Introduction
Atherosclerosis is a chronic degenerative arterial
condition, and is a leading cause of most cerebrovascular and
cardiovascular diseases via its complex and progressive effects on
the arterial wall (1,2). Atherosclerosis is characterized by
the abnormal accumulation of fibrous components and lipids in the
intima, resulting in arterial wall thickening and a reduction in
the size of the vascular cavity (1,3).
Vascular inflammation in atherosclerosis is accompanied by an
accumulation of cholesterol, lipids, calcium and cellular debris
within the vessel wall intima (4). This deposition can result in plaque
production, revascularization, acute and chronic lumen obstruction,
blood flow abnormalities and reduced oxygen supply to the target
organs (5). The mechanisms
underlying the pathology of atherosclerosis are complex, but mainly
consist of endothelial cell dysfunction, macrophage polarization,
inflammation and the immune response (1).
Inflammation is involved in all stages of
atherosclerosis, ranging from endothelial cell injury to the
ultimate rupture of plaques (3).
Furthermore, it has been reported that chronic inflammation may be
an independent risk factor for atherosclerosis (6). Critical factors that can contribute
to the early stages of atherosclerosis and plaque development are
pro-inflammatory cytokines, including interleukin-6 (IL-6),
interferon (IFN)-α, IFN-γ and Toll-like receptor 4 (TLR4) (7). Notably, nuclear factor-κB (NF-κB)
activation is required for the regulation of a number of genes
involved in the inflammatory response of cells that are critical to
atherogenesis. The activation of NF-κB results in the subsequent
transcription of pro-inflammatory genes, including intercellular
adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1
(VCAM-1), IL-1β and TNF-α; therefore, pro-inflammatory genes are
principally regulated by NF-κB (8).
The signal transducer and activator of transcription
(STAT) family of transcription factors can activate key mediators
of cytokine responses; within this family, STAT3 is a principal
transcription factor associated with immunity and inflammation
(9). Notably, STAT3 has a crucial
regulatory role in cell survival and proliferation; its activation
has been detected in numerous human tumors, including melanoma,
head and neck squamous cell carcinoma, multiple myeloma, mantle
cell lymphoma, glioma, and colon, lung, breast, pancreas and
prostate cancer (10,11). In addition, STAT3 has been
reported to be associated with various cardiovascular diseases,
including arteriosclerosis, cardiac hypertrophy and heart failure
(6). STAT3 may serve a crucial
role in all of these diseases through endothelial cell dysfunction,
macrophage polarization, inflammation and the immune response, thus
indicating that STAT3 may be a potential new target of
atherosclerosis therapies (1).
Therefore, inhibiting NF-κB and STAT3 expression may prevent the
formation of atherosclerosis and slow its progression. Therefore,
the present study attempted to suppress atherosclerosis by blocking
the NF-κB and STAT3 transcription factors to regulate
inflammation.
Decoy oligodeoxynucleotides (ODNs) inhibit target
gene expression through the consensus binding-site sequences of
target transcription factors (12). Consequently, decoy ODNs bind to
sequence-specific transcription factors and limit gene expression
by interfering with transcription in vitro and in
vivo (13). Transfection of
cis-element double-stranded ODNs attenuates authentic cis-trans
interactions, resulting in the removal of trans factors from
endogenous cis elements and the subsequent modulation of gene
expression (13). Gene therapy
based on decoy ODNs may be useful for treating a number of
diseases, including glomerulonephritis, myocardial infarction and
rheumatoid arthritis, and may prevent acute rejection after renal
transplantation (14). Our
previous study demonstrated the anti-atherosclerotic effects of
NF-κB decoy ODNs on pro-inflammatory cytokines and adhesion
molecules in a mouse model of lipopolysaccharide (LPS)-induced
atherosclerosis (15).
Furthermore, our previous study demonstrated the effect of SREBP
decoy ODNs on a mouse model of non-alcoholic fatty liver disease in
high-fat diet-induced hyperlipidemia (12). However, to the best of our
knowledge, the effect of STAT3/NF-κB decoy ODNs on an animal model
of atherosclerosis has not yet been investigated.
The present study hypothesized that STAT3/NF-κB
decoy ODNs may suppress the development of atherosclerosis in a
mouse model by simultaneously inhibiting the STAT/NF-κB signaling
pathway and its related inflammatory reaction. The aim of this
study was to determine the useful functions and possible underlying
molecular mechanisms of STAT3/NF-κB decoy ODNs in a mouse model of
bacterial endotoxin LPS-induced atherosclerosis.
Materials and methods
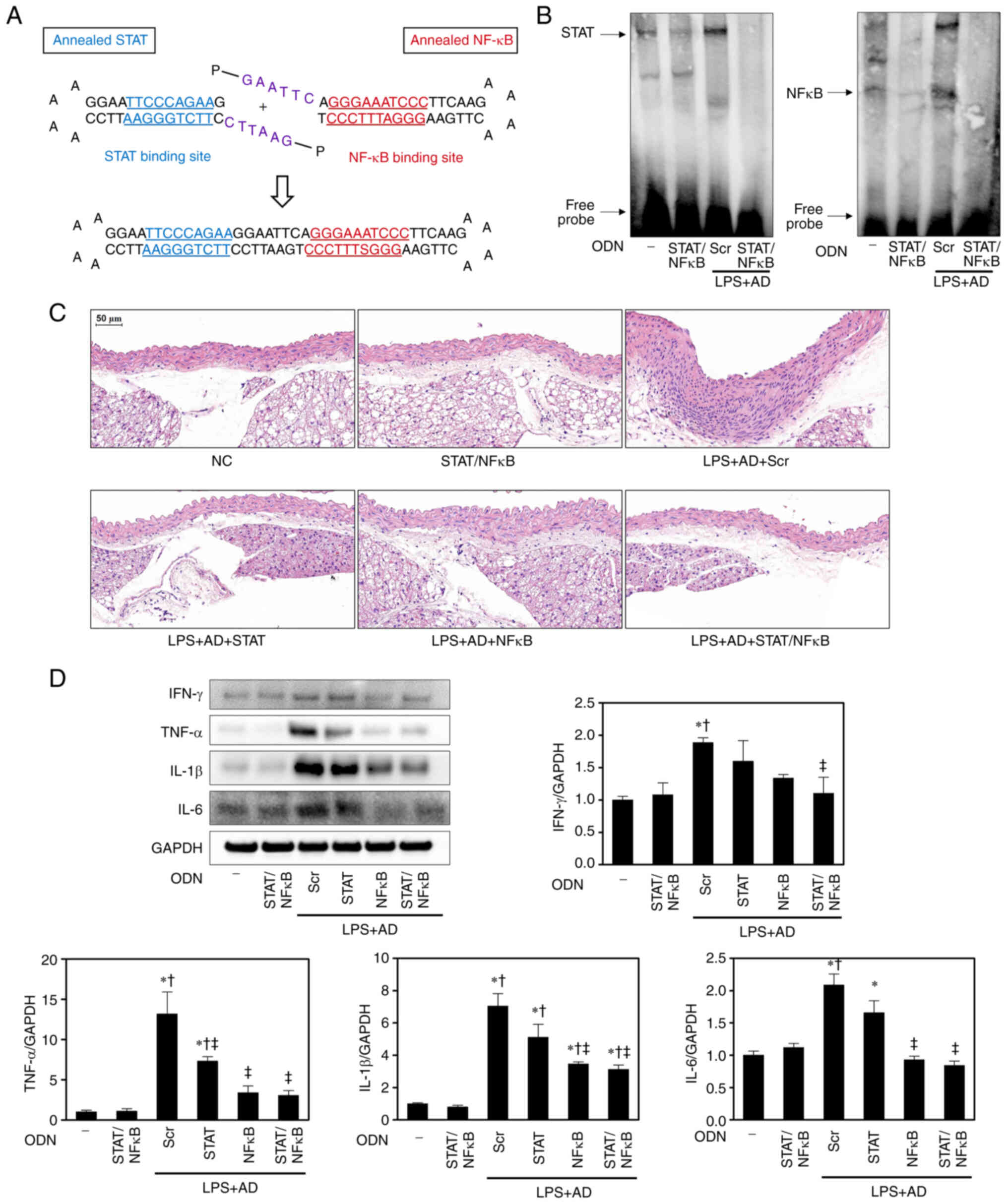
Construction and synthesis of decoy
ODNs
Synthetic decoy ODNs were synthesized by Macrogen,
Inc. using the following ODN sequences (the target sites of the
consensus-binding sequences are underlined): STAT3 decoy ODN,
5′-GAA TTC CTT CTG GGA
ATT CCA AAA GGA ATT CCC AGA AG-3′; NF-κB decoy ODN, 5′-GAA
TTC AGG GAAATC CCT
TCA AGA AAA CTT GAA GGG
ATT TCC CT-3′; and scramble (Scr) ODN, 5′-GAA TTC AAT TCA
GGG TAC GGC AAA AAA TTG CCG TAC CCT GAA TT-3′. Subsequently, STAT3,
NF-κB, STAT3/NF-κB decoy ODNs and Scr decoy ODN were annealed for 6
h, decreasing the temperature from 80 to 25°C during this time. To
obtain a covalent ligation for ring-type decoy ODNs, each ODN was
mixed with T4 DNA ligase (Takara Bio, Inc.) and incubated for 18 h
at 16°C. These decoy ODNs were predicted to form a covalently
ligated ring-type structure (Fig.
1A).
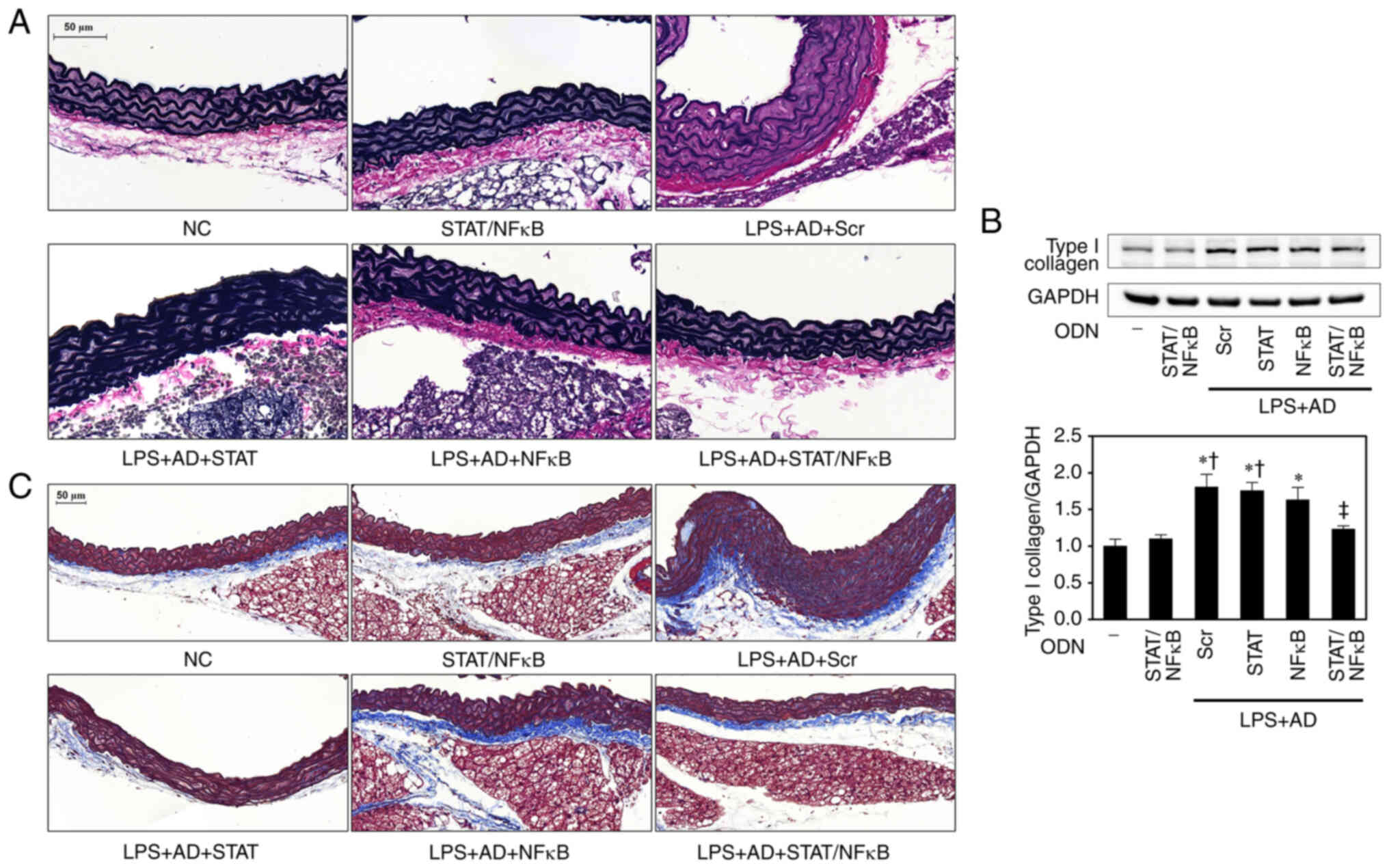
 | Figure 1STAT/NF-κB decoy ODNs suppress
histological changes and inflammation in atherosclerotic mice
aortae. (A) Structure of STAT/NF-κB synthetic decoy ODNs. (B)
Electrophoretic mobility shift assay was performed to analyze the
effects of STAT/NF-κB synthetic decoy ODNs on STAT and
NF-κB-binding activity in atherosclerotic mice (n=5). (C)
Histopathological alterations were determined by hematoxylin and
eosin staining; representative images from each group are shown
(n=5). Scale bar, 50 µm. (D) Western blotting was performed
to detect the expression levels of inflammatory cytokines in aorta
tissues. The graph summarizes the semi-quantification of protein
expression normalized to GAPDH (n=3). *P<0.05 vs. NC
group; †P<0.05 vs. STAT/NF-κB group;
‡P<0.05 vs. LPS + AD + Scr group. AD, atherogenic
diet; IFN-γ, interferon-γ; IL, interleukin; NC, normal control;
NF-κB, nuclear factor-κB; ODN, oligode-oxynucleotide; LPS,
lipopolysaccharide; Scr, scramble; STAT, signal transducer and
activator of transcription. |
Atherosclerosis model
A total of 60, male C57BL/6 mice (age, 6 weeks;
weight, 20-25 g; Samtaco Bio Korea Co., Ltd.) were housed in a room
at a controlled temperature of 22±2°C and a humidity of 55%, under
a 12-h light/dark cycle. The mice were given free access to water
and food. Atherosclerotic injuries were induced via intraperitoneal
injection of LPS from Escherichia coli O111:B4 (2 mg/kg body
weight; dissolved in 200 µl PBS; MilliporeSigma) once a week
for 8 weeks. Simultaneously, the mice were fed an atherogenic diet
(AD; 21.2% milkfat, 1.25% cholesterol, 0.5% cholic acid; DooYeol
Biotech.). Decoy ODNs (10 µg) were administered every 2
weeks for 8 weeks via an injection into the tail vein using a Trans
IT in vivo gene delivery system (Mirus Bio.). The mice were
divided into the following six groups (n=10 mice/group): i)
Untreated group [normal control (NC)], ii) STAT3/NF-κB decoy
ODN-treated group (STAT/NF-κB) fed a standard diet (cat. no. 2018S;
Envigo), iii) Scr decoy ODN-treated group injected with LPS and fed
an AD (LPS + AD + Scr), iv) STAT3 decoy ODN-treated group injected
with LPS and fed an AD (LPS + AD + STAT), v) NF-κB decoy
ODN-treated group injected with LPS and fed an AD (LPS + AD +
NF-κB), vi) STAT3/NF-κB decoy ODN-treated group injected with LPS
and fed an AD (LPS + AD + STAT/NF-κB). All mice were anesthetized
with 2-3% isoflurane inhalation (Ifran; HANA Pharm Co., Ltd.) using
an RC2 Rodent Circuit Controller (VetEquip, Inc.) before tail vein
injection. At the end of each treatment period, blood was collected
by cardiac puncture from the mice, and the mice were euthanized by
asphyxiation with 60-70% CO2 of the cage volume/min.
Subsequently, their aorta and heart tissues were excised for
subsequent experiments. The humane endpoints in the present study
were as follows: i) The animals showed hypothermia (<37°C) in
the absence of anesthesia; ii) the animal lost 20% of its original
weight; iii) the animal exhibited loss of ability to ambulate
(unable to access food or water). All animals reaching these
endpoints were euthanized with 60-70% CO2 of the cage
volume/min. Death verification included the absence of heartbeat,
breathing or respiration. All animal protocols were approved by the
Institutional Animal Care and Use Committee of the Catholic
University of Daegu (Daegu, South Korea; approval no. DC
IAFCR-181204-27Y). All procedures performed in experiments
involving animals were in accordance with the ethical standards of
the institution or practice at which the studies were
conducted.
Electrophoretic mobility shift assay
(EMSA)
Nuclear extract fractionation was performed on the
abdominal aorta tissues of mice using an NE-PER™ Nuclear and
Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions based on standard
protocols. Subsequently, a Lightshift Chemiluminescent EMSA kit
(cat. no. 20148; Thermo Fisher Scientific, Inc.) was performed to
analyze STAT3 and NF-κB DNA binding activation. ODNs containing the
consensus STAT3 and NF-κB binding sites (STAT3, forward 5′-GAT CCT
TCT GGG AAT TCC TAG ATC-3′, reverse 5′-GAT CTA GGA ATT CCC AGA AGG
ATC-3′; NF-κB, forward 5′-CTT GAA GGG ATT TCC CTG GCT-3′, reverse
5′-AGC CAG GGA AAT CCC TTC AAG-3′) and 3′ end-labeled with biotin
were used as probes. Biotin-labeled probes were synthesized by
Macrogen, Inc. The probes (20 fmol) were subjected to a
denaturation step at 90°C for 1 min followed by annealing at room
temperature for 30 min. Then, for the binding reaction, the binding
reaction components were added in the order listed according to the
manufacturer's instructions. At this time, 10 µg nuclear
extract was added to binding reactions and incubated for 20 min at
room temperature. The nuclear protein concentration (1-10
µg/µl) was determined using the Bradford protein
assay (Bio-Rad Laboratories, Inc.). The Novex™ TBE gels 4-12% (cat.
no. EC62352; Thermo Fisher Scientific, Inc.) were used for
pre-electrophoresis for 30-60 min at 100 V. The sample complexes
were separated by electrophoresis on gels using 0.5X TBE (cat. no.
LC6675; Thermo Fisher Scientific, Inc.) as a running buffer until
the bromophenol blue dye has migrated ~3/4 down the length of the
gel. After electrophoresis, the gels were transferred to nylon
membranes and detected using the Streptavidin-Horseradish
Peroxidase Conjugate and the Chemiluminescent Substrate (cat. no.
89880; Thermo Fisher Scientific, Inc.), according to manufacturer's
instructions. The signal intensity was detected using an image
analyzer (ChemiDoc™XRS+; Bio-Rad Laboratories, Inc.).
Atherosclerosis lesion analyses
All abdominal aorta and heart tissue specimens were
fixed in 10% formalin for 24 h at room temperature. Thereafter,
sections of the aorta and heart were dehydrated in graded ethanol,
cleared in xylene and embedded in paraffin. The sections (4
µm) were mounted on glass slides, rehydrated in distilled
water and stained with hematoxylin and eosin (H&E),
Verhoeff-Van Gieson and Masson's trichrome based on standard
protocols. The tissue sections were deparaffinized and stained with
hematoxylin at room temperature for 8 min and then with eosin at
room temperature for 5 min. For the Verhoeff-Van Gieson stain, the
tissue sections were deparaffinized and stained with alcoholic
hematoxylin, 10% ferric chloride and Weigert's iodine mixed
solution for 5 min at room temperature. After differentiating in 2%
ferric chloride for 1-2 min and treating with sodium thiosulfate
for 1 min, the sections were counterstained with Van Gieson's
solution for 40 sec. For the Masson's trichrome stain, the tissue
sections were deparaffinized and refixed in Bouin's solution for 30
min at 60°C. After being stained at room temperature with Weigert's
hematoxylin and a Biebrich scarlet-acid fuchsin each for 10 min,
the sections were treated in phosphomolybdic–phosphotungstic acid
for 10 min and finally stained with aniline blue for 10 min. All
slides were examined under a Pannoramic MIDI slide scanner
(3DHISTECH Kft.).
Western blot analysis
The aorta tissue specimens were lysed in protein
lysis buffer (CelLytic™ M; MilliporeSigma) for 20 min on ice and
were then centrifuged at 13,800 × g for 20 min at 4°C. Nuclear and
cytosolic protein samples were prepared from the aorta tissue using
NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
supernatant was collected and the protein concentration was
measured using the Bradford protein assay (Bio-Rad Laboratories,
Inc.). The protein samples were then loaded on precast gradient SDS
polyacrylamide gels (Bolt™ 4-12% Bis-Tris Plus Gels; Thermo Fisher
Scientific, Inc.) and transferred to nitrocellulose membranes (GE
Healthcare) using a Bolt™ Mini Blot Module and Mini Gel Tank
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
recommendations. The membranes were blocked for 1 h at room
temperature in 5% bovine serum albumin (MilliporeSigma) and
incubated overnight with primary anti-bodies at 4°C. After primary
antibody incubation, horseradish peroxidase (HRP)-conjugated
secondary antibodies were used to incubate the membranes for 2 h at
room temperature. The primary antibodies used in the present study
were as follows: Anti-IFN-γ (1:1,000; cat. no. ab9657), anti-TNF-α
(1:1,000; cat. no. ab1793), anti-IL-6 (1:1,000; cat. no. ab208113),
anti-ATP-binding cassette transporter A1 (ABCA1; 1:500; cat. no.
ab18180), anti-monocyte chemoattractant protein-1 (MCP-1; 1:1,000;
cat. no. ab21396), anti-fibronectin (1:1,000; cat. no. ab2413),
anti-α-smooth muscle actin (α-SMA; 1:1,000; cat. no. ab5694) (all
from Abcam), anti-IL-1β (1:1,000; cat. no. sc-32294), anti-COL1A2
(1:500; cat. no. sc-8788), anti-TLR4 (1:1,000; cat. no. sc-10741),
anti-JAK2 (1:1,000; cat. no. sc-294) (all from Santa Cruz
Biotechnology, Inc.), anti-ICAM-1 (1:1,000; cat. no. AF796),
anti-VCAM-1 (1:1,000; cat. no. AF643) (both from R&D Systems,
Inc.), anti-phosphorylated (P)-JAK2 (1:1,000; cat. no. 8082),
anti-P-IκB (1:1,000; cat. no. 2859), anti-IκB (1:1,000; cat. no.
9242), anti-NF-κB (1:1,000; cat. no. 8242), anti-P-NF-κB (1:1,000;
cat. no. 3033), anti-STAT3 (1:1,000; cat. no. 9139), anti-P-STAT3
(1:1,000; cat. no. 9145), anti-GAPDH (1:2,000; cat. no. 2118) (all
from Cell Signaling Technology, Inc.) and anti-Lamin B1 (1:1,000;
cat. no. 332000; Invitrogen; Thermo Fisher Scientific, Inc.). The
secondary antibodies used in the present study were as follows:
Anti-mouse (1:1,000; cat. no. 7076) and anti-rabbit (1:1,000; cat.
no. 7074) (both from Cell Signaling Technology, Inc.). After
washing, the membranes were visualized using enhanced
chemiluminescence detection reagents (SuperSignal™ West Femto
Maximum Sensitivity Substrate; cat. no. 34096; Thermo Fisher
Scientific, Inc.) for 1 min. The signal intensity was detected
using an image analyzer (ChemiDoc™XRS+; Bio-Rad Laboratories, Inc.)
and was semi-quantified with Image Lab software version 5.1
(Bio-Rad Laboratories, Inc.).
Immunohistochemical (IHC) staining
Paraffin-embedded tissue sections (4 µm) were
placed in a BOND-MAX Fully Automated IHC Staining System slide
stainer (Leica Microsystems, Inc.) according to the following
protocol. First, tissues were deparaffinized and pre-treated with
the Epitope Retrieval Solution 2 (EDTA-buffer pH 8.8) at 98°C for
20 min. After washing steps, peroxidase blocking was carried out
for 10 min using the BOND Polymer Refine Detection Kit (cat. no.
DS9800; Leica Microsystems, Inc.). Tissues were washed again and
then incubated with the primary antibodies (1:100) for 30 min at
room temperature. The following primary antibodies were used:
Anti-ICAM-1, anti-VCAM-1 (both from R&D Systems) and
anti-monocyte + macrophage (MOMA-2; cat. no. ab33451; Abcam). The
tissue sections were then incubated with post-primary for 8 min and
were then incubated with polymer for 8 min and developed with
DAB-Chromogen for 10 min at room temperature. Hematoxylin was used
as the counterstain for 5 min at room temperature. The slides were
examined using a Pannoramic MIDI slide scanner.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Statistical significance was assessed using one-way
analysis of variance with Tukey's multiple comparison test using
GraphPad Prism 5.0 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Construction of decoy ODNs
The present study first designed STAT3, NF-κB and
STAT3/NF-κB decoy ODNs (Fig. 1A).
Decoy ODNs containing the DNA-binding consensus sequences of
transcription factors selectively inhibit STAT3 and NF-κB by
binding to the DNA-binding domains. To identify the functions of
STAT3/NF-κB decoy ODNs in controlling STAT3 and NF-κB expression,
EMSA was performed with nuclear extracts obtained from the aorta
tissues of mice with LPS/AD-induced atherosclerosis and treated
with Scr and STAT3/NF-κB decoy ODNs. Compared with in NC mice, LPS
+ AD + Scr atherosclerotic mice exhibited increased STAT3 and NF-κB
DNA-binding activity. However, the LPS + AD + STAT/NF-κB treatment
groups exhibited reduced STAT3 and NF-κB DNA-binding activity
compared with the LPS + AD + Scr group (Fig. 1B). These results indicated that
STAT3/NF-κB decoy ODNs effectively decreased STAT and NF-κB
expression.
STAT3/NF-κB decoy ODNs attenuate
morphological changes and inflammation in atherosclerotic mice
aortae
To investigate the effect of STAT3/NF-κB decoy ODNs
on atherosclerotic mice, H&E histological analysis was
performed. A marked increase in the thickness of the adventitia and
media, and noticeable cellular infiltration in the adventitial
layers of the aortae were detected in the LPS + AD + Scr group
compared with in the NC group. Histological analysis showed that
these histological changes were markedly decreased in the LPS + AD
+ STAT/NF-κB groups (Fig. 1C).
Furthermore, no differences in body weight were determined among
the groups (data not shown).
To examine the mechanism of action of STAT3/NF-κB
decoy ODNs, the protein expression levels of IFN-γ, TNF-α, IL-1β
and IL-6 were measured in the aorta tissues of atherosclerotic mice
using western blot analysis (Fig.
1D). Notably, the expression levels of IFN-γ, TNF-α, IL-1β and
IL-6 were significantly higher in the LPS + AD + Scr group than
those in the NC and STAT/NF-κB groups. However, in the LPS + AD +
STAT/NF-κB group, the increase in these protein expression levels
was returned to a near-NC level. These observations suggested that
STAT3/NF-κB decoy ODNs can inhibit atherosclerotic morphological
changes and inflammation.
STAT3/NF-κB decoy ODNs diminish
structural damage in the aortae of atherosclerotic mice
The tissue sections were subjected to histological
analysis with Verhoeff-Van Gieson and Masson trichrome staining in
order to characterize the abdominal aortic lesions. Besides
cellular components, the extracellular matrix (ECM) of
atherosclerotic plaque serves a relevant role in the initiation and
subsequent progression of atherosclerosis. Major ECM structural and
signaling components include elastin and collagen (16). Verhoeff-Van Gieson staining of
elastin revealed no elastin fiber fragmentation in the NC and
STAT/NF-κB groups. The elastin content in the medial layer was
diminished and fragmented in the LPS + AD + Scr atherosclerosis
group, perhaps because of the increased medial area or thinning of
the elastin fibers; however, the elastin fibers were less disrupted
in the LPS + AD + STAT/NF-κB mice compared with that in the LPS +
AD + Scr mice (Fig. 2A). In
addition, based on Masson's trichrome staining, a marked increase
in collagen deposition in the aortae of LPS + AD + Scr group mice
was detected, whereas this was markedly reduced in LPS + AD +
STAT/NF-κB mice (Fig. 2C).
Western blot analysis confirmed these results, showing that the
protein expression levels of COL1A2 were increased in the LPS + AD
+ Scr group compared with those in the NC group, but were
significantly decreased following treatment with STAT3/NF-κB decoy
ODNs (Fig. 2B). Elastin is one of
the dominant ECM proteins and collagen accumulation is
characteristic of atherosclerotic plaques (16,17). These results suggested that
increased ECM components in the LPS + AD + Scr mice may be
attenuated by STAT3/NF-κB decoy ODNs.
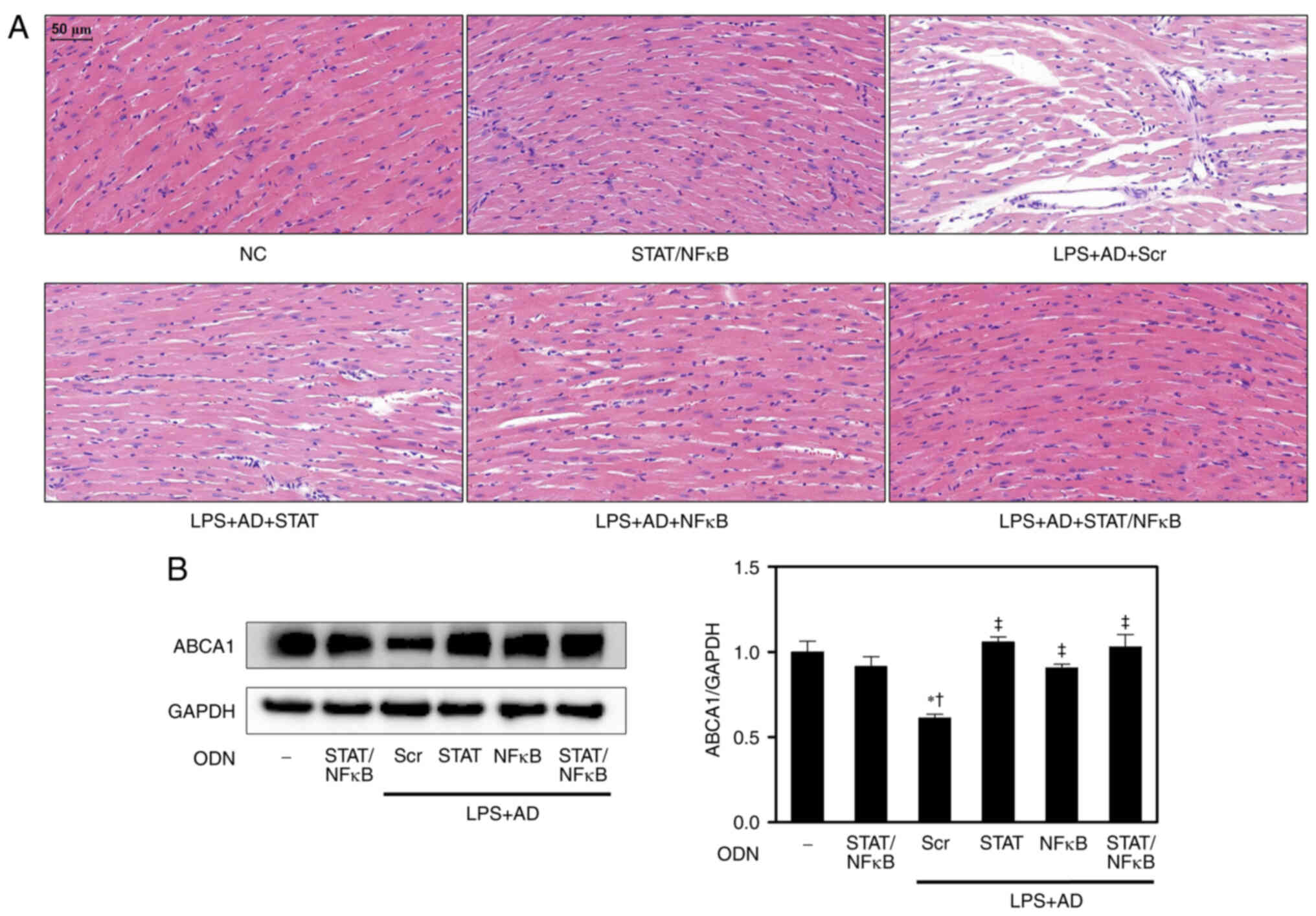
STAT3/NF-κB decoy ODNs inhibit
morphological changes in the hearts of atherosclerotic mice and
regulate cholesterol metabolism
To further investigate the cardioprotective effects
of STAT3/NF-κB decoy ODNs, their effects on heart morphology were
assessed. H&E staining of heart tissues revealed that
myocardial fibers in the NC group were normal and ordered. There
were no broken fibers and the nuclei of the myocardial cells were
regular. In addition, no marked changes were observed between the
NC and STAT/NF-κB groups. The LPS + AD + Scr mice displayed
structural abnormalities, with disordered, extensively collapsed
and degenerated muscle fibers; however, these histological changes
were markedly alleviated by treatment with STAT3/NF-κB decoy ODNs
(Fig. 3A).
ABCA1 serves a central role in the early stages of
the reverse cholesterol transport pathway by mediating lipid efflux
from macrophages and is important for maintaining cellular
cholesterol homeostasis (18,19). Therefore, the present study
explored the effects and potential mechanisms of STAT3/NF-κB decoy
ODNs on ABCA1 expression in atherosclerotic mice. As shown in
Fig. 3B, STAT3, NF-κB and
STAT3/NF-κB decoy ODNs significantly increased the protein
expression levels of ABCA1 compared with those in the LPS + AD +
Scr group. Given that ABCA1 expression can be suppressed by
pro-inflammatory stimuli via the NF-κB signaling pathway,
STAT3/NF-κB decoy ODNs may regulate ABCA1 expression and
cholesterol metabolism.
Effects of STAT3/NF-κB decoy ODNs on the
expression levels of adhesion molecules in atherosclerotic mice
aortae
The expression of ICAM-1 and VCAM-1 in endothelial
cells is the earliest known event in the initiation and progression
of atherosclerosis (20). To
identify the function of STAT3/NF-κB decoy ODNs in ICAM-1 and
VCAM-1 expression, IHC staining was performed. IHC staining
demonstrated that ICAM-1 and VCAM-1 were expressed in some
endothelial cells over lesions and in the endothelia adjacent to
the lesions in the LPS + AD + Scr mice, whereas the changes in
ICAM-1 and VCAM-1 expression were markedly decreased in the LPS +
AD + STAT/NF-κB mice (Fig. 4A and
C). Western blot analysis further confirmed that ICAM-1 and
VCAM-1 expression levels in the aortae of LPS + AD + Scr
atherosclerotic mice were higher than those in the NC and
STAT/NF-κB groups, whereas these expression levels were decreased
in the LPS + AD + STAT/NF-κB mice (Fig. 4B).
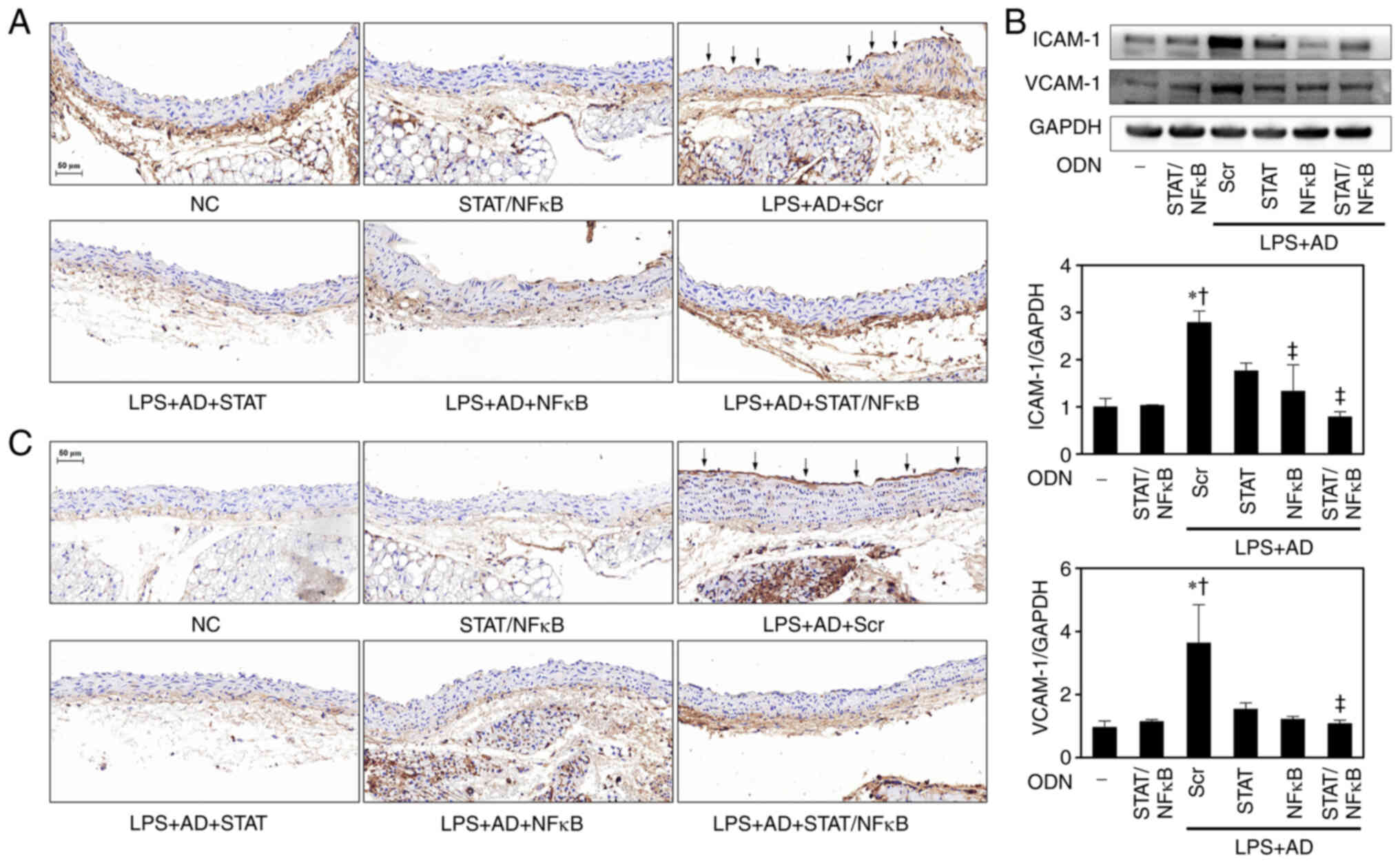 | Figure 4STAT/NF-κB decoy ODNs alleviate
atherosclerotic mice aortae injury. Immunohistochemistry images of
(A) ICAM-1. Representative images from each group are shown (n=5).
Arrows indicate areas of respective adhesion molecule expression.
Scale bar, 50 µm. (B) Western blotting was performed to
detect the protein expression levels of adhesion molecules in aorta
tissues. The graph summarizes the semi-quantification of molecules
of protein expression normalized to GAPDH (n=3).
*P<0.05 vs. NC group; †P<0.05 vs.
STAT/NF-κB group; ‡P<0.05 vs. LPS + AD + Scr group.
(C) Immunohistochemistry images of VCAM-1. Representative images
from each group are shown (n=5). Arrows indicate areas of
respective adhesion molecule expression. Scale bar, 50 µm.
AD, atherogenic diet; ICAM-1, intercellular adhesion molecule-1;
NC, normal control; NF-κB, nuclear factor-κB; ODN,
oligodeoxynucleotide; LPS, lipopolysaccharide; Scr, scramble; STAT,
signal transducer and activator of transcription; VCAM-1, vascular
cell adhesion molecule-1. |
Effects of STAT3/NF-κB decoy ODNs on the
expression of fibrosis-related proteins in atherosclerotic
mice
To determine whether STAT3/NF-κB decoy ODNs may act
on fibrosis, the expression levels of fibrosis-related proteins
were detected in aortic lesions. Vascular fibrosis involves the
excess accumulation of ECM proteins, such as collagen, proteoglycan
and fibronectin, in the arterial wall, leading to decreased luminal
diameter and increased vascular stiffness, which are
characteristics of atherosclerosis (21). Western blot analysis detected the
expression levels of the ECM-related marker fibronectin and the
contractile smooth muscle cell (SMC)-related marker α-SMA. In the
LPS + AD + Scr group the aortic protein expression levels of
fibronectin and α-SMA were significantly increased compared with
those in the NC and STAT/NF-κB groups. By contrast, the expression
levels of fibronectin and α-SMA were significantly decreased in the
LPS + AD + STAT/NF-κB mice compared with those in the LPS + AD +
Scr group (Fig. 5A). MCP-1 serves
a crucial role in initiating atherosclerosis by recruiting
macrophages and monocytes to the vessel walls (22). The results of the present study
showed that the expression levels of MCP-1 were increased in the
LPS + AD + Scr group compared with those in the NC and STAT/NF-κB
groups. Conversely, the expression levels of MCP-1 were inhibited
in the LPS + AD + STAT/NF-κB group (Fig. 5A).
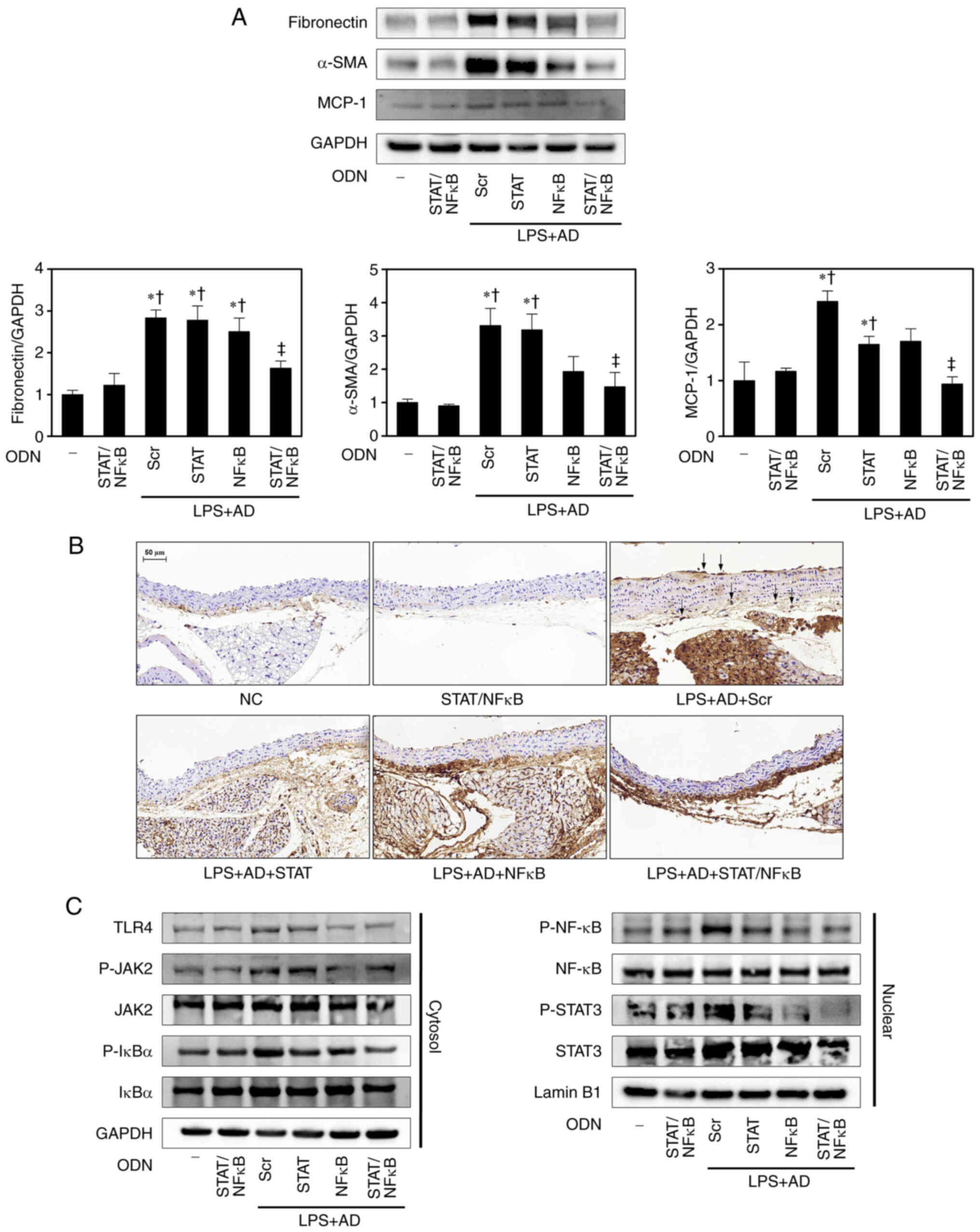 | Figure 5STAT/NF-κB decoy ODNs inhibit the
expression levels of fibrosis-related proteins and the STAT/NF-κB
pathway in atherosclerotic mice. (A) Western blotting was performed
to detect the protein expression levels of fibronectin, α-SMA and
MCP-1. The graph summarizes the semi-quantification of molecules of
protein expression normalized to GAPDH (n=3). *P<0.05
vs. NC group; †P<0.05 vs. STAT/NF-κB group;
‡P<0.05 vs. LPS + AD + Scr group. (B)
Immunohistochemistry images of the macrophage marker MOMA-2.
Representative images from each group are shown (n=5). Arrows
indicate areas of MOMA-2-positive cells. Scale bar, 50 µm.
(C) Western blotting was performed to detect the protein expression
levels of proteins in the STAT and NF-κB pathways (n=3). α-SMA,
α-smooth muscle actin; AD, atherogenic diet; MCP-1, monocyte
chemoattractant protein-1; NC, normal control; NF-κB, nuclear
factor-κB; ODN, oligodeoxynucleotide; LPS, lipopolysaccharide; P-,
phosphorylated; Scr, scramble; STAT, signal transducer and
activator of transcription; TLR4, Toll-like receptor 4. |
IHC staining was also performed with the common
macrophage marker MOMA-2 (Fig.
5B). The progressive deposition of inflammatory cells, such as
macrophages, promotes plaque vulnerability leading to enlarged
unstable plaques covered by a thin fibrous cap (23). MOMA-2-positive cells were not
detected in the NC and STAT/NF-κB groups, but were observed in the
LPS + AD + Scr group. Moreover, macrophage accumulation was
markedly reduced in the LPS + AD + STAT/NF-κB atherosclerotic
lesions.
STAT3/NF-κB decoy ODNs inhibit activation
of the JAK/STAT and TLR4/NF-κB pathways in LPS-induced
inflammation
Numerous inflammatory signaling pathways that
contribute to the pathogenesis of atherosclerosis are modulated by
the transcription factor NF-κB, which is a master regulator of
innate and adaptive immune responses (24). In addition, TLR4 serves a crucial
role in the initiation of an innate immune response through
activation of inflammatory cells via the NF-κB-dependent pathway
(24). Therefore, the present
study examined the effects of STAT3/NF-κB decoy ODNs on
LPS-stimulated inflammation via the TLR4/NF-κB signaling pathway in
atherosclerotic mice. Western blot analysis indicated that the
protein expression levels of TLR4 were increased in the LPS + AD +
Scr group compared with those in the NC group; however, were
decreased following treatment with STAT3/NF-κB decoy ODNs (Fig. 5C). Because the activity of NF-κB
is regulated by IκBα, the effect of STAT3/NF-κB decoy ODNs on the
phosphorylation of IκBα was assessed. As shown in Fig. 5C, in a mouse model of
atherosclerosis, the expression levels of P-IκBα were increased in
the cytoplasm and the expression levels of P-NF-κB were increased
in the nucleus. In the LPS + AD + STAT/NF-κB mice it was indicated
that that STAT3/NF-κB decoy ODNs may inhibit NF-κB activation by
inhibiting P-IκBα. These results suggested that STAT3/NF-κB decoy
ODNs may markedly inhibit the LPS-activated TLR4/NF-κB signaling
pathway in a mouse model of atherosclerosis.
The JAK/STAT signaling pathway has a vital role in
various important cellular responses, such as inflammation,
metabolism, cell proliferation and gene transcription (25). Western blot analysis was performed
to detect the protein expression levels of JAK2 and STAT3 in the
aortae. As shown in Fig. 5C, the
LPS + AD + Scr group exhibited markedly enhanced P-JAK2 and P-STAT3
expression compared with that in the NC group. By contrast, the LPS
+ AD + STAT/NF-κB groups exhibited decreased P-JAK2 and P-STAT3
expression levels compared with those in the LPS + AD + Scr group.
Taken together, these findings suggested that STAT3/NF-κB decoy
ODNs may suppress inflammation in atherosclerotic mice by
effectively inhibiting the STAT/NF-κB signaling pathway.
Discussion
The present study explored the anti-atherosclerotic
effects and molecular mechanisms of STAT3/NF-κB decoy ODNs in
LPS-induced atherosclerotic mice. The results revealed that
STAT3/NF-κB decoy ODNs can attenuate the development of
atherosclerosis by promoting ABCA1-mediated cholesterol efflux, and
reducing pro-inflammatory cytokine secretion and fibrosis-related
protein expression via suppression of the STAT/NF-κB pathway.
Atherosclerosis is characterized by the progressive
formation of vascular lesions caused by immoderate lipid deposition
and a chronic inflammatory response within the arterial walls,
subsequently leading to ischemic cardiovascular and cerebrovascular
diseases (26). ABCA1 can
regulate foam cell formation via the export of excessive
cholesterol from lipid-loaded macrophages, and maintains cellular
lipid and cholesterol homeostasis (27). Although its key role is to
maintain lipid homeostasis by controlling cellular cholesterol and
phospholipid efflux, ABCA1 has gradually been recognized as having
anti-inflammatory functions in various diseases in which
inflammation is an underlying pathogenic mechanism (28). ABCA1 expression in can promote the
export of cellular cholesterol to the extracellular acceptor
protein apolipoprotein-A1 (29).
ABCA1 expression has been reported to be decreased in
atherosclerosis, which is associated with an aggravated
intracellular cholesterol accumulation, and enhanced foam cell
generation and formation (30).
In the present study, ABCA1 expression was significantly decreased
in the arteriosclerotic mice, whereas it was significantly
increased by treatment with STAT3/NF-κB decoy ODNs.
Atherosclerosis is a chronic inflammatory condition
that is characterized by the accumulation of lipids, SMC
proliferation, cell apoptosis of SMCs, T lymphocytes and
macrophages, necrosis, fibrosis and local inflammation (31,32). The LPS from gram-negative bacteria
used in the present study is a natural ligand of TLR4 that
stimulates macrophage signaling via MyD88-dependent TLR signaling
(8,33). TLR4 stimulation activates the
NF-κB transcription factor and pro-inflammatory proteins, which
promote inflammation and initiate atherogenesis, as well as the
destabilization of atherosclerotic plaques (34). The transcription factor NF-κB
directly targets inflammation by increasing the formation of
inflammatory cytokines, chemokines and adhesion molecules, and it
can also regulate cell proliferation, differentiation, apoptosis
and morphogenesis (24). The
present study revealed that STAT3/NF-κB decoy ODNs not only
inhibited the NF-κB signaling pathway but also regulated TLR4
expression. Furthermore, STAT3/NF-κB decoy ODNs were shown to
inhibit the expression levels of the pro-inflammatory cytokines
IFN-γ, TNF-α, IL-1β and IL-6, as well as the adhesion molecules
VCAM-1 and ICAM-1 in atherosclerotic mice. In view of these
results, STAT3/NF-κB decoy ODNs may be considered effective for the
prevention and treatment of atherosclerosis, reducing inflammation
through inhibition of the NF-κB signaling pathway.
The STAT family consists of seven members (STAT1, 2,
3, 4, 5A, 5B, 6) that transduce signals from various extracellular
stimuli initiated by different cytokine families (35). STAT proteins exist in the
cytoplasm in a latent form and are stimulated by tyrosine
phosphorylation, which occurs when the JAK and Src families are
activated (9). P-STATs form homo-
or heterodimers that translocate to the nuclei and modulate the
transcription of numerous target genes (36). Within the STAT family, STAT3 is an
important transcription factor in both immunity and inflammation
(37,38). It has previously been reported
that STAT3 serves a crucial role in various diseases, including
cancer, myocardial ischemic injury, cerebral stroke and obesity
(1). In addition, it has been
suggested that STAT3 may have a critical role in all pathological
mechanisms of atherosclerosis, indicating that STAT3 could be a
novel target of atherosclerosis therapies (1). The JAK/STAT intracellular pathway is
essential in the regulation of leukocyte recruitment, foam cell
formation, and the proliferation and migration of vascular SMCs
(VSMCs), which are principal features of atherosclerosis (39-41). JAK2, STAT1 and STAT3 inhibition
can reduce lesion size and neointimal hyperplasia (42). Based on these findings, it was
observed in the present study that LPS-induced atherosclerotic mice
fed an AD and treated with a STAT3/NF-κB decoy ODN exhibited
reduced P-JAK2 and P-STAT3 protein expression levels. In
conclusion, the present study may contribute to the application of
STAT as a novel target for the treatment of atherosclerosis. The
present study highlights the essential role of STAT3 in
atherosclerosis and indicates that STAT3 inhibitors may be
potential therapeutic agents for atherosclerosis.
The vascular inflammatory response includes complex
interactions between inflammatory cells (lymphocytes, neutrophils,
monocytes and macrophages), endothelial cells, VSMCs and the ECM
(43). During chronic
inflammation, ECM proteins and their fragments can regulate the
migration of several types of cell, including endothelial cells,
SMCs and monocyte/macrophages, which are well known to contribute
to various stages of atherosclerosis (44). In particular, macrophages serve a
decisive role at all stages of the progression of atherosclerotic
lesions (45). Macrophages are
the first inflammatory cells to invade atherosclerotic lesions, and
secrete a wide range of cytokines and chemokines. NF-κB is a key
transcription factor of macrophages that is required for inducing
numerous inflammatory genes, including those encoding TNF-α, IL-1β,
IL-6, IL-12p40 and cyclooxygenase-2 (46). Therefore, inhibiting inflammation
and lipid deposition in macrophages may be an attractive
therapeutic strategy for preventing atherosclerosis (47). In the present study, it was
revealed that macrophage activation induced by an AD and LPS in
atherosclerotic mice was markedly reduced by STAT/NF-κB decoy
ODN.
In summary, the present study demonstrated that
STAT/NF-κB decoy ODNs can suppress STAT and NF-κB signaling pathway
activation in aortic tissues, and reduce VCAM-1, ICAM-1 and
inflammatory cytokine expression. The present results also
indicated that STAT/NF-κB decoy ODNs can mitigate atherosclerosis
progression by magnifying ABCA1-mediated cholesterol efflux and
relieving inflammation via inhibition of the TLR4 and STAT/NF-κB
pathways. These results provide novel insights into the
antiatherogenic molecular mechanism of STAT/NF-κB decoy ODNs and
thus provide a novel method to prevent atherosclerosis.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HJA, JBL and KKP contributed to the design of the
study and wrote the paper. HJA, MGG, HG and SB carried out the
experiments and analyzed the results. HJA and JL analyzed the data
and edited the manuscript. HJA and KKP confirm the authenticity of
all the raw data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the
Institutional Animal Care and Use Committee of the Catholic
University of Daegu (Daegu, South Korea; approval no. DCIAFCR-181
204-27Y).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This research was supported by the Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education (grant no.
NRF-2021R1I1A1A01058291). This work was supported by the grant of
Daegu Catholic University Medical Center (grant no.
RD-22-0052).
References
|
1
|
Chen Q, Lv J, Yang W, Xu B, Wang Z, Yu Z,
Wu J, Yang Y and Han Y: Targeted inhibition of STAT3 as a potential
treatment strategy for atherosclerosis. Theranostics. 9:6424–6442.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Szelag M, Piaszyk-Borychowska A,
Plens-Galaska M, Wesoly J and Bluyssen HA: Targeted inhibition of
STATs and IRFs as a potential treatment strategy in cardiovascular
disease. Oncotarget. 7:48788–48812. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xue F, Nie X, Shi J, Liu Q, Wang Z, Li X,
Zhou J, Su J, Xue M, Chen WD and Wang YD: Quercetin inhibits
LPS-induced inflammation and ox-LDL-induced lipid deposition. Front
Pharmacol. 8:402017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sikorski K, Czerwoniec A, Bujnicki JM,
Wesoly J and Bluyssen HA: STAT1 as a novel therapeutical target in
pro-atherogenic signal integration of IFNγ, TLR4 and IL-6 in
vascular disease. Cytokine Growth Factor Rev. 22:211–219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shah PK and Lecis D: Inflammation in
atherosclerotic cardiovascular disease. F1000. Res.
8:F10002019.
|
|
7
|
Fatkhullina AR, Peshkova IO and Koltsova
EK: The role of cytokines in the development of atherosclerosis.
Biochemistry (Mosc). 81:1358–1370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang S, Zou J, Li P, Zheng X and Feng D:
Curcumin protects against atherosclerosis in apolipoprotein
e-knockout mice by inhibiting toll-like receptor 4 expression. J
Agric Food Chem. 66:449–456. 2018. View Article : Google Scholar
|
|
9
|
Imbaby S, Matsuda N, Tomita K, Hattori K,
Palikhe S, Yokoo H and Hattori Y: Beneficial effect of STAT3 decoy
oligodeoxynucleotide transfection on organ injury and mortality in
mice with cecal ligation and puncture-induced sepsis. Sci Rep.
10:153162020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bromberg JF: Activation of STAT proteins
and growth control. Bioessays. 23:161–169. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bromberg J: Stat proteins and oncogenesis.
J Clin Invest. 109:1139–1142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
An HJ, Kim JY, Gwon MG, Gu H, Kim HJ, Leem
J, Youn SW and Park KK: Beneficial effects of SREBP decoy
oligodeoxy-nucleotide in an animal model of hyperlipidemia. Int J
Mol Sci. 21:5522020. View Article : Google Scholar
|
|
13
|
Morishita R, Higaki J, Tomita N and
Ogihara T: Application of transcription factor 'decoy' strategy as
means of gene therapy and study of gene expression in
cardiovascular disease. Circ Res. 82:1023–1028. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomita N, Ogihara T and Morishita R:
Transcription factors as molecular targets: Molecular mechanisms of
decoy ODN and their design. Curr Drug Targets. 4:603–608. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SJ, Park JH, Kim KH, Lee WR, Lee S,
Kwon OC, Kim KS and Park KK: Effect of NF-kappaB decoy
oligodeoxynucleotide on LPS/high-fat diet-induced atherosclerosis
in an animal model. Basic Clin Pharmacol Toxicol. 107:925–930.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reimann C, Brangsch J, Colletini F, Walter
T, Hamm B, Botnar RM and Makowski MR: Molecular imaging of the
extracellular matrix in the context of atherosclerosis. Adv Drug
Deliv Rev. 113:49–60. 2017. View Article : Google Scholar
|
|
17
|
Brasselet C, Durand E, Addad F, Zen AAH,
Smeets MB, Laurent-Maquin D, Bouthors S, Bellon G, de Kleijn D,
Godeau G, et al: Collagen and elastin cross-linking: A mechanism of
constrictive remodeling after arterial injury. Am J Physiol Heart
Circ Physiol. 289:H2228–H2233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang ST, Kreutzberger AJB, Lee J,
Kiessling V and Tamm LK: The role of cholesterol in membrane
fusion. Chem Phys Lipids. 199:136–143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Singaraja RR, Fievet C, Castro G, James
ER, Hennuyer N, Clee SM, Bissada N, Choy JC, Fruchart JC, McManus
BM, et al: Increased ABCA1 activity protects against
atherosclerosis. J Clin Invest. 110:35–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Q, Zhang M, Liang B, Shirwany N, Zhu
Y and Zou MH: Activation of AMP-activated protein kinase is
required for berberine-induced reduction of atherosclerosis in
mice: The role of uncoupling protein 2. PLoS One. 6:e254362011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lan TH, Huang XQ and Tan HM: Vascular
fibrosis in atherosclerosis. Cardiovasc Pathol. 22:401–407. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harrington JR: The role of MCP-1 in
atherosclerosis. Stem Cells. 18:65–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu W, Park SH, Meng Z, Wang F and Zhou C:
Deficiency of adipocyte IKKbeta affects atherosclerotic plaque
vulnerability in obese LDLR deficient mice. J Am Heart Assoc.
8:e0120092019. View Article : Google Scholar
|
|
24
|
Liu T, Zhang L, Joo D and Sun SC:
NF-kappaB signaling in inflammation. Signal Transduct Target Ther.
2:170232017. View Article : Google Scholar
|
|
25
|
Usui F, Shirasuna K, Kimura H, Tatsumi K,
Kawashima A, Karasawa T, Hida S, Sagara J, Taniguchi S and
Takahashi M: Critical role of caspase-1 in vascular inflammation
and development of atherosclerosis in Western diet-fed
apolipoprotein E-deficient mice. Biochem Biophys Res Commun.
425:162–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spagnoli LG, Bonanno E, Sangiorgi G and
Mauriello A: Role of inflammation in atherosclerosis. J Nucl Med.
48:1800–1815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schumacher T and Benndorf RA: ABC
transport proteins in cardiovascular disease-A brief summary.
Molecules. 22:5892017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He P, Gelissen IC and Ammit AJ: Regulation
of ATP binding cassette transporter A1 (ABCA1) expression:
Cholesterol-dependent and-independent signaling pathways with
relevance to inflammatory lung disease. Respir Res. 21:2502020.
View Article : Google Scholar
|
|
29
|
Miller NE: HDL metabolism and its role in
lipid transport. Eur Heart J. 11(Suppl H): 1–3. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Favari E, Chroni A, Tietge UJ, Zanotti I,
Escola-Gil JC and Bernini F: Cholesterol efflux and reverse
cholesterol transport. Handb Exp Pharmacol. 224:181–206. 2015.
View Article : Google Scholar
|
|
31
|
Taleb S: Inflammation in atherosclerosis.
Arch Cardiovasc Dis. 109:708–715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang X, Xue C, Xu Q, Zhang Y, Li H, Li F,
Liu Y and Guo C: Caprylic acid suppresses inflammation via
TLR4/NF-kappaB signaling and improves atherosclerosis in
ApoE-deficient mice. Nutr Metab (Lond). 16:402019. View Article : Google Scholar
|
|
33
|
Yu M, Zhou H, Zhao J, Xiao N, Roychowdhury
S, Schmitt D, Hu B, Ransohoff RM, Harding CV, Hise AG, et al:
MyD88-dependent interplay between myeloid and endothelial cells in
the initiation and progression of obesity-associated inflammatory
diseases. J Exp Med. 211:887–907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
den Dekker WK, Cheng C, Pasterkamp G and
Duckers HJ: Toll like receptor 4 in atherosclerosis and plaque
destabilization. Atherosclerosis. 209:314–320. 2010. View Article : Google Scholar
|
|
35
|
Darnell JE Jr: The JAK-STAT pathway:
Summary of initial studies and recent advances. Recent Prog Horm
Res. 51:391–403; discussion 403-394. 1996.PubMed/NCBI
|
|
36
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hillmer EJ, Zhang H, Li HS and Watowich
SS: STAT3 signaling in immunity. Cytokine Growth Factor Rev.
31:1–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miklossy G, Hilliard TS and Turkson J:
Therapeutic modulators of STAT signalling for human diseases. Nat
Rev Drug Discov. 12:611–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marrero MB: Introduction to JAK/STAT
signaling and the vasculature. Vascul Pharmacol. 43:307–309. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tamiya T, Kashiwagi I, Takahashi R,
Yasukawa H and Yoshimura A: Suppressors of cytokine signaling
(SOCS) proteins and JAK/STAT pathways: Regulation of T-cell
inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol.
31:980–985. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tian J, Liu Y, Liu Y, Chen K and Lyu S:
Cellular and molecular mechanisms of diabetic atherosclerosis:
Herbal medicines as a potential therapeutic approach. Oxid Med Cell
Longev. 2017:90808692017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sprague AH and Khalil RA: Inflammatory
cytokines in vascular dysfunction and vascular disease. Biochem
Pharmacol. 78:539–552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Raines EW: The extracellular matrix can
regulate vascular cell migration, proliferation, and survival:
Relationships to vascular disease. Int J Exp Pathol. 81:173–182.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bobryshev YV, Ivanova EA, Chistiakov DA,
Nikiforov NG and Orekhov AN: Macrophages and their role in
atherosclerosis: Pathophysiology and transcriptome analysis. Biomed
Res Int. 2016:95824302016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang N, Liang H and Zen K: Molecular
mechanisms that influence the macrophage m1-m2 polarization
balance. Front Immunol. 5:6142014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rubinow KB, Wall VZ, Nelson J, Mar D,
Bomsztyk K, Askari B, Lai MA, Smith KD, Han MS, Vivekanandan-Giri
A, et al: Acyl-CoA synthetase 1 is induced by Gram-negative
bacteria and lipopolysaccharide and is required for phospholipid
turnover in stimulated macrophages. J Biol Chem. 288:9957–9970.
2013. View Article : Google Scholar : PubMed/NCBI
|