Introduction
Influenza A virus (IAV) is a principal pathogen of
infectious respiratory diseases (1) and a common trigger for a number of
other respiratory diseases, such as acute exacerbation of chronic
obstructive pulmonary disease (AECOPD) (2). Severe IAV infection contributes
significantly to substantial morbidity and mortality, and is
responsible for health and economic burdens experienced worldwide
(3,4). Numerous airway inflammatory
responses are induced by respiratory viruses, leading to increased
proinflammatory cytokines and the consequent development of
cytokine storms (5). It is well
known that the proinflammatory agents TNF-α and nitric oxide can
interfere with cell cycle progression in different somatic cells,
and it has been reported that TNF-α-generated oxidative stress can
reduce cellular proliferation and induce cell cycle arrest
(6,7). Notably, the effect of TNF-α on
L-cells is cytostatic, which manifests as cell cycle arrest in the
G2 stage, thus indicating that the cell cycle is
dependent upon TNF cytotoxicity (8). While such data indicate that
inflammation and the cell cycle may be linked, the molecular
mechanisms that mediate IAV-induced airway inflammatory responses
remain unclear.
Raf kinase inhibitor protein (RKIP) is a member of
the phosphatidylethanolamine-binding protein (PEBP) family, and is
a highly conserved small molecule protein that is primarily located
in the cytoplasm and plasma membrane (9). By binding specifically to Raf-1
kinase, RKIP modulates crucial intracellular signaling networks
that include the Raf/MEK/ERK, NF-κB and GSK-3β signaling cascades
(1,3,10-12). Several studies have revealed that
RKIP participates in the regulation of a variety of physiological
and pathological processes, such as cellular differentiation, cell
cycle progression, apoptosis (11,13-15), autophagy, ferroptosis (16,17) and inflammation (18,19). The expression of RKIP has been
reported to be enhanced by didymin, which results in inhibition of
the MAPK and NF-κB pathways, and contributes to the
anti-inflammatory effects of RKIP (20). It has been demonstrated that RKIP
serves a negative role in regulating NLR family pyrin
domain-containing (NLRP)1, NLRP3 and NLR family CARD
domain-containing 4 inflammasome activation, and that it is a
potential therapeutic target for the treatment of
inflammasome-related diseases (21). It has also been reported that RKIP
occupies a critical role in the cell cycle of numerous types of
cancer (22). Although there are
a considerable number of published reports linking RKIP to various
intracellular signaling networks that control cellular growth
(23-26), the effects exerted by RKIP on
inflammation and cell cycle progression in airway epithelial cells
following IAV infection have not yet been reported.
The present study investigated the detailed
mechanisms underlying RKIP action in IAV-induced airway
inflammation by adopting primary human bronchial epithelial (pNHBE)
cells, cell lines and mouse models.
Materials and methods
Reagents and materials
Locostatin (cat. no. T8823) was purchased from
Shanghai Topscience Co., Ltd. SCH772984 (cat. no. S7101) was
purchased from Selleck Chemicals. RKIP (cat. no. ab76582) and
β-actin (cat. no. ab8227) antibodies were purchased from Abcam.
NLRP3 (cat. no. WL02635), ERK1/2 (cat. no. WL01864) and
phosphorylated (p)-ERK1/2 (Thr202/Tyr204; cat. no. WLP1512)
antibodies were purchased from Wanleibio Co., Ltd. CDK4 antibody
(D9G3E; cat. no. 12790) was obtained from Cell Signaling
Technology, Inc.
Animal experiments
C57BL/6 mice (female; age, 6 weeks; weight, 20-25 g;
n=24) were purchased from the Animal Service Unit of Anhui Medical
University. Mice were maintained at a controlled temperature
(22-25°C) and humidity (50-60%), under a 12-h light/dark cycle, and
provided with free access to water and food. In the present study,
mice were challenged with IAV to induce airway inflammation and
treated with locostatin. Mice were randomly allocated to the
following four groups (n=6/group): i) The negative control (NC)
group: IAV(−) + locostatin(−); ii) IAV(+) + locostatin(−) group;
iii) IAV(−) + locostatin(+) group; and iv) IAV(+) + locostatin(+)
group. Locostatin (10 mg/kg) pretreatment was conducted for 4 h via
intraperitoneal injection before 100 PFU of IAV was administered in
100 µl through oropharyngeal aspiration and the NC group was
treated with 0.9% saline. After 7 days, mice were anesthetized with
pentobarbital (70 mg/kg), and after euthanizing the mice, the
serum, BALF and lung tissues were collected for further analysis.
Blood was drawn by enucleation of the eyeball, followed by
centrifugation at 3,000 × g for 5 min at 4°C, and the serum was
collected for subsequent experiments. To collect BALF, a needle was
inserted into the trachea and fixed. The lungs were lavaged with 1
ml cold PBS three times to collect the BALF, followed by
centrifugation at 500 × g for 10 min at 4°C; the supernatants were
collected for subsequent experiments. Mice health and behavior were
monitored daily. During the experiment, any mice that were unable
to drink or eat, had difficulty breathing or had lost 20% of their
body weight were regarded as having reached the humane endpoints
and were immediately euthanized. None of the mice reached the
aforementioned endpoints in this experiment and thus none were
euthanized before the end of the experiment. Every effort was made
to minimize discomfort, distress, pain or injury to the mice. The
total duration of the experiment was 7 days. After the experiment,
all remaining animals were euthanized by cervical dislocation and
death was verified by respiratory arrest, cardiac arrest and
dilated pupils. All protocols were reviewed and approved by the
Animal Ethics Committee of Anhui Medical University.
Cell isolation and culture
The pNHBE cells were isolated from the normal
bronchial tissues of patients with lung carcinoma in situ
(n=3; 2 women, 1 man; age, 48-52 years) as determined by senior
pathologists. The bronchial tissues were cut at a site >3 cm
distant from the edge of the lung carcinoma according to methods
modified from previous studies (27-30) between August 2020 and January
2021, and were cultured in bronchial epithelial cell growth medium
(Lonza Group, Ltd.). The present study was approved by the
Biomedical Ethics Committee of Anhui Medical University (approval
no. 20200070; Hefei, China). All of the participants were informed
of the purpose of the study and provided written informed consent
in accordance with the ethical requirements. Human airway
epithelial BEAS-2B cells were purchased from Shanghai Fuheng
Biotechnology Co., Ltd. (cat. no. FH0319) and cultured in RPMI-1640
medium (Hyclone; Cytiva) supplemented with 10% fetal bovine serum
(cat. no. A6901FBS-500; Invigentech, Inc.), 100 U/ml penicillin and
100 ng/ml streptomycin (Absin Bioscience, Inc.) at 37°C in a
humidified atmosphere containing 5% CO2/95% air.
Diseased human bronchial epithelial (DHBE) cells, which are
immortalized cells from the bronchial tube of a patient with
chronic obstructive pulmonary disease, were purchased from Otwo
Biotech (Shenzhen), Inc. (cat. no. HTX2551) and were cultured
identically to BEAS-2B cells. Subsequently, BEAS-2B, DHBE and pNHBE
cells were infected with IAV [H3N2; strain was kindly provided by
Professor Yan Liu (Department of Microbiology, Anhui Medical
University, Hefei, China), multiplicity of infection (MOI), 2] for
48 h at 37°C prior to subsequent experiments. Locostatin (50 nM) or
SCH772984 (1 µM) was used to pretreat DHBE cells 4 h prior
to infection with IAV at 37°C.
Western blotting
Proteins from cells or the right lung tissues of
mice were harvested using RIPA lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) and the BCA kit (cat. no.
P0010; Beyotime Institute of Biotechnology) was used to determine
the protein concentration. Equal amounts of protein (20 µg)
were separated by SDS-PAGE on 12.5% gels (cat. no. PG113, Shanghai
Epizyme Biomedical Technology Co., Ltd.) and transferred to 0.45
µm PVDF membranes (cat. no. IPVH00010; MilliporeSigma).
Subsequently, 5% skim milk powder (cat. no. BS102-500 g; Biosharp
Life Sciences) was used to block the membranes for 1 h at room
temperature and 1X TBS-0.05% Tween 20 (TBST; cat. no. 9005-64-5;
neoFroxx GmbH) was used to wash the membranes three times (10
min/wash). Subsequently, the membranes were incubated with primary
anti-RKIP (1:1,000), anti-CDK4 (1:1,000), anti-NLRP3 (1:1,500),
anti-ERK1/2 (1:500), anti-p-ERK1/2 (1:300) and anti-β-actin
(1:3,000) anti-bodies at 4°C overnight. After washing with 1X TBST
three times (10 min/wash), the membranes were incubated with a
secondary anti-rabbit IgG, HRP-linked antibody (1:2,500; cat. no.
7074; Cell Signaling Technology, Inc.) for 1 h at room temperature.
Signals were detected using the Omni-ECL™ Femto Light
Chemiluminescence Kit (cat. no. SQ201; Shangh ai Epizyme Biomedical
Technology Co., Ltd.) and Tanon 5200 Multi Chemiluminescent Imaging
System (Tanon Science and Technology Co., Ltd.). Densitometric
analysis was performed using ImageJ 1.53e software (National
Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from the cells and mice right lung
tissues using Total RNA Isolation Reagent (cat. no. YY101; Shangh
ai Epizyme Biomedical Technology Co., Ltd.) and reversed
transcribed to cDNA using the Hifair® Ⅲ 1st Strand cDNA
Synthesis SuperMix for qPCR (gDNA digester plus) kit (cat. no.
11141ES60; Shanghai Yeasen Biotechnology Co., Ltd.) according to
the manufacturer's protocol. qPCR was performed using 2X S6
Universal SYBR qPCR Mix (cat. no. Q204; EnzyArtisan) under the
following conditions: 95°C for 30 sec, followed by 45 cycles at
95°C for 10 sec and 60°C for 30 sec. The primer sequences are
listed in Table I. The expression
levels of target mRNA were calculated using the 2−ΔΔCq
method (31) relative to the
reference gene (β-actin).
 | Table IPrimers used for reverse
transcription-quantitative PCR. |
Table I
Primers used for reverse
transcription-quantitative PCR.
| Primer | Sequence,
5′-3′ |
|---|
| RKIP (human) F |
GCTCTACACCTTGGTCCTGACA |
| RKIP (human) R |
AATCGGAGAGGACTGTGCCACT |
| IL-1β (human)
F |
CCACAGACCTTCCAGGAGAATG |
| IL-1β (human)
R |
GTGCAGTTCAGTGATCGTACAGG |
| IL-18 (human)
F |
AGCAAGGAATTGTCTCCCAG |
| IL-18 (human)
R |
GAAGCGATCTGGAAGGTCTG |
| β-actin (human)
F |
CACCATTGGCAATGAGCGGTTC |
| β-actin (human)
R |
AGGTCTTTGCGGATGTCCACGT |
| IL-1β (mouse)
F |
TGGACCTTCCAGGATGAGGACA |
| IL-1β (mouse)
R |
GTTCATCTCGGAGCCTGTAGTG |
| IL-18 (mouse)
F |
AGGGTTTGTGTTCCAGAAAGATG |
| IL-18 (mouse)
R |
AGCCTCGGGTATTCTGTTATGG |
| β-actin (mouse)
F |
CATTGCTGACAGGATGCAGAAGG |
| β-actin (mouse)
R |
TGCTGGAAGGTGGACAGTGAGG |
Transduction with RKIP lentivirus
A lentiviral vector overexpressing the RKIP gene
(LV5-PEBP1) and its empty vector (LV5NC) were commercially
constructed and provided by Shanghai GenePharma Co., Ltd. (3rd
generation; cat. no. LV2021-7006). The sequence (Fig. S1) and the shuttle plasmid
(Fig. S2) were synthesized by
Shanghai GenePharma Co., Ltd. Briefly, 5 µg overexpression
vector and the packing vectors, 3 µg PG-P1-VSVG, 2 µg
PG-P2-REV and 6 µg PG-P3-RRE, were mixed for 20 min at room
temperature to form the transfection mixture and were then
co-transfected into 293T cells (80% confluence; cat. no. SCSP-502;
Cell Bank of the Chinese Academy of Sciences) using RNAi-Mate (cat.
no. G04001; Shanghai GenePharma Co., Ltd.) at 37°C for 6 h.
Subsequently, the transfection complex was removed and fresh medium
was added to the cells. The viral supernatant was harvested after
72 h and centrifuged at 20,000 × g for 2 h at 4°C. The titer of
lentivirus was determined by counting GFP-positive cells to
determine successfully infected 293T cells. The NC lentivirus was
constructed using the same method. Subsequently, BEAS-2B cells were
transduced with lentivirus (MOI, 10) and polybrene (5 µg/ml)
for 24 h at 37°C in a humidified atmosphere containing 5%
CO2/95% air, after which, fresh medium was added to the
cells. After 3 days, transduction efficiency was observed. The time
interval between transfection and subsequent experiments was ≥10
days. BEAS-2B cells infected with the lentivirus were selected with
2 µg/ml puromycin (cat. no. GCD0289949; Shanghai GeneChem
Co., Ltd.).
Immunofluorescence staining
Cells were plated in 24-well culture plates after 48
h of IAV infection. The cells were then washed with cold
phosphate-buffered saline, fixed with 4% paraformaldehyde for 20
min at room temperature and permeabilized with 0.1% Triton X-100
for 15 min at room temperature. After being blocked with 5% BSA
(cat. no. 9048-46-8; neoFroxx GmbH) at room temperature for 30 min,
the cells were incubated with RKIP antibody (1:100) overnight at
4°C and were then incubated with Alexa Fluor® 594 goat
anti-rabbit IgG H&L (1:500; cat. no. ab150080; Abcam) for 1 h
at room temperature. DAPI (cat. no. C1002; Beyotime Institute of
Biotechnology) was applied for 15 min in the dark at room
temperature to visualize the nuclei, and the cells were detected
under a laser confocal microscope (Zeiss LSM880; Carl Zeiss
AG).
ELISA
The levels of inflammatory cytokines IL-1β and IL-18
in cellular supernatants were examined using IL-1β (cat. no.
ml058059) and IL-18 (cat. no. ml058055) ELISA kits (all from
Shanghai Enzyme-linked Biotechnology Co., Ltd.). The levels of
IL-1β and IL-18 in the serum and bronchoalveolar lavage fluid
(BALF) of mice were measured using IL-1β (cat. no. ml301814) and
IL-18 (cat. no. ml002294) (all from Shanghai Enzyme-linked
Biotechnology Co., Ltd.). ELISA kits were performed according to
the manufacturer's protocols.
Cell cycle assay
Cells (1×105/well) were plated in 12-well
plates and were fixed in 70% ethanol at 4°C overnight. The cells
were then stained with propidium iodide/RNase A (cat. no. C1052;
Beyotime Institute of Biotechnology) at 37°C for 30 min in the
dark. Cell cycle progression was assessed using a NovoCyte flow
cytometer (Agilent Technologies, Inc.) and FlowJo software (FlowJo
X 10.0.7r2; FlowJo LLC).
Cell viability assay
Cells were plated at a density of 5×103
in 96-well plates. After cells were infected with IAV (MOI, 2) for
48 h at 37°C in a humidified atmosphere containing 5%
CO2, cell viability was assessed using a Cell Counting
Kit-8 (CCK-8) kit (cat. no. C0037; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
Briefly, 10 µl CCK-8 reagent was added to each well, and the
cells were incubated at 37°C for 2 h. Absorbance was measured at
450 nm to evaluate cell viability.
5-Ethynyl-2′-deoxyuridine (EdU)
assay
Cells (2×104/well) were cultured in
24-well plates after IAV infection for 48 h and an EdU kit (cat.
no. C0075S; Beyotime Institute of Biotechnology) was used to detect
the degree of DNA damage according to the manufacturer's protocol.
Briefly, 10 µM EdU was used to incubate cells at 37°C for 2
h to label them. Subsequently, cells were fixed in 4%
paraformaldehyde at room temperature for 20 min and permeabilized
with 0.1% Triton X-100 at room temperature for 15 min. Next, cells
were dyed with reaction solution for 30 min at room temperature in
the dark and the Hoechst was applied for 15 min at room temperature
in dark. The results were analyzed using a Leica fluorescence
microscope (Leica DM6B; Leica Microsystems, Inc.).
Hematoxylin and eosin (H&E)
staining
Lung tissues harvested in animal experiments were
fixed in 4% paraformaldehyde at room temperature for 48 h, embedded
in paraffin, cut into 5-µm sections and stained using a
H&E stain kit (cat. no. G1120; Beijing Solarbio Science &
Technology Co., Ltd.). Briefly, the sections were stained with
hematoxylin solution for 10 min at room temperature and washed in
running tap water for 5 min, after which, differentiation solution
was added for 10 sec at room temperature. The sections were
subsequently stained with eosin solution for 10 sec at room
temperature, dehydrated in alcohol (75, 85, 95 and 100%; each for
2-3 sec at room temperature) and rinsed in 100% alcohol for 1 min
at room temperature. After clearing with xylene and sealing with
neutral balsam, the sections were scanned using a Pannoramic Whole
Slide Scanner (Pannoramic Desk; 3DHISTECH Kft.) and viewed with
Caseviewer 2.2 (3DHISTECH Kft.). Histological scores of
inflammation in the lungs of IAV-induced mice were semi-quantified
using the following scoring system: 0, no inflammation; 1, only
moderate peribronchial inflammation; 2, <10% inflamed lung
tissue; 3, 10-25% inflamed lung tissue; 4, 26-50% inflamed lung
tissue; 5, >50% inflamed lung tissue (32). Scoring was performed independently
by two investigators, whose scores were averaged.
Immunohistochemistry (IHC)
Briefly, lung tissues were fixed, embedded and
sectioned as performed prior to H&E staining. The sections were
then deparaffinized in xylene, rehydrated in a graded series of
alcohol and subjected to antigen retrieval by microwaving (650 W)
for 12 min in sodium citrate buffer (pH 6.0). The endogenous
peroxidase activity was quenched by incubating the sections in 3%
H2O2 for 15 min at room temperature.
Subsequently, the lung tissue sections were blocked with 5% BSA for
30 min at room temperature and then incubated with RKIP primary
antibody (1:250) at 4°C overnight, after which they were incubated
with a goat anti-rabbit IgG H&L (HRP) secondary antibody
(1:1,000; cat. no. ab97051; Abcam) at room temperature for 1 h.
After DAB staining for 25 sec at room temperature, the sections
were counterstained with hematoxylin and dyed in 1% ammonia
solution for 30 sec at room temperature. The sections were scanned
using a Pannoramic Whole Slide Scanner (Pannoramic Desk) and viewed
with Caseviewer 2.2.
Statistical analysis
For in vitro experiments, each measurement
was obtained by three independent experiments. The data are
presented as the mean ± SEM and statistical analyses were conducted
using SPSS 23.0 (IBM, Corp.). Differences between two groups were
analyzed using unpaired Student's t-test, whereas differences among
three or more groups were assessed by one-way ANOVA and Tukey's
post hoc test. The histological score was presented as median and
IQR, and was statistically analyzed using Kruskal-Wallis and Dunn's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of IAV infection on the
expression of RKIP in BEAS-2B, DHBE and pNHBE cells
BEAS-2B and DHBE cells were exposed to IAV (MOI, 2)
for 48 h and the expression levels of RKIP were detected. Notably,
the expression levels of RKIP were significantly decreased
following IAV infection in both BEAS-2B cells (P=0.0083; Fig. 1A and B) and DHBE cells
(P<0.0001; Fig. 1E and F), as
assessed by western blotting. Subsequently, inflammatory cytokines
were assessed using RT-qPCR and ELISA; the results revealed that
IL-1β and IL-18 were significantly increased in response to IAV
(MOI, 2) infection for 48 h in BEAS-2B cells, as determined by
RT-qPCR (P<0.0001 for IL-1β, P=0.0081 for IL-18; Fig. 1C) and ELISA (P<0.0001 for
IL-1β, P=0.0035 for IL-18; Fig.
1D). Similarly, IL-1β and IL-18 levels were elevated in DHBE
cells in response to IAV, as determined using RT-qPCR (P=0.0010 for
IL-1β, P=0.0029 for IL-18; Fig.
1G) and ELISA (P=0.0016 for IL-1β, P=0.0005 for IL-18; Fig. 1H). To further confirm this result,
pNHBE cells were employed to detect the levels of RKIP and
inflammatory cytokines. As shown in Fig. 1I and J (P=0.0100), RKIP was
significantly reduced in pNHBE cells after IAV infection (MOI, 2),
as determined by western blotting; by contrast, IL-1β and IL-18
were increased, as revealed by RT-qPCR (P=0.0015 for IL-1β,
P=0.0004 for IL-18; Fig. 1K) and
ELISA (P=0.0005 for IL-1β, P= 0.0034 for IL-18; Fig. 1L). These results indicated that
IAV infection significantly attenuated RKIP expression in BEAS-2B,
DHBE and pNHBE cells, and that the airway inflammatory cytokines,
IL-1β and IL-18, were significantly elevated in response to IAV
infection.
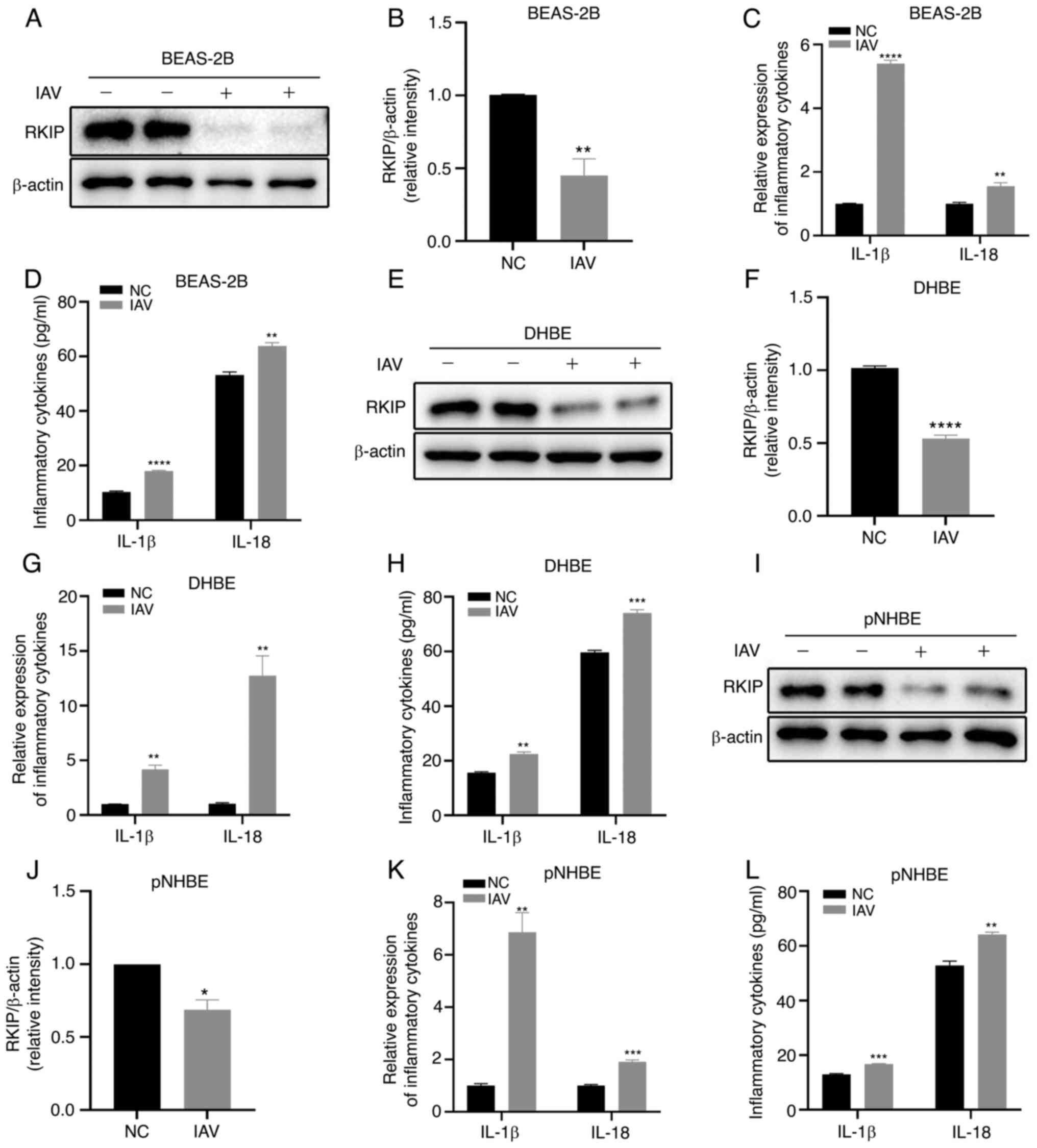 | Figure 1Effects of IAV infection
(multiplicity of infection, 2) for 48 h on BEAS-2B, DHBE and pNHBE
cells. (A and B) Protein expression levels of RKIP in BEAS-2B
cells. (C) mRNA expression levels of IL-1β and IL-18 in BEAS-2B
cells were detected by RT-qPCR. (D) Levels of IL-1β and IL-18 in
BEAS-2B cells were detected by ELISA. (E and F) Protein expression
levels of RKIP in DHBE cells. (G) mRNA expression levels of IL-1β
and IL-18 in DHBE cells. (H) Levels of IL-1β and IL-18 in DHBE
cells were detected by ELISA. (I and J) Protein expression levels
of RKIP in pNHBE cells were detected by western blotting. (K) mRNA
expression levels of IL-1β and IL-18 in pNHBE cells were detected
by RT-qPCR. (L) Levels of IL-1β and IL-18 in pNHBE cells were
detected by ELISA. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 vs. NC.
DHBE, diseased human bronchial epithelial; IAV, influenza A virus;
NC, negative control; pNHBE, primary human bronchial epithelial;
RKIP, Raf kinase inhibitor protein; RT-qPCR, reverse
transcription-quantitative PCR. |
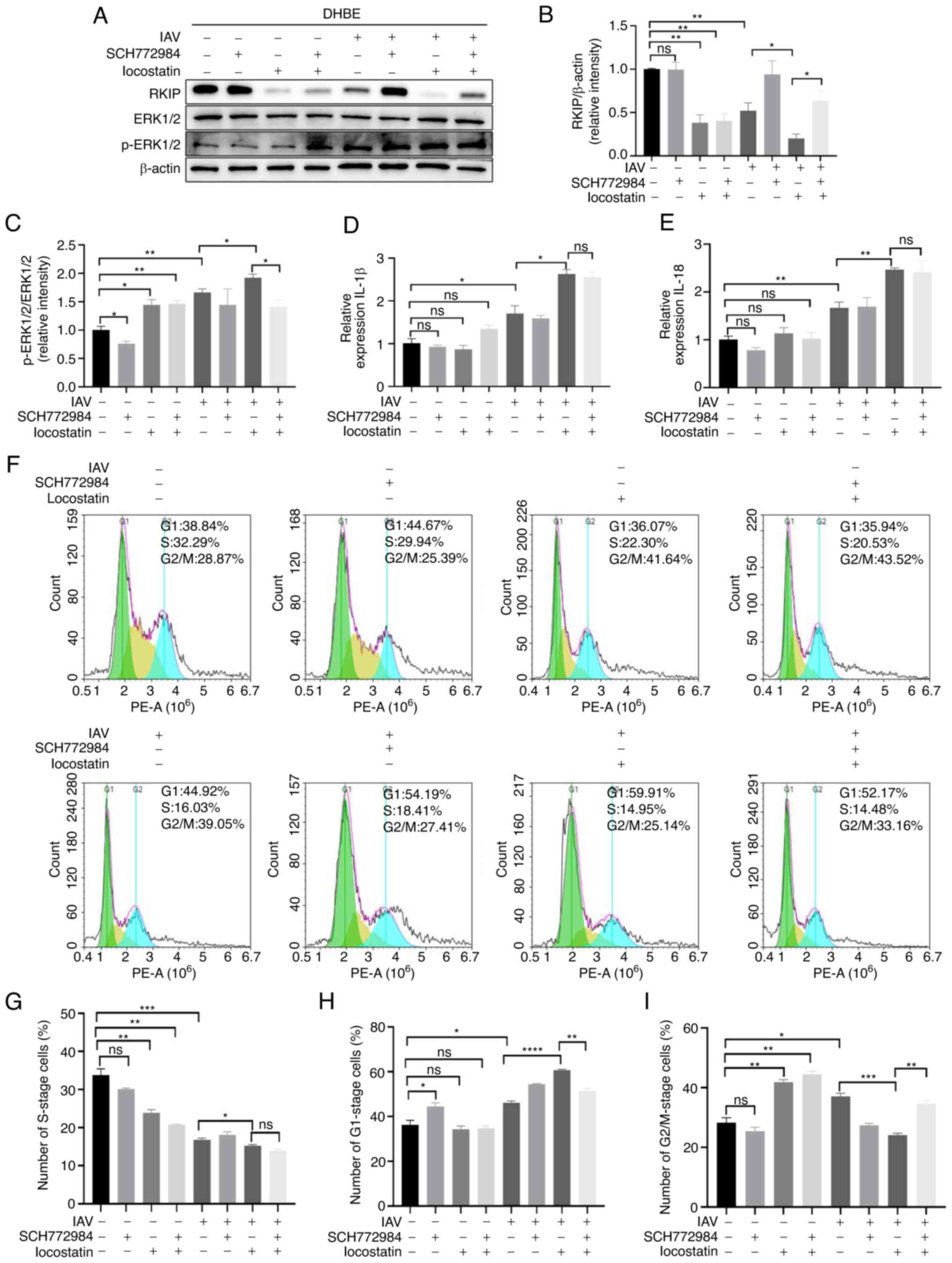
RKIP inhibition by locostatin augments
inflammatory responses induced by IAV in airway epithelial
cells
To investigate whether airway inflammation induced
by IAV was regulated by RKIP, the DHBE cells were pretreated for 4
h before IAV infection with locostatin. Locostatin is a small
molecule that covalently binds RKIP, which results in a
protein-protein interaction where locostatin inhibits and abrogates
the ability of RKIP to bind; locostatin also inhibits Raf-1 kinase
specifically to inhibit the expression of RKIP (33,34). The present study ascertained that
the expression of RKIP was significantly reduced in the IAV(+) +
locostatin(+) group compared with in the IAV(+) + locostatin(−)
group, as determined using western blotting (P=0.0003; Fig. 2A and B) and immunofluorescence
(Fig. 2J). In addition, the mRNA
expression levels of the inflammatory cytokines IL-1β and IL-18
were detected by RT-qPCR and the cellular supernatant levels were
detected by ELISA. As shown in Fig.
2F and G, the mRNA expression levels of the cytokines were
elevated in the IAV(+) + locostatin(−) group compared with in the
NC group (P=0.0107 for IL-1β, P=0.0016 for IL-18) and were more
markedly increased in the IAV(+) + locostatin(+) group compared
with in the IAV(+) + locostatin(−) group (P=0.0004 for IL-1β,
P=0.0046 for IL-18). As shown in Fig.
2H and I, ELISA exhibited similar trends to RT-qPCR; IL-1β and
IL-18 were significantly elevated in the IAV(+) + locostatin(−)
group compared with in the NC group (P=0.0002 for IL-1β, P=0.0002
for IL-18) and were more markedly increased in the IAV(+) +
locostatin(+) group compared with in the IAV(+) + locostatin(−)
group (P=0.0010 for IL-1β, P=0.0198 for IL-18). Furthermore, the
protein expression levels of NLRP3 also exhibited a similar trend,
as determined using western blotting [P=0.0002 for IAV(+) +
locostatin(−) group vs. IAV(−) + locostatin(−) group; P=0.0094 for
IAV(+) + locostatin(+) group vs. IAV(+) + locostatin(−) group;
Fig. 2A and C). To further
confirm the effect of RKIP on cell cycle progression in DHBE cells,
western blotting was performed and revealed that CDK4 was
downregulated after IAV infection compared with in the NC group
(P=0.0489), and the same trend was observed in the IAV(+) +
locostatin(+) group compared with in the IAV(+) + locostatin(−)
group (P=0.0052) (Fig. 2A and D).
Additionally, the EdU (Fig. 2K and
L) and CCK-8 (Fig. 2M) assays
confirmed that there was enhanced DNA damage and reduced cell
viability in the IAV(+) + locostatin(+) group compared with in the
IAV(+) + locostatin(−) group (P=0.0351 for EdU, P<0.0001 for
CCK-8) and the IAV(+) + locostatin(−) group compared with in the NC
group (P=0.0021 for EdU, P= 0.0007 for CCK-8). Furthermore, the
cell cycle was analyzed using flow cytometry. As shown in Fig. 2N and O, there was a significant
inhibition in the number of cells at S phase in the IAV(+) +
locostatin(−) group compared with in the NC group (P=0.0004).
Furthermore, the G1 phase of cell cycle was arrested
(P=0.0488) and the ratio of S phase was decreased in the IAV(+) +
locostatin(+) group compared with in the IAV(+) + locostatin(−)
group (P=0.0021). Furthermore, the protein expression levels of
ERK1/2 and p-ERK1/2 were detected using western blotting (Fig. 2A and E). The results suggested
that the ratio of pERK1/2/ERK1/2 was increased when comparing the
IAV(+) + locostatin(−) group with the NC group (P=0.0106), and the
IAV(+) + locostatin(+) group with the IAV(+) + locostatin(−) group
(P=0.0224). These results suggested that the inhibition of RKIP by
locostatin may increase the inflammatory response and suppress cell
cycle progression during the inflammatory response induced by
IAV.
 | Figure 2RKIP inhibition by locostatin (50 nM)
augments inflammatory responses induced by IAV infection
(multiplicity of infection, 2) for 48 h in DHBE cells. (A) Protein
expression levels of RKIP, ERK1/2, p-ERK1/2, CDK4 and NLRP3 were
detected by western blotting. Densitometric analysis of (B) RKIP,
(C) NLRP3, (D) CDK4 and (E) p-ERK1/2 normalized to ERK1/2 in the
different groups. (F and G) mRNA expression levels of the
inflammatory cytokines IL-1β and IL-18 were measured by reverse
transcription-quantitative PCR. (H and I) Levels of the
inflammatory cytokines IL-1β and IL-18 were measured by ELISA. (J)
Immunofluorescence staining was performed to detect the expression
of RKIP. RKIP (red) and DAPI (blue); scale bars, 20 µm;
magnification, ×200. (K and L) EdU assay was used to detect cells
synthesizing DNA in the S phase of the cell cycle. Scale bar, 50
µm; magnification, ×50. (M) Cell Counting Kit-8 was used to
detect the viability of DHBE cells. (N and O) Cell cycle was
detected by flow cytometry. *P<0.05,
**P<0.01 and ***P<0.001 vs. NC;
#P<0.05, ##P<0.01,
###P<0.001 and ####P<0.0001 vs. IAV(+)
+ locostatin(-). DHBE, diseased human bronchial epithelial; EdU,
5-ethynyl-2'-deoxyuridine; IAV, influenza A virus; NC, negative
control; NLRP3, NLR family pyrin domain-containing 3; p,
phosphorylated; RKIP, Raf kinase inhibitor protein. |
RKIP alleviates the inflammatory response
in BEAS-2B cells after IAV infection
To further confirm that RKIP regulated inflammation
and cell cycle progression after IAV infection, RKIP overexpression
(RKIP-OE) was induced in BEAS-2B cells by lentiviral transduction.
As shown in Fig. 3B, the mRNA
expression levels of RKIP were significantly upregulated after
transduction, as determined by RT-qPCR (P=0.0021). As shown in
Fig. 3A, C and K, the expression
levels of RKIP were markedly reduced after IAV infection, as
detected by western blotting (P=0.0037) and immunofluorescence.
Furthermore, changes in the inflammatory protein NLRP3 were
determined through western blotting and it was demonstrated that
RKIP-OE significantly alleviated the inflammatory response after
IAV infection (P=0.0323; Fig. 3A and
D). Furthermore, the inflammatory cytokines, IL-1β and IL-18,
were assessed and it was revealed that their levels were
significantly increased in the IAV(+) + RKIP-OE(−) group compared
with in the NC group by RT-qPCR (P=0.0057 for IL-1β, P=0.0061 for
IL-18; Fig. 3G and H) and ELISA
(P=0.0008 for IL-1β, P=0.0009 for IL-18; Fig. 3I and J). By contrast, the levels
of IL-1β and IL-18 were significantly inhibited in the IAV(+) +
RKIP-OE(+) group compared with in the IAV(+) + RKIP-OE(−) group, as
determined by RT-qPCR (P=0.0337 for IL-1β, P= 0.0287 for IL-18;
Fig. 3G and H) and ELISA
(P=0.0110 for IL-1β, P=0.0006 for IL-18; Fig. 3I and J). The present study also
assessed CDK4 expression, as it is related to the cell cycle; the
western blotting results revealed that CDK4 was upregulated when
RKIP was overexpressed after IAV infection (P=0.0212; Fig. 3A and E). In addition, cell cycle
progression was impaired after IAV infection in BEAS-2B cells;
however, RKIP-OE reversed this effect, as reflected by the results
of EdU (P=0.0236; Fig. 3L and M)
and CCK-8 (P=0.0090; Fig. 3N)
assays. Flow cytometry of cell cycle kinetics suggested that the
G1 phase of cell cycle was arrested (P= 0.0011) and the
ratio of S stage was decreased in the IAV(+) + RKIP-OE(−) group
compared with in the NC group (P<0.0001; Fig. 3O and P), and that the percentage
of cells in S phase that was reduced after IAV infection was
restored when RKIP was overexpressed (P=0.0048; Fig. 3O and P). RKIP-OE significantly
mitigated the production of inflammatory cytokines and provided
recovery from cell cycle arrest in BEAS-2B cells following IAV
infection. Furthermore, overexpression of RKIP decreased the ratio
of p-ERK1/2/ERK1/2 induced by IAV infection, as determined by
western blotting (P=0.0403; Fig. 3A
and F). All of these results revealed that OE of RKIP
alleviated the inflammatory response and restored cell cycle
progression in BEAS-2B cells infected with IAV.
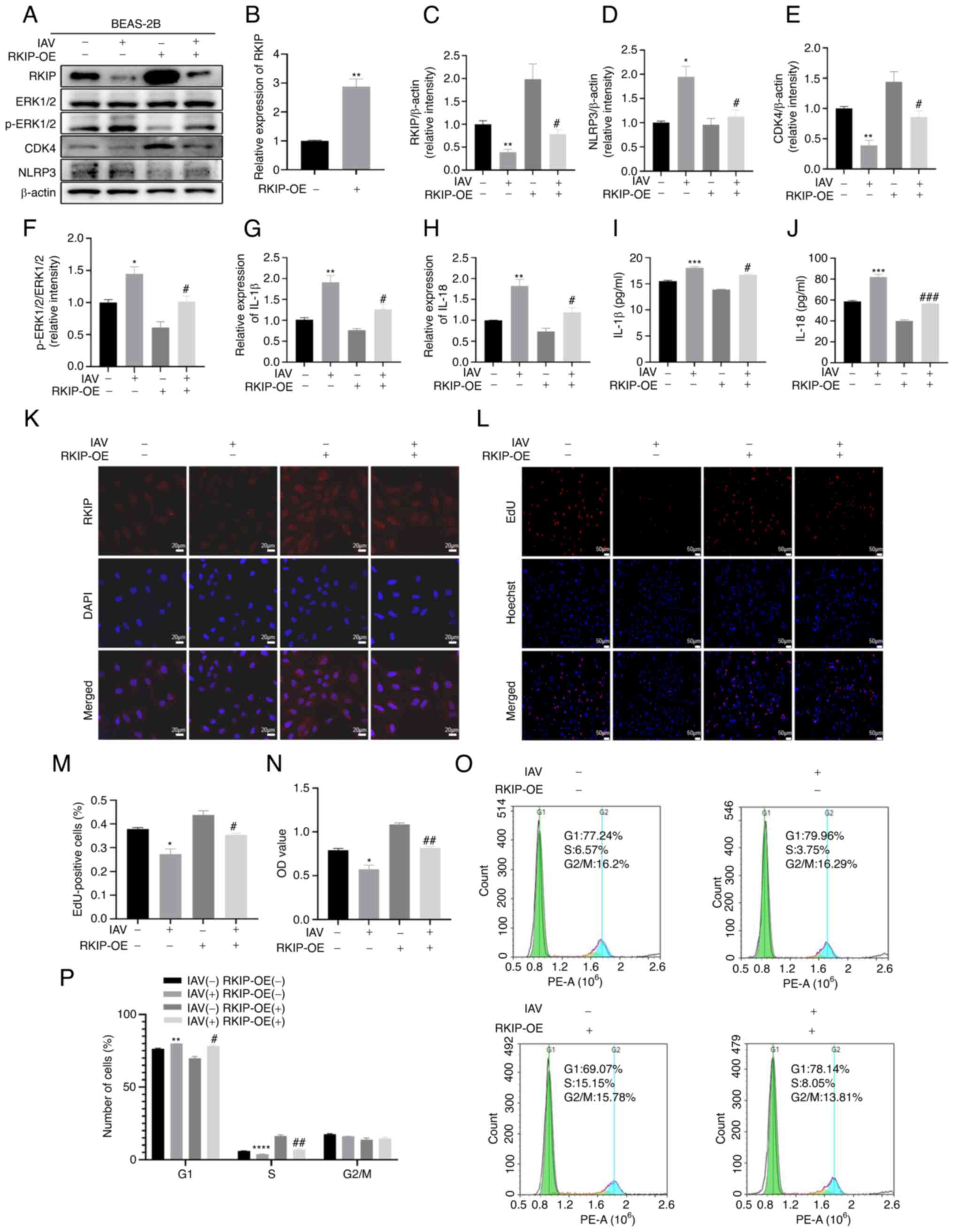 | Figure 3RKIP alleviates the inflammatory
response in BEAS-2B cells after IAV infection. (A) Protein
expression levels of RKIP, ERK1/2, p-ERK1/2, CDK4 and NLRP3 in
BEAS-2B cells were detected by western blotting. Densitometric
analysis of (C) RKIP, (D) NLRP3, (E) CDK4 and (F) p-ERK1/2
normalized to ERK1/2 in the different groups. (B) Efficiency of
RKIP-OE lentivirus transduction was verified in BEAS-2B cells by
RT-qPCR. (G and H) mRNA expression levels of the inflammatory
cytokines IL-1β and IL-18 were measured by RT-qPCR. (I and J)
Levels of the inflammatory cytokines IL-1β and IL-18 were measured
by ELISA. (K) Immunofluorescence staining was performed to detect
the expression of RKIP. RKIP (red) and DAPI (blue); scale bar, 20
µm; magnification, ×200. (L and M) EdU assay was used to
detect the cells synthesizing DNA in the S-phase of the cell cycle.
Scale bar, 50 µm; magnification, ×50. (N) Cell Counting
Kit-8 was used to detect the viability of BEAS-2B cells. (O and P)
Cell cycle was detected by flow cytometry. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001 vs. NC; #P<0.05,
##P<0.01 and ###P<0.001 vs. IAV(+) +
RKIP-OE(-). EdU, 5-ethynyl-2'-deoxyuridine; IAV, influenza A virus;
NC, negative control; NLRP3, NLR family pyrin domain-containing 3;
OE, overexpression; p, phosphorylated; RKIP, Raf kinase inhibitor
protein; RT-qPCR, reverse transcription-quantitative PCR. |
RKIP regulates the inflammatory response
via the ERK/MAPK pathway
To investigate whether RKIP regulated the
inflammatory response via the ERK/MAPK pathway, the specific
ERK/MAPK-pathway inhibitor SCH772984 was used to treat cells after
challenging them with or without IAV. The protein expression levels
of RKIP, ERK1/2 and p-ERK1/2 were determined by western blotting,
and the results suggested that SCH772984 significantly inhibited
the ERK/MAPK pathway as p-ERK1/2/ERK1/2 was reduced in response to
the IAV(−) + SCH772984(+) + locostatin(−) group compared with the
IAV(−) + SCH772984(−) + locostatin(−) group (P=0.0416; Fig. 4A and C). In addition, SCH772984
reversed activation of the ERK/MAPK pathway induced by IAV and RKIP
inhibition, as determined by comparing the IAV(+) + SCH772984(+) +
locostatin(+) group with the IAV(+) + SCH772984(−) + locostatin(+)
group (P=0.0182; Fig. 4A and C).
Following IAV infection, IL-1β and IL-18 levels were elevated
compared with in the control group (P=0.0297 for IL-1β, P= 0.0093
for IL-18), and their levels were significantly increased in the
IAV(+) + SCH772984(−) + locostatin(+) group compared with in the
IAV(+) + SCH772984(−) + locostatin(−) group (P=0.0122 for IL-1β,
P=0.0033 for IL-18), as determined using RT-qPCR (Fig. 4D and E). In addition, when the
ERK/MAPK pathway was inhibited by SCH772984, inhibiting RKIP did
not induce a significant change in the inflammatory cytokines IL-1β
and IL-18, as determined by RT-qPCR (Fig. 4D and E). Cells were arrested in
G1 phase (P=0.0106; Fig.
4H) and the proportion of cells in S phase was decreased after
IAV infection (P=0.0006; Fig. 4G)
when comparing the IAV(+) + SCH772984(−) + locostatin(−) group with
the IAV(−) + SCH772984(−) + locostatin(−) group, and locostatin
aggravated the effects of IAV infection (P<0.0001 for
G1 phase, P=0.0325 for S phase) when comparing the
IAV(+) + SCH772984(−) + locostatin(+) group with the IAV(+) +
SCH772984(−) + locostatin(−) group. However, there was no
significant difference in the percentage of cells in S phase in the
IAV(+) + SCH772984(+) + locostatin(+) group compared with in the
IAV(+) + SCH772984(−) + locostatin(+) group, as determined using
flow cytometry (P>0.05; Fig. 4F
and G). These results suggested that the percentage of cells in
S phase were not influenced when SCH772984 was used to inhibit the
ERK/MAPK pathway, even after inhibiting RKIP. As shown in Fig. 4F, H and I, there were significant
differences in the number of cells at G1 (P=0.0016) and
G2/M (P=0.0010) phases in the IAV(+) + SCH772984(+) +
locostatin(+) group compared with in the IAV(+) + SCH772984(−) +
locostatin(+) group. This confirmed that RKIP regulated
inflammatory cytokine levels and the S phase of cell cycle
progression via the ERK/MAPK signal transduction pathway.
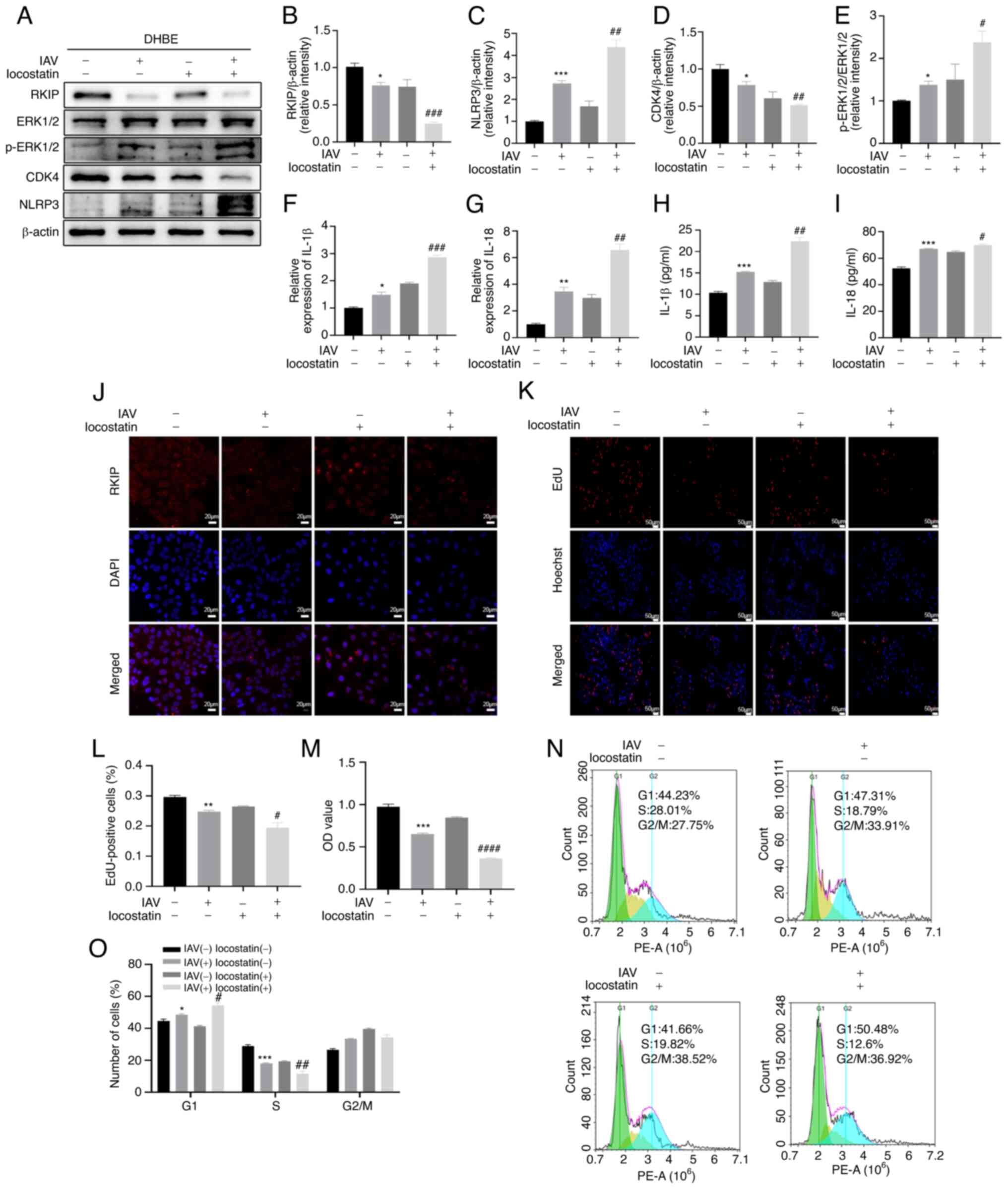
Inhibition of RKIP aggravates the airway
inflammatory response in vivo
To further confirm the effects of RKIP in
vivo, a mouse model infected with IAV was implemented. In our
previous study (29), a model of
airway inflammation was established and changes in the inflammatory
cytokines IL-1β and IL-18, related protein expression and
histopathology were analyzed 7 days after IAV infection. BALF and
serum were collected to examine the IL-1β and IL-18 levels, and it
was revealed that the levels of these two cytokines were
significantly increased in the BALF (P=0.0006 for IL-1β, P=0.0009
for IL-18; Fig. 5H and I) and
serum (P=0.0009 for IL-1β, P<0.0001 for IL-18; Fig. 5J and K) of the IAV(+) +
locostatin(−) group compared with in the NC group, as determined by
ELISA. The levels of IL-1β and IL-18 were also significantly
elevated in the BALF (P=0.0064 for IL-1β, P=0.0042 for IL-18;
Fig. 5H and I) and serum
(P=0.0049 for IL-1β, P=0.0003 for IL-18; Fig. 5J and K) in the IAV(+) +
locostatin(+) group compared with in the IAV(+) + locostatin(−)
group, as determined using ELISA. Furthermore, the mRNA expression
levels of IL-1β and IL-18 were detected in lung tissues, and it was
revealed that trends in IL-1β and IL-18 lung tissue expression were
similar to those in BALF and serum (Fig. 5F and G). These findings indicated
that the expression levels of IL-1β and IL-18 were augmented after
IAV infection (P= 0.0100 for IL-1β, P=0.0179 for IL-18), and that
the inhibition of RKIP exacerbated the production of IL-1β and
IL-18 after IAV infection (P=0.0134 for IL-1β, P=0.0006 for IL-18).
In addition, the protein expression levels of NLRP3 were detected
using western blotting. As shown in Fig. 5A and C, IAV infection increased
the expression levels of NLRP3 (P=0.0004) and locostatin
exacerbated this increase (P=0.0360). IAV-induced histopathological
changes in the lung were also assessed. As shown in Fig. 5L and M, more severe infiltration
of inflammation was observed in IAV-infected mice compared with in
the control group (P=0.0113). Furthermore, locostatin increased the
pulmonary inflammation of mice infected with IAV (P=0.048). To
further determine the expression of RKIP in lung tissues from
C57BL/6 mice, the expression levels of RIKP were assessed by
western blotting and IHC. As shown in Fig. 5A, B, N and O, RKIP was
significantly attenuated in response to IAV (P<0.0001 for
western blotting, P=0.0026 for IHC) and locostatin (P=0.0169 for
western blotting, P=0.0057 for IHC) further decreased RKIP
expression. ERK1/2 and p-ERK1/2 expression levels were also
detected, in order to determine whether the ERK/MAPK pathway was
activated, and CDK4 expression was measured to assess the cell
cycle, using western blotting (Fig.
5A, D and E); notably, IAV activated the ERK/MAPK pathway
(P=0.0017) and the expression of CDK4 was significantly decreased
(P=0.0374) compared with in the control group. Furthermore, RKIP
inhibition by locostatin aggravated these effects (P=0.0464 for
p-ERK1/2/ERK1/2, P=0.0090 for CDK4/β-actin), thus suggesting that
RKIP may be a negative regulator of IAV-induced airway inflammatory
responses.
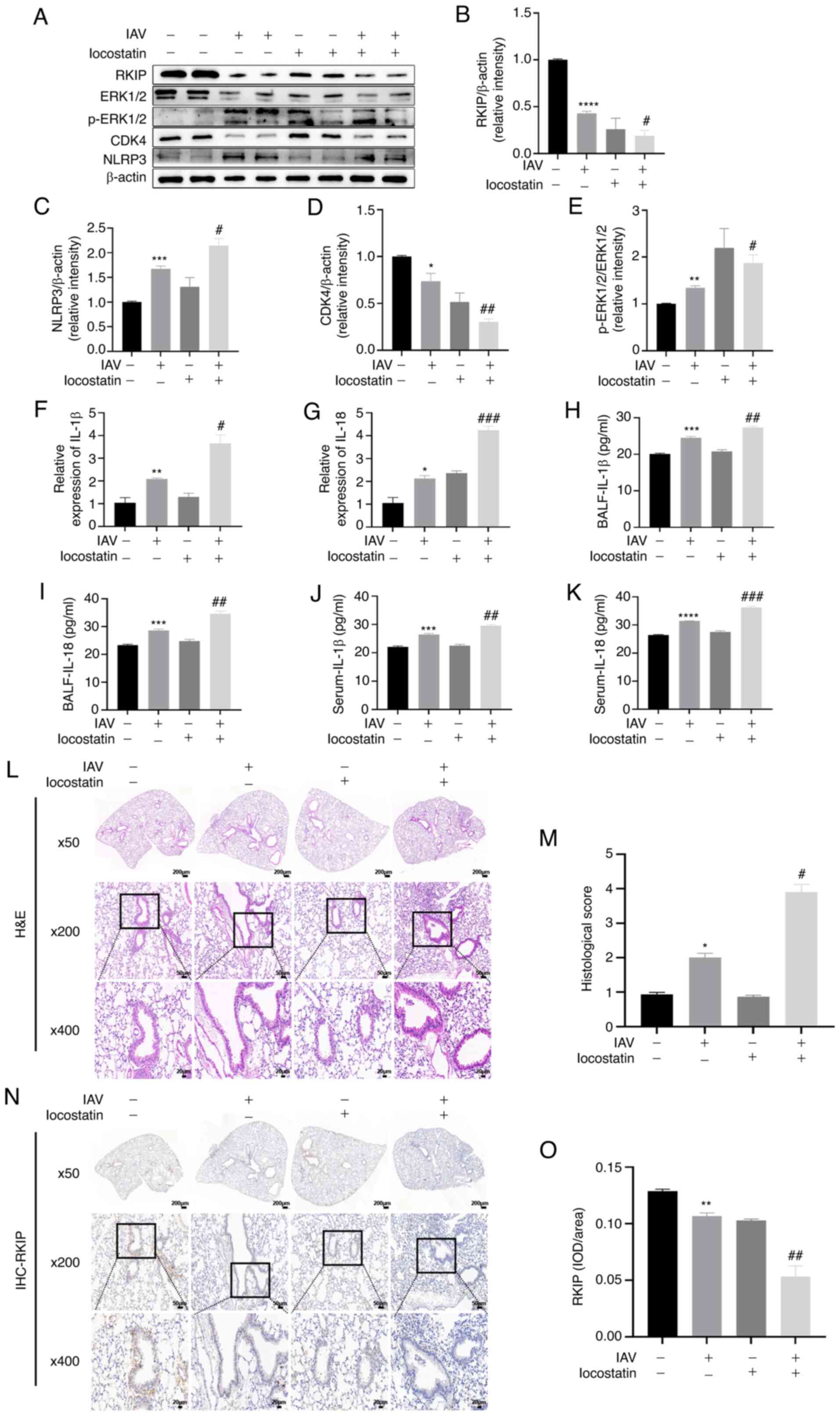 | Figure 5Inhibition of RKIP aggravates the
airway inflammatory response in vivo. After locostatin (10
mg/kg) was administered by intraperitoneal injection, mice were
treated with 0.9% saline or 100 µl IAV in saline via
oropharyngeal aspiration. (A) Lung tissues were harvested to detect
the protein expression levels of RKIP, ERK1/2, p-ERK1/2, NLRP3 and
CDK4 by western blotting. Densitometric analysis of (B) RKIP, (C)
NLRP3, (D) CDK4 and (E) p-ERK1/2 normalized to ERK1/2 in the
different groups. (F and G) Lung tissues were harvested to detect
the mRNA expression levels of IL-1β and IL-18 by reverse
transcription-quantitative PCR. (H and I) BALF and (J and K) serum
were harvested to examine the levels of the inflammatory cytokines
IL-1β and IL-18 by ELISA. (L and M) H&E staining was used to
evaluate the semi-quantitative scoring of inflammation in lung
tissue. Scale bar, 200 µm for magnification ×50; 50
µm for magnification ×200; 20 µm for magnification
×400. (N and O) IHC was performed to localize RKIP in bronchial
epithelium. Scale bar, 200 µm for magnification ×50; 50
µm for magnification ×200; 20 µm for magnification
×400. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 vs. NC;
#P<0.05, ##P<0.01 and
###P<0.001 vs. IAV(+) + locostatin(-). BALF,
bronchoalveolar lavage; H&E, hematoxylin and eosin; IAV,
influenza A virus; IHC, immunohistochemistry; IOD, integrated
optical density; NLRP3, NLR family pyrin domain-containing 3; p,
phosphorylated; RKIP, Raf kinase inhibitor protein. |
Discussion
The present study demonstrated that RKIP functions
as an inhibitory mediator of IAV-induced airway inflammatory
response, and that it acts via the ERK/MAPK pathway. RKIP-OE
significantly mitigated the production of inflammatory cytokines
and reversed the cell cycle arrest triggered by IAV infection. To
the best of our knowledge, the present study is the first to
demonstrate that the RKIP-mediated, IAV-induced airway inflammatory
response is conducted via the ERK/MAPK pathway in vitro and
in vivo.
While RKIP has been reported to be involved in
numerous disease processes (17,21,35), the role of RKIP in the airway
inflammatory response induced by IAV remains to be elucidated. The
present study ascertained that the expression of RKIP was
downregulated in BEAS-2B, DHBE and pNHBE cells following IAV
infection, whereas the levels of the inflammatory cytokines IL-1β
and IL-18 were elevated. These results suggested that RKIP was
critical to the airway inflammatory response following IAV
infection.
The present study further investigated the role of
RKIP in airway inflammatory responses following IAV infection;
notably, it was observed that the expression of RKIP was
specifically downregulated by locostatin and that this then
exacerbated airway inflammation. The viability and proliferation of
cells also slowed with the inhibition of RKIP expression suggesting
that RKIP was involved in cell cycle progression. Furthermore, the
percentage of cells in S phase of cell cycle was significantly
decreased after IAV infection and inhibition of RKIP expression
further diminished the proportion of cells in the S phase. Previous
reports have also demonstrated the involvement of RKIP in cell
cycle progression (36) and have
shown that cell cycle arrest occurs following influenza virus
infection (37). For example,
similar to the aforementioned findings, airway inflammation was
worsened and the cell cycle was arrested after influenza virus
infection in these previous studies. Inhibiting the expression of
RKIP also aggravated the inflammatory response. The present study
further strengthened the hypothesis that RKIP was indeed protective
against the production of cytokines and recovered cell cycle
progression. Such data may greatly enhance the search for drugs
that can potentially ameliorate IAV-induced inflammatory
diseases.
The present results additionally revealed that
airway inflammation was significantly suppressed and that cell
cycle arrest was reversed with RKIP-OE in vitro. Congruent
with our previous study, a mouse model of airway inflammation
induced by IAV was successfully constructed (29,38), and revealed that inhibition of
RKIP enhanced the production of cytokines and the expression of
CDK4, which is closely to cell cycle progression, thus suggesting
that downregulation of RKIP arrested the cell cycle after IAV
infection in vivo. These results were similar to the in
vitro outcomes of the present study.
It has previously been reported that reducing RKIP
expression may alleviate liver fibrosis (39), and that inhibition of RKIP could
lead to an improvement in hepatic fibrosis (40). These previous studies suggested
that RKIP may have different roles in different organs. Although
the role of RKIP is controversial with respect to different organs
and organ systems, the present study confirmed that RKIP serves a
protective role in airway inflammation and cell cycle progression
following IAV infection.
Emerging evidence has suggested that the ERK/MAPK
pathway is critically involved in numerous pathophysiological
processes, including cell proliferation, stress, inflammatory
responses, differentiation and apoptosis (18,41,42). However, whether RKIP mediates the
IAV-induced airway inflammatory response via the ERK/MAPK signal
transduction pathway remains unclear. The present study
demonstrated that this pathway was activated following IAV
infection; notably, in DHBE cells, RKIP inhibition did not further
increase the production of IL-1β and IL-18 and cell cycle arrest
when the ERK/MAPK pathway was inhibited by SCH772984 after IAV
infection. These findings indicated that RKIP could protect airway
epithelial cells against an inflammatory response induced by IAV
via the ERK/MAPK pathway, and suggested that ERK/MAPK may be a
potential pathway that mediates the anti-viral effect of RKIP. In
addition, it has been consistently reported that RKIP functions as
an anti-viral agent in innate immunity (43). These findings collectively reveal
that RKIP constitutes a promising target for anti-viral treatment
modalities, with IAV the principal pathogen in emerging respiratory
infectious diseases that lack an effective therapy. The present
results might therefore be of relevance in the future development
of targeted treatment approaches in IAV-induced inflammatory
diseases.
It has previously been demonstrated that airway
inflammation is exacerbated and the cell cycle blocked after
influenza virus infection (37,44), and a previous study reported that
cell cycle arrest promoted viral replication to increase
inflammation (44). The present
study elucidated the role of RKIP in promoting recovery of the cell
cycle after its arrest to alleviate the inflammatory responses
induced by IAV in airway epithelial cells. Collectively, these
findings suggested that the cell cycle is tightly linked to
inflammatory diseases. Moreover, it has been indicated that
inflammation is a critical component of tumor progression (45), and it is well known that the cell
cycle is accelerated in tumor progression. Therefore, it was
hypothesized that the cell cycle changes dynamically during the
progression of inflammation-related diseases. With the development
of inflammatory-related diseases, inflammation could progress from
acute to chronic, and chronic inflammation could lead to cancer.
During this progression, the cell cycle may be initially arrested
followed by its acceleration. It may be hypothesized that this
approach could constitute a mechanism underlying inflammation in
cancer transformation.
The present study investigated the airway
inflammatory response induced by IAV. It is well known that IAV
infection can induce several inflammatory cytokines, including
IL-1β (46), IL-18 (47), TNF-α (48), IL-6 (49), IL-8 (50) and IL-10 (51). The present study mainly measured
the levels of inflammation by IL-1β and IL-18; however, the other
inflammatory cytokines were not assessed. In future studies, we aim
to further explore the pathways related to other inflammatory
cytokines and the potential molecular mechanism of IAV-induced
AECOPD.
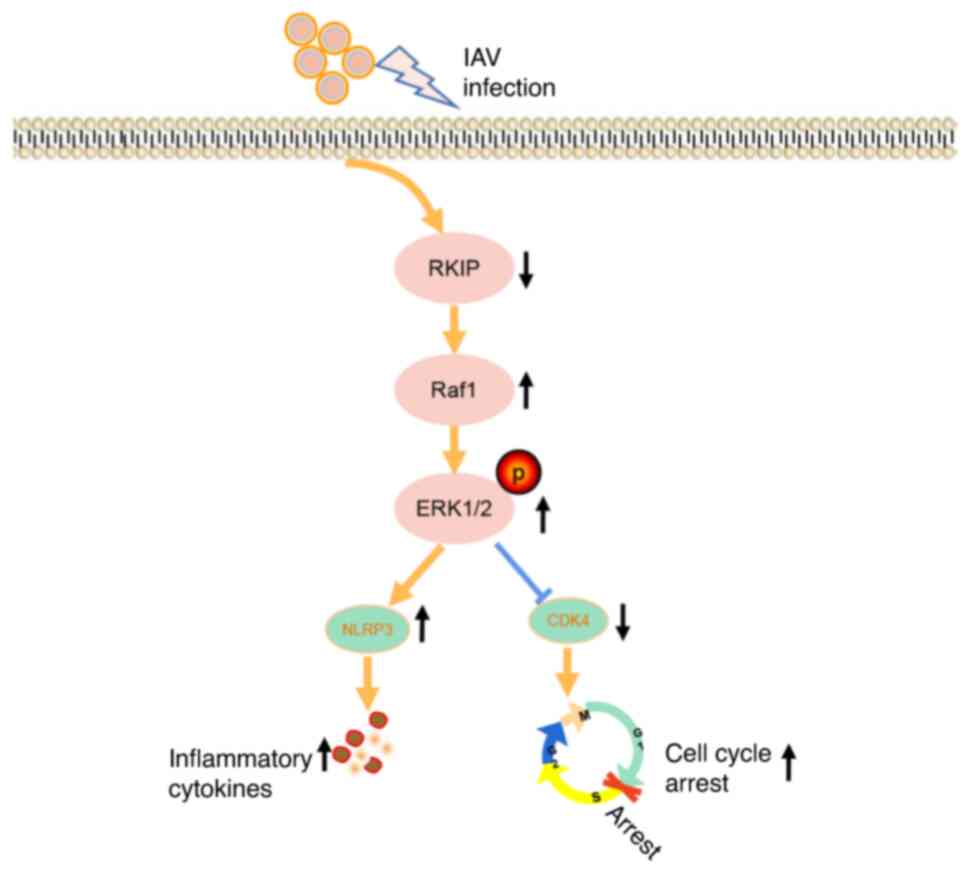
In conclusion, the present study demonstrated that
the expression of RKIP was significantly diminished following IAV
infection, and that RKIP served a protective role in the
alleviation of airway inflammation and in the recovery of the cell
cycle after IAV infection through the activities of the ERK/MAPK
pathway (Fig. 6). These actions
may constitute a novel treatment option for respiratory diseases
that involve IAV infection.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GHF and YW designed the study and revised the paper.
JJY and SLW conducted the experiments, wrote the paper and analyzed
the data. YYW, DWZ and LS assisted with the experiments. HMW, JLS
and LY assisted with the data analysis and critically reviewed the
manuscript. GHF and JJY confirmed the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All studies involving animals were approved by the
Animal Ethics Committee of Anhui Medical University (approval no.
LLSC20200117) in accordance with ethical principles. All of the
participants provided written informed consent and this study was
approved by the Biomedical Ethics Committee of Anhui Medical
University (approval no. 20200070).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82070047), the Subject Construction
Project of Anhui Medical University (grant no. 2021lcxk001) and the
Basic and Clinical Collaborative Research Promotion Program of
Anhui Medical University (grant no. 2020xkjT061).
Abbreviations:
|
AECOPD
|
acute exacerbation of chronic
obstructive pulmonary disease
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
CCK-8
|
Cell Counting Kit-8
|
|
DHBE
|
diseased human bronchial
epithelial
|
|
EdU
|
5-ethynyl-2'-deoxyuridine
|
|
H&E
|
hematoxylin and eosin
|
|
IAV
|
influenza A virus
|
|
MOI
|
multiplicity of infection
|
|
NLRP
|
NLR family pyrin domain-containing
|
|
PEBP
|
phosphatidylethanolamine-binding
protein
|
|
pNHBE
|
primary human bronchial epithelial
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
RKIP
|
Raf kinase inhibitor protein
|
References
|
1
|
Ellul MA, Benjamin L, Singh B, Lant S,
Michael BD, Easton A, Kneen R, Defres S, Sejvar J and Solomon T:
Neurological associations of COVID-19. Lancet Neurol. 19:767–783.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu X, Cai T, Fan L, Liang Z, Du Q, Wang Q,
Yang Z, Vlahos R, Wu L and Lin L: The traditional herbal
formulation, Jianpiyifei II, reduces pulmonary inflammation induced
by influenza A virus and cigarette smoke in mice. Clin Sci (Lond).
135:1733–1750. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prigge AD, Ma R, Coates BM, Singer BD and
Ridge KM: Age-dependent differences in T-Cell responses to
influenza A virus. Am J Respir Cell Mol Biol. 63:415–423. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi HJ, Lim CH, Song JH, Baek SH and Kwon
DH: Antiviral activity of raoulic acid from Raoulia australis
against Picornaviruses. Phytomedicine. 16:35–39. 2009. View Article : Google Scholar
|
|
5
|
Callon D, Berri F, Lebreil AL, Fornes P
and Andreoletti L: Coinfection of parvovirus B19 with influenza
A/H1N1 causes fulminant myocarditis and pneumonia. An autopsy case
report. Pathogens. 10:9582021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sasaki M, Ikeda H, Sato Y and Nakanuma Y:
Proinflammatory cytokine-induced cellular senescence of biliary
epithelial cells is mediated via oxidative stress and activation of
ATM pathway: A culture study. Free Radic Res. 42:625–632. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang C, Hein TW, Wang W, Ren Y, Shipley
RD and Kuo L: Activation of JNK and xanthine oxidase by TNF-alpha
impairs nitric oxide-mediated dilation of coronary arterioles. J
Mol Cell Cardiol. 40:247–257. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darzynkiewicz Z, Williamson B, Carswell EA
and Old LJ: Cell cycle-specific effects of tumor necrosis factor.
Cancer Res. 44:83–90. 1984.PubMed/NCBI
|
|
9
|
Al-Mulla F, Bitar MS, Taqi Z and Yeung KC:
RKIP: Much more than Raf kinase inhibitory protein. J Cell Physiol.
228:1688–1702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iuliano AD, Roguski KM, Chang HH,
Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D,
Cowling BJ, et al: Estimates of global seasonal
influenza-associated respiratory mortality: A modelling study.
Lancet. 391:1285–1300. 2018. View Article : Google Scholar :
|
|
11
|
Yeung K, Janosch P, McFerran B, Rose DW,
Mischak H, Sedivy JM and Kolch W: Mechanism of suppression of the
Raf/MEK/extracellular signal-regulated kinase pathway by the raf
kinase inhibitor protein. Mol Cell Biol. 20:3079–3085. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng L, Imamoto A and Rosner MR: Raf
kinase inhibitory protein (RKIP): A physiological regulator and
future therapeutic target. Expert Opin Ther Targets. 12:1275–1287.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vandamme D, Herrero A, Al-Mulla F and
Kolch W: Regulation of the MAPK pathway by raf kinase inhibitory
protein. Crit Rev Oncog. 19:405–415. 2014. View Article : Google Scholar
|
|
14
|
Yesilkanal AE and Rosner MR: Raf kinase
inhibitory protein (RKIP) as a metastasis suppressor: Regulation of
signaling networks in cancer. Crit Rev Oncog. 19:447–454. 2014.
View Article : Google Scholar
|
|
15
|
Klysik J, Theroux SJ, Sedivy JM, Moffit JS
and Boekelheide K: Signaling crossroads: the function of Raf kinase
inhibitory protein in cancer, the central nervous system and
reproduction. Cell Signal. 20:1–9. 2008. View Article : Google Scholar :
|
|
16
|
Noh HS, Hah YS, Zada S, Ha JH, Sim G,
Hwang JS, Lai TH, Nguyen HQ, Park JY, Kim HJ, et al: PEBP1, a RAF
kinase inhibitory protein, negatively regulates starvation-induced
autophagy by direct interaction with LC3. Autophagy. 12:2183–2196.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wenzel SE, Tyurina YY, Zhao J, St Croix
CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA,
Amoscato AA, et al: PEBP1 wardens ferroptosis by enabling
lipoxygenase generation of lipid death signals. Cell. 171:628–641
e26. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaminska B: MAPK signalling pathways as
molecular targets for anti-inflammatory therapy-from molecular
mechanisms to therapeutic benefits. Biochim Biophys Acta.
1754:253–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu T, Zhang L, Joo D and Sun SC: NF-κB
signaling in inflammation. Signal Transduct Target Ther.
2:170232017. View Article : Google Scholar
|
|
20
|
Huang Q, Bai F, Nie J, Lu S, Lu C, Zhu X,
Zhuo L and Lin X: Didymin ameliorates hepatic injury through
inhibition of MAPK and NF-κB pathways by up-regulating RKIP
expression. Int Immunopharmacol. 42:130–138. 2017. View Article : Google Scholar
|
|
21
|
Qin Q, Liu H, Shou J, Jiang Y, Yu H and
Wang X: The inhibitor effect of RKIP on inflammasome activation and
inflammasome-dependent diseases. Cell Mol Immunol. 18:992–1004.
2021. View Article : Google Scholar :
|
|
22
|
Mansoori B, Mohammadi A, Ditzel HJ, Duijf
PHG, Khaze V, Gjerstorff MF and Baradaran B: HMGA2 as a critical
regulator in cancer development. Genes (Basel). 12:2692021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang L, Fu Z, Binkley C, Giordano T,
Burant CF, Logsdon CD and Simeone DM: Raf kinase inhibitory protein
inhibits beta-cell proliferation. Surgery. 136:708–715. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma J, Li F, Liu L, Cui D, Wu X, Jiang X
and Jiang H: Raf kinase inhibitor protein inhibits cell
proliferation but promotes cell migration in rat hepatic stellate
cells. Liver Int. 29:567–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pnueli L, Gutfinger T, Hareven D, Ben-Naim
O, Ron N, Adir N and Lifschitz E: Tomato SP-interacting proteins
define a conserved signaling system that regulates shoot
architecture and flowering. Plant Cell. 13:2687–2702. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akaishi J, Onda M, Asaka S, Okamoto J,
Miyamoto S, Nagahama M, Ito K, Kwanami O and Shimizu K:
Growth-suppressive function of phosphatidylethanolamine-binding
protein in anaplastic thyroid cancer. Anticancer Res. 26:4437–4442.
2006.
|
|
27
|
Fulcher ML, Gabriel S, Burns KA, Yankaskas
JR and Randell SH: Well-differentiated human airway epithelial cell
cultures. Methods Mol Med. 107:183–206. 2005.
|
|
28
|
Yamaya M, Nishimura H, Hatachi Y, Yoshida
M, Fujiwara H, Asada M, Nakayama K, Yasuda H, Deng X, Sasaki T, et
al: Procaterol inhibits rhinovirus infection in primary cultures of
human tracheal epithelial cells. Eur J Pharmacol. 650:431–444.
2011. View Article : Google Scholar
|
|
29
|
Guo Y, Tu YH, Wu X, Ji S, Shen JL, Wu HM
and Fei GH: ResolvinD1 protects the airway barrier against injury
induced by influenza A virus through the Nrf2 pathway. Front Cell
Infect Microbiol. 10:6164752020. View Article : Google Scholar
|
|
30
|
Wei YY, Zhang DW, Ye JJ, Lan QX, Ji S, Sun
L, Li F and Fei GH: Interleukin-6 neutralizing antibody attenuates
the hypersecretion of airway mucus via inducing the nuclear
translocation of Nrf2 in chronic obstructive pulmonary disease.
Biomed Pharmacother. 152:1132442022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Kajon AE, Gigliotti AP and Harrod KS:
Acute inflammatory response and remodeling of airway epithelium
after subspecies B1 human adenovirus infection of the mouse lower
respiratory tract. J Med Virol. 71:233–244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mc Henry KT, Ankala SV, Ghosh AK and
Fenteany G: A non-antibacterial oxazolidinone derivative that
inhibits epithelial cell sheet migration. Chembiochem. 3:1105–1111.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu S, Mc Henry KT, Lane WS and Fenteany
G: A chemical inhibitor reveals the role of Raf kinase inhibitor
protein in cell migration. Chem Biol. 12:981–991. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin W, Ma C, Su F, Jiang Y, Lai R, Zhang
T, Sun K, Fan L, Cai Z, Li Z, et al: Raf kinase inhibitor protein
mediates intestinal epithelial cell apoptosis and promotes IBDs in
humans and mice. Gut. 66:597–610. 2017. View Article : Google Scholar
|
|
36
|
al-Mulla F, Bitar MS, Taqi Z, Rath O and
Kolch W: RAF kinase inhibitory protein (RKIP) modulates cell cycle
kinetics and motility. Mol Biosyst. 7:928–941. 2011. View Article : Google Scholar
|
|
37
|
Zhu L, Zhao W, Lu J, Li S, Zhou K, Jiang
W, Duan X, Fu L, Yu B, Cai KQ, et al: Influenza virus matrix
protein M1 interacts with SLD5 to block host cell cycle. Cell
Microbiol. 21:e130382019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ji S, Bai Q, Wu X, Zhang DW, Wang S, Shen
JL and Fei GH: Unique synergistic antiviral effects of Shufeng
Jiedu Capsule and oseltamivir in influenza A viral-induced acute
exacerbation of chronic obstructive pulmonary disease. Biomed
Pharmacother. 121:1096522020. View Article : Google Scholar
|
|
39
|
Ma J, Qiu Y, Wang M, Zhang M, Zhao X and
Jiang H: Locostatin alleviates liver fibrosis induced by carbon
tetrachloride in mice. Dig Dis Sci. 64:2570–2580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin X, Bai F, Nie J, Lu S, Lu C, Zhu X,
Wei J, Lu Z and Huang Q: Didymin alleviates hepatic fibrosis
through inhibiting ERK and PI3K/Akt pathways via regulation of raf
kinase inhibitor protein. Cell Physiol Biochem. 40:1422–1432. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pearson G, Robinson F, Gibson TB, Xu BE,
Karandikar M, Berman K and Cobb MH: Mitogen-activated protein (MAP)
kinase pathways: Regulation and physiological functions. Endocr
Rev. 22:153–183. 2001.PubMed/NCBI
|
|
42
|
Wang X, Xing Y, Tang Z, Tang Y, Shen J and
Zhang F: Thioredoxin-2 impacts the inflammatory response via
suppression of NF-κB and MAPK signaling in sepsis shock. Biochem
Biophys Res Commun. 524:876–882. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gu M, Liu Z, Lai R, Liu S, Lin W, Ouyang
C, Ye S, Huang H and Wang X: RKIP and TBK1 form a positive feedback
loop to promote type I interferon production in innate immunity.
EMBO J. 35:2553–2565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ho HT, Peischard S, Strutz-Seebohm N and
Seebohm G: Virus-host interactions of enteroviruses and parvovirus
B19 in myocarditis. Cell Physiol Biochem. 55:679–703. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Andrejeva G and Rathmell JC: Similarities
and distinctions of cancer and immune metabolism in inflammation
and tumors. Cell Metab. 26:49–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McAuley JL, Tate MD, MacKenzie-Kludas CJ,
Pinar A, Zeng W, Stutz A, Latz E, Brown LE and Mansell A:
Activation of the NLRP3 inflammasome by IAV virulence protein
PB1-F2 contributes to severe pathophysiology and disease. PLoS
Pathog. 9:e10033922013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ichinohe T, Pang IK, Kumamoto Y, Peaper
DR, Ho JH, Murray TcS and Iwasaki A: Microbiota regulates immune
defense against respiratory tract influenza A virus infection. Proc
Natl Acad Sci USA. 108:5354–5359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu H, Chelvanambi S, Poirier C, Saliba J,
March KL, Clauss M and Bogatcheva NV: EMAPII monoclonal antibody
ameliorates influenza A virus-induced lung injury. Mol Ther.
26:2060–2069. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou J, Wang D, Gao R, Zhao B, Song J, Qi
X, Zhang Y, Shi Y, Yang L, Zhu W, et al: Biological features of
novel avian influenza A (H7N9) virus. Nature. 499:500–503. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ito Y, Correll K, Zemans RL, Leslie CC,
Murphy RC and Mason RJ: Influenza induces IL-8 and GM-CSF secretion
by human alveolar epithelial cells through HGF/c-Met and TGF-α/EGFR
signaling. Am J Physiol Lung Cell Mol Physiol. 308:L1178–L1188.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ring S, Eggers L, Behrends J, Wutkowski A,
Schwudke D, Kröger A, Hierweger AM, Hölscher C, Gabriel G and
Schneider BE: Blocking IL-10 receptor signaling ameliorates
Mycobacterium tuberculosis infection during influenza-induced
exacerbation. JCI Insight. 5:e1265332019. View Article : Google Scholar : PubMed/NCBI
|