Introduction
Osteoporosis is a systemic bone disease
characterized by a low bone mass and the deterioration of bone
tissue microstructure (1). With
the aging population, osteoporosis has become one of the major
public health concerns worldwide, and osteoporotic fractures have
become a main cause of mortality and disability among the elderly
(2,3). The maintenance of bone homeostasis
requires the balance between bone resorption and bone formation.
After the loss of estrogen, this balance is broken, resulting in
bone loss, which is the pathogenic mechanism of osteoporosis
(4,5). This imbalance may be due to the
anti-apoptosis of osteoclasts and the pro-apoptosis of osteoblasts
caused by oxidative stress, which is induced by estrogen
deficiency, accelerating the rate of bone remodeling (6). Recent research has demonstrated that
oxidative stress plays a critical role in the progression of
osteoporosis (7). It has been
shown that estrogen is an anti-oxidant, and the increase in
oxidation-related biomarkers in post-menopausal women has proven
that post-menopausal women with osteoporosis are in a state of
oxidative stress (8). Reactive
oxygen species (ROS) were previously considered to be exclusively
toxic, causing oxidative stress and tissue damage (9). However, it has been proven that a
low level of ROS has a signal transduction effect (10). Excessive and abnormal levels of
ROS cause molecular damage and initiate the apoptotic process,
which is produced mainly due to mitochondrial dysfunction (11). Therefore, the study of
osteoporotic drugs targeting mitochondrial dysfunction induced by
oxidative stress has promising application prospects.
Over the past 30 years, a range of drugs for the
treatment of post-menopausal osteoporosis have been developed and
shown to be effective (12). At
present, the first-line treatment drugs include bisphosphonates,
denosumab, raloxifene and teriparatide (13). However, the rare, yet severe
side-effects of bisphosphonates and denosumab are atypical femoral
fractures and osteonecrosis of the jaw, which limit their
application (14). The long-term
use of raloxifene increases the incidence of thromboembolism, and
teriparatide can be used for up to 2 years due to its
cerebrovascular and central nervous side-effects (3,15).
New drug therapies are still being explored (16). Metformin has been used for the
treatment of type 2 diabetes for >60 years and is characterized
by its safety and low cost (17).
At present, the application of metformin in other fields is also
being explored, such as for cardiovascular disease, tumors and even
aging, with its application involving different mechanisms roles
through various signaling pathways (18). It has been demonstrated that
metformin exerts a therapeutic effect on osteoporosis induced by
diabetes; however, this has focused on the effects of metformin on
bone metabolism in a high glucose state (19). However, to date, at least to the
best of our knowledge, there are few studies available on whether
metformin can reverse post-menopausal osteoporosis induced by
oxidative stress (20). At the
same time, it is unclear whether metformin can improve
pre-osteoblast oxidative stress by reversing mitochondrial
dysfunction.
In the present study, a pre-osteoblast model of
oxidative stress was constructed to explore the therapeutic effects
and mechanisms of action of metformin in post-menopausal
osteoporosis. A previous study demonstrated that glycogen synthase
kinase (GSK)-3β improved cardiac toxicity caused by ROS and calcium
ions (Ca2+) by regulating the mitochondrial permeability
transition pore (mPTP) (21).
Under conditions of oxidative stress, GSK-3β activates and opens
the mPTP, thus inducing calcium overload. It was hypothesized that
metformin inactivates GSK-3β via EGFR, which decreases the
intracellular Ca2+ concentration to attenuate
pre-osteoblast apoptosis induced by oxidative stress. To the best
of our knowledge, through bioinformatics analyses and experimental
verification, the present study, for the first time, examined the
entire treatment axis of metformin in post-menopausal osteoporosis.
The findings presented herein demonstrate that metformin has
potential for use as a safe and effective drug for the treatment of
osteoporosis.
Materials and methods
Reagents, cells and cell culture
The MC3T3-E1 cells (GNM15), mouse pre-osteoblast
precursors, were obtained The Cell Bank of Type Culture Collection
of The Chinese Academy of Sciences. Metformin was obtained from
Dalian Meilun Biotechnology Co., Ltd. Primary antibodies against
caspase-3 (cat. no. ab184787), Bcl-2 (cat. no. ab182858), BAX (cat.
no. ab32503), EGFR (cat. no. ab52894), phosphorylated (p-)EGFR
(cat. no. ab52894) and horseradish peroxidase-conjugated
anti-rabbit secondary antibodies (cat. no. ab288151) were obtained
from Abcam. Primary antibodies against GSK-3β (cat. no. 12456),
p-GSK-3β (cat. no. 5558), cleaved caspase-3 (cat. no. 9661), and
β-actin (cat. no. 4970) were obtained from Cell Signaling
Technology, Inc.
The MC3T3-E1 cells were maintained in α-MEM
(HyClone; USA) supplemented with 10% fetal bovine serum (HyClone;
Cytiva), 100 U/ml penicillin and 100 mg/ml streptomycin (Corning,
Inc.). The cells were cultured in a humidified incubator with 5%
CO2 at 37°C.
Oxidative damage was induced with 0.2 mM hydrogen
peroxide (H2O2; 7722-84-1, MilliporeSigma)
added to the medium. Following 6 h of treatment, metformin at a
concentration of 0.2 mM was added to achieve the optimal effects on
the reversal of apoptosis.
Cell viability assay
A Cell Counting Kit-8 (CCK-8) colorimetric assay
(APExBIO Technology LLC) was applied to measure cell proliferation
and viability. MC3T3-E1 Subclone 14 growth medium (Procell Life
Science & Technology Co., Ltd.) containing 89% MEM, 10% FBS, 1%
P/S solution. The MC3T3-E1 cells were plated in 100 µl of
growth medium/well in 96-well plates (Corning, Inc.) at
5×103 cells/well. The cells were exposed to 0.2 mM
H2O2 for 6 h and then treated with metformin
at 0.2 mM for 48 h at 37°C. CCK-8 reagent (10 µl/well) was
then added to the supernatant followed by incubation for 1 h at
37°C. The optical density (OD) representing cell viability was
measured at 450 nm using an absorbance microplate reader (BioTek
Instruments, Inc.).
Apoptosis assay
The apoptosis assay of the MC3T3-E1 cells was
performed using a FITC Annexin V Apoptosis Detection kit I (BD
Biosciences). A total of 2×105 cells were harvested
following treatment with metformin, washed with pre-cooled PBS and
centrifuged at 200 × g for 5 min at 4°C. The cells were resuspended
in binding buffer and incubated at room temperature with 5
µl of Annexin V-FITC for 15 min and then with 5 µl of
PI for 15 min. Finally, the sample was detected using a flow
cytometer, CytoFLEX, (Beckman Coulter, Inc.) and analyzed using
CytExpert 2.3 software (Beckman Coulter, Inc.).
Fluorescence inverted microscopy and flow
cytometry
JC-1, Fluo-4, DCFH-DA and MitoSOX Red staining (as
described below) was examined using a fluorescence inverted
microscope (Nikon Eclipse Ti, Nikon Corporation) and a FACScan flow
cytometer (Beckman Coulter, Inc.), respectively. A fluorescence
inverted microscope was used to observe the intracellular
localization and relative qualitative analysis of the markers.
Subsequently, flow cytometry was used to detect the average
fluorescence intensity of the fluorescence channel where the marker
was located via absolute quantitative analysis.
Measurement of mitochondrial membrane
potential
The mitochondrial membrane potential of the MC3T3-E1
cells was examined using a mitochondrial membrane potential assay
kit with JC-1 (Beyotime Institute of Biotechnology). A total of
2×105 cells were mixed well with 1 ml of JC-1 dye
working solution and then incubated at 37°C for 20 min. After
washing twice with JC-1 dyeing buffer, the sample was detected
using a FACScan flow cytometer. The red fluorescence at 590 nm was
accepted via the PE channel, while the green fluorescence at 520 nm
was accepted via the FITC channel. The red/green fluorescence ratio
was used to represent the mitochondrial membrane potential level.
The decrease in the red/green ratio indicated the openness of the
mPTP.
Detection of the intracellular calcium
concentration
The concentration of intracellular Ca2+
in the MC3T3-E1 cells was examined using Fluo-4 AM (Beyotime
Institute of Biotechnology). The cells inoculated in 24-well plates
were washed three times with PBS to ensure that the medium was
completely removed. Subsequently, the cells were completely covered
with 300 µl Fluo-4 AM working solution and incubated at 37°C
for 30 min. After washing twice with PBS, the cells were incubated
for a further 30 min to ensure that the Fluo-4 AM in the cells was
completely transformed into Fluo-4 with fluorescence. The
fluorescence intensity of intracellular Fluo-4 was measured, which
represented the concentration of Ca2+. Bay K8644 (Dalian
Meilun Biology Technology Co., Ltd.), as a highly selective L-type
calcium channel agonist, was used to increase cytoplasmic calcium
concentration. Bay K8644 at a concentration of 10 µM was
added to the MC3T3-E1 cells with metformin and incubated for 48 h
at 37°C.
Detection of ROS
The MC3T3-E1 cells were inoculated in 24-well plates
and examined using a ROS assay kit (Beyotime Institute of
Biotechnology) following exposure to H2O2 and
treatment with metformin. DCFH-DA was diluted with serum-free
culture medium at a 1:1,000 ratio to prepare the working solution.
The cells to be tested were covered with diluted DCFH-DA and
incubated in a cell incubator at 37°C for 30 min. The fluorescence
intensity of DCFH-DA was measured to represent the content of
intracellular ROS.
Detection of mitochondrial
superoxide
Superoxide in the mitochondria was detected using
the MitoSOX Red mitochondrial superoxide indicator (Shanghai Yeasen
Biotechnology Co., Ltd.). The dry powder of the probe was diluted
with DMSO as the storage solution, which was diluted 1,000-fold
with PBS to yield a 5-µm MitoSOX Red working solution. The
MC3T3-E1 cells were inoculated in a 24-well plate and washed with
PBS at 37°C. Subsequently, 300 µl of working solution were
added to each well and incubated at 37°C away from light for 10
min. The fluorescence intensity of MitoSOX Red was measured to
represent the content of superoxide in the mitochondria.
Cellular immunochemical analysis
The MC3T3-E1 cells were first treated with 0.1%
Triton X-100 (Beyotime Institute of Biotechnology) to increase the
cell membrane permeability and then fixed with 4% paraformaldehyde
at room temperature for 30 min. The fixed cell climbing tablets
were covered by sufficient primary antibody of GSK-3β diluted at
1/200 and incubated at 4°C away from light overnight. Following
thorough cleaning with PBST, the cells were incubated in
fluorescently labeled secondary antibody (Alexa Fluor®
594, cat. no. ab150080, Abcam) diluted at 1/200 for 1 h in the
dark. Finally, the cells were treated with DAPI (Beyotime Institute
of Biotechnology) to stain the nuclei for 5 min at room temperature
and observed under a fluorescence microscope (Nikon Eclipse Ti,
Nikon Corporation).
Enrichment analysis of Gene Ontology (GO)
and Kyoto Encyclopedia of Genes and Genes (KEGG) pathways
The present study analyzed the transcriptome data
(GSE198254) of metformin reversing osteoporosis in pre-osteoblasts
(22). Gene Set Enrichment
Analysis (GSEA) of differentially expressed genes (DEGs) was
carried out using clusterProfiler R statistical software (R, ×64,
4.1.3). The GO database was used to annotate genes from three
aspects: Biological process (BP), molecular function (MF) and
cellular component (CC). At the same time, the KEGG database was
used to analyze the top enrichment pathway involved in the effects
of metformin against osteoporosis induced by oxidative stress. The
ggplot2 package (http://ggplot2.tidyverse.org) was used to visualize
the results.
Western blot analysis
The cells to be tested were treated with
radioimmunoprecipitation assay (RIPA) buffer (Beyotime Institute of
Biotechnology) on ice for 30 min and then lysed under ultrasound to
obtain complete lysate. The pyrolysis product was centrifuged at
4°C for 10 min at 10,000 × g, and the supernatant was then
collected. A BCA assay kit (Beyotime Institute of Biotechnology)
was used to detect the protein concentration. Electrophoresis was
performed using 10% SDS-PAGE and the proteins were then transferred
onto polyvinylidene difluoride membranes (PVDF). After blocking
with 5% BSA on a shaking table at room temperature for 1 h, the
membranes were incubated with the primary antibodies overnight.
After cleaning with TBST, the membranes were incubated with the
secondary antibody for 1 h at 4°C. Primary antibodies against
GSK-3β, p-GSK-3β, cleaved caspase-3, caspase-3, Bcl-2, BAX, EGFR,
p-EGFR, β-actin were diluted at 1/3,000. Horseradish
peroxidase-conjugated anti-rabbit secondary antibodies was diluted
at 1/2,000. β-actin (43 kDa) was used as the standard for protein
levels. The protein bands that were fully immersed in the enhanced
chemiluminescence (ECL) were visualized using the chemiluminescence
imaging system (Analytik Jena Ag). Finally, the quantified optical
density was calculated using ImageJ v1.8.0 software (National
Institutes of Health).
siRNA transfection
GSK-3β-small interfering RNAs (siRNAs) were
purchased from Synbio-tech Suzhou Co., Ltd. Three siRNAs were
transfected at the same time, and the content of GSK-3β was
detected using western blot analysis to reflect the transfection
efficiency. MC3T3-E1 cells transfected with negative siRNAs was as
a negative control. GSK-3β-si-1 was selected for use in subsequent
experiments as the content of GSK-3β was the lowest. The MC3T3-E1
cells were inoculated into six-well plates at a cell density of 50%
when transfected 24 h later. siRNA at 50 nM and Lipofectamine™ 3000
reagent (Thermo Fisher Scientific, Inc.) were prepared in
antibiotic-free Opti-MEM (Thermo Fisher Scientific, Inc.) and then
mixed at room temperature for 20 min. Finally, the cells were
treated with siRNA transfection reagent for 6 h, then change fresh
medium for incubation for 48 h in the 37°C incubator. The siRNA
sequences are listed in Table
I.
 | Table IRNA interference sequences used for
transfection. |
Table I
RNA interference sequences used for
transfection.
| Name | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| GSK-3β-si-1 |
GAAAGUUAGCAGAGAUAAATT |
UUUAUCUCUGCUAACUUUUCTT |
| GSK-3β-si-2 |
AGAAAGUUCUACAGGACAATT |
UUGUCCUGUAGAACUUUCUTT |
| GSK-3β-si-3 C |
AAAUUAUACAGAAUUCAATT |
UUGAAUUCUGUAUAAUUUGTT |
Statistical analysis
An unpaired Student's t-test was used to examine
relative protein expression in cells transfected with siRNA against
GSK-3β and one-way ANOVA followed by Tukey's post hoc test was used
to determine significant differences between multiple treatment
groups. P<0.05 was considered to indicate a statistically
significant difference. All experiments were repeated three times
and analyzed using SPSS 19.0 software (IBM Corp).
Results
Metformin reverses pre-osteoblast
mitochondrial dysfunction by attenuating mPTP opening
Based on a previous study by the authors, 0.2 mM was
determined to be the optimal concentration of
H2O2 to induce oxidative stress in
pre-osteoblasts, which reached the maximum apoptotic rate (22). In addition, 0.2 mM metformin was
shown to have the strongest ability to reverse oxidative apoptosis
(22). The present study first
examined the mechanisms by which metformin prevents pre-osteoblast
apoptosis. The transcriptome data (GSE198254) of ovariectomized
(OVX) mice treated with and without metformin were analyzed and
GSEA was performed using the R software package (clusterProfiler)
(23). Functional enrichment (GO)
analysis revealed that in the CC category, DEGs were mainly
enriched in mitochondrial components (mitochondrial inner membrane
and mitochondrial protein-containing complex). In the MF category,
DEGs were closely related to transcriptional regulation
(transcription coregulator activity and catalytic activity, acting
on RNA). In the BP category, DEGs were mainly enriched in protein
modification (negative regulation of protein modification process
and regulation of protein stability) (Fig. 1A). To further examine the effects
and pathways through which metformin affects mitochondrial
function, the openness of the mPTP was examined. With a decrease in
mitochondrial membrane potential, the JC-1 monomer enters the
cytoplasm through the open mPTP, while it exists in the form of a
polymer within healthy mitochondria (24). JC-1 was observed in the cytoplasm
using fluorescence microscopy and it was found that the ratio of
red/green fluorescence was decreased in MC3T3-E1 cells exposed to
H2O2. Similarly, this effect was attenuated
by metformin, suggesting that metformin prevented mitochondrial
dysfunction by controlling mPTP opening (Fig. 1B and C). During this process, the
level of intracellular ROS was consistent with the opening of the
mPTP (Fig. 1D and E).
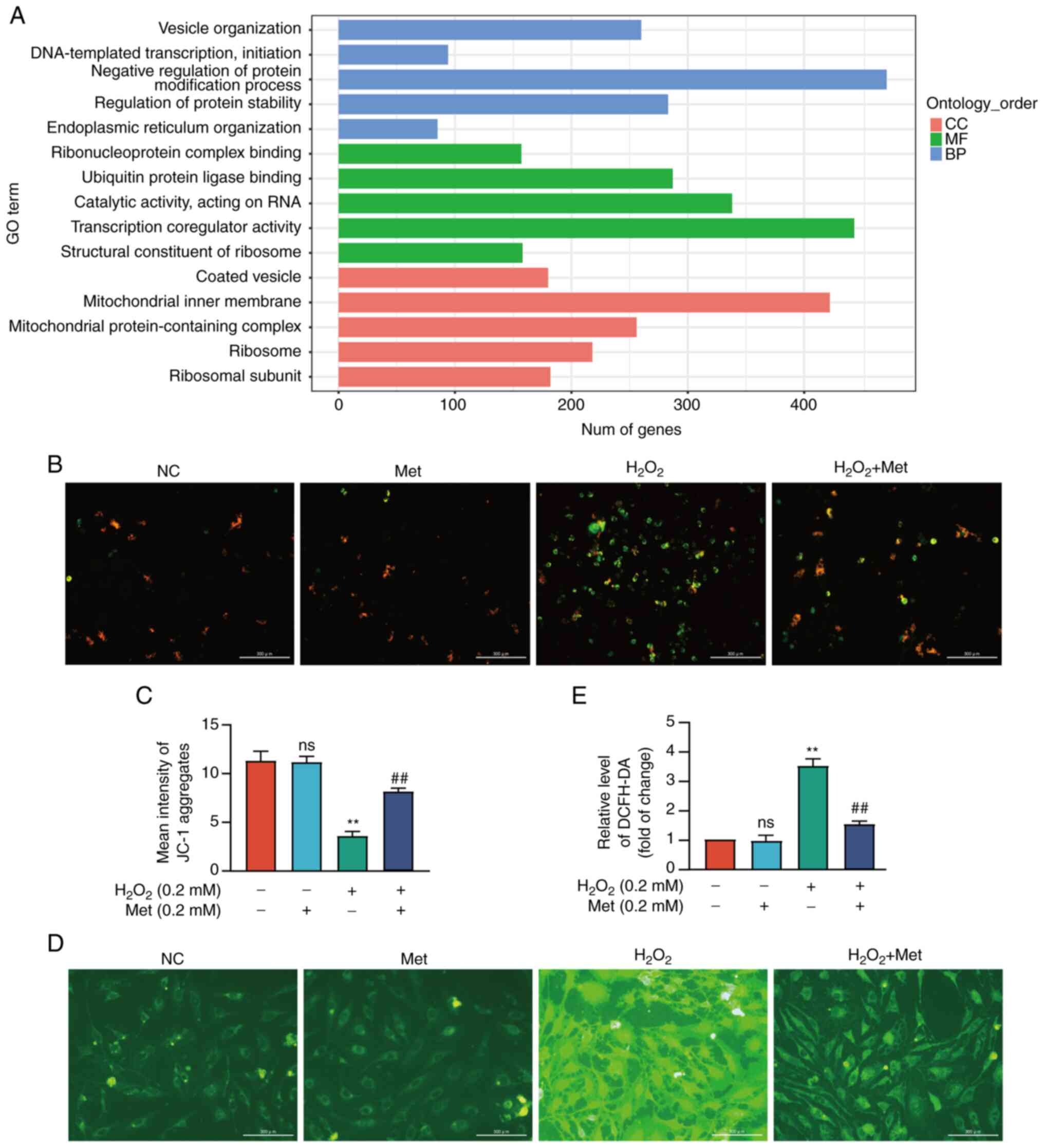 | Figure 1Metformin reverses pre-osteoblast
mitochondrial apoptosis by attenuating mitochondrial permeability
transition pore opening. (A) Metformin in the process of
cytological components, molecular biological functions and
biological processes through GO analysis. (B) The number of JC-1
monomers and JC-1 aggregates in MC3T3-E1 cells with or without
metformin treatment was observed. (C) The fluorescence intensity of
JC-1 aggregates was detected using a full-wavelength
multifunctional microplate reader. The fluorescence intensity
decreased in the H2O2 group, and metformin
group reversed this process. (D) The intracellular ROS level was
observed using a fluorescence electron microscope. (E) The average
fluorescence intensity was detected using flow cytometry. ROS
generation increased in H2O2 group and
decreased in the metformin group. The experiment was conducted
three times. The data are expressed as the mean ± SD.
**P<0.01, compared with the control cells;
##P<0.01, compared with the
H2O2 group. ns, not significant; GO, Gene
Ontology; CC, cellular component; MF, molecular function; BP,
biological process; ROS, reactive oxygen species; Met, metformin;
H2O2, hydrogen peroxide. |
Metformin reverses pre-osteoblast
mitochondrial apoptosis by reducing the cytoplasmic calcium
concentration through the mPTP
The present study further examined whether metformin
regulates apoptosis through calcium. It was found that in the
H2O2 group, the concentration of
Ca+ increased significantly, and metformin reversed this
effect. Following the addition of the calcium channel agonist, Bay
K8644, the calcium concentration increased compared with that in
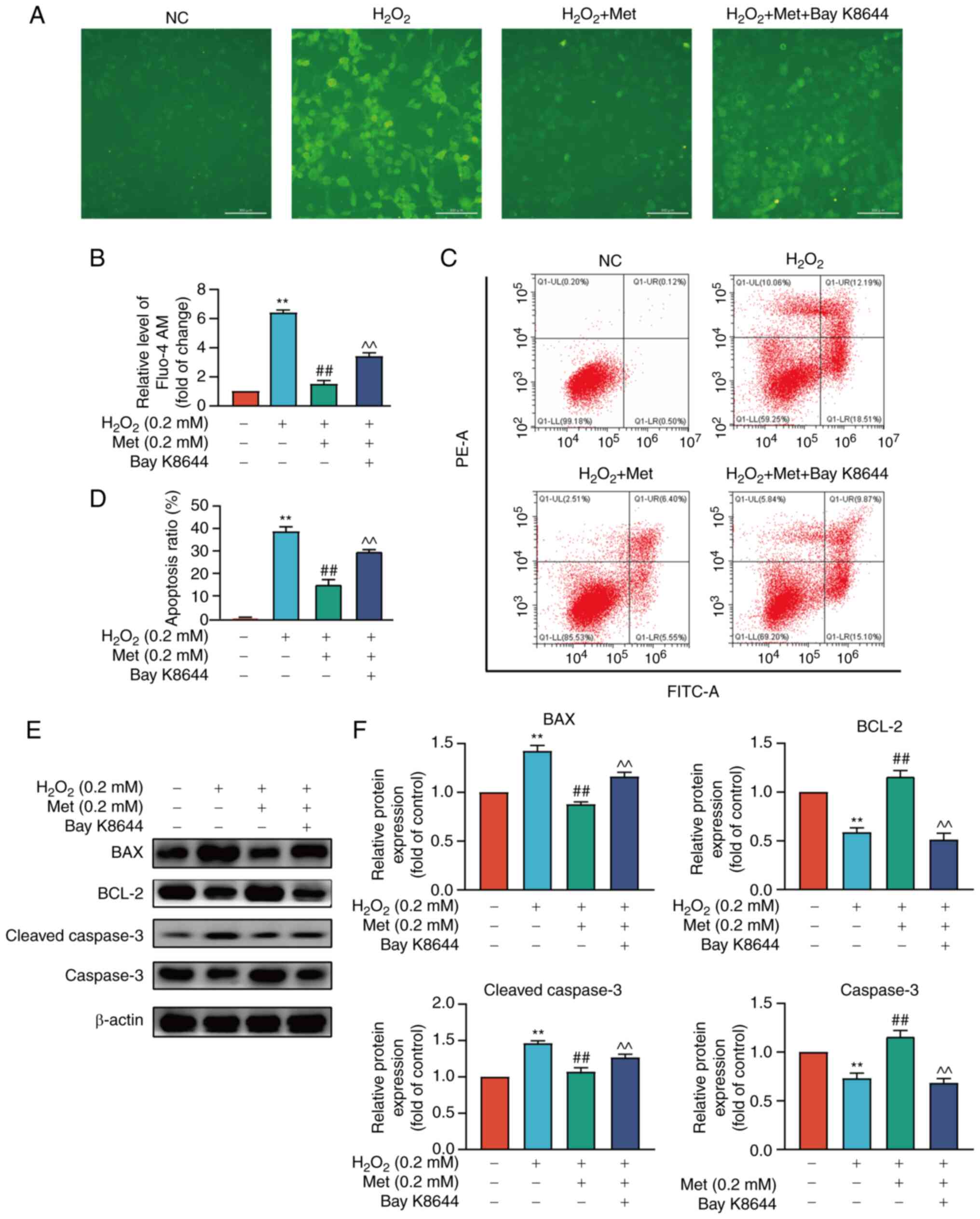
the H2O2 + metformin group (Fig. 2A and B). Furthermore, the
apoptotic rate of MC3T3-E1 cells was measured during this process.
In the H2O2 group, the apoptotic rate
increased, metformin reversed pre-osteoblast apoptosis, and Bay
K8644 attenuated the protective effects of metformin (Fig. 2C and D). The results of western
blot analysis also revealed that the levels of mitochondrial
apoptosis-related proteins were altered: The levels of BAX and
cleaved caspase-3 were increased, while those of BCL-2 were
decreased in the cell model of oxidative stress induced by
H2O2. These changes were reversed by
metformin and Bay K8644 attenuated the effects of metformin
(Fig. 2E and F).
Metformin attenuates pre-osteoblast
apoptosis induced by oxidative stress through GSK-3β
To examine the mechanisms through which metformin
ameliorates mitochondrial dysfunction and attenuates apoptosis,
GSK-3β was selected from the genes involved in the negative
regulation of protein modification, as determined using GO
enrichment analysis; GSK-3β is involved in regulating the mPTP in
mitochondria (25). GSEA revealed
that metformin promoted the negative regulation of protein
modification (Fig. S1). The
expression of GSK-3β was observed using fluorescence inverted
microscopy. Contrary what was initially expected, metformin and
oxidative stress did not markedly increase the levels of GSK-3β.
However, it was found that the fluorescence in the control group
was evenly distributed, while there was localized fluorescence in
the H2O2 and metformin groups (Fig. 3A). It was hypothesized that GSK-3β
may accumulate in organelles under conditions of oxidative stress.
To further clarify the role of GSK-3β in the metformin-mediated
reversal of pre-osteoblast apoptosis induced by oxidative stress,
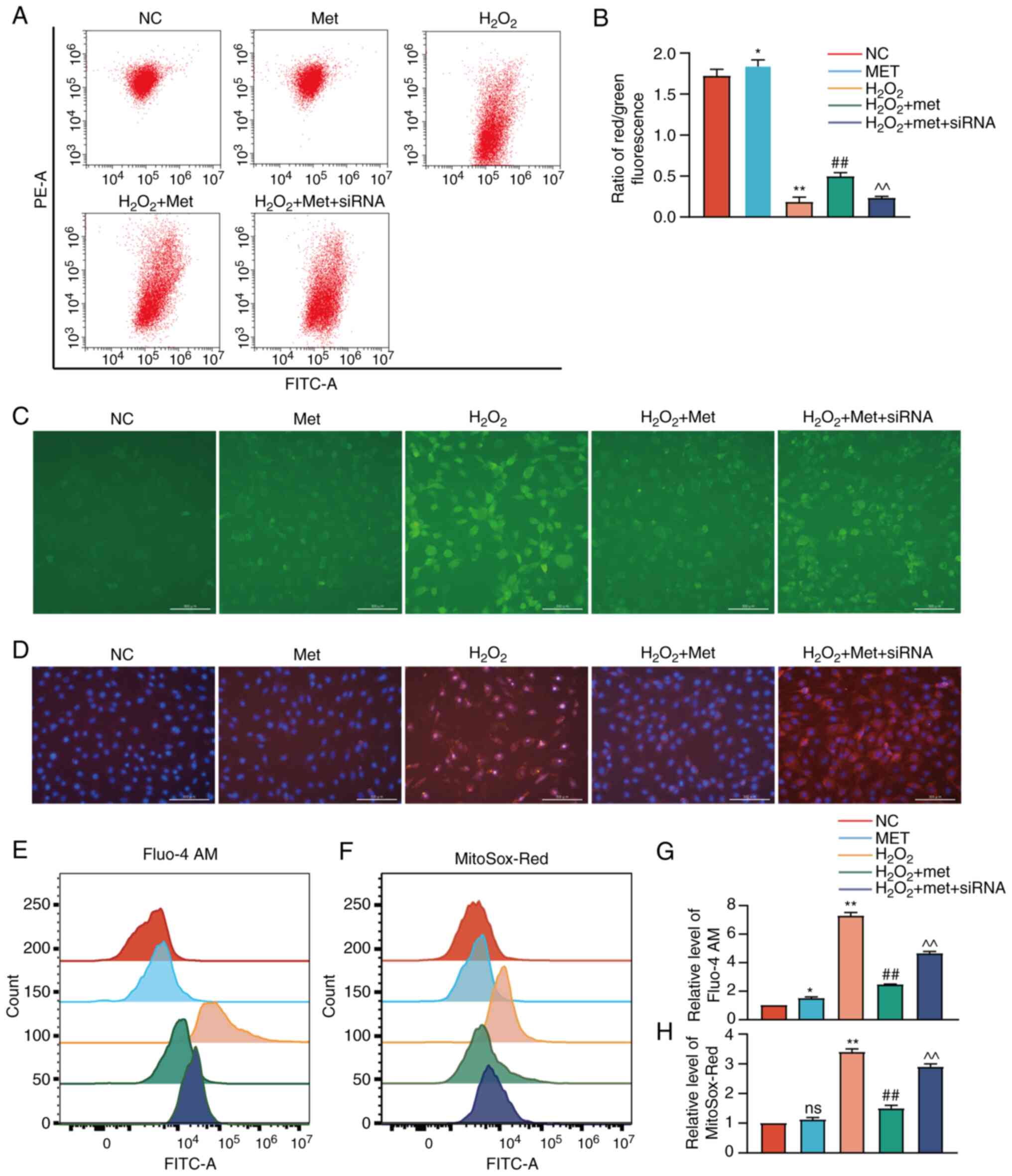
GSK-3β was knocked down using siRNA (Fig. 3B). It was found that the knockdown
of GSK-3β prevented metformin from reversing apoptosis (Fig. 3C). The levels of ROS in the
cytoplasm were also measured (Fig. 3D
and E), which confirmed the reversal of oxidative stress by
metformin and the loss of the antioxidant effects following the
knockdown of GSK-3β. Finally, the expression of mitochondrial
apoptosis marker proteins was detected using western blot analysis
(Fig. 3F). The results revealed
that the BAX and cleaved caspase-3 expression levels were higher in
the metformin treatment group following GSK-3β knockdown than in
the group treated with metformin and not subjected to GSK-3β
knockdown, while BCL-2 expression was lower. These results thus
revealed that GSK-3β was involved in the anti-apoptotic effects of
metformin by improving mitochondrial dysfunction, although this
effect was not related to the expression of GSK-3β. It was
hypothesized that metformin may protect pre-osteoblast mitochondria
and may prevent apoptosis by regulating the activity of GSK-3β.
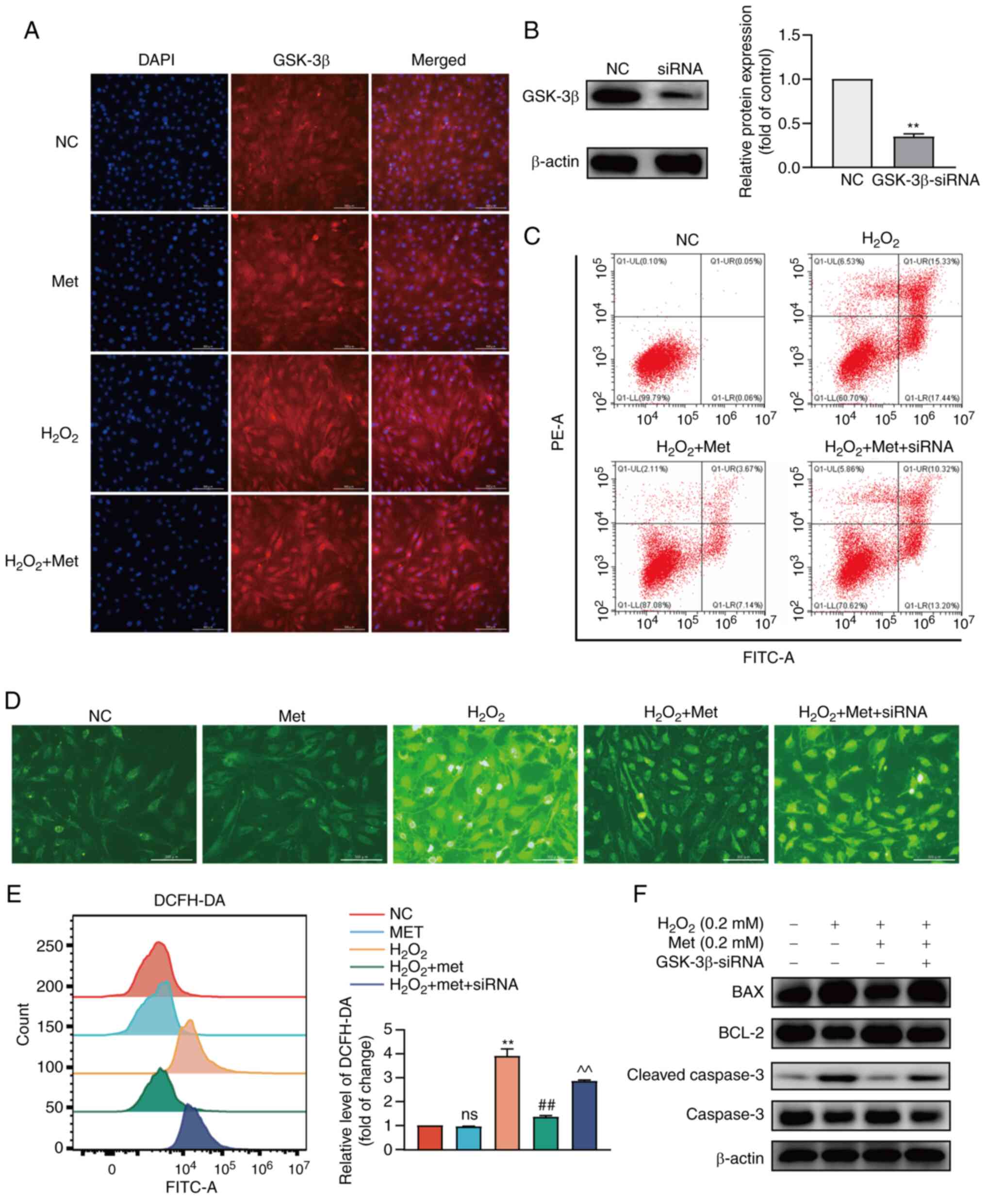 | Figure 3Metformin improves pre-osteoblast
apoptosis induced by oxidative stress through GSK-3β. (A) The
localization and expression of GSK-3β was observed in MC3T3-E1
cells and GSK-3β was in a more aggregated state. (B) The silencing
efficiency of GSK-3β was detected at the protein level. (C) The
apoptotic rate after the silencing of GSK-3β was detected. The
apoptotic rate of the H2O2 group was 32.77%,
that of the H2O2 + metformin group was
10.81%, and that of the H2O2 + metformin +
siRNA group was 23.52%. (D) The level of ROS in the cytoplasm was
detected before and after treatment with metformin and
siRNA-GSK-3β. (E) The fluorescence intensity of ROS was detected
using flow cytometry. (F) The expression of mitochondrial
apoptosis-related proteins after the silencing of GSK-3β was
detected again. The experiments were performed three times. Data
are presented as the mean ± SD. **P<0.01, compared
with the control cells; ##P<0.01, compared with the
H2O2 group; ^^P<0.01, compared
with the H2O2 + Met group. ns, not
significant; ROS, reactive oxygen species; Met, metformin;
H2O2, hydrogen peroxide; GSK-3β, glycogen
synthase kinase 3β. |
Metformin inhibits mPTP opening and
Ca2+-mediated mitochondrial dysfunction through
GSK-3β
Subsequently, the present study observed changes in
mitochondrial membrane potential to determine the openness of the
mPTP (Fig. 4A and B).
Furthermore, changes in the Ca2+ concentration in the
cytoplasm during this process were measured (Fig. 4C, E and G). The results revealed
that H2O2 induced oxidative stress, mPTP
opening and an increase in the Ca2+ concentration in
MC3T3-E1 cells; these effects were reversed by metformin. Following
the knockdown of GSK-3β, metformin could not reverse mPTP opening
or the increase in Ca2+ concentrations. Furthermore, the
level of superoxide in mitochondria was measured to determine
mitochondrial dysfunction (Fig. 4D, F
and H). It was found that H2O2 increased
the mitochondrial superoxide levels and metformin decreased the
superoxide levels; the superoxide levels increased following the
knockdown of GSK-3β. This finding was consistent with the
hypothesis that metformin inhibited mPTP opening, weakened the
cytoplasmic calcium influx, attenuated mitochondrial superoxide
damage to the mitochondria and reversed apoptosis via GSK-3β.
Metformin reverses pre-osteoblast
apoptosis induced by oxidative stress by phosphorylating GSK-3β via
the EGFR pathway
Finally, the present study examined whether the
regulation of GSK-3β by metformin was mediated by the level of
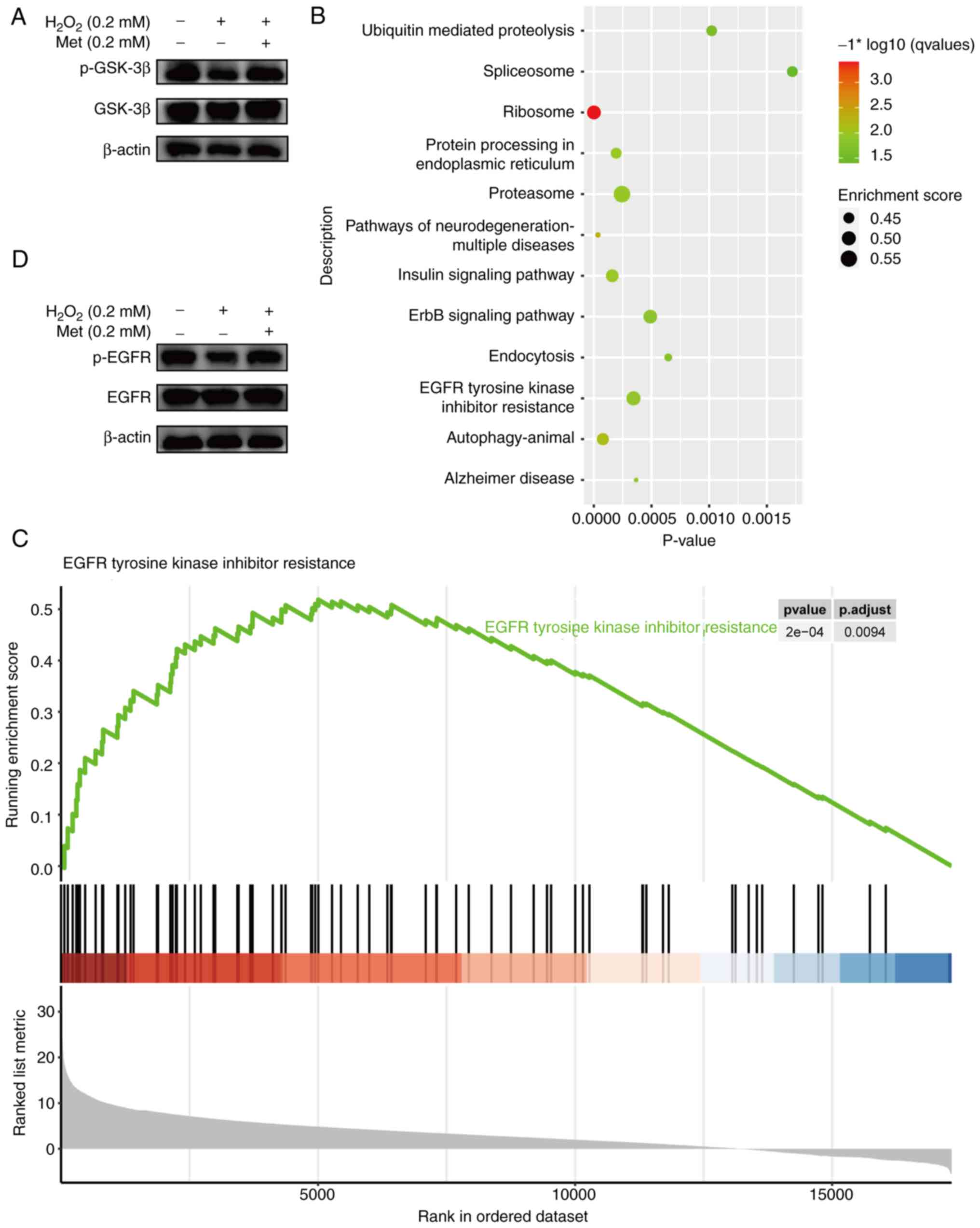
phosphorylation. The results of western blot analysis revealed that
metformin markedly increased GSK-3β phosphorylation, which was
consistent with the antioxidant level (Fig. 5A). The results of pathway
enrichment analysis (KEGG) revealed that the DEGs were closely
related to the ErbB signaling pathway and EGFR tyrosine kinase
inhibitor resistance (Fig. 5B).
It was found that the metformin group had an upregulated EGFR
tyrosine kinase inhibitor resistance pathway (Fig. 5C). ErbB is a member of the EGFR
family (26). Moreover, some
studies have shown that EGFR is involved in the regulation of
GSK-3β activity in tumors (27).
Therefore, it was hypothesized that metformin can affect
pre-osteoblast apoptosis through the EGFR pathway. The present
study measured the expression of EGFR and p-EGFR in each group
using western blot analysis. The results revealed that oxidative
stress inhibited the phosphorylation of EGFR, while metformin
reversed this change (Fig. 5D).
These results indicate that metformin affects the activity of
GSK-3β through the EGFR pathway and improves mitochondrial
dysfunction to prevent MC3T3-E1 apoptosis caused by oxidative
stress.
Discussion
Metformin is one of the most widely used oral
hypoglycemic drugs in clinic practice, and its safety has been
verified (28). At the same time,
metformin exhibits prominent anti-inflammatory potential and
regulates cell aging, apoptosis and macromolecular damage (29). Following menopause, the body is in
a state of high oxidation, which is the main factor leading to bone
aging and osteoporosis (30).
Through the construction of a pre-osteoblast oxidative stress
model, the present study confirmed that metformin improved the
oxidation state of MC3T3-E1 cells and reversed the occurrence of
apoptosis by reversing mitochondrial dysfunction, clarifying the
underlying mechanisms. Metformin has been shown to be beneficial in
attenuating the hallmarks of aging (31). Mitochondria are the key organelles
regulating cellular life and participate in the functional
regulation of energy metabolism, calcium signal transduction, cell
homeostasis and apoptosis (32).
An increasing number of studies have proven that apoptosis is
attributed to the internal killing mechanism of mitochondria and is
the key factor in osteoporosis (33). The present study further proposed
that the mPTP plays a major role. mPTP is a group of non-specific
pores formed by protein complexes between the inner and outer
membranes of the mitochondria, which maintains the proton barrier
and energy metabolism balance of the mitochondria (34). At the same time, calcium, as a
second messenger, plays a precise regulatory role in pre-osteoblast
apoptosis induced by oxidative stress (35). During oxidative stress, calcium
accumulates in the cytoplasmic matrix from the endoplasmic
reticulum or extracellular matrix, causing calcium overload
(36,37). The present study found that
calcium overload occurred during oxidative stress, while metformin
decreased the calcium levels. The addition of Bay K8644 increased
the level of intracellular calcium and attenuated the ability of
metformin to reverse pre-osteoblast apoptosis. It was suggested
that metformin regulates apoptosis by regulating mPTP opening and
controlling the Ca2+ influx in MC3T3-E1 cells.
The present study then investigated the mechanism
through which metformin regulates mPTP opening. GSK-3β is a Ser/Thr
kinase widely present in the cytoplasmic matrix, nucleus and
mitochondria. Originally, it was identified as a rate-limiting
enzyme for glycogen synthesis. Subsequently, it was found to affect
a variety of cellular processes, including embryonic development,
cell growth, differentiation, apoptosis and the stress response
(38,39). The mPTP is composed of the ion
channel VDAC of the mitochondrial outer membrane, the mitochondrial
inner membrane transporter (ANT) and cyclophilin D (CypD) (40). Among these, CypD is a functional
regulator of mPTP, which triggers the conformational change of mPTP
by combining with ANT (41).
Recent research has found that CypD is also a phosphorylation
substrate of GSK-3β. The phosphorylation of CypD increases binding
with ANT, which increases the sensitivity of mPTP to
Ca2+ and reduces the threshold of mPTP opening (42). The present study found that in
pre-osteoblasts exposed to H2O2, the total
amount of GSK-3β remained unaltered, although the level of p-GSK-3β
decreased, accompanied by the opening of the mPTP and increases in
Ca2+, intracellular ROS and mitochondrial superoxide. It
has been shown that GSK-3β is active in the physiological state and
inactivated following phosphorylation (43). This is consistent with the results
obtained herein, in that following treatment with metformin, GSK-3β
phosphorylation increased, and inactivated GSK-3β decreased the
binding ability of CypD to ANT, thereby inhibiting mPTP
opening.
Although GSK-3β plays a role in the intracellular
mitochondria, the mechanisms through which metformin communicates
extracellular signals to GSK-3β warrants further clarification
(44). The present study further
explored the enzyme-mediated pathway by which metformin regulates
GSK-3β from the extracellular space to the mitochondria. GSEA of GO
and KEGG was carried out on the transcriptome data of OVX and (OVX
+ Met) osteoporosis model mice. It was found that metformin was
related to the composition of the mitochondria (mitochondrial inner
membrane, mitochondrial protein-containing complex) in the
cytological components. GSK-3β was involved in upregulating the
negative expression of protein modification in biological
processes, which also suggest that metformin might have inhibited
the opening of mPTP by modifying CypD. It was found that DEGs were
highly enriched in the EGFR signaling pathway and upregulated the
phosphorylation of EGFR by tyrosine kinase in KEGG. EGFR signaling
is closely related to life processes, such as angiogenesis, tumor
invasion, metastasis and apoptosis (45). At the same time, it has been found
that EGFR-specific inhibitors reduce the number of bone marrow
mesenchymal progenitor cells and lead to cortical bone degeneration
(46). The present study further
confirmed that metformin significantly increased EGFR
phosphorylation in pre-osteoblasts exposed to oxidative stressed
and that EGFR was the membrane receptor of metformin on
pre-osteoblasts. It is suggested that metformin can regulate the
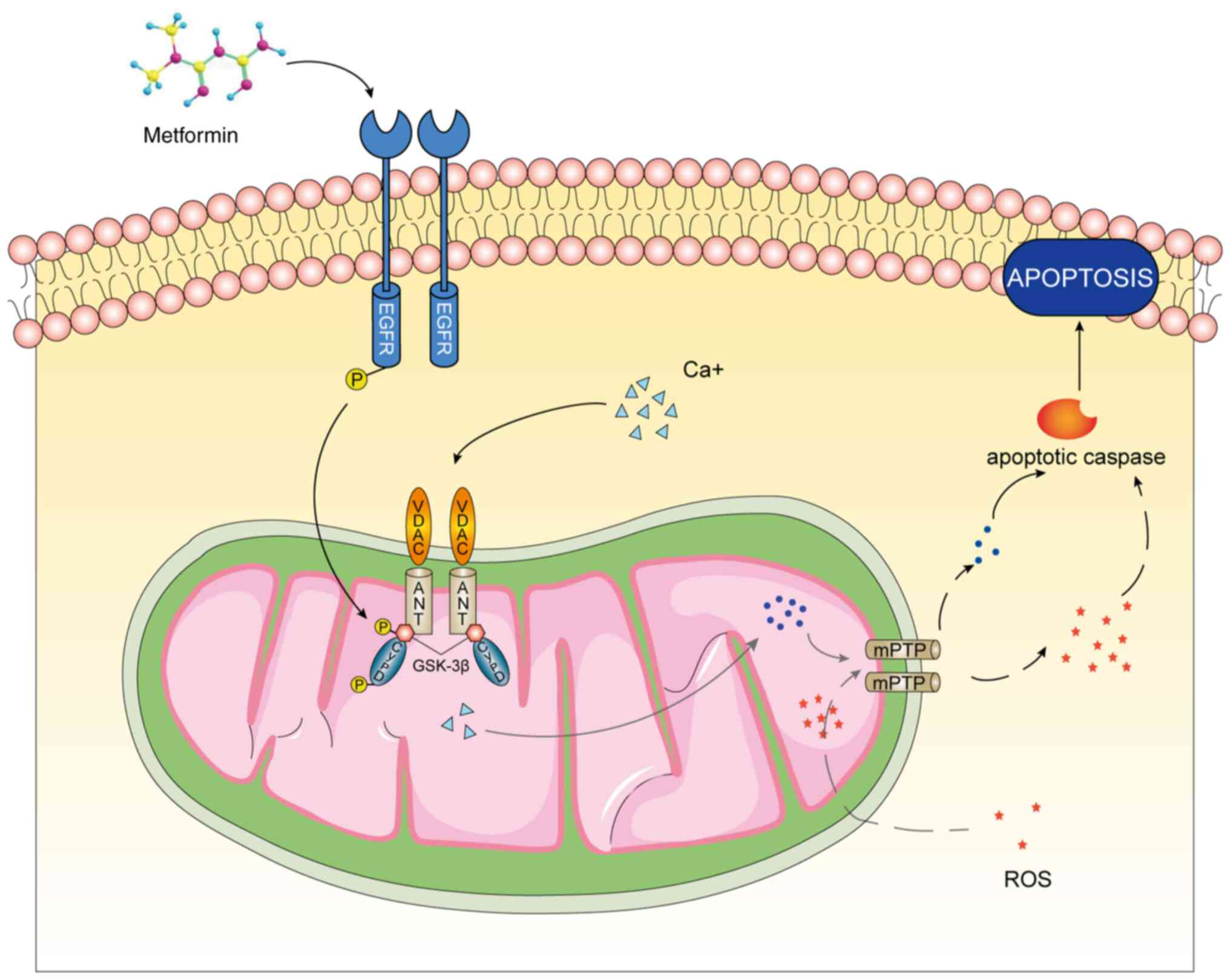
mPTP of MC3T3-E1 through the EGFR/GSK-3β/calcium axis and reverse
osteoporosis induced by oxidative stress (Fig. 6).
The present study first proposed the therapeutic
effects of GSK-3β on osteoporosis induced by oxidative stress by
regulating mPTP. At the same time, the EGFR/GSK-3β/calcium axis
plays a crucial role in this process. As the direct use of mPTP
inhibitors will produce various side-effects in patients (47), GSK-3β will be another target for
the treatment of osteoporosis in the future. However, a limitation
of the present study is that GSK-3β has a wide range of substrates
and targeting only the mitochondria can reduce side-effects and
improve the effects. It was found that GSK-3β in the cytoplasm
underwent mitochondrial translocation in response to oxidative
stress and metformin treatment. This may be related to the
N-terminal domain of GSK-3β, which may be used as a target sequence
of the mitochondria (48). In the
future, further research on the structure of GSK-3β is required to
realize the targeted inhibition of the mitochondria, which will be
the key to the treatment of diseases caused by oxidative
stress.
In conclusion, the present study demonstrated that
metformin reversed oxidative stress-induced mitochondrial
dysfunction in pre-osteoblasts via the EGFR/GSK-3β/calcium pathway.
Moreover, GSK-3β has potential for use as a key target for the
treatment of osteoporosis in the future. A limitation of the
present study however, was that although the concentration of 0.2
mM H2O2 was too high for clinical use,
MC3T3-E1 cells reached the maximum apoptotic rate at the
concentration. Herein, 0.2 mM was used to establish the
osteoporosis model at the cellular level.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FC was involved in data curation and in the study
methodology, as well as in the writing of the original draft. KY
was involved in data curation and in the study methodology, as well
as in the provision of software. SQ was involved in the
conceptualization of the study, and in the writing of the original
draft. JL was involved in the investigative aspects of the study
and in data curation. WJ was involved in the investigative aspects
of the study and in the provision of software. LT was involved in
the conceptualization of the study, as well as in data validation,
and in the writing, reviewing and editing of the manuscript. YZ was
involved in funding acquisition, in the provision of resources,
project administration, in the writing of the manuscript, and in
the conception and design of the study. All authors have approved
the final manuscript and confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81472044) and the Science and
Technology Planning Project in Shenyang (grant no.
20-205-2-045).
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
GSK-3β
|
glycogen synthase kinase 3β
|
|
mPTP
|
mitochondrial permeability transition
pore
|
References
|
1
|
Bandeira L and Bilezikian JP: Novel
therapies for postmenopausal osteoporosis. Endocrinol Metab Clin
North Am. 46:207–219. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kanis JA, Cooper C, Rizzoli R and
Reginster JY; Scientific Advisory Board of the European Society for
Clinical and Economic Aspects of Osteoporosis (ESCEO) and the
Committees of Scientific Advisors and National Societies of the
International Osteoporosis Foundation (IOF): European guidance for
the diagnosis and management of osteoporosis in postmenopausal
women. Osteoporos Int. 30:3–44. 2019. View Article : Google Scholar
|
|
3
|
Black DM and Rosen CJ: Clinical practice.
Postmenopausal osteoporosis. N Engl J Med. 374:254–262. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kespohl B, Schumertl T, Bertrand J, Lokau
J and Garbers C: The cytokine interleukin-11 crucially links bone
formation, remodeling and resorption. Cytokine Growth Factor Rev.
60:18–27. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Appelman-Dijkstra NM and Papapoulos SE:
Modulating bone resorption and bone formation in opposite
directions in the treatment of postmenopausal osteoporosis. Drugs.
75:1049–1058. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Li X, Liu D, Hamamura K, Wan Q, Na
S, Yokota H and Zhang P: eIF2α signaling regulates autophagy of
osteoblasts and the development of osteoclasts in OVX mice. Cell
Death Dis. 10:9212019. View Article : Google Scholar
|
|
7
|
Zhao F, Guo L, Wang X and Zhang Y:
Correlation of oxidative stress-related biomarkers with
postmenopausal osteoporosis: A systematic review and meta-analysis.
Arch Osteoporos. 16:42021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohamad NV, Ima-Nirwana S and Chin KY: Are
oxidative stress and inflammation mediators of bone loss due to
estrogen deficiency? A review of current evidence. Endocr Metab
Immune Disord Drug Targets. 20:1478–1487. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vatner SF, Zhang J, Oydanich M, Berkman T,
Naftalovich R and Vatner DE: Healthful aging mediated by inhibition
of oxidative stress. Ageing Res Rev. 64:1011942020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang FS, Wu RW, Chen YS, Ko JY, Jahr H and
Lian WS: Biophysical modulation of the mitochondrial metabolism and
redox in bone homeostasis and osteoporosis: How biophysics converts
into bioenergetics. Antioxidants (Basel). 10:13942021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khosla S and Hofbauer LC: Osteoporosis
treatment: Recent developments and ongoing challenges. Lancet
Diabetes Endocrinol. 5:898–907. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arceo-Mendoza RM and Camacho PM:
Postmenopausal osteoporosis: Latest guidelines. Endocrinol Metab
Clin North Am. 50:167–178. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ensrud KE: Bisphosphonates for
postmenopausal osteoporosis. JAMA. 325:962021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Buckley L, Guyatt G, Fink HA, Cannon M,
Grossman J, Hansen KE, Humphrey MB, Lane NE, Magrey M, Miller M, et
al: 2017 American college of rheumatology guideline for the
prevention and treatment of glucocorticoid-induced osteoporosis.
Arthritis Rheumatol. 69:1521–1537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reid IR: A broader strategy for
osteoporosis interventions. Nat Rev Endocrinol. 16:333–339. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lv Z and Guo Y: Metformin and its benefits
for various diseases. Front Endocrinol (Lausanne). 11:1912020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Foretz M, Guigas B, Bertrand L, Pollak M
and Viollet B: Metformin: From mechanisms of action to therapies.
Cell Metab. 20:953–966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tseng CH: Metformin use is associated with
a lower risk of osteoporosis/vertebral fracture in Taiwanese
patients with type 2 diabetes mellitus. Eur J Endocrinol.
184:299–310. 2021. View Article : Google Scholar
|
|
20
|
Jiating L, Buyun J and Yinchang Z: Role of
metformin on osteoblast differentiation in type 2 diabetes. Biomed
Res Int. 2019:92039342019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Sun Q, Jiang Q, Jiang Y, Zhang Y,
Cao J, Lu L, Li C, Wei P, Wang Q and Wang Y: Cryptotanshinone
ameliorates doxorubicin-induced cardiotoxicity by targeting
Akt-GSK-3β-mPTP pathway in vitro. Molecules. 26:14602021.
View Article : Google Scholar
|
|
22
|
Yang K, Cao F, Qiu S, Jiang W, Tao L and
Zhu Y: Metformin promotes differentiation and attenuates
H2O2-induced oxidative damage of osteoblasts via the
PI3K/AKT/Nrf2/HO-1 pathway. Front Pharmacol. 13:8298302022.
View Article : Google Scholar :
|
|
23
|
Yang K, Pei L, Zhou S, Tao L and Zhu Y:
Metformin attenuates H2O2-induced osteoblast
apoptosis by regulating SIRT3 via the PI3K/AKT pathway. Exp Ther
Med. 22:13162021. View Article : Google Scholar
|
|
24
|
Bai J, Xie N, Hou Y, Chen X, Hu Y, Zhang
Y, Meng X, Wang X and Tang C: The enhanced mitochondrial
dysfunction by cantleyoside confines inflammatory response and
promotes apoptosis of human HFLS-RA cell line via AMPK/Sirt 1/NF-κB
pathway activation. Biomed Pharmacother. 149:1128472022. View Article : Google Scholar
|
|
25
|
Zhang R, Li G, Zhang Q, Tang Q, Huang J,
Hu C, Liu Y, Wang Q, Liu W, Gao N and Zhou S: Hirsutine induces
mPTP-dependent apoptosis through ROCK1/PTEN/PI3K/GSK3β pathway in
human lung cancer cells. Cell Death Dis. 9:5982018. View Article : Google Scholar
|
|
26
|
Roskoski R Jr: Small molecule inhibitors
targeting the EGFR/ErbB family of protein-tyrosine kinases in human
cancers. Pharmacol Res. 139:395–411. 2019. View Article : Google Scholar
|
|
27
|
Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H,
Ding Q and Xia J: Amphiregulin confers regulatory T cell
suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3
axis. J Biol Chem. 291:21085–21095. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanchez-Rangel E and Inzucchi SE:
Metformin: Clinical use in type 2 diabetes. Diabetologia.
60:1586–1593. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cameron AR, Morrison VL, Levin D, Mohan M,
Forteath C, Beall C, McNeilly AD, Balfour DJ, Savinko T, Wong AK,
et al: Anti-inflammatory effects of metformin irrespective of
diabetes status. Circ Res. 119:652–665. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang TC, Duthie GG, Aucott LS and
Macdonald HM: Vitamin E homologues α- and γ-tocopherol are not
associated with bone turnover markers or bone mineral density in
peri-menopausal and post-menopausal women. Osteoporos Int.
27:2281–2290. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kulkarni AS, Gubbi S and Barzilai N:
Benefits of metformin in attenuating the hallmarks of aging. Cell
Metab. 32:15–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang T, Liu Q, Gao W, Sehgal SA and Wu H:
The multifaceted regulation of mitophagy by endogenous metabolites.
Autophagy. 18:1216–1239. 2022. View Article : Google Scholar :
|
|
33
|
Abate M, Festa A, Falco M, Lombardi A,
Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia
M and Misso G: Mitochondria as playmakers of apoptosis, autophagy
and senescence. Semin Cell Dev Biol. 98:139–153. 2020. View Article : Google Scholar
|
|
34
|
Panel M, Ghaleh B and Morin D:
Mitochondria and aging: A role for the mitochondrial transition
pore? Aging Cell. 17:e127932018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Y, Yang M, Fan J, Peng Y, Deng L, Ding
Y, Yang R, Zhou J, Miao D and Fu Q: Deficiency of osteoblastic
Arl6ip5 impaired osteoblast differentiation and enhanced
osteoclastogenesis via disturbance of ER calcium homeostasis and
induction of ER stress-mediated apoptosis. Cell Death Dis.
5:e14642014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S,
Li Y, Zhou H and Chen Y: Ripk3 promotes ER stress-induced
necroptosis in cardiac IR injury: A mechanism involving calcium
overload/XO/ROS/mPTP pathway. Redox Biol. 16:157–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
NavaneethaKrishnan S, Rosales JL and Lee
KY: mPTP opening caused by Cdk5 loss is due to increased
mitochondrial Ca2+ uptake. Oncogene. 39:2797–2806. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nagini S, Sophia J and Mishra R: Glycogen
synthase kinases: Moonlighting proteins with theranostic potential
in cancer. Semin Cancer Biol. 56:25–36. 2019. View Article : Google Scholar
|
|
39
|
Lal H, Ahmad F, Woodgett J and Force T:
The GSK-3 family as therapeutic target for myocardial diseases.
Circ Res. 116:138–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Morciano G, Naumova N, Koprowski P,
Valente S, Sardão VA, Potes Y, Rimessi A, Wieckowski MR and
Oliveira PJ: The mitochondrial permeability transition pore: An
evolving concept critical for cell life and death = The GSK-3
family as therapeutic target for myocardial diseases. Biol Rev Camb
Philos Soc. 96:2489–2521. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Karch J, Bround MJ, Khalil H, Sargent MA,
Latchman N, Terada N, Peixoto PM and Molkentin JD: Inhibition of
mitochondrial permeability transition by deletion of the ANT family
and CypD. Sci Adv. 5:eaaw45972019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang H, Ai J, Shopit A, Niu M, Ahmed N,
Tesfaldet T, Tang Z, Li X, Jamalat Y, Chu P, et al: Protection of
pancreatic β-cell by phosphocreatine through mitochondrial
improvement via the regulation of dual AKT/IRS-1/GSK-3β and
STAT3/Cyp-D signaling pathways. Cell Biol Toxicol. 38:531–551.
2022. View Article : Google Scholar
|
|
43
|
Hu Z, Crump SM, Zhang P and Abbott GW:
Kcne2 deletion attenuates acute post-ischaemia/reperfusion
myocardial infarction. Cardiovasc Res. 110:227–237. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ding L and Billadeau DD: Glycogen synthase
kinase-3β: A novel therapeutic target for pancreatic cancer. Expert
Opin Ther Targets. 24:417–426. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim DH, Triet HM and Ryu SH: Regulation of
EGFR activation and signaling by lipids on the plasma membrane.
Prog Lipid Res. 83:1011152021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu G, Xie Y, Su J, Qin H, Wu H, Li K, Yu
B and Zhang X: The role of EGFR signaling in age-related
osteoporosis in mouse cortical bone. FASEB J. 33:11137–11147. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Phukan S, Babu VS, Kannoji A, Hariharan R
and Balaji VN: GSK3beta: Role in therapeutic landscape and
development of modulators. Br J Pharmacol. 160:1–19. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Z, Lv Z, Zhang W, Guo M and Li C: A
novel β-catenin from Apostichopus japonicus mediates Vibrio
splendidus-induced inflammatory-like response. Int J Biol Macromol.
156:730–739. 2020. View Article : Google Scholar : PubMed/NCBI
|