Introduction
Acute lung injury (ALI) is characterized by the
exudation of large quantities of inflammatory fluid, the
infiltration of inflammatory cells, tje severe destruction of
alveolar structures, remodeling, lung hemorrhage, edema, fibrosis
and decreased alveolar compliance. Acute respiratory distress
syndrome (ARDS) is an extreme form of ALI, with a fatality rate as
high as 30-40% (1). Pyroptosis is
a form of inflammatory programmed cell death (2) triggered by pathogenic infection, and
causes local as well as systemic inflammation or septic shock
(3). The pyroptosis pathways
include the caspase-1-dependent classical pathway and
caspase-11/4/5 dependent non-canonical pathway. Pyroptosis,
characterized by pore formation in the plasma membrane, cell
swelling and the rupture of the plasma membrane, occurs rapidly and
is accompanied by the release of multiple pro-inflammatory factors
(4). Gasdermins (GSDMs), the
family of pore-forming proteins that include GSDM A, B, C, D, E and
autosomal recessive deafness type 59 (DFNB59) (4), play a critical role in pyroptosis,
which is also referred to as GSDM-mediated necrosis (3,4).
Of these GSDM proteins, the cleavage of GSDMD, known as the
pyroptosis execution protein, has been extensively studied. After
receiving inflammatory stimuli from the cell, the
nucleotide-binding and oligomerization domain-like receptor protein
3 (NLRP3) inflammasome activates caspase-1, which then cleaves
pro-interleukin (IL)-1β, pro-IL-18 and GSDMD. The cleavage
separates the N-terminal pore-forming domain of GSDMD from its
C-terminal repressive domain. The pore-forming domain oligomerizes
and creates holes in the cytoplasmic membrane, resulting in the
release of cellular contents, which induce a robust inflammatory
response (3-5).
Alveolar macrophages (AMs) account for ~95% of
alveolar leukocytes (6) and
affect the development of both infection-induced and non-infectious
ALI by synthesizing and releasing several inflammatory mediators
(7,8). Previous studies have confirmed that
AM cell death significantly affects the progression of lung
inflammation (9-11). In a previous study, a
lipopolysaccharide (LPS)-induced septic lung injury survey revealed
that LPS activated the NLRP3 inflammasome through the
TLR4/MyD88/NF-ĸB signaling pathway and promoted the secretion of
IL-1β by AM; LPS enhanced the sensitivity of AM to IL-1β, resulting
in AM pyroptosis (12). That
previous study also demonstrated that AM pyroptosis induced the
migration of neutrophils into the lungs, increased IL-6, tumor
necrosis factor (TNF)-α, and IL-1β production in alveoli, and
aggravated the histological manifestations of lung injury (12). Therefore, inhibiting AM pyroptosis
may be a promising therapeutic strategy for reducing ALI.
Heat shock protein (HSP)90 is a well-known molecular
chaperone belonging to the HSP family. As a client protein of
HSP90, the assembly and stability of the NLRP3 inflammasome are
affected by HSP90 (13,14). HSP90 binds NLRP3, which regulates
inflammasome activation and IL-1β secretion (13,15,16). Without HSP90, NLRP3 can be
misfolded and degraded by proteases (17,18). HSP90 inhibitors have previously
been shown to reduce the expression of HSP90 in a time- and
dose-dependent manner and to significantly attenuate the LPS- and
nigericin-induced pyroptosis of human monocytic leukemia (THP-1)
cells (2), suggesting that HSP90
is a promising target for the treatment of inflammation. Compared
to other HSP90 inhibitors, tanespimycin (17-AAG) has a lower
cytotoxic effect; it significantly reduces the production of active
caspase-1 and inhibits NLRP3 expression (2).
Irisin is a muscle- or lipid-derived cytokine
expressed in the heart, brain, ovaries, testes, kidneys, stomach
and liver (19,20), but not in the lungs. Irisin,
produced by the hydrolysis of fibronectin type III
repeat-containing protein (FNDC5), reduces oxidative stress and has
neuroprotective, anti-inflammatory and anticancer properties. It
also plays a biological role in several cellular signaling pathways
(20). FNDC5/irisin reduces
inflammation and M1 macrophage polarization by regulating the 5′
AMP-activated protein kinase pathway (21). As demonstrated in a previous
study, following irisin pre-treatment, the stimulation of RAW264.7
macrophages with LPS significantly decreased T-lymphoid receptor
and myeloid differentiation primary response protein 88 levels. It
also decreased the phosphorylation of nuclear factor κB (NF-κB) and
inhibited the release of critical pro-inflammatory cytokines, such
as IL-6, TNF-α and IL-1β (22).
Shao et al (23) found
that irisin inhibited the activation of NF-κB and phosphorylated
(p-) NF-κB by LPS, and attenuated inflammatory responses and
pathological changes in cells within an LPS-induced mouse lung
injury model, indicating that irisin may be a treatment agent for
ALI (23).
The present study aimed to examine the protective
role of irisin in ALI and to elucidate its underlying mechanisms of
action, in an aim to identify novel targets for the treatment of
ALI/ARDS.
Materials and methods
Reagents
Irisin was purchased from Cayman Chemical Company.
LPS (Escherichia coli 0111: B4) was purchased from
MilliporeSigma. MCC950 (cat. no. HY-12815) and
17-N-allylamino-17-demethoxygeldanamycin (17-AAG, cat. no.
M2320-01) were purchased from MedChemExpress. The
immunohistochemistry kit (cat. no. PV-9000) was purchased from
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd. Ethanol and
dimethyl benzene were purchased from Tianjin Yongda Chemical
Reagent Co., Ltd. The hematoxylin and eosin (H&E) Stain Kit
(cat. no. G1120) and paraffin wax were purchased from Beijing
Solarbio Science & Technology Co., Ltd. Primers used for
reverse transcription-quantitative PCR (RT-qPCR) primers were
synthesized by Sangon Biotech Co., Ltd.
Animals
C57BL/6J mice (n=48, 24 male and 24 female mice;
aged 6-8 weeks and weighing 18-22 g) were purchased from Beijing
Weitong Lihua Experimental Animal Technology Co. Ltd. All mice were
raised and maintained at the Hebei Medical University Animal Care
Center (Shijiazhuang, China) under a standard SPF facility. Animal
experimental and handling procedures were approved by the Ethics
Committee of the Second Hospital of Hebei Medical University
(Approval no: 2022-AE008, 28.2.2022). All efforts were made to
minimize the suffering of the animals, and conformed with the
National Institutes of Health guide for the care and use of
laboratory animals (24). All
experiments were performed under standard growth conditions as
follows: 40-60% relative humidity, room temperature (23-25°C) and a
12-h light/dark cycle, with 5 mice per cage with cleaned bedding
and ad libitum access to food and water. The mice were
allowed to acclimatize to the laboratory conditions for at least 7
days prior to the commencement of the experiments. Humane endpoints
associated with infection in the experiment were compliant with the
Guidelines for the Assessment of Humane Endpoints in Animal
Experiments (RB/T 173-2018, in Chinese), as follows: The animal
body temperature decreases by >4-6°C; animal weight loss of
10-20%; reduced animal activity, lethargy and other physiological
and behavioral changes up to 24 h. None of the mice used in the
experiment exhibited any of these endpoints.
Establishment of mouse model of
LPS-induced of ALI and mouse grouping
The mice (n=48) were randomly divided into eight
groups as follows: i) The control (Con; 0.05 ml of 0.9% saline
injected intratracheally); ii) low LPS (LPS; 2 mg/kg LPS injected
intratracheally); iii) high LPS (LPS-H; 4 mg/kg LPS injected
intratracheally); iv) LPS + low irisin (LPS + IR-L; 0.25 mg/kg
irisin injected in the caudal vein 0.5 h before 2 mg/kg LPS
administration); v) LPS + high irisin (LPS + IR; 0.5 mg/kg irisin
injected 0.5 h prior to the 2 mg/kg LPS administration); vi) low
MCC950 (LPS + M-L; 25 mg/kg of MCC950 injected intraperitoneally 12
h before the 2 mg/kg LPS administration); vii) high MCC950 (LPS +
M; 50 mg/kg of MCC950 injected intraperitoneally 12 h before the 2
mg/kg LPS administration); and viii) LPS + dexamethasone (Dex, CSPC
Pharmaceutical Group Ltd; LPS + Dex; 0.5 mg/kg of Dex injected
intraperitoneally 0.5 h before the 2 mg/kg LPS administration).
MCC950, an inhibitor of the NLRP3 inflammasome and Dex were used as
the positive controls.
Preparation of bronchoalveolar lavage
fluid (BALF)
After 24 h, all mice (n=48) were euthanized by deep
anesthesia with pentobarbital sodium and subjected to rapid
bloodletting. The mice were weighed before anesthesia and the
average weight was 20.7 g. A 1% pentobarbital sodium solution (35
mg/kg) was prepared and the mice were injected intraperitoneally
for deep anesthesia. The orbital artery of the mouse was then
selected to perform rapid bloodletting, completing the euthanasia
of the mice (the bloodletting volume was 0.7-1 ml). If the mouse
did not die after bleeding, it was euthanized by cervical
dislocation as soon as possible. The criteria for confirming the
death of the mice were as follows: No breathing, no pulse and no
heartbeat in the thoracic cavity of the mouse for >5 min; the
corneal reflex of the animal disappeared; the pupils dilated; and
the nerve reflexes disappeared. Blood samples were then collected
for use in further experimental procedures. The mice were
immobilized on the operating table and the skin and muscles were
cut in the front of the neck of the mice to expose the trachea. A
22G Disposable Vein Detaining Needle (BD Biosciences) was inserted
into the primary airway, and 500 µl saline solution were
then injected in three batches. BALF was collected by injection and
withdrawal three times. The BALF samples were mixed, and 50
µl of the mixed sample were used to count the white blood
cell (WBC) number using a hemocytometer (Countess 3, Thermo Fisher
Scientific, Inc.). The remaining BALF was maintained at −80°C for
use in cytokine analysis for cell counting and detecting cytokine
levels.
Histopathological evaluation and
immunohistochemistry
Fresh tissue samples were collected from the
inferior lobe of the left lung and fixed with 4% paraformaldehyde
for 24 h. The samples were dehydrated using a series of ethanol,
cleared using dimethyl benzene, and embedded in paraffin wax.
Paraffin-embedded sections with a thickness of 4 µm were cut
and stained with hematoxylin and eosin staining for 1 min at room
temperature. The pathological changes in the lung tissues were
observed using a light microscope (Zeiss Scope.A1; Zeiss AG; ×200
magnification). FNDC5 expression was detected using
immunohistochemistry. Briefly, following deparaffinization and
rehydration, antigen retrieval was performed by heat-mediated
antigen retrieval with sodium citrate buffer (cat. no. DNS-0811,
MXB Biotechnologies, pH 9.0) at 98°C for 15 min. Subsequently, the
sections were stained with anti-FNDC5 (1:100; cat. no. BS-8486R,
BIOSS) primary antibody at 4°C overnight. Subsequently, at room
temperature, the sections were incubated with biotin-labeled goat
anti-rabbit IgG (1:50; cat. no. SP KIT-C2, MXB Biotechnologies) for
30 min, followed by a reaction with diaminobenzidine (cat. no.
DAB-2031, MXB Biotechnologies) for 3-5 min. Images were observed
under an optical microscope (Zeiss Scope.A1; Zeiss AG; ×200
magnification).
Cells and cell culture
The mouse AM-derived cell line, MH-S, was purchased
from ATCC (cat. no. BFN60807584) and cultured in RPMI-1640 medium
(MilliporeSigma). The cells were supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin solution (Thermo Fisher Scientific, Inc.)
and incubated at 37°C under a 5% CO2 and 95%
O2 atmosphere.
Cell proliferation assay
The MH-S cells were plated in 96-well plates at a
density of 1×104 cells per well for 24 h and then grown
for a further 24 h in various concentrations of LPS (0.01, 0.1, 1,
10 and 100 µg/ml), irisin (100, 200 and 400 ng/ml), or
17-AAG (50, 100 and 200 ng/ml). 17-AAG, an inhibitor of the HSP90
inflammasome, was used as a positive control. Cell viability was
estimated using the Cell Counting Kit-8 assay (Dalian Meilun
Biotechnology, Co., Ltd.) according to the manufacturer's
instructions.
Morphological changes in the MH-S
cells
The MH-S cells in the logarithmic growth phase were
seeded in 6-well plates at 2×105 cells per well. The
cells were divided into four groups as follows: The control (Con;
saline solution 10 µl), LPS (LPS 10 µg/ml), LPS +
irisin (LPS + IR; 10 µg/ml LPS and 200 ng/ml IR) and the LPS
+ 17-AAG (LPS + 17; 10 µg/ml LPS and 100 ng/ml 17-AAG)
groups were set up. Irisin and 17-AAG were combined with serum-free
RPMI-1640 medium and added to the cells 2 h prior to LPS
stimulation. Morphological changes in the cells were observed using
the PE Perkin Elmer Operetta CLS system 4 h following LPS
stimulation.
Cell pyroptosis assay
To evaluate cell pyroptosis, the MH-S cells were
plated in six-well plates (2×105 cells per well) for 24
h. The cells were treated and grouped as described above. Following
4 h of LPS treatment, the MH-S cells were washed thrice with PBS
after being incubated with Calcein AM and 7-aminoactinomycin D
(7-AAD) using the Live/Membrane Damage Cell Staining kit (cat. no.
BB-41272, BestBio), following the manufacturer's instructions. The
cells were incubated in the dark at 4°C for 15-30 min, washed with
PBS, and then analyzed under a fluorescence microscope (Zeiss Axio
Vert.A1, Zeiss AG; ×400 magnification).
Immunofluorescence assays
The lung tissue sections were dewaxed, and cell
samples were fixed with 4% paraformaldehyde for 15 min. The samples
were permeabilized with 0.3% Triton X-100 for 10 min and blocked
using 1% bovine serum albumin (BSA) for 1 h at room temperature.
Subsequently, the samples were incubated overnight at 4°C with
anti-CD68 (1:100), anti-CD206 (1:200), or anti-inducible nitric
oxide synthase (iNOS, 1:200) primary antibodies. The same method
was applied as that described above for the immunofluorescence
experiments of macrophage pyroptosis (CD80, 1:100; and GSDMD,
1:200). Following incubation, the samples were washed twice with
PBS and incubated in the dark for 2 h with fluorescein-labeled
secondary antibodies. Finally, the nuclei were stained with
4′,6-diamidino-2-phenylindole (DAPI; RS0029, Report Biotech, China)
for 5 min at room temperature. Antibodies against CD68 (cat. no.
EM1706-11) and CD80 (cat. no. M1007-10) were purchased from HUABIO.
Antibodies against CD206 (cat. no. 18704-1-AP), iNOS (cat. no.
22226-1-AP) and GSDMD (cat. no. 20770-1-AP) were purchased from
Proteintech Group, Inc. Secondary antibodies used for
immunofluorescence were purchased from the Beyotime Institute of
Biotechnology (Alexa Fluor 488-labeled goat anti-mouse IgG, cat.
no. A0428; Alexa Fluor 647-labeled goat anti-rabbit IgG, cat. no.
A0468), diluted at a ratio of 1:500. Immunofluorescence images were
captured using an Axio Vert.A1 fluorescence microscope (Zeiss AG;
×400 magnification).
Enzyme-linked immunosorbent assay
(ELISA)
For the in vivo experiments, BALF and blood
samples were centrifuged at 3,500 × g at 4°C for 10 min and the
cell culture supernatant was collected by in vitro
centrifugation. The TNF-α (EMC102a, Neobioscience Technology Co.,
Ltd.), IL-1β (EMC001b, Neobioscience Technology Co., Ltd.) and
IL-18 [EK218, Hangzhou Multisciences (Lianke) Biotech Co., Ltd.]
levels were measured using respective ELISA kits. According to the
manufacturer's instructions, the optical density of the samples was
measured at 450 nm (OD450) using a TECAN SPARK
microplate reader (Tecan Group, Ltd.).
RT-qPCR
Total RNA was extracted from the samples in each
group using RNA extraction reagents (Nanjing Novizan Medical
Technology Co., Ltd.), following the manufacturer's instructions.
RNA was quantified using a spectrophotometer and then reverse
transcribed into cDNA using PrimeScript™ RT Master Mix kit (cat.
no. RR036A, Takara Biotechnology Co., Ltd.). qPCR was performed
using PrimeSTAR® Max DNA Polymerase (Takara
Biotechnology Co., Ltd.) on an RT-PCR system (Agilent Technologies,
Inc.). The samples were amplified using the following thermocycling
conditions: Initial denaturation at 95°C for 30 sec; followed by 40
cycles of denaturation at 95°C for 5 sec, annealing at 55°C for 30
sec and elongation at 72°C for 30 sec; and a final extension at
72°C for 10 min. The relative mRNA expression levels of the target
genes were calculated as the fold change of the control using the
2−ΔΔCq method (25).
GAPDH primers, obtained from Sangon Biotech (Shanghai) Co., Ltd.,
were used as the internal reference. The forward and reverse
primers used are listed in Table
I.
 | Table IPrimers used for the reverse
transcription-quantitative polymerase chain reaction in the present
study. |
Table I
Primers used for the reverse
transcription-quantitative polymerase chain reaction in the present
study.
| Gene | Forward primer
(5′→3′) | Reverse primer
(3′→5′) |
|---|
| NLRP3 |
GCCGTCTACGTCTTCTTCCTTTCC |
CATCCGCAGCCAGTGAACAGAG |
| HSP90 |
CTCAGTCTGGAGATGAGATGAC |
CAGTCATATACACCACCTCGAA |
|
Caspase-1 |
AGAGGATTTCTTAACGGATGCA |
TCACAAGACCAGGCATATTCTT |
| GSDMD |
CTAGCTAAGGCTCTGGAGACAA |
GATTCTTTTCATCCCAGCAGTC |
| IL-1β |
CACTACAGGCTCCGAGATGAACAAC |
TGTCGTTGCTTGGTTCTCCTTGTAC |
| IL-18 |
AGACCTGGAATCAGACAACTTT |
TCAGTCATATCCTCGAACACAG |
| TNF-α |
ATGTCTCAGCCTCTTCTCATTC |
GCTTGTCACTCGAATTTTGAGA |
| IL-10 |
CAAGGCAGTGGAGCAGGTGAAG |
CGCTTTGGTGAGTAGACAGAGGTC |
| IL-6 |
TAGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
| CD206 |
CCTATGAAAATTGGGCTTACGG |
CTGACAAATCCAGTTGTTGAGG |
| iNOS |
ATCTTGGAGCGAGTTGTGGATTGTC |
TAGGTGAGGGCTTGGCTGAGTG |
| GAPDH |
GGTTGTCTCCTGCGACTTCA |
TGGTCCAGGGTTTCTTACTCC |
Analysis of caspase activity in vivo and
in vitro
FLIVO (FAM-FLIVO, cat. no. #980) and FLICA
(FAM-FLICA, cat. no. #91) probes were obtained from ImmunoChemistry
Technologies, LLC. FAM-FLICA is a kit that includes Hoechst and
propidium iodide (PI). These are non-cytotoxic
fluorescently-labeled inhibitors of caspases that covalently bind
active caspases and can be used to quantify caspase activity in
vivo and in vitro and to detect pyroptosis (26,27). The FLIVO and FLICA probes were
dissolved in 50 µl DMSO and used according to the
manufacturer's instructions. To evaluate pyroptosis in living
animals, the fluorescent green probe, FLIVO, was injected into the
tail vein of the mice (n=18) 24 h after the LPS intervention. The
FLIVO reagent was allowed to circulate in the mouse systems for 30
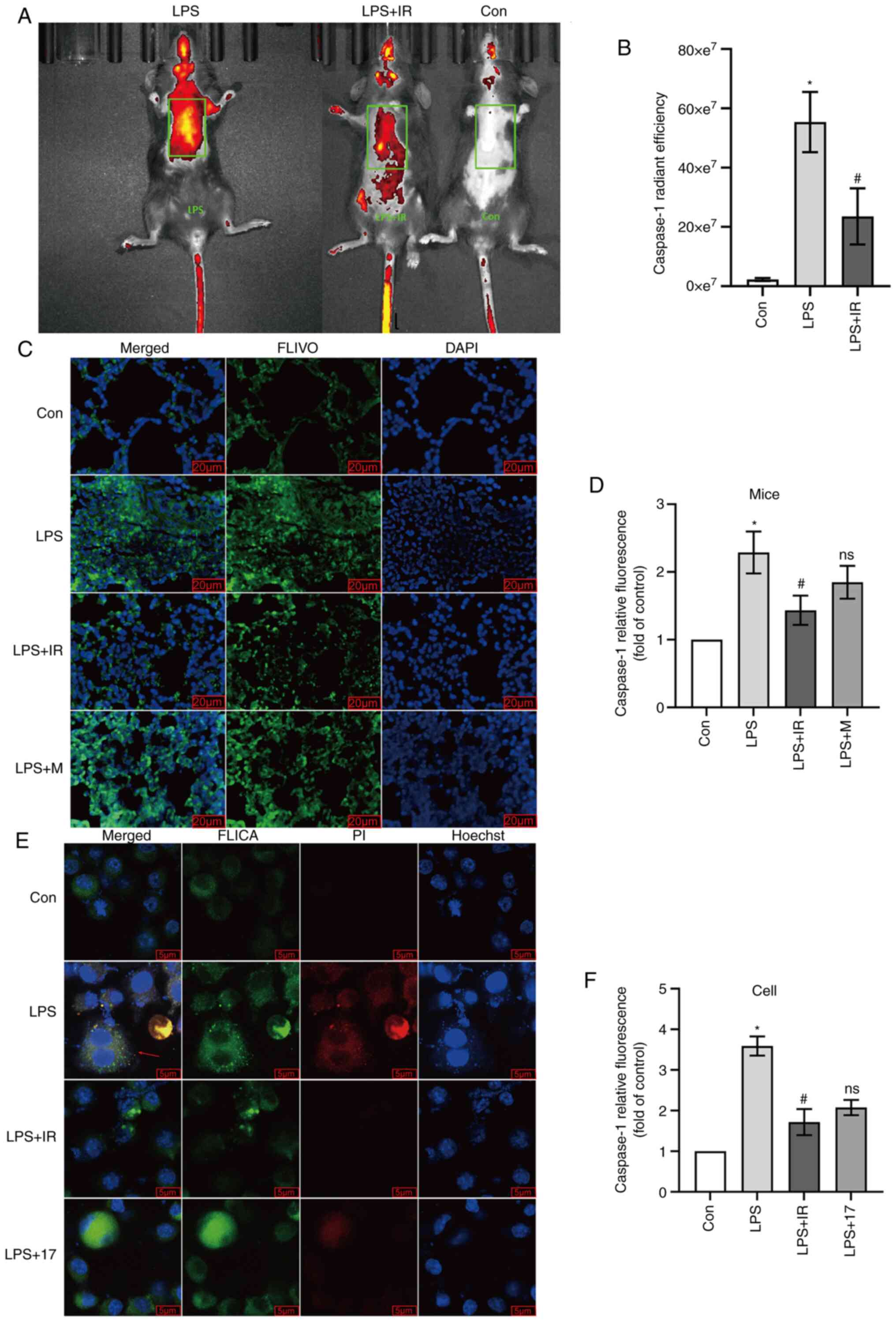
min prior to the analysis (28).
The mice were then anesthetized using isoflurane (inhalation
concentration: 4-5% for induction and 1-3% for maintenance) and
fluorescent images were captured using an IVIS Lumina LT Series III
instrument (PE, PerkinElmer, Inc.).
To evaluate pyroptosis in vitro, the MH-S
cells were plated on 25 mm poly-L-lysine-coated coverslips at
3×105 cells per coverslip and incubated with 10
µg/ml LPS for 4 h at 37°C to induce pyroptosis. The cells
were fixed with 4% paraformaldehyde for 15 min at room temperature,
labeled with FLICA, and incubated for 60 min at 37°C; Subsequently,
the cells were washed, stained with Hoechst stain, and incubated
for 5 min at room temperature; Finally, the cells were stained with
propidium iodide (PI) for 15 min. Images were acquired using a
Zeiss two-photon laser confocal microscope (LSM-NLO880, Zeiss AG;
×400 magnification).
Protein extraction
The cell samples and lung tissues were lysed using
radioimmunoprecipitation assay lysis buffer (Beijing Solarbio
Science & Technology Co., Ltd.) containing a
phenylmethanesulfonyl fluoride and protein phosphatase inhibitor
mixture (Beijing Solarbio Science & Technology Co., Ltd.). The
solution was centrifuged at 14,000 × g at 4°C for 20 min, and
proteins in the supernatant were quantified using the NanoDrop
2000c spectrophotometer (Thermo Fisher Scientific, Inc.). Proteins
were combined with 5X sodium dodecyl-sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) loading buffer containing dithiothreitol
(P1040; Beijing Solarbio Science & Technology Co., Ltd.) and
boiled for 10 min at 100°C. The mixture was then frozen at −80°C
until further use.
Western blot analysis
Equal quantities of protein (40 µg/lane) were
separated electrophoretically using SDS-PAGE (10-12% gels) and then
transferred onto polyvinylidene fluoride membranes (MerckMillipore
ISEQ00010, Merck KGaA). The membranes were blocked for 30 min at
25°C with QuickBlock blocking solution (Beyotime Institute of
Biotechnology). Subsequently, the membranes were incubated
overnight at 4°C with primary antibodies (diluted 1:1,000 in
QuickBlock™ Primary Antibody Dilution Buffer, Beyotime Institute of
Biotechnology) and then with horseradish peroxidase-conjugated
secondary antibodies (diluted 1:10,000 in Tris-buffered saline and
Tween-20) for 2 h at 25°C. The expression levels of the target
proteins were normalized to those of β-actin, which was used as an
internal control. Images were digitally acquired using Amersham
Imager 600 (GE Healthcare; Cytiva) chemiluminescence image analysis
system. The protein concentration was determined using ImageJ 1.8.0
software (National Institutes of Health).
The primary antibodies used for western blotting
were as follows: NLRP3 (cat. no. D4D8T), pro-caspase-1 (cat. no.
E2Z1C), cleaved caspase-1 (cat. no. Asp296), IL-1β (cat. no. 3A6)
were purchased from Cell Signaling Technology, Inc.; GSDMD (cat.
no. AF4012) was purchased from Affinity Biosciences; HSP90 (cat.
no. A5027) was purchased from ABclonal Biotech Co., Ltd.; β-actin
(cat. no. 20536-1-AP); was purchased from Proteintech Group, Inc.
Secondary antibodies used for western blotting were obtained from
the Beyotime Institute of Biotechnology, (HRP-conjugated goat
anti-rabbit IgG, cat. no. ZA0277; HRP-conjugated goat anti-mouse
IgG, cat. no. A0286), diluted at a ratio of 1:10,000.
Statistical analysis
All experiments were performed at least thrice.
Statistical analyses were performed using GraphPad Prism 9 software
(GraphPad Software, Inc.). The results are expressed as the mean ±
standard deviation (SD). Bartlett's test was performed to test the
homogeneity of variance. The Student's unpaired t-test was used to
compare the means of two independent samples. One-way analysis of
variance (ANOVA) was performed to compare the differences among
groups, and Tukey's post hoc test was used to compare the
differences between the two groups following ANOVA. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
Irisin protects against LPS-induced ALI
in mice
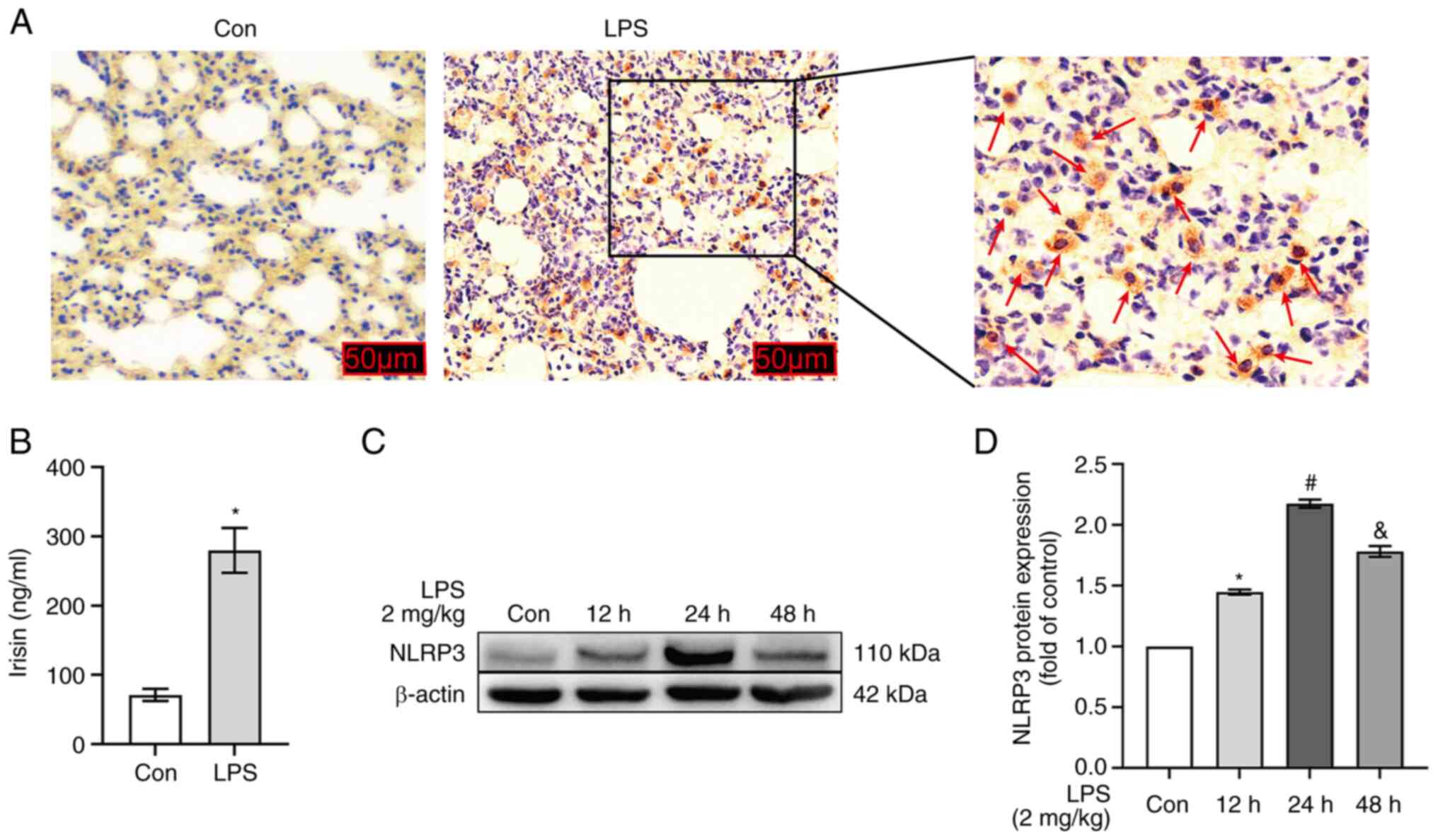
Immunohistochemical analysis revealed that
FNDC5/irisin was expressed in the damaged lung tissue, but not in
normal lung tissue following LPS stimulation (2 mg/kg; Fig. 1A). At the same time, the serum
irisin content in mice was detected using an ELISA test kit. The
results revealed that LPS significantly increased the serum irisin
content in the mice (P<0.05; Fig.
1B). It was hypothesized that the expression of FNDC5/irisin
may be driven by the inflammatory responses in the lung tissues.
Therefore, the protein expression of NLPR3 was examined at
different time periods following LPS stimulation to determine the
optimal intervention time. The expression of NLRP3 increased after
LPS intervention for 12 and 48 h. However, NLRP3 expression was
more significantly upregulated after 24 h of LPS stimulation (2
mg/kg; P<0.05; Fig. 1C and
D).
To verify the anti-inflammatory effects of irisin in
animal models of ALI, the indicators of inflammatory damage were
examined in mice treated with various drugs. Stimulation with LPS
alone significantly damaged the lung tissue through inflammatory
cell infiltration, pulmonary interstitial hemorrhage and edema,
alveolar wall thickening and diffused alveolar injury compared with
the control group. These changes were more evident in the LPS-H (4
mg/kg) stimulation group than those in the LPS (2 mg/kg) group
(P<0.05; Fig. 2). Exogenous
irisin treatment at 0.25 mg/kg effectively alleviated these
inflammatory reactions, reduced lung tissue damage and considerably
decreased the infiltration of inflammatory cells. However, irisin
treatment at 0.5 mg/kg had significant effects compared to the LPS
group (P<0.05; Fig. 2). It has
been have confirmed that MCC950 (50 mg/kg) can effectively inhibit
LPS-induced neutrophil infiltration in the lungs of mice, identical
to Dex (0.5 mg/kg) (29). The
present study also demonstrated that high-dose irisin reduced lung
inflammation similar to Dex (0.5 mg/kg) and MCCC950 (50 mg/kg)
(P>0.05, Fig. 2B).
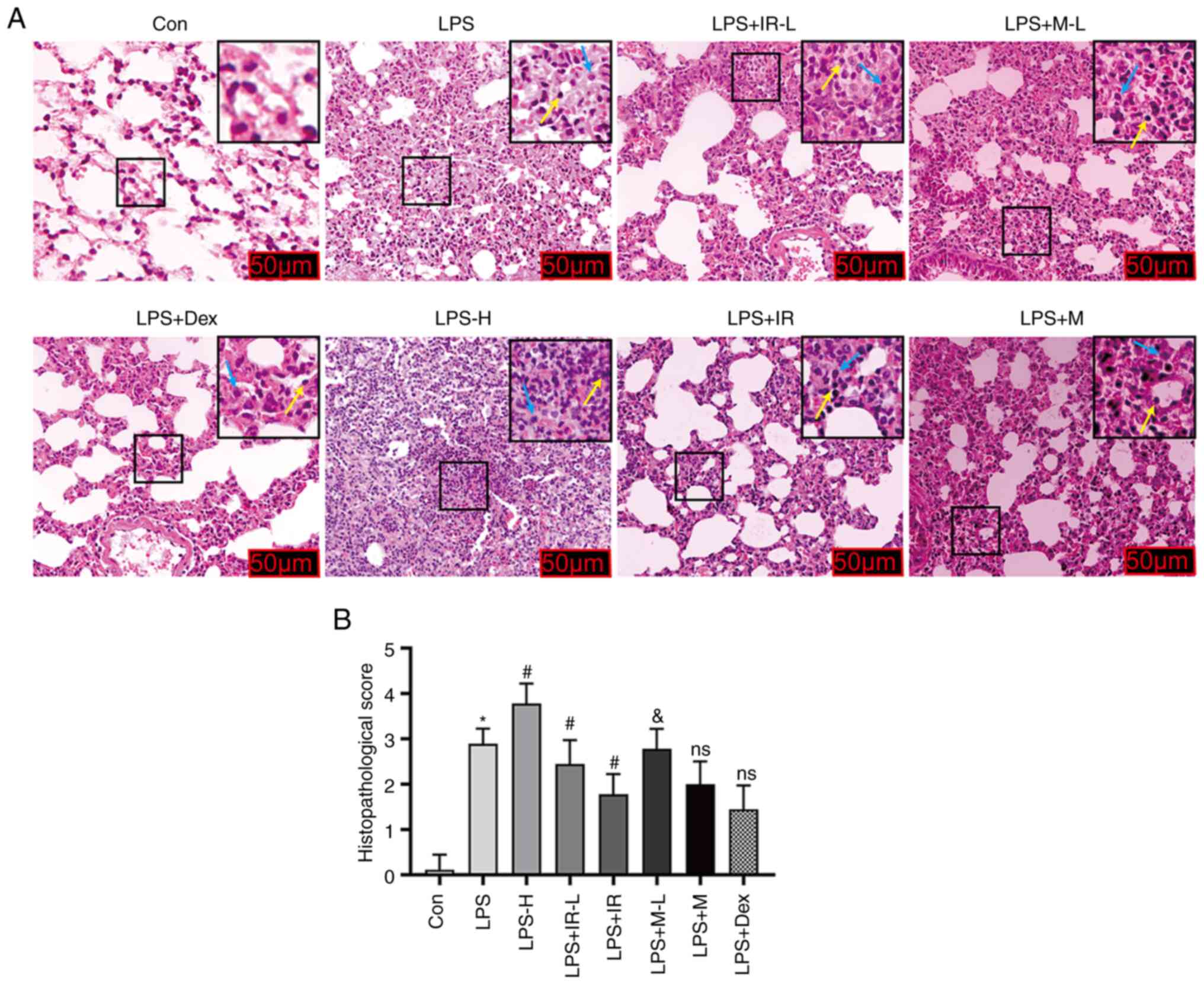 | Figure 2Irisin protects against LPS-induced
ALI in mice (magnification, ×200). C57BL/6J mice (weighing 18-22 g)
aged 6-8 weeks, were randomly assigned to the Con, LPS (LPS 2
mg/kg), LPS-H (LPS 4 mg/kg), LPS + IR-L (LPS 2 mg/kg, irisin 0.25
mg/kg), LPS + IR (LPS 2 mg/kg, irisin 0.5 mg/kg), LPS + M-L (LPS 2
mg/kg, MCC950 25 mg/kg), LPS + M (LPS 2 mg/kg, MCC950 50 mg/kg),
and LPS + Dex (LPS 2 mg/kg, dexamethasone 0.5 mg/kg) groups. (A)
Lung tissue samples stained with hematoxylin and eosin after 24 h
of LPS treatment. Yellow and blue arrows indicate the infiltration
of inflammatory cells (lymphocyte and macrophage). (B) The
pathological damage scoring of ALI. The data are expressed as the
mean ± SD, n=6. *P<0.05 vs. Con group;
#P<0.05 vs. LPS group; &P<0.05 vs.
LPS + IR group; ns, not significant vs. LPS + IR group. LPS,
lipopolysaccharide; ALI, acute lung injury; Con, control. |
Subsequently, the BALF of mice was analyzed to
confirm the suppressive effects of irisin on the LPS-induced
inflammatory response. The total number of cells, protein
concentrations and neutrophil numbers in BALF in the LPS (2 mg/kg)
group increased considerably compared with the control group
(P<0.05; Fig. 3A-C), but
decreased markedly in the LPS + IR group or LPS + MCC950 group. The
quantitative data revealed that irisin (0.5 mg/kg) exerted an
anti-inflammatory effect equivalent to that of MCC950 (50 mg/kg)
(Fig. 3A-C).
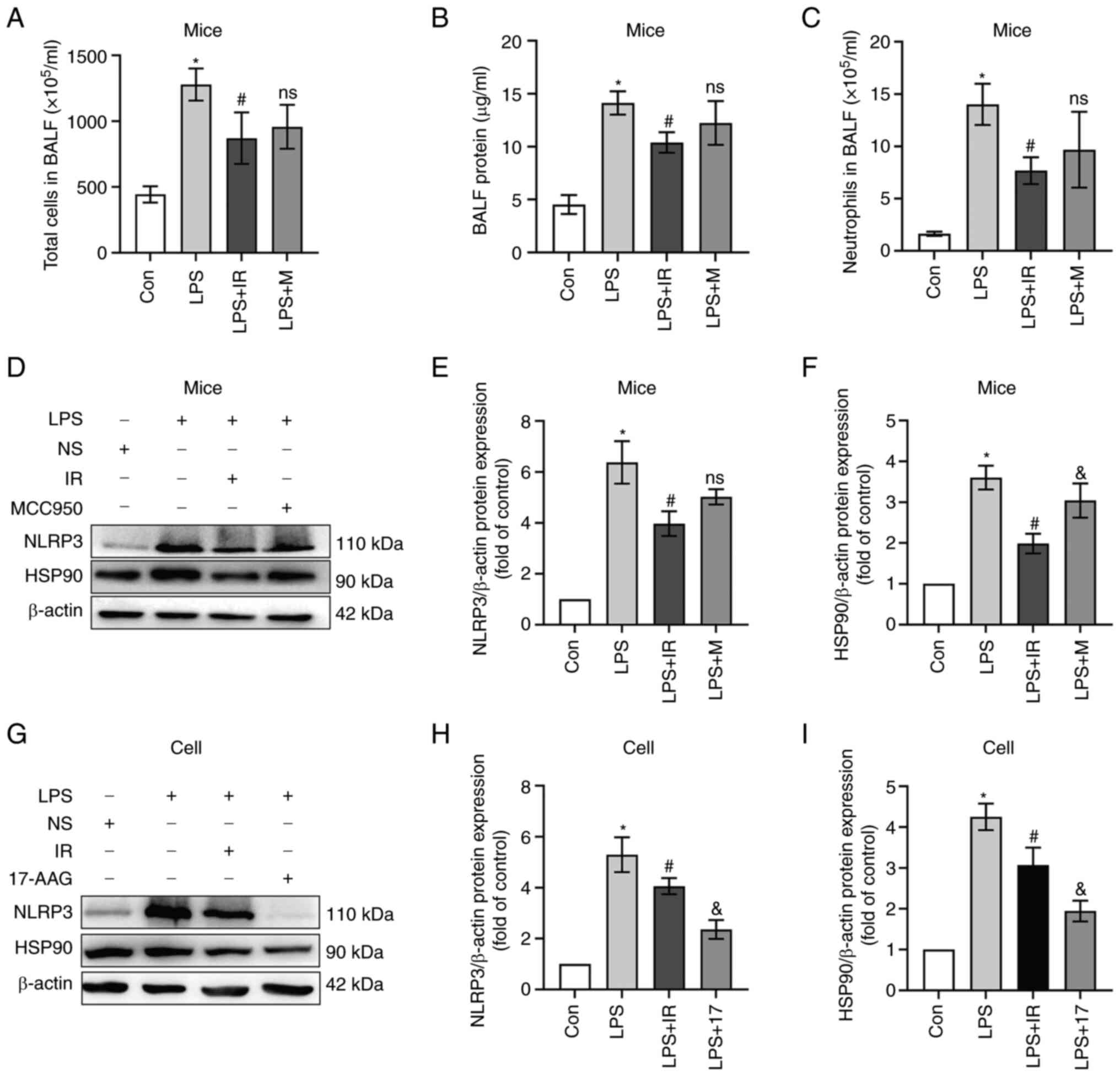 | Figure 3Irisin attenuates the LPS-induced
inflammatory response. Mice were randomly assigned to the control,
LPS (2 mg/kg), LPS + IR (LPS 2 mg/kg, irisin 0.5 mg/kg), and LPS +
MCC950 (LPS 2 mg/kg, MCC950 50 mg/kg) groups (n=6). MH-S cells
(n=3) were co-treated with IR (200 ng/ml) or 17-AAG (100 ng/ml) and
LPS (10 µg/ml) for 4 h. Western blotting was used to measure
NLRP3 and HSP90 protein expression. (A) Total number of cells, (B)
proteins, and (C) neutrophils in the specimens of BALF. (D-I)
Irisin inhibited the activation of the NLRP3 inflammasome and the
expression of HSP90 in mice and MH-S cells. The data are expressed
as the mean ± SD. *P<0.05 vs. Con group;
#P<0.05 vs. LPS group; &P<0.05 vs.
LPS + IR group; ns, not significant vs. LPS + IR group. LPS,
lipopolysaccharide; BALF, bronchoalveolar lavage fluid; Con,
control; NLRP3, nucleotide-binding and oligomerization domain-like
receptor protein 3; HSP90, heat shock protein 90; 17-AAG,
17-N-allylamino-17-demethoxygeldanamycin. |
NLRP3 is often used as an essential indicator of the
inflammatory response. In contrast to the control group, the mRNA
and protein expression levels of NLRP3 in mice exposed to LPS were
significantly increased (P<0.05; Fig. 3D and E), and this increase was
considerably reduced by irisin pre-treatment (P<0.05; Fig. 3D and E). HSP90 is a molecular
chaperone of NLRP3 that plays an essential role in inflammasome
assembly and stability. In vivo, the expression of HSP90 was
upregulated by LPS treatment, while irisin exerted the opposite
effect, and MCC950 had no regulatory effect on HSP90 (P>0.05;
Fig. 3F). Based on this result,
it was hypothesized that the mechanism through which irisin
attenuates inflammatory lung injury in mice differs from that of
MCC950. To examine this hypothesis and elucidate the underlying
mechanisms of the anti-inflammatory effect of irisin, an in
vitro experiment was performed using 17-AAG (an inhibitor of
HSP90)-treated cells as the positive control (Fig. 3G-I). It was found irisin exerted a
similar effect to that of 17-AGG in reducing inflammasome
production.
Irisin protects against LPS-induced cell
damage
CCK-8 assays were used to determine the effects of
various concentrations of LPS, irisin and 17-AAG on cell viability.
LPS stimulation at 100 µg/ml for 24 h decreased cell
viability (P<0.05; Fig. 4A),
while LPS stimulation at 10 µg/ml for 4 h did not affect
cell viability (P>0.05; Fig.
4B). Therefore, 10 µg/ml LPS was used in subsequent
experiments. Irisin at <400 ng/ml had no significant toxic
effects on the MH-S cells (P>0.05; Fig. 4C). The cell numbers decreased with
the increasing concentrations of 17-AAG, and 100 ng/ml was selected
as the final concentration for the intervention (P>.05; Fig. 4D). The intervention of MH-S cells
with LPS for different periods of time revealed that LPS (10
µg/ml) stimulation significantly upregulated NLPR3
expression in the cells treated for 4 h compared to those in the
untreated cells or the cells treated for 2 h (P<0.05; Fig. 4E and F).
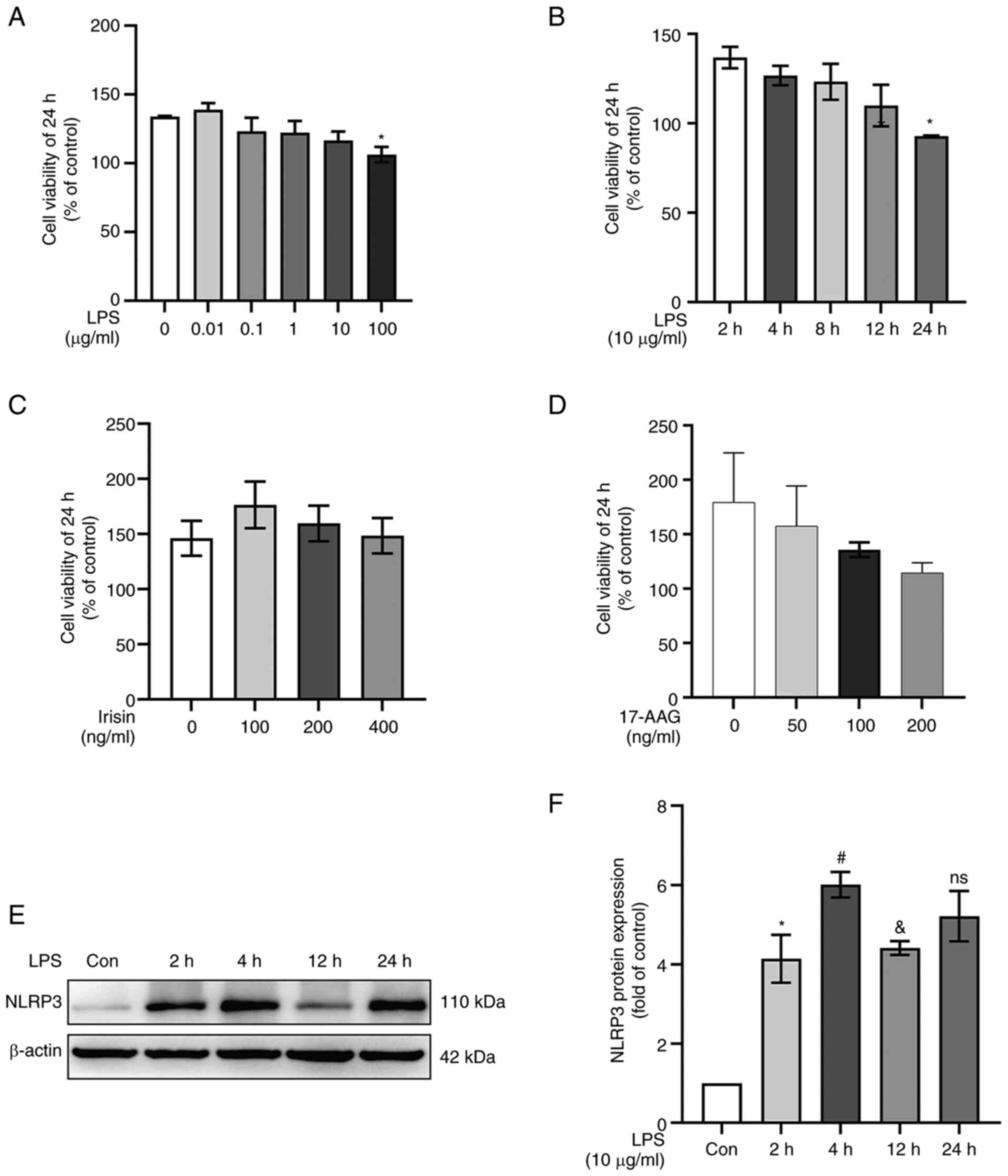 | Figure 4Irisin protects against LPS-induced
cell damage. The data are expressed as mean ± SD, n=3. (A) MH-S
cells were treated with various concentrations (0, 0.01, 0.1, 1, 10
and 100 µg/ml) of LPS for 24 h and analyzed using CCK-8
assay. *P<0.05 vs. 0 µg/ml group. (B) CCK-8
assay results of LPS (10 µg/ml)-treated MH-S cells measured
at different time points. *P<0.05 vs. 2 h group. (C
and D) CCK-8 assay to detect the cytotoxicity of irisin (0, 100,
200 and 400 ng/ml) and 17-AAG (0, 50, 100 and 200 ng/ml) in MH-S
cells. (E and F) Quantitative analysis of NLRP3 protein expression
at different time points of LPS stimulation (10 µg/ml).
*P<0.05 vs. Con group; #P<0.05 vs. 2 h
group; &P<0.05 vs. 4 h group; ns, not significant
vs. 4 h group. LPS, lipopolysaccharide; Con, control; 17-AAG,
17-N-allylamino-17-demethoxygeldanamycin. |
Irisin inhibits the release of
pro-inflammatory cytokines by regulating the polarization of
AMs
To evaluate the inhibitory effects of irisin on the
release of pro-inflammatory cytokines in vivo and in
vitro, ELISA kits were used to detect IL-1β, IL-18 and TNF-α
secretion in the BALF and serum of mice and MH-S cell culture
supernatants. The results revealed that the IL-1β, IL-18 and TNF-α
levels significantly increased in the LPS group compared with the
control group (P<0.05; Fig.
5). Compared with the LPS group, irisin effectively decreased
the IL-1β and IL-18 levels in BALF (P<0.05; Fig. 5A and B), serum (P<0.05,
Fig. 5A and B) and cell culture
supernatants (P<0.05 Fig. 5A and
B). MCC950 also had similar suppressive effects on the levels
of IL-1β and IL-18 in BALF (P<0.05, Fig. 5A and B), serum (P<0.05,
Fig. 5A and B). On the contrary,
irisin effectively suppressed the release of TNF-α (P<0.05,
Fig. 5C), while MCC950 did not
(Fig. 5C). In vitro,
17-AAG considerably reduced the LPS-induced release of IL-1β and
IL-18 (P<0.05 Fig. 5A and B),
and slightly decreased the levels of TNF-α (P<0.05, Fig. 5C).
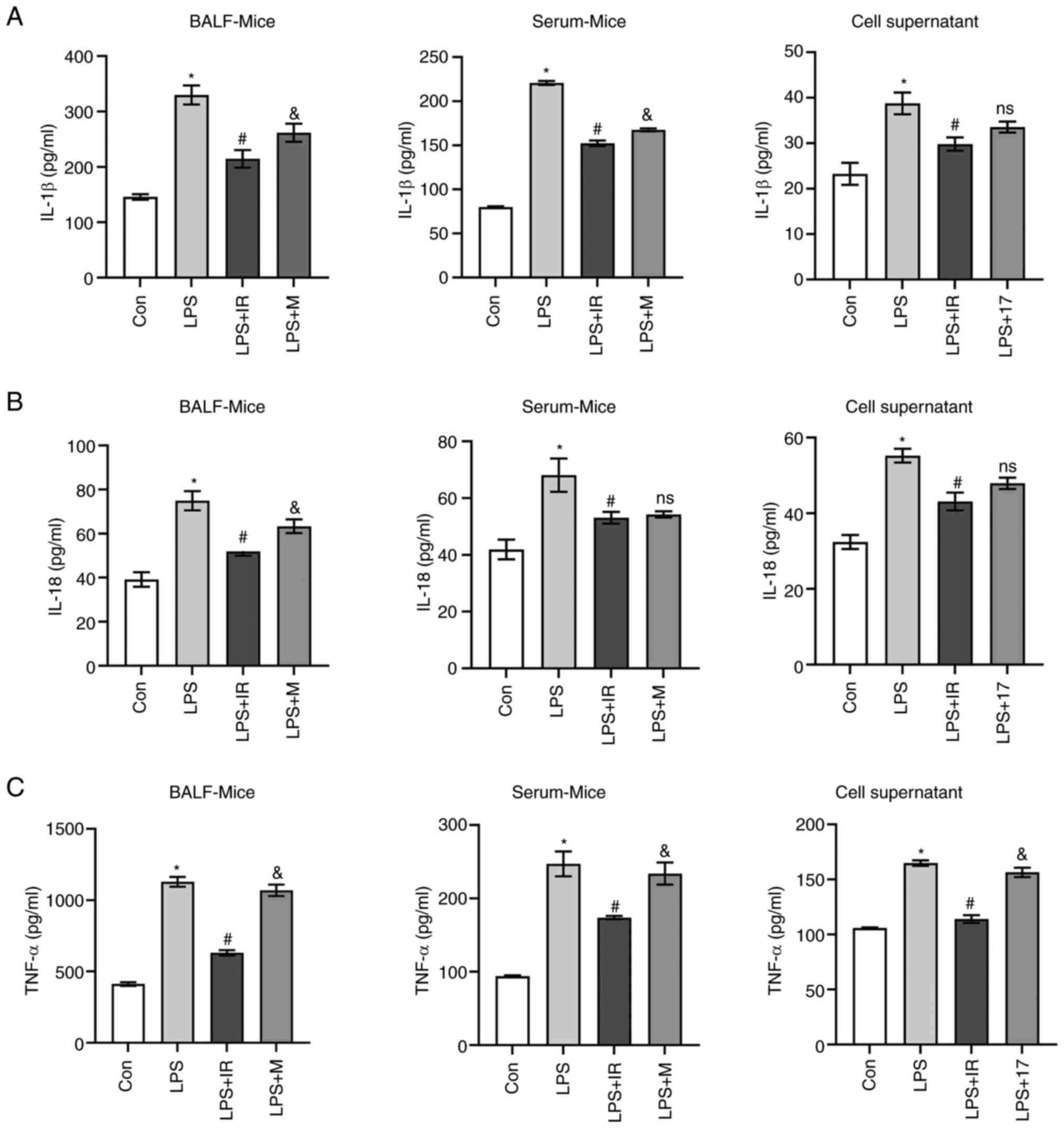 | Figure 5Irisin inhibits the release of
pro-inflammatory cytokines. Mice were randomly assigned to the
control, LPS (2 mg/kg), LPS + IR (LPS 2 mg/kg, irisin 0.5 mg/kg),
and LPS + MCC950 (LPS 2 mg/kg, MCC950 50 mg/kg) groups (n=6).
Following treatment, serum and BALF samples were collected. MH-S
cells were co-cultured with LPS (10 µg/ml) and IR (200
ng/ml) or 17-AAG (100 ng/ml) for 4 h (n=3). ELISA was used to
detect IL-1β, IL-18, and TNF-α secretion in (A) BALF, (B) serum,
and (C) cell supernatant samples. The data are expressed as the
mean ± SD. *P<0.05 vs. Con group;
#P<0.05 vs. LPS group; &P<0.05 vs.
LPS + IR group; ns, not significant vs. LPS + IR group. LPS,
lipopolysaccharide; Con, control; 17-AAG,
17-N-allylamino-17-demethoxygeldanamycin; IL, interleukin; TNF-α,
tumor necrosis factor α. |
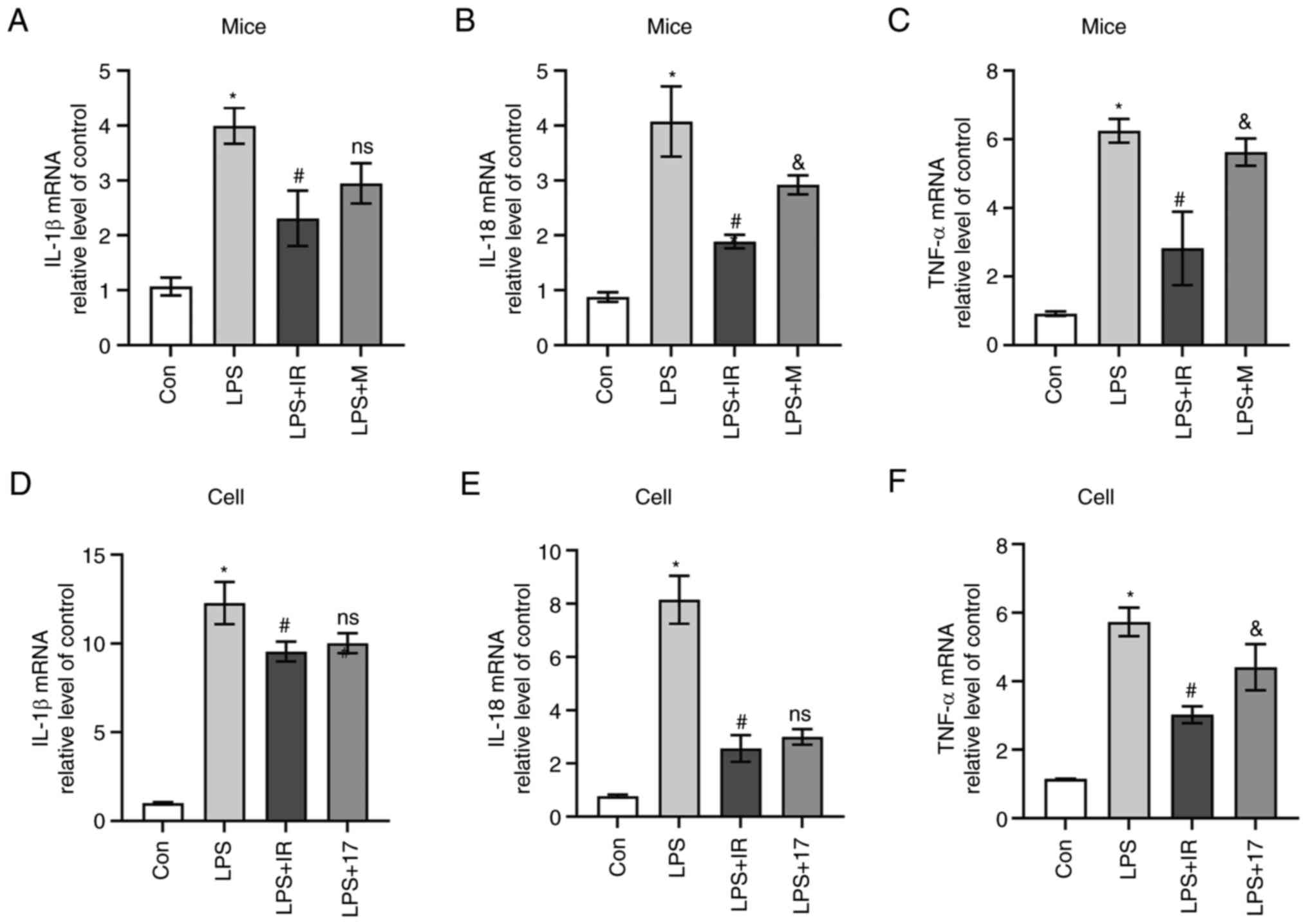
RT-qPCR was used to detect the mRNA expression of
inflammatory factors in mice and MH-S cells. The results revealed
that compared with the control group, LPS significantly induced the
release of IL-1β, IL-18 and TNF-α in lung tissue and MH-S cells
(P<0.05, Fig. 6). Compared
with the LPS group, irisin reduced the release of IL-1β(P<0.05,
Fig. 6A), IL-18 (P<0.05,
Fig. 6B) and TNF-α (P<0.05,
Fig. 6C) in the lung tissue. The
mRNA levels of these inflammatory factors were also reduced in
vitro (P<0.05, Fig. 6D-F)
in the irisin-treated cells compared with those in the
LPS-stimulated cells. Furthermore, the mRNA expression of IL-1β
(P<0.05, Fig. 6A) and IL-18
(P<0.05, Fig. 6B) in the
MCC950-treated mice was lower compared with that in the in
LPS-exposed mice; however, no marked inhibitory effect of MCC950
was observed on TNF-α levels compared with the LPS group
(P>0.05, Fig. 6C). The
suppressive effects of 17-AAG on IL-1β (P<0.05, Fig. 6D) and IL-18 (P<0.05 Fig. 6E) levels were similar to those of
irisin; however, the suppressive effects of irisin on TNF-α levels
were more prominent than those of 17-AAG (P<0.05, Fig. 6F).
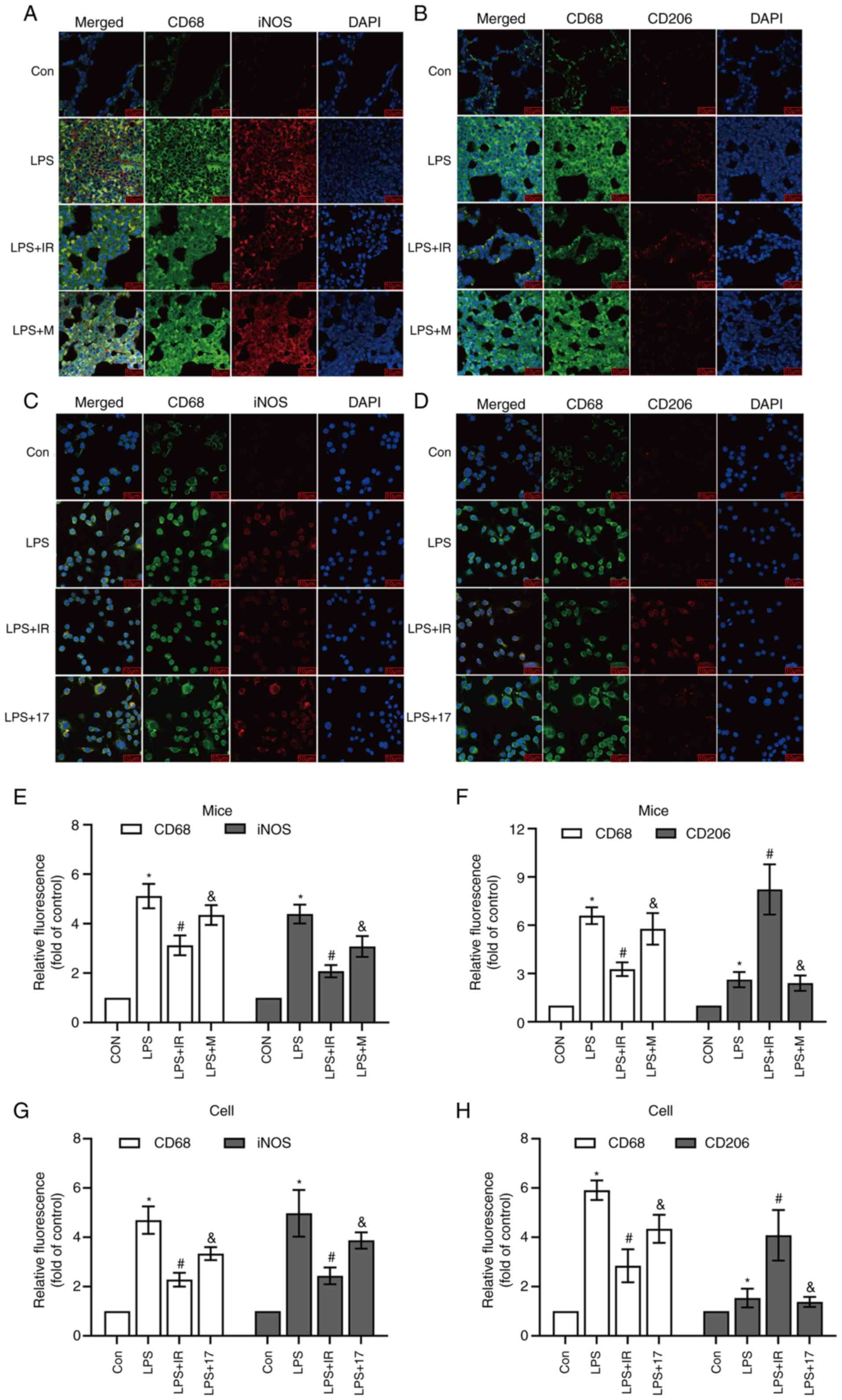
Subsequently, iNOS and CD206 antibodies were used as
biomarkers for AMs to evaluate macrophage polarization in
vivo and in vitro. Immunofluorescence co-staining with
CD68 and iNOS confirmed that LPS promoted the transformation of AMs
to the M1 type in vivo and in vitro (iNOS, P<0.05;
Fig. 7E and G) compared with the
control group. Irisin pre-treatment increased the numbers of M2
macrophages compared with the LPS group (CD206, P<0.05; Fig. 7F and H). This may be due to a
significantly increased co-localization and reduced differentiation
of macrophages to the M1 type (iNOS, P<0.05; Fig. 7B, E and G). Compared with irisin,
LPS, MCC950 and 17-AAG had a lesser effect on the regulation of
M2-type macrophages (Fig. 7).
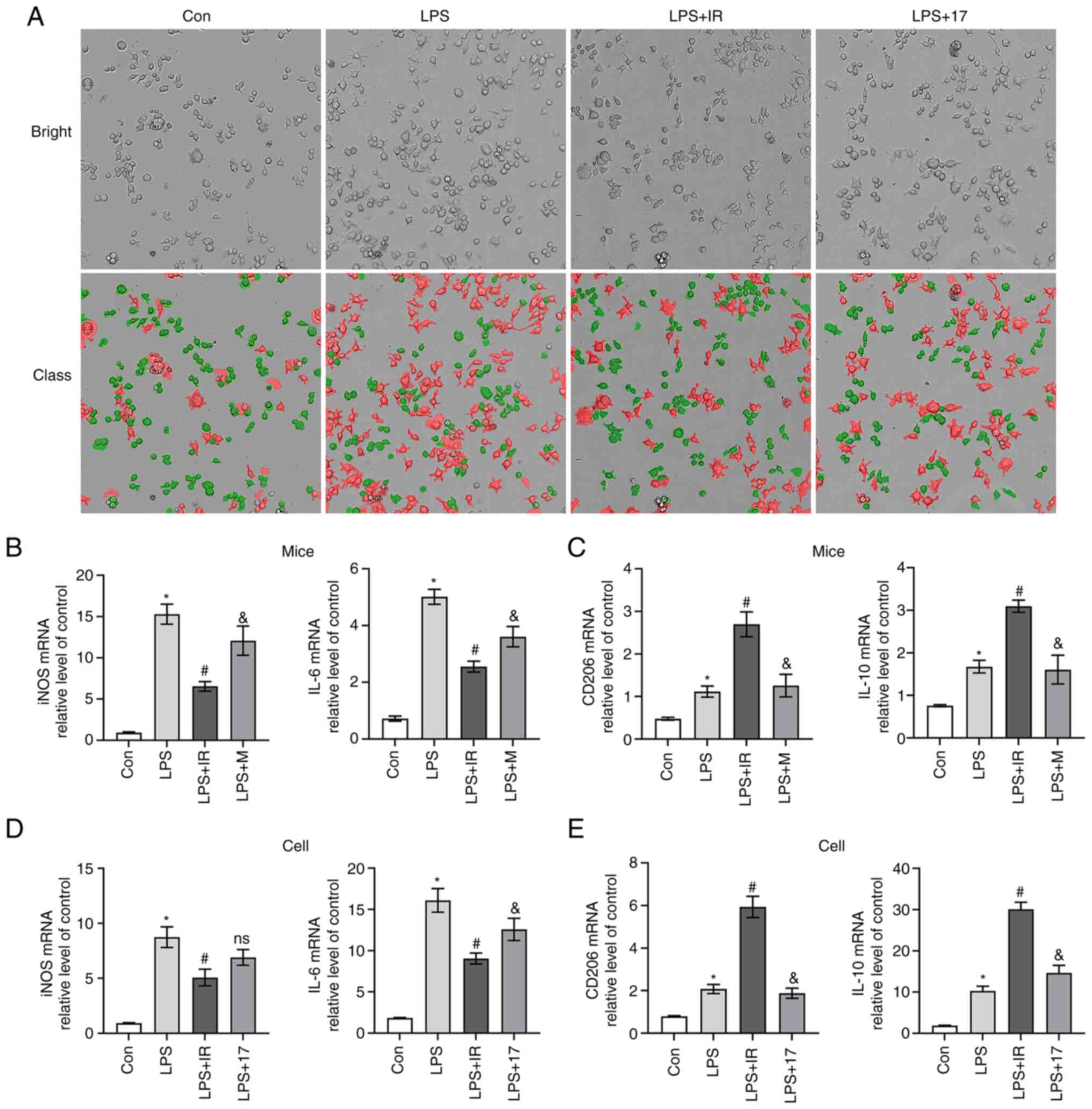
The visualization of cell morphology using Operetta
CLS revealed that the MH-S cells in the control group had an oval
shape with complete cell membranes (30). Cell morphology tracking technology
revealed that numerous cells had irregular shapes, increased
protrusions, longer pseudopodia, cell swelling or vesicle-like
changes and ruptured cell membranes following LPS (10 µg/ml)
stimulation. Irisin protected the cells from morphological changes,
whereas 17-AAG did not (Fig. 8A).
The results of RT-qPCR revealed that the mRNA expression levels of
the M1 macrophage biomarkers, iNOS and IL-6, in vivo and
in vitro were significantly higher in the LPS group than in
the control group (P<0.05; Fig. 8B
and D). Compared with the Con group, LPS increased the levels
of the M2 macrophage biomarkers, CD206 and IL-10, in mouse lung
tissue and cells (P<0.05; Fig. 8C
and E). Macrophages can self-regulate the phenotype, which may
be due to the polarization of macrophages themselves to LPS-induced
inflammatory responses. Irisin reversed the elevated expression of
iNOS and IL-6 induced by LPS, promoting the high expression of
CD206 and IL-10 in vivo and in vitro (P<0.05;
Fig. 8B-E).
Irisin inhibits macrophage
pyroptosis
The destroyed integrity of the cell membrane
characterizes pyroptosis. Pyroptotic cells exhibit swelling, and
numerous bubble-like protrusions appear on the surface of the
cellular membrane before its rupture. Still, DNA damage and
chromatin condensation occur after the break of the cell membrane
(31). It has been found that
pyroptotic cells are permeable to 7-AAD and PI due to the low
weight of these dyes (32). The
present study used the Calcein AM/7-AAD double staining method to
distinguish living cells from cells with damaged membranes. When
pyroptosis occurs in cells, the cell membranes are damaged and do
not have cell activity, and thus they can be stained red. Some
cells in the early stages of pyroptosis can be double-stained by
Calcein AM and 7-AAD, and the cells are stained green and red
simultaneously. As shown in Fig.
9A, the majority of the cells in the LPS group were colored
red, proving that the cell membrane was damaged, and pyroptosis had
occurred. The number of red-stained cells in the irisin and 17-AAG
groups was relatively reduced, demonstrating that the number of
pyroptotic cells was decreased.
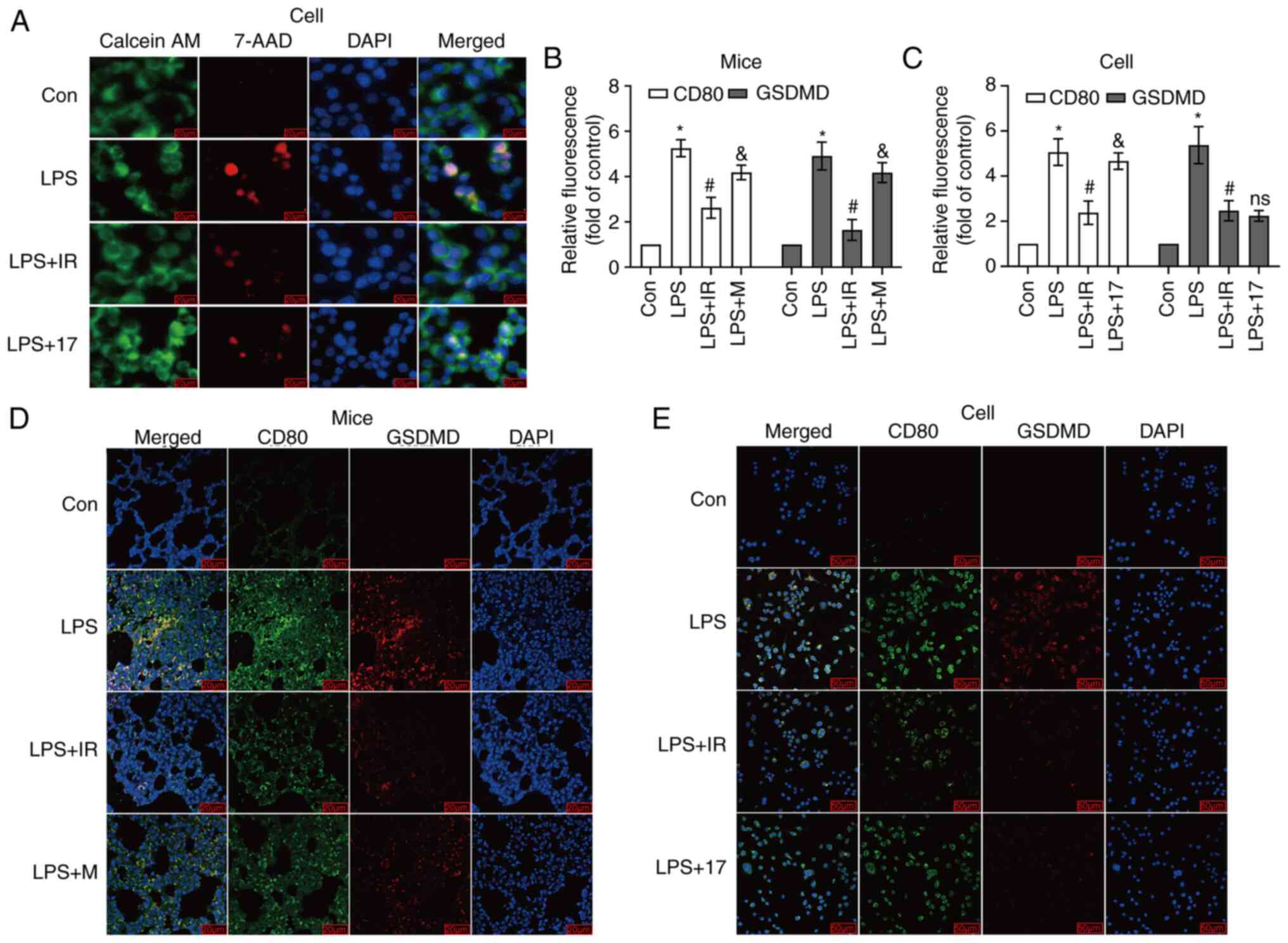 | Figure 9(A) Calcein AM/7-AAD double staining
detected pyroptosis (n=3, magnification, ×400). Blue color
indicates DAPI staining; green color indicates Calcein AM staining;
red color indicates 7-AAD staining. The cells stained in green and
red represent ongoing pyroptosis. The experimental groupings are as
those described in Fig. 5. (B and
C) Quantitative analysis of fluorescence intensity using ImageJ
software. (D and E) Immunofluorescence co-staining with the M1-type
macrophage biomarker, CD80, and pyroptotic executive protein
antibody (GSDMD) in mice (n=6, magnification, ×400) and MH-S cell
(n=3, magnification, ×200). Blue color indicates DAPI staining;
green color indicates CD80 staining; red color indicates GSDMD
staining. The data are expressed as the mean ± SD.
*P<0.05 vs. Con group; #P<0.05 vs. LPS
group; &P<0.05 vs. LPS + IR group; ns, not
significant vs. LPS + IR group. LPS, lipopolysaccharide; Con,
control; M, MCC950; IR, irisin; 17, 17-AAG
(17-N-allylamino-17-demethoxygeldanamycin); iNOS, inducible nitric
oxide synthase; GSDMD, gasdermin D. |
Immunofluorescence experiments were also performed
using the M1 macrophage-specific antibody, CD80 (green
fluorescence), and the pyroptotic executive protein antibody, GSDMD
(red fluorescence). More green fluorescent particles were located
in M1 macrophages specifically labeled with CD80 antibody (Fig. 9D and E). The results revealed that
in vivo and in vitro, M1 macrophages (green
fluorescence) in the LPS group were stained red by the GSDMD
antibody. Compared with LPS, irisin reduced the occurrence of
pyroptosis (P<0.05; Fig. 9B and
C). Therefore, it was deemed that M1 macrophages undergo
pyroptosis.
Irisin inhibits caspase-1 activity
The production of IL-1β and IL-18 is associated with
pyroptosis, and caspase-1 is a core factor in the initiation of
pyroptosis (14). The present
study used FLIVO to detect caspase-1 enzyme activity in mice. LPS
significantly enhanced the fluorescence of caspase-1, which was
evenly distributed in multiple lung lobes and was substantially
more potent than that in the control group (P<0.05; Fig. 10A and B). Compared with the LPS
group, fewer FLIVO fluorophores and a lower fluorescence intensity
in the lung lobes of mice were observed in the irisin group,
confirming that irisin reduced caspase-1 activity (P<0.05;
Fig. 10A and B). The results of
the immunofluorescence staining of frozen sections of mouse lung
tissue were consistent with the results of the in vivo
experiments, confirming that irisin inhibited the activity of
caspase-1 in mice (P<0.05; Fig.
10C and D). The results of the in vivo experiments
revealed that MCC950 did not play a role in inhibiting the activity
of caspase-1 (Fig. 10C and
D).
For the in vitro experiments, FLICA was
selected as the detection reagent for the intracellular activity of
the caspase-1 enzyme. At the same time, the PI staining of the
cells with damaged membranes confirmed that cells at an early stage
of pyroptosis (stained in both green and red) had damaged
membranes, but were not completely dead. The results of
immunofluorescence confocal staining revealed that the activity of
caspase-1 in the LPS group was significantly higher than that in
the control group (P<0.05; Fig.
10E and F). Irisin effectively reduced the LPS-induced
activation of caspase-1 (P<0.05; Fig. 10E and F). 17-AAG exerted a
similar effect as irisin (P>0.05 vs. irisin group; Fig. 10E and F). As shown in Fig. 10E (reds arrow), in the cells
double-stained with FLICA and PI, the integrity of the cell
membrane was disrupted, suggesting that the cell was undergoing
pyroptosis (32).
Irisin inhibits caspase-1-mediated
pyroptosis by regulating the HSP90/NLRP3 signaling pathway
LPS significantly increased the expression and
release of HSP90, NLRP3 and cleaved caspase-1 in mice, while
upregulating the GSDMD and IL-1β levels (P<0.05 Fig. 11A and C-H). Similar results were
observed when the MH-S cells were analyzed (P<0.05 Fig. 11B and I-N). This confirmed that
the LPS-induced pyroptosis of murine AMs amplifies the inflammatory
response. The results of western blot analysis (Fig. 11) and RT-qPCR (Fig. 12) revealed that irisin
significantly inhibited the LPS-induced overexpression of HSP90 and
NLRP3. It also reduced pro-caspase-1 activity and decreased the
expression of cleaved caspase-1, GSDMD and IL-1β, indicating the
inhibition of pyroptosis.
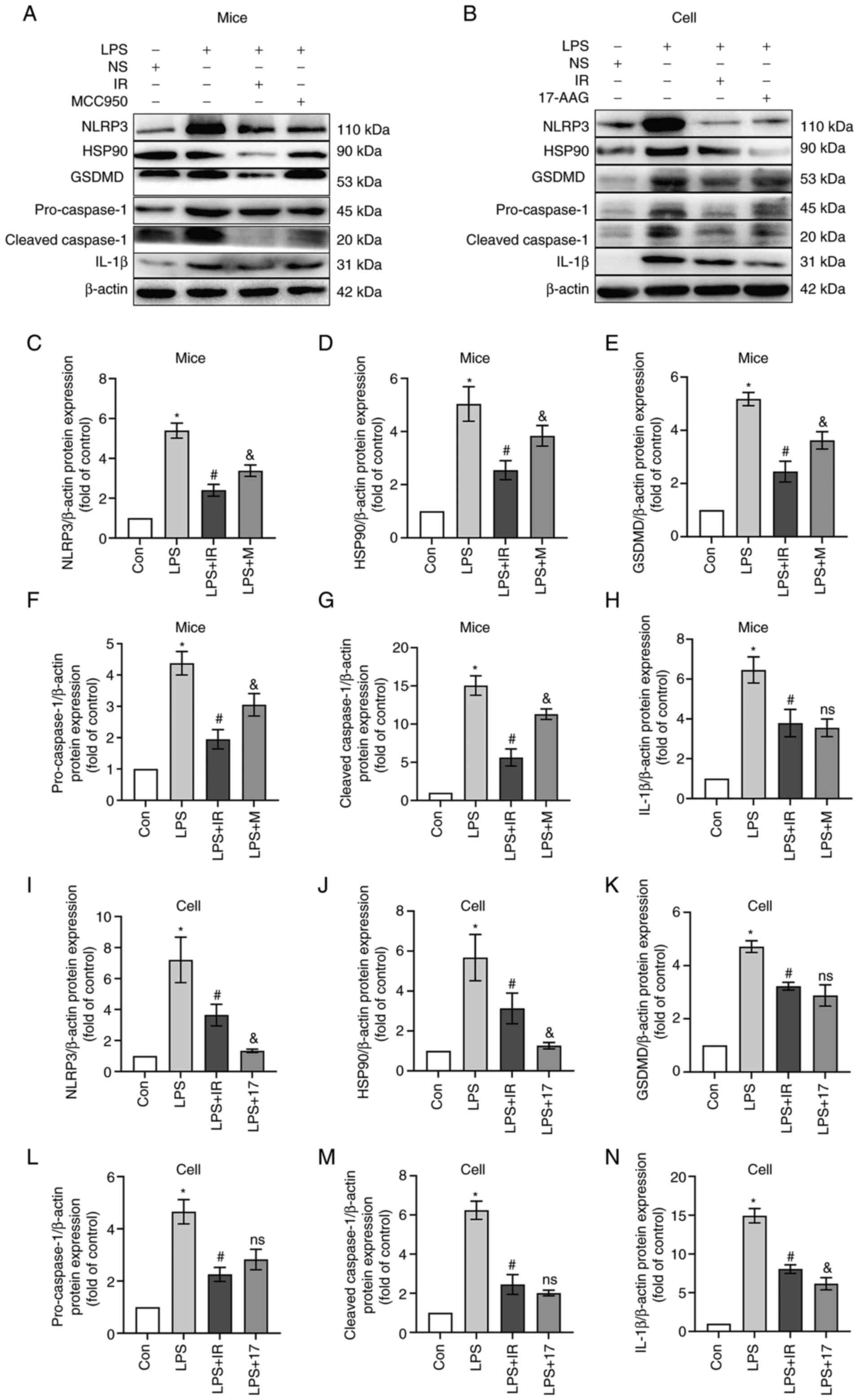 | Figure 11Irisin inhibits caspase-1-mediated
pyroptosis by regulating the HSP90/NLRP3 signaling pathway. The
grouping of mice and cells is the same as in Fig. 5. (A and B) After drug treatment,
proteins were extracted from (A) lung tissue and (B) MH-S cells and
subjected to western blotting to determine the expression levels of
NLRP3, HSP90, GSDMD, caspase-1, cleaved caspase-1 and IL-1β
proteins. (C-N) Relative quantitative analysis of protein
expression. The data are expressed as the mean ±
SD.*P<0.05 vs. Con group; #P<0.05 vs.
LPS group; &P<0.05 vs. LPS + IR group; ns, not
significant vs. LPS + IR group. LPS, lipopolysaccharide; Con,
control; NLRP3, nucleotide-binding and oligomerization domain-like
receptor protein 3; HSP90, heat shock protein 90; GSDMD, gasdermin
D; IL, interleukin; M, MCC950; IR, irisin; 17, 17-AAG
(17-N-allylamino-17-demethoxygeldanamycin). |
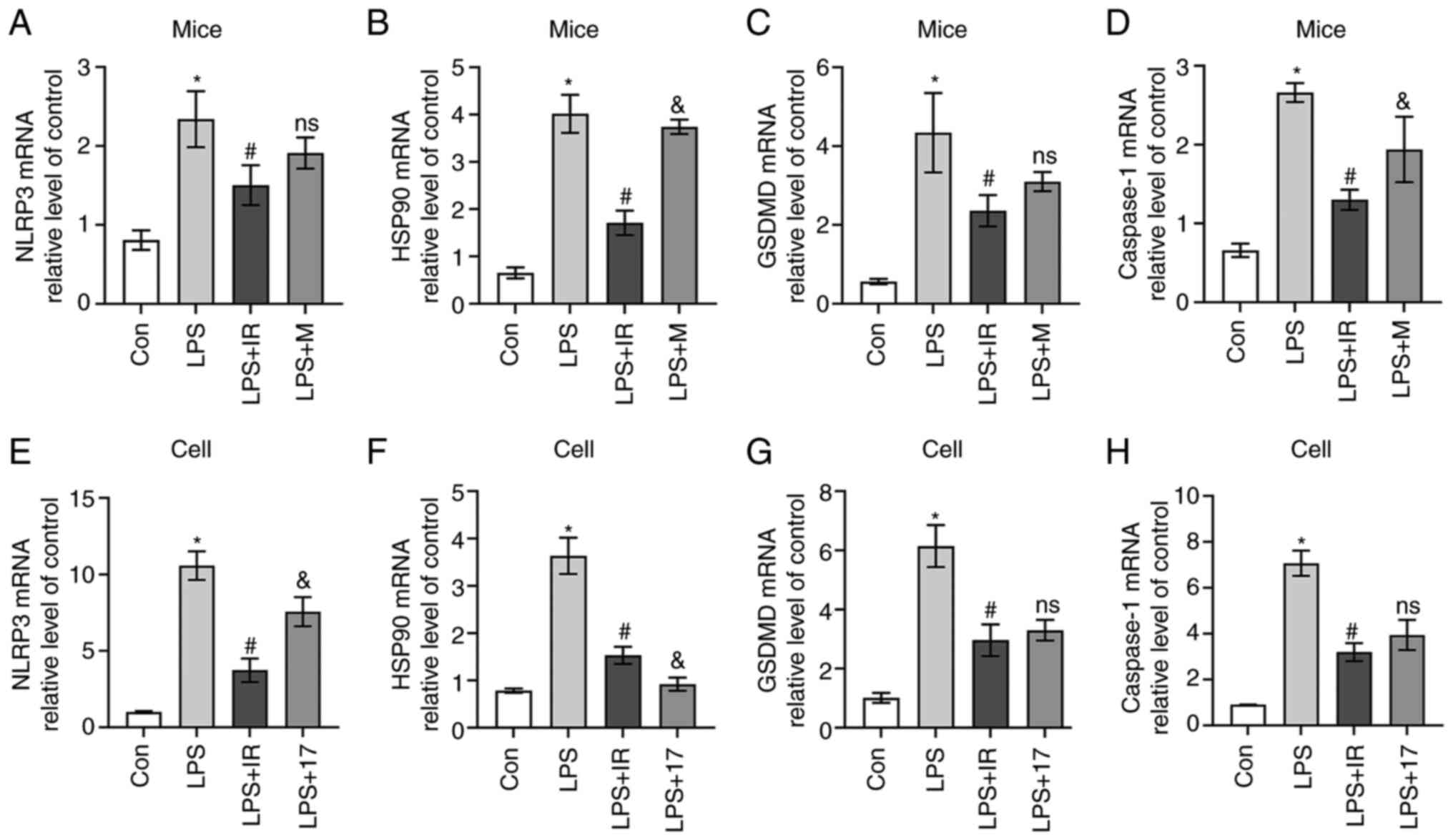 | Figure 12RT-qPCR analysis of (A and E) NLRP3,
(B and F) HSP90, (C and G) GSDMD, and (D and H) Caspase-1 mRNA
expression in vivo (n=6, A-D) and in vivo (n=3, E-H).
The data are expressed as the mean ± SD.*P<0.05 vs.
Con group; #P<0.05 vs. LPS group;
&P<0.05 vs. LPS + IR group; ns, not significant
vs. LPS + IR group. LPS, lipopolysaccharide; Con, control; M,
MCC950; IR, irisin; 17, 17-AAG
(17-N-allylamino-17-demethoxygeldanamycin); GSDMD, gasdermin D;
NLRP3, nucleotide-binding and oligomerization domain-like receptor
protein 3; HSP90, heat shock protein 90. |
As shown in Figs.
11 and 12, in vivo,
MCC950 reduced the expression of cleaved caspase-1 but had no
noticeable inhibitory effect on the overexpression of GSDMD. In
vitro, 17-AAG inhibited the levels of HSP90 and NLRP3, and
decreased GSDMD expression. Irisin played a role similar to that of
17-AAG, confirming that irisin can regulate the HSP90/NLRP3
signaling pathway and reduce LPS-induced caspase-1-dependent
macrophage pyroptosis.
Discussion
The novel coronavirus disease 2019 (COVID-19) has
swept the world since 2019 and is still affecting human health
across the globe. One of the major complications of COVID-19,
ALI/ARDS, is responsible for the death of patients. A previous
study demonstrated that AM death activated multiple signaling
pathways upon lung injury and led to the release inflammatory
mediators, such as IL-1β, IL-6, IL-18 and TNF-α (33). Inflammatory cell infiltration
destroys the alveolar epithelium and pulmonary microvascular
barrier, accelerates lung inflammation and aggravates lung tissue
damage (34). Therefore,
regulating AM death may be a potential therapeutic strategy for
controlling ALI/ARDS. In the present study, both in vitro
cell experiments and in vivo mouse models were used. It was
demonstrated that irisin, a myokine with anti-inflammatory effects
that regulate cell signaling pathways, protects against LPS-induced
cell damage and ALI in mice. It inhibited the release of
pro-inflammatory cytokines by regulating the polarization of AM, AM
pyroptosis, and caspase-1-mediated pyroptosis.
Irisin is the product of FNDC5 hydrolysis. Unlike
previous studies (19,20), the present study found that FNDC5
was expressed in the lungs of mice with LPS-induced inflammatory
injury, and it was hypothesized that irisin may have a positive
anti-inflammatory effect. Inflammatory cell infiltration in mouse
lung tissue was significantly reduced following treatment with
irisin, confirming that irisin can improve LPS-induced ALI.
Macrophages are divided into two subgroups: M1 and M2 (35). M1-type macrophages play a role in
inflammatory activation, while M2-type macrophages are involved in
the resolution of inflammation (36). In the early stages of ALI, high
levels of pro-inflammatory factors, such as IL-1β, TNF-α and iNOS
in the body induce M1-type macrophages to continuously expand the
inflammatory response. Over time, IL-4 and IL-13 induce the
production of M2-type macrophages. They express and produce the
anti-inflammatory factors, IL-10 and IL-1Rα, which play a role in
tissue damage repair (37). The
present study confirmed that irisin pre-treatment significantly
increased the transcription and release of CD206 and IL-10 compared
to LPS pre-treatment. It reduced IL-1β, IL-18 and TNF-α expression
in vivo and in vitro, indicating that irisin can
regulate the transformation of AMs into the M2 type, and can play a
role in inhibiting inflammation and repairing tissue damage.
Pyroptosis is also known as programmed necrotic
death. Unlike the apoptotic pathway, the pyroptotic pathway often
exhibits features of necrosis and amplifies inflammatory signaling
by altering the permeability of the cell membrane to release the
cellular contents (38,39). To examine the permeability of the
cell membrane, the present study used CalceinAM/7-AAD double
staining. Calcein-AM can easily penetrate the membrane of living
cells, fluorescently label living cells, and emit uniform green
solid fluorescence in living cells. It has the characteristics of
low cytotoxicity and can reduce damage to cells. 7-ADD is a
non-permeable fluorescent nuclear dye that cannot penetrate the
cell membrane of normal living cells, but can penetrate cells with
membrane damage and can embed into the DNA of necrotic cells to
form highly fluorescent adducts that emit red fluorescence.
Therefore, 7-ADD only stains cells with damaged membranes (40). The present study found that the
majority of the cells in the LPS group were colored red, proving
that the cell membrane is damaged and pyroptosis had occurred.
Considering that this experiment is not specific, the present study
further applied FLICA dyes in subsequent cell experiments to verify
that caspase-1 was activated and pyroptosis had occurred. The
principal steps of pyroptosis involve the activation of caspase-1
mediated via the inflammasome complex to cleave GSDMD, leading to
the perforation of the cell membrane followed by cell swelling,
rupture and release of cellular contents (41). Caspase-1 cuts pro-IL-1β or
pro-IL-18 to promote the production and release of IL-1β and IL-18,
respectively (42). The staining
of live and membrane-damaged cells revealed that LPS induced the
pyroptosis of MH-S cells, in which membrane-damaged cells appeared
red due to staining with 7-AAD. Simultaneously, ELISA assays
revealed that the IL-1β and IL-18 levels in the culture supernatant
of the LPS group were significantly higher than those of the
control group, confirming that the MH-S cells had undergone
pyroptosis. The present study performed a preliminary exploration
of the types of macrophages that undergo pyroptosis. The in
vivo and in vitro immunofluorescence experiments
revealed that the majority of the cells labeled by CD80 (specific
antibody for M1 macrophages, green fluorescence) in the LPS group
were also stained red (GSDMD antibody). Accordingly, it was
hypothesized that M1 macrophages undergo pyroptosis under the
stimulation of LPS. Irisin reduced the occurrence of
pyroptosis.
FLIVO and FLICA were used to bind activated
caspase-1 in vivo and in vitro, after which caspase
activity was quantified to detect pyroptosis. The results indicated
that irisin reduced caspase-1 activity and pyroptosis in
vivo and in vitro. NLRP3 is the promoter of pyroptosis.
It was found that NLRP3 was intuitively increased with the number
of dead cells. It was hypothesized that it could be explained by
the following reasons: i) Dead cells release more NLRP3, indicating
that other pathways may promote the expression of NLRP3; ii)
following LPS intervention, a specific reaction against pyroptosis
occurred in the cells, which caused the reduction of NLRP3 in the
cells at 12 h of LPS intervention. Luo et al (43) found that the expression of NLRP3
was upregulated in NR8383AM cells exposed to LPS at 6 h; the
presentation of NLRP3 was decreased at 12 h of LPS intervention.
The mRNA of NLRP3 was increased when exposed to LPS for 24 h
compared with that at 12 h (43).
The results of the present study were similar to these. The
expression of NLRP3 was increased following LPS intervention in the
MH-S cells at 4 h, but was reduced at 12 h. Still, the expression
of NLRP3 was increased after 24 h of LPS intervention. It was
hypothesized that there may be some mechanisms against the
activation of the NLRP3 inflammasome in the cell, which is worthy
of further exploration. NLRP3 plays a critical role in the
initiation of pyroptosis; hence, the present study used MCC950, an
inhibitor of NLRP3, as a novel positive control to assess the
effects of irisin in vivo. The characteristics of the
mechanisms of action of irisin and MCC950 were compared. MCC950
inhibited LPS-induced neutrophil infiltration in lung tissue to a
similar extent as Dex, an anti-inflammatory and immunosuppressant
synthetic glucocorticoid, and decreased IL-1β and IL-18 protein
expression levels (2). In a
previous study, MCC950 inhibited the secretion of IL-1β in murine
bone marrow-derived macrophages and NLRP3-induced
apoptosis-associated speck-like protein containing a caspase
recruitment domain oligomerization (44). The results of the present study
confirmed that irisin and MCC950 downregulated the expression of
IL-1β and IL-18 in mouse serum and BALF. Additionally, irisin
inhibited TNF-α production, whereas MCC950 did not. Western blot
analysis and RT-qPCR revealed that irisin significantly inhibited
the activation of HSP90, reduced the production and release of
NLRP3, and inhibited downstream caspase-1 and GSDMD expression,
indicating a different mechanism of action for irisin and
MCC950.
HSP90, involved in the signaling pathway associated
with the pyroptosis of AMs, is a potential target for
anti-inflammatory drugs (2).
Furthermore, the regulation of the stability of NLRP3 and the
modulation of inflammasome activation by HSP90 is associated with
IL-1β secretion and pyroptosis (15). Therefore, it was considered that
irisin inhibits the activation of HSP90 and interrupts the assembly
of NLRP3, thus inhibiting pyroptosis and exerting anti-inflammatory
effects. To examine the association between irisin and HSP90 in
vitro, 17-AAG was used as a positive control. The results
confirmed that 17-AAG, an inhibitor of HSP90, reduced the stability
of NLRP3. Reportedly, it reduces the cleavage of caspase-1 and
GSDMD in macrophages, thereby inhibiting pyroptosis (2). In vitro experiments revealed
that irisin and 17-AAG were more effective in inhibiting the
activation and release of caspase-1 and significantly reduced the
expression of GSDMD, IL-1β, and IL-18 following the co-incubation
of MH-S cells with LPS. However, the decrease in TNF-α levels
induced by 17-AAG treatment was not as pronounced as that induced
by irisin, suggesting that irisin reduced the production and
release of TNF-α through other signaling pathways. However, further
studies are required to elucidate the underlying pathways and
elucidate the precise mechanisms of action of irisin.
The present study has certain limitations. Targeted
experiments to examine the expression of irisin in the lungs were
not conducted. The authors plan to design experiments to further
explore the mechanisms through which FNDC5/irisin is expressed in
injured lung tissue, as well as its clinical application in
ALI.
In conclusion, the pyroptosis of AMs produces and
releases pro-inflammatory cytokines in LPS-induced ALI. Irisin acts
as an HSP90 inhibitor, regulating AM pyroptosis and exerting
anti-inflammatory effects. Collectively, irisin regulates
macrophage typing and inhibits the pyroptosis of AMs by regulating
the HSP90/NLRP3/caspase-1/GSDMD pathway, downregulating the
expression of inflammasomes and the release of pro-inflammatory
cytokines. Irisin can be used as a novel treatment for ALI, and
HSP90 may be a novel target with which inhibit the occurrence of
pyroptosis.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors participated in the design and
interpretation of the study, in the analysis of the data and in the
review of the manuscript. ZH, JM and AM designed the study,
interpreted the data and revised the manuscript critically. ZH and
JM conducted the majority of the experiments, performed the
statistical analysis and wrote the manuscript. RJ, YH and GY
conducted parts of the experiments. All authors have read and
approved the final manuscript. YH and AM confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The animal experimental and handling procedures were
approved by the Ethics Committee of the Second Hospital of Hebei
Medical University (Approval no. 2022-AE008, 2.28.2022). All
experimental processes were carried out according to the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the Hebei Key
Laboratory of Vascular Homeostasis and the Hebei Collaborative
Innovation Center for Cardio-Cerebrovascular Disease, Shijiazhuang,
China for providing all the facilities to conduct the study.
Funding
The present study was supported by the Natural Science
Foundation of Hebei Province (grant no. H2019206263); the Key
R&D Program of Hebei Province (grant no. 19277760D); and the
Hebei Province Applied Basic Research Program (grant no.
15967753D).
References
|
1
|
Wheeler AP and Bernard GR: Acute lung
injury and the acute respiratory distress syndrome: A clinical
review. Lancet. 369:1553–1564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou Z, Li X, Qian Y, Liu C, Huang X and
Fu M: Heat shock protein 90 inhibitors suppress pyroptosis in THP-1
cells. Biochem J. 477:3923–3934. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kovacs SB and Miao EA: Gasdermins:
Effectors of pyroptosis. Trends Cell Biol. 27:673–684. 2017.
View Article : Google Scholar :
|
|
4
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan EK and Fan J: Regulation of alveolar
macrophage death in acute lung inflammation. Respir Res. 19:502018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martin WJ II, Wu M and Pasula R: A novel
approach to restore lung immunity during systemic
immunosuppression. Trans Am Clin Climatol Assoc. 116:221–227.
2005.
|
|
7
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gordon S, Plüddemann A and Martinez
Estrada F: Macrophage heterogeneity in tissues: Phenotypic
diversity and functions. Immunol Rev. 262:36–55. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu
Z, Li Y, Scott MJ, Xiao G, Li S, et al: Macrophage endocytosis of
high-mobility group box 1 triggers pyroptosis. Cell Death Differ.
21:1229–1239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Scott MJ, Fan EK, Li Y, Liu J, Xiao
G, Li S, Billiar TR, Wilson MA, Jiang Y and Fan J: Tissue damage
negatively regulates LPS-induced macrophage necroptosis. Cell Death
Differ. 23:1428–1447. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Zhao Y, Zhang P, Li Y, Yang Y,
Yang Y, Zhu J, Song X, Jiang G and Fan J: Hemorrhagic shock primes
for lung vascular endothelial cell pyroptosis: Role in pulmonary
inflammation following LPS. Cell Death Dis. 7:e23632016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He X, Qian Y, Li Z, Fan EK, Li Y, Wu L,
Billiar TR, Wilson MA, Shi X and Fan J: TLR4-upregulated IL-1β and
IL-1RI promote alveolar macrophage pyroptosis and lung inflammation
through an autocrine mechanism. Sci Rep. 6:316632016. View Article : Google Scholar
|
|
13
|
Mayor A, Martinon F, De Smedt T, Pétrilli
V and Tschopp J: A crucial function of SGT1 and HSP90 in
inflammasome activity links mammalian and plant innate immune
responses. Nat Immunol. 8:497–503. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Davis BK, Wen H and Ting JP: The
inflammasome NLRs in immunity, inflammation, and associated
diseases. Annu Rev Immunol. 29:707–735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piippo N, Korhonen E, Hytti M, Skottman H,
Kinnunen K, Josifovska N, Petrovski G, Kaarniranta K and Kauppinen
A: Hsp90 inhibition as a means to inhibit activation of the NLRP3
inflammasome. Sci Rep. 8:67202018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang M, Liu L, Lin X, Wang Y, Li Y, Guo
Q, Li S, Sun Y, Tao X, Zhang D, et al: A translocation pathway for
vesicle-mediated unconventional protein secretion. Cell.
181:637–652.e615. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu X and Rapoport TA: Mechanistic insights
into ER-associated protein degradation. Curr Opin Cell Biol.
53:22–28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ruggiano A, Foresti O and Carvalho P:
Quality control: ER-associated degradation: Protein quality control
and beyond. J Cell Biol. 204:869–879. 2014. View Article : Google Scholar
|
|
19
|
Huh JY, Panagiotou G, Mougios V,
Brinkoetter M, Vamvini MT, Schneider BE and Mantzoros CS: FNDC5 and
irisin in humans: I. Predictors of circulating concentrations in
serum and plasma and II. mRNA expression and circulating
concentrations in response to weight loss and exercise. Metabolism.
61:1725–1738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rabiee F, Lachinani L, Ghaedi S,
Nasr-Esfahani MH, Megraw TL and Ghaedi K: New insights into the
cellular activities of Fndc5/Irisin and its signaling pathways.
Cell Biosci. 10:512020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong XQ, Geng Z, Zhou B, Zhang F, Han Y,
Zhou YB, Wang JJ, Gao XY, Chen Q, Li YH, et al: FNDC5 attenuates
adipose tissue inflammation and insulin resistance via
AMPK-mediated macrophage polarization in obesity. Metabolism.
83:31–41. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mazur-Bialy AI, Pocheć E and Zarawski M:
Anti-inflammatory properties of irisin, mediator of physical
activity, are connected with TLR4/MyD88 signaling pathway
activation. Int J Mol Sci. 18:7012017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shao L, Meng D, Yang F, Song H and Tang D:
Irisin-mediated protective effect on LPS-induced acute lung injury
via suppressing inflammation and apoptosis of alveolar epithelial
cells. Biochem Biophys Res Commun. 487:194–200. 2017. View Article : Google Scholar
|
|
24
|
National Research Council (NRC): Institute
for laboratory animal research: Guide for the care and use of
laboratory animals. 8th edition. National Academies Press;
Washington, DC: 2011
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Griffin RJ, Williams BW, Bischof JC, Olin
M, Johnson GL and Lee BW: Use of a fluorescently labeled
poly-caspase inhibitor for in vivo detection of apoptosis related
to vascular-targeting agent arsenic trioxide for cancer therapy.
Technol Cancer Res Treat. 6:651–654. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cursio R, Colosetti P, Auberger P and
Gugenheim J: Liver apoptosis following normothermic
ischemia-reperfusion: In vivo evaluation of caspase activity by
FLIVO assay in rats. Transplant Proc. 40:2038–2041. 2008.
View Article : Google Scholar
|
|
28
|
Gill SE, Rohan M and Mehta S: Role of
pulmonary microvascular endothelial cell apoptosis in murine
sepsis-induced lung injury in vivo. Respir Res. 16:1092015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang L, Lei W, Zhang S and Yao L: MCC950,
a NLRP3 inhibitor, ameliorates lipopolysaccharide-induced lung
inflammation in mice. Bioorg Med Chem. 30:1159542021. View Article : Google Scholar
|
|
30
|
Yokoyama S, Cai Y, Murata M, Tomita T,
Yoneda M, Xu L, Pilon AL, Cachau RE and Kimura S: A novel pathway
of LPS uptake through syndecan-1 leading to pyroptotic cell death.
Elife. 7:e378542018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, He WT, Hu L, Li J, Fang Y, Wang X,
Xu X, Wang Z, Huang K and Han J: Pyroptosis is driven by
non-selective gasdermin-D pore and its morphology is different from
MLKL channel-mediated necroptosis. Cell Res. 26:1007–1020. 2016.
View Article : Google Scholar :
|
|
32
|
Fink SL and Cookson BT:
Caspase-1-dependent pore formation during pyroptosis leads to
osmotic lysis of infected host macrophages. Cell Microbiol.
8:1812–1825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv H, Liu Q, Wen Z, Feng H, Deng X and Ci
X: Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute
lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox
Biol. 12:311–324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hughes KT and Beasley MB: Pulmonary
manifestations of acute lung injury: More than just diffuse
alveolar damage. Arch Pathol Lab Med. 141:916–922. 2017. View Article : Google Scholar
|
|
35
|
Nahrendorf M and Swirski FK: Abandoning
M1/M2 for a network model of macrophage function. Circ Res.
119:414–417. 2016. View Article : Google Scholar :
|
|
36
|
Jiang R, Xu J, Zhang Y, Zhu X, Liu J and
Tan Y: Ligustrazine alleviate acute lung injury through suppressing
pyroptosis and apoptosis of alveolar macrophages. Front Pharmacol.
12:6805122021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Benoit M, Desnues B and Mege JL:
Macrophage polarization in bacterial infections. J Immunol.
181:3733–3739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Man SM, Karki R and Kanneganti TD:
Molecular mechanisms and functions of pyroptosis, inflammatory
caspases and inflammasomes in infectious diseases. Immunol Rev.
277:61–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Frank D and Vince JE: Pyroptosis versus
necroptosis: Similarities, differences, and crosstalk. Cell Death
Differ. 26:99–114. 2019. View Article : Google Scholar
|
|
40
|
Sun X, Sun J, Dong B, Huang G, Zhang L,
Zhou W, Lv J, Zhang X, Liu M, Xu L, et al: Noninvasive temperature
monitoring for dual-modal tumor therapy based on lanthanide-doped
up-conversion nanocomposites. Biomaterials. 201:42–52. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He WT, Wan H, Hu L, Chen P, Wang X, Huang
Z, Yang ZH, Zhong CQ and Han J: Gasdermin D is an executor of
pyroptosis and required for interleukin-1β secretion. Cell Res.
25:1285–1298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Luo D, Dai W, Feng X, Ding C, Shao Q, Xiao
R, Zhao N, Peng W, Yang Y, Cui Y, et al: Suppression of lncRNA
NLRP3 inhibits NLRP3-triggered inflammatory responses in early
acute lung injury. Cell Death Dis. 12:8982021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Coll RC, Robertson AA, Chae JJ, Higgins
SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG,
Stutz A, et al: A small-molecule inhibitor of the NLRP3
inflammasome for the treatment of inflammatory diseases. Nat Med.
21:248–255. 2015. View Article : Google Scholar : PubMed/NCBI
|