Introduction
Pancreatic cancer is recognized as one of the most
prevalent malignancies, with an increasing incidence rate worldwide
(4.9 per 100.00 person/years, in 2020) (1). In addition to hereditary risk
factors leading to the development of pancreatic cancer, there are
environmental factors, such as tobacco use, alcohol consumption,
chronic pancreatitis, obesity and diabetes mellitus. Furthermore,
the lack of feasible screening tests and the asymptomatic early
stage complicates the detection of the disease and decreases the
survival rate (the 7th leading cause of cancer death, data from
2018) (2).
Similar to pancreatic cancer, the incidence rate of
hepatocellular carcinoma (HCC) is among the highest (9.5 per
100.000 person/years in 2020); however, it differs in different
regions of the world (Western Europe, 5.4 per 100.000
person/years), with most cases in Eastern Asia (17.8 per 100.000
person/years) (1). In addition
to ethnicity, important risk factors are cirrhosis, chronic
hepatitis infections and an unhealthy lifestyle (3). Furthermore, both pancreatic cancer
and HCC are characterized by frequent chemoresistance, which is
associated with unfavorable prognoses (4,5).
The development of chemoresistance mechanisms are particularly
important in patients with HCC and unresectable tumors, where
chemotherapy remains the first choice of treatment (6-8).
Due to the limited treatment options, there is a high requirement
for new therapeutic agents, which has not been met.
The adenosine receptor (AdoR) family of G
protein-coupled receptors (GPCRs) were identified as key modulators
of myocardial and neuronal cell functions, immune system, and as
regulators of carcinogenesis (9,10). The A3 adenosine
receptor subtype (A3AR) is highly expressed in tumor
tissue, including pancreatic cancer and HCC (11-13); therefore, A3AR is a
promising target for anticancer therapy. A3AR was also
found to be a prognostic tumor marker in colorectal cancer by Gessi
et al (14).
The first highly selective A3AR agonist,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
(2-Cl-IB-MECA), was synthesized more than two decades ago (15). Initially, 2-Cl-IB-MECA-mediated
cardiovascular effects were demonstrated in rats (16) then, 2-Cl-IB-MECA was recognized
as a neuromodulator (17), a
cardio-protectant (18), a pain
reliever (19), an
anti-inflammatory agent (20),
an immuno-modulatory agent (21), and an anticancer agent (22).
Presently, 2-Cl-IB-MECA is undergoing clinical
trials for treating non-alcoholic steatohepatitis (NASH) and HCC,
showing no dose-limiting toxicity or serious drug-related side
effects, while being well tolerated by patients (23,24), and also showing an improvement in
moderate hepatic dysfunction (25,26). Another A3AR agonist,
N(6)-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
(IB-MECA), successfully passed Phase III clinical trials for
treating psoriasis with promising results (27), and a follow-up Phase III clinical
trial for treating moderate-to-severe plaque psoriasis is currently
recruiting patients (NCT03168256). Currently, 2-Cl-IB-MECA is used
as a reference A3AR for both in vitro (28) and in vivo studies
(29) and new N6-derivatized
analogues are being developed (30,31). In addition, novel avenues of GPCR
signaling are emerging, such as biased agonism, allostery and
receptor oligomerization (32,33). These observations indicate that
adenosine analogues and 2-Cl-IB-MECA are particularly useful
pharmacological tools in both experimental and clinical
settings.
The therapeutic potential of 2-Cl-IB-MECA in the
human JoPaca-1 pancreatic cancer and the Hep-3B HCC cell lines was
investigated in the present study. The JoPaca-1 cell line,
established in 2012, is strongly tumorigenic and manifests a highly
aggressive heterogenic phenotype, that is similar to primary
pancreatic tumors (34).
Notably, adenosine signaling in pancreas pathologies is
surprisingly understudied (35).
The Hep-3B HCC cell line is also tumorigenic, expressing
mesenchymal proteins that are associated with
epithelial-mesenchymal transition (EMT) and is notably responsive
to Smoothened (SMO) antagonists targeting the Hedgehog pathway (Hh)
(36). Adenosinergic signaling
was found to be a relevant anticancer mechanism against resistant
tumors in vitro (37,38) and in vivo (39). Furthermore, an increasing number
of studies demonstrated the effectiveness of 2-Cl-IB-MECA in
resistant tumor types (40-42). In addition, the modulatory role
of 2-Cl-IB-MECA in P-glycoprotein (P-gp) chemoresistance in
leukemia cells (43) and the
interaction of novel A3AR agonists with P-gp (44) have been found.
As a result, the ability of 2-Cl-IB-MECA to induce
cytotoxicity in tumor cells; to effect the PI3K/AKT/NF-κB,
Wnt/β-catenin, and Sonic hedgehog (Shh)/Patched
(Ptch)/Glioma-associated oncogene homolog zinc finger protein (Gli)
signaling pathways; to modulate the expression and function of the
multidrug-resistance-associated protein 1 (MRP1) and P-gp
multi-drug transporters; and to enhance the cytotoxic effects of
conventional chemotherapeutic drugs in JoPaca-1 pancreatic and
Hep-3B HCC cancer cell lines was investigated in the present study.
In addition, a specific A3AR antagonist,
N-[9-Chloro-2-(2-furanyl)(1,2,4)-triazolo(1,5-c)
quinazolin-5-yl]benzene acetamide (MRS 1220) was used to
distinguish between A3AR-dependent and
A3AR-independent effects. Such knowledge could lead to
the selection of suitable patient cohorts and maximization of their
benefits from the therapy.
Materials and methods
Materials
The reagents and chemicals were purchased as
follows: MRS 1220 (Tocris Bioscience); 0.9% w/v sodium chloride
intravenous infusion (B. Braun Melsungen AG); GlutaMAX™, TrypLE™
Express Enzyme (Thermo Fisher Scientific, Inc.), Geneticin, Zeocin
Selection Reagent (Gibco; Thermo Fisher Scientific, Inc.), Pierce™
BCA Protein Assay kit and Ham's F-12K (Kaighn's) medium (Thermo
Fisher Scientific, Inc.); Iscove's modified Dulbecco's medium
(IMDM), Eagle's minimum essential medium (EMEM) and RPMI-1640,
McCoy's 5A modified medium (Lonza Group Ltd.); Ham's F12 and
DMEM/F-12 (HyClone; Cytiva); ethanol (VWR International, LLC); Cell
wash reagent, Permeabilizing solution 2 and Fc receptor saturation
reagent (BD Biosciences); FBS (Capricorn Scientific GmbH);
Coelenterazine h (Biotium, Inc.); CellTiter 96 AQueous
Non-Radioactive Cell Proliferation Assay (Promega Corporation);
paraformaldehyde (Merck KGaA); glycine (AMSBIO LLC); Amersham™ ECL™
Prime Western blotting detection reagent and Amersham
nitrocellulose membrane 0.2 μM (Cytiva); Britelite plus
Luminescence Reporter Gene Assay System (PerkinElmer, Inc.);
fluorouracil (5-FU) and gemcitabine (Accord Healthcare Ltd.);
carboplatin (Fresenius Kabi Asia-Pacific, Ltd.); doxorubicin
(Pharmagen GmbH); 5′-N-ethylcarboxamidoadenosine (NECA),
2-Cl-IB-MECA, dimethyl sulfoxide (DMSO), BSA, EDTA, PI,
5-Bromo-2′-deoxyuridine (BrdU), 5-Bromouridine (BrU), sodium
tetraborate decahydrate, nonyl phenoxypolyethoxylethanol (NP-40),
RNase A, Fraction V, daunorubicin, puromycin and all other
chemicals were purchased from Sigma-Aldrich (Merck KGaA). All
compounds used for treatments were of ≥98% purity. For all
experiments, DMSO was used as vehicle for MRS 1220, NECA and
2-Cl-IB-MECA, while 0.9% w/v sodium chloride intravenous infusion
was used as a vehicle for 5-FU, gemcitabine, carboplatin and
doxorubicin.
The following primary antibodies were used:
A3AR (cat. no. ab203298; 1:500), phosphorylated pAKT
Ser473 (cat. no. ab192623; 1:500), pAKT
Thr308 (cat. no. ab38449; 1:500), NF-κB (cat. no.
ab16502; 1:1,000), β-catenin (cat. no. ab32572; 1:2,500), Gli1
(cat. no. ab134906; 1:500), Ptch1 (cat. no. ab53715; 1:1,000),
c-Myc (cat. no. ab32072; 1:1,000) all from Abcam; ERK1/2 (cat. no.
9102; 1:1,000), pERK1/2 Thr202/Tyr204 (cat.
no. 20G11; 1:1,000), AKT (cat. no. 9272S; 1:1,000), pNF-κB
Ser536 (cat. no. 3033; 1:1,000), GSK-3b (cat. no. 27C10;
1:1,000), pGSK-3b Ser9 (cat. no. 9336; 1:1,000), cyclin
D1 (cat. no. 2978; 1:500), pc-Myc Ser62 (cat. no.
13748S; 1:1,000), pc-Myc Thr58 (cat. no. 46650S;
1:1,000) from Cell Signaling Technology, Inc.; MRP1 (cat. no.
801-012-C250; 1:250) from Enzo Life Sciences, Inc.; p-histone H3
Ser10 (cat. no. 06-570; 1:500) from Merck KGaA; β-actin
(cat. no. A2228; 1:3,000), IgG1 isotype control unconjugated (cat.
no. M5284; 1:140), P-gp (cat. no. P7965; 1:250) from Sigma-Aldrich
(Merck KGaA); and anti-BrdU (cat. no. 11-286-C025; 1:250) from
EXBIO Praha, a.s. Secondary antibodies conjugated with HRP
anti-rabbit IgG (cat. no. A-0545; 1:10,000) and anti-mouse IgG
(cat. no. A-2304; 1:10,000), and anti-mouse-IgG-FITC (cat. no.
F2883; 1:250) were purchased from Sigma-Aldrich (Merck KGaA).
Anti-rabbit-IgG-Alexa Fluor 488 (cat. no. A-11070; 1:500) was
purchased from Thermo Fisher Scientific, Inc.
The JoPaca-1 pancreatic carcinoma and Hep-3B HCC
cell lines were purchased from German Collection of Microorganisms
and Cell Cultures. The A549 lung carcinoma, BJ non-tumor
fibroblast, BT-549 and MDA-MB-231 breast cancer, CCRF-CEM leukemia,
non-tumor MRC-5 fibroblast, U2OS osteosarcoma, HCT116 colon
carcinoma and HT-29 colorectal cancer cell lines were purchased
from the American Type Culture Collection. The human ES-012-A
A3AR aequorin cell line was purchased from Perkin Elmer,
Inc., while the parental K562 leukemia and the HCT116
p53−/− cell lines were purchased from Horizon Discovery
Ltd. The CEM-DNR (resistant to daunorubicin) and K562-TAX
(resistant to paclitaxel) cell lines were established in our
laboratory (45).
The β-catenin reporter cell lines were prepared as
follows: The U2OS, HCT 116 and HT-29 cell lines were stably
transduced at multiplicity of infection, 5, at 37°C for 16 h with
commercially available lentiviral particles [Cignal Lenti T cell
factor/lymphoid enhancer factor (TCF/LEF) Reporter Assays; cat. no.
CLS-018L; Qiagen GmbH] carrying the TCF/LEF responsive element
coupled to firefly luciferase reporter gene. The following three
models of the Wnt/β-catenin pathway were established using
antibiotic selection in the presence of 1 μg/ml puromycin:
Wild-type (derived from the U2OS cell line;
β-cateninWT); constitutively active with deletion of
Ser45 in β-catenin, where this single nucleotide variant
prevents β-catenin proteasomal degradation (derived from the HCT116
cell line; β-cateninmut); and constitutively active with
a loss-of-function mutation in the APC gene (derived from the HT-29
cell line; APCmut). Monoclonal cell lines were generated
from polyclonal population after one-week selection in presence of
1 μg/ml puromycin using single cell sorting (FACSAria II
SORP; BD Biosciences) and maintained in the corresponding media
containing 1 μg/ml puromycin. Before the experiments,
specific reporter activity for each clonal cell line was verified
by stimulation with the Wnt3a ligand and small interfering
(si)RNA-mediated downregulation of β-catenin. The experiments were
conducted within one month from the transduction.
Cell culture
The cell lines were maintained and sub-cultured
according to manufacturer's instructions under the following
conditions: JoPaca-1 (IMDM; 20% FBS); K562 (IMDM; 10% FBS); Hep-3B
(EMEM; 10% FBS; GlutaMAX™); MRC-5 and BJ (EMEM; 10% FBS); K562-TAX,
CCRF-CEM, CEM-DNR, BT-549 and MDA-MB-231 (RPMI-1640; 10% FBS); A549
(Ham's F-12K; 10% FBS); HCT116 and HCT116 p53−/− (RPMI;
10% FBS); U2OS and HT-29 (McCoy's 5A modified medium; 10% FBS);
A3AR reporter ES-012-A (Ham's F-12; 10% FBS; GlutaMAX™;
0.4 mg/ml geneticin; and 0.25 mg/ml zeocin); β-catenin reporter
cell lines (McCoy's 5A modified medium; 10% FBS; 1 μg/ml
puromycin). The cell lines were authenticated using short tandem
repeat DNA profiling analysis (PowerPlex 18D; Promega Corporation),
analyzed every two weeks for mycoplasma contamination (reverse
transcription-quantitative PCR), and maintained in T-150 flasks at
37°C in a humidified incubator with 5% CO2 and 100%
humidity. For the experiments, the cells were placed in fresh
medium, with no antibiotics at densities specified for each
method.
A3AR functional assay
The A3AR functional assay used was
performed as previously described (46) with modifications. Briefly,
ES-012-A is an AequoScreen reporter cell line stably expressing
A3AR, the photoprotein aequorin, and the promiscuous
Gα16 protein subunit. The ES-012-A cell line was non-enzymatically
harvested with PBS/EDTA and incubated in DMEM/F12 medium with 5
μM coelenterazine for 4 h at room temperature (RT) in the
dark to reconstitute the active form of aequorin. Meanwhile,
384-well microplates with pre-diluted 2-Cl-IB-MECA (50 μM-1
pM) and MRS 1220 (10 μM-0.01 pM) were prepared using the
acoustic liquid handler ECHO 550 (LabCyte, Inc.).
Coelenterazine-loaded cells (5×103 cells
per well) were dispensed using a cell suspension system and the
FLIPR tetra multimodal reader (Molecular Devices, LLC.). The
resulting luminescence was recorded immediately. The cells were
incubated within the instrument for another 15 min at RT and the
antagonistic response, represented by a drop in luminescence
signal, was measured directly after the addition of the reference
agonist NECA, at 80% of maximal effective concentration
(EC80). Raw data (area under the curve; AUC) were
normalized to the percentage of the reference agonist NECA response
(arbitrarily set at 100%). Normalized response in percentage was
plotted against concentration, and EC50 (for agonist)
and IC50 (for antagonist) were calculated. The data used
for analysis were the result of four independent experiments.
Cytotoxicity assay
The cytotoxicity assay was performed as previously
described (47). Briefly, the
cell lines were seeded into 384-well microplates, at specific
densities for each cell line (0.9-2.5×103 cells per
well). After 24 h, all the tested compounds were added to the
microplates using the acoustic liquid handler ECHO 550. Either
vehicle, 2-Cl-IB-MECA, MRS 1220 or 2-Cl-IB-MECA in combination with
0.1 μM fixed concentration of MRS 1220 were analyzed at a
concentration range 100-0.2 μM in the JoPaca-1 and Hep-3B
cell lines. The vehicle volume was equal to the maximal
concentration of treatment. In the other tumor and non-tumor cell
lines, 2-Cl-IB-MECA was analyzed at 0.02-50 μM.
In the β-catenin reporter cell lines, 2-Cl-IB-MECA
was analyzed at a single concentration of 50 μM. In the
synergy combination experiments, 2-Cl-IB-MECA was analyzed at a
concentration range from 1.56 to 50 μM in the JoPaca-1 cell
line and from 0.78 to 25 μM in the Hep-3B cell line. The
chemotherapeutic agents were used at concentrations as follows:
5-FU and carboplatin (0.78-200 μM), doxorubicin (0.1-25
μM), gemcitabine (0.2-50 μM). For the 2-Cl-IB-MECA
and MRS 1220 combination experiments, MRS 1220 was added to the
cells 30 min prior to 2-Cl-IB-MECA.
The treated cells were incubated at 37°C with 5%
CO2 and 100% humidity for 24 and 48 h (β-catenin
reporters) or 72 h (other cell lines). After incubation, MTS
solution and phenazine methosulfate (PMS), an electron coupling
reagent, was prepared according to manufacturer's instructions and
dispensed to the microplates using a Multidrop Combi reagent
dispenser (Thermo Fisher Scientific, Inc.). The final concentration
of MTS/PMS in the wells was 235 μg/ml. All the cell lines
were allowed to metabolize the MTS/PMS reagent into formazan for
1.5-3 h at 37°C with 5% CO2 and 100% humidity.
Absorbance of the formazan product was measured at 490 nm using an
EnVision plate reader (Perkin Elmer, Inc.) and, after subtracting
the blank, the cytotoxic (anti-proliferative) effects were
expressed as percentage of inhibition. The data used for analysis
were the result of three independent experiments.
For the enhanced effect of combined treatments in
synergy experiments, the effects were expressed as the fraction
affected (FA) normalized to cells treated with vehicle (DMSO for
2-Cl-IB-MECA and MRS 1220; 0.9% w/v sodium chloride intravenous
infusion for chemotherapeutic agents), and was calculated using the
following equation: FA=1-(sample-blank/control-blank), where the
sample is the value from a single well treated with a compound, the
control is the average absorbance of the cells treated with vehicle
and the blank is the average absorbance of the background without
cells.
The effects of two drug treatments were assessed
using the CalcuSyn software (version 2.0; Biosoft), as a
combination index (CI) and the Chou-Talalay method (48,49). The FA was used as data input,
where CI <0.9 was quantitatively defined as synergism, CI
>0.9 and <1.1 as an additive effect, and CI >1.1 as
antagonism.
Cell cycle analysis
Cell cycle analysis was performed as previously
described (50). The JoPaca-1
and Hep-3B cell lines were seeded 24 h before treatment in 6-well
multi dishes, at 2.7- and 2.2×105 cells per well,
respectively. The JoPaca-1 and Hep-3B cells were treated for 24 h
with either vehicle, 20 nM or 20 μM 2-Cl-IB-MECA using the
ECHO 550 dispenser. Both adherent and detached cell fractions were
subsequently harvested using TrypLE reagent on ice, washed with
PBS, fixed with 70% ethanol at −20°C added drop by drop, and
immediately transferred to −20°C for another 24 h. The fixed cells
were washed with citrate buffer to enhance extraction of the
fragmented DNA (the subG1 population), stained with 600
μl PI (50 μg/ml) for 30 min at 37°C in the dark,
followed by incubation of samples with added 500 μl RNAse (2
mg/ml) for another 15 min under the same conditions. Before adding
PI, half of each sample was separated and incubated for 1 h with an
antibody against p-histone H3 Ser10 (pH3) at RT in the
dark. The samples were washed with PBS containing 1% FBS, then
incubated with fluorescent secondary antibody, Alexa Fluor 488 for
another 30 min at RT in the dark, washed with PBS containing 1% FBS
and stained with PI as aforementioned.
PI and Alexa Fluor 488 fluorescence intensities in
relative fluorescence units (RFU) were then measured using a flow
cytometer (FACS Calibur; BD Imunocytometry Systems) with a 488 nm
single beam argon laser, with two types of settings for
fluorescence measurement of PI (linear for cell cycle and pH3 and
logarithmic for subG1). Analysis of the cell cycle
phases was performed using ModFit LT (version 2.0; Verity Software
House, Inc.) and quantifications of the subG1 and
pH3-marked mitotic cells were performed using CellQuest software
(version 3.3; BD Immunocytometry Systems). Each sample consisted of
at least 1×104 cells in compliance with the required
parameters from three independent experiments.
DNA synthesis
The level of DNA synthesis was quantified by
incorporating BrdU into the DNA from the JoPaca-1 and Hep-3B cell
lines according to a previous study (50). Briefly, the cells were seeded 24
h before treatment in 6-well multi dishes at 2.7×105 and
2.2×105 cells per well for the JoPaca-1 and Hep-3B
cells, respectively, to achieve an approximate density of 60% at
the time of treatment. The cell lines were treated with either
vehicle, 20 nM or 20 μM 2-Cl-IB-MECA, or with the
combination of 2-Cl-IB-MECA (20 nM or 20 μM) and MRS 1220
(0.1 μM) for 24 h, for the JoPaca-1 and Hep-3B cells,
respectively. For the combination experiments, MRS 1220 was added
to the cells 30 min before 2-Cl-IB-MECA. The cells were then
labelled with 1 μM BrdU solution at 37°C with 5%
CO2 and 100% humidity for 60 min before harvesting.
The cells were harvested with TrypLE reagent, washed
with PBS, fixed with 70% ethanol at −20°C, added dropwise and
immediately transferred to −20°C for 24 h. The samples were then
incubated with 2 M HCl/Triton X-100 solution for 30 min at RT. The
DNA denaturing reaction was neutralized with 0.1 M sodium
tetraborate decahydrate. The cells were then washed with PBS
containing 1% BSA and 0.5% Tween-20 and stained with primary
anti-BrdU antibody for 30 min at RT in the dark. After washing with
PBS, the cells were incubated with secondary anti-mouse-IgG-FITC
antibody for 30 min at RT in the dark. The stained samples were
washed with PBS one last time and incubated with 0.5 mg/m RNAse A
for 15 min, followed by 0.1 mg/ml PI for another 15 min both at RT
in the dark.
The samples were measured using a FACSCalibur flow
cytometer and a 488 nm argon laser. Each sample included at least
1×104 cells in compliance with the required parameters
from three independent experiments. The rate of DNA synthesis was
analyzed using CellQuest software (version 3.3; BD Immunocytometry
Systems).
RNA synthesis
RNA synthesis was quantified by incorporating BrU
into RNA in the JoPaca-1 and Hep-3B cell lines according to a
previous study (50). In short,
the cells were seeded 24 h before treatment in 6-well multi dishes
at 2.7×105 and 2.2×105 cells per well for the
JoPaca-1 and Hep-3B cell lines, respectively, to achieve ~60%
density at the time of treatment. The concentrations and
combinations of the compounds added to the cells were identical to
the DNA synthesis experiment, as aforementioned. The cells were
incubated for 24 h. Before harvesting, the cells were labelled with
1.5 μM BrU solution for 90 min at 37°C with 5%
CO2 and 100% humidity. The cells were harvested using
TrypLE reagent followed by fixation in 1% paraformaldehyde, with
0.05% NP-40 in PBS for 15 min on a rotating wheel at RT. The
samples were stored overnight at 4°C. Subsequently, the cells were
washed with PBS, containing 1% glycine, with PBS alone, then
stained with unconjugated anti-BrdU antibody for 45 min at RT,
which cross-reacts with the BrU incorporated into the cells. After
washing again with PBS containing 0.1% NP-40 and 0.1% BSA, the cell
suspension was fluorescently labelled by incubation with secondary
anti-mouse-IgG-FITC antibody for 30 min at RT in the dark. The
labelled samples were washed with PBS containing 0.1% NP-40 and
0.1% BSA and fixed with 1% paraformaldehyde and 0.05% NP-40 in PBS
for 15 min at RT in the dark and, subsequently, incubated for 1 h
at 4°C. After the last wash with PBS, the cells were incubated with
0.5 mg/ml RNAse A for 15 min at RT, followed by 0.1 mg/ml PI for
another 15 min at RT in the dark. BrU-labelled samples were
analyzed as previously described for DNA staining and the data used
was from three independent experiments.
Western blot analysis
The JoPaca-1 and Hep-3B cell lines were seeded in
6-well multi dishes, at densities of 2.7×105 and
2.2×105 cells, respectively, and treated with either
vehicle (24 h), 20 μM 2-Cl-IB-MECA, or 0.1 μM MRS
1220 for 30 min prior to 2-Cl-IB-MECA, for 24 and 48 h. The cells
were harvested on ice using a scraper and lysed with ice-cold RIPA
buffer (150 μM NaCl, 1% NP-40, 0.5% sodium deoxycholate,
0.1% SDS, 50 μM Tris-HCl pH 8.0 and 1 μM EDTA) for 30
min, then sonicated at 35 kHz for 1 min in a 4°C ultrasonic water
bath. Next, the cell debris were discarded using centrifugation at
14,000 × g for 15 min at 4°C. The protein concentrations were
determined using a BCA protein assay. After 5X concentrated Laemmli
buffer was added, the protein samples were incubated at 70°C for 10
min. Equal concentrations (20 μg) of each sample were
separated using 7.5 or 12% SDS-PAGE. The resolved proteins were
transferred to nitrocellulose membranes using the semi-dry blot
method (Trans-Blot Turbo Transfer System; Bio-Rad Laboratories,
Inc.). The membranes were blocked with 5% BSA in TBS, containing
0.1% Tween-20 for 1 h at RT, then incubated overnight at 4°C with
the primary antibodies. Subsequently, the membranes were washed in
TBS, containing 0.1% Tween-20 and incubated for 1 h at RT with HRP
anti-rabbit IgG or anti-mouse IgG secondary anti-bodies. Next, the
proteins were visualized using ECL substrate and a ChemiDoc MP
Imaging System (Bio-Rad Laboratories, Inc.). The immunoblot images
were processed using Image Lab v6.1 (Bio-Rad Laboratories, Inc.)
and, if relevant, band densities were analyzed using ImageJ
software (51). Relative
intensities were calculated using β-actin as the loading control
and expressed as fold change. In the same manner, A3AR
endogenous protein expression was analyzed using western blot
analysis in the MRC-5, JoPaca-1 and Hep-3B cell lines. The data
used for analysis were the result of three independent
experiments.
β-catenin reporter assay
The β-cateninWT, β-cateninmut,
and APCmut reporter cell lines were seeded onto 384-well
microplates, at a density of 2.5×103 cells per well.
After incubation for 24 h at 37°C with 5% CO2 and 100%
humidity, all analyzed compounds were added to the microplates, at
the indicated concentrations as aforementioned using an acoustic
liquid handler ECHO 550 and the treated cells were incubated for
specific durations (1, 3, 6, 12, 24 and 48 h).
Immediately after the addition of the Britelite plus
reagent, the luminescence signal proportional to the
transcriptional activity of the reporters was measured using an
EnVision plate reader. After subtracting the blank, β-catenin
transcriptional activity was calculated as a percentage of
inhibition, defined similarly to the MTS assay. The data used for
analysis were from three independent experiments.
P-gp expression
he JoPaca-1 and Hep-3B cell lines were seeded in
6-well multi dishes, at 2.7×105 and 2.2×105
cells per well, respectively. The cells at ~60% density were
treated with either vehicle or 20 μM 2-Cl-IB-MECA 24 h
later. After another 24 h, the cells were collected using TrypLE
reagent, centrifuged at 500 × g for 5 min, at RT and the pellet was
washed with PBS. The cells were fixed with cold methanol (−20°C;
100%), added dropwise, then the cells were stored at −20°C for a
minimum of 24 h before analysis.
Subsequently, the cells were washed with rinsing
buffer, composed of Cell wash reagent, 0.5% BSA, 0.1% NP-40, then
permeabilized with Permeabilizing solution 2 for 10 min, and washed
again with rinsing buffer. A saturation reagent that blocks Fc
receptors to prevent unspecific binding of the antibody (Cell wash
reagent, 0.5% BSA and Fc receptor saturation reagent) was added to
the cells, then incubated for 10 min at RT, and washed twice more
with Cell wash reagent containing 0.5% BSA and 0.1% NP-40. The
samples were divided into two tubes: One for P-gp expression
analysis (monoclonal anti-P-gp antibody) and one for the isotype
control (IgG1 isotype control; unconjugated); both tubes were
incubated with the antibodies for 30 min at RT in the dark.
Following which, all the samples were washed with rinsing buffer
and incubated with FITC-conjugated secondary antibody
anti-mouse-IgG for 30 min at RT, washed once more, then stored at
4°C for 15 min.
Median fluorescence intensities (MFI) for both
samples and isotype controls were measured with a 488 nm argon
laser and a FACS Calibur flow cytometer, where the isotype controls
defined the intensity threshold at a median of 10 relative
fluorescence units. MFI raw data were the result of three
independent experiments and analyzed using CellQuest (version 3.3;
BD Immunocytometry Systems).
P-gp efflux assay
The K562-TAX cell line, derived from K562 chronic
myelogenous leukemia cell line, was developed at the Institute of
Molecular and Translational Medicine, as a cell line that is
resistant to paclitaxel, with high expression of the P-gp
transporter and low expression levels of MRP1 and lung resistance
protein (LRP) transporters (45).
The P-gp efflux assay was conducted as previously
described (52). Briefly, the
K562-TAX cells were seeded in 6-well multi dishes at
2.5×105 and allowed to proliferate for 24 h. The next
day, the cells were treated with 1 μM daunorubicin
(fluorescent) for 60 min at 37°C in the dark. The cells were washed
twice with complete medium and incubated with 2-Cl-IB-MECA
(0.78-100 μM), either alone or following 0.1 μM MRS
1220 pre-treatment for 30 min at 37°C, for another 60 min under the
same conditions as aforementioned. Following which, the K562-TAX
cells were harvested using a scraper and fluorescence intensity was
measured immediately using a FACS Calibur flow cytometer and a 488
nm argon laser.
Each sample comprised at least 1×104
cells, that were compliant with the required parameters. The raw
data were the result of three independent experiments and were
measured as MFI. MFI for the vehicle was set to 10 relative
fluorescence units. MFI comparisons were analyzed using CellQuest
(version 3.3; BD Immunocytometry Systems).
Statistical analysis
The results are expressed as the mean ± SD, with the
number of independent replicates (n) indicated for each experiment.
Quantification of the results was performed using GraphPad Prism v9
for Windows (GraphPad Software, Inc.), including IC50
values for the JoPaca-1 and Hep-3B cell lines. The IC50
values in the MTS experiment using the other tumor and non-tumor
cell lines were calculated using Dotmatics software (version 5.5;
Dotmatics, Ltd.). Statistical analyses were performed using R
statistical software, v3.5.0 (R Foundation for Statistical
Computing; www.r-project.org). The data was
checked for normality using the Shapiro-Wilk test. Statistical
differences between treatment group and the reference group
(vehicle) were analyzed using a Student's one-sample t-test. An
unpaired Student's two-sample t-test was used for comparison of
treatments. ANOVA followed by Dunn's post hoc test was used to
analyze 2-Cl-IB-MECA at 24 and 48 h and the reference group (MRS
1220 at 24 h). A one-way ANOVA test, followed by Dunn's post hoc
test was used to analyze the 2-Cl-IB-MECA group at 24 and 48 h and
the vehicle as the reference group, for phosphorylated/total
protein. In case of comparison of more than two treatment
conditions, ANOVA followed by Tukey's post hoc test was used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
2-Cl-IB-MECA shows cytotoxicity for
cancer cell lines and agonistic activity on A3AR
Previous studies have shown both pro- and antitumor
effects of 2-Cl-IB-MECA in vitro and in vivo at
either sub-micromolar or micromolar concentrations (reviewed in
53). To determine the overall cytotoxic ability of 2-Cl-IB-MECA
(Fig. 1A), the effects of
2-Cl-IB-MECA against a panel of tumor and non-tumor cell lines was
performed. Using a MTS assay, the cells were incubated with
2-Cl-IB-MECA for 72 h and the cellular response was analyzed using
different concentrations (0.2-100 μM for the JoPaca-1 and
Hep-3B cell lines and 0.02-50 μM for the other cell lines).
Cytotoxicity against 2-Cl-IB-MECA was found in micromolar
concentrations across various tumor cell lines, of different
histogenetic origin, including JoPaca-1 and the most sensitive cell
line, Hep-3B, with their respective IC50 values of
25.26±1.6 and 10.68±1.1 μM (Table I). Furthermore, the non-tumor
fibroblast cell lines, MRC-5 and BJ cells remained metabolically
active despite increasing 2-Cl-IB-MECA concentrations, with
IC50 values >50 μM.
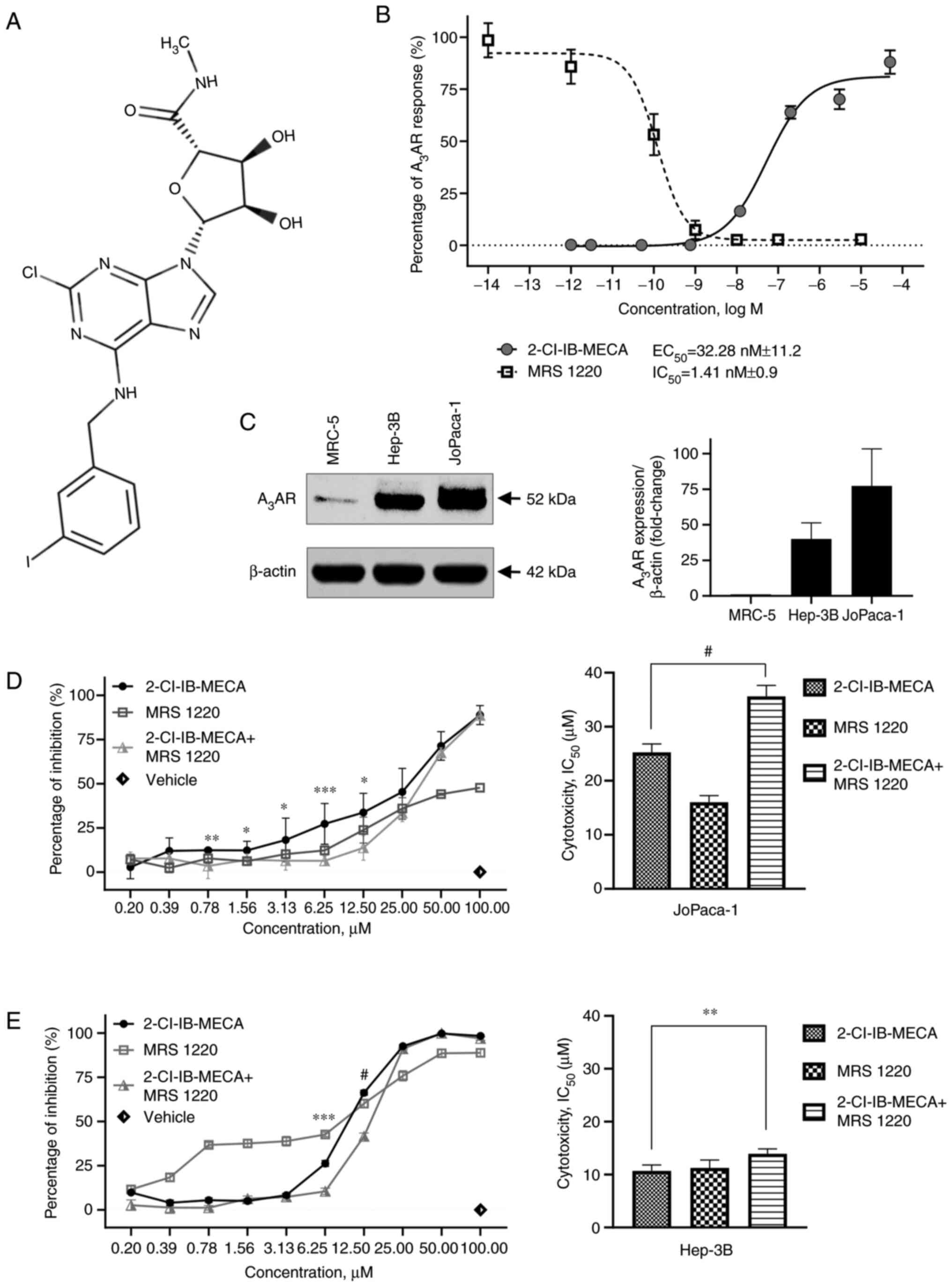 | Figure 12-Cl-IB-MECA demonstrates
A3AR agonistic activity and cytotoxic effects on cancer
cells. (A) Structure of 2-Cl-IB-MECA. (B) Functional assay was
performed with the reporter cell line stably expressing
A3AR. The cells were treated with the indicated
concentrations of 2-Cl-IB-MECA or MRS 1220, and the relative
luminescence response for each independent experiment was
normalized to the maximal response of a reference agonist NECA.
EC50 and IC50 values were calculated. n=4.
(C) Comparison of A3AR expression in the tested cell
lines was analyzed using western blot. A3AR relative
signal intensities were calculated using densitometry. n=3.
Cytotoxicity of 2-Cl-IB-MECA and MRS 1220 alone, and in combination
in the (D) JoPaca-1 and (E) Hep-3B cancer cell lines were analyzed
using a MTS assay after 72 h. The cells were treated at the
indicated concentrations of 2-Cl-IB-MECA, MRS 1220 or 2-Cl-IB-MECA
after 30-min pre-treatment with 0.1 μM MRS 1220. The values
are expressed as the percentage of inhibition (left) and
cytotoxicity IC50 (right). n=3. The data are represented
as the mean ± SD. The results were analyzed using a Student's
two-tailed t-test. *P<0.05, **P<0.01,
***P<0.005, #P<0.001 for 2-Cl-IB-MECA
vs. 2-Cl-IB-MECA + MRS 1220. 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl) (1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
 | Table ICytotoxicity of 2-Cl-IB-MECA in tumor
and non-tumor cell lines. |
Table I
Cytotoxicity of 2-Cl-IB-MECA in tumor
and non-tumor cell lines.
| Cell line | IC50,
μM | SD |
|---|
| A549 | 45.51 | 5.73 |
| BT-549 | 12.48 | 0.67 |
| CCRF-CEM | 21.39 | 1.05 |
| CEM-DNR | 26.72 | 5.35 |
| HCT116 | 33.73 | 3.07 |
| HCT116
p53−/− | 31.50 | 3.27 |
| Hep-3B | 10.68 | 1.14 |
| JoPaca-1 | 25.26 | 1.57 |
| K562 | 24.85 | 3.28 |
| K562-TAX | 29.23 | 2.78 |
| MDA-MB-231 | 23.89 | 4.14 |
| U2OS | 42.78 | 7.69 |
| BJa | >50 | - |
| MRC-5a | >50 | - |
Next, the compounds, 2-Cl-IB-MECA and MRS 1220 were
both analyzed on reporter cell lines expressing A3AR.
The functional assay revealed A3AR activation by
2-Cl-IB-MECA (EC50, 32.28±11.2 nM), and its inhibition
by MRS 1220 (IC50, 1.41±0.9 nM) at nanomolar
concentrations (Fig. 1B).
To identify whether any possible effects could be
A3AR related, western blot analysis was performed to
analyze A3AR protein expression (Fig. 1C). By contrast to the non-tumor
MRC-5 fibroblasts, the results showed the presence of
A3AR in both the JoPaca-1 and Hep-3B tumor cell lines
analyzed.
To address the nature of the effects of 2-Cl-IB-MECA
on human pancreatic cancer and HCC cells more precisely, the
JoPaca-1 and Hep-3B cell lines were treated with increasing
concentrations of 2-Cl-IB-MECA and MRS 1220 alone, and in
combination for 72 h. MRS 1220 is a specific A3AR
antagonist (54,55) and MRS 1220 intrinsic cytotoxicity
as well as its ability to counteract the effect of the
A3AR agonist 2-Cl-IB-MECA was analyzed in the cancer
cells. Notably, pre-treatment of the cells with 0.1 μM MRS
1220 led to significantly decreased 2-Cl-IB-MECA cytotoxicity in
both cell lines. MRS 1220 alone at sub-micromolar concentrations
were not cytotoxic for either cell line (Fig. 1D and E). Taken together,
2-Cl-IB-MECA inhibited the proliferation of the cancer cell lines,
including JoPaca-1 and HEP-3B cancer cells at micromolar
concentrations and its effect in the JoPaca-1 and HEP-3B cell lines
was reduced by MRS 1220. Therefore, 20 nM (the
A3AR-activating concentration) and 20 μM
(considerable cytotoxicity-inducing concentration) of 2-Cl-IB-MECA
was used for further experiments.
2-Cl-IB-MECA downregulates cell cycle
progression by increasing the number of cells in the G1
phase and induces cell death
Previous studies showed that the N6-adenosine
analogue 2-Cl-IB-MECA reduces the cell cycle in other cancer cell
types, such as NPA thyroid carcinoma and A375 melanoma cells
(12,56). Flow cytometry analysis was
performed to analyze cell cycle progression, including the
subG1 population, histone H3 phosphorylation, and
changes in RNA and DNA synthesis, to further assess a possible
anticancer mechanism of 2-Cl-IB-MECA. Based on the cytotoxicity
experiments, the JoPaca-1 and Hep-3B cell lines were each treated
with either vehicle, 20 nM or 20 μM of 2-Cl-IB-MECA for 24
h. In DNA and RNA synthesis experiments, the cells were pre-treated
with 0.1 μM MRS 1220 for 30 min prior to the addition of
2-Cl-IB-MECA.
Only 20 μM 2-Cl-IB-MECA affected the cell
cycle, showing an increase in the number of cells in the
G1 phase. In addition, there was a simultaneous increase
in the subG1 population, representing cells undergoing
apoptosis (Fig. 2A-C). The
Hep-3B cell line was more responsive compared with that in the
JoPaca-1 cell line (Fig. 2B). In
concordance with the block at the G1 transition point,
there was a reduction in the number of cells in S phase. Indeed,
the reduction in the number of cells in the S phase was further
accentuated by the drop in the cell population actively
incorporating BrdU and BrU, indicating an inhibition of DNA
(Fig. 2E), and RNA synthesis
(Fig. 2F), respectively.
Notably, pre-treatment with 0.1 μM MRS 1220 significantly
reversed the effects of 2-Cl-IB-MECA on BrdU (in the JoPaca-1 cell
line at 20 nM and 20 μM of 2-Cl-IB-MECA) and BrU
incorporation (in the Hep-3B cell line at 20 nM and 20 μM of
2-Cl-IB-MECA). There was also decrease in RNA synthesis in the
JoPaca-1 cell line following treatment with 0.1 μM MRS 1220,
suggesting a possible intrinsic effect of MRS 1220 alone.
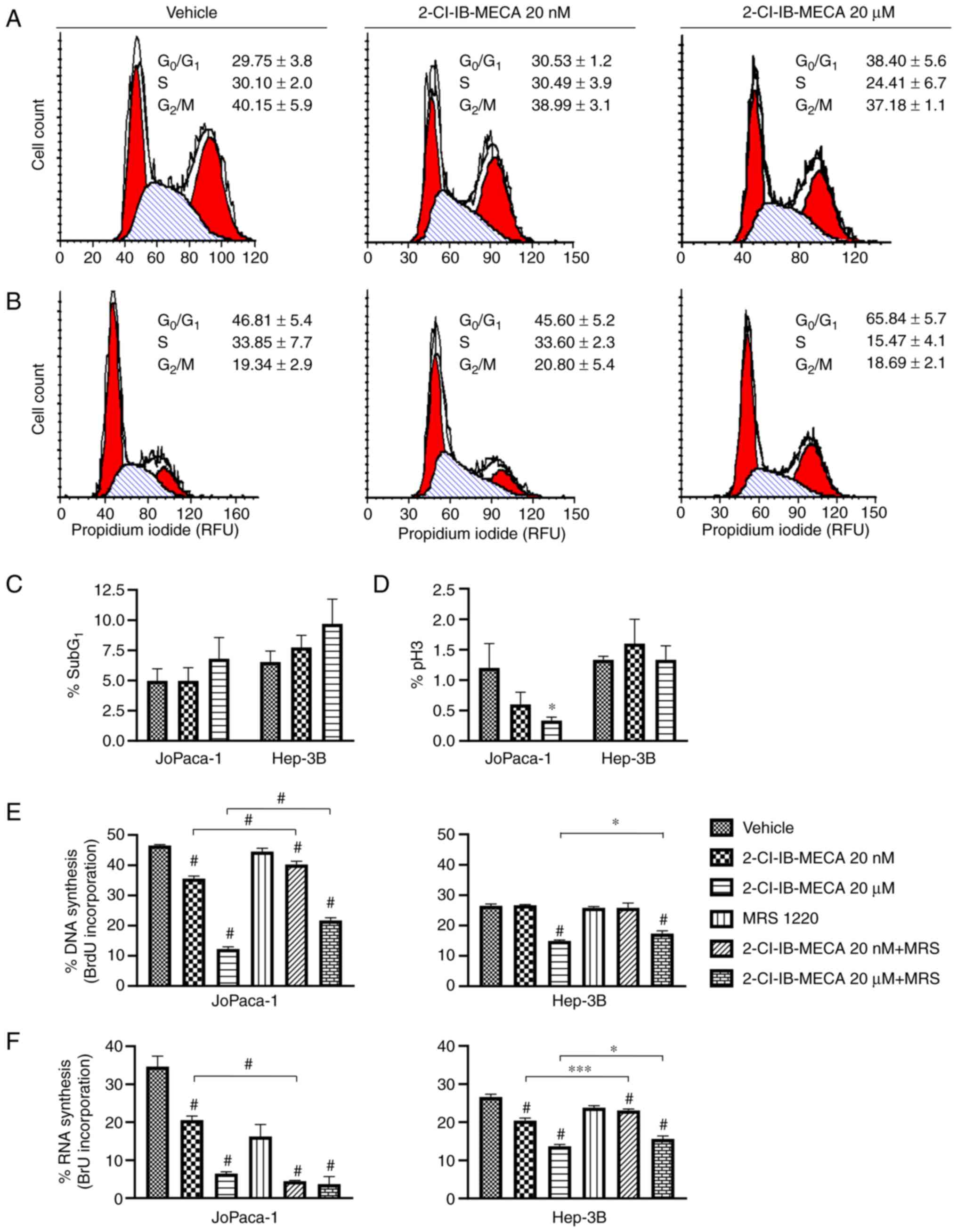 | Figure 22-Cl-IB-MECA treatment leads to the
alteration of cell cycle progression in the JoPaca-1 and Hep-3B
cell lines. Cell cycle analysis of (A) JoPaca-1 and (B) Hep-3B cell
lines following 2-Cl-IB-MECA treatment. The cells were treated with
vehicle (DMSO) or the indicated concentrations of 2-Cl-IB-MECA (20
nM or 20 μM) for 24 h. Cell cycle phase distribution
(G0/G1, 1st red peak; S, stripes;
G2/M, 2nd red peak) was quantified as RFU. (C)
quantification of subG1 cells and (D) cell population
positive for pH3 at Ser10 were analyzed separately. n=3.
All data are expressed as a percentage of the whole. (E) BrdU and
(F) BrU incorporation representing the percentage of the cells
actively synthesizing DNA and RNA for JoPaca-1 (left) and Hep-3B
(right) cell lines, treated with vehicle (DMSO), 2-Cl-IB-MECA (20
nM and 20 μM), or 30 min pre-treatment with 0.1 μM
MRS 1220 followed by 2-Cl-IB-MECA (20 nM and 20 μM),
respectively. n=3. Data was analyzed using one-way ANOVA with
Tukey's post hoc multiple comparison test. All the data are
represented as the mean ± SD. *P<0.05,
***P<0.005, #P<0.001 vs. vehicle,
2-Cl-IB-MECA vs. 2-Cl-IB-MECA + MRS 1220. RFU, relative
fluorescence units; pH3, phosphorylated histone H3; BrdU,
5-Bromo-2′-deoxyuridine; BrU, 5-Bromouridine; 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl)(1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
With respect to the JoPaca-1 cell line, the minor
changes in the number of cells in the G2/M phase were
mirrored by the decrease in the mitosis marker histone H3
phosphorylated at Ser10 (Fig. 2D).
2-Cl-IB-MECA regulates the proliferation
signaling pathways
To further understand the mechanism of 2-Cl-IB-MECA
in the pancreatic and HCC cell lines, the protein expression level
of three essential pathways, previously indicated to be affected,
either directly or indirectly, by AdoRs and their modulators
(57), was analyzed. The
JoPaca-1 and Hep-3B cell lines were treated with vehicle or 20
μM 2-Cl-IB-MECA for 24 and 48 h. The cells were pre-treated
with 0.1 μM MRS 1220 for 30 min prior to the addition of
2-Cl-IB-MECA, to confirm whether the effects were A3AR
mediated.
First, A3AR protein expression level was
analyzed after 24 and 48 h of 2-Cl-IB-MECA treatment. There was a
decrease in A3AR protein expression level in the Hep-3B
cell line, while there were no changes in the JoPaca-1 cell line
(Fig. 3). Then, the expression
level of proteins in the PI3K/AKT/NF-κB pathway were analyzed
(53). It was found in both cell
lines that the proliferation-related proteins, AKT and NF-κB, were
phosphorylated, at Ser473 and Ser536,
respectively, in a time-dependent manner, after 2-Cl-IB-MECA
treatment alone. Surprisingly, AKT kinase was dephosphorylated at
Thr308 following 2-Cl-IB-MECA treatment for 24 and 48 h
in the JoPaca-1 cell line, and after 48 h in the Hep-3B cell. line.
Furthermore, the results demonstrated that MRS 1220 pre-treatment
did not prevent 2-Cl-IB-MECA from most of its effects at 24 and 48
h, with the exception of AKT at Thr308 and pNF-κB at
Ser536 (Fig. 3).
Notably, there were differences between the JoPaca-1 and Hep-3B
cell lines in the expression and phosphorylation of ERK1/2. The
Hep-3B cell line showed a decrease in the expression level of
ERK1/2 and pERK1/2 following treatment for 48 h with 2-Cl-IB-MECA
and in combination with MRS 1220; however, total and phosphorylated
ERK1/2 protein expression level in the JoPaca-1 cell line was
increased in the same experimental groups in a time-dependent
manner.
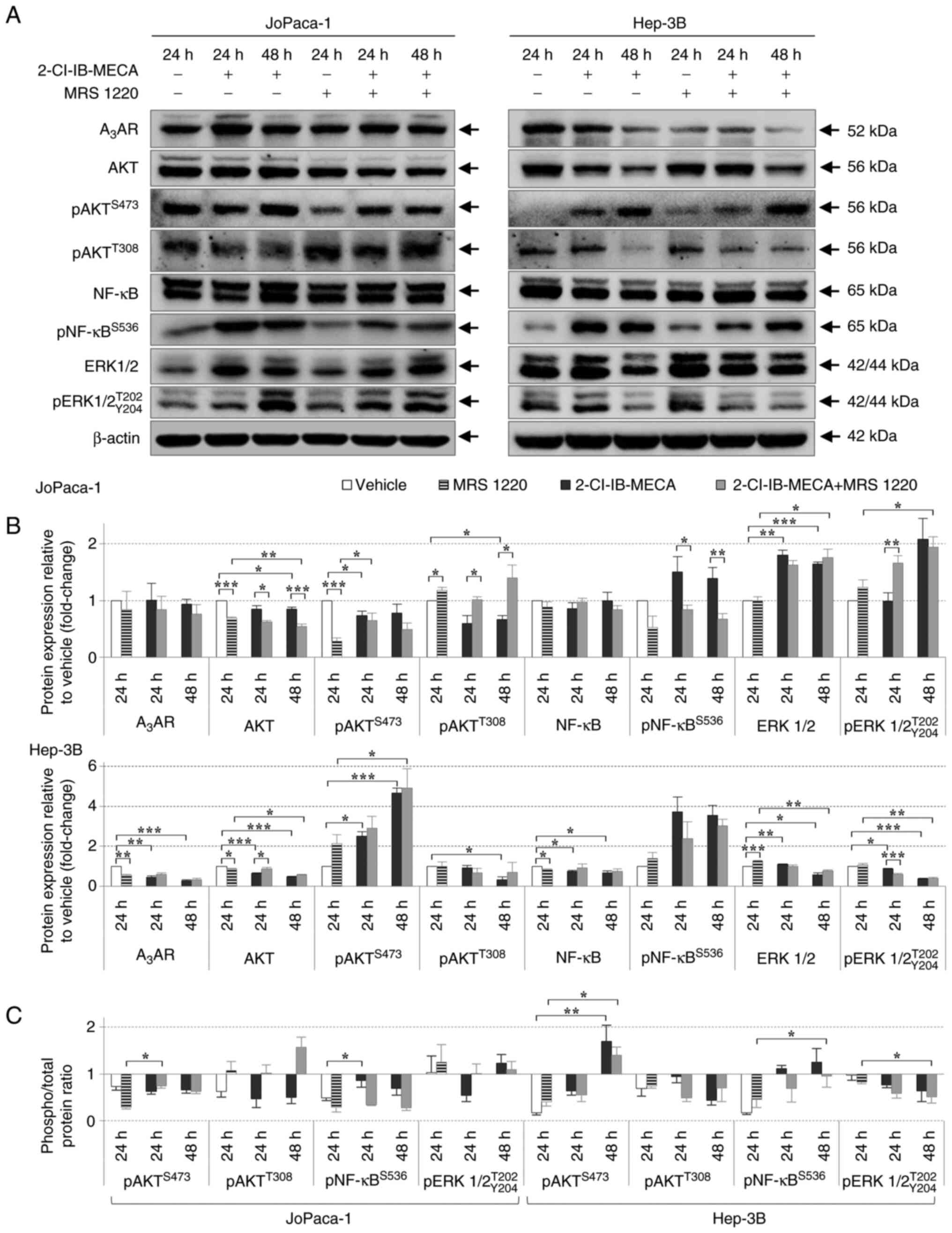 | Figure 32-Cl-IB-MECA regulates key components
of the PI3K/AKT/NF-κB proliferation signaling pathway in the
JoPaca-1 and Hep-3B cell lines. (A) Western blot analysis of the
PI3K/AKT/NF-κB pathway-related proteins in the JoPaca-1 (left) and
Hep-3B (right) cells treated either with vehicle (DMSO), 0.1
μM MRS 1220 for 24 h, 2-Cl-IB-MECA (20 μM) for 24 and
48 h, or 30 min MRS 1220 (0.1 μM) pre-treatment and
2-Cl-IB-MECA (20 μM) for 24 and 48 h. Representative blots
from 3 replicates is shown. (B) Quantitative analysis of protein
expression level. Protein relative intensities were calculated
using densitometry analysis and expressed as fold change relative
to vehicle. The data are presented as the mean ± SD. n=3. The data
were analyzed using Student's one-sample t-test (treatment vs.
vehicle), two-sample t-test and one-way ANOVA with Dunn's test
(2-Cl-IB-MECA vs. 2-Cl-IB-MECA + MRS 1220). (C) Ratio of
phosphorylated vs. total protein for JoPaca-1 (left) and Hep-3B
(right) cell lines. Statistical significance was analyzed with
one-way ANOVA with Dunn's test. Symbols indicate vehicle vs.
2-Cl-IB-MECA and vehicle vs. 2-Cl-IB-MECA + MRS 1220.
*P<0.05, **P<0.01,
***P<0.005. A3AR, A3 adenosine
receptor; p, phosphorylated; 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl) (1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
Another signaling pathway that has been associated
with several types of cancer, including pancreatic and HCC, is the
Wnt/β-catenin pathway (58,59). Therefore, the effects of
2-Cl-IB-MECA on the protein expression level of β-catenin and
GSK-3β kinase, two main molecules in the Wnt signaling pathway were
analyzed (Fig. 4). The β-catenin
protein expression levels were decreased in both cell lines
following 2-Cl-IB-MECA treatment for 24 and 48 h. Surprisingly,
GSK-3β kinase expression did not change distinctly in either cell
line within the same experimental group as β-catenin. In addition,
its phosphorylated form, pGSK-3β, remained unaltered in the
JoPaca-1 cell line, but was increased in the Hep-3B cell line after
2-Cl-IB-MECA treatment at 48 h, suggesting that kinase inactivation
only occurs in the Hep-3B cell line. Notably, these effects on the
components of the Wnt/β-catenin signaling pathway were partially
abrogated by the A3AR antagonist, MRS 1220.
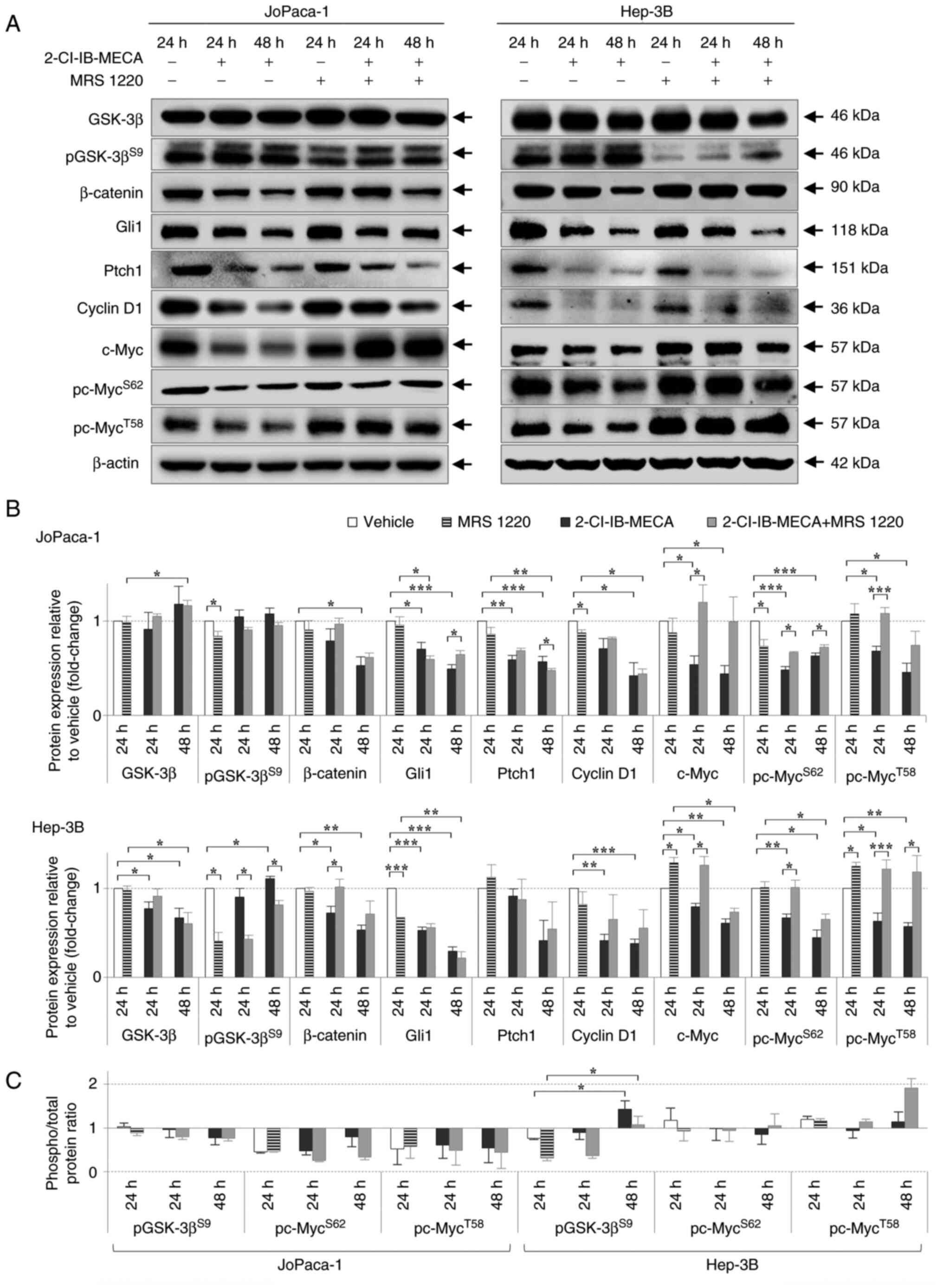 | Figure 42-Cl-IB-MECA regulates key components
of the Wnt/β-catenin and Shh/Ptch/Gli proliferation signaling
pathways in the JoPaca-1 and Hep-3B cell lines. (A) Western blot
analysis of the Wnt/β-catenin and Shh/Ptch/Gli pathway-related
proteins in the JoPaca-1 (left) and Hep-3B (right) cell lines
treated either with vehicle (DMSO), 0.1 μM MRS 1220 for 24
h, 2-Cl-IB-MECA (20 μM) for 24 and 48 h, or 30 min of MRS
1220 (0.1 μM) pre-treatment and 2-Cl-IB-MECA (20 μM)
for 24 and 48 h. Representative image from 3 replicates is shown.
(B) Quantitative analysis of protein expression level. Protein
relative intensities were calculated using densitometry and
expressed as fold change relative to vehicle. The data are
presented as the mean ± SD. n=3. The data was analyzed using
Student's one-sample t-test (treatment vs. vehicle), two-sample
t-test and one-way ANOVA with Dunn's test (2-Cl-IB-MECA vs.
2-Cl-IB-MECA + MRS 1220). (C) Ratio of phosphorylated vs. total
protein for the JoPaca-1 (left) and Hep-3B (right) cell lines.
Statistical significance was analyzed with one-way ANOVA with
Dunn's test. Symbols indicate vehicle vs. 2-Cl-IB-MECA and vehicle
vs. 2-Cl-IB-MECA + MRS 1220. *P<0.05,
**P<0.01, ***P<0.005. Shh, Sonic
Hedgehog; Ptch1, Patched; Gli, glioma-associated oncogene homolog
zinc finger protein; GSK-3β, glycogen synthase kinase-3β; p,
phosphorylated; 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl)(1,2,4)-triazolo(1,5-c)
quinazolin-5-yl]benzene acetamide. |
The Shh/Ptch/Gli axis was identified as another
relevant pathway for bypassing cell death for malignant pancreatic
and liver cells (59-61). The essential component of the Shh
pathway, the Gli1 transcription factor, further regulates the
expression level of cyclin D1, c-Myc, and Ptch1 receptor, another
key molecule of the Shh pathway. In the JoPaca-1 and Hep-3B cell
lines, 2-Cl-IB-MECA-mediated the decrease in the protein expression
level of both Gli1 and Ptch1 in a time-dependent manner.
Furthermore, the protein expression level of cyclin D1 and c-Myc,
together with the c-Myc phosphorylated forms, was reduced following
treatment with 2-Cl-IB-MECA alone for 24 and 48 h. These effects
were, however, partially reversed by MRS 1220 (Fig. 4).
Furthermore, β-catenin transcriptional activity
upon 2-Cl-IB-MECA treatment was analyzed using a reporter system at
various time points (Fig. 5).
β-catenin can bind to the TCF/LEF transcription factor, triggering
subsequent transcriptional activity of Wnt/β-catenin pathway target
genes, e.g. c-Myc and cyclin D1 (62,63). Therefore, three reporter systems
were designed to assess the transcriptional activity of: The WT
Wnt/β-catenin pathway (β-cateninWT); a β-catenin
mutation that prevents its proteasomal degradation
(β-cateninmut); and a loss of function of the APC gene,
which is critical for binding β-catenin in a complex with AXIN and,
thus, regulates β-catenin availability for transcription
(APCmut). The β-cateninWT (Fig. 5A), β-cateninmut and
APCmut (Fig. 5B)
reporter cell lines were treated with either vehicle, 50 μM
2-Cl-IB-MECA or 0.1 μM MRS 1220 for 30 min followed by 50
μM 2-Cl-IB-MECA. MTS assays were performed in parallel for
24 and 48 h to distinguish any decline in reporter activities from
simple cytotoxic effects. The results indicated early inhibition of
β-catenin transcription. WT transcriptional activity was inhibited
by 75% after 6 h (Fig. 5A).
Furthermore, 2-Cl-IB-MECA was more effective in the mutated
reporter systems, where 2-Cl-IB-MECA completely abrogated β-catenin
transcription within 12 h (Fig.
5B). This early decline in transcriptional activity was not
primarily caused by inhibited proliferation and the degree of MRS
1220 antagonism against 2-Cl-IB-MECA was insignificant in this
case.
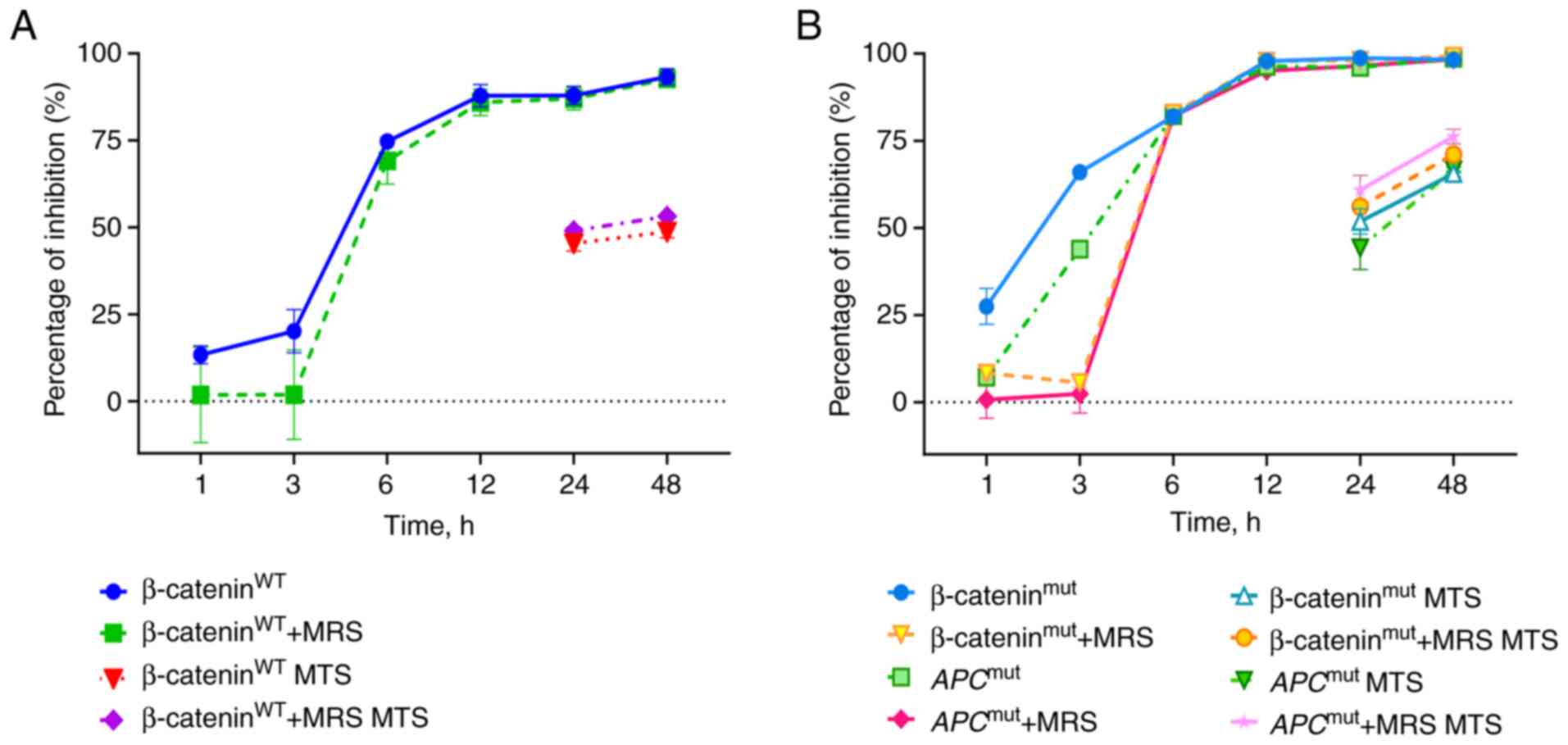 | Figure 52-Cl-IB-MECA downregulates β-catenin
transcriptional activity in WT and mutated Wnt/β-catenin reporters.
(A) Reporter assay of β-catenin transcriptional activity in
β-cateninWT and (B) mutated (β-cateninmut and
APCmut) reporter system after treatment with vehicle
(DMSO), 2-Cl-IB-MECA (50 μM), or 30 min MRS 1220 (0.1
μM) pre-treatment and 2-Cl-IB-MECA (50 μM) over time.
For cytotoxicity comparisons of reporter activities, percentage of
inhibition from a MTS assay, run in parallel, are included.
Normalized data are shown as mean ± SD. n=3. 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl)(1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide; WT, wild-type; mut, mutated. |
2-Cl-IB-MECA reduces xenobiotic
transporter MRP1 and P-gp protein expression and P-gp function
Multidrug resistance (MDR) can seriously hinder
anticancer treatment. Recently, some studies have reported an
association between AdoRs and MDR (37,43). Therefore, MRP1 and P-gp protein
expression level was analyzed in both the pancreatic and HCC cell
lines. In addition to analyzing the protein expression levels, P-gp
activity was also analyzed using a functional test of K562-TAX
overexpressing P-gp protein.
To analyze the protein expression level, the cells
were treated with either vehicle or 20 μM 2-Cl-IB-MECA for
24 h. The results indicated the reduction of MRP1 protein
expression level after treatment with 2-Cl-IB-MECA, with a more
significant effect in the Hep-3B cell line (Fig. 6A and B). P-gp expression was
analyzed using flow cytometry under the same conditions. Similarly,
P-gp overall expression was significantly attenuated by 20
μM 2-Cl-IB-MECA after 24 h (Fig. 6C and D). Of note, the Hep-3B cell
line had higher endogenous P-gp levels.
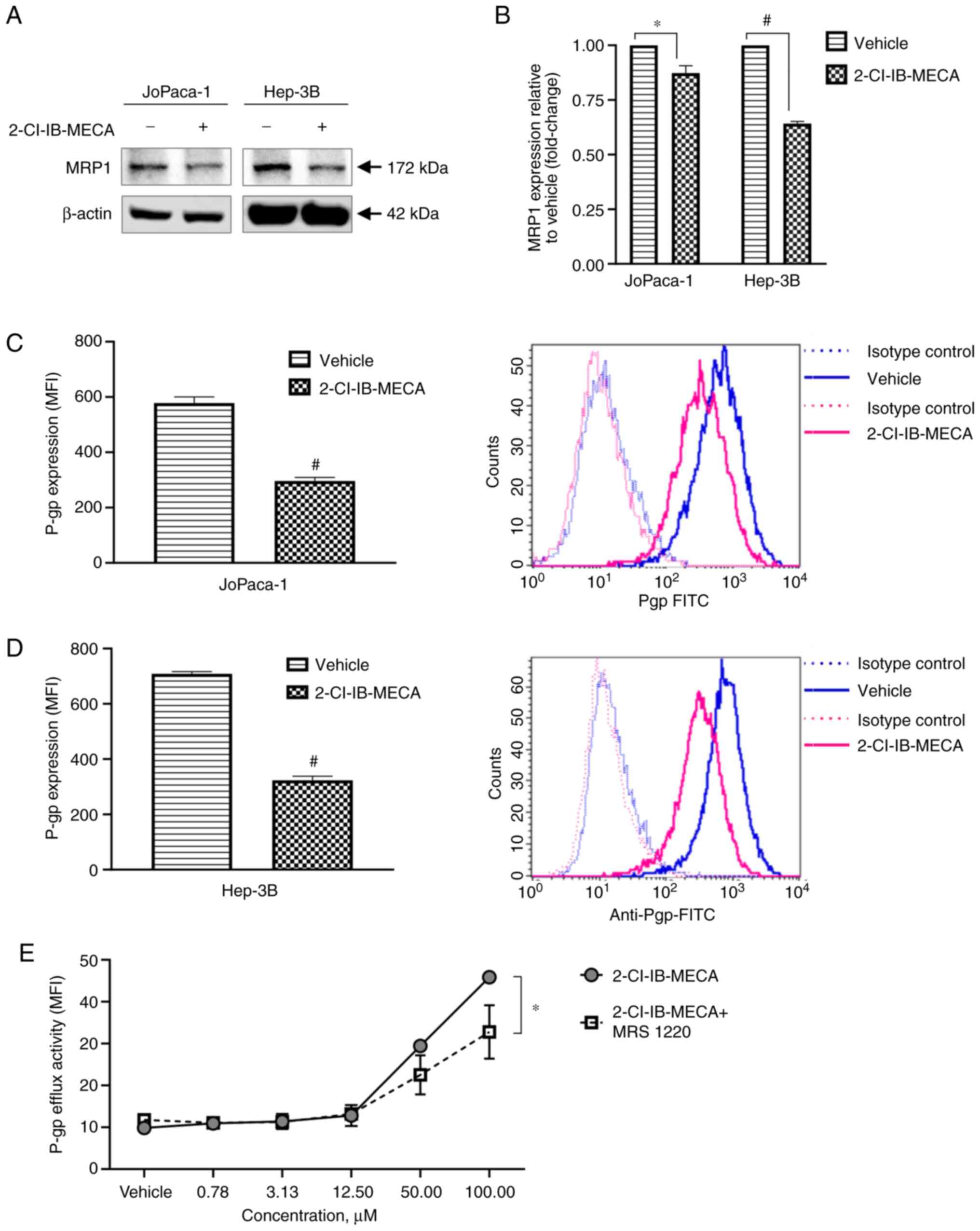 | Figure 62-Cl-IB-MECA deregulates multidrug
resistance proteins, MRP1 and P-gp protein expression level and
P-gp function. (A) Western blot analysis of MRP1 protein expression
levels in the JoPaca-1 and Hep-3B cells treated either with vehicle
(DMSO) or 2-Cl-IB-MECA (20 μM) for 24 h. (B) Densitometry
analysis from western blot analysis. n=3. The data was analyzed
using a Student's one-sample t-test, and one representative image
is shown. *P<0.05, #P<0.001 vs.
vehicle. Flow cytometry analysis of P-gp total protein expression
after vehicle (DMSO) or 2-Cl-IB-MECA (20 μM) treatment for
24 h in the (C) JoPaca-1 and (D) Hep-3B cells. The data was
analyzed using a Student's two-sample t-test, and one
representative histogram with overlays of isotype controls and
treatment is shown. #P<0.001 vs. vehicle. (E)
Quantification of P-gp efflux function after treatment with
vehicle, different concentrations of 2-Cl-IB-MECA for 1 h or 0.1
μM MRS 1220 for 30 min followed by 2-Cl-IB-MECA at different
concentrations for 1 h in the K562-TAX cell lines overexpressing
P-gp. Fluorescence intensity of daunorubicin detained or released
from cells was measured and data were quantified as mean ± SD. n=3.
The data were analyzed using Student's two-sample t-test.
*P<0.05. MFI, median fluorescence intensity; MRP1,
multidrug resistance-associated protein 1; P-gp, P-glycoprotein;
2-Cl-IB-MECA, 2-chlo
ro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS 1220,
N-[9-Chloro-2-(2-furanyl)(1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
Reducing P-gp expression could be a powerful tool
against chemoresistance in cancer cells. Nonetheless, it was also
investigated whether 2-Cl-IB-MECA could diminish the P-gp efflux
function to any extent. Chemotherapeutic agent daunorubicin serves
as the P-gp substrate and its intracellular uptake reflects P-gp
ability to actively transport this xenobiotic from the cells
(64). Therefore, P-gp
functional ability to actively effuse daunorubicin when treated
with 2-Cl-IB-MECA was analyzed. For this purpose, the K562-TAX cell
line, with high P-gp expression, was used (45). The K562-TAX cells were incubated
with 1 μM daunorubicin for 1 h prior to treatment with
either vehicle or 2-Cl-IB-MECA (0.78-100 μM) for an
additional 1 h. For pre-treatment with MRS 1220, 0.1 μM MRS
1220 was added 30 min before the 1 h incubation with 2-Cl-IB-MECA.
Within 1 h of treatment, 2-Cl-IB-MECA, at high micromolar
concentrations was able to increase P-gp efflux activity (Fig. 6E). This functional interference
suggests that 2-Cl-IB-MECA is a direct substrate of P-gp.
Surprisingly, the negative effects of 2-Cl-IB-MECA on P-gp function
were partially abolished by A3AR antagonist MRS
1220.
2-Cl-IB-MECA enhances anticancer effects
of clinically relevant drugs
The aforementioned results showed that 2-Cl-IB-MECA
mediated the inhibition of key components of the Wnt/β-catenin and
Shh/Ptch/Gli pathways, together with disruption of the xenobiotic
efflux system. These are two interlinked mechanisms in which cancer
cells can resist pharmacotherapy (60,61,65). Therefore, we hypothesized the
beneficial effects of 2-Cl-IB-MECA could be part of a combined
cancer treatment. First, cellular proliferation after treatment was
normalized to vehicle as FA, then the derived analysis of the
combination effect of two compounds was calculated. The resulting
CI values for respective concentration ratios are shown in Fig. 7. The outcomes of treatment
combinations were analyzed: 2-Cl-IB-MECA (1.56-50 μM for the
JoPaca-1 cell line and 0.78-25 μM for the Hep-3B cell line)
and each of the following four current conventional
chemotherapeutic agents: 5-FU (0.78-200 μM), carboplatin
(0.78-200 μM), doxorubicin (0.1-25 μM) and
gemcitabine (0.2-50 μM).
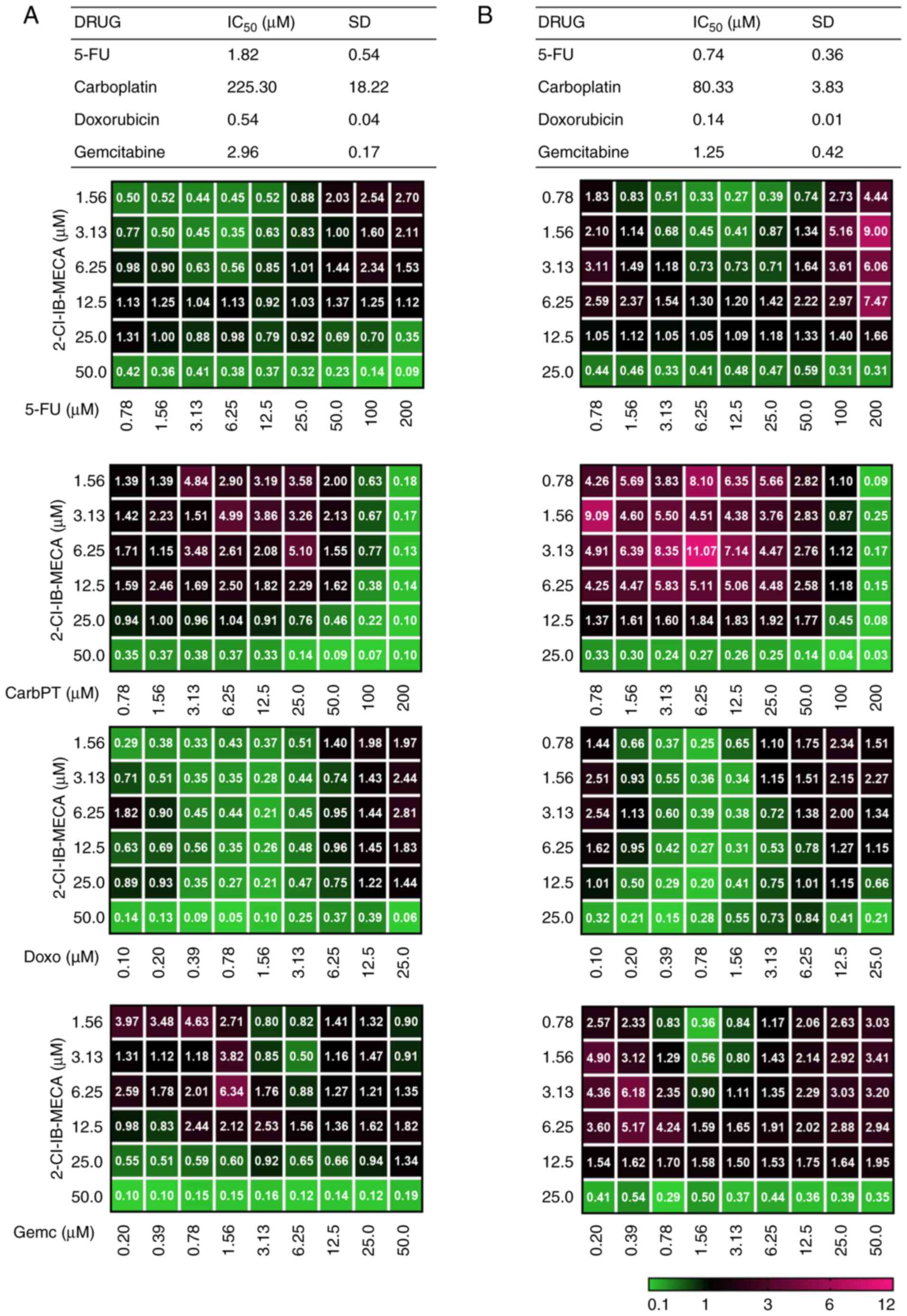 | Figure 72-Cl-IB-MECA mediates the enhancement
of cytotoxicity of conventional chemotherapeutic agents in the
JoPaca-1 and Hep-3B cell lines. (A) JoPaca-1 and (B) Hep-3B cells
were treated with each of the following chemotherapeutic agents
alone and in concurrent combination with 2-Cl-IB-MECA (1.56-50
μM for the JoPaca-1 cell line; 0.78-25 μM for the
Hep-3B cell line) for 72 h: 5-FU (0.78-200 μM), CarbPT
(0.78-200 μM), Doxo (0.1-25 μM) and Gemc (0.2-50
μM). Cytotoxicity was analyzed using a MTS assay, followed
by IC50 and CI calculations based on the Chou-Talalay
method and using the CalcuSyn software. The cytotoxic
IC50 results for each compound alone are expressed as
the mean ± SD. (n=3). The results for combination treatments are
represented as mean CI (n=3) and are color coded (green indicates
high synergy and pink indicates no synergy). 5-FU, fluorouracil;
CarbPT, carboplatin; Doxo, doxorubicin; Gemc, gemcitabine; CI,
combination index; 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl) (1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
The JoPaca-1 and Hep-3B cell lines were treated
with each compound alone and in either a concurrent or sequential
combination comprising 2-Cl-IB-MECA and one of the four
chemotherapeutics for 72 h. For sequential combinations,
2-Cl-IB-MECA was added to the cells first, followed 16 h later by
one of the chemotherapeutics, or vice versa. Possible synergistic
or additive effects of 2-Cl-IB-MECA on cancer cell sensitivity to
the aforementioned drugs were analyzed using a MTS assay. Endpoint
analysis of cytotoxicity was performed 72 h after concurrent or
sequential treatment and the CI was calculated. The degree of
synergism, additive effect or antagonism was evaluated based on the
CI method (48) and color coded
(green indicates the highest synergy, while pink indicates no
synergy).
In the JoPaca-1 cell line, synergistic effects for
2-Cl-IB-MECA concurrent combinations with carboplatin, doxorubicin
and 5-FU were found at several concentration ratios (green color).
For gemcitabine, synergy was found only if cells were concurrently
treated with 2-Cl-IB-MECA at concentrations ≥IC50
(Fig. 7A). The Hep-3B cell line
was more sensitive to 2-Cl-IB-MECA alone; therefore, the highest
concentration of 2-Cl-IB-MECA was 25 μM (Fig. 7B). Similar to the JoPaca-1 cell
line, synergy was found in the Hep-3B cell line treated with
multiple, independent, concomitant combinations of 2-Cl-IB-MECA
with either carboplatin or doxorubicin, whereas 5-FU and
gemcitabine showed synergistic effects exclusively at high
2-Cl-IB-MECA concentrations (~IC75) and low 2-Cl-IB-MECA
concentrations with 5-FU and gemcitabine at ~IC50.
Discussion
In the present study, the antitumor mechanism of
2-Cl-IB-MECA in the JoPaca-1 and Hep-3B cell lines was analyzed
(Fig. 8). The JoPaca-1 cell line
is an established cellular model for pancreatic carcinoma with high
tumorigenic potential; it shows clonal heterogeneity and
aggressiveness, thus resembling primary tumors (34). To the best of our knowledge, the
present study is the first to characterize the effect of
2-Cl-IB-MECA in the JoPaca-1 cell line in vitro. On the
contrary, the Hep-3B HCC cell line is a well-defined tumorigenic
cell line that has features of non-differentiated cells prone to
EMT (36).
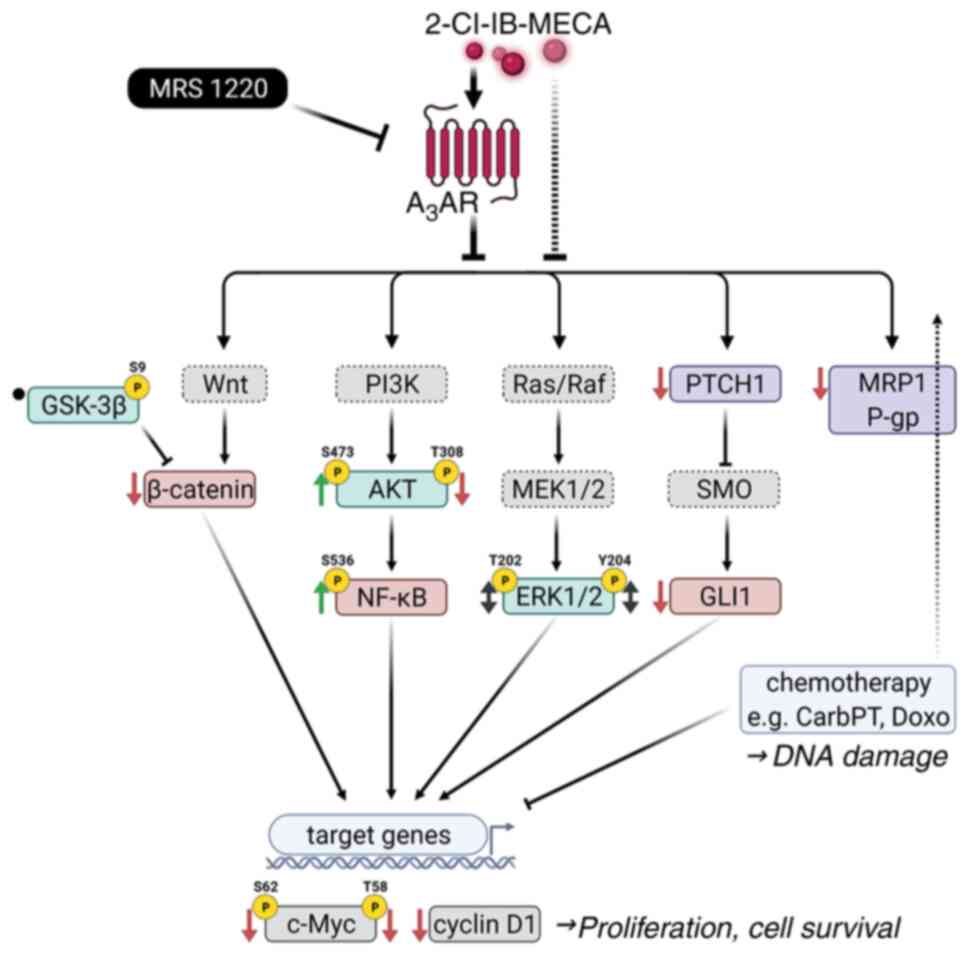 | Figure 8Schematic summary of the effect of
2-Cl-IB-MECA on intracellular signaling pathways. 2-Cl-IB-MECA
modulates key components of the PI3K/AKT/NF-κB,
Ras/Raf/MEK1/2/ERK1/2, Wnt/β-catenin, and Shh/Ptch/Gli signaling
pathways together with MRD proteins, MRP1 and P-gp, leading to
inhibition of JoPaca-1 and Hep-3B cancer cell proliferation and
survival. In cooperation with chemotherapeutic agents, carboplatin
and doxorubicin, 2-Cl-IB-MECA synergistically enhances their
cytotoxicity. Studied proteins are indicated using solid line
frames, while the others are indicated using dashed line frames.
Observed effects of 2-Cl-IB-MECA treatment are designated by ↓
(inhibition/downregulation), ↑ (activation/upregulation), ↔
(context-dependent upregulation/downregulation), and • (no
changes). A3AR, A3 adenosine receptor;
CarbPT, carboplatin; Doxo, doxorubicin; Gli1, Glioma-associated
oncogene homolog zinc finger protein 1; GSK-3β, glycogen synthase
kinase 3β; MRP1, multidrug-resistance-associated protein 1; P-gp,
P-glycoprotein; Ptch1, Patched 1; Shh, Sonic hedgehog; SMO,
Smoothened; p, phosphorylated; 2-Cl-IB-MECA,
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS
1220, N-[9-Chloro-2-(2-furanyl) (1,2,4)-triazolo(1,5-c)quinazolin-5-yl]benzene
acetamide. |
Initially, a MTS assay was performed using twelve
tumor and two non-tumor cell lines to analyze the general cytotoxic
effect of 2-Cl-IB-MECA against several tumor types. 2-Cl-IB-MECA
was cytotoxic not only for the JoPaca-1 and Hep-3B cell lines, but
also for other types of cancer cells in vitro. From the
results using the JoPaca-1 and Hep-3B cell lines, the
cytotoxicity-inducing concentration of 2-Cl-IB-MECA was used for
further experiments. Most importantly, the detrimental effects of
2-Cl-IB-MECA (Table I; Fig. 1D and E) were selective for cancer
cells. The exact mechanism of 2-Cl-IB-MECA selectivity toward tumor
cells has not yet been revealed; however, the results are
consistent with previous findings from other tumor models (11), where the overexpression of
A3AR in tumor vs. adjacent tissue was analyzed with
respect to the responsiveness of cells to A3AR-targeted
therapy. The results from the present study showed that
A3AR protein expression level was exclusive to cancer
cells (Fig. 1C) and the higher
IC50 values with respect to 2-Cl-IB-MECA in combination
with MRS1220 (Fig. 1D and E)
also corroborate this possibility. Next, A3AR functional
assay was performed to determine the activating and inhibiting
concentrations of 2-Cl-IB-MECA and MRS 1220, respectively, for use
throughout the study (Fig. 1B).
The results of which are consistent with previous studies (15,66).
Studies show that 2-Cl-IB-MECA and its predecessor,
IB-MECA disrupt cell cycle progression (56,67). In the present study, 2-Cl-IB-MECA
induced cell cycle arrest in the G0/G1 phase
with an associated decrease in the number of cells in the S phase
(Fig. 2A and B) and inhibited
DNA, and RNA synthesis (Fig. 2E and
F). Furthermore, inhibition of DNA and RNA synthesis was
previously ascribed to an AdoR-independent anticancer mechanism in
other adenosine analogues, e.g. clofarabine, fludarabine and
8-Cl-Ado (9). MRS 1220 was found
to significantly reverse 2-Cl-IB-MECA-mediated inhibition of DNA
and RNA synthesis in the present study. Notably, 20 μM
2-Cl-IB-MECA was cytotoxic for at least 50% of the cell population
(Fig. 1D and E); however, the
number of apoptotic cells in the subG1 population were
inconspicuous (Fig. 2C),
suggesting another more prevalent type of cell death in this
system.
2-Cl-IB-MECA has been described as a partially
biased signaling molecule (68).
Biased agonism could be particularly useful for eliciting the sole
therapeutic effect while reducing risks of side effects (29,30). A3AR is known to
undergo rapid desensitization and internalization, and is
thereafter degraded or recycled back to the surface to mediate its
signaling (69). The observed
differences in the protein expression levels of A3AR
between the JoPaca-1 and Hep-3B cell line after 2-Cl-IB-MECA
stimulation might contribute to the engagement of distinct
molecular pathways (Fig. 3).
The effects of 2-Cl-IB-MECA on three important
proliferative pathways, that are often deregulated in cancer, were
investigated to reveal the predominant signaling cascade that is
downregulated by 2-Cl-IB-MECA in the JoPaca-1 and Hep-3B cell line.
A3AR is a GPCR interchangeably associated with
Gi/o and Gq proteins, depending on various
factors (10). As such,
A3AR could mediate signaling either via the
αi/o, αq or β/γ subunits of the G protein.
The β/γ G protein subunit activates the PI3K/AKT pathway which, in
turn, leads to the phosphorylation of NF-κB at Ser536,
enhanced nuclear localization, and transactivation, presumably
leading to pro-survival and inflammatory signals (70). Phosphorylation of AKT at
Ser473 also leads to cross-activation of ERK1/2
signaling. The MAP kinases ERK1/2 could again be activated via the
Gαq protein subunit of the GPCR and protein kinase C, if
the activation of this pathway is a predominant event. Furthermore,
the protein kinase C pathway may also promote NF-κB stimulation
(71). The apparent and
surprising activation of AKT kinase and NF-κB in the present study
(Fig. 3) could be possibly
explained by the endoplasmic reticulum (ER) stress response pathway
(72). AKT kinase and NF-κB
could be activated during the unfolded protein response, an event
during which the ER cannot meet the high protein production demand
and cannot ensure their proper folding (73,74). Phosphorylation of both residues
is important for, but not reciprocally dependent on, high AKT
kinase activity (75). Notably,
Vincent et al (76)
showed association between phosphorylation of AKT at
Thr308 and proliferation of human non-small cell lung
carcinoma samples. No association was observed for phosphorylation
at Ser473. A decrease in AKT kinase phosphorylation at
Thr308 was found after 2-Cl-IB-MECA treatment (Fig. 3), which aligns with the overall
decrease in proliferation in the JoPaca-1 and Hep-3B cell lines. In
addition, a study by Yung et al (77) demonstrated increased
Ser473 phosphorylation alongside decreased
Thr308 AKT residues that were modulated by ER stress in
choriocarcinoma cells. In addition, previous descriptions of the ER
stress response in tumor cells related to treatments with adenosine
(78) and its analogue AICAR
(79) further supports the
notion.
When an agonist binds, A3AR, as a
Gαi-coupled receptor, inhibits adenylyl cyclase activity
and, thus, protein kinase A, which would otherwise block GSK-3β
kinase function (80). If the
Gαi subunit was the preferred route, the total GSK-3β
protein expression level should increase and pGSK-3β (inactive
form) levels should reduce. However, neither GSK-3β protein
accumulation nor pGSK-3β reduction was observed (Fig. 4). This observation contradicts
previous reports of GSK-3β kinase upregulation or activation by
2-Cl-IB-MECA (13,20) or its predecessor IB-MECA
(81). Still, both cell lines
showed decreased β-catenin expression after 2-Cl-IB-MECA treatment
(Fig. 4). Furthermore,
functional assays revealed for the first time early blockage of
β-catenin binding to the TCF/LEF transcription factor and, hence,
the inhibition of its transcriptional activity in reporter cell
lines with 2-Cl-IB-MECA treatment (Fig. 5). The effects were more evident
within reporter systems representing the defective Wnt/β-catenin
pathway (Fig. 5B). This finding
could be particularly useful for HCC, which often exhibits
β-catenin mutations (58,82).
In addition, MRS 1220 pre-treatment had limited effect on the
reporter systems.
The Shh/Ptch/Gli signaling pathway, another
evolutionarily conserved pathway, was also analyzed. Treatment with
2-Cl-IB-MECA resulted in decreased expression of both the
transcription factor Gli1 and the receptor Ptch1 (Fig. 4), two key molecules in the Shh
signaling pathway. Notably, cyclin D1 and c-Myc are downstream
targets, where several proliferation pathways, including
Shh/Ptch/Gli and Wnt/β-catenin, converge. Among other molecules,
activated NF-κB was previously shown to increase cyclin D1 and
c-Myc expression to stimulate proliferation (83,84). In the present study, cyclin D1
and c-Myc protein expression level, including c-Myc phosphorylated
forms, were significantly downregulated (Fig. 4) despite AKT and NF-κB
phosphorylation (Fig. 3),
indicating that neither AKT nor NF-κB is a predominant antitumor
target for 2-Cl-IB-MECA. Instead, β-catenin impaired
transcriptional activity and deregulated protein expression of
Gli1/Ptch1 are more likely to be the first steps, as the reporter
assay and western blot data suggest. Furthermore, the results from
the present study indicate reduction of the Wnt/β-catenin pathway
upon 2-Cl-IB-MECA treatment, that is independent of GSK-3β kinase
status.
Regulation of upstream molecules (AKT, ERK and
GSK-3β) could be explained by stimulation of A3AR as a
GPCR; however, MRS 1220 only partially reduced the effect of
2-Cl-IB-MECA on the protein expression levels of pAKT
Thr308, pNF-κB, pGSK-3β, β-catenin and c-Myc. Modulation
of the expression level of pAKT Ser473, pERK1/2 and
cyclin D1 was less dependent on MRS 1220 pre-treatment, suggesting
that an A3AR-independent mechanism also plays a
role.
An increasing number of studies promote the use of
adenosine analogues against invasive cancer cells with stem-like or
chemoresistant phenotype (55,56,85). So far, only a few studies suggest
any association between 2-Cl-IB-MECA and MDR-associated
transporters (43,86). Therefore, the potential impact of
2-Cl-IB-MECA on MDR proteins, MRP1 and P-gp in the JoPaca-1 and
Hep-3B cell lines was investigated. Treatment with 2-Cl-IB-MECA
revealed decrease in the protein expression level of both MRP1 and
P-gp (Fig. 6A-D), possibly
offering a new tool for reducing chemoresistance in cancer cells.
Furthermore, for the first time, it was found that 2-Cl-IB-MECA
could also block the P-gp efflux (Fig. 6E). Notably, MRS 1220 was able to
partially counteract this effect. The observed changes in MRP1 and
P-gp protein expression levels are consistent with recent findings
associating MRP1 and P-gp transporters, chemoresistance and the Hh
transduction pathway (61,65), and could complement the
downregulation of the Shh/Ptch/Gli axis in the JoPaca-1 and Hep-3B
cell lines treated with 2-Cl-IB-MECA. Previous research has also
associated β-catenin activity with P-gp expression (87). Collectively, the results from the
present study indicate that 2-Cl-IB-MECA could downregulate MRP1
and P-gp expression, and P-gp involvement in the transport of
chemotherapeutics, as well as xenobiotics in general via the
recruitment of A3AR.
On the other hand, evidence for the enhancement of
chemotherapeutic effects of clinically relevant drugs, that are
mediated by A3AR agonists, in vitro and in
vivo, is sparse. The use of 2-Cl-IB-MECA, together with
cyclophosphamide, inhibited growth of B16-F10 melanoma cells and
reduced the number of metastatic foci in murine lung (40). In addition, HCT116 colon
carcinoma cells, as well as tumor-bearing mice, were successfully
treated with a combination of the 2-Cl-IB-MECA predecessor,
IB-MECA, with 5-FU (84). Soares
et al (41,42) showed that 2-Cl-IB-MECA
potentiated paclitaxel cytotoxicity in human melanoma cells.
Notably, they did not find 2-Cl-IB-MECA-elicited effects to be
associated with A3AR.
Therefore, possible synergistic/additive effects of
2-Cl-IB-MECA were analyzed when used with the following therapeutic
agents from current anticancer protocols for HCC and pancreatic
cancer: 5-FU, carboplatin, doxorubicin and gemcitabine. It was
confirmed that 2-Cl-IB-MECA also facilitated the cytotoxic effects
of carboplatin and doxorubicin in the JoPaca-1 (Fig. 7A) and Hep-3B (Fig. 7B) cell lines at several
concentration ratios. By contrast, the combination of 2-Cl-IB-MECA
with either 5-FU or gemcitabine showed synergy only if a high
concentration of 2-Cl-IB-MECA was used. Surprisingly, synergistic
effects were more profound in the JoPaca-1 cell line, which are
generally more chemoresistant (34). Furthermore, the chemosensitizing
effects of 2-Cl-IB-MECA were not enhanced with sequential treatment
combinations (data not shown). Studies have shown that P-gp and
MRP1 overexpression accompanies the acquired chemoresistance of the
HCC cell lines, Hep-3B and HepG2 to doxorubicin in vitro.
Buschauer et al (88)
suggested that this phenomenon possibly occurs after transarterial
chemoembolization followed by doxorubicin chemotherapy that targets
residual HCC cells. Recently, inhibition of the P-gp transporter
improved doxorubicin anticancer effects in HCC cells (89). In addition, improved
chemosensitivity to gemcitabine was previously shown in a murine
model of pancreatic ductal adenocarcinoma after co-targeting the Hh
signaling pathway (60). Taken
together, the results from the present study on the
chemosensitizing effect of 2-Cl-IB-MECA on cancer cell lines in
vitro further support the potential of 2-Cl-IB-MECA for
combination therapy for pancreatic and liver cancer.
Whether the effects of the adenosine analogue
2-Cl-IB-MECA are mediated by A3AR is an unrelenting
question. The dependency of 2-Cl-IB-MECA on the A3AR
receptor was analyzed for its cytotoxic effect by first showing
that the JoPaca-1 and Hep-3B cells expressed A3AR. In
the present study, the A3-specific antagonist, MRS 1220
was used as a pharmacological tool for successful blocking of the
receptor function and established the antagonist concentration that
reliably blocked A3AR activity while remaining non-toxic
for the cells (0.1 μM). Indeed, MRS 1220 reduced
2-Cl-IB-MECA cytotoxicity towards cancer cell lines. However, there
is always a possibility of off-target effects of a pharmacological
inhibitor and further analyses are warranted. Notably, the
responsiveness of a biological model to A3AR
pharmacological treatments depends on the specific equilibrium
between receptor, ligand (agonist) and any other modulator
(antagonist) competing for the binding site, especially in an
artificial system (90,91). This phenomenon could explain the
results from some studies demonstrating A3AR-dependent
(13,56) and -independent effects (92,93), as well as some studies supporting
the effects of 0.1 μM MRS 1220 (54,55). Nonetheless, the cacophony of
findings regarding MRS 1220 does not reduce the potential of
2-Cl-IB-MECA or novel derivatives arising from the adenosine
structure. Furthermore, co-existing mechanisms that are dependent
and independent of the adenosine receptor were described for other
adenosine analogues (9,81,94).
Collectively, the results from the present study
provides further insights into 2-Cl-IB-MECA decreased proliferation
of the HCC and pancreatic cancer cells (Fig. 8). 2-Cl-IB-MECA negatively
regulated the Wnt/β-catenin and Shh/Ptch/Gli pathways, and for the
first time, it was shown that β-catenin transcriptional activity
was reduced. The results from the present study also highlight the
effect of 2-Cl-IB-MECA on MRP1 and P-gp transporters, and its
potential for combination treatment with chemotherapeutic agents.
Nonetheless, there are limitations of in vitro studies
utilizing pharmacological tools and these finding should be further
verified in knock-out models or animal studies.
Acknowledgments
The structure of 2-Cl-IB-MECA in Fig. 1 was created using MarvinSketch
(ChemAxon). The schematic in Fig.
8 was created using BioRender.com.
Funding
The present study was supported by the European Regional
Development Fund (Project ENOCH no.
CZ.02.1.01/0.0/0.0/16_019/0000868), the Czech Ministry of
Education, Youth and Sports (grant nos. EATRIS-CZ, LM2018133 and
CZ-OPENSCREEN, LM2018130), the Czech Science Foundation (grant no.
GACR 19-08124S) and the Technology Agency of the Czech Republic:
Czech National Centres of Competence, project 'PerMed' Personalized
Medicine - Diagnostics and Therapy (grant no. TN01000013).
Availability of data and materials
All the data generated and/or analyzed during this
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JK, PD and MH conceptualized the study. JK and PD
wrote the manuscript. JK, KL and AK designed and performed the
experiments. JK, JV and PK analyzed the data. JK, PD and MH revised
and edited the manuscript. PD and MH acquired the funding. JK and
PD confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
2-Cl-IB-MECA
|
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
|
|
5-FU
|
fluorouracil
|
|
A3AR
|
A3 adenosine receptor
|
|
AdoR
|
adenosine receptor
|
|
AUC
|
area under the curve
|
|
BrdU
|
5-bromo-2′-deoxyuridine
|
|
BrU
|
5-bromouridine
|
|
CI
|
combination index
|
|
EMEM
|
Eagle's minimum essential medium
|
|
EMT
|
epithelial-mesenchymal transition
|
|
FA
|
fraction affected
|
|
Gli1
|
glioma-associated oncogene homolog
zinc finger protein 1
|
|
GPCR
|
G protein-coupled receptor
|
|
HCC
|
hepatocellular carcinoma
|
|
Hh
|
hedgehog
|
|
IB-MECA
|
N(6)-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
|
|
IMDM
|
Iscove's modified Dulbecco's
medium
|
|
LRP
|
lung resistance protein
|
|
MDR
|
multidrug resistance
|
|
MFI
|
median fluorescence intensity
|
|
MRP1
|
multidrug resistance-associated
protein 1
|
|
MRS 1220
|
N-[9-Chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazolin-5-yl]benzene
acetamide
|
|
NECA
|
5′-N-ethylcarboxamidoadenosine
|
|
NP-40
|
nonyl phenoxypolyethoxylethanol
|
|
P-gp
|
P-glycoprotein
|
|
pH3
|
phosphorylated histone H3
|
|
PMS
|
phenazine methosulfate
|
|
Ptch1
|
patched 1
|
|
RFU
|
relative fluorescence units
|
|
Shh
|
Sonic hedgehog
|
|
SMO
|
smoothened
|
|
TCF/LEF
|
T cell factor/lymphoid enhancer
factor
|
References
|
1
|
Ferlay J, Ervik M, Lam F, Colombet M, Mery
L, Piñeros M, Znaor A, Soerjomataram I and Bray F: Global cancer
observatory: Cancer today. Lyon, France: International Agency for
Research on Cancer; 2020, Available from: https://gco.iarc.fr/today. Accessed November 24,
2021.
|
|
2
|
Rawla P, Sunkara T and Gaduputi V:
Epidemiology of pancreatic cancer: Global trends, etiology and risk
factors. World J Oncol. 10:10–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marquardt JU, Gomez-Quiroz L, Arreguin
Camacho LO, Pinna F, Lee YH, Kitade M, Domínguez MP, Castven D,
Breuhahn K, Conner EA, et al: Curcumin effectively inhibits
oncogenic NF-κB signaling and restrains stemness features in liver
cancer. J Hepatol. 63:661–669. 2015. View Article : Google Scholar :
|
|
5
|
Zeng SY, Pottler M, Lan B, Grutzmann R,
Pilarsky C and Yang H: Chemoresistance in pancreatic cancer. Int J
Mol Sci. 20:45042019. View Article : Google Scholar
|
|
6
|
Adamska A and Falasca M: ATP-binding
cassette transporters in progression and clinical outcome of
pancreatic cancer: What is the way forward? World J Gastroenterol.
24:3222–3238. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lohitesh K, Chowdhury R and Mukherjee S:
Resistance a major hindrance to chemotherapy in hepatocellular
carcinoma: An insight. Cancer Cell Int. 18:442018. View Article : Google Scholar
|
|
8
|
Liu A, Wu Q, Peng D, Ares I, Anadón A,
Lopez-Torres B, Martínez-Larrañaga MR, Wang X and Martínez MA: A
novel strategy for the diagnosis, prognosis, treatment, and
chemoresistance of hepatocellular carcinoma: DNA methylation. Med
Res Rev. 40:1973–2018. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Man S, Lu Y, Yin L, Cheng X and Ma L:
Potential and promising anticancer drugs from adenosine and its
analogs. Drug Discov Today. 26:1490–1500. 2021. View Article : Google Scholar
|
|
10
|
Fredholm BB, IJzerman AP, Jacobson KA,
Linden J and Müller CE: International union of basic and clinical
pharmacology. LXXXI. Nomenclature and classification of adenosine
receptors-an update. Pharmacol Rev. 63:1–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Madi L, Ochaion A, Rath-Wolfson L,
Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F
and Fishman P: The A3 adenosine receptor is highly expressed in
tumor versus normal cells: Potential target for tumor growth
inhibition. Clin Cancer Res. 10:4472–4479. 2004. View Article : Google Scholar
|
|
12
|
Morello S, Petrella A, Festa M, Popolo A,
Monaco M, Vuttariello E, Chiappetta G, Parente L and Pinto A:
Cl-IB-MECA inhibits human thyroid cancer cell proliferation
independently of A3 adenosine receptor activation. Cancer Biol
Ther. 7:278–284. 2008. View Article : Google Scholar
|
|
13
|
Bar-Yehuda S, Stemmer SM, Madi L, Castel
D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L
and Fishman P: The A3 adenosine receptor agonist CF102 induces
apoptosis of hepatocellular carcinoma via de-regulation of the Wnt
and NF-kappaB signal transduction pathways. Int J Oncol.
33:287–295. 2008.PubMed/NCBI
|
|
14
|
Gessi S, Cattabriga E, Avitabile A, Gafa'
R, Lanza G, Cavazzini L, Bianchi N, Gambari R, Feo C, Liboni A, et
al: Elevated expression of A3 adenosine receptors in human
colorectal cancer is reflected in peripheral blood cells. Clin
Cancer Res. 10:5895–5901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles
GL and Jacobson KA: 2-Substitution of
N6-benzyladenosine-5′-uronamides enhances selectivity for A3
adenosine receptors. J Med Chem. 37:3614–3621. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van Schaick EA, Jacobson KA, Kim HO,
Ijzerman AP and Danhof M: Hemodynamic effects and histamine release
elicited by the selective adenosine A3 receptor agonist
2-Cl-IB-MECA in conscious rats. Eur J Pharmacol. 308:311–314. 1996.
View Article : Google Scholar
|
|
17
|
Wittendorp MC, Biber K and Boddeke HWGM:
CL-IB-MECA induced release of CCL2 by astrocytes: Possible role for
the adenosine A3 receptor? Naunyn-Schmiedeb Arch Pharmacol.
369:R1782004.
|
|
18
|
Ge ZD, Peart JN, Kreckler LM, Wan TC,
Jacobson MA, Gross GJ and Auchampach JA: Cl-IB-MECA
[2-chloro-N6-(3-iodobenzyl) adenosine-5′-N-methylcarboxamide]
reduces ischemia/reperfusion injury in mice by activating the A3
adenosine receptor. J Pharmacol Exp Ther. 319:1200–1210. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Coppi E, Cherchi F, Fusco I, Failli P,
Vona A, Dettori I, Gaviano L, Lucarini E, Jacobson KA, Tosh DK, et
al: Adenosine A3 receptor activation inhibits pronociceptive N-type
Ca2+ currents and cell excitability in dorsal root
ganglion neurons. Pain. 160:1103–1118. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cohen S, Stemmer SM, Zozulya G, Ochaion A,
Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA and
Fishman P: CF102 an A3 adenosine receptor agonist mediates
anti-tumor and anti-inflammatory effects in the liver. J Cell
Physiol. 226:2438–2447. 2011. View Article : Google Scholar
|
|
21
|
Morello S, Sorrentino R, Montinaro A,
Luciano A, Maiolino P, Ngkelo A, Arra C, Adcock IM and Pinto A:
NK1.1 cells and CD8 T cells mediate the antitumor activity of
Cl-IB-MECA in a mouse melanoma model. Neoplasia. 13:365–373. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bar Yehuda S, Stemmer SM, Madi L, Castel
D, Ochaion A, Cohen S, Barer F, Perez-Liz G, Del Valle L and
Fishman P: Effect of CF102 on growth suppression and apoptosis in
an orthotopic model of hepatocellular carcinoma. J Clin Oncol.
26(Suppl 15): S221132008. View Article : Google Scholar
|
|
23
|
Safadi R, Braun M, Francis A, Milgrom Y,
Massarwa M, Hakimian D, Hazou W, Issachar A, Harpaz Z, Farbstein M,
et al: Randomised clinical trial: A phase 2 double-blind study of
namodenoson in non-alcoholic fatty liver disease and
steatohepatitis. Aliment Pharmacol Ther. 54:1405–1415. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stemmer SM, Manojlovic NS, Marinca MV,
Petrov P, Cherciu N, Ganea D, Ciuleanu TE, Puscas IA, Beg MS,
Purcell WT, et al: A phase II, randomized, double-blind,
placebo-controlled trial evaluating efficacy and safety of
namodenoson (CF102), an A3 adenosine receptor agonist (A3AR), as a
second-line treatment in patients with Child-Pugh B (CPB) advanced
hepatocellular carcinoma (HCC). J Clin Oncol. 37(Suppl 15):
S25032019. View Article : Google Scholar
|
|
25
|
Stemmer SM, Manojlovic NS, Marinca MV,
Petrov P, Cherciu N, Ganea D, Ciuleanu TE, Pusca IA, Beg MS,
Purcell WT, et al: Namodenoson in advanced hepatocellular carcinoma
and Child-Pugh B cirrhosis: Randomized placebo-controlled clinical
trial. Cancers (Basel). 13:1872021. View Article : Google Scholar
|
|
26
|
Ohana G, Cohen S, Rath-Wolfson L and
Fishman P: A3 adenosine receptor agonist, CF102, protects against
hepatic ischemia/reperfusion injury following partial hepatectomy.
Mol Med Rep. 14:4335–4341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
David M, Gospodinov DK, Gheorghe N, Mateev
GS, Rusinova MV, Hristakieva E, Solovastru LG, Patel RV,
Giurcaneanu C, Hitova MC, et al: Treatment of plaque-type psoriasis
with oral CF101: Data from a phase II/III multicenter, randomized,
controlled trial. J Drugs Dermatol. 15:931–938. 2016.PubMed/NCBI
|
|
28
|
Storme J, Tosh DK, Gao ZG, Jacobson KA and
Stove CP: Probing structure-activity relationship in β-arrestin2
recruitment of diversely substituted adenosine derivatives. Biochem
Pharmacol. 158:103–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suresh RR, Jain S, Chen Z, Tosh DK, Ma Y,
Podszun MC, Rotman Y, Salvemini D and Jacobson KA: Design and in
vivo activity of A3 adenosine receptor agonist prodrugs.
Purinergic Signal. 16:367–377. 2020. View Article : Google Scholar :
|
|
30
|
Pottie E, Tosh DK, Gao ZG, Jacobson KA and
Stove CP: Assessment of biased agonism at the A3
adenosine receptor using β-arrestin and miniGαi
recruitment assays. Biochem Pharmacol. 177:1139342020. View Article : Google Scholar
|
|
31
|
Kim SJ, Min HY, Chung HJ, Park EJ, Hong
JY, Kang YJ, Shin DH, Jeong LS and Lee SK: Inhibition of cell
proliferation through cell cycle arrest and apoptosis by
thio-Cl-IB-MECA, a novel A3 adenosine receptor agonist, in human
lung cancer cells. Cancer Lett. 264:309–315. 2008. View Article : Google Scholar
|
|
32
|
Baltos JA, Paoletta S, Nguyen AT, Gregory
KJ, Tosh DK, Christopoulos A, Jacobson KA and May LT:
Structure-activity analysis of biased agonism at the human
adenosine A3 receptor. Mol Pharmacol. 90:12–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vecchio EA, Baltos JA, Nguyen ATN,
Christopoulos A, White PJ and May LT: New paradigms in adenosine
receptor pharmacology: Allostery, oligomerization and biased
agonism. Br J Pharmacol. 175:4036–4046. 2018. View Article : Google Scholar :
|
|
34
|
Fredebohm J, Boettcher M, Eisen C, Gaidaμ
M, Heller A, Keleg S, Tost J, Greulich-Bode KM, Hotz-Wagenblatt A,
Lathrop M, et al: Establishment and characterization of a highly
tumourigenic and cancer stem cell enriched pancreatic cancer cell
line as a well defined model system. PLoS One. 7:e485032012.
View Article : Google Scholar
|
|
35
|
Novak I, Yu H, Magni L and Deshar G:
Purinergic signaling in pancreas-from physiology to therapeutic
strategies in pancreatic cancer. Int J Mol Sci. 21:87812020.
View Article : Google Scholar
|
|
36
|
Qiu GH, Xie X, Xu F, Shi XH, Wang Y and
Deng L: Distinctive pharmacological differences between liver
cancer cell lines HepG2 and Hep3B. Cytotechnology. 67:1–12. 2015.
View Article : Google Scholar :
|
|
37
|
Torres A, Vargas Y, Uribe D, Jaramillo C,
Gleisner A, Salazar-Onfray F, López MN, Melo R, Oyarzún C, San
Martín R and Quezada C: Adenosine A3 receptor elicits
chemoresistance mediated by multiple resistance-associated
protein-1 in human glioblastoma stem-like cells. Oncotarget.
7:67373–67386. 2016. View Article : Google Scholar
|
|
38
|
Torres Á, Erices JI, Sanchez F, Ehrenfeld
P, Turchi L, Virolle T, Uribe D, Niechi I, Spichiger C, Rocha JD,
et al: Extracellular adenosine promotes cell migration/invasion of
glioblastoma stem-like cells through A3 Adenosine
Receptor activation under hypoxia. Cancer Lett. 446:112–122. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Montraveta A, Xargay-Torrent S,
López-Guerra M, Rosich L, Pérez-Galán P, Salaverria I, Beà S, Kalko
SG, de Frias M, Campàs C, et al: Synergistic anti-tumor activity of
acadesine (AICAR) in combination with the anti-CD20 monoclonal
antibody rituximab in in vivo and in vitro models of mantle cell
lymphoma. Oncotarget. 5:726–739. 2014. View Article : Google Scholar :
|
|
40
|
Fishman P, Bar-Yehuda S, Barer F, Madi L,
Multani AS and Pathak S: The A3 adenosine receptor as a new target
for cancer therapy and chemoprotection. Exp Cell Res. 269:230–236.
2001. View Article : Google Scholar
|
|
41
|
Soares AS, Costa VM, Diniz C and Fresco P:
The combination of Cl-IB-MECA with paclitaxel: A new
anti-metastatic therapeutic strategy for melanoma. Cancer Chemother
Pharmacol. 74:847–860. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Soares AS, Costa VM, Diniz C and Fresco P:
Potentiation of cytotoxicity of paclitaxel in combination with
Cl-IB-MECA in human C32 metastatic melanoma cells: A new possible
therapeutic strategy for melanoma. Biomed Pharmacother. 67:777–789.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mlejnek P, Dolezel P and Kosztyu P:
P-glycoprotein mediates resistance to A3 adenosine receptor agonist
2-chloro-N6-(3-io dobenzyl)-adenosine-5′-n-methyluronamide in human
leukemia cells. J Cell Physiol. 227:676–685. 2012. View Article : Google Scholar
|
|
44
|
Abel B, Tosh DK, Durell SR, Murakami M,
Vahedi S, Jacobson KA and Ambudkar SV: Evidence for the interaction
of A3 adenosine receptor agonists at the drug-binding
site(s) of human P-glycoprotein (ABCB1). Mol Pharmacol. 96:180–192.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Noskova V, Dzubak P, Kuzmina G, Ludkova A,
Stehlik D, Trojanec R, Janostakova A, Korinkova G, Mihal V and
Hajduch M: In vitro chemoresistance profile and expression/function
of MDR associated proteins in resistant cell lines derived from
CCRF-CEM, K562, A549 and MDA MB 231 parental cells. Neoplasma.
49:418–425. 2002.
|
|
46
|
Le Poul E, Hisada S, Mizuguchi Y, Dupriez
VJ, Burgeon E and Detheux M: Adaptation of aequorin functional
assay to high throughput screening. J Biomol Screen. 7:57–65. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Borková L, Frydrych I, Jakubcová N, Adámek
R, Lišková B, Gurská S, Medvedíková M, Hajdúch M and Urban M:
Synthesis and biological evaluation of triterpenoid thiazoles
derived from betulonic acid, dihydrobetulonic acid, and ursonic
acid. Eur J Med Chem. 185:1118062020. View Article : Google Scholar
|
|
48
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar
|
|
50
|
Bourderioux A, Naus P, Perlíková P, Pohl
R, Pichová I, Votruba I, Dzubák P, Konecný P, Hajdúch M, Stray KM,
et al: Synthesis and significant cytostatic activity of
7-hetaryl-7-deazaadenosines. J Med Chem. 54:5498–5507. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH image to imageJ: 25 Years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dzubák P, Hajdúch M, Gazák R, Svobodová A,
Psotová J, Walterová D, Sedmera P and Kren V: New derivatives of
silybin and 2,3-dehydrosilybin and their cytotoxic and
P-glycoprotein modulatory activity. Bioorg Med Chem. 14:3793–3810.
2006. View Article : Google Scholar
|
|
53
|
Borea PA, Varani K, Vincenzi F, Baraldi
PG, Tabrizi MA, Merighi S and Gessi S: The A3 adenosine receptor:
History and perspectives. Pharmacol Rev. 67:74–102. 2015.
View Article : Google Scholar
|
|
54
|
Laudadio MA and Psarropoulou C: The A3
adenosine receptor agonist 2-Cl-IB-MECA facilitates epileptiform
discharges in the CA3 area of immature rat hippocampal slices.
Epilepsy Res. 59:83–94. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jafari SM, Panjehpour M, Aghaei M,
Joshaghani HR and Enderami SE: A3 adenosine receptor agonist
inhibited survival of breast cancer stem cells via GLI-1 and ERK1/2
pathway. J Cell Biochem. 118:2909–2920. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Merighi S, Benini A, Mirandola P, Gessi S,
Varani K, Leung E, Maclennan S and Borea PA: A3 adenosine receptor
activation inhibits cell proliferation via phosphatidylinositol
3-kinase/Akt-dependent inhibition of the extracellular
signal-regulated kinase 1/2 phosphorylation in A375 human melanoma
cells. J Biol Chem. 280:19516–19526. 2005. View Article : Google Scholar
|
|
57
|
Borea PA, Gessi S, Merighi S, Vincenzi F
and Varani K: Pharmacology of adenosine receptors: The state of the
art. Physiol Rev. 98:1591–1625. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Haines K, Sarabia SF, Alvarez KR,
Tomlinson G, Vasudevan SA, Heczey AA, Roy A, Finegold MJ, Parsons
DW, Plon SE, et al: Characterization of pediatric hepatocellular
carcinoma reveals genomic heterogeneity and diverse signaling
pathway activation. Pediatr Blood Cancer. 66:e277452019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jones S, Zhang XS, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ding J, Zhou XT, Zou HY and Wu J: Hedgehog
signaling pathway affects the sensitivity of hepatoma cells to drug
therapy through the ABCC1 transporter. Lab Invest. 97:819–832.
2017. View Article : Google Scholar
|
|
62
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
64
|
Spoelstra EC, Westerhoff HV, Pinedo HM,
Dekker H and Lankelma J: The multidrug-resistance-reverser
verapamil interferes with cellular P-glycoprotein-mediated pumping
of daunorubicin as a non-competing substrate. Eur J Biochem.
221:363–373. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Queiroz KCS, Ruela-de-Sousa RR, Fuhler GM,
Aberson HL, Ferreira CV, Peppelenbosch MP and Spek CA: Hedgehog
signaling maintains chemoresistance in myeloid leukemic cells.
Oncogene. 29:6314–6322. 2010. View Article : Google Scholar
|
|
66
|
Jacobson KA: Adenosine A3 receptors: Novel
ligands and paradoxical effects. Trends Pharmacol Sci. 19:184–191.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Aghaei M, Panjehpour M, Karami-Tehrani F
and Salami S: Molecular mechanisms of A3 adenosine receptor-induced
G1 cell cycle arrest and apoptosis in androgen-dependent and
independent prostate cancer cell lines: Involvement of intrinsic
pathway. J Cancer Res Clin Oncol. 137:1511–1523. 2011. View Article : Google Scholar
|
|
68
|
Gao ZG and Jacobson KA: Translocation of
arrestin induced by human A3 adenosine receptor ligands in an
engineered cell line: Comparison with G protein-dependent pathways.
Purinergic Signal. 4:S78–S79. 2008.
|
|
69
|
Mundell S and Kelly E: Adenosine receptor
desensitization and trafficking. Biochim Biophys Acta.
1808:1319–1328. 2011. View Article : Google Scholar
|
|
70
|
Hu J, Nakano H, Sakurai H and Colburn NH:
Insufficient p65 phosphorylation at S536 specifically contributes
to the lack of NF-kappaB activation and transformation in resistant
JB6 cells. Carcinogenesis. 25:1991–2003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16(Suppl 2):
S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hasnain SZ, Lourie R, Das I, Chen AC and
McGuckin MA: The interplay between endoplasmic reticulum stress and
inflammation. Immunol Cell Biol. 90:260–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tam AB, Mercado EL, Hoffmann A and Niwa M:
ER stress activates NF-κB by integrating functions of basal IKK
activity, IRE1 and PERK. PLoS One. 7:e450782012. View Article : Google Scholar
|
|
74
|
Makhov P, Naito S, Haifler M, Kutikov A,
Boumber Y, Uzzo RG and Kolenko VM: The convergent roles of NF-κB
and ER stress in sunitinib-mediated expression of pro-tumorigenic
cytokines and refractory phenotype in renal cell carcinoma. Cell
Death Dis. 9:3742018. View Article : Google Scholar
|
|
75
|
Alessi DR, Andjelkovic M, Caudwell B, Cron
P, Morrice N, Cohen P and Hemmings BA: Mechanism of activation of
protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551. 1996.
View Article : Google Scholar
|
|
76
|
Vincent EE, Elder DJ, Thomas EC, Phillips
L, Morgan C, Pawade J, Sohail M, May MT, Hetzel MR and Tavaré JM:
Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt
protein kinase activity in human non-small cell lung cancer. Br J
Cancer. 104:1755–1761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yung HW, Charnock-Jones DS and Burton GJ:
Regulation of AKT phosphorylation at Ser473 and Thr308 by
endoplasmic reticulum stress modulates substrate specificity in a
severity dependent manner. PLoS One. 6:e178942011. View Article : Google Scholar :
|
|
78
|
Wu LF, Wei BL, Guo YT, Ye YQ, Li GP, Pu ZJ
and Feng JL: Apoptosis induced by adenosine involves endoplasmic
reticulum stress in EC109 cells. Int J Mol Med. 30:797–804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nie J, Liu A, Tan Q, Zhao K, Hu K, Li Y,
Yan B and Zhou L: AICAR activates ER stress-dependent apoptosis in
gallbladder cancer cells. Biochem Biophys Res Commun. 482:246–252.
2017. View Article : Google Scholar
|
|
80
|
Ding L and Billadeau DD: Glycogen synthase
kinase-3β: A novel therapeutic target for pancreatic cancer. Expert
Opin Ther Targets. 24:417–426. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fishman P, Bar Yehuda S, Stemmer SM and
Madi L: CF101 enhances the apoptotic effect of chemotherapy on
colon and pancreatic carcinoma cell lines: Molecular mechanisms
involved. J Clin Oncol. 22(Suppl 14): S31732004. View Article : Google Scholar
|
|
82
|
Kwee SA and Tiirikainen M: Beta-catenin
activation and immunotherapy resistance in hepatocellular
carcinoma: Mechanisms and biomarkers. Hepatoma Res.
7:82021.PubMed/NCBI
|
|
83
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-kappaB function in growth control:
Regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999. View Article : Google Scholar
|
|
84
|
Bar-Yehuda S, Madi L, Silberman D, Gery S,
Shkapenuk M and Fishman P: CF101, an agonist to the A3 adenosine
receptor, enhances the chemotherapeutic effect of 5-fluorouracil in
a colon carcinoma murine model. Neoplasia. 7:85–90. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Varani K, Vincenzi F, Targa M, Paradiso B,
Parrilli A, Fini M, Lanza G and Borea PA: The stimulation of A(3)
adenosine receptors reduces bone-residing breast cancer in a rat
preclinical model. Eur J Cancer. 49:482–491. 2013. View Article : Google Scholar
|
|
86
|
Frydrych I, Dolezel P and Mlejnek P:
P-glycoprotein overexpression confers resistance to A3 adenosine
receptor agonists
2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide
(Cl-IB-MECA) in human leukemia cells. Purinergic Signal. 4(Suppl
1): S1–S210. 2008.
|
|
87
|
Lim JC, Kania KD, Wijesuriya H, Chawla S,
Sethi JK, Pulaski L, Romero IA, Couraud PO, Weksler BB, Hladky SB
and Barrand MA: Activation of beta-catenin signalling by GSK-3
inhibition increases p-glycoprotein expression in brain endothelial
cells. J Neurochem. 106:1855–1865. 2008.PubMed/NCBI
|
|
88
|
Buschauer S, Koch A, Wiggermann P, Müller
M and Hellerbrand C: Hepatocellular carcinoma cells surviving
doxorubicin treatment exhibit increased migratory potential and
resistance to doxorubicin re-treatment in vitro. Oncol Lett.
15:4635–4640. 2018.
|
|
89
|
Yin W, Xiang D, Wang T, Zhang Y, Pham CV,
Zhou S, Jiang G, Hou Y, Zhu Y, Han Y, et al: The inhibition of
ABCB1/MDR1 or ABCG2/BCRP enables doxorubicin to eliminate liver
cancer stem cells. Sci Rep. 11:107912021. View Article : Google Scholar :
|
|
90
|
Hoare SRJ: The problems of applying
classical pharmacology analysis to modern in vitro drug discovery
assays: Slow binding kinetics and high target concentration. SLAS
Discov. 26:835–850. 2021.PubMed/NCBI
|
|
91
|
Fredholm BB: Adenosine receptors as drug
targets. Exp Cell Res. 316:1284–1288. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kim SG, Ravi G, Hoffmann C, Jung YJ, Kim
M, Chen A and Jacobson KA: p53-Independent induction of Fas and
apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA.
Biochem Pharmacol. 63:871–880. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mlejnek P, Dolezel P and Frydrych I:
Effects of synthetic A3 adenosine receptor agonists on cell
proliferation and viability are receptor independent at micromolar
concentrations. J Physiol Biochem. 69:405–417. 2013. View Article : Google Scholar
|
|
94
|
Jajoo S, Mukherjea D, Watabe K and
Ramkumar V: Adenosine A(3) receptor suppresses prostate cancer
metastasis by inhibiting NADPH oxidase activity. Neoplasia.
11:1132–1145. 2009. View Article : Google Scholar :
|














