Introduction
Vascular calcification (VC) is characterized by the
abnormal deposition of minerals in the wall of blood vessels and is
commonly observed in patients with chronic kidney disease (CKD)
(1). VC can be divided into
intima calcification and media calcification according to the
calcified site, of which the VC characteristic in patients with CKD
mostly involves the tunica media (2). Calcification of the tunica media
leads to an increase in vascular stiffness and decreased
compliance, resulting in higher pulse pressure, left ventricular
hypertrophy, and changes in left ventricular pressure and coronary
perfusion (3). It has been
demonstrated that VC is associated with an increase in all-cause
mortality, especially from cardiovascular causes, such as ischemic
heart disease (4-6). Although several medications may ease
the progression of VC, there is still no proven treatment for it in
clinical practice (7). Therefore,
early prevention and treatment of VC have important clinical
significance for reducing cardiovascular complications and
improving the prognosis of patients with CKD.
VC is considered to be an active, tunable
physiological process that involves abnormalities in calcium (Ca)
and phosphorus metabolism and differentiation of vascular smooth
muscle cells (VSMCs) (1,2,8).
Intima calcification usually starts from atherosclerotic plaques,
while early medial calcification begins at focal medial
calcification and gradually expands to whole-vessel calcification
(2). VC has been previously
considered to occur at the end stage of CKD (9); however, epidemiological studies have
suggested that VC could also be observed even at the early stage of
CKD, which is mainly characterized by abnormal blood biochemistry
(10-12). How these abnormal signals are
transmitted to VSMCs in the tunica media through the intima of
vessels and then initiate the whole process of VC remains to be
elucidated.
Exosomes, a type of extracellular vesicle (EV) with
a diameter of 30-200 nm, can carry bioactive molecules, including
proteins, nucleic acids and lipids, and serve a role in signal
transmission between adjacent cells or distal cells, which can be
widely found in body fluids, including blood and urine (13). Exosomes have been reported to
participate in VC through various mechanisms (14). Pan et al (15) added exosomes extracted from a VC
mouse model to VSMCs and found that they also began to express
calcification-related proteins, suggesting that the calcification
signal could be transmitted between VSMCs through exosomes. Li
et al (16) found that
exosomes secreted by high glucose-stimulated vascular endothelial
cells (ECs) promoted aging and Ca deposition in VSMCs, suggesting
that exosomes may serve a role in diabetes-related VC. The
interplay between ECs and VSMCs is the main driver of vascular
pathological changes, which also serve a key role in the process of
VC (14,17). Most studies have focused on the
effects of pathological stimuli on ECs, which compose the inner
layer of blood vessels, since they are directly stimulated by
factors such as hyperglycemia (16,18-20). Liu et al (19) reported that the effect of ECs on
VSMC calcification was achieved by secreting soluble factors such
as cholestane-3β, 5α and 6β-triol, demonstrating the roles of ECs
in VC modulated by oxysterols in atherosclerotic plaques.
Hyperglycemia-stimulated ECs release exosomal Notch3 and versican,
which can be taken up by VSMCs and linked to their
calcification/senescence, indicating the role of exosomes in
extracellular communication (16,20). Therefore, we hypothesized that
abnormal biochemical factors in the early stage of CKD, especially
higher phosphorus, could stimulate vascular ECs and transmit this
abnormal signal to the tunica media through exosomes, initiating
the process of VC in CKD.
The present study explored the role and mechanism of
exosomes derived from high phosphorus (HP)-stimulated HUVECs in
regulating VSMC calcification. The aims of the present study were
to investigate whether HP-HUVEC-Exos could promote VSMC
calcification and to determine which contents of HP-HUVEC-Exos may
serve a role in regulating calcification and its potential
signaling pathway.
Materials and methods
Cell culture and transfection
Human aortic VSMCs were purchased from American Type
Culture Collection (ATCC-CRL-1999) and HUVECs were purchased from
iCell Bioscience, Inc. VSMCs were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (MilliporeSigma)
and 1% penicillin/streptomycin. HUVECs were cultured in ECM medium
(ScienCell Research Laboratories, Inc.) with 10% FBS
(MilliporeSigma) and 1% penicillin/streptomycin. Cells were
cultured at 37°C in a humidified atmosphere with 5% CO2
and passaged every 2-3 days.
For cell transfection, STAT1 small interfering RNA
(si-STAT1; cat. no. stB0000954A) and si-negative control (si-NC;
cat. no. siN0000001-1-5; the sequence is not publicly available)
oligos (Guangzhou RiboBio Co., Ltd.), and STAT1 overexpression
(o/e) and its negative control (NC) plasmids based on the backbone
of pCMV3-C-HA (Sino Biological, Inc.) were transfected into VSMCs
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) at a final concentration of 50 nM. Briefly, VSMCs were plated
at a density of 5×105 cells in 6-well plates and
cultured at 37°C with 5% CO2. When they reached 50%
confluence, cells were transfected with specific siRNA or plasmids
with Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. After 6 h of
transfection with opti-MEM at 37°C with 5% CO2, the DMEM
containing 10% FBS was replaced. After 24 h following transfection,
the cells were used for subsequent experimentation. The target
sequences of si-STAT1 are listed in Table SI.
For some experiments verifying whether Wnt/β-catenin
pathway was involved in VSMC calcification, VSMCs were transfected
with the overexpression plasmids of STAT1 (STAT1 o/e group) and its
controls (STAT1-NC group), si-STAT1 (si-STAT1 group) and its
controls (si-NC group) under NP conditions at 37°C with 5%
CO2. After the knockdown or overexpression of STAT1 by
siRNA or plasmid, VSMCs were cultured in DMEM with 3 mM phosphorus
for 7 days at 37°C with 5% CO2 to induce calcification.
For the last 3 days of this period, lithium chloride (LiCl; 5
mmol/l; Sigma-Aldrich; Merck KGaA) was added to the medium of the
si-STAT1 group (si-STAT1 + LiCl group) to activate the
Wnt/β-catenin pathway and Dickkopf-1 (Dkk-1; 100 ng/ml;
MedChemExpress) was added to the medium of the STAT1 o/e group
(STAT1 o/e + Dkk-1 group) to inhibit the Wnt/β-catenin pathway at
37°C with 5% CO2.
Isolation of exosomes from HUVECs
HUVECs were cultured in DMEM with the addition of
NaH2PO4 for the HP (3 mM; pH=7.4) group or
without NaH2PO4 for the no phosphorus (NP)
group for 48 h at 37°C in a humidified atmosphere with 5%
CO2 for exosome isolation. NaH2PO4
(MilliporeSigma) was used to increase the concentration of
phosphorus in the medium. Before extraction of exosomes from
HUVECs, the cells were cultured with medium supplemented with 10%
exosome-depleted FBS for 48 h at 37°C with 5% CO2
(C38010100; Shanghai VivaCell Biosciences, Ltd.).
Exosomes were concentrated using an ExoQuick-TC
Exosome Precipitation Solution kit (EXOTC10A; System Biosciences,
LLC). This method has been widely used before and has been proven
to collect exosomes effectively (21-23). Briefly, the supernatant was
centrifuged at room temperature as follows: 300 × g for 10 min,
2,000 × g for 30 min and 10,000 × g for 30 min. An Amicon Ultra15
Centrifugal Filter Unit (100 kDa; MilliporeSigma) was used to
concentrate the supernatant. The ultrafiltration liquid and exosome
isolation reagents were mixed at a 5:1 ratio and incubated at 4°C
for ~16 h. Finally, the mixture was centrifuged at room temperature
at 1,500 × g for 30 min, and the exosome pellets were resuspended
in 200 µl PBS. Protein quantification of exosomes was
performed using a BCA kit (Beyotime Institute of
Biotechnology).
Identification of exosomes
Transmission electron microscopy (TEM) was used to
observe the morphology of exosomes. Briefly, exosomes were fixed
with equal volumes of 1% phosphotungstic acid (pH=7.4) at room
temperature for 30 min. After rinsing, 10 µl of the sample
was loaded onto a bronze net with film and left for 10 min at room
temperature. Then, 10 µl uranyl dioxyacetate was added to
the bronze net to precipitate for 10 min at room temperature, and
the floating liquid was absorbed by filter paper at room
temperature. Finally, images were observed under a Hitachi HT7760
transmission electron microscope (Hitachi, Ltd.) at 100 kV. The
size distribution of the exosomes was measured by dynamic light
scattering using nanoparticle tracking analysis (NTA; Zetasizer
Nano ZS; Malvern Instruments, Inc.). The expression levels of the
exosomal surface marker proteins CD63 (ab68418; Abcam) and CD9
(ab223052; Abcam) were analyzed by western blot analysis, which was
performed as described subsequently.
Exosome uptake by VSMCs
Exosomes were labeled with a PKH26 Red Fluorescent
Cell Linker kit (UR52302; Umibio Science and Technology Group)
according to the manufacturer's instructions. Briefly, labeling dye
was added to the exosomes resuspended in PBS and incubated at room
temperature with shaking for 10 min. Then, the tube was spun at
100,000 × g for 17 min at room temperature, and the exosome pellet
was resuspended in PBS. Labeled exosomes were incubated with VSMCs
for 0 and 24 h at 37°C with 5% CO2, and then the cells
were fixed with 4% paraformaldehyde at room temperature for 10 min
and washed three times with PBS. DAPI (Invitrogen; Thermo Fisher
Scientific, Inc.) was added at room temperature for 5 min. After
washing the cells with PBS three times, the staining signals were
analyzed with ZEN 2012 microscopy software (blue edition; Carl
Zeiss AG) using confocal microscopy (DMI600B; Leica Microsystems
GmbH).
Measurement of VSMC calcification
To induce calcification, VSMCs were co-cultured with
HUVEC-Exos for 14 days (10,000 cells were treated with 5 µg
HUVEC-Exos continuously). For the control group, VSMCs were
cultured with normal DMEM without the addition of HUVEC-Exos.
Alizarin Red S staining, alkaline phosphatase (ALP) activity assays
and Ca content measurements were conducted to determine the
calcification condition.
Alizarin Red S staining was conducted to assess VSMC
calcification. Cells were washed twice with PBS, fixed in 4%
paraformaldehyde for 30 min at room temperature and then stained
with 0.2% Alizarin Red (pH=8.3; Beijing Solarbio Science &
Technology Co., Ltd.) for 20 min at room temperature. Subsequently,
the cells were washed with PBS, and mineralized nodules were
assessed and images were captured using a light microscope (Carl
Zeiss AG) and analyzed with ZEN 2012 microscopy software (blue
edition; Carl Zeiss AG).
ALP activity was detected using an ALP kit purchased
from Beyotime Institute of Biotechnology (P0321S) according to the
manufacturer's instructions. Spectrophotometric measurement of
p-nitrophenol release was utilized. ALP activity was normalized to
the total protein content of the cell lysate.
The Ca content was quantified using a Ca content kit
obtained from Nanjing Jiancheng Bioengineering Institute (C004-2-1)
according to the manufacturer's instructions.
The expression levels and mRNA levels of
runt-related transcription factor 2 (Runx2) and osteopontin (OPN)
were detected using western blotting at the protein level and
reverse transcription-quantitative PCR (RT-qPCR) at the gene level.
All experiments were performed as described subsequently.
RT-qPCR
Total RNA was extracted from cells using a total RNA
isolation kit (Vazyme Biotech Co., Ltd.) according to the
manufacturer's instructions. Complementary DNA was synthesized
using HiScript II Q Select RT SuperMix for qPCR (Vazyme Biotech
Co., Ltd.) according to the manufacturer's instructions. For
RT-qPCR analysis, GAPDH was used as the reference. The mRNA levels
of Runx2, OPN, Wnt-3a and β-catenin were analyzed using RT-qPCR and
SYBR Green Supermix (Vazyme Biotech Co., Ltd.). The thermocycling
conditions were: i) Incubation step of 30 sec at 90°C; ii) 40
cycles of 10 sec at 95°C and 30 sec at 60°C; and iii) 15 sec at
95°C, 60 sec at 60°C and 15 sec at 95°C. The primers were
synthesized by TsingKe Biological Technology, and the sequences are
listed in Table I. All treatments
and conditions were performed in triplicate to calculate the
statistical significance, and the results were calculated using the
2−ΔΔCq method (24).
 | Table ISequences of the primers for reverse
transcription-quantitative PCR. |
Table I
Sequences of the primers for reverse
transcription-quantitative PCR.
| Human gene | Sequence
(5′-3′) |
|---|
| F-OPN |
AATCTCCTAGCCCCACAGACC |
| R-OPN |
CCACACTATCACCTCGGCCA |
| F-Runx2 |
GCGGTGCAAACTTTCTCCAG |
| R-Runx2 |
TGTCACTGTGCTGAAGAGGC |
| F-GAPDH |
AATGGGCAGCCGTTAGGAAA |
| R-GAPDH |
GCGCCCAATACGACCAAATC |
| F-STAT1 |
CAAGTGTTATGGGACCGCAC |
| R-STAT1 |
CTCTCATTCACATCTCTCAACTTCA |
| F-Wnt-3a |
GTGTTCCACTGGTGCTGCTA |
| R-Wnt-3a |
CCCTGCCTTCAGGTAGGAGT |
| F-β-catenin |
CTGAGGAGCAGCTTCAGTCC |
| R-β-catenin |
GGCCATGTCCAACTCCATCA |
Western blot analysis
Total protein was extracted from cells using
radioimmune precipitation assay lysis buffer (Beyotime Institute of
Biotechnology) supplemented with protease inhibitor cocktail and
phosphatase inhibitor cocktail (Bimake). The protein concentrations
were determined using a BCA protein assay kit (Biosharp Life
Sciences) and 30 µg protein was loaded per lane. Equal
amounts of protein lysate were separated by 10-12% SDS-PAGE in
Tris/SDS buffer and then transferred onto polyvinylidene difluoride
membranes (MilliporeSigma). The membranes were blocked in 5% nonfat
milk (w/v) in Tris-buffered saline with 0.1% Tween-20 for 1 h at
room temperature and incubated with the corresponding primary
antibodies at 4°C overnight. After washing, the membranes were
further incubated with HRP-conjugated secondary antibodies
(1:10,000) at room temperature for 1 h. The immunoreactive bands
were evaluated to visualize the expression of designated proteins
using the chemiluminescence detection system through the peroxidase
reaction, and the images of the bands were recorded with the Chemi
Doc MP imaging system (Bio-Rad Laboratories, Inc.). The
chemiluminescence substrate reagent (cat. no. 4AW011-100) was
purchase from Beijing 4A Biotech Co., Ltd. GAPDH was used as the
internal loading control. The films were analyzed with Image Lab
software 6.0.1 (Bio-Rad Laboratories, Inc.). All experiments were
repeated at least three times. The primary antibodies used in the
present study were as follows: Anti-GAPDH (ab8245; Abcam; 1:2,000),
anti-CD63 (ab217345; Abcam; 1:1,000), anti-CD9 (ab223052; Abcam;
1:1,000), anti-OPN (ab8245; Abcam; 1:1,000), anti-Runx2 (A11753;
ABclonal Biotech Co., Ltd.; 1:1,000), anti-STAT1 (T55227; Abmart
Pharmaceutical Technology Co., Ltd.; 1:2,000), anti-Wnt-3a (M63350;
Abmart Pharmaceutical Technology Co., Ltd.; 1:1,000) and
anti-β-catenin (A19657; ABclonal Biotech Co., Ltd.; 1:1,000). The
secondary antibodies were as follows: Goat Anti-Mouse IgG H&L
(HRP; ab205719; Abcam; 1:10,000) and Goat Anti-Rabbit IgG H&L
(HRP; ab205718; Abcam; 1:10,000).
Proteomics analysis
The HUVEC-Exos samples were processed for
label-free-based quantitative proteomic analysis by Shanghai
Huaying Biomedical Technology Co., Ltd. The proteomic content of
HUVEC-Exos in HP and NP conditions was compared using
high-performance liquid chromatography tandem mass spectrometry at
room temperature. Briefly, 2 µg samples (2-3 µl in
volume) for each group were taken and separated by Easy-NLC1000
(Thermo Fisher Scientific, Inc.) using an analytical column (C18;
1.9 µm; 75 µm x 20 cm; Thermo Fisher Scientific,
Inc.) at a mobile solution flow rate of 200 nl/min. The mobile
phase A was 0.1% formic acid (FA) in water (v/v) and mobile phase B
was 0.1% FA in acetonitrile (v/v). The analysis time for each
sample was 120 min and gradient elution was used (v/v; 0-5 min,
2-8% mobile phase B; 5-90 min, 8-24% mobile phase B; 90-110 min,
24-32% mobile phase B; 110-115 min, 32-90% mobile phase B; 115-120
min, 90% mobile phase B). Since label-free quantitative methods
were used, no internal standards were used in the present study.
The mass spectrometer was an Orbitrap Fusion Lumos (Thermo Fisher
Scientific, Inc.). Tandem mass spectrometry was performed using
Data Dependent Acquisition mode. The full sweep resolution was
60,000 full width at half maxima, the mass/charge ratio range was
set to M/Z 350-1,600, and the collision energy was set to 30% in
higher energy collisioninduced dissociation mode. All raw files
were converted into mgf files and searched against the Swissprot
database using Proteome Discoverer2.4 (Sequent HT; Thermo Fisher
Scientific, Inc.). Taxonomy was selected as Homo sapiens. Enzyme
was selected as trypsin. Fixed modifications were set as
Carbamidomethyl (C). Variable modifications were set as Oxidation
(M), Acetyl (Protein N-term), Met-loss+Acetyl (M), Met-loss (M).
Max Missed Cleavages was 2. Target FDR (Strict) was set as 0.01,
and target FDR (Relaxed) was set as 0.05. Min. peptide Length was
set as 6. Mass Tolerance was 10 ppm. For normalizing the different
protein abundances in different experiments, the protein precursor
intensity was calculated and then median-normalized. All further
analyses were based on the normalized results. Differentially
expressed proteins were identified with a cutoff of absolute fold
change ≥4. For each category, a two-tailed Fisher's exact test was
employed to test the enrichment of the differentially expressed
proteins against all identified proteins. A two-tailed P-value
<0.05 was considered significant. The mass spectrometry
proteomics data have been deposited to the ProteomeXchange
Consortium (http://proteomecentral.proteomexchange.org) via the
iProX (http://www.iprox.org) partner repository
with the dataset identifier PXD036925.
Statistical analysis
The results of the experiments are presented as the
mean ± SD, and the analysis was performed using SPSS software
(version 21.0; IBM Corp.). One-way ANOVA followed by Tukey's post
hoc test was employed to evaluate the significance of differences
among multiple groups. All experiments were repeated at least three
times, and representative experimental data are shown in the
figures. P<0.05 was considered to indicate a statistically
significant difference.
Results
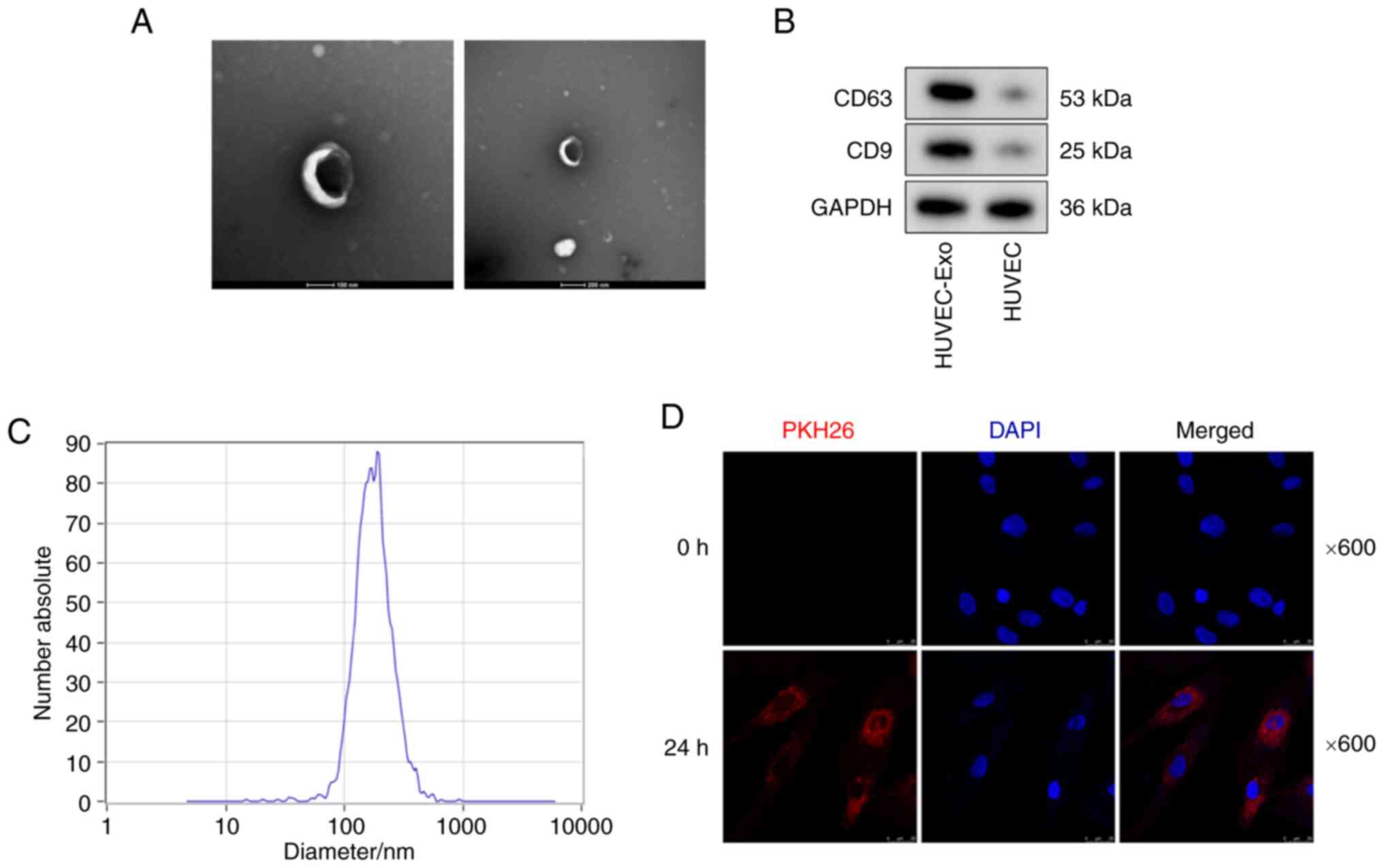
Identification of exosomes
Exosomes were isolated from the supernatant of
HUVECs. NTA and TEM were used to characterize the vesicles. TEM
indicated that these vesicles had a typical bilayer structural
morphology (Fig. 1A). Western
blot analysis further indicated the presence of exosome markers
(including CD9 and CD63) in the HUVEC-Exo group, while almost no
expression of CD9 and CD63 was observed in the HUVEC group
(Fig. 1B). NTA demonstrated that
these vesicles had a diameter of 80-170 nm (Fig. 1C). The typical bilayer structural
morphology, expression of exosome markers (CD9 and CD63) and the
diameter of 80-170 nm confirmed that these vesicles were
exosomes.
HP-HUVEC-Exos induce VSMC
calcification
To explore the ability of HUVEC-Exos to regulate
VSMC calcification, VSMCs were co-cultured with HP-HUVEC-Exos and
NP-HUVEC-Exos. For the control (Ctrl) group, VSMCs were cultured
with normal DMEM in the absence of HUVEC-exosomes.
First, the present study examined whether HUVEC-Exos
could be taken up by VSMCs. Exosomes were labelled with PKH26, and
HUVECs were incubated with the labeled exosomes for 0 and 24 h.
Fluorescence microscopy revealed that PKH26-labeled HUVEC-Exos
could be taken up by VSMCs (Fig.
1D). VSMCs were further incubated with HP-HUVEC-Exos,
NP-HUVEC-Exos or without exosomes. Compared with VSMCs treated with
NP-HUVEC-Exos and without exosomes, HP-HUVEC-Exos could induce VSMC
calcification. The protein expression levels of both Runx2 and OPN
were significantly upregulated in the HP-HUVEC-Exos group compared
with the other two groups (Fig.
2A), as were the Runx2 and OPN mRNA levels (Fig. 2B). The results of ALP activity and
Ca content measurements also revealed that HP-HUVEC-Exos could
induce the calcification of VSMCs (Fig. 2C and D). In addition, Alizarin Red
S staining showed that mineralized nodules were greatly increased
in VSMCs treated with HP-HUVEC-Exo for 14 days (Fig. 2E). The present results suggested
that exosomes derived from HP-stimulated HUVECs could promote the
calcification of VSMCs.
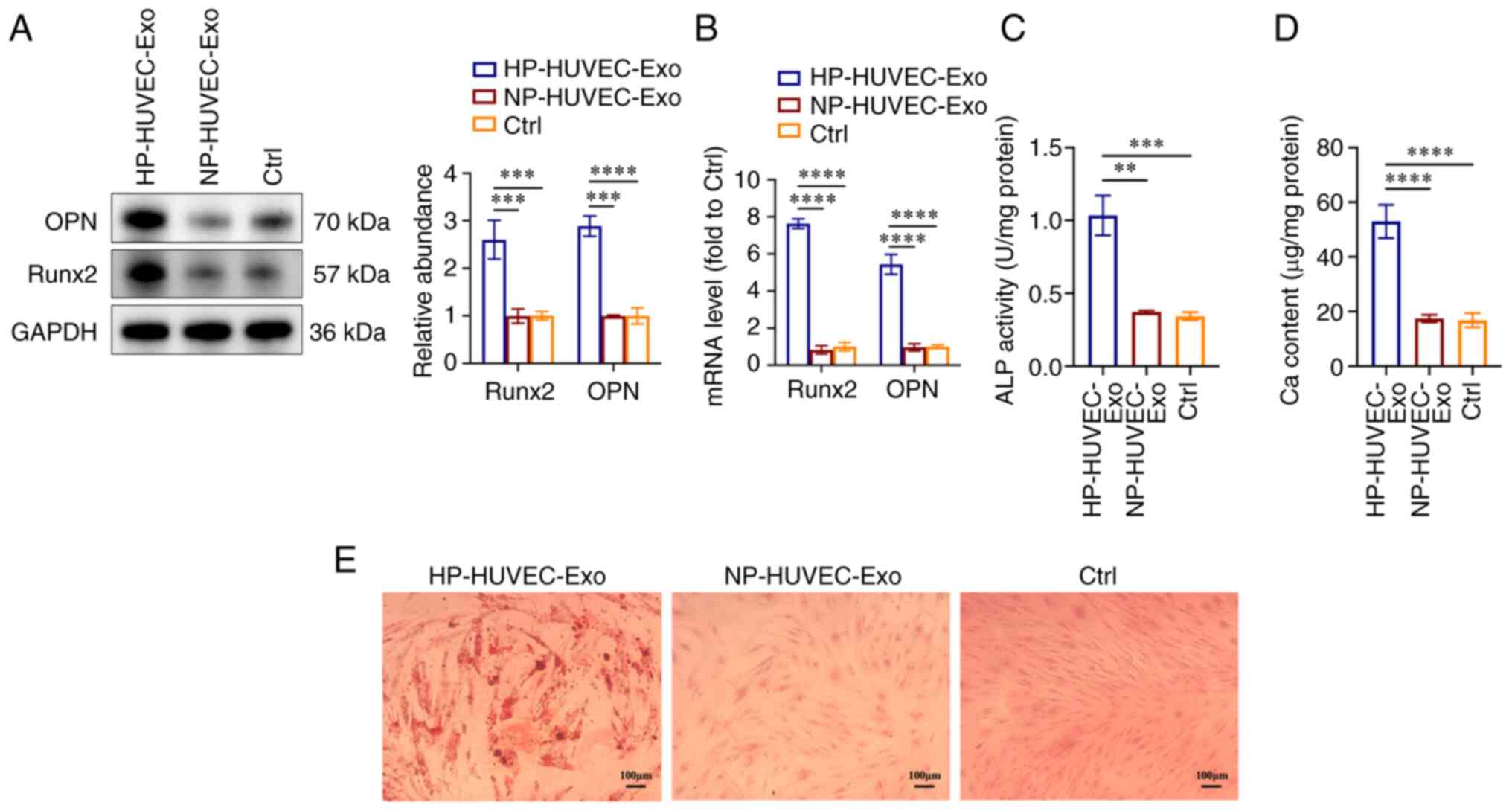 | Figure 2HP-stimulated HUVEC-Exos promote VSMC
calcification. (A) Expression levels of OPN and Runx2 in VSMCs were
measured by western blot analysis. (B) Reverse
transcription-quantitative PCR analysis of OPN and Runx2 expression
in VSMCs. (C) ALP activity assays were performed. (D) Ca content in
VSMCs was measured. (E) VSMCs were stained for mineralization using
Alizarin red S. Scale bar, 100 µm. **P<0.01,
***P<0.001, ****P<0.0001 compared with
the Ctrl group. ALP, alkaline phosphatase; Ca, calcium; Ctrl,
control; HP, high phosphorus; HUVEC-Exos, exosomes from HUVECs; NP,
no phosphorus; OPN, osteopontin; Runx2, runt-related transcription
factor 2; VSMC, vascular smooth muscle cell. |
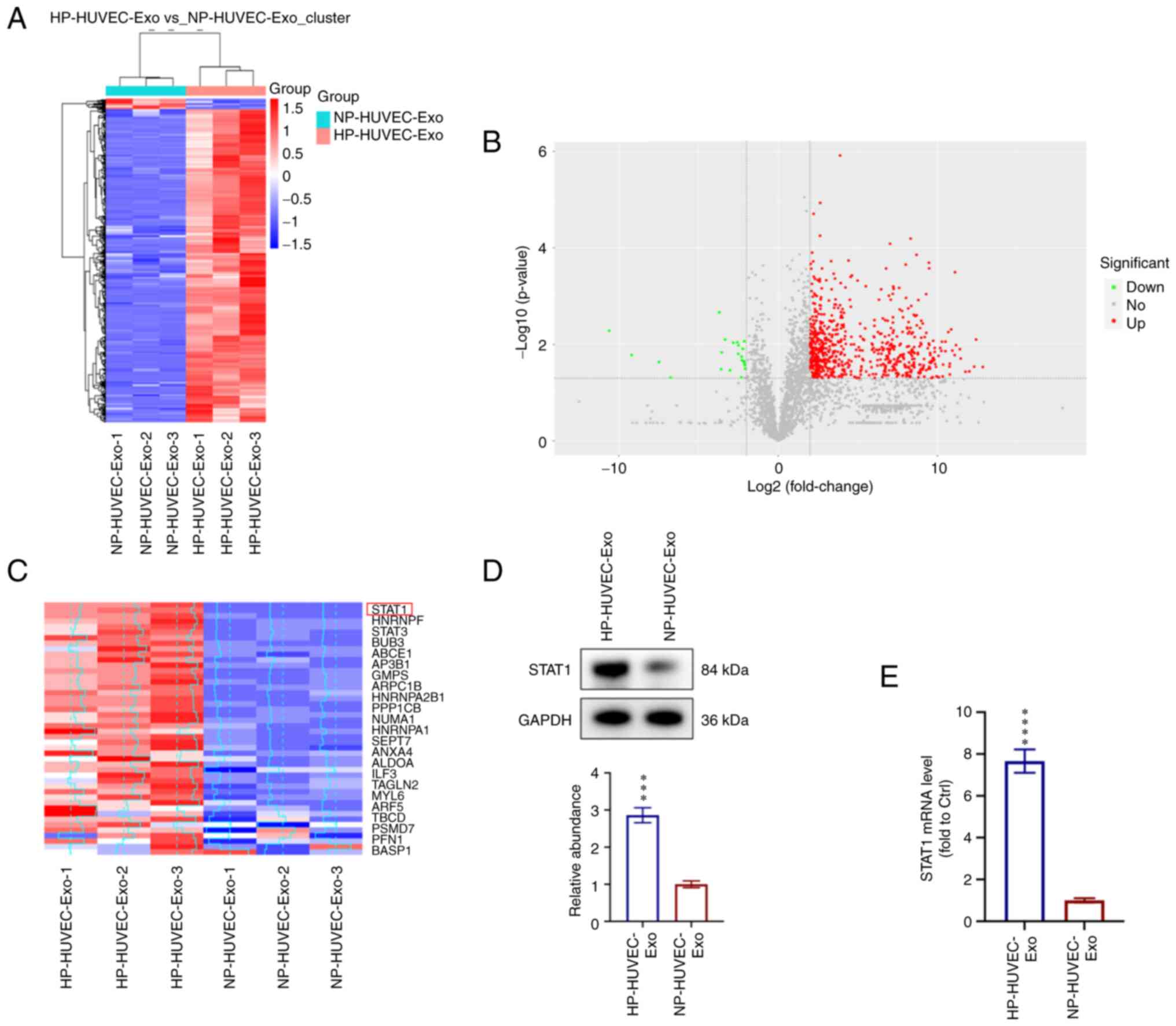
STAT1 is enriched in HP-HUVEC-Exos and
involved in regulating VSMC calcification
To explore the molecular mechanisms of HP-HUVEC-Exos
in VSMC calcification, proteomic analysis using label-free
technology was conducted to detect the differentially expressed
proteins between HP-HUVEC-Exos and NP-HUVEC-Exos. A total of 719
differentially expressed proteins were identified, among which 697
proteins were significantly upregulated and 22 proteins were
downregulated in HP-HUVEC-Exos compared with NP-HUVEC-Exos
(Fig. 3A and B). Based on the
results of proteomic analysis, STAT1 expression was significantly
different between HP-HUVEC-Exos and NP-HUVEC-Exos. The present
study focused on the different expression and potential role of
STAT1 protein in VSMC calcification (Fig. 3C). HP treatment was associated
with a significant upregulation of STAT1 in HUVEC-Exos.
Furthermore, western blot analysis confirmed the enrichment of
STAT1 in HP-HUVEC-Exos (Fig. 3D).
In addition, STAT1 mRNA expression was significantly upregulated in
VSMCs treated with HP-HUVEC-Exos compared with those treated with
NP-HUVEC-Exos (Fig. 3E).
To further investigate the role of STAT1 in VSMC
calcification, fludarabine (an inhibitor of STAT1) was used to
inhibit STAT1 expression in VSMCs. Compared with the HP-HUVEC-Exos
group, both western blotting and RT-qPCR analyses showed that the
expression levels of Runx2 and OPN were significantly downregulated
in the HP-HUVEC-Exos + fludarabine group (Fig. 4A and B). Alizarin Red S staining
demonstrated that mineralized nodules were greatly decreased in
VSMCs treated with HP-HUVEC-Exos + fludarabine compared with VSMCs
treated with HP-HUVEC-Exos (Fig.
4C). The results of ALP activity and Ca content measurements
also demonstrated that the inhibition of STAT1 by fludarabine
alleviated HP-HUVEC-Exo-induced VSMC calcification (Fig. 4D and E). Overall, the ability of
HP-HUVEC-Exos to induce VSMC calcification was decreased when STAT1
was suppressed.
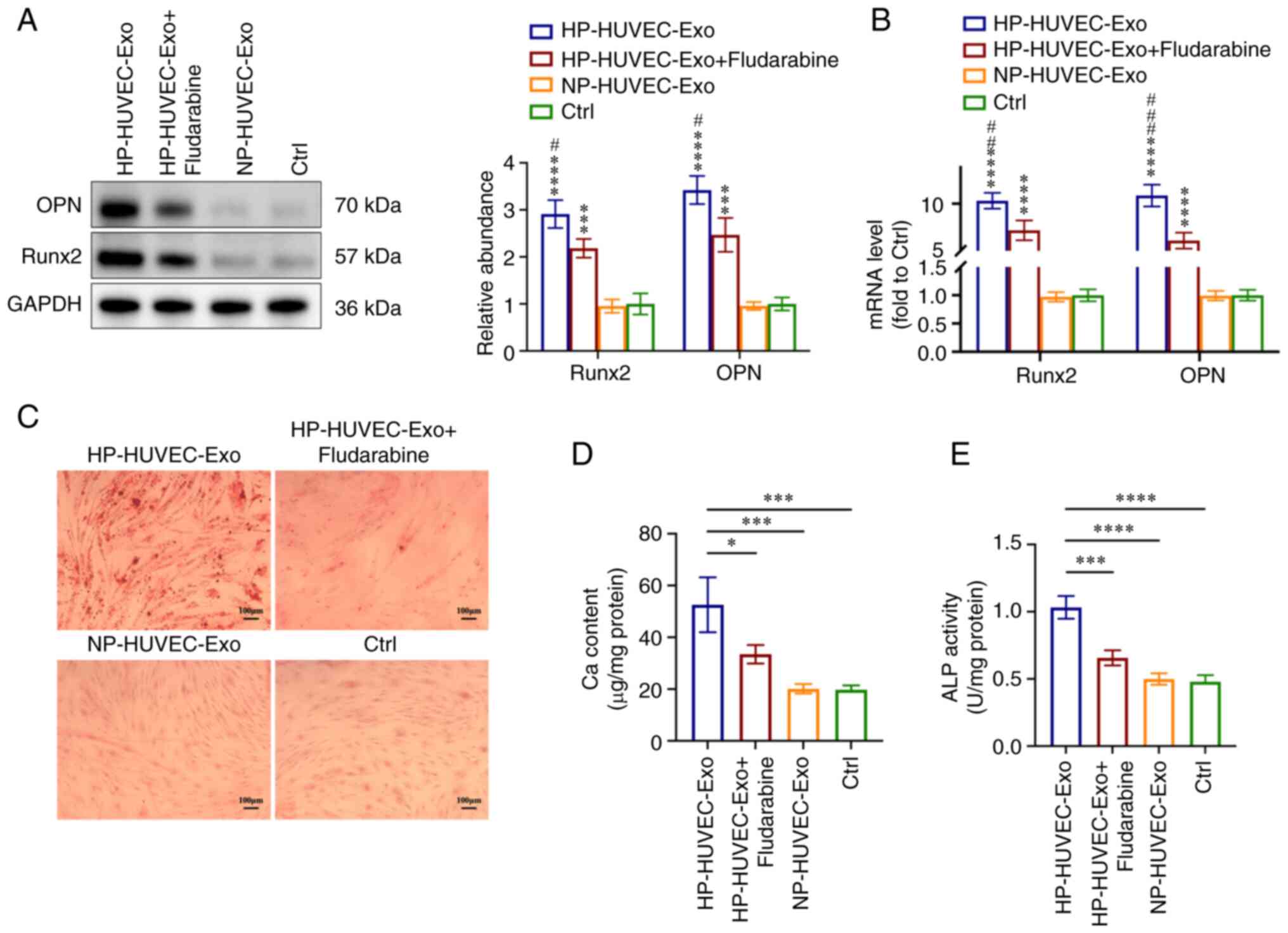 | Figure 4STAT1 is involved in
HP-HUVEC-Exos-induced VSMC calcification. VSMCs were treated with
HP-HUVEC-Exos, HP-HUVEC-Exos + fludarabine, NP-HUVEC-Exos or fresh
conditioned medium (Ctrl group) for 14 days. (A) Expression levels
of OPN and Runx2 in VSMCs were measured by western blot analysis.
(B) Reverse transcription-quantitative PCR analysis of OPN and
Runx2 expression in VSMCs. (C) Alizarin Red S staining revealed
mineralization. Scale bar, 100 µm. (D) Ca content in VSMCs
was measured. (E) ALP activity was tested. *P<0.05,
***P<0.001, ****P<0.0001 compared with
the Ctrl group. #P<0.05, ##P<0.01,
###P<0.001 compared with the HP-HUVEC-Exos +
fludarabine group. ALP, alkaline phosphatase; Ca, calcium; Ctrl,
control; HP, high phosphorus; HUVEC-Exos, exosomes from HUVECs; NP,
no phosphorus; OPN, osteopontin; Runx2, runt-related transcription
factor 2; VSMC, vascular smooth muscle cell. |
Inhibition of STAT1 alleviates HP-induced
VSMC calcification via the Wnt/β-catenin pathway
The present study further explored the role of STAT1
in VSMC calcification. First, VSMCs were transfected with si-STAT1
to suppress STAT1 expression and with si-NC. The knockdown effect
of si-STAT1 on the protein levels of STAT1 was measured by western
blotting (Fig. 5A). Based on the
downregulation effect, si-STAT1-01 was selected for further
experiments. VSMCs were incubated with HP (3 mM) to induce
calcification or fresh conditional medium (Ctrl) simultaneously.
When the expression of STAT1 was knocked down, the effect of HP on
VSMC calcification was almost abolished, as evidenced by the
decreased expression levels of OPN and Runx2 in the HP + si-STAT1
group compared with the HP group, which were analyzed by western
blotting and RT-qPCR (Fig. 5B and
C). Furthermore, the mineralized nodules were also reduced when
STAT1 was downregulated in the HP + si-STAT1 group compared with
the HP group (Fig. 5D). The Ca
content and ALP activity measurements also revealed that the
inhibition of STAT1 alleviated HP-induced VSMC calcification
(Fig. 5E and F).
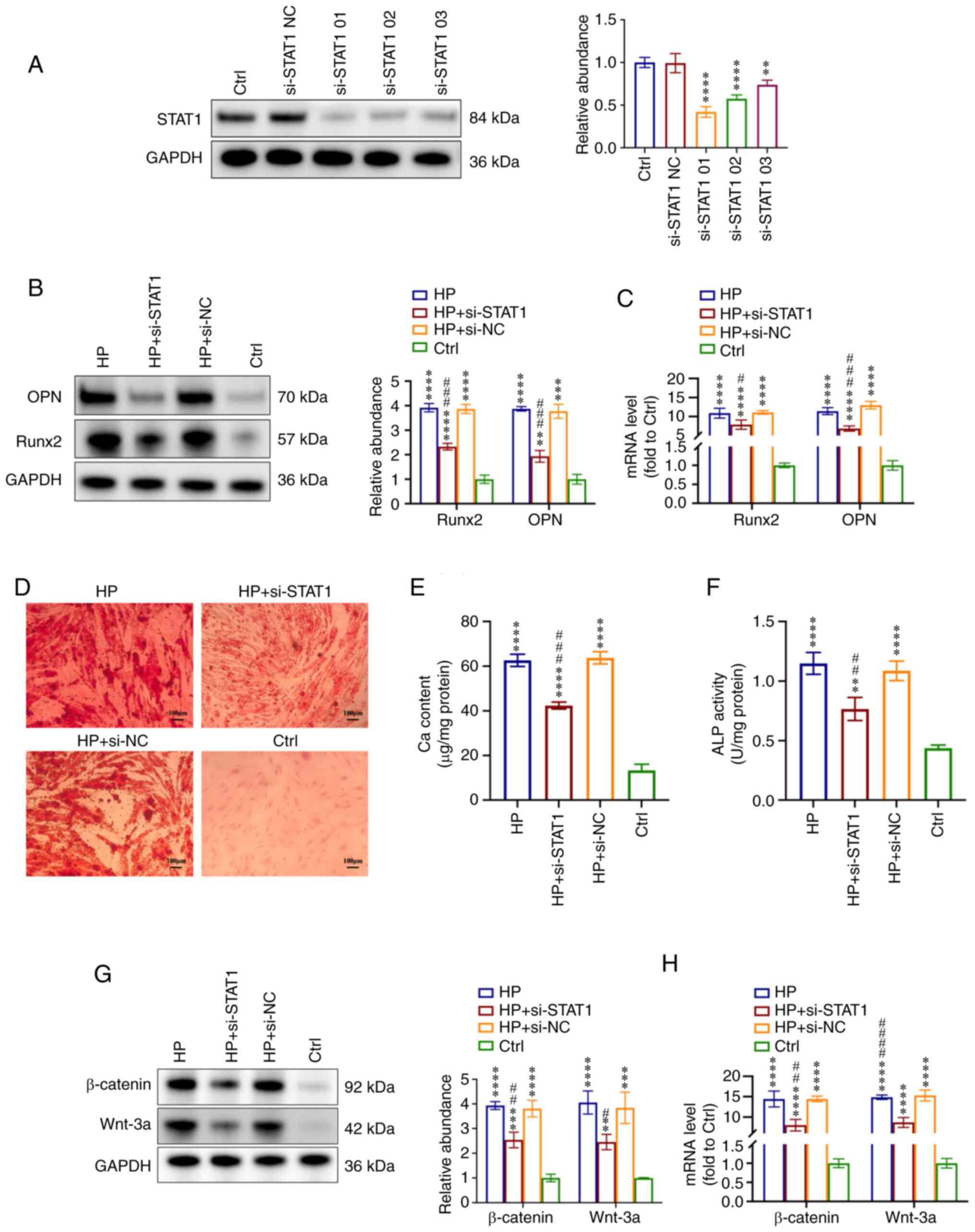 | Figure 5STAT1 inhibition alleviates
HP-induced VSMC calcification via the Wnt/β-catenin signaling
pathway. VSMCs were treated with si-STAT1 and si-NC and then
incubated with HP (3 mM) to induce calcification or fresh
conditioned medium (Ctrl) for 7 days. (A) Effect of si-STAT1 on
protein levels of STAT1. Based on the knockdown effect, si-STAT1-01
was selected for further experiments. (B) Expression levels of OPN
and Runx2 were measured by western blot analysis in VSMCs. (C)
RT-qPCR analysis of OPN and Runx2 mRNA expression in VSMCs. (D)
Alizarin Red S staining indicated the mineralization. Scale bar,
100 µm. (E) Ca content in VSMCs was measured. (F) ALP
activity was tested. (G) Expression levels of Wnt-3a and β-catenin
were measured by western blot analysis. (H) RT-qPCR analysis of
Wnt-3a and β-catenin expression in VSMCs. **P<0.01,
***P<0.001, ****P<0.0001 compared with
the Ctrl group. #P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001 compared with
the HP group. ALP, alkaline phosphatase; Ca, calcium; Ctrl,
control; HP, high phosphorus; NC, negative control; OPN,
osteopontin; RT-qPCR, reverse transcription-quantitative PCR;
Runx2, runt-related transcription factor 2; si, small interfering
RNA; VSMC, vascular smooth muscle cell. |
In addition, the expression levels of Wnt/β-catenin
pathway-related proteins were detected. Wnt-3a and β-catenin were
significantly upregulated in calcification VSMCs (HP, HP + si-STAT1
and HP + si-NC groups) compared with the Ctrl group, and the
inhibition of STAT1 (HP + si-STAT1 group) also suppressed the
expression of Wnt-3a and β-catenin both at the protein and mRNA
levels compared with the HP group (Fig. 5G and H). These results indicated
that STAT1 was involved in HP-induced VSMC calcification, and the
activation of the Wnt/β-catenin pathway may be the potential
mechanism.
Wnt/β-catenin signaling pathway is
involved in STAT1-mediated VSMC calcification
To verify whether STAT1 affected VSMC calcification
via the Wnt/β-catenin pathway, VSMCs were transfected with the
overexpression plasmids of STAT1 (STAT1 o/e group) and its controls
(STAT1-NC group) under NP conditions. Western blot analysis
demonstrated the overexpression of STAT1 in VSMCs after
transfection (Fig. 6A).
Subsequently, an activator or inhibitor of the Wnt/β-catenin
signaling pathway was used for the treatment of HP-treated VSMCs.
Briefly, STAT1-overexpressing VSMCs were treated with 3 mM
phosphorus for 7 days, with or without 100 ng/ml Dkk-1 (Wnt
inhibitor) added for the last 3 days of this period. si-STAT1 VSMCs
were treated in the presence of 3 mM phosphorus for 7 days, with or
without 5 mmol/l LiCl (Wnt activator) added for the last 3 days of
this period. The LiCl-treated si-STAT1 group (si-STAT1 + LiCl
group) exhibited increased expression levels of OPN, Runx2, Wnt-3a
and β-catenin, while the Dkk-1-treated STAT1 o/e group (STAT1 o/e +
DKK-1) displayed decreased expression at both the protein and gene
levels (Fig. 6B and C). The Ca
deposition in cultured VSMCs was detected by Alizarin Red S
staining. The LiCl-treated si-STAT1 group (si-STAT1 + LiCl group)
exhibited more mineralized nodules compared with the si-STAT1 and
si-NC groups, and the Dkk-1-treated STAT1 o/e group (STAT1 o/e +
Dkk-1 group) exhibited fewer mineralized nodules compared with the
STAT1 o/e group and STAT1-NC group (Fig. 6D). Similarly, the ALP and Ca
contents were increased in the LiCl-treated si-STAT1 group
(si-STAT1 + LiCl group) compared with si-STAT1 group and si-NC
group, while they were decreased in the Dkk-1-treated STAT1 o/e
group (STAT1 o/e + Dkk-1 group) compared with the STAT1 o/e group
(Fig. 6E and F). These results
suggested that LiCl reversed the protective effect of STAT1
inhibition on VSMC calcification and Dkk-1 alleviated the effect of
STAT1 overexpression in HP-induced VSMC calcification.
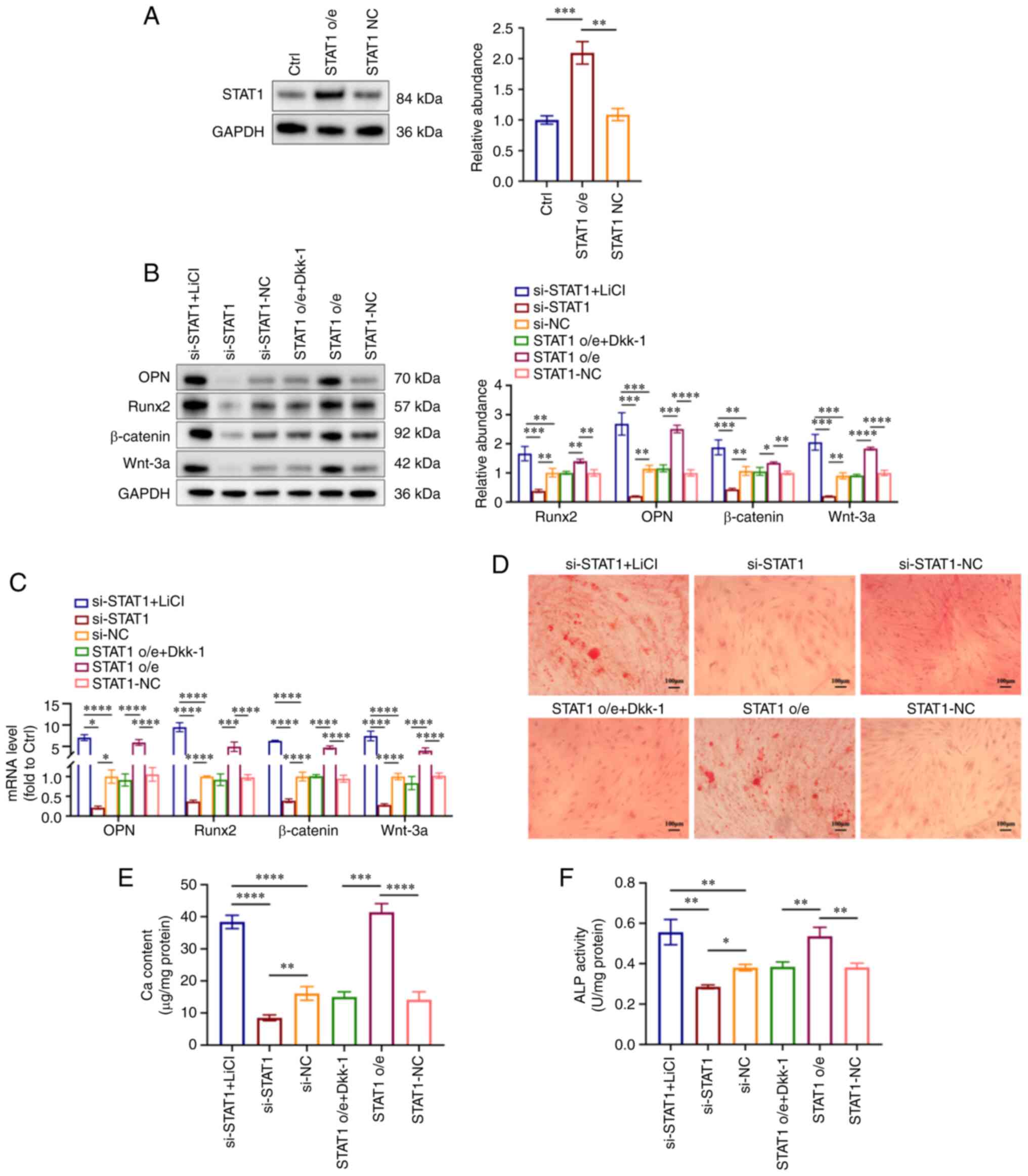 | Figure 6Wnt/β-catenin signaling pathway is
involved in STAT1-mediated VSMC calcification. STAT1-overexpressing
VSMCs were treated with 3 mM phosphorus for 7 days, with or without
100 ng/ml Dkk-1 added for the last 3 days of this period. si-STAT1
VSMCs were treated in the presence of 3 mM phosphorus for 7 days,
with or without 5 mmol/l LiCl for the last 3 days of this period.
(A) Expression levels of STAT1 in VSMCs were measured by western
blot analysis. (B) Expression levels of OPN, Runx2, Wnt-3a and
β-catenin were measured by western blot analysis. (C) Reverse
transcription-quantitative PCR analysis of OPN, Runx2, Wnt-3a and
β-catenin in VSMCs. (D) Mineralization was examined using Alizarin
Red S staining. Scale bar, 100 µm. (E) Ca content in VSMCs
was measured. (F) ALP activity was tested. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. ALP, alkaline phosphatase; Ca, calcium;
Ctrl, control; NC, negative control; o/e, overexpression; OPN,
osteopontin; Runx2, runt-related transcription factor 2; si, small
interfering RNA; VSMC, vascular smooth muscle cell; Dkk-1,
Dickkopf-1; LiCl, lithium chloride. |
Taken together, the present results indicated that
exosomal STAT1 derived from HP-treated HUVECs promoted VSMC
calcification by activating the Wnt/β-catenin signaling
pathway.
Discussion
VC is associated with both cardiovascular and
all-cause mortality in patients with CKD, and CKD-related VC
commonly starts from the tunica media (1,10).
However, the exact mechanism of how the calcification signal in
circulating blood is transferred to the tunica media remains
unknown. It is also unknown whether exosomes can mediate the
interaction between ECs and VSMCs in a HP environment. In the
present study, exosomes derived from HP-treated HUVECs could
promote VSMC calcification. Higher expression of STAT1 was observed
in HP-HUVEC-Exo compared with NP-HUVEC-Exo. Subsequently, si-STAT1,
fludarabine or overexpression plasmid of STAT1 were used to
knockdown, inhibit or upregulate STAT1 expression, respectively,
and the promoting or inhibitory effects on calcification were
observed. Finally, the present study further revealed that the
Wnt-3a/β-catenin pathway may be the potential mechanism. In
summary, the present study revealed that HP-stimulated HUVECs
secreted exosomes, which carried the STAT1 protein to VSMCs, thus
promoting the calcification of VSMCs via activation of the
Wnt/β-catenin signaling pathway. The present findings demonstrated
that exosomal STAT1 from ECs in HP environments may contribute to
the calcification of VSMCs, indicating a potential role of exosomes
in intercellular communication between ECs and VSMCs in CKD-related
VC.
Exosomes are EVs derived from cells and serve as
important intercellular message transporters (25,26). Previous studies have demonstrated
that exosomes participate in the process of VC (27,28). Kapustin et al (13) reported the co-location of CD63 (a
biomarker of exosomes) and the calcification site in the vessel
wall, indicating the involvement of exosomes in VC. Exosomes can
participate in VC formation by acting as mineralization sites for
minerals such as Ca and phosphorus (29,30). Kapustin et al (31) observed that the mineralization of
VSMC-derived matrix vesicles was a pathological response to
disturbed intracellular Ca homeostasis that led to calcification
inhibitor depletion and the formation of annexin
A6/phosphatidylserine nucleation complexes. It also regulates the
phenotypic conversion of VSMCs from a contractile to a synthetic
state, thus contributing to vascular pathologies, including VC,
restenosis and atherosclerosis (7). Sortilin has been reported as a key
transport factor that regulates VSMC calcification; it can be
transformed into exosomes with calcification potential and
participate in the formation of microcalcification (32). In addition, the cargos of
exosomes, including RNA, cytokines, proteins and lipids, could be
transported between cells via exosomes, thus serving an important
role in intercellular communication during the process of VC
(33). Xu et al (34) reported that exosomes from
melatonin-treated VSMCs could alleviate the calcification and aging
of VSMCs in a paracrine manner through an exosomal microRNA
(miRNA/miR)-204/miR-211 cluster by targeting bone morphogenetic
protein-2. Guo et al (35)
investigated the mechanisms of bone marrow mesenchymal stem cell
(BMSC)-derived exosomes in attenuating VC. They found that
BMSC-derived exosomes alleviated HP-induced calcification in human
aortic VSMCs by modifying miRNA profiles, and the mTOR, MAPK and
Wnt signaling pathways were involved in this process (35). Similar to our study, Li et
al (16) reported that
exosomes from hyperglycemia-stimulated vascular ECs promoted the
calcification and senescence of VSMCs through the transport of
versican by inducing mitochondrial dysfunction. Exosomal Notch3
from high glucose-stimulated ECs has also been reported to promote
VSMC calcification and aging, and the mTOR signaling pathway is
closely related to the Notch3 protein and involved in regulating
calcification and aging (20).
The present results for PKH26-labeled exosomes demonstrated that
exosomes from HUVECs were mainly located in the cytoplasm of VSMCs.
Similarly, Li et al (36)
also reported that exosomal circRTN4 from mesenchymal stem cells
could alleviate sepsis-induced myocardial injury, and their
fluorescence in situ hybridization assay results
demonstrated the location of circRTN4 in the cytoplasm of
cardiomyocytes. Although the details of exosome localization in the
cytoplasm need to be further explored, the present study revealed
that exosomes could serve as an important cargo transporter between
HUVECs and VSMCs and are thus involved in the process of VC.
In addition, the present study demonstrated that
STAT1 was enriched in exosomes derived from HP-stimulated HUVECs
and served a key role in VC. STAT family members are potential
cytoplasmic transcription factors that mediate a variety of
biological responses, including cell proliferation, survival,
apoptosis and differentiation (37). STAT1 can be activated by a variety
of extracellular stimulators, such as Janus kinase, and then enters
the nucleus and regulates the expression of target genes (38,39). STAT1 may also be involved in the
development of VC (40-42). Smyth et al (40) reported that patients with a
gain-of-function STAT1 mutation were more likely to have
significant VC, in which progressive calcification of the aorta and
aortic valve can even lead to constricted obstruction at the
calcified site. Kuniga et al (41) revealed that the expression of
STAT1 in VSMCs could be activated by adding monocyte-expressed
urokinase, and the activation of STAT1 inhibited the proliferation
of VSMCs. In another in vitro experiment, Demyanets et
al (42) treated VSMCs with
the STAT1 activator oncostatin-M (OSM). They found that OSM
promoted the production and activation of STAT1 and then led to the
loss of VSMC contractile phenotype, which is a typical feature of
osteogenic VSMCs (42). Further
studies have demonstrated that the overexpression of STAT1 reduces
VSMC contractile gene expression, thus leading to a reduced
contractile phenotype and promoting VSMC dedifferentiation
(43). Regarding cellular
signaling communication between cells, Cossetti et al
(44) revealed that interferon-γ
(IFN-γ) could bind to EVs through the activation of STAT1 in target
cells by IFN-γ receptor 1 (IFNGR1), indicating the role of STAT1 in
intercellular communication. In addition, endogenous STAT1 and
IFNGR1 in target cells were indispensable for the activation of
STAT1 signaling via EV-related IFN-γ/IFNGR1 complexes,
demonstrating the role of STAT1 in cellular signaling regulated by
EVs (44). Cai et al
(45) reported that exosomal
miRNA-221 promoted the polarization of M1 macrophages through the
upregulation of STAT1 and STAT3, thus suggesting a novel crosstalk
signaling pathway between mammary epithelial cells and macrophages
during inflammation. Similar to previous studies (40-43), it was also observed that high
expression of STAT1 was involved in the calcification of VSMCs,
suggesting that the inhibition of STAT1 may represent a novel
therapeutic target for the control of vascular diseases such as
calcification, atherosclerosis and restenosis.
The present results also demonstrated that vascular
endothelial dysfunction may contribute to the calcification
mechanism of vessels. The present study demonstrated that the
initial endothelial dysfunction caused by HP could serve a key role
in the progression of VC by releasing EVs. Under pathological
stimuli, ECs secrete exosomes and transfer protein cargos, thus
leading to the osteochondrogenic transdifferentiation of VSMCs
followed by the formation of micro- and macrocalcification in the
vessel wall (13,14,25). In vivo and in vitro
studies have suggested that EVs serve a central role in promoting
cellular dysfunction by directly interacting with the endothelium
(46,47). EVs reduce nitric oxide (NO)
bioavailability, inhibit endothelial NO synthase and activate ERK
signaling under pathological conditions to impair vasorelaxation
(48,49). There was a similar effect of
circulating EVs on ECs in animal models that mimic diabetes-induced
endothelial dysfunction, although the mechanisms were different
(50). A higher level of arginase
1 (Arg1) was detected in circulating EVs from diabetic mice than in
those from normoglycemic mice. It could reduce L-arginine
availability, which is essential to NO production, by converting it
to urea and ornithine (50).
Therefore, circulating EVs transfer Arg1 to ECs, thus reducing NO
and impairing vasorelaxation (50). In warfarin-treated rats,
compromised basal NO availability/increased vessel tone was
observed during the arterial media calcification process, and the
L-N-nitro-arginine methyl ester-induced further deterioration of
endothelial function was associated with the calcification process,
indicating that the loss of endothelial function was associated
with early stages of arterial media calcification development
(51). In addition, the function
of the endothelium is related to the activation of platelets and
other biological processes (47).
Grześk et al (52)
compared the effects of low- and high-dose aspirin co-administered
with ticagrelor on the reactivity of VSMCs and found that high-dose
aspirin impaired the anticontractile effect of ticagrelor on
ADP-induced VSMC contraction in a rat model. Additionally, a
previous study also demonstrated the responses of VSMC contraction
to phenylephrine, angiotensin II and mastoparan-7 modulated by
endothelial function (53).
Endothelium dysfunction serves a key role in the progression of VC;
however, how it impacted VSMCs through exosomes in the present
study still requires further research.
The Wnt/β-catenin pathway is widely reported to be
associated with VC (54). Gaur
et al (55) reported that
Runx2 was a target gene of Wnt signaling, and the activation of
Runx2 stimulated osteoblast differentiation and bone formation.
Similarly, another study suggested that HP could activate
Wnt/β-catenin signaling and activated Wnt/β-catenin signaling
promoted VSMCs osteogenic transdifferentiation and calcification by
directly modulating Runx2 gene expression (56). In our previous study, CKD rats
showed more significant VC than rats in control group, and the
expression levels of Wnt-3a and β-catenin at calcified sites were
increased, which was positively associated with the tissue
calcification score, suggesting that the activation of
Wnt/β-catenin signaling was involved in VC in CKD (57). Wu et al (58) reported that miR-708-5p was
inhibited and pituitary specific transcription factor-1 (Pit-1) was
upregulated in HP-induced VC. Further experiments demonstrated that
miR-708-5p could inactivate the Wnt8b/β-catenin pathway by
targeting Pit-1 to alleviate HP-induced VC (58). Cong et al (59) found that related transcriptional
enhancer factor could ameliorate β-glycerolphosphate-induced
calcification and osteoblastic differentiation of VSMCs by
inhibiting the Wnt/β-catenin signaling pathway. The interplay
between STAT1 and the Wnt/β-catenin signaling pathway has also been
reported before. Zhao et al (60) revealed that the downregulation of
STAT1 could weaken the aggressiveness of glioblastoma cells through
inhibition of epithelial-mesenchymal transition both in vivo
and in vitro, which is mediated via the Wnt/β-catenin
signaling pathway. Yuan et al (61) demonstrated that STAT1 could be
recruited into the promoter of β-catenin to activate its
expression, and this effect was regulated by IFN-γ. In epithelial
ovarian cancer, STAT1 could promote the upregulation of long
non-coding RNA LINC00958, thus accelerating tumorigenesis by
modulating Wnt/β-catenin signaling (62). A similar cross-link of STAT1 and
Wnt/β-catenin signaling was also observed in the present study. The
overexpression of STAT1 promoted VSMC calcification, and the
downregulation of STAT1 alleviated calcification. These effects
were attenuated when Dkk-1 or LiCl was added to the VSMCs,
suggesting the interplay of STAT1 and the Wnt/β-catenin signaling
pathway in VC.
The present study first demonstrated that exosomes
derived from HP-treated HUVECs could promote VSMC calcification.
Then, the different protein cargos in exosomes were determined and
the different expression of STAT1 between the HP-HUVEC-Exo and
NP-HUVEC-Exo groups was further observed. Subsequently, si-STAT1,
fludarabine or overexpression plasmid of STAT1 were used to
knockdown, inhibit or upregulate STAT1 expression, respectively,
and the promoting or inhibitory effects on calcification were
observed. Finally, the present study further explored the potential
signal pathway and found that the pro-calcified effect caused by
HP-HUVEC-Exo may be associated with the Wnt-3a/β-catenin pathway.
However, the inhibition of STAT1 in HUVECs or exosomes would
further confirm the results, which should be performed in the
future. Another limitation is that all experiments were conducted
in vitro, and thus, further in vivo studies are still
required.
In conclusion, exosomes enriched with STAT1 derived
from HP-treated HUVECs could be transmitted to VSMCs, thus
promoting the calcification of VSMCs by activating the
Wnt/β-catenin signaling pathway, indicating that exosomal STAT1
might be a novel therapeutic direction in CKD-related VC. This
finding highlights a novel method of intercellular communication
between ECs and VSMCs and provides insights into the mechanism of
VC in patients with CKD.
Supplementary Data
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the iProX repository, https://www.iprox.cn/page/project.html?id=IPX0005088000.
All other datasets used and/or analyzed during the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
ZQ, YL, JL and LJ performed the experiments and data
analysis. ZZ, KC, QY and SC provided technical support and
materials, and interpreted data. ZQ and RL contributed to
manuscript writing. RL and BS contributed to the design of the
experiments and revised the article. ZQ and RL confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Ke Hu (West China
School of Medicine, Sichuan University, Chengdu, China) and Dr
Yawen Zhang (West China School of Medicine, Sichuan University,
Chengdu, China) for their technical support in experiments and
manuscript preparation.
Funding
This work was supported by the Natural Science Foundation of
Sichuan, China (grant no. 2022NSFSC1353), National Natural Science
Foundation of China (grant no. 82000702), Sichuan Science and
Technology Program (grant no. 2022YFS0147), Science and Technology
Achievement Transformation Fund of West China Hospital of Sichuan
University (grant no. CGZH19006), Med-X Innovation Programme of
Med-X Center for Materials of Sichuan University (grant no.
MCM202101), 1.3.5 project for disciplines of excellence from West
China Hospital of Sichuan University (grant no. ZYJC21010) and Med+
Biomaterial Institute of West China Hospital/West China School of
Medicine of Sichuan University (grant no. ZYME20001). The funding
sources had no role in the design, analysis and interpretation of
the data or the preparation, approval or decision to submit the
manuscript for review.
References
|
1
|
Ren SC, Mao N, Yi S, Ma X, Zou JQ, Tang X
and Fan JM: Vascular calcification in chronic kidney disease: An
update and perspective. Aging Dis. 13:673–697. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nelson AJ, Raggi P, Wolf M, Gold AM,
Chertow GM and Roe MT: Targeting vascular calcification in chronic
kidney disease. JACC Basic Transl Sci. 5:398–412. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denker M, Boyle S, Anderson AH, Appel LJ,
Chen J, Fink JC, Flack J, Go AS, Horwitz E, Hsu CY, et al: Chronic
renal insufficiency cohort study (CRIC): Overview and summary of
selected findings. Clin J Am Soc Nephrol. 10:2073–2083. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dube P, DeRiso A, Patel M, Battepati D,
Khatib-Shahidi B, Sharma H, Gupta R, Malhotra D, Dworkin L, Haller
S and Kennedy D: Vascular calcification in chronic kidney disease:
Diversity in the vessel wall. Biomedicines. 9:4042021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Düsing P, Zietzer A, Goody PR, Hosen MR,
Kurts C, Nickenig G and Jansen F: Vascular pathologies in chronic
kidney disease: Pathophysiological mechanisms and novel therapeutic
approaches. J Mol Med (Berl). 99:335–348. 2021. View Article : Google Scholar
|
|
6
|
Wang XR, Zhang JJ, Xu XX and Wu YG:
Prevalence of coronary artery calcification and its association
with mortality, cardiovascular events in patients with chronic
kidney disease: A systematic review and meta-analysis. Ren Fail.
41:244–256. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lanzer P, Boehm M, Sorribas V, Thiriet M,
Janzen J, Zeller T, St Hilaire C and Shanahan C: Medial vascular
calcification revisited: Review and perspectives. Eur Heart J.
35:1515–1525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamada S and Giachelli CM: Vascular
calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho.
Bone. 100:87–93. 2017. View Article : Google Scholar :
|
|
9
|
Raggi P: Cardiovascular calcification in
end stage renal disease. Contrib Nephrol. 149:272–278. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen J, Budoff MJ, Reilly MP, Yang W,
Rosas SE, Rahman M, Zhang X, Roy JA, Lustigova E, Nessel L, et al:
Coronary artery calcification and risk of cardiovascular disease
and death among patients with chronic kidney disease. JAMA Cardiol.
2:635–643. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fang Y, Ginsberg C, Sugatani T,
Monier-Faugere MC, Malluche H and Hruska KA: Early chronic kidney
disease-mineral bone disorder stimulates vascular calcification.
Kidney Int. 85:142–150. 2014. View Article : Google Scholar
|
|
12
|
Toussaint ND, Pedagogos E, Tan SJ, Badve
SV, Hawley CM, Perkovic V and Elder GJ: Phosphate in early chronic
kidney disease: Associations with clinical outcomes and a target to
reduce cardiovascular risk. Nephrology (Carlton). 17:433–444. 2012.
View Article : Google Scholar
|
|
13
|
Kapustin AN, Chatrou ML, Drozdov I, Zheng
Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT,
Alvarez-Hernandez D, et al: Vascular smooth muscle cell
calcification is mediated by regulated exosome secretion. Circ Res.
116:1312–1323. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin Z, Liao R, Xiong Y, Jiang L, Li J,
Wang L, Han M, Sun S, Geng J, Yang Q, et al: A narrative review of
exosomes in vascular calcification. Ann Transl Med. 9:5792021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pan W, Liang J, Tang H, Fang X, Wang F,
Ding Y, Huang H and Zhang H: Differentially expressed microRNA
profiles in exosomes from vascular smooth muscle cells associated
with coronary artery calcification. Int J Biochem Cell Biol.
118:1056452020. View Article : Google Scholar
|
|
16
|
Li S, Zhan JK, Wang YJ, Lin X, Zhong JY,
Wang Y, Tan P, He JY, Cui XJ, Chen YY, et al: Exosomes from
hyperglycemia-stimulated vascular endothelial cells contain
versican that regulate calcification/senescence in vascular smooth
muscle cells. Cell Biosci. 9:12019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang A, Guo G, Yu Y and Yao L: The roles
of collagen in chronic kidney disease and vascular calcification. J
Mol Med (Berl). 99:75–92. 2021. View Article : Google Scholar
|
|
18
|
Song X, Yang B, Qiu F, Jia M and Fu G:
High glucose and free fatty acids induce endothelial progenitor
cell senescence via PGC-1α/SIRT1 signaling pathway. Cell Biol Int.
41:1146–1159. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu H, Yuan L, Xu S and Wang K:
Endothelial cell and macro-phage regulation of vascular smooth
muscle cell calcification modulated by cholestane-3beta, 5alpha,
6beta-triol. Cell Biol Int. 31:900–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin X, Li S, Wang YJ, Wang Y, Zhong JY, He
JY, Cui XJ, Zhan JK and Liu YS: Exosomal Notch3 from high
glucose-stimulated endothelial cells regulates vascular smooth
muscle cells calcification/aging. Life Sci. 232:1165822019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo Z, Sun Y, Qi B, Lin J, Chen Y, Xu Y
and Chen J: Human bone marrow mesenchymal stem cell-derived
extracellular vesicles inhibit shoulder stiffness via let-7a/Tgfbr1
axis. Bioact Mater. 17:344–359. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang C, Wang XY, Zhang P, He TC, Han JH,
Zhang R, Lin J, Fan J, Lu L, Zhu WW, et al: Cancer-derived exosomal
HSPC111 promotes colorectal cancer liver metastasis by
reprogramming lipid metabolism in cancer-associated fibroblasts.
Cell Death Dis. 13:572022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JH, Song J, Kim IG, You G, Kim H, Ahh
JH and Mok H: Exosome-mediated delivery of transforming growth
factor-β receptor 1 kinase inhibitors and toll-like receptor 7/8
agonists for combination therapy of tumors. Acta Biomater.
141:354–363. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Yang W, Zou B, Hou Y, Yan W, Chen T and Qu
S: Extracellular vesicles in vascular calcification. Clin Chim
Acta. 499:118–122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kalluri R and LeBleu VS: The biology,
function, and biomedical applications of exosomes. Science.
367:eaau69772020. View Article : Google Scholar :
|
|
27
|
Bano S, Tandon S and Tandon C: Emerging
role of exosomes in arterial and renal calcification. Hum Exp
Toxicol. 40:1385–1402. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liberman M and Marti LC: Vascular
calcification regulation by exosomes in the vascular wall. Adv Exp
Med Biol. 998:151–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bobryshev YV, Killingsworth MC, Huynh TG,
Lord RS, Grabs AJ and Valenzuela SM: Are calcifying matrix vesicles
in atherosclerotic lesions of cellular origin? Basic Res Cardiol.
102:133–143. 2007. View Article : Google Scholar
|
|
30
|
Bommanavar S, Hosmani J, Togoo RA, Baeshen
HA, Raj AT, Patil S, Bhandi S and Birkhed D: Role of matrix
vesicles and crystal ghosts in bio-mineralization. J Bone Miner
Metab. 38:759–764. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kapustin AN, Davies JD, Reynolds JL,
McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot
D, Mayr M and Shanahan CM: Calcium regulates key components of
vascular smooth muscle cell-derived matrix vesicles to enhance
mineralization. Circ Res. 109:e1–e12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goettsch C, Hutcheson JD, Aikawa M, Iwata
H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, et
al: Sortilin mediates vascular calcification via its recruitment
into extracellular vesicles. J Clin Invest. 126:1323–1336. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bardeesi ASA, Gao J, Zhang K, Yu S, Wei M,
Liu P and Huang H: A novel role of cellular interactions in
vascular calcification. J Transl Med. 15:952017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu F, Zhong JY, Lin X, Shan SK, Guo B,
Zheng MH, Wang Y, Li F, Cui RR, Wu F, et al: Melatonin alleviates
vascular calcification and ageing through exosomal miR-204/miR-211
cluster in a paracrine manner. J Pineal Res. 68:e126312020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo Y, Bao S, Guo W, Diao Z, Wang L, Han
X, Guo W and Liu W: Bone marrow mesenchymal stem cell-derived
exosomes alleviate high phosphorus-induced vascular smooth muscle
cells calcification by modifying microRNA profiles. Funct Integr
Genomics. 19:633–643. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li J, Jiang R, Hou Y and Lin A:
Mesenchymal stem cells-derived exosomes prevent sepsis-induced
myocardial injury by a CircRTN4/miR-497-5p/MG-53 pathway. Biochem
Biophys Res Commun. 618:133–140. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim HS and Lee MS: STAT1 as a key
modulator of cell death. Cell Signal. 19:454–465. 2007. View Article : Google Scholar
|
|
38
|
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou
X, Ma H, Wei D and Sun S: The role of JAK/STAT signaling pathway
and its inhibitors in diseases. Int Immunopharmacol. 80:1062102020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dodington DW, Desai HR and Woo M:
JAK/STAT-emerging players in metabolism. Trends Endocrinol Metab.
29:55–65. 2018. View Article : Google Scholar
|
|
40
|
Smyth AE, Kaleviste E, Snow A, Kisand K,
McMahon CJ, Cant AJ and Leahy TR: Aortic calcification in a patient
with a gain-of-function STAT1 mutation. J Clin Immunol. 38:468–470.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kunigal S, Kusch A, Tkachuk N, Tkachuk S,
Jerke U, Haller H and Dumler I: Monocyte-expressed urokinase
inhibits vascular smooth muscle cell growth by activating Stat1.
Blood. 102:4377–4383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Demyanets S, Kaun C, Rychli K,
Pfaffenberger S, Kastl SP, Hohensinner PJ, Rega G, Katsaros KM,
Afonyushkin T, Bochkov VN, et al: Oncostatin M-enhanced vascular
endothelial growth factor expression in human vascular smooth
muscle cells involves PI3K-, p38 MAPK-, Erk1/2- and
STAT1/STAT3-dependent pathways and is attenuated by interferon-γ.
Basic Res Cardiol. 106:217–231. 2011. View Article : Google Scholar
|
|
43
|
Kirchmer MN, Franco A, Albasanz-Puig A,
Murray J, Yagi M, Gao L, Dong ZM and Wijelath ES: Modulation of
vascular smooth muscle cell phenotype by STAT-1 and STAT-3.
Atherosclerosis. 234:169–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cossetti C, Iraci N, Mercer TR, Leonardi
T, Alpi E, Drago D, Alfaro-Cervello C, Saini HK, Davis MP,
Schaeffer J, et al: Extracellular vesicles from neural stem cells
transfer IFN-γ via Ifngr1 to activate Stat1 signaling in target
cells. Mol Cell. 56:193–204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cai M, Shi Y, Zheng T, Hu S, Du K, Ren A,
Jia X, Chen S, Wang J and Lai S: Mammary epithelial cell derived
exosomal MiR-221 mediates M1 macrophage polarization via
SOCS1/STATs to promote inflammatory response. Int Immunopharmacol.
83:1064932020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Buffolo F, Monticone S, Camussi G and
Aikawa E: Role of extracellular vesicles in the pathogenesis of
vascular damage. Hypertension. 79:863–873. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Godo S and Shimokawa H: Endothelial
functions. Arterioscler Thromb Vasc Biol. 37:e108–e114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Taguchi K, Hida M, Narimatsu H, Matsumoto
T and Kobayashi T: Glucose and angiotensin II-derived endothelial
extracellular vesicles regulate endothelial dysfunction via ERK1/2
activation. Pflugers Arch. 469:293–302. 2017. View Article : Google Scholar
|
|
49
|
Brodsky SV, Zhang F, Nasjletti A and
Goligorsky MS: Endothelium-derived microparticles impair
endothelial function in vitro. Am J Physiol Heart Circ Physiol.
286:H1910–H1915. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang H, Liu J, Qu D, Wang L, Wong CM, Lau
CW, Huang Y, Wang YF, Huang H, Xia Y, et al: Serum exosomes mediate
delivery of arginase 1 as a novel mechanism for endothelial
dysfunction in diabetes. Proc Natl Acad Sci USA. 115:E6927–E6936.
2018.PubMed/NCBI
|
|
51
|
Van den Bergh G, Van den Branden A,
Opdebeeck B, Fransen P, Neven E, De Meyer GRY, D'Haese PC and
Verhulst A: Endothelial dysfunction aggravates arterial media
calcification in warfarin administered rats. FASEB J.
36:e223152022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Grześk G, Kozinski M, Tantry US, Wicinski
M, Fabiszak T, Navarese EP, Grzesk E, Jeong YH, Gurbel PA and
Kubica J: High-dose, but not low-dose, aspirin impairs
anticontractile effect of ticagrelor following ADP stimulation in
rat tail artery smooth muscle cells. Biomed Res Int.
2013:9282712013. View Article : Google Scholar
|
|
53
|
Bosman M, Krüger DN, Favere K, Wesley CD,
Neutel CHG, Van Asbroeck B, Diebels OR, Faes B, Schenk TJ, Martinet
W, et al: Doxorubicin impairs smooth muscle cell contraction: Novel
insights in vascular toxicity. Int J Mol Sci. 22:128122021.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bundy K, Boone J and Simpson CL: Wnt
signaling in vascular calcification. Front Cardiovasc Med.
8:7084702021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gaur T, Lengner CJ, Hovhannisyan H, Bhat
RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS
and Lian JB: Canonical WNT signaling promotes osteogenesis by
directly stimulating Runx2 gene expression. J Biol Chem.
280:33132–33140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J
and He W: WNT/β-catenin signaling promotes VSMCs to osteogenic
trans-differentiation and calcification through directly modulating
Runx2 gene expression. Exp Cell Res. 345:206–217. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liao R, Wang L, Li J, Sun S, Xiong Y, Li
Y, Han M, Jiang H, Anil M and Su B: Vascular calcification is
associated with Wnt-signaling pathway and blood pressure
variability in chronic kidney disease rats. Nephrology (Carlton).
25:264–272. 2020. View Article : Google Scholar
|
|
58
|
Wu N, Liu GB, Zhang YM, Wang Y, Zeng HT
and Xiang H: MiR-708-5p/Pit-1 axis mediates high phosphate-induced
calcification in vascular smooth muscle cells via Wnt8b/β-catenin
pathway. Kaohsiung J Med Sci. 38:653–661. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cong J, Cheng B, Liu J and He P: RTEF-1
inhibits vascular smooth muscle cell calcification through
regulating Wnt/β-catenin signaling pathway. Calcif Tissue Int.
109:203–214. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhao L, Li X, Su J, Wang Gong F, Lu J and
Wei Y: STAT1 determines aggressiveness of glioblastoma both in vivo
and in vitro through wnt/β-catenin signalling pathway. Cell Biochem
Funct. 38:630–641. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yuan X, He F, Zheng F, Xu Y and Zou J:
Interferon-gamma facilitates neurogenesis by activating
Wnt/β-catenin cell signaling pathway via promotion of STAT1
regulation of the β-catenin promoter. Neuroscience. 448:219–233.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xie M, Fu Q, Wang PP and Cui YL:
STAT1-induced upregulation lncRNA LINC00958 accelerates the
epithelial ovarian cancer tumorigenesis by regulating Wnt/β-catenin
signaling. Dis Markers. 2021:14050452021. View Article : Google Scholar
|