGAG | MIMAT0000450 | 0.445735219 |
| hsa-miR-484 |
ATCGGGAGGGGACTGA | MIMAT0002174 | 0.442227863 |
|
hsa-miR-125a-5p |
TCACAGGTTAAAGGGTCTC | MIMAT0000443 | 0.430950242 |
| hsa-miR-503-5p |
CTGCAGAACTGTTCCCGC | MIMAT0002874 | 0.41623705 |
|
hsa-miR-193b-3p |
AGCGGGACTTTGAGGG | MIMAT0002819 | 0.400527954 |
| hsa-miR-7-1-3p |
TATGGCAGACTGTGATTTG | MIMAT0004553 | 0.385026073 |
| hsa-miR-21-3p |
ACAGCCCATCGACTG | MIMAT0004494 | 0.275550498 |
|
hsa-miR-29b-1-5p |
TCTAAACCACCATATGAAACCAG | MIMAT0004514 | 0.154495867 |
| Table IIIUpregulated miRNA expression in human
bronchial epithelial cells following EGCG treatment (EGCG+CSE vs.
CSE). |
Table III
Upregulated miRNA expression in human
bronchial epithelial cells following EGCG treatment (EGCG+CSE vs.
CSE).
| Systematic
name |
active_sequence |
miRBase_accession_No | Fold change |
|---|
|
hsa-miR-1229-5p | CGCTCTCCCCCAA | MIMAT0022942 | 9.622524326 |
| hsa-miR-1246 |
CCTGCTCCAAAAATCC | MIMAT0005898 | 8.609823167 |
| hsa-miR-1260a |
TGGTGGCAGAGGTGG | MIMAT0005911 | 8.014158049 |
| hsa-miR-1260b |
ATGGTGGCAGTGGTG | MIMAT0015041 | 7.831567982 |
| hsa-miR-1290 |
TCCCTGATCCAAAAATCC | MIMAT0005880 | 6.888659175 |
| hsa-miR-1973 |
TATGCTACCTTTGCACG | MIMAT0009448 | 5.991243388 |
| hsa-miR-198 |
GAACCTATCTCCCCTC | MIMAT0000228 | 5.868299699 |
| hsa-miR-3135b | CACCACTGCACTCG | MIMAT0018985 | 5.285732155 |
| hsa-miR-320c |
ACCCTCTCAACCCAG | MIMAT0005793 | 5.078379085 |
| hsa-miR-331-3p |
TTCTAGGATAGGCCCAGGG | MIMAT0000760 | 4.69258682 |
|
hsa-miR-3663-3p | GCGCCCGGCCT | MIMAT0018085 | 4.148186871 |
|
hsa-miR-3679-5p | TCCCCTTCCCTGCC | MIMAT0018104 | 3.898141597 |
| hsa-miR-4298 | CTGCCTCCTCCTCC | MIMAT0016852 | 3.563938448 |
| hsa-miR-4459 | CTCCACCTCCTCCG | MIMAT0018981 | 3.006691394 |
| hsa-miR-4466 | CCCCGCCGGCC | MIMAT0018993 | 2.973974082 |
|
hsa-miR-4485-3p |
TTAGGGTACCGCGGC | MIMAT0019019 | 2.965321858 |
|
hsa-miR-4485-5p | TCACTGGGCAGGCG | MIMAT0032116 | 2.89697668 |
| hsa-miR-4497 | GCCCAGCCGTCC | MIMAT0019032 | 2.842534905 |
| hsa-miR-4530 | CGCTCCCGTCCTG | MIMAT0019069 | 2.833925499 |
|
hsa-miR-4685-5p |
AACCTTGCCCCACTC | MIMAT0019771 | 2.779881775 |
| hsa-miR-4698 |
TGGGGTCTTCCTCTAC | MIMAT0019793 | 2.628778688 |
| hsa-miR-4741 | AGCCGACCCCTCC | MIMAT0019871 | 2.502211839 |
|
hsa-miR-4793-5p | CCTCTGCCCTGTGG | MIMAT0019965 | 2.495999429 |
|
hsa-miR-4800-5p |
TCCTTCCTTCCTCGG | MIMAT0019978 | 2.410802371 |
| hsa-miR-483-5p |
CTCCCTTCTTTCCTC | MIMAT0004761 | 2.397856092 |
| hsa-miR-5100 | AGAGGCACCGCTGG | MIMAT0022259 | 2.392975383 |
|
hsa-miR-5585-3p |
ACCTGTAGTCCCAGCT | MIMAT0022286 | 2.309263021 |
| hsa-miR-5787 | ACCTCCCCGCGC | MIMAT0023252 | 2.286325308 |
| hsa-miR-6085 | TGTGCTCCCCCAGC | MIMAT0023710 | 2.23497638 |
|
hsa-miR-6510-5p |
GACTCCTCTCTCTCCC | MIMAT0025476 | 2.217240609 |
|
hsa-miR-6785-5p |
CACCATCATCCACGC | MIMAT0027470 | 2.211828087 |
|
hsa-miR-6821-5p | CCCCGCCTCGAG | MIMAT0027542 | 2.177540381 |
|
hsa-miR-6867-5p |
TCCCTTCTTCCTCTACA | MIMAT0027634 | 2.170855211 |
|
hsa-miR-6891-5p | CCCCTCATCCCCC | MIMAT0027682 | 2.139695303 |
|
hsa-miR-7107-5p | CCCTTCCTCCTCCC | MIMAT0028111 | 2.084862808 |
|
hsa-miR-7108-5p | CCACCCGCCTGC | MIMAT0028113 | 2.025528729 |
| Table IVmiRNA expression of HBE cells
downregulated by EGCG treatment (EGCG+CSE vs. CSE). |
Table IV
miRNA expression of HBE cells
downregulated by EGCG treatment (EGCG+CSE vs. CSE).
| Systematic
name |
active_sequence |
miRBase_accession_No | Fold change |
|---|
| hsa-miR-7110-5p
C | TCTCTCTCCCCACA | MIMAT0028117 | 0.491007589 |
|
hsa-miR-7114-5p | ACAGGCACCCCACT | MIMAT0028125 | 0.489623515 |
| hsa-miR-7150 |
TACCTCTCCCCCTGC | MIMAT0028211 | 0.486650133 |
| hsa-miR-762 | GCTCGGCCCCGG | MIMAT0010313 | 0.481613982 |
|
hsa-miR-7847-3p | GCCTCCTCCTCGTC | MIMAT0030422 | 0.424841654 |
| hsa-miR-8063 | AAGCCCCGACTCCT | MIMAT0030990 | 0.383320641 |
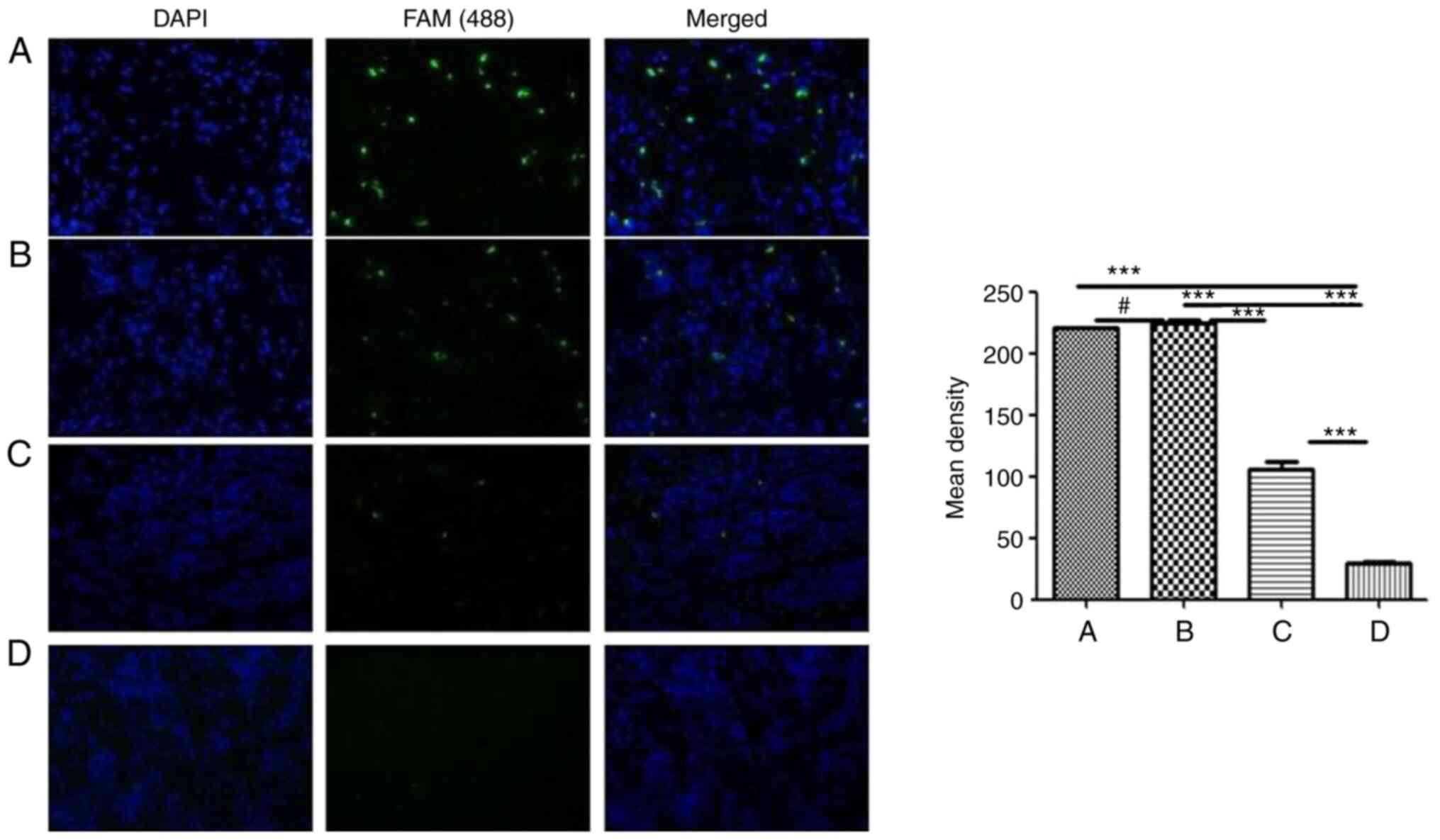
These differentially expressed miRNAs were initially
screened in HBE cells treated with EGCG and CSE. Non-small cell
lung cancer, para-cancer, atypical hyperplasia and inflammatory
tissues were collected and evaluated. miRNA levels were assessed by
fluorescence in situ hybridization. This was performed to
understand the clinical significance of the miRNA expression
changes observed in the CSE-treated HBE cells. For example,
miRNA-7114-5p was highly expressed in non-small cell lung cancer
and atypical hyperplasia, with low expression in para-carcinoma and
inflammatory tissues (Fig. 3).
The difference was statistically significant (P<0.05).
Expression levels of TGF-β, CYP1A1 and
NF-κB in non-small cell lung carcinoma tissues
Based on the original data, the functional miRNAs
regulated by EGCG were screened using functional acquisition/loss
analysis. A potential miRNA target gene had to be predicted by at
least two predictive software (47-50). Targeted signaling pathways were
predicted by the signal pathway analysis software 'Ingenuity
Pathway Analysis' (http://www.ingenuity.com) (51,52). The bioinformatic analysis
revealed that the CSE-mediated damage response of HBE cells
involved multiple signaling pathways. The 'TGF-β signaling
pathway', 'NOD-like receptor signaling pathway', 'cytochrome P450
pathway', 'p53 signaling pathway' and 'ErbB signaling pathway' were
all targeted. The results of signaling pathway prediction were
compared between the EGCG-treated group and the non-EGCG treated
groups (Fig. S1). The
protective effect of EGCG against CSE-induced damage was mediated
by the NF-κB, TGF-β, p53, Bcl-2 and CYP signaling pathways.
Conversely, in clinical pathological samples, TGF-β and CYP1A1 were
revealed to be overexpressed in non-small cell lung carcinoma
tissue and in atypical hyperplasia tissue, with low expression in
para-lung carcinoma and chronic inflammatory lung tissue. NF-κB, on
the other hand, had low expression levels in non-small cell lung
carcinoma and atypical hyperplasia tissue, while being
overexpressed in para-lung carcinoma and chronic inflammatory lung
tissue (Fig. 4).
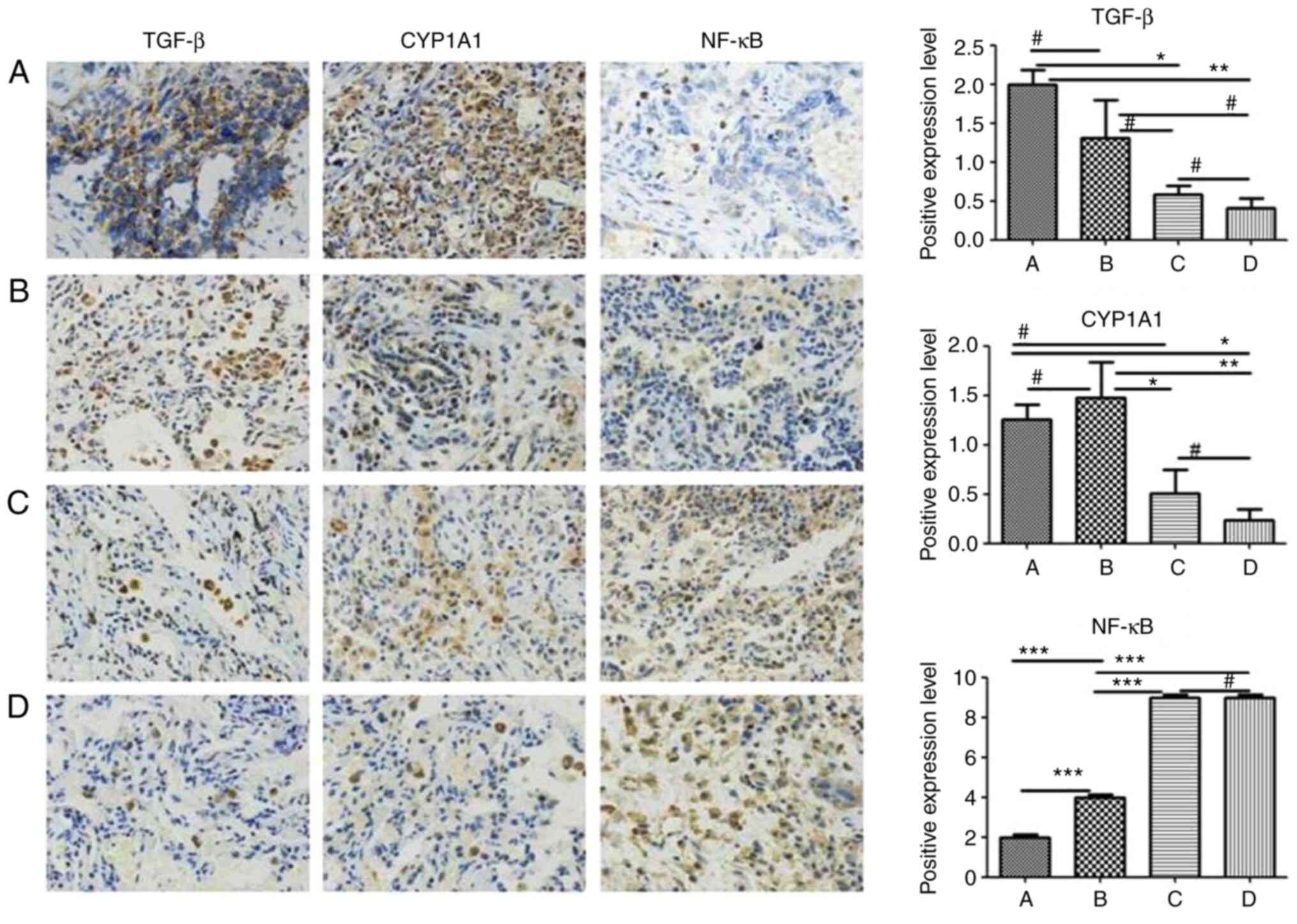 | Figure 4Expression of TGF-β, CYP1A1 and NF-κB
in non-small cell lung carcinoma tissues. The samples were detected
by immunohistochemistry and stained by DAB at a magnification of
×100. The expression of TGF-β and CYP1A1 in non-small cell lung
carcinoma and lung atypical hyperplasia tissues was significantly
higher than in para-carcinoma and chronic inflammatory tissues.
Conversely, the expression of NF-κB in non-small cell carcinoma and
in lung atypical hyperplasia tissues was significantly lower than
in para-carcinoma and chronic inflammatory tissues. (A) In
non-small cell lung carcinoma tissue, TGF-β and CYP1A1 proteins
were highly expressed, with NF-κB expressed at a low level. (B) In
lung atypical hyperplasia tissue, TGF-βand CYP1A1 proteins were
strongly expressed but NF-κB was weakly expressed. (C) In para-lung
carcinoma tissues, TGF-β and CYP1A1 proteins were expressed at low
levels, with NF-κB protein expressed at a high level. (D) In
chronic inflammatory lung tissue, TGF-β and CYP1A1 proteins were
weakly expressed, while NF-κB was strongly expressed.
#P>0.05, *P<0.05,
**P<0.01 and ***P<0.001. TGF,
transforming growth factor; NF-κB, nuclear factor-κB. |
Bronchial epithelium lesions induced by
CSE in rats and protection of EGCG
As in the previous experiment, the amount of water
the rats drank varied slightly across the different experimental
stages. However, the water consumed by the two groups of rats was
roughly equal, with the water consumed by each rat averaging
between 20 and 30 ml. Other conditions, such as food consumption,
body weight and activity levels of the rats, were not significantly
different between the EGCG-treated group and the EGCG-non-treated
group. In the animal experiment, the rat lungs were stained with
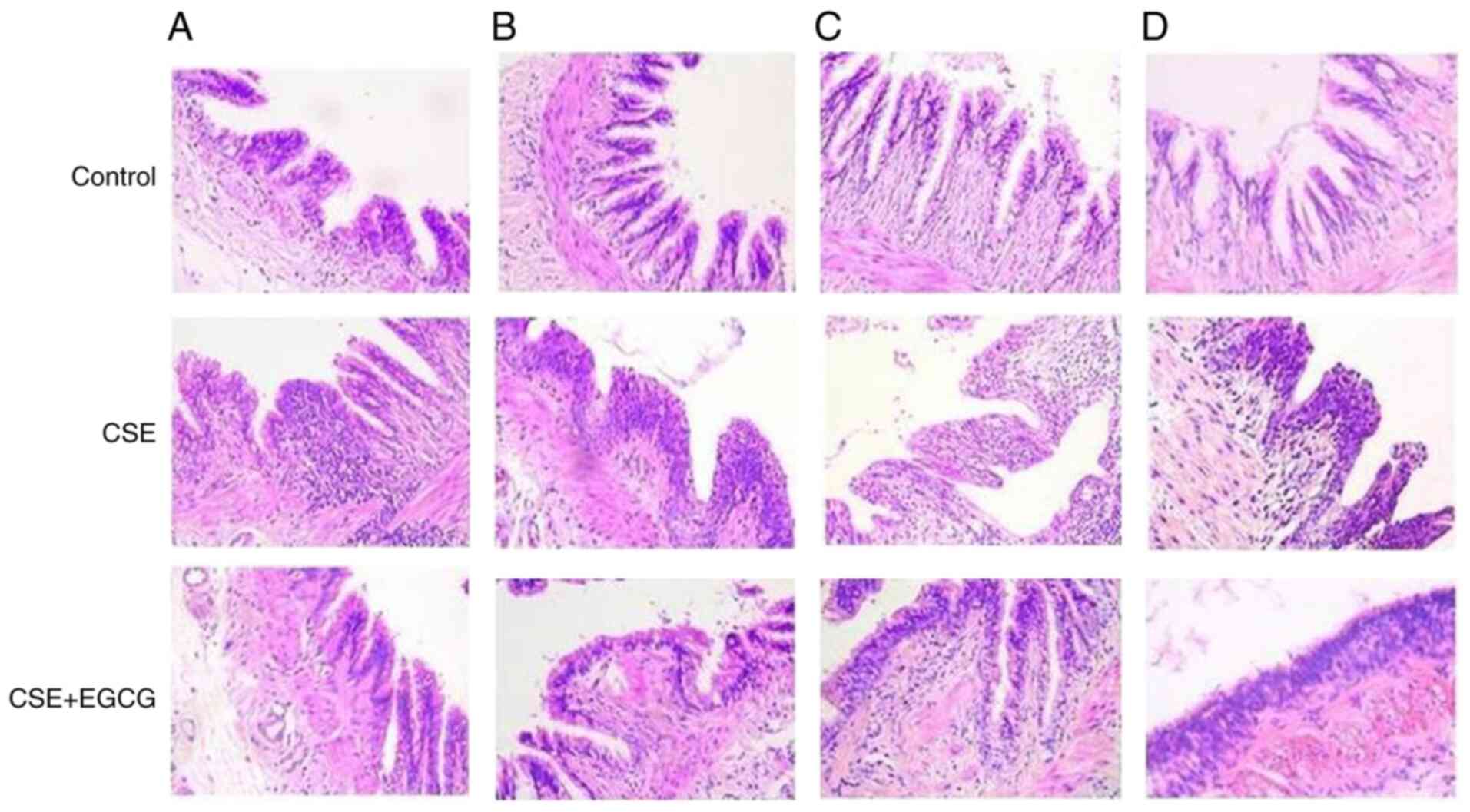
H&E before being assessed by two independent pathologists.
After 4 weeks of passive smoke inhalation to the end of the
experiment, chronic pulmonary inflammation and BE hyperplasia was
evident. EGCG treatment significantly alleviated these CSE-induced
lesions. There was no evidence of disease in the lungs of the
control group (Fig. 5).
Effect of EGCG on CSE-induced
benzopyrene-DNA adduct formation in rat lungs
This experiment used immunofluorescence
histochemistry to detect pulmonary benzopyrene-DNA adducts induced
by CSE. After four weeks, there was fluorescence; this indicated
the presence of benzopyrene-DNA adducts in the lung tissue of rats
treated with CSE. The level of fluorescence increased gradually
until the experimental endpoint. This demonstrated an increase in
the number of benzo pyrene-DNA adducts in the lung tissue and BE
cells of CSE-treated rats. Rats treated with CSE+EGCG revealed
markedly weaker staining, suggesting they developed significantly
fewer benzopyrene-DNA adducts compared with the CSE only treatment
group. The control group developed no benzopyrene-DNA adducts
across the full experiment (Fig.
6).
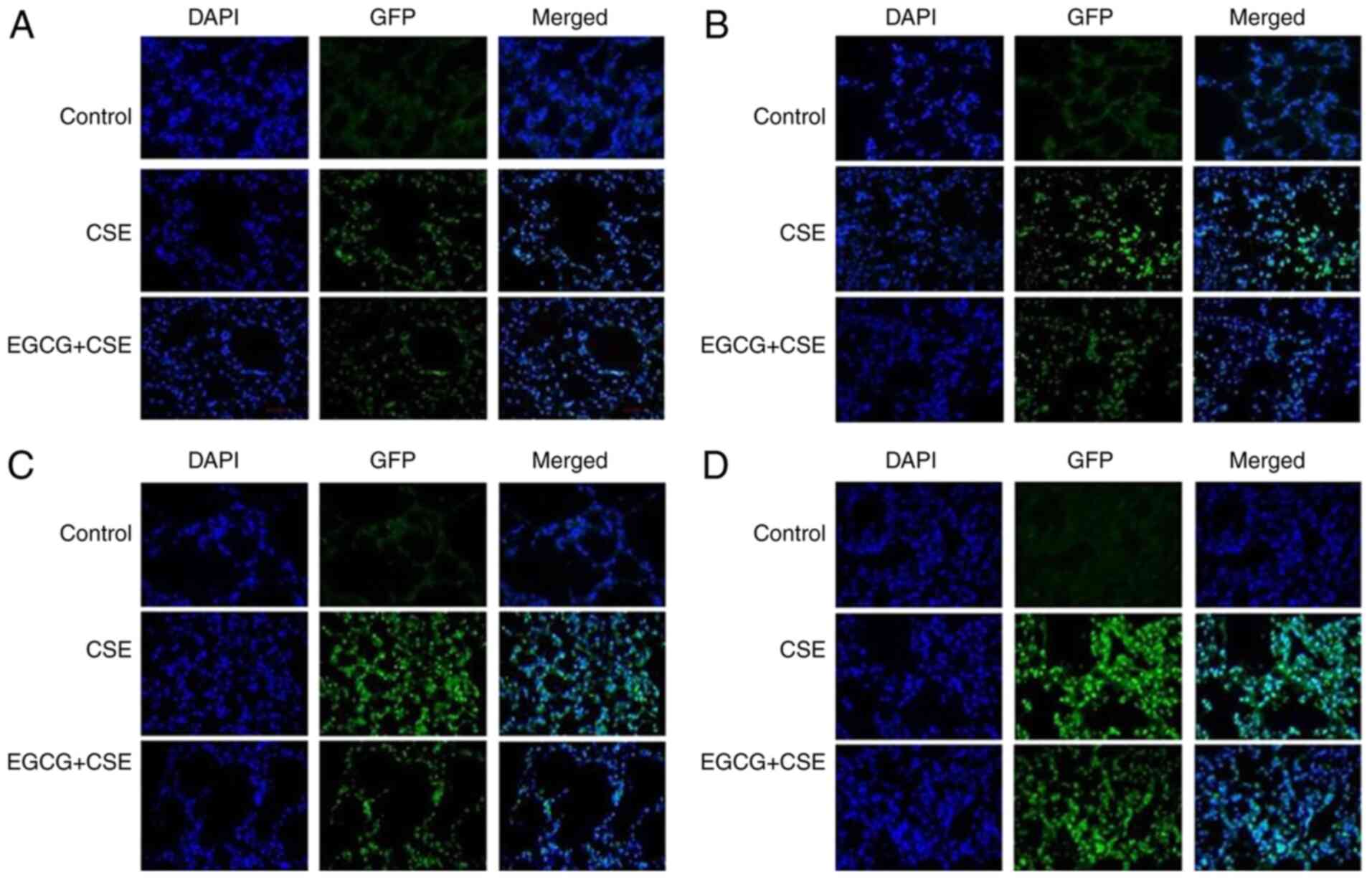 | Figure 6Pulmonary benzopyrene-DNA adducts in
rats treated with CSE and EGCG+CSE, detected by immunofluorescence
histochemistry. (A) After four weeks, there was weak fluorescence
in the lungs of rats exposed to CSE, indicating the formation of
benzopyrene DNA-adducts. The fluorescence levels were lower in the
CSE+EGCG treatment group, while there was no fluorescence in the
control group. (B) After eight weeks of CSE treatment, the level of
fluorescence increased in the CSE only group, indicating an
increase in the number of benzopyrene DNA-adducts. There was
markedly lower fluorescence in the CSE+EGCG group. No aberrancies
were observed in the control group. (C) After 12 weeks,
fluorescence levels increased in the CSE only group, while EGCG
intervention markedly reduced the formation of the DNA-adducts. The
control group exhibited no fluorescence. (D) After 16 weeks, the
fluorescence levels had further increased in the CSE only group
lungs. It was markedly lower in the CSE+EGCG group. The control
group remained normal. CSE, cigarette smoke extract; EGCG,
epigallocatechin-3-gallate. |
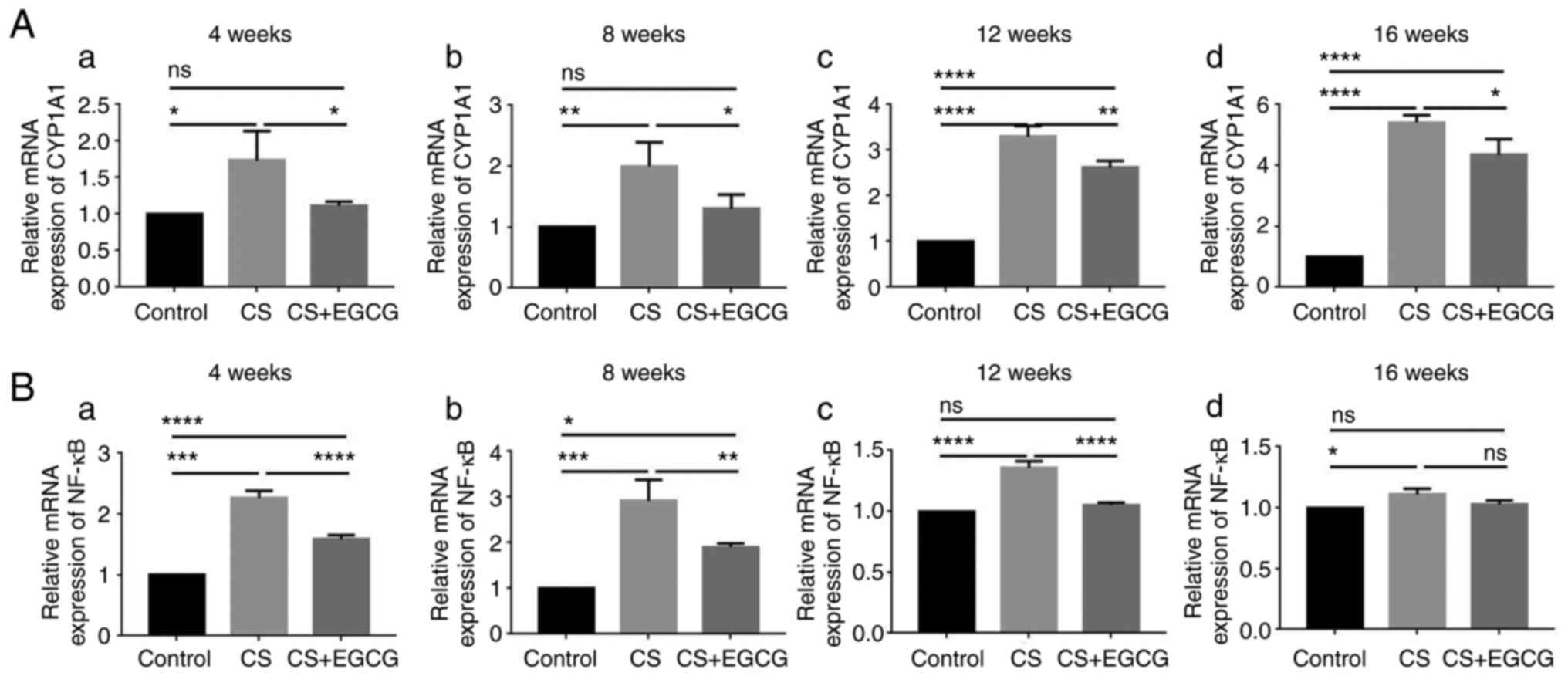
Effects of EGCG on CYP1A1 and NF-κB mRNA
expression induced by CSE in rat lung tissues
The mRNA expression levels of the target genes
CYP1A1 and NF-κB were detected by RT-qPCR. CYP1A1 mRNA expression
in the lung tissue of rats exposed to CS began to increase after
four weeks of exposure. There was a gradual increase in the
expression of mRNA across the full experiment, with the highest
expression levels after 16 weeks of CS exposure. The mRNA level of
CYP1A1was significantly lower in the EGCG-treated group across all
time-points, when compared with the smoking only group (Fig. 7) (P<0.05). The NF-κB
expression levels, following CS exposure, differed from that of
CYP1A1. The level of NF-κB mRNA expression was increased after the
fourth week. NF-κB expression was highest after 8 weeks of CS
exposure. Following this, the expression level decreased. This
pattern was also mirrored in the rats exposed to CS and treated
with EGCG. At four and eight weeks, EGCG intervention led to
significantly lower NF-κB expression compared with rats exposed to
CS only (Fig. 7)
(P<0.01).
Effects of EGCG on CYP1A1, NF-κB protein
expression induced by CSE in rat lung tissues
Western blotting was used to detect the protein
expression of CYP1A1 and NF-κB. Both CYPA1 and NF-κB proteins were
at low levels in untreated rat lung tissue. After four weeks of
treatment, CYP1A1 and NF-κB protein expression levels were
increased in lung tissue treated with CS. Over the 16-week
observation period, CYP1A1 protein expression levels increased in
the lungs of rats treated with CS. Unlike CYP1A1, NF-κB protein
expression in the lung tissues of rats treated with CS started to
decrease after twelve weeks. Over the course of 16 weeks, EGCG
downregulated the overexpression of CYP1A1 and NF-κB proteins in
lung tissues of rats treated with CS (Fig. 8).
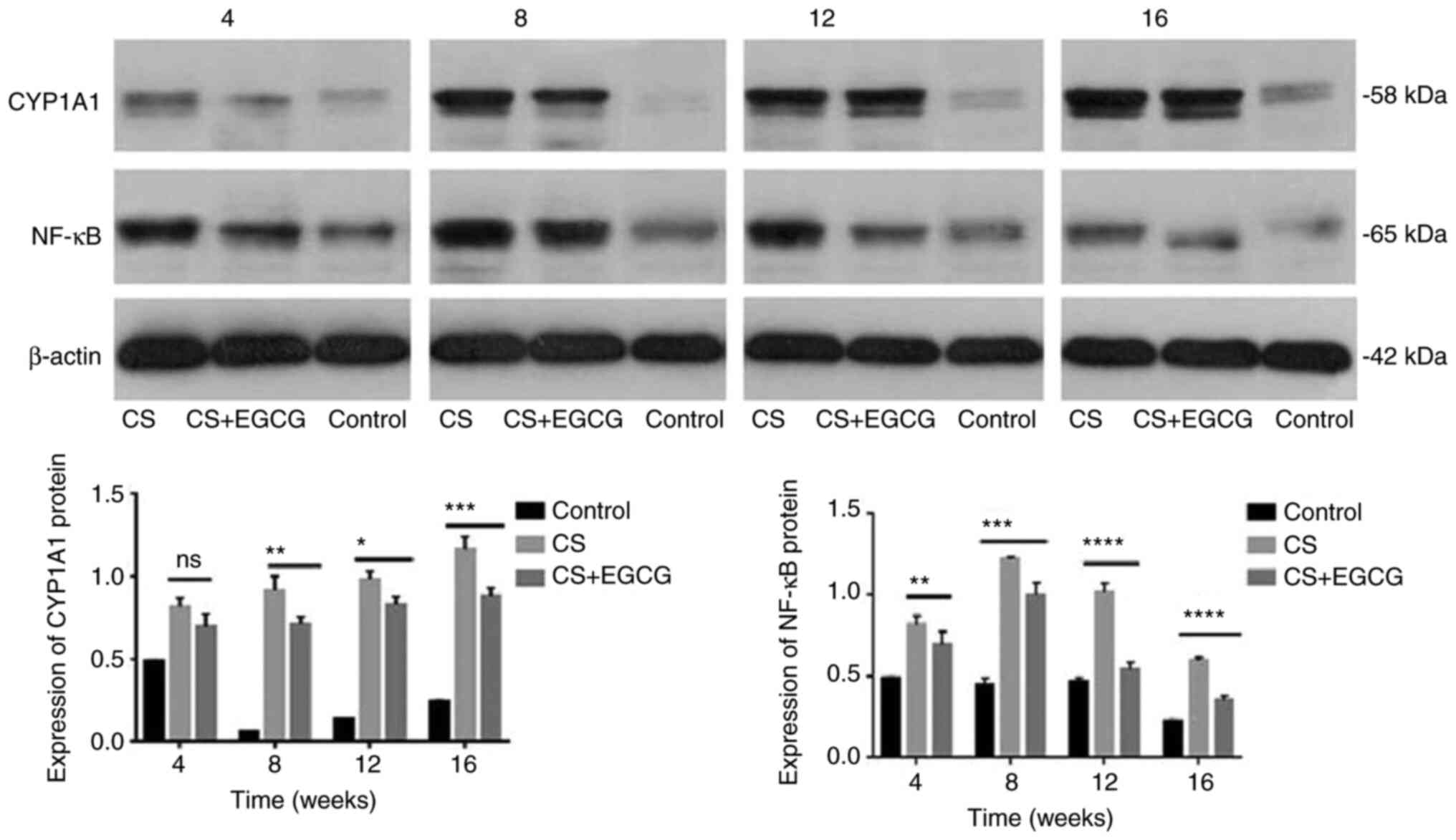 | Figure 8Protein expression of target genes
CYP1A1 and NF-κB in rat lung tissues detected by western blotting.
In the control group, the lung tissue of the rats revealed low
expression of CYP1A1. After 4 weeks of CS, the expression of CYP1A1
protein in rat lung tissues increased. From week 8 to week 16,
there was significant overexpression of CYP1A1 protein in the rat
lungs exposed to CS. EGCG treatment significantly downregulated the
overexpression of CYP1A1 protein in rat lung tissues induced by CS.
NF-κB was weakly expressed in the lung tissue of control rats.
Between weeks 4 and 12, CS increased the protein expression of
NF-κB in rat lung tissue, while EGCG administration downregulated
this expression. However, NF-κB overexpression then decreased until
the experimental endpoint in both treatment groups.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. NF-κB,
nuclear factor-κB; EGCG, epigallocatechin-3-gallate; CS, cigarette
smoke. |
Discussion
EGCG inhibits benzopyrene-DNA adducts and
alleviates smoking-induced precancerous lesions in bronchial
epithelium
Lung cancer is a chronic disease that does not
develop immediately upon exposure to a carcinogen (53,54), with chronic exposure to CS
significantly increasing the risk of developing disease. It may
take a long time between the initial carcinogen exposure to the
onset of lung cancer (6).
Tobacco contains carcinogenic substances such as benzopyrene. After
smoking, benzopyrene binds to the DNA of epithelial cells and forms
DNA adducts; this is considered to be a key step in the initiation
of lung cancer (55,56). If the accumulation of
benzopyrene-DNA adducts induced by cigarette smoking can be
prevented, it may be possible to reduce the carcinogenic effect of
benzopyrene. In the present study, there was significant formation
of benzopyrene-DNA adducts in human BE cells treated with CSE.
These results indicated the presence of active benzopyrene in CS.
Smoking increases the risk of cancer developing in the epithelial
cells of the trachea (57).
However, the present study has revealed that epithelial cells
exposed to CS in combination with EGCG, had a significant reduction
in the amount of benzopyrene-DNA adducts formed. These results
provided direct evidence that the green tea extract is protective
against the formation of potentially cancerous adducts. In further
in vivo experiments, it was observed that significant
bronchial inflammation developed in rats after 4 weeks of CS
inhalation, which was accompanied by the detection of
benzopyrene-DNA adducts. This inflammation led to the development
of BE atypical hyperplasia and heteroplasms in these rats.
Importantly, these precancerous lesions were significantly reduced
by the consumption of water containing 0.3% EGCG. Thus, it could be
concluded that the green tea extract EGCG is protective against
precancerous lesions and that EGCG interrupts a series of processes
in the development of lung cancer, preventing carcinogenesis by
mediating the damage caused by carcinogens found in CS.
Effects of EGCG on the expression of
CYP1A1 in the prevention of smoke-induced bronchial epithelial
lesions
As above-mentioned, lung cancer is a chronic
disease, with the malignant transformation of BE cells being a slow
process. There are numerous genes (such as NLRP3, Nrf2, IL-1β,
caspase-1 and K-ras) involved in this transformation (58-60). It has been proposed that EGCG may
interact in a protective manner with these numerous genes (61). However, the number of genes which
play a key role in BE cell carcinogenesis may be few. Therefore, it
was crucial to identify these genes and whether they may be
modulated by the green tea extract EGCG. For the most part,
aberrant gene expression was related to changes caused by the
developing malignancy and not the carcinogenesis itself. Moreover,
the expression of various genes and the activation of signaling
pathways are regulated by miRNAs. Molecular changes, such as the
dysregulation of miRNA expression, have been linked to tobacco
smoking in lung cancer (35). In
the present study, in human epithelial cells, it was observed that
several miRNAs had upregulated expression following CSE treatment,
with multiple miRNAs also being downregulated. Not only did the use
of EGCG as a therapeutic inhibit the upregulation of potentially
cancerous miRNAs following CS exposure, it also upregulated novel
miRNAs that may play a protective role. Not all of the target genes
of these miRNAs are related to carcinogenesis, however there are
several genes which have been previously linked to the development
of lung cancer (5,10,11,13,21,27). TGF-β, CYP1A1 and NF-κB are all
targets genes of these miRNAs; their expression may also be
affected by the intervention of EGCG. TGF-β and CYP1A1 were
overexpressed in lung cancer tissues, while NF-κB was weakly
expressed in atypical hyperplasia tissue and highly expressed in
inflammatory tissues. It was indicated that the aberrant expression
of TGF-β and CYP1A1 was closely related to smoking-induced lung
cancer, while the aberrant expression of NF-κB may be more related
to smoking-induced inflammation.
In the present study, rats exposed to CS had an
initial bronchial/lung inflammatory response, followed by atypical
proliferation and dysplasia of the bronchial epithelium. In the
early inflammatory response to smoking, the expression of CYP1A1
and NF-κB was significantly upregulated; EGCG treatment
significantly inhibited the overexpression of CYP1A1 and NF-κB. In
the later experimental stages, the expression of NF-κB in the lung
tissue from rats in the experimental group was not significantly
different to that of the tissue identified in the control. This
result further supported the hypothesis that NF-κB expression may
be more correlated with the initial inflammation of the lung,
rather than the development of tumors. In the CS treatment
experiment, the expression of CYP1A1 in the lung tissue of the
smoke-exposed rats was high, gradually increasing throughout the
experiment. EGCG treatment consistently inhibited the
overexpression of CYP1A1 induced by smoking in rat lung tissues.
These results indicated that the mechanism of smoking-induced BE
carcinogenesis was most probably related to the aberrant expression
of CYP1A1; EGCG may therefore block smoking-induced BE
carcinogenesis by regulating the function of CYP1A1. Compared with
CYP1A1, the aberrant expression of NF-κB may be more closely
associated with inflammation rather than with the carcinogenesis of
BE cells.
CYP1A1 encodes the cytochrome P450 1A1. This protein
is a major enzyme that activates polycyclic aromatic (pahs)
carcinogens. Activated CYP1A1 can turn organic substances such as
polyaromatic hydrocarbons into cytotoxins and other carcinogens,
increasing the risk of cancer (62). Tobacco contains a large amount of
benzopyrene, which is inhaled into the lungs when smoking.
Benzopyrene is firstly epoxidized by CYP1A1, which is then
hydrolyzed by epoxide hydrolase to form a dihydroxyl compound. This
is then oxidized by CYP1A1 to form a diol epoxide. Diol epoxides
have significant carcinogenic and mutagenic effects. These reactive
metabolites produce DNA adducts, resulting in DNA mutations, gene
expression profile alterations and tumorigenesis (62). Inhibition of CYP1A1-catalyzed
benzopyrene metabolism may reduce the risk of smoking-induced lung
cancer. In this in vivo experiment, it was observed that
EGCG could inhibit the overexpression of CYP1A1 induced by smoking
exposure. This therefore inhibits the metabolic mechanism of DNA
adducts involving CYP1A1; subsequently preventing the development
of atypical hyperplasia and heterogeneous precancerous lesions of
bronchial epithelium. In addition to smoking, cooking, air
pollution, automobile exhaust fumes and numerous other
environmental sources may produce benzopyrene (62-65). There are still numerous
opportunities for people to be exposed to environmental
benzopyrene. Drinking green tea is therefore recommended as it is
rich in EGCG which will inhibit the development of disease
associated with benzopyrene exposure.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
available from the corresponding author on reasonable request.
Authors' contributions
All of the authors made a significant contribution
to the conception, study design, execution, acquisition of data,
analysis. QG, NC and FC confirmed the authenticity of all the raw
data. Moreover, QG presided over the project design, experiment
execution, data acquisition and analysis, as well as paper writing
and revision. FC was in charge of the in vivo experiment. NC
performed the in vitro experiment and bioinformatics
analysis. JW was responsible for the acquisition and analysis of
histopathological data. ZL and XD participated in the data
acquisition, sorting and analysis. All of the authors provided
final approval of the version to be published and agree to be
accountable for all aspects of the work.
Ethics approval and consent to
participate
This research protocol was approved by the Medical
Ethics Committee of Xiangya Hospital of Central South University
(approval no. 201703133) and all patients provided written informed
consent before surgery. All of the animals were treated humanely
and in compliance with the Animal Welfare Act of America. The
experiment was approved by the Ethics Department of Xiangya
Hospital, Central South University (Changsha, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the staff of the
Laboratory of Respiratory Medicine and the Department of Pathology
at Xiangya Hospital affiliated to the Central South University, for
their support.
Abbreviations:
|
BE
|
bronchial epithelial
|
|
EGCG
|
epigallocatechin-3-gallate
|
|
HBE
|
human bronchial epithelial
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
CSE
|
cigarette smoke extract
|
References
|
1
|
Tu CY, Cheng FJ, Chen CM, Wang SL, Hsiao
YC, Chen CH, Hsia TC, He YH, Wang BW, Hsieh IS, et al: Cigarette
smoke enhances oncogene addiction to c-MET and desensitizes
EGFR-expressing non-small cell lung cancer to EGFR TKIs. Mol Oncol.
12:705–723. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raja R, Sahasrabuddhe NA, Radhakrishnan A,
Syed N, Solanki HS, Puttamallesh VN, Balaji SA, Nanjappa V, Datta
KK, Babu N, et al: Chronic exposure to cigarette smoke leads to
activation of p21 (RAC1)-activated kinase 6 (PAK6) in non-small
cell lung cancer cells. Oncotarget. 7:61229–61245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thai P, Statt S, Chen CH, Liang E,
Campbell C and Wu R: Characterization of a novel long noncoding
RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer
cell lines. Am J Respir Cell Mol Biol. 49:204–211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Billatos E, Faiz A, Gesthalter Y, LeClerc
A, Alekseyev YO, Xiao X, Liu G, Ten Hacken NHT, Heijink IH, Timens
W, et al: Impact of acute exposure to cigarette smoke on airway
gene expression. Physiol Genomics. 50:705–713. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gu Q, Hu C, Chen N and Qu J: A comparison
between lung carcinoma and a subcutaneous malignant tumor induced
in rats by a 3,4-benzopyrene injection. Int J Clin Exp Pathol.
11:3934–3942. 2018.PubMed/NCBI
|
|
6
|
Sá VK, Rocha TP, Moreira A, Soares FA,
Takagaki T, Carvalho L, Nicholson AG and Capelozzi VL:
Hyaluronidases and hyaluronan synthases expression is inversely
correlated with malignancy in lung/bronchial pre-neoplastic and
neoplastic lesions, affecting prognosis. Braz J Med Biol Res.
48:1039–1047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rushing BR and Selim MI: Aflatoxin B1: A
review on metabolism, toxicity, occurrence in food, occupational
exposure, and detoxification methods. Food Chem Toxicol.
124:81–100. 2019. View Article : Google Scholar
|
|
8
|
Gavish M, Cohen S and Nagler R: Cigarette
smoke effects on TSPO and VDAC expression in a cellular lung cancer
model. Eur J Cancer Prev. 25:361–367. 2016. View Article : Google Scholar
|
|
9
|
Nagler R, Cohen S and Gavish M: The effect
of cigarette smoke on the translocator protein (TSPO) in cultured
lung cancer cells. J Cell Biochem. 116:2786–2792. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng X, Meng C, Yang B, Zhao L, Sun X, Su
Y, Liu H, Fan F, Liu X and Jia L: AP-2α downregulation by cigarette
smoke condensate is counteracted by p53 in human lung cancer cells.
Int J Mol Med. 34:1094–1100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Filosto S, Becker CR and Goldkorn T:
Cigarette smoke induces aberrant EGF receptor activation that
mediates lung cancer development and resistance to tyrosine kinase
inhibitors. Mol Cancer Ther. 11:795–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Faiz A, Heijink IH, Vermeulen CJ, Guryev
V, van den Berge M, Nawijn MC and Pouwels SD: Cigarette smoke
exposure decreases CFLAR expression in the bronchial epithelium,
augmenting susceptibility for lung epithelial cell death and DAMP
release. Sci Rep. 8:124262018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu Q, Hu C, Chen Q and Xia Y: Tea
polyphenols prevent lung from preneoplastic lesions and effect p53
and bcl-2 gene expression in rat lung tissues. Int J Clin Exp
Pathol. 6:1523–1531. 2013.PubMed/NCBI
|
|
14
|
Sundar IK and Rahman I: Gene expression
profiling of epigenetic chromatin modification enzymes and histone
marks by cigarette smoke: Implications for COPD and lung cancer. Am
J Physiol Lung Cell Mol Physiol. 311:L1245–L1258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hayakawa S, Ohishi T, Miyoshi N, Oishi Y,
Nakamura Y and Isemura M: Anti-cancer effects of green tea
epigallocatchin-3-gallate and coffee chlorogenic acid. Molecules.
25:45532020. View Article : Google Scholar :
|
|
16
|
Singh BN, Shankar S and Srivastava RK:
Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms,
perspectives and clinical applications. Biochem Pharmacol.
82:1807–1821. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Chen W, Tu G, Chen X, Lu Y, Wu L
and Zheng D: Enhanced chemotherapeutic efficacy of
PLGA-encapsulated epigallocatechin gallate (EGCG) against human
lung cancer. Int J Nanomedicine. 15:4417–4429. 2020.PubMed/NCBI
|
|
18
|
Tang N, Wu Y, Zhou B, Wang B and Yu R:
Green tea, black tea consumption and risk of lung cancer: A
meta-analysis. Lung Cancer. 65:274–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fritz H, Seely D, Kennedy DA, Fernandes R,
Cooley K and Fergusson D: Green tea and lung cancer: A systematic
review. Integr Cancer Ther. 12:7–24. 2013. View Article : Google Scholar
|
|
20
|
Lu Y, Yao R, Yan Y, Wang Y, Hara Y, Lubet
RA and You M: A gene expression signature that can predict green
tea exposure and chemopreventive efficacy of lung cancer in mice.
Cancer Res. 66:1956–1963. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu Q, Hu C, Chen Q, Xia Y, Feng J and Yang
H: Development of a rat model by 3,4-benzopyrene intra-pulmonary
injection and evaluation of the effect of green tea drinking on p53
and bcl-2 expression in lung carcinoma. Cancer Detect Prev.
32:444–451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou H, Chen JX, Yang CS, Yang MQ, Deng Y
and Wang H: Gene regulation mediated by microRNAs in response to
green tea polyphenol EGCG in mouse lung cancer. BMC Genomics.
15(Suppl 11): S32014. View Article : Google Scholar
|
|
23
|
Huang J, Chen S, Shi Y, Li CH, Wang XJ, Li
FJ, Wang CH, Meng QH, Zhong JN, Liu M and Wang ZM: Epigallocatechin
gallate from green tea exhibits potent anticancer effects in A-549
non-small lung cancer cells by inducing apoptosis, cell cycle
arrest and inhibition of cell migration. J BUON. 22:1422–1427.
2017.
|
|
24
|
Oya Y, Mondal A, Rawangkan A, Umsumarng S,
Iida K, Watanabe T, Kanno M, Suzuki K, Li Z, Kagechika H, et al:
Down-regulation of histone deacetylase 4, -5 and -6 as a mechanism
of synergistic enhancement of apoptosis in human lung cancer cells
treated with the combination of a synthetic retinoid, Am80 and
green tea catechin. J Nutr Biochem. 42:7–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Milligan SA, Burke P, Coleman DT, Bigelow
RL, Steffan JJ, Carroll JL, Williams BJ and Cardelli JA: The green
tea polyphenol EGCG potentiates the antiproliferative activity of
c-Met and epidermal growth factor receptor inhibitors in non-small
cell lung cancer cells. Clin Cancer Res. 15:4885–4894. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li M, Li JJ, Gu QH, An J, Cao LM, Yang HP
and Hu CP: EGCG induces lung cancer A549 cell apoptosis by
regulating Ku70 acetylation. Oncol Rep. 35:2339–2347. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L, Xie J, Gan R, Wu Z, Luo H, Chen
X, Lu Y, Wu L and Zheng D: Synergistic inhibition of lung cancer
cells by EGCG and NF-κB inhibitor BAY11-7082. J Cancer.
10:6543–6556. 2019. View Article : Google Scholar :
|
|
28
|
Jiang P, Xu C, Zhang P, Ren J, Mageed F,
Wu X, Chen L, Zeb F, Feng Q and Li S: Epigallocatechin-3-gallate
inhibits self-renewal ability of lung cancer stem-like cells
through inhibition of CLOCK. Int J Mol Med. 46:2216–2224. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deng YT and Lin JK: EGCG inhibits the
invasion of highly invasive CL1-5 lung cancer cells through
suppressing MMP-2 expression via JNK signaling and induces G2/M
arrest. J Agric Food Chem. 59:13318–13327. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gu JJ, Qiao KS, Sun P, Chen P and Li Q:
Study of EGCG induced apoptosis in lung cancer cells by inhibiting
PI3K/Akt signaling pathway. Eur Rev Med Pharmacol Sci.
22:4557–4563. 2018.PubMed/NCBI
|
|
31
|
Yu C, Jiao Y, Xue J, Zhang Q, Yang H, Xing
L, Chen G, Wu J, Zhang S, Zhu W and Cao J: Metformin sensitizes
non-small cell lung cancer cells to an epigallocatechin-3-gallate
(EGCG) treatment by suppressing the Nrf2/HO-1 signaling pathway.
Int J Biol Sci. 13:1560–1569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu J, Jiang Y, Yang X, Wang S, Xie C, Li
X, Li Y, Chen Y, Wang X, Meng Y, et al: Wnt/β-catenin pathway
mediates (-)-Epigallocatechin-3-gallate (EGCG) inhibition of lung
cancer stem cells. Biochem Biophys Res Commun. 482:15–21. 2017.
View Article : Google Scholar
|
|
33
|
Wei R, Wirkus J, Yang Z, Machuca J,
Esparza Y and Mackenzie GG: EGCG sensitizes
chemotherapeutic-induced cytotoxicity by targeting the ERK pathway
in multiple cancer cell lines. Arch Biochem Biophys.
692:1085462020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen A, Jiang P, Zeb F, Wu X, Xu C, Chen L
and Feng Q: EGCG regulates CTR1 expression through its
pro-oxidative property in non-small-cell lung cancer cells. J Cell
Physiol. 235:7970–7981. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Doukas SG, Vageli DP, Lazopoulos G,
Spandidos DA, Sasaki CT and Tsatsakis A: The effect of NNK, a
tobacco smoke carcinogen, on the miRNA and mismatch dna repair
expression profiles in lung and head and neck squamous cancer
cells. Cells. 9:10312020. View Article : Google Scholar :
|
|
36
|
Hu DL, Wang G, Yu J, Zhang LH, Huang YF,
Wang D and Zhou HH: Epigallocatechin-3-gallate modulates long
non-coding RNA and mRNA expression profiles in lung cancer cells.
Mol Med Rep. 19:1509–1520. 2019.PubMed/NCBI
|
|
37
|
Bhardwaj V and Mandal AKA: Next-generation
sequencing reveals the role of epigallocatechin-3-gallate in
regulating putative novel and known microRNAs which target the MAPK
pathway in non-small-cell lung cancer a549 cells. Molecules.
24:3682019. View Article : Google Scholar :
|
|
38
|
Chen Y, Pan Y, Ji Y, Sheng L and Du X:
Network analysis of differentially expressed smoking-associated
mRNAs, lncRNAs and miRNAs reveals key regulators in
smoking-associated lung cancer. Exp Ther Med. 16:4991–5002.
2018.PubMed/NCBI
|
|
39
|
Torkashvand J, Farzadkia M, Sobhi HR and
Esrafili A: Littered cigarette butt as a well-known hazardous
waste: A comprehensive systematic review. J Hazard Mater.
383:1212422020. View Article : Google Scholar
|
|
40
|
Hecht SS: Approaches to chemoprevention of
lung cancer based on carcinogens in tobacco smoke. Environ Health
Perspect. 105(Suppl 4): S955–S963. 1997.
|
|
41
|
Hoffmann D, Rivenson A, Chung FL and Hecht
SS: Nicotine-derived N-nitrosamines (TSNA) and their relevance in
tobacco carcinogenesis. Crit Rev Toxicol. 21:305–311. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li GX, Chen YK, Hou Z, Xiao H, Jin H, Lu
G, Lee MJ, Liu B, Guan F, Yang Z, et al: Pro-oxidative activities
and dose-response relationship of (-)-epigallocatechin-3-gallate in
the inhibition of lung cancer cell growth: A comparative study in
vivo and in vitro. Carcinogenesis. 31:902–910. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thomassen DG, Chen BT, Mauderly JL,
Johnson NF and Griffith WC: Inhaled cigarette smoke induces
preneoplastic changes in rat tracheal epithelial cells.
Carcinogenesis. 10:2359–2361. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bjermer L, Cai Y, Nilsson K, Hellström S
and Henriksson R: Tobacco smoke exposure suppresses
radiation-induced inflammation in the lung: A study of
bronchoalveolar lavage and ultrastructural morphology in the rat.
Eur Respir J. 6:1173–1180. 1993.PubMed/NCBI
|
|
45
|
Xue Y, Harris E, Wang W and Baybutt RC:
Vitamin A depletion induced by cigarette smoke is associated with
an increase in lung cancer-related markers in rats. J Biomed Sci.
22:842015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
47
|
Yuan C, Xiang L, Bai R, Cao K, Gao Y,
Jiang X, Zhang N, Gong Y and Xie C: MiR-195 restrains lung
adenocarcinoma by regulating CD4+ T cell activation via
the CCDC88C/Wnt signaling pathway: A study based on the cancer
genome atlas (TCGA), gene expression omnibus (GEO) and
bioinformatic analysis. Ann Transl Med. 7:2632019. View Article : Google Scholar
|
|
48
|
Chen Y and Wang X: miRDB: An online
database for prediction of functional microRNA targets. Nucleic
Acids Res. 48D:D127–D131. 2020. View Article : Google Scholar
|
|
49
|
Vlachos IS, Zagganas K, Paraskevopoulou
MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T and
Hatzigeorgiou AG: DIANA-miRPath v3.0: Deciphering microRNA function
with experimental support. Nucleic Acids Res. 43W:W460–W466. 2015.
View Article : Google Scholar
|
|
50
|
Freis A, Keller A, Ludwig N, Meese E,
Jauckus J, Rehnitz J, Capp E, Strowitzki T and Germeyer A: Altered
miRNA-profile dependent on ART outcome in early pregnancy targets
Wnt-pathway. Reproduction. 154:799–805. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang C, Wu R, Sargsyan D, Zheng M, Li S,
Yin R, Su S, Raskin I and Kong AN: CpG methyl-seq and RNA-seq
epigenomic and transcriptomic studies on the preventive effects of
Moringa isothiocyanate in mouse epidermal JB6 cells induced by the
tumor promoter TPA. J Nutr Biochem. 68:69–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao X, Liang M, Li X, Qiu X and Cui L:
Identification of key genes and pathways associated with osteogenic
differentiation of adipose stem cells. J Cell Physiol.
233:9777–9785. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Heinrich U, Muhle H, Takenaka S, Ernst H,
Fuhst R, Mohr U, Pott F and Stöber W: Chronic effects on the
respiratory tract of hamsters, mice and rats after long-term
inhalation of high concentrations of filtered and unfiltered diesel
engine emissions. J Appl Toxicol. 6:383–395. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Melnick RL, Huff JE, Roycroft JH, Chou BJ
and Miller RA: Inhalation toxicology and carcinogenicity of
1,3-butadiene in B6C3F1 mice following 65 weeks of exposure.
Environ Health Perspect. 86:27–36. 1990.PubMed/NCBI
|
|
55
|
Ceppi M, Munnia A, Cellai F, Bruzzone M
and Peluso MEM: Linking the generation of DNA adducts to lung
cancer. Toxicology. 390:160–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Munnia A, Giese RW, Polvani S, Galli A,
Cellai F and Peluso MEM: Bulky DNA adducts, tobacco smoking,
genetic susceptibility, and lung cancer risk. Adv Clin Chem.
81:231–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Avino P, Scungio M, Stabile L, Cortellessa
G, Buonanno G and Manigrasso M: Second-hand aerosol from tobacco
and electronic cigarettes: Evaluation of the smoker emission rates
and doses and lung cancer risk of passive smokers and vapers. Sci
Total Environ. 642:137–147. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duan S, Wang N, Huang L, Shao H, Zhang P,
Wang W, Wu Y, Wang J, Liu H, Zhang Q and Feng F: NLRP3 inflammasome
activation involved in LPS and coal tar pitch extract-induced
malignant transformation of human bronchial epithelial cells.
Environ Toxicol. 34:585–593. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vaz M, Hwang SY, Kagiampakis I, Phallen J,
Patil A, O'Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu
VE, et al: Chronic cigarette smoke-induced epigenomic changes
precede sensitization of bronchial epithelial cells to single-step
transformation by KRAS mutations. Cancer Cell. 32:360–376.e6. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen C, Jiang X, Gu S and Zhang Z:
MicroRNA-155 regulates arsenite-induced malignant transformation by
targeting Nrf2-mediated oxidative damage in human bronchial
epithelial cells. Toxicol Lett. 278:38–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fujiki H, Watanabe T, Sueoka E, Rawangkan
A and Suganuma M: Cancer prevention with green tea and its
principal constituent, EGCG: From early investigations to current
focus on human cancer stem cells. Mol Cells. 41:73–82.
2018.PubMed/NCBI
|
|
62
|
Moorthy B, Chu C and Carlin DJ: Polycyclic
aromatic hydrocarbons: From metabolism to lung cancer. Toxicol Sci.
145:5–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lawther PJ and Waller RE: Coal fires,
industrial emissions and motor vehicles as sources of environmental
carcinogens. IARC Sci Publ. 6:27–40. 1976.
|
|
64
|
Perera F: Carcinogenicity of airborne fine
particulate benzo(a) pyrene: An appraisal of the evidence and the
need for control. Environ Health Perspect. 42:163–185. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Knize MG, Salmon CP, Pais P and Felton JS:
Food heating and the formation of heterocyclic aromatic amine and
polycyclic aromatic hydrocarbon mutagens/carcinogens. Adv Exp Med
Biol. 459:179–193. 1999. View Article : Google Scholar : PubMed/NCBI
|