Introduction
The receptor for advanced glycation end products
(RAGE) protein is a type of innate immune receptor protein, which
is encoded by the advanced glycosylation end-product specific
receptor (AGER) gene and plays a crucial role in a number of
inflammatory diseases. For example, RAGE is involved in the
inflammatory response during acute respiratory distress syndrome
(ARDS) (1). RAGE is a type 1
transmembrane glycoprotein, belonging to the Ig receptor
superfamily due to the existence of multiple immunoglobulins
(Ig)-like domains in the extracellular domain. The extracellular
domain contains three Ig domains, including the V-type domain, the
C1 domain and the C2 domain. It has been shown that ligand binding
mainly occurs in the V-type domain (2). The V-shaped structure of its
extracellular ligand binding part can bind to the Gla domain of
prothrombin (F2) under normal physiological conditions (3,4).
Under normal physiological conditions, the expression level of RAGE
is low; however, due to the accumulation of RAGE ligands, the
expression of RAGE is upregulated under inflammatory conditions
(5). A normal RAGE expression is
critical for maintaining a normal cell phenotype and tissue
structure, ensuring efficient gas exchange function (diffusion of
oxygen and carbon dioxide) in alveolar cells (6). RAGE is widely expressed on various
cell surfaces; however, the expression level of RAGE is low in
normal tissues. In adults, RAGE is mainly expressed in ATI cells
and exists in the form of sRAGE in gas exchange in the interstitial
space of capillary alveolar membrane (7,8).
In the case of severe lung injury, manifested as acute lung injury
or ARDS, the loss of alveolar-capillary membrane integrity, or
changes in capillary permeability, resulting a massive outflow of
protein-rich edematous fluid from the alveolar cavity. RAGE is a
key mediator of lung oxidative stress, the activation of alveolar
macrophages and emphysema following cigarette smoke stimulation
(9). Macrophages release
cytokines, tumor necrosis factor A and interleukin, and trigger
MAPK cascade reaction, leading to alveolar coagulation induction
and pulmonary coagulopathy (10-12). High mobility group protein-1
(HMGB1), also known as amphoteric protein, is a highly conserved
nuclear protein. Consisting of two DNA associative domains (a-box
and B-box) and A negatively charged C-terminal (13,14), HMGB1 is an inflammatory-related
protein that plays a critical role in numerous diseases (15,16). Although the list of HMGB1
receptors is extensive, there are only two receptor systems, and
RAGE has been fully identified as one of the HMGB1 receptors
(17). HMGB1 can function both
inside and outside the cell. Under pathological conditions, HMGB1
is passively released from infected, injured and necrotic cells or
is actively released from activated immune cells (18,19), and HMGB1 released into the
extracellular tissues functions as a damage-associated molecular
pattern protein (DAMP) by binding to its receptors (20). In vitro cell studies have
demonstrated that extracellular HMGB1 binds to receptors to produce
inflammatory cytokines mainly mediated by RAGE (21). It has been suggested that HMGB1
binding with RAGE can mediate the production of various
inflammatory mediators, such as tumor necrosis factor α (TNF-α) and
interleukin (IL)-1β, by activating the downstream inflammatory
pathways, MAPK and NF-кB (22).
The phosphorylation of IкB is a key regulator of the NF-кB pathway
during mechanical reactions, which enables NF-кB to be transferred
to the nucleus and activate target genes (23). In addition, the p65 protein is one
of the transcription factors in the NF-кB/Rel family and contains a
C-terminal activation domain, which is crucial for inducing the
expression of target genes in the NF-кB pathway (24). These data indicated that the
activation of the HMGB1/RAGE pathway amplifies the inflammatory
response in vivo and plays a critical role in the
pathogenesis of inflammatory diseases.
Prothrombin (also known as coagulation factor II,
F2) is a multifunctional serine protease and a well-known clotting
factor. The Gla domain of human F2 is an N-terminal region composed
of 46 amino acids, with 10 G-carboxyl glutamate residues (Gla). The
Gla domain is highly conserved among species and is present at the
N-terminus of proteins that undergo the vitamin K-dependent
g-carboxylation of glutamate residues (25,26). It is produced at the site of
vascular injury and plays a key role in tissue repair and lung
inflammation (27). It has been
demonstrated that the levels of thrombin are increased in the
bronchoalveolar lavage fluid of patients with ARDS and pneumonia
(28). F2 can induce neutrophil
migration and accumulation in mouse airways (29). It has been suggested that thrombin
activates protease-activated receptor (PAR)1, PAR3 and PAR4,
activating g-protein-coupled signaling pathways by cleaving
extracellular N-terminal domains and subsequently inducing cellular
responses (30) It has been
demonstrated that the RAGE ligand-binding domains, VC1 and sRAGE,
bind to F2 through the Gla domain (4). In lung and airway chambers, thrombin
regulates tissue repair by altering vascular permeability,
stimulating protease secretion, and promoting fibroblast and smooth
muscle cell adhesion, diffusion and proliferation (31,32).
It has been demonstrated that in in vivo
models, exposure to chronic hypoxia upregulates RAGE expression
(33). However, in another study,
in an in vitro model of acute hypoxia in a cancer-derived
lung epithelial cell line, hypoxia increased the expression of
total genes and RAGE genes (34).
In an in vitro model using pancreatic tumor cells, RAGE
expression was found to be independent of hypoxia regulation by
hypoxia-inducible factor (HIF)-1α; instead, the role of NF-κB was
identified (35). In addition,
HMGB1 has been confirmed to participate in tissue inflammation in a
hypoxic environment, resulting in the release of various
inflammatory mediators and the recruitment of macrophages. HMGB1 is
transferred from the nucleus to the cytoplasm and secreted to the
extracellular space to participate in the process of angiogenesis.
In vitro experiments have demonstrated that HMGB1 stimulates
the expression of HIF-1α and VEGF in perforated intervertebral disc
cells, and promotes endothelial cell migration and tube formation.
It is suggested that the HMGB1/HIF-1α/VEGF pathway may be involved
(36).
Plateau pulmonary dysfunction and plateau pulmonary
edema and lung injury are inseparable from the respiratory system,
and studies on the respiratory system are mainly concentrated on
different causes of acute lung injury (such as high oxygen, low
oxygen, smoke, etc.) and asthma, pulmonary fibrosis and other
diseases. Studies have suggested that the association between acute
lung injury, pulmonary edema, asthma and pulmonary cystic fibrosis
may be related to the abnormal expression and function of
HMGB1-RAGE (37,38). Currently, there are limited
studies available on lung injury induced by altitude hypoxia
through the HMGB1/RAGE pathway (5,39),
and only a previous study demonstrated that the HMGB1/RAGE pathway
may be involved in hypoxia-induced kidney injury (40). Some studies have demonstrated that
RAGE and F2 may be closely combined, which is closely related to
the coagulation process and related diseases (3,4).
In the altitude environment, low pressure and low oxygen can lead
to local hemorrhaging, including the capillary congestion of the
alveolar interstitium and abnormal vasoconstriction, etc.
Therefore, the present study examined whether F2 may be involved in
lung injury induced by high-altitude hypoxia. In the present study,
pathological analysis, immunofluorescence staining, western blot
analysis and other experimental methods were used to observe a rat
model of lung injury and a NR8383 (rat alveolar macrophage) in
vitro model exposed to hypoxia (altitude of 6,000 m) for 3
days. The aim was to investigate the effects of the F2/Rho and
HMGB1/RAGE/NF-кB pathways on high-altitude hypoxia-induced lung
injury in rats, and to provide a theoretical basis for the clinical
intervention of the F2/Rho and HMGB1/RAGE/NF-кB pathways in the
treatment of hypoxia-induced lung injury. The findings presented
herein provide insight into the pathogenesis of high-altitude
hypoxia-induced lung injury at the level of signal transduction
pathways, and provide new hypotheses and perspectives for the
diagnosis and treatment of high-altitude hypoxia-induced lung
injury in the future.
Materials and methods
Chemicals, reagents and antibodies
The rat TNF-α ELISA kit (cat. no. MM-0180R1), rat
IL-6 ELISA kit (cat. no. MM-0190R1), rat IL-1β ELISA kit (cat. no.
MM-0047R1), rat FII ELISA kit (cat. no. MM-0366R1) and rat RAGE
ELISA kit (cat. no. MM-70614R1) were obtained from Jiangsu Meimian
Industrial Co., Ltd.
Anti-HMGB1 (cat. no. ab190377) and anti-mouse IgG
H&L (Alexa Fluor 647) (cat. no. ab150115) antibodies were
obtained from Abcam. Anti-RAGE (cat. no. orb622096), anti-F2 (cat.
no. orb539413) and anti-rabbit IgG (H&L; CF488A; cat. no.
orb216207-CF488A) were from Biorbyt, Ltd.; anti-p38 (cat. no.
R25239), anti-phosphorylated (p-) p38 (cat. no. 310091),
anti-NF-κB-p65 (cat. no. 340830), anti-p-NF-κBp-p65 (cat. no.
340830), anti-IκBα (cat. no. 380682), anti-p-IκBα (cat. no.
R24672), anti-RhoA (cat. no. 346086), anti-ROCK1 (cat. no. R25607),
anti-β-actin (cat. no. 380624) and anti-rabbit IgG (H&L;
HRP-conjugated; cat. no. 511203) were from Chengdu Zen Bioscience
Co., Ltd. The p38MAPK inhibitor, SB203580 (cat. no. HY-10256), was
obtained from MedChemExpress.
Animal experiments
A total of 16 healthy male Sprague-Dawley (SD) rats
weighing 200-220 g (8 weeks old) were provided by the Beijing
Weitonglihua Experimental Animal Center (animal production license
no. SCXK-2021-0011). The animal experiments were approved by the
Ethics Committee of Beijing Institute of Radiation Medicine Animal
Center (approval no. IACUC-DWZX-2021-605). All animal studies
complied with the ARRIVE guidelines and the AVMA euthanasia
guidelines 2020. The rats in the normal control group (n=8) were
raised under normal temperature (~25°C) and normal oxygen
conditions (the oxygen content was about ~21%). The rats in the
model group were placed in a low-pressure oxygen chamber simulating
a plateau environment at an altitude of 6,000 m (the oxygen
concentration was maintained at 10±0.5%, n=8); the altitude was
increased to 6,000 m above sea level at a uniform speed of 10
m/sec, the ambient temperature was maintained at 20-24°C, the
relative humidity was ~40%, and the light/dark cycle was alternated
for 12/12 h. During this time period, the animals were provided
with free access to water and food. Following 72 h of continuous
exposure to hypoxia, the altitude was decreased to plain height at
a uniform speed of 10 m/sec. The rats in each group were
anesthetized intraperitoneally with 1% pentobarbital sodium at a
dose of 40 mg/kg (the anesthetic took effect at ~10-15 min. The
rats did not move, and the respiration was stable and even. The
four toes of the rats were lightly clamped with tweezers, and there
was no reaction); blood was obtained from the abdominal aorta, part
of the blood was placed in the vacuum blood collection vessel
containing EDTA for peripheral blood detection, and the target
molecules were detected after the serum was separated from the rest
of the blood; Part of the blood was placed in an EP tube containing
sodium citrate anticoagulant. The URIT-610 Coagulation Analyzer
(URIT Medical Electronic Co., Ltd.) was used to measure the four
coagulation factors: PT, TT, APTT and FIB. Part of the blood was
placed in an EP tube containing EDTA anticoagulant, and the white
blood cells, red blood cells, hemoglobin and hematocrit in
peripheral blood images were measured using a blood cell analyzer
(XN-1000V, Sysmex). the left lungs were fixed with 4%
paraformaldehyde for subsequent pathological detection; The
remaining lung tissues were placed into the cryopreservation tube,
placed in liquid nitrogen, and then transferred to the refrigerator
at -80°C for freezing, followed by analyses using ELISA, western
blot analysis and other experiments.
Hematoxylin and eosin (H&E)
staining
Briefly, the lung tissues were fixed in 4%
paraformaldehyde for 24 h and then embedded in paraffin. The
embedded tissues were cut into 4-µm-thick sections, dewaxed
and dehydrated and stained with H&E. The sections were fixed in
4% paraformaldehyde (G0002; Wuhan Servicebio Technology Co., Ltd.)
and 3pc hydrogen peroxide was used as a disinfectant (G1101; Wuhan
Servicebio Technology Co., Ltd.). The sections were then stained
with hematoxylin dye solution (G1004; Wuhan Servicebio Technology
Co., Ltd.) and the dye solution was discarded after 2 min. The
sections were then rinsed with water and % hydrochloric ethanol
(PH1813; PHYGENE Ltd.) was added for 2 min. The sections were
rinsed with water for 30 sec and 0.5% eosin dye solution was then
added for 3 min followed by rinsing with water. The sections were
observed under a light microscope (Olympus Corporation).
Immunohistochemistry
In brief, rat lung tissue was fixed in 4%
paraformaldehyde (G0002, Wuhan Servicebio Technology Co., Ltd.) at
25°C for 1 week, paraffin-embedded sections were dehydrated (G1128,
Wuhan Servicebio Technology Co., Ltd.), antigen repair was
performed, endogenous peroxidase was blocked, serum was sealed: The
sections were placed in citric acid antigenic repair buffer [citric
acid (pH 6.0) Antigen Repair Solution G1202, Wuhan Servicebio
Technology Co., Ltd.] for antigenic repair. After natural cooling,
the slides were placed in PBS (C10010500BT, Gibco; Thermo Fisher
Scientific, Inc.; pH 7.4) and washed by shaking on a decolorizing
shaker three times, 5 min each time. The slices were then placed in
3% hydrogen peroxide solution (G1101, Wuhan Servicebio Technology
Co., Ltd.) and incubated at room temperature and away from light
for 25 min. The slides were placed in PBS (pH 7.4) and washed three
times on a decolorizing shakable bed, 5 min each time. The tissue
was uniformly covered with 3% BSA (G5001, Wuhan Servicebio
Technology Co., Ltd.) in the chemical ring and closed at room
temperature for 30 min. The primary antibodies [HMGB1 (cat. no.
R22773, Zen-Bio, Inc.; 1:1,000), RAGE (cat. no. ab216329, Abcam
(1:1,000:)] were then added followed by incubation in a wet box at
4°C overnight. The secondary antibody [goat anti-rabbit IgG H&L
(HRP); cat. no. 511203, Zen-Bio, Inc.] was then added followed by
incubation at room temperature for 50 min, color staining with DAB
(Histochemistry kit DAB color developing agent; cat. no. G1211,
Wuhan Servicebio Technology Co., Ltd.; washing three times at room
temperature for 5 min each time), the cell nuclei were re-stained
with hematoxylin, and the sections were then dehydrated and sealed
(Super Clean Fast Drying Tablet Sealing Glue; cat. no. G1404, Wuhan
Servicebio Technology Co., Ltd.). Finally, microscopic examination
(Imaging system Nikon DS-U3 Nikon, Microscope Nikon E100; Nikon
Corporation) and image collection and analysis using ImageJ
software (V1.8.0; National Institutes of Health) were carried
out.
Immunoprecipitation
The immunoprecipitation method was used to examine
whether F2 and RAGE protein can produce immunoprecipitation. The
Abbkine universal immunocoprecipitation kit (Abbkine Scientific
Co., Ltd. KTD104-CN) was used to perform the experiments. Lung
tissue proteins were first extracted and homogenized using a
high-throughput ball mill. Following homogenization, the protein
concentrations were determined using the BCA method, followed by
the steps provided with the immunoprecipitation kit, denaturing
elution method, and finally western blot analysis.
ELISA
These experiments were performed according to the
protocols provided by the kit manufacturers. All reagents and
working fluids were prepared and maintained at room temperature.
The 96-well enzyme-labeled plates were prepared. The standard
solution and samples were added, with three repeated wells for each
sample. HRP was added, mixed and incubated at 37°C for 30 min. The
plates were washed and dried. The color reagent was added to each
well. The termination fluid (included in the ELISA kit) was then
added. The optical density value of each well was measured at 450
nm (Multi-label enzyme marker VICTOR X PerkinElmer, Inc.). A
standard curve was calculated and the concentration of each
specimen was calculated.
Cells, cell culture and exposure to
hypoxia
Rat pulmonary macrophages were cultured in F12k
medium (Procell Life Science & Technology Co., Ltd.) at 37°C
and a 5% CO2 saturation humidity. When the cell density
reached 80%, it was sub-cultured. The cells in the logarithmic
growth stage were prepared into a single cell suspension, and the
cells were then divided into the normal control group (C) and
hypoxia group (H). CCK-8 assay [CK04; Dongren chemical technology
(Shanghai) Co. Ltd.] was used to detect the cell viability after
12, 24 and 48 h of hypoxia. The cells at 24 h were more stable;
thus, a hypoxia incubator (Bugbox M, Baker Ruskinn) was used, and
the hypoxia culture gas was set to 5% CO2, 1%
O2, 94% N2, and the time was set to
continuous hypoxia for 24 h in a Bugbox M anaerobic/microaerobic
workstation. In order to confirm the effects of hypoxia, ELISA was
used to detect the content of certain inflammatory factors in the
culture medium of cells in each group following 24 h of culture
under different conditions, such as TNF-α, IL-6 and IL-1β. The
expression levels of HMGB1, RAGE, p38MAPK and (NF-кB) p65 were
detected using western blot analysis, and RAGE and HMGB1 were
detected using the immunofluorescence single labeling method.
Immunofluorometric assay
In brief, the NR8383 cells were fixed with 4%
paraformaldehyde, infiltrated with 1% Triton X-100 for 20 min, and
then incubated at room temperature for 1 h with sufficient blocking
solution [QuickBlock™ Blocking Buffer for Immunol Staining (P0260),
Beyotime Institute of Biotechnology]. The blocking solution was
then discarded, and the primary antibodies (HMGB1, cat. no.
ab190377 Abcam; 1:200; RAGE, cat. no. AF309, Affinity Biosciences;
1:200) were added followed by overnight incubation at 4°C. After
washing off the primary antibody with Immunol Staining Wash Buffer
(P0106, Beyotime Institute of Biotechnology), the secondary
antibodies [goat anti-rabbit IgG (H+L) Fluor 594-conjugated
antibody, cat. no. S0006, Affinity Biosciences, 1:500; goat
anti-mouse IgG H&L (Alexa Fluor 647), cat. no. ab150115, Abcam
1:500] were added, followed by incubation at room temperature for 1
h. After washing off the secondary antibody, the nuclei were
stained with DAPI (Beyotime Institute of Biotechnology) at room
temperature for 15 min and anti-fluorescence quenching agent was
then added. The sections were then observed and images were
collected under a fluorescence microscope (Ts2R, Nikon
Corporation). The results of immunofluorescence staining were
evaluated using ImageJ software (V1.8.0; National Institutes of
Health).
Transfection
The NR8383 cells in good growth state were digested
[Pancreatic Enzyme with Phenol Red with EDTA 0.25% 500 ml cell
digestive juice (25200056, Gibco; Thermo Fisher Scientific, Inc.]
and collected. After counting, 8×105 cells/well were
absorbed and inoculated into six-well plate. Each group had three
multiple wells. Following culture for 24 h, transfection with
siRAGE (Tsingke Biotechnology Co., Ltd.; concentration of 20 nM)
was performed. The cells were randomly divided into four groups as
follows: The control group, control + siRAGE group, hypoxia +
siRAGE group and the hypoxia group. The siRNA was diluted using
Opti-MEM, the transfection reagent [LabFect SP suspension cell
small nucleic acid transfection reagent (T1024, Beijing Lablead
Biotech Co., Ltd.)] was mixed upside down, the siRNA and the
transfection reagent were mixed, and the transfection compound was
added into the six-well plate. After 24 h, western blot analysis
was performed to detect the transfection efficiency of siRAGE, and
to detect whether the knockdown of RAGE had any effect on the
expression of other related proteins. The siRNA sequences were as
follows: Target sequence sense, 5′-GAC CAA CUC UCU CCU GUA UTT-3′
and antisense, 5′-AUA CAG GAG AGU UGG UCT T-3′; and negative
control sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and antisense, ACG
UGA CAC GUU CGG AAT T-3′.
Pre-treatment with p38MAPK inhibitor
In order to investigate the association between the
p38MAPK and NF-κB signaling pathways following the exposure of
NR8383 cells to anoxia, the NR8383 rat lung macrophages were
cultured with F12k at 37°C and 5% CO2 saturation and
humidity. The NR8383 cells were transferred to a 15-ml centrifuge
tube and centrifuged at 1,000 rpm for 5 min at 37°C, and the
supernatant was discarded. The cells were inoculated into six-well
plates and divided into the control group, hypoxia group and the
normal inhibition group (control + SB203580) and hypoxia inhibition
group (hypoxia + SB203580). The cells were pre-treated with the
p38MAPK inhibitor, SB203580 (10 µM), for 2 h, and the normal
control and normal inhibition groups were cultured under normal
oxygen conditions for 24 h. The expression levels of RAGE, p38MAPK,
p-p38MAPK, IκBα, p-IκBα, (NF-κB) p65 and p-(NF-κB) p65 were
examined using western blot analysis in the cells in the hypoxia
group and hypoxia inhibition group cultured at 1% O2 for
24 h.
Western blot analysis
RIPA buffer (CW2333, CWBio) was used to lyse total
proteins from lung or cells that had been frozen in liquid nitrogen
(Thermo Fisher Scientific, Inc.). Tissue/cell debris was removed
following a brief centrifugation at 13,000 rpm, and the supernatant
was collected. Total protein was examined using a BCA Protein Assay
kit as per the manufacturer's guidelines (Thermo Fisher Scientific,
Inc.). The protein (30 µg) was then separated using sodium
dodecyl sulfate-polyacrylamide gel electrophoresis in a 10%
polyacrylamide gel and electrotransferred onto polyvinylidene
difluoride membranes. The membranes were then incubated in a
blocking solution (5% skim milk powder) at room temperature for 1
h. This was followed by incubation with primary antibodies [RAGE
(1:1,000), HMGB1 (1:1,000), NF-κB-p65 (1:1,000), p38MAPK (1:1,000),
prothrombin (F2) (1:1,000), ROCK1 (1:2,000), RhoA (1:2,000), IKBα
(1:2,000) and β-actin (1:3,000)] overnight at 4°C. The membranes
were incubated with goat anti-rabbit or goat anti-mouse
immunoglobulin peroxidase conjugated secondary antibodies for 1 h
at room temperature. The membranes were developed using an enhanced
chemiluminescence detection system (ImageQuant™ 500 imaging system;
Cytiva). Data analysis was performed using ImageJ software (V1.8.0;
National Institutes of Health).
CCK-8 assay
The NR8383 cells were seeded directly into 96-well
culture plates at 8×103 cells/well and cultured in 100
µl complete DMEM (CL-0172, Procell Life Science &
Technology Co., Ltd.) with hypoxia for 12, 24 and 48 h. After 2 h,
10 µl CCK-8 solution [cat. no. 40203ES76, Yeasen
Biotechnology (Shanghai) Co., Ltd.] were added to each well and
incubated at 37°C for 1.5 h. By using an ELX-800 spectrometer
reader (BioTek Instruments, Inc.), the absorbance was measured at
450 nm.
Statistical analysis
One-way ANOVA was used with Tukey's post hoc test
for parametric data and the Kruskal-Wallis test with Dunn's
multiple comparisons for non-parametric tubular damage data. For
continuous variables, parametric variables are expressed as the
mean ± SD, and non-parametric variables are expressed as the median
and interquartile (25 and 75th percentile) range. P-values <0.05
were considered to indicate statistically significant differences.
GraphPad Prism 9 was used for all statistical analyses (GraphPad
Software, Inc.).
Results
Routine blood testing and blood
coagulation tests
The white blood cell count (WBC), red blood cell
count (RBC), hemoglobin (HGB) and hematocrit (HCT) levels of the
rats in the model of hypoxia were significantly increased compared
with those of the control group (Fig.
1A-D).
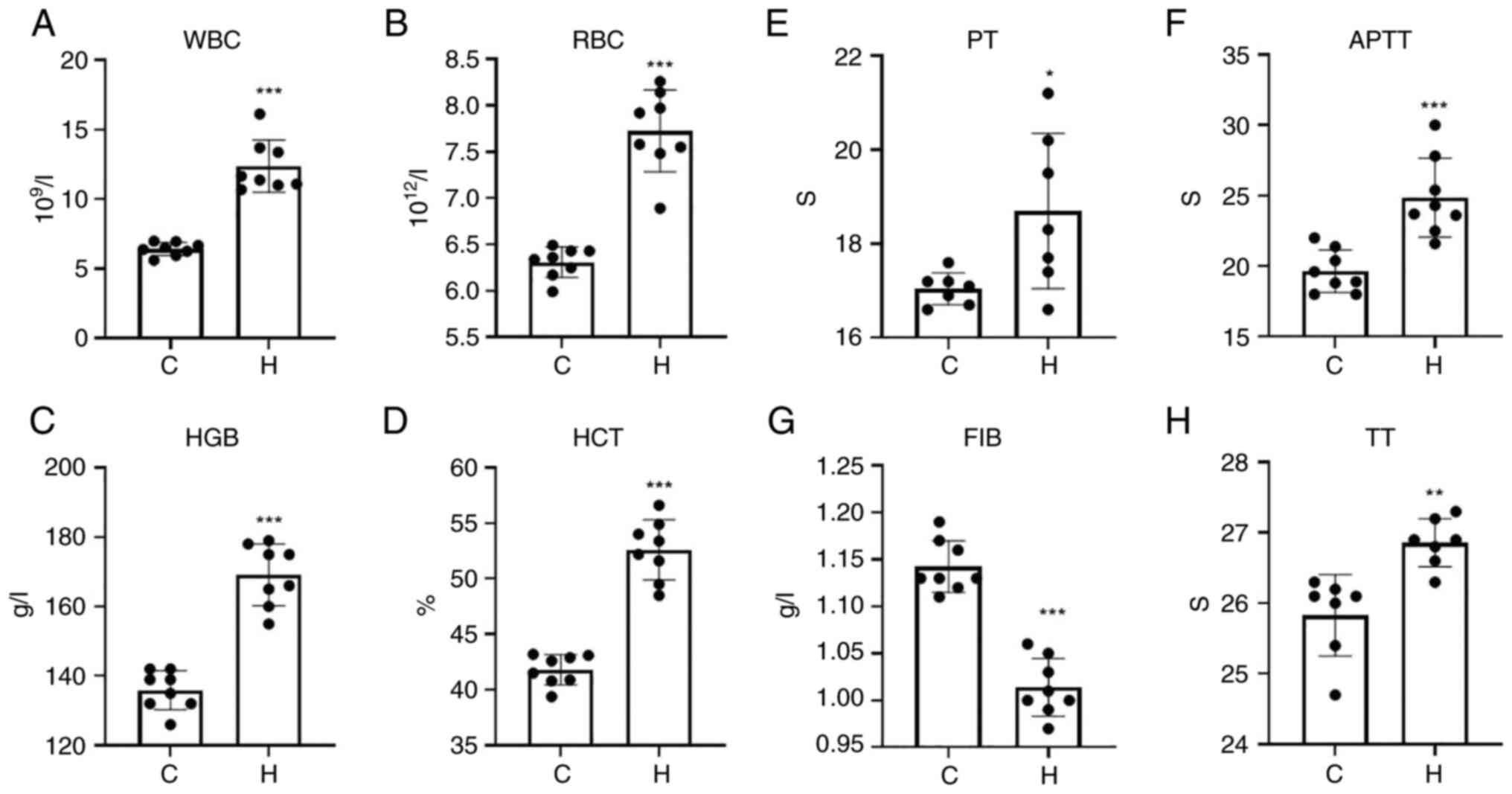 | Figure 1WBC, RBC, HGB and HCT levels in the
hypoxia and control groups, and time changes of PT, APTT, FIB and
TT. (A) WBC; (B) RBC; (C) HGB; (D) HCT; (E) PT; (F) APTT; (G) FIB,
fibrinogen; (H) TT. Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01 and
***P<0.001, vs. the control group. WBC, white blood
cell count; RBC, red blood cell count; HGB, hemoglobin; HCT,
hematocrit; PT, prothrombin time; APTT, activated partial
thromboplastin time; FIB, fibrinogen; TT, thrombin time; C,
control; H, hypoxia. |
The four coagulation tests, including prothrombin
time (PT), activated partial thromboplastin time (APTT), thrombin
time (TT) and fibrinogen (FIB), were used to determine normal
coagulation functions. PT mainly reflects the status of the
exogenous coagulation system. APTT mainly reflects the status of
the endogenous coagulation system. TT mainly reflects the time of
conversion of fibrinogen to fibrin and FIB mainly reflects the
amount of fibrinogen. The results of the four coagulation indices
are presented in Fig. 1E-H. The
PT, APTT and TT of the hypoxia group was significantly prolonged,
and the content of FIB was significantly decreased. The abnormal
coagulation function of the hypoxia group was affected by PT, APTT,
TT and FIB, which may indicate that SD rats exposed to hypoxia
exhibit bleeding phenomena and abnormal coagulation.
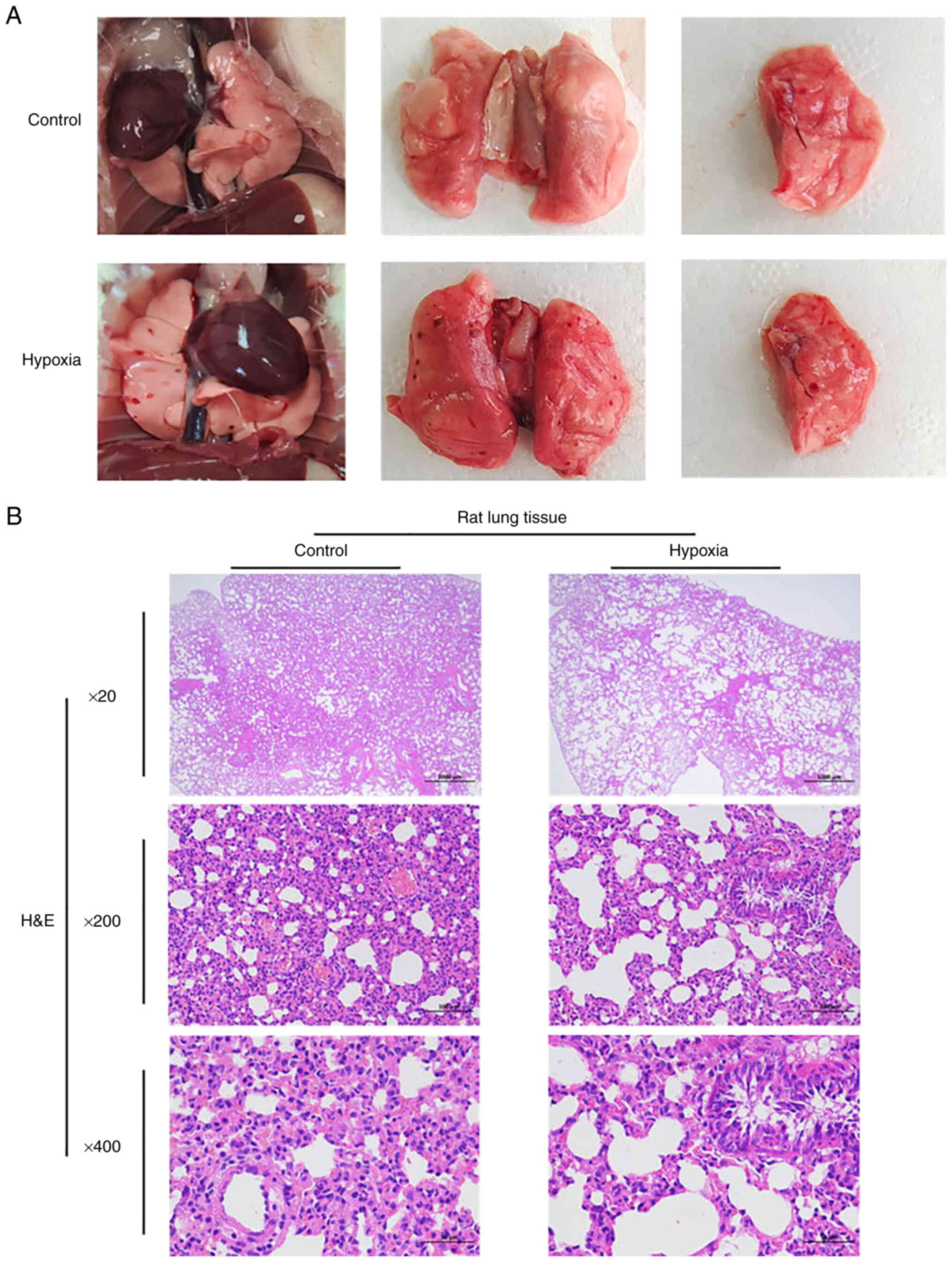
Ecchymosis and bleeding appear in the
lungs of rats following acute hypoxia; acute hypoxia can lead to
lung injury
The anatomical functions of the rats in the two
groups were recorded in real-time. During the dissection of rats in
the hypoxia group, congestion and bleeding of internal organs was
common, and ecchymosis occurred in the lungs of the rats in the
hypoxia group compared with the control group (Fig. 2A). In general, H&E staining
revealed that compared with the control group, the rats in the
hypoxia group had lung injury. The lung tissue of the rats in the
hypoxia group exhibited the watery degeneration of bronchial
epithelial cells, loose cytoplasm and light staining; perivascular
edema was observed, with a small amount of lymphocyte infiltration.
Emphysema was also observed locally, and the alveolar wall became
narrow and broken, forming a large cystic cavity. H&E staining
revealed a certain degree of lung injury in the hypoxia group, as
illustrated in Fig. 2B.
The F2/Rho and HMGB1/RAGE/NF-кB signaling
pathways may be involved in hypoxia-induced lung injury in
rats
It has been shown that the combination of HMGB1 and
RAGE can stimulate the downstream signaling pathways, p38MAPK and
ERK. p38MAPK plays a crucial role in the regulation of stress,
hypoxia and inflammation, and is considered to be the meeting point
and common channel of multiple signaling pathways (41); the binding of the extracellular
ligand HMGB1 to the RAGE receptor, in turn activates the NF-κB
pathway and leads to the production of related proinflammatory
cytokines, such as TNF-α (42).
The present study demonstrated that the rats in the hypoxia group
exposed to hypoxia simulation at an altitude of 6,000 m exhibited
inflammatory injury in the lung tissue. The results of ELISA of rat
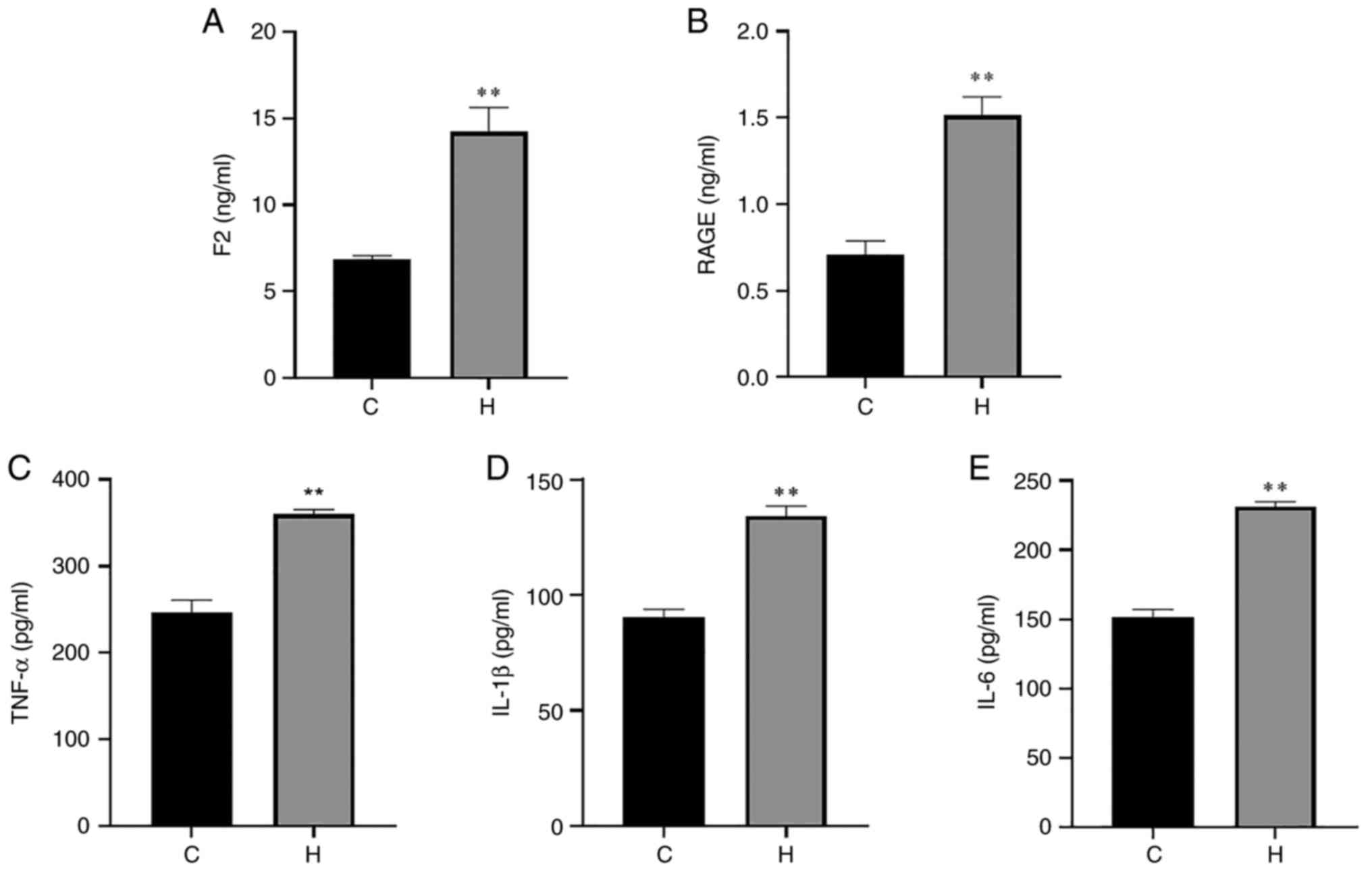
serum and lung tissue are presented in Figs. 3 and 4. Compared with the control group, the
levels of F2, RAGE and TNF-α in serum in the hypoxia group were
increased, with a highly significant difference; the levels of
IL-1β and IL-6 were also significantly increased (Fig. 3). The contents of F2, RAGE, TNF-α,
IL-1β and IL-6 in the lung tissue of rats in the hypoxia group were
also significantly higher than those in the control group (Fig. 4). The lung tissues of the rats
were used for western blot analysis. The levels of HMGB1/RAGE and
F2/Rho pathway-related proteins and p38 MAPK/NF-кB pathway-related
proteins were detected. When comparing protein expression of in the
lung tissue in the two groups, the levels of F2, ROCK1 and RhoA
were found to be increased in the hypoxia group (Fig. 5A and B). The Rho/ROCK pathway is
related to a number of physiological functions, such as vascular
and tissue permeability, tissue contraction and growth (43). It has been shown that RAGE/ROCK1
pathway can play a role in the early changes of human pulmonary
microvascular endothelial cell barrier permeability induced by
HMGB1 (44). After HMGB1
activates the RhoA/ROCK1 pathway through RAGE, it can participate
in the mechanism of early microvascular endothelial cell barrier
permeability disorder (44).
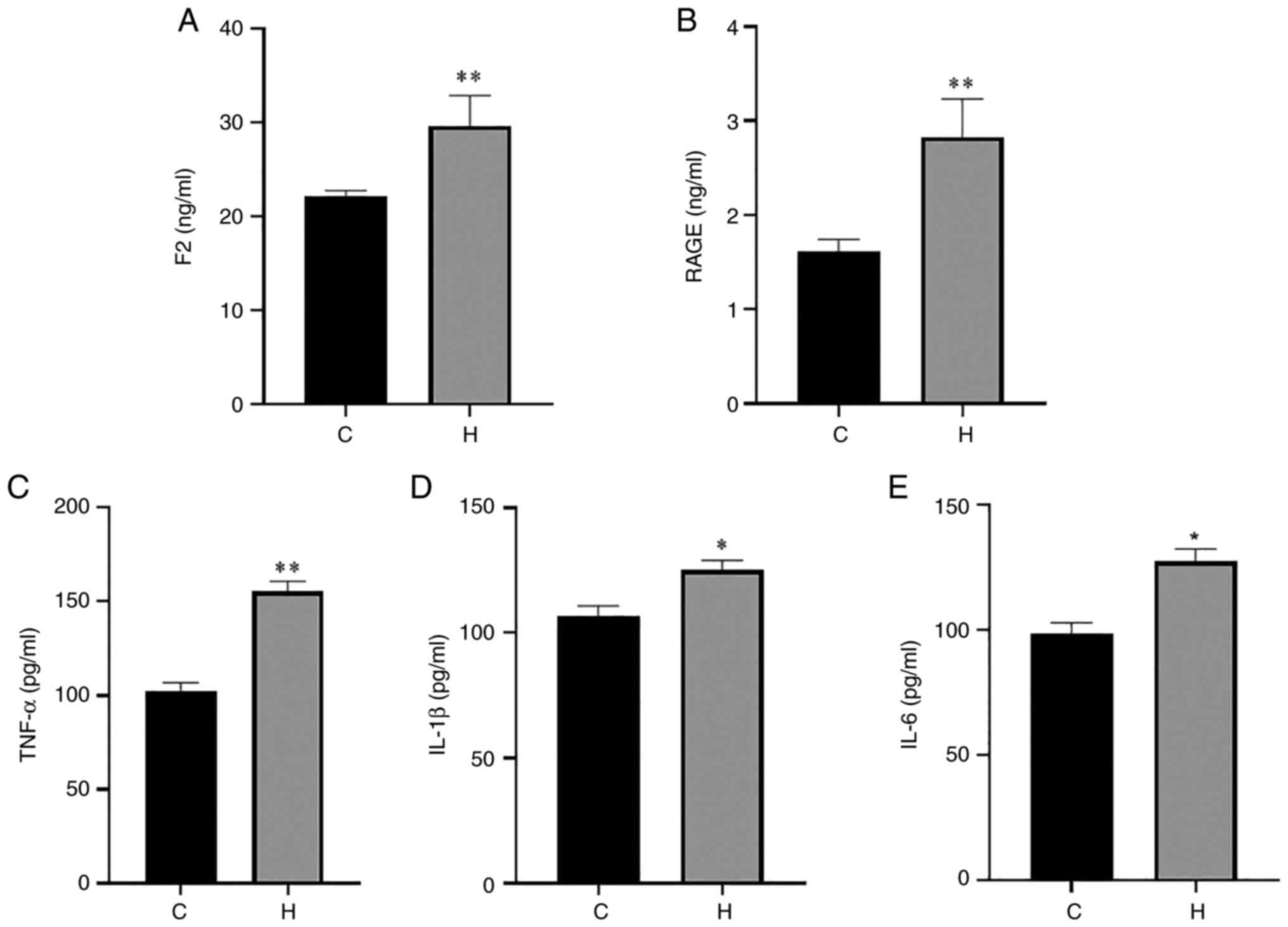 | Figure 3Contents of F2, RAGE, TNF-α, IL-1β
and IL-6 in the serum of rats in the control and hypoxia groups.
(A) The content of F2; (B) the content of RAGE; (C) the content of
TNF-α; (D) the content of IL-1β; (E) the content of IL-6. Data are
presented as the mean ± standard deviation. *P<0.05
and **P<0.01, vs. the control group. C, control; H,
hypoxia; F2, prothrombin; RAGE, receptor for advanced glycation end
products; TNF-α, tumor necrosis factor α; IL, interleukin. |
 | Figure 4Contents of F2, RAGE, TNF-α, IL-1β
and IL-6 in the lungs of rats in the control and hypoxia groups.
(A) The content of F2; (B) the content of RAGE; (C) the contents of
TNF-α; (D) the content of IL-1β; (E) the contents of IL-6. Data are
presented as the mean ± standard deviation. **P<0.01,
vs. the control group. C, control; H, hypoxia; F2, prothrombin;
RAGE, receptor for advanced glycation end products; TNF-α, tumor
necrosis factor α; IL, interleukin. |
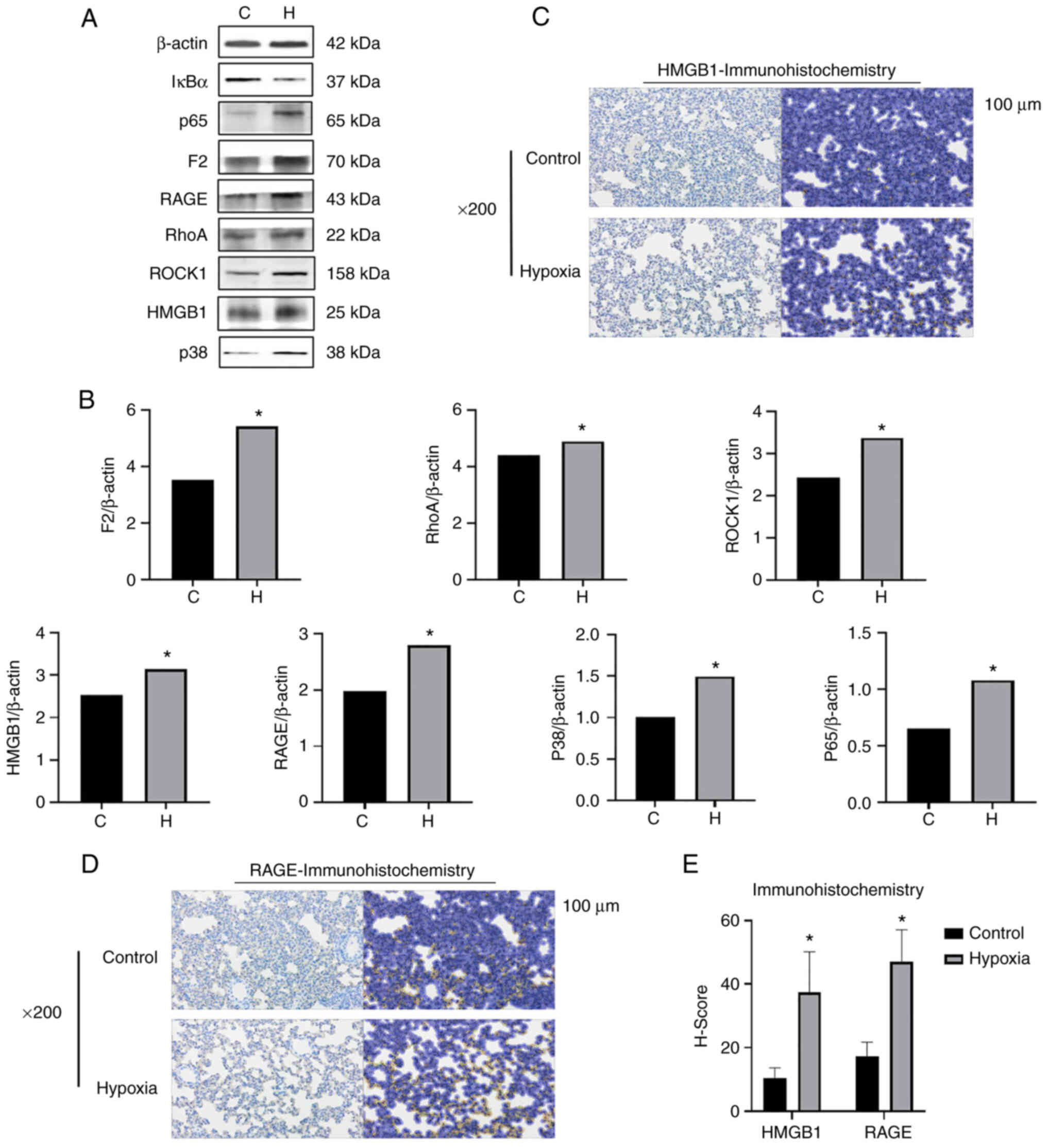 | Figure 5The expression of F2/Rho,
HMGB1/RAGE/p38MAPK/NF-κB signal pathway-related proteins increases
in the lung tissues of rats simulated high-altitude hypoxia. (A)
Western blot analysis was performed to examined the protein
expression levels of F2, ROCK1, RhoA, HMGB1, RAGE, p38 MAPK and
(NF-κB) p65 in the rat lung tissue. (B) Relative protein expression
was normalized to that of the respective control, β-actin. (C-E)
Immunohistochemistry of RAGE and HMGB1 in lung tissues of rats. The
staining results of the images revealed that the expression of RAGE
and HMGB1 in the lung tissues of rats exposed to hypoxia was
significantly increased compared with that of the control rats. The
left and right dark and bright images in each group is the
comparison of the results before and after molecular staining. The
positive results in the bright pictures refer to the immunostaining
of HMGB1 and RAGE. *P<0.05 vs. the control group. F2,
prothrombin; RAGE, receptor for advanced glycation end products;
HMGB1, high mobility group protein-1; C, control; H, hypoxia. |
In addition, it has been shown that RAGE and F2 can
closely bind under normal physiological conditions, participate in
coagulation and complement processes, and play a role in certain
inflammatory and coagulation disorders (4). In the present study, the expression
of F2, RhoA, ROCK1 and HMGB1/RAGE in the lung tissue of rats in the
hypoxia group was found to be increased (Fig. 5A and B), which may indicate that
high-altitude hypoxia can increase vascular permeability in rat
lung tissue. The activation of HMGB1/RAGE may trigger an immune
response and a pulmonary inflammatory response that causes the
pulmonary microvascular endothelial cells to maintain the
endothelial barrier between the microvascular lumen and the lung
matrix, resulting in abnormal permeability and contraction of
pulmonary microvessels. The abnormal increase in the expression of
F2, ROCK1 and RhoA, combined with the lung pathological results of
the present study, suggest that hypoxia can lead to pulmonary
microvascular permeability and endothelial dysfunction, resulting
in local exudative bleeding in the lungs. The protein expression
levels of HMGB1, RAGE, p38MAPK and (NF-кB) p65 were significantly
increased in the lung tissue of rats in the hypoxia group (Fig. 5A and B). Moreover, the results of
the immunohistochemical analyses of RAGE and HMGB1 in the rat lung
tissues revealed that the expression of RAGE and HMGB1 in the lung
tissues of rats in the hypoxia group was significantly increased
compared with those of the control group, indicating that the
HMGB1/RAGE inflammatory axis in the rat lung tissues was activated
and was involved in their inflammatory responses under hypoxic
conditions (Fig. 5C-E). The
abnormal increase in the expression of p38 and p65, as well as the
abnormal increase in the levels of the proinflammatory factors,
TNF-α, IL-1β and IL-6, in the aforementioned ELISA experiments,
suggest that the HMGB1/RAGE and p38MAPK/NF-κB signaling pathways
are associated with hypoxia-induced inflammatory injury in rat lung
tissue.
The F2/Rho and HMGB1/RAGE/NF-кB signaling
pathways may be involved in the hypoxia-induced inflammatory injury
to NR8383 cells
HMGB1 is considered a DAMP, which is passively
leaked by cells or actively secreted by stimulated macrophages and
monocytes due to its loose binding to chromatin. The detection of
RAGE in immune cells suggested the biological function of RAGE in
the occurrence and development of inflammation (45). Alveolar macrophages play a crucial
role in the innate immune response as the first line of defense
against external stimuli and the invasion of pathogenic
microorganisms in lung tissue and coordinate other immune cells
(46). Thus, to further determine
whether HMGB1-RAGE/NF-кB is involved in pulmonary inflammatory
injury during hypoxia, in the present study, rat alveolar
macrophage NR8383 cells were cultured and exposed to hypoxia. The
NR8383 cells were randomly divided into two groups as follows: The
control group and 1% O2 hypoxia group. An
immunofluorescence single-labeling experiment was performed on
cells in each group, and the content of pro-inflammatory factors
(TNF-α, IL-1β and IL-6) in the culture medium was detected after 24
h of cell culture using ELISA and the protein expression level of
HMGB1-RAGE, F2/Rho, p38MAPK and (NF-кB) p65 were detected using
western blot analysis. The viability of macrophages under hypoxic
conditions at different time points was examined synchronously. It
was found that the viability of the macrophages in the hypoxia
group decreased compared with the control group at 24 h, and the
cells did not appear polarized at this time. The results
immunofluorescence staining revealed that following exposure to
hypoxia, compared with the control group, the cells in the hypoxia
group exhibited an increased fluorescence intensity of HMGB1 and
RAGE (Fig. 6B-D). The results of
ELISA revealed that there was a significant increase in the content
of TNF-α, IL-1β and IL-6 in the hypoxia group compared with the
control group (Fig. 6E-G). The
results of western blot analysis also demonstrated that the
expression levels of F2, ROCK1, RhoA, HMGB1, RAGE, p38MAPK and
(NF-κB) p65 in hypoxia group were significantly higher than those
in the control group, and the expression of p65 and other proteins
significantly increased in the hypoxia group (Fig. 7). The results of the cell hypoxia
experiment in the present study are consistent with those obtained
for the animal experiments, indicating that the F2/Rho,
HMGB1/RAGE/p38MAPK/NF-кB signaling pathways play a role in lung
injury induced by hypoxia.
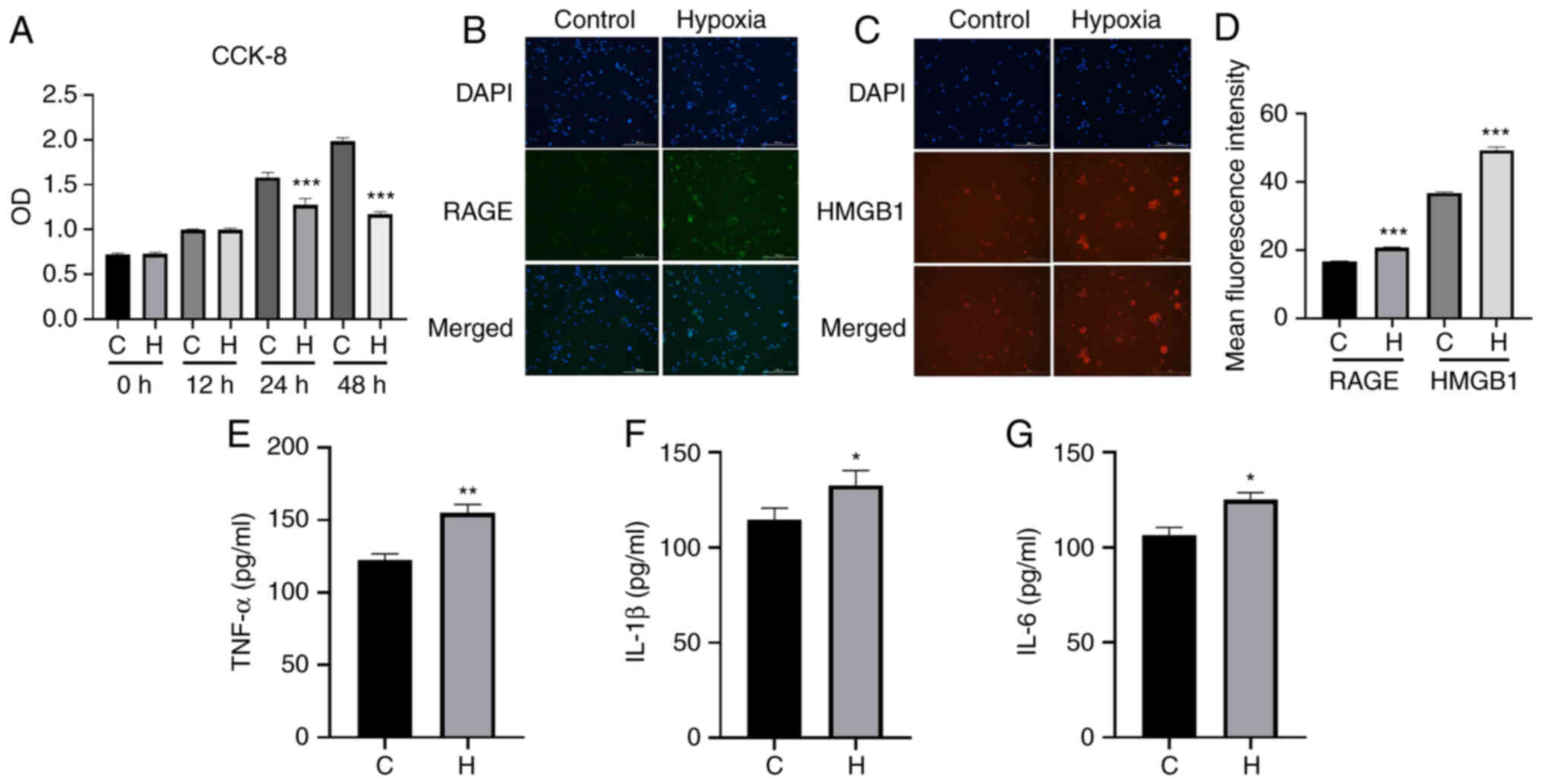 | Figure 6Results of immunofluorescence
staining of HMGB1/RAGE and ELISA of the inflammatory factors,
TNF-α, IL-1β and IL-6, in the two groups of NR8383 cells. (A) From
0 to 24 h, cells in both the control and the anoxic group exhibited
an increasing trend in viability; from 24 to 48 h, the cells in the
control and the model group continued to exhibit cell growth,
although the growth of the cells in the anoxic group was slower,
and using an image cell analyzer, the cells at 24 h were more
stable than those at the other time points. Therefore, the final
anoxic conditions of NR8383 cells were 1% oxygen, 5% carbon dioxide
and 94% nitrogen for 24 h. (B) The fluorescence intensity of HMGB1
protein in the control and hypoxia group. (C) The fluorescence
intensity of RAGE protein in the control and hypoxia group. HMGB1
staining is red, DAPI staining is blue, RAGE staining is green. (D)
The mean fluorescence intensity of RAGE and HMGB1 is presented.
(E-G) ELISA was used to assess the contents of (E) TNF-α, (F) IL-1β
and (G) IL-6 in the control and hypoxia group (24 h of culture).
Magnification, ×40 and scale bar, 200 µm. Data are presented
as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001, vs. the
control group. C, control; H, hypoxia; RAGE, receptor for advanced
glycation end products; HMGB1, high mobility group protein-1;
TNF-α, tumor necrosis factor α; IL, interleukin. |
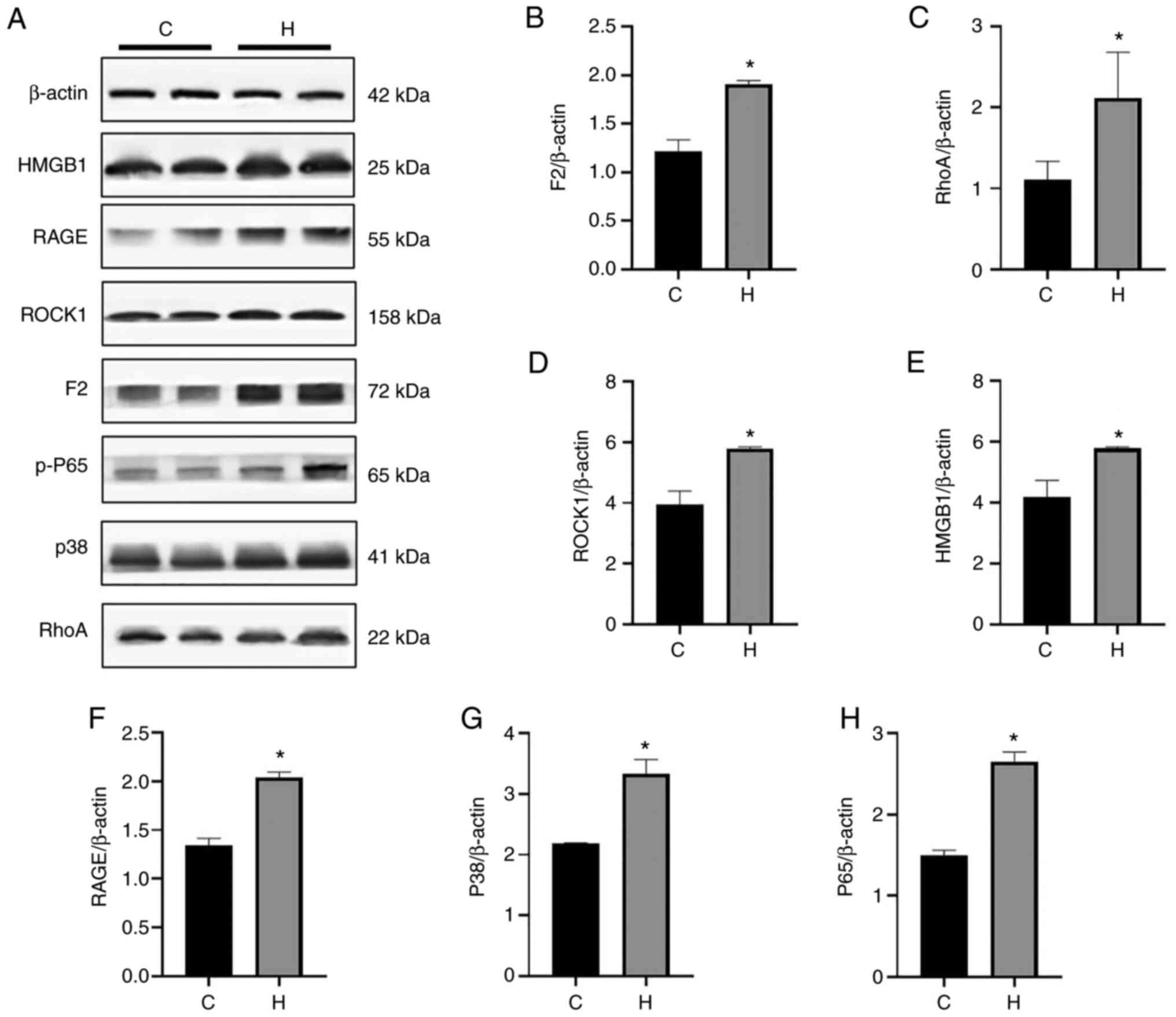 | Figure 7The expression of F2/Rho,
HMGB1/RAGE/p38MAPK/NF-κB signaling pathway-related proteins
increased in the NR8383 cells with exposed to hypoxia. (A) Western
blot analysis was performed to examine the protein expression
levels of F2, ROCK1, RhoA, HMGB1, RAGE, p38MAPK and (NF-κB) p65.
(B-H) Relative protein expression was normalized to that of the
respective control, β-actin. Protein expression exhibited variable
changes; *P<0.05, vs. the control group. C, control;
H, hypoxia; RAGE, receptor for advanced glycation end products;
HMGB1, high mobility group protein-1. |
Immunoprecipitation of F2 and RAGE
reveals a direct interaction between the two proteins
To confirm whether F2 interacts with RAGE, the
present study performed an immunoprecipitation assay to detect the
association between F2 and RAGE. The experimental procedure was
performed according to the instructions provided with the
respective the kit. Protein denaturation was carried out, and
western blot analysis was then used to detect the expression levels
of the F2 and RAGE proteins, and the results of interaction between
the two proteins were obtained (Fig.
8). When comparing the western blot band positions of the
control and hypoxia groups, the bands corresponding to the
co-expression of the F2-RAGE interaction in the two-bead hypoxia
and two-bead control groups were less prominent than those of F2
and more prominent than those of RAGE. Moreover, the expression of
interacting co-expressed proteins in the two-bead hypoxia group was
higher than that in the two-bead control group. This result also
indicated that the protein expression levels of F2 and RAGE
increased under hypoxic conditions (Fig. 8). The interaction between F2 and
RAGE was confirmed by immunoprecipitation, which provides reliable
evidence for the involvement of the F2/RhoA and RAGE/HMGB1
signaling pathways in the mechanisms of lung injury at a high
altitude.
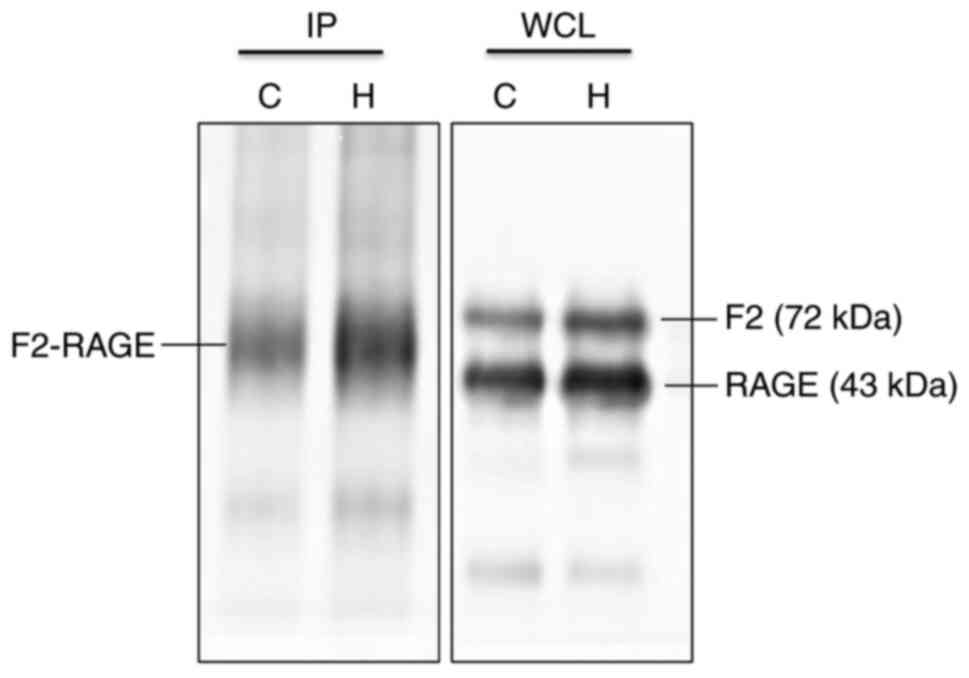 | Figure 8Immunoprecipitation of F2 and RAGE
reveals a direct interaction between the two proteins. In whole
cell lysate group, F2 and RAGE were expressed at the molecular
weight of normal protein (F2, 72 kDa; in the immunoprecipitation
group, F2 and RAGE were co-expressed at the same location (~55
kDa). Comparing the expression of F2 and RAGE proteins in the two
groups, it was found that there was a direct interaction between
the two proteins. IP, immunoprecipitation; WCL, whole cell lysate;
C, control; H, hypoxia; RAGE, receptor for advanced glycation end
products; HMGB1, high mobility group protein-1; F2,
prothrombin. |
Expression of HMGB1 and the
phosphorylated protein of NF-кB are affected by hypoxia following
RAGE gene knockdown
The cells were randomly divided into four groups as
follows: The control, control + siRAGE, hypoxia + siRAGE and
hypoxia groups, and cultured according to the culture conditions.
First of all, according to the experimental results show in
Fig. 9A and B, the control siRNA
+ hypoxia group and the siRAGE + hypoxia group were compared, and
it was found that the control siRNA had no effect on RAGE
expression following hypoxia; however, siRAGE decreased RAGE
expression under hypoxic conditions. It was found that the
expression levels of HMGB1, RAGE, p-NF-κB and p-p38 in the cells in
the hypoxia group were significantly higher than those in the
control group (Fig. 9C and D).
The protein expression level of RAGE was significantly decreased in
the cells exposed to hypoxia and RAGE gene knockdown, and the
expression level of HMGB1 was also significantly decreased.
Moreover, the protein expression levels of p38MAPK were also
significantly affected. Therefore, the HMGB1/RAGE/p38MAPK/NF-кB
inflammatory signaling pathway may be activated in cells exposed to
hypoxia (Fig. 9).
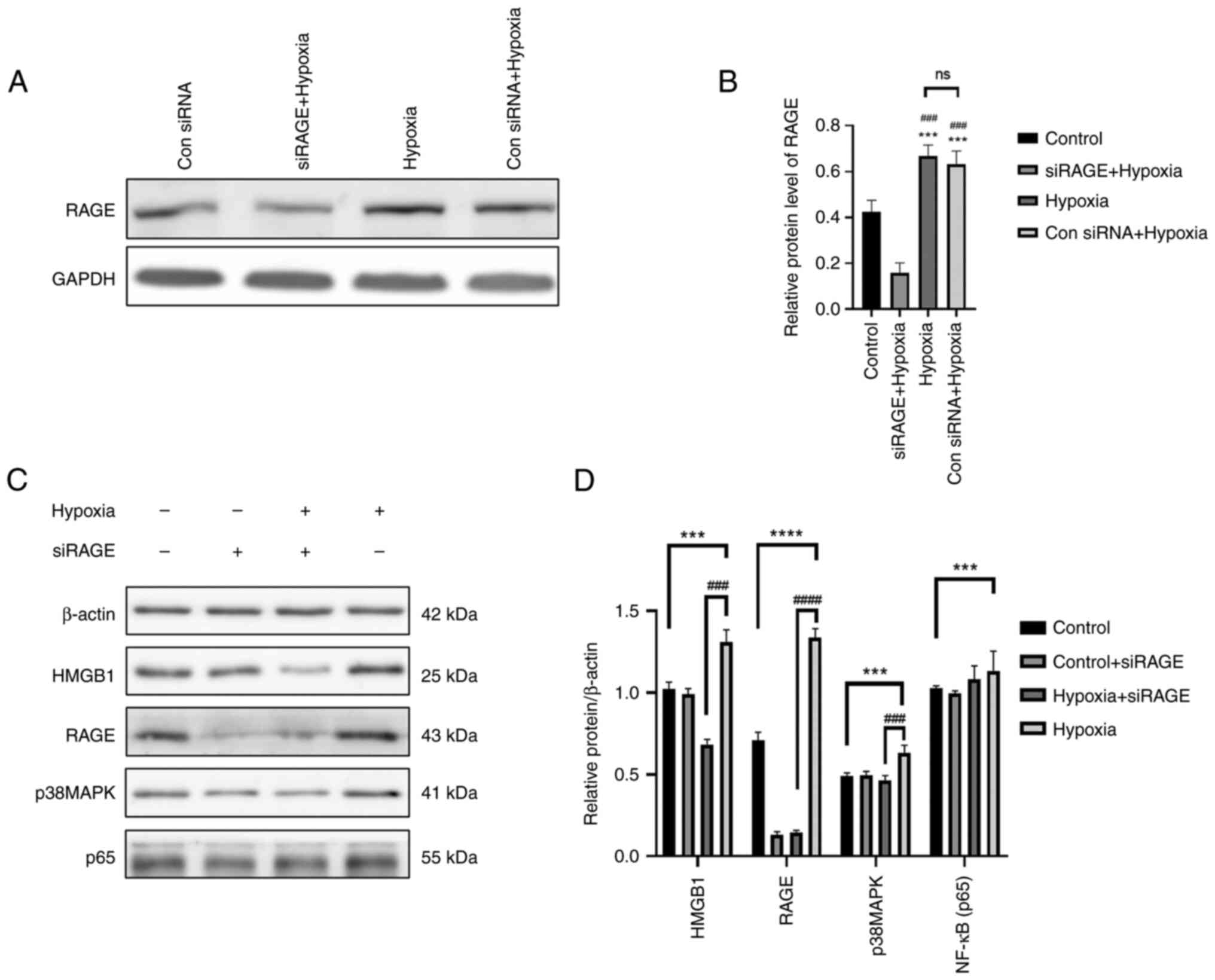 | Figure 9The cells were randomly divided into
four groups as follows: The control group, control + siRAGE group,
hypoxia + siRAGE group and hypoxia group, and cultured according to
the culture conditions (the concentration of siRAGE was 100 nM).
(A) Western blot analysis was performed to examine the protein
expression levels of RAGE in the different groups. (B) The relative
protein expression gray scale statistics of RAGE. (C) Western blot
analysis was performed to examine the protein expression levels of
RAGE, HMGB1, p-p38, p-p65, HIF-1α and β-actin in NR8383 cells in
the control, control + siRAGE, hypoxia + siRAGE and hypoxia groups.
(D) The relative protein expression was normalized to that of the
respective control, β-actin. Data are presented as the mean ±
standard deviation. ***P<0.001 and
****P<0.0001, comparison between control group and
hypoxia group; ###P<0.001 and
####P<0.0001, comparison between the hypoxia group
and hypoxia + siRAGE group; ns, not significant; RAGE, receptor for
advanced glycation end products; HMGB1, high mobility group
protein-1. |
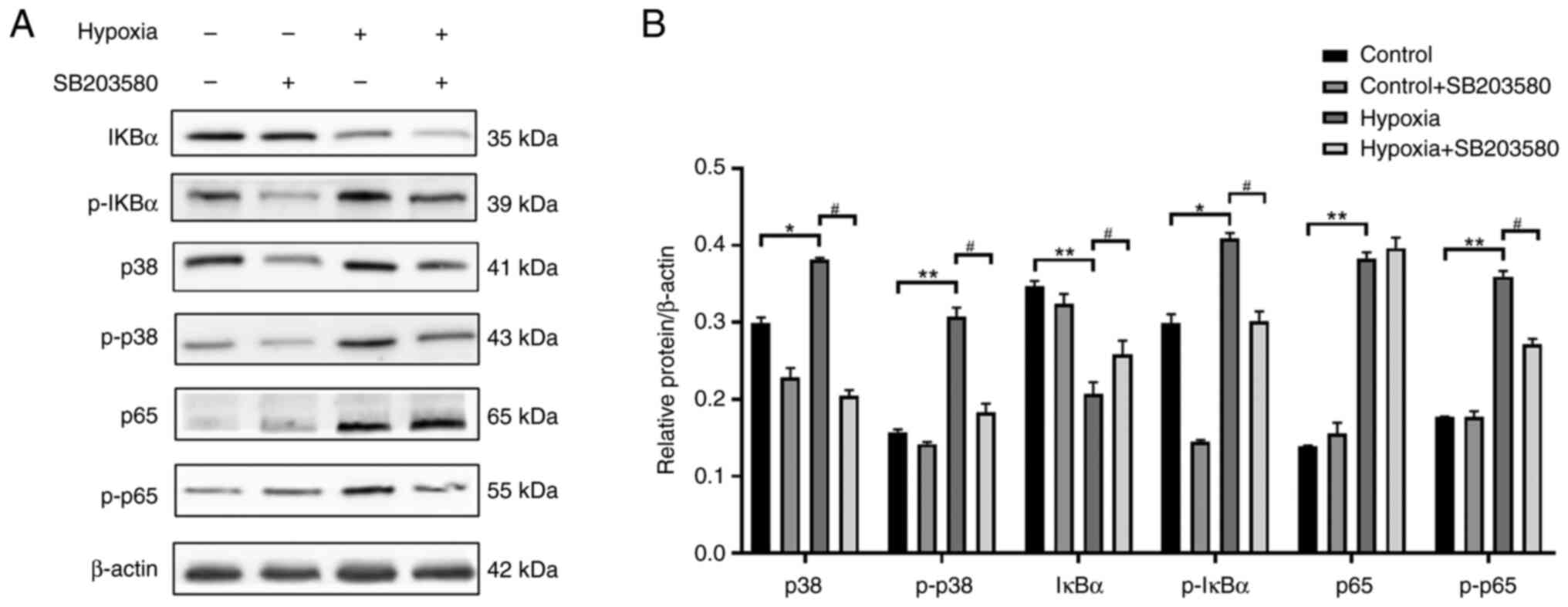
Pre-treatment with p38MAPK inhibitor
suppresses the phosphorylation of (NF-кB) p65 in NR8383 cells
cultured under hypoxic conditions
Rat lung NR8383 macrophages were cultured with F12k
at 37°C and 5%CO2 saturation and humidity. When the cell
density reached 80%, the cells were sub-cultured. To explore the
association between the p38MAPK and NF-κB signaling pathways, the
cells were pre-treated with SB-203580 (10 µM), a p38MAPK
inhibitor, for 2 h. The cells in the logarithmic growth stage were
then prepared into cell suspension, and divided into the normal
control, hypoxia, normal inhibition (control + SB203580 group) and
the hypoxia inhibition group (hypoxia + SB203580 group) groups. The
cells in the normal control and normal inhibition groups were
cultured under normoxic conditions for 24 h, and those in the
hypoxia and hypoxia inhibition groups were cultured under
conditions of 1% O2 for 24 h. The expression levels of
p38MAPK, p-p38MAPK, IκBα, p-IκBα, (NF-κB) p65 and p-(NF-κB) p65
were detected using western blot analysis (Fig. 10). As shown in Fig. 10, p38MAPK protein was inhibited
after hypoxia in pretreated cells treated with p38MAPK inhibitor,
and the protein expressions of p-p38MAPK, p-IKB α, and p-NF-κBp65
were also decreased. There was no significant difference in the
protein expression of NF-κBp65
Discussion
Hypoxia is a potent pro-inflammatory stimulus in
systemic organs (47). The
responses to high altitude acute hypoxia in the lungs apparently
allow for the development of an inflammatory phenotype. A key
pathological feature of acute lung injury is local alveolar
hypoxia, which leads to an inflammatory pulmonary phenotype in high
altitude regions. Lung inflammation may actually contribute to the
pathogenesis and progression of lung injury (48). Additional studies have
demonstrated that alveolar hypoxia contributes to an inflammatory
phenotype in the lungs, both in humans and in rodents exposed to
hypoxia (49-51). These damaging inflammatory effects
in the lungs may be not merely a consequence of the disease
process, but may actively contribute to progressive lung injury
after the disease process is initially established (49,52). A high altitude is a natural
hypoxic chamber. Due to its unique geographical environment of low
pressure and hypoxia, various diseases are caused under these
conditions. Acute hypoxia may directly cause respiratory
discomfort. In severe cases, it can result in profound lung injury,
pulmonary edema and ARDS. Hypoxia-induced inflammatory responses
lead to the recruitment of immune cells, the activation of
downstream signaling pathways, and the induction of proinflammatory
cytokines and chemokines (53,54). In addition, low concentrations of
oxygen prolong neutrophil survival and increase the permeability of
endothelial and vascular cells (55,56). It should be noted that
inflammation is a main cause of hypoxia. Inflamed tissue is
exacerbated by hypoxia due to cellular infiltration, edema
formation and microthrombosis, which in combination, lengthen the
diffusion distance between capillaries and metabolically active
cells. The consequent reduction in oxygen delivery rate reduces
cellular PO2, which induces inflammation and further
promotes the spread of a destructive immune response (57). Hypoxia and inflammation are
mutually causal, ultimately leading to lung damage.
Previous studies have demonstrated that the
activation of the HMGB1/RAGE signaling pathway may be related to
lung diseases. It has been found that RAGE knockdown, the exogenous
administration of sRAGE and anti-HMGB1 antibody can alleviate lung
injury in a mouse model of pulmonary ischemia/reperfusion (39). Following stimulation with hypoxia,
RAGE and HMGB1 form the HMGB1/RAGE axis, which interacts with
various molecules on the cell membrane to induce an inflammatory
response (58). The released
HMGB1 can mediate the release of a large number of pro-inflammatory
mediators such as TNF-α, IL-6, IL-1β and IL8 through the activation
of p38MAPKand NF-κB signaling pathways by RAGE, and promote the
inflammatory response (59-61). Therefore, in the present study,
western blot analysis was used to detect the proteins related to
NF-κB and p38MAPK signaling pathways that may be activated by RAGE
and HMGB1, and the protein expression levels of NF-κB p65 and
p38MAPK in the hypoxia group were significantly increased (Fig. 5). In addition, in the present
study, an in vitro cell hypoxia model was established by
placing NR8383 cells in a Bugbox M anaerobic/microaerobic
workstation under continuous hypoxia for 24 h. The expression
levels of TNF-α, IL-6 and IL-1β were significantly increased
following exposure to hypoxia. The results of the cellular
immunofluorescence assay revealed that the fluorescence intensity
of RAGE and HMGB1 increased in cells following exposure to hypoxia
(Fig. 6). Western blot analysis
revealed that the levels of RAGE, HMGB1, F2, RhoA, ROCK1, p38MAPK
and NF-κBp65 were significantly increased in the cells following
exposure to hypoxia (Fig. 7). The
results of the in vitro experiments were consistent with
those obtained for the animal experiments. Research has
demonstrated that RAGE can directly activate p38MAPK and NF-κB upon
binding to its ligands (62).
Therefore, the present study examined whether there was a link
between the p38MAPK and NF-κB pathways. With the aid of the p38MAPK
inhibitor, SB203580, the protein expression levels of p38, p-p38,
IкBα, p-IкBα, p65 and p-p65 were detected. It was found that the
expression levels of p-IкBα, p38, p-p38 and p-p65 were upregulated,
whereas the expression levels of p-IкBα, p38, p-p38, and p-p65 were
downregulated (Fig. 10),
indicating that the NF-κB and p38MAPK signaling pathways were
activated following hypoxia and that there was an association
between them. Following hypoxia, the signal of IκBα was weakened,
which is due to the phosphorylation of IκBα under the stimulation
of hypoxia. It has been demonstrated when stimulated by external
factors, IKBα is phosphorylated, and NF-κB is dissociated from it
and is transferred to the nucleus, where it acts as a transcription
factor to regulate downstream genes (such as adhesion molecules,
cytokines, etc.) and mediate signaling cascade (63).
Coagulation and inflammation are closely related to
the development of vascular disease. Tissue factor is the primary
initiator of the extrinsic pathway of blood coagulation, and it
plays a central role by activating coagulation factors to induce a
pro-inflammatory response, thereby initiating coagulation and
downstream cellular signaling pathways. Not only does inflammation
activate coagulation, but coagulation in turn perpetuates the
inflammatory response (64).
Research has indicated that anti-coagulation therapy not only
reduces the activation of coagulation, but also inhibits
inflammation, which underscores the interaction between coagulation
activation and cytokine release in vivo (65). Inflammation can enhance blood
coagulation through tissue factor-mediated thrombin production.
These tissue factors are upregulated on monocytes, macrophages and
endothelial cells (66). After
cells are stimulated to induce hypoxia, they will selectively
activate NF-κB p65, which further activates the transcription of
pro-inflammatory factors (TNF-α, IL-1 and IL-6) genes, resulting in
an uncontrolled inflammatory response in the lungs. The
inflammatory response will increase the permeability of
capillaries, damage the pulmonary air-blood barrier, cause
pulmonary edema, and then lead to pathological changes such as
pulmonary fibrosis (67).
In the present study, the results of the hemogram
revealed that the red blood cells, hemoglobin and hematocrit in the
blood of rats in the hypoxia group were significantly higher than
those in the control group, suggesting that hypoxia may lead to
hemorrhaging in the body. The number of white blood cells was
significantly increased, which may be stimulated by hypoxia to
produce inflammation or due to abnormal bleeding in vivo
following hypoxia. Whether the inflammation and hemorrhage caused
by hypoxia occur at the same time or exist in a sequence is not
clear at present and requires further investigation. Coagulation
indicators (PT, APTT, TT and FIB) reflect coagulation functions.
The PT, APTT and TT of the rats in the hypoxia group was
significantly prolonged, and blood coagulation disorders appeared,
which may indicate that the microvessels and tissues in rats were
bleeding under the hypoxic stimulation. After bleeding, the
coagulation system in the body could not normally function on the
bleeding point, resulting in abnormal coagulation functions.
Prothrombin is synthesized in the liver and then reaches the whole
body through the blood to be hydrolyzed into thrombin (F2), which
exerts coagulation effects. In the ELISA of serum and lung tissue
of the rats in the hypoxia group, the level of F2 was significantly
increased. Combined with the results of coagulation index analysis,
F2 could not be hydrolyzed properly under hypoxic stimulation due
to coagulation disorders caused by abnormal bleeding in
vivo, resulting in the abnormal increase of F2 in blood and
lung tissue of the rats. Similarly, the expression of TNF-α, IL-1β
and IL-6 in the blood and lung tissue of the rats in the hypoxia
group was significantly increased, which further indicated that
hypoxic stimulation led to the development of inflammation.
Therefore, the present study considers whether inflammatory injury
and coagulation disorders will occur in the body under hypoxic
conditions, and whether there is an association between the two.
The results of the present study prove that hypoxia can activate
the inflammatory signaling pathways of HMGB1 and RAGE, thus
activating the downstream signaling pathways, p38MAPK and NF-κB; in
addition, the PT, APTT and TT in the coagulation test indexes
increased, indicating that hypoxia can lead to coagulation
abnormalities. The results of the immunoprecipitation experiment
revealed the interaction between F2 and RAGE, which may indicate
that the association between coagulation disorders and the
inflammatory response can be derived from the association between
F2 and RAGE.
On the whole, the present study explored some of the
molecular mechanisms of lung injury due to hypoxia, and provides a
new direction for the targeting of therapeutic interventions.
However, the findings obtained herein need to be validated by
further pharmacological intervention studies. In addition, the
present study has some limitations. For example, the approach
cannot exclude other possible molecular mechanisms of
hypoxia-induced lung injury.
In conclusion, the present study explored the
activation of related inflammatory pathways and coagulation
function pathways in lung injury induced by acute hypoxia at high
altitude using animal and cell models. The simultaneous activation
of the HMGB1/RAGE/NF-κB and F2/Rho pathways, as well as the
interaction of inflammation-related proteins and
coagulation-related proteins play a critical role in lung injury
induced by acute hypoxia at a high altitude. The results of the
present study provide new insight into the molecular mechanisms of
acute hypoxia at high altitude, and may aid the development of
novel treatment strategies.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JG carried out the animal and cell experiments. ZZ
designed the technical route and the study content, and was a major
contributor to the writing of the manuscript. JYY and YXG analyzed
the results of all the experiments. YG participated in the design
of the whole research protocol and experimental methods,
coordinated all the experimental programs and provided guidance. JG
and ZZ confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study did not involve human subjects.
The animals were provided by the Beijing Weitonglihua Experimental
Animal Center (Beijing, China; animal production license no.
SCXK-2021-0011). The animal experiments were approved by the Ethics
Committee of the Animal Center (approval no. IACUC-DWZX-2021-605).
All animal studies complied with the ARRIVE guidelines and the AVMA
euthanasia guidelines 2020.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Logistics and Health
Research Project (grant no. 2021ZY057) and the Innovation Team and
Talents Cultivation Program of National Administration of
Traditional Chinese Medicine (grant no. ZYYCXTD-D-202207).
References
|
1
|
Yu SL, Wong CK, Szeto CC, Li EK, Cai Z and
Tam LS: Members of the receptor for advanced glycation end products
axis as potential therapeutic targets in patients with lupus
nephritis. Lupus. 24:675–686. 2015. View Article : Google Scholar
|
|
2
|
Hudson BI and Lippman ME: Targeting RAGE
signaling in inflammatory disease. Ann Rev Med. 69:349–364. 2018.
View Article : Google Scholar
|
|
3
|
Rao NV, Argyle B, Xu X, Reynolds PR,
Walenga JM, Prechel M, Prestwich GD, MacArthur RB, Walters BB,
Hoidal JR and Kennedy TP: Low anticoagulant heparin targets
multiple sites of inflammation, suppresses heparin-induced
thrombocytopenia, and inhibits interaction of RAGE with its
ligands. Am J Physiol Cell Physiol. 299:C97–C110. 2010. View Article : Google Scholar
|
|
4
|
Degani G, Altomare A, Digiovanni S, Arosio
B, Fritz G, Raucci A, Aldini G and Popolo L: Prothrombin is a
binding partner of the human receptor of advanced glycation end
products. J Biol Chem. 295:12498–12511. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C, Dong H, Chen F, Wang Y, Ma J and
Wang G: The HMGB1-RAGE/TLR-TNF-α signaling pathway may contribute
to kidney injury induced by hypoxia. Exp Ther Med. 17:17–26.
2019.
|
|
6
|
Sokolova E and Reiser G: A novel
therapeutic target in various lung diseases: Airway proteases and
protease-activated receptors. Pharmacol Ther. 115:70–83. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Demling N, Ehrhardt C, Kasper M, Laue M,
Knels L and Rieber EP: Promotion of cell adherence and spreading: A
novel function of RAGE, the highly selective differentiation marker
of human alveolar epithelial type I cells. Cell Tissue Res.
323:475–488. 2006. View Article : Google Scholar
|
|
8
|
Lizotte PP, Hanford LE, Enghild JJ,
Nozik-Grayck E, Giles BL and Oury TD: Developmental expression of
the receptor for advanced glycation end-products (RAGE) and its
response to hyperoxia in the neonatal rat lung. BMC Dev Biol.
7:152007. View Article : Google Scholar
|
|
9
|
Gross CM, Kellner M, Wang T, Lu Q, Sun X,
Zemskov EA, Noonepalle S, Kangath A, Kumar S, Gonzalez-Garay M, et
al: LPS-induced acute lung injury involves NF-κB-mediated
downregulation of SOX18. Am J Respir Cell Mol Biol. 58:614–624.
2018. View Article : Google Scholar
|
|
10
|
Johnson ER and Matthay MA: Acute lung
injury: Epidemiology, pathogenesis, and treatment. J Aerosol Med
Pulm Drug Deliv. 23:243–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blank R and Napolitano LM: Epidemiology of
ARDS and ALI. Crit Care Clin. 27:439–458. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Camprubí-Rimblas M, Tantinyà N, Bringué J,
Guillamat-Prats R and Artigas A: Anticoagulant therapy in acute
respiratory distress syndrome. Ann Transl Med. 6:362018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Geering B, Gurzeler U, Federzoni E,
Kaufmann T and Simon HU: A novel TNFR1-triggered apoptosis pathway
mediated by class IA PI3Ks in neutrophils. Blood. 117:5953–5962.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Itakura E, Huang RR, Wen DR, Paul E,
Wünsch PH and Cochran AJ: IL-10 expression by primary tumor cells
correlates with melanoma progression from radial to vertical growth
phase and development of metastatic competence. Mod Pathol.
24:801–809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao XJ, Qu YY, Liu XW, Zhu M, Ma CY, Jiao
YL, Cui B, Chen ZJ and Zhao YR: Immune complexes induce TNF-α and
BAFF production from U937 cells by HMGB1 and RAGE. Eur Rev Med
Pharmacol Sci. 21:1810–1819. 2017.PubMed/NCBI
|
|
16
|
Fritz G: RAGE: A single receptor fits
multiple ligands. Trends Biochem Sci. 36:625–632. 2011. View Article : Google Scholar
|
|
17
|
Bangert A, Andrassy M, Müller AM,
Bockstahler M, Fischer A, Volz CH, Leib C, Göser S, Korkmaz-Icöz S,
Zittrich S, et al: Critical role of RAGE and HMGB1 in inflammatory
heart disease. Proc Natl Acad Sci USA. 113:E155–E164. 2016.
View Article : Google Scholar
|
|
18
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sanders A, Delker DA, Huecksteadt T, Beck
E, Wuren T, Chen Y, Zhang Y, Hazel MW and Hoidal JR: RAGE is a
critical mediator of pulmonary oxidative stress, alveolar
macrophage activation and emphysema in response to cigarette smoke.
Sci Rep. 9:2312019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pilzweger C and Holdenrieder S:
Circulating HMGB1 and RAGE as clinical biomarkers in malignant and
autoimmune diseases. Diagnostics (Basel). 5:219–253. 2015.
View Article : Google Scholar
|
|
21
|
Zhang QY, Wu LQ, Zhang T, Han YF and Lin
X: Autophagy-mediated HMGB1 release promotes gastric cancer cell
survival via RAGE activation of extracellular signal-regulated
kinases 1/2. Oncol Rep. 33:1630–1638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ségal-Bendirdjian E and Geli V:
Non-canonical roles of telomerase: Unraveling the imbroglio. Front
Cell Dev Biol. 7:3322019. View Article : Google Scholar
|
|
23
|
Ghosh S and Hayden M: New regulators of
NF-kappa B in inflammation. Nat Rev Immunol. 8:837–848. 2008.
View Article : Google Scholar
|
|
24
|
Shen Y, Xie X, Li Z, Huang Y, Ma L, Shen
X, Liu Y and Zhao Y: Interleukin-17-induced expression of monocyte
chemoattractant protein-1 in cardiac myocytes requires nuclear
factor κB through the phosphorylation of p65. Microbiol Immunol.
61:280–286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu A, Sun H, Raymond RM Jr, Furie BC,
Furie B, Bronstein M, Kaufman RJ, Westrick R and Ginsburg D: Fatal
hemorrhage in mice lacking gamma-glutamyl carboxylase. Blood.
109:5270–5275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Innerhofer P and Kienast J: Principles of
perioperative coagulopathy. Best Pract Res Clin Anaesthesiol.
24:1–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Terada M, Kelly EA and Jarjour NN:
Increased thrombin activity after allergen challenge: A potential
link to airway remodeling? Am J Respir Crit Care Med. 169:373–377.
2004. View Article : Google Scholar
|
|
28
|
Bartko J, Schoergenhofer C, Schwameis M,
Buchtele N, Wojta J, Schabbauer G, Stiebellehner L and Jilma B:
Dexamethasone inhibits endotoxin-induced coagulopathy in human
lungs. J Thromb Haemost. 14:2471–2477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stroo I, Ding C, Novak A, Yang J, Roelofs
JJTH, Meijers JCM, Revenko AS, van't Veer C, Zeerleder S, Crosby JR
and van der Poll T: Inhibition of the extrinsic or intrinsic
coagulation pathway during pneumonia-derived sepsis. Am J Physiol
Lung Cell Mol Physiol. 315:L799–L809. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steinhoff M, Buddenkotte J, Shpacovitch V,
Rattenholl A, Moormann C, Vergnolle N, Luger TA and Hollenberg MD:
Proteinase-activated receptors: Transducers of proteinase-mediated
signaling in inflammation and immune response. Endocrine Rev.
26:1–43. 2005. View Article : Google Scholar
|
|
31
|
Bar-Shavit R, Benezra M, Sabbah V, Bode W
and Vlodavsky I: Thrombin as a multifunctional protein: Induction
of cell adhesion and proliferation. Am J Respir Cell Mol Biol.
6:123–130. 1992. View Article : Google Scholar
|
|
32
|
Ellis CA, Malik AB, Gilchrist A, Hamm H,
Sandoval R, Voyno-Yasenetskaya T and Tiruppathi CP: Thrombin
induces proteinase-activated receptor-1 gene expression in
endothelial cells via activation of Gi-linked Ras/mitogen-activated
protein kinase pathway. J Biol Chem. 274:13718–13727. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gopal P, Gosker HR, de Theije CC, Eurlings
IM, Sell DR, Monnier VM and Reynaert P: Effect of chronic hypoxia
on RAGE and its soluble forms in lungs and plasma of mice. Biochim
Biophys Acta. 1852:992–1000. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Wu R, Zhao S, Cheng H, Ji P, Yu M
and Tian Z: RAGE/NF-κB pathway mediates lipopolysaccharide-induced
inflammation in alveolar type I epithelial cells isolated from
neonate rats. Inflammation. 37:1623–1629. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alexiou P, Chatzopoulou M, Pegklidou K and
Demopoulos VJ: A multi-ligand receptor unveiling novel insights in
health and disease. Curr Med Chem. 17:2232–2252. 2010. View Article : Google Scholar
|
|
36
|
Feng Y, Ke J, Cao P, Deng M, Li J, Cai H,
Meng Q, Li Y and Long X: HMGB1-induced angiogenesis in perforated
disc cells of human temporomandibular joint. J Cell Mol Med.
22:1283–1291. 2018.
|
|
37
|
He F, Gu L, Cai N, Ni J, Liu Y, Zhang Q
and Wu C: The HMGB1-RAGE axis induces apoptosis in acute
respiratory distress syndrome through PERK/eIF2α/ATF4-mediated
endoplasmic reticulum stress. Inflamm Res. 71:1245–1260. 2022.
View Article : Google Scholar
|
|
38
|
Sharma AK, LaPar DJ, Stone ML, Zhao Y,
Kron IL and Laubach VE: Receptor for advanced glycation end
products (RAGE) on iNKT cells mediates lung ischemia-reperfusion
injury. Am J Transplant. 13:2255–2267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mi L, Zhang Y, Xu Y, Zheng X, Zhang X,
Wang Z, Xue M and Jin X: HMGB1/RAGE pro-inflammatory axis promotes
vascular endothelial cell apoptosis in limb ischemia/reperfusion
injury. Biomed Pharmacother. 116:1090052019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Andersson U, Yang H and Harris H:
High-mobility group box 1 protein (HMGB1) operates as an alarmin
outside as well as inside cells. Semin Immunol. 38:40–48. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar
|
|
42
|
Heinbockel L, Sánchez-Gómez S, de Tejada
GM, Dömming S, Brandenburg J, Kaconis Y, Hornef M, Dupont A,
Marwitz S, Goldmann T, et al: Preclinical investigations reveal the
broad-spectrum neutralizing activity of peptide Pep19-2.5 on
bacterial pathogenicity factors. Antimicrob Agents Chemother.
57:1480–1487. 2013. View Article : Google Scholar :
|
|
43
|
Entezari M, Javdan M, Antoine DJ, Morrow
DM, Sitapara RA, Patel V, Wang M, Sharma L, Gorasiya S, Zur M, et
al: Inhibition of extracellular HMGB1 attenuates hyperoxia-induced
inflammatory acute lung injury. Redox Biol. 2:314–322. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao MJ, Jiang HR, Sun JW, Wang ZA, Hu B,
Zhu CR, Yin XH, Chen MM, Ma XC, Zhao WD and Luan ZG: Roles of
RAGE/ROCK1 pathway in HMGB1-induced early changes in barrier
permeability of human pulmonary microvascular endothelial cell.
Front Immunol. 12:6970712021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Paudel YN, Angelopoulou E, Piperi C,
Balasubramaniam V, Othman I and Shaikh MF: Enlightening the role of
high mobility group box 1 (HMGB1) in inflammation: Updates on
receptor signalling. Eur J Pharmacol. 858:1724872019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Joshi N, Walter JM and Misharin AV:
Alveolar macrophages. Cell Immunol. 330:86–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gonzalez NC and Wood JG: Alveolar
hypoxia-induced systemic inflammation: What low PO(2) does and does
not do. Adv Exp Med Biol. 662:27–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fröhlich S, Boylan J and McLoughlin P:
Hypoxia-induced inflammation in the lung: A potential therapeutic
target in acute lung injury? Am J Respir Cell Mol Biol. 48:271–279.
2013. View Article : Google Scholar
|
|
49
|
Minamino T, Christou H, Hsieh CM, Liu Y,
Dhawan V, Abraham NG, Perrella MA, Mitsialis SA and Kourembanas S:
Targeted expression of heme oxygenase-1 prevents the pulmonary
inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci
USA. 98:8798–8803. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vergadi E, Chang MS, Lee C, Liang OD, Liu
X, Fernandez-Gonzalez A, Mitsialis SA and Kourembanas S: Early
macrophage recruitment and alternative activation are critical for
the later development of hypoxia-induced pulmonary hypertension.
Circulation. 123:1986–1995. 2011. View Article : Google Scholar :
|
|
51
|
Carpenter TC and Stenmark KR: Hypoxia
decreases lung neprilysin expression and increases pulmonary
vascular leak. Am J Physiol. 281:L941–L948. 2001.
|
|
52
|
Wang M and Cheong KL: Preparation,
structural characterisation, and bioactivities of fructans: A
review. Molecules. 28:16132023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen GY and Nuñez G: Sterile inflammation:
Sensing and reacting to damage. Nat Rev. 10:826–837. 2010.
|
|
54
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar
|
|
55
|
Walmsley SR, Print C, Farahi N,
Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM,
Cowburn AS, Johnson N and Chilvers ER: Hypoxia-induced neutrophil
survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J
Exp Med. 201:105–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Eckle T, Faigle M, Grenz A, Laucher S,
Thompson LF and Eltzschig HK: A2B adenosine receptor dampens
hypoxia-induced vascular leak. Blood. 111:2024–2035. 2008.
View Article : Google Scholar
|
|
57
|
Eltzschig HK and Carmeliet P: Hypoxia and
inflammation. New Engl J Med. 364:656–665. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhu S, Li W, Ward MF, Sama AE and Wang H:
High mobility group box 1 protein as a potential drug target for
infection- and injury-elicited inflammation. Inflamm Allergy Drug
Targets. 9:60–72. 2010. View Article : Google Scholar :
|
|
59
|
Smolarczyk R, Cichoń T, Jarosz M and Szala
S: HMGB1-its role in tumor progression and anticancer therapy.
Postepy Hig Med Dosw (Online). 66:913–920. 2012.In Polish.
View Article : Google Scholar
|
|
60
|
Yamada Y, Fujii T, Ishijima R, Tachibana
H, Yokoue N, Takasawa R and Tanuma S: The release of high mobility
group box 1 in apoptosis is triggered by nucleosomal DNA
fragmentation. Arch Biochem Biophys. 506:188–193. 2011. View Article : Google Scholar
|
|
61
|
Zhang Y, Zhang M, Wang CY and Shen A:
Ketamine alleviates LPS induced lung injury by inhibiting
HMGB1-RAGE level. Eur Rev Med Pharmacol Sci. 22:1830–1836.
2018.PubMed/NCBI
|
|
62
|
Chavakis T, Bierhaus A and Nawroth PP:
RAGE (receptor for advanced glycation end products): A central
player in the inflammatory response. Microbes Infect. 6:1219–1225.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang L, Li JY, Zhang XZ, Liu L, Wan ZM, Li
RX and Guo Y: Involvement of p38mapk/nf-κb signaling pathways in
osteoblasts differentiation in response to mechanical stretch. Ann
Biomed Eng. 40:1884–1894. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ott I: Soluble tissue factor emerges from
inflammation. Circ Res. 96:1217–1218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Erlich JH, Boyle EM, Labriola J, Kovacich
JC, Santucci RA, Fearns C, Morgan EN, Yun W, Luther T, Kojikawa O,
et al: Inhibition of the tissue factor-thrombin pathway limits
infarct size after myocardial ischemia-reperfusion injury by
reducing inflammation. Am J Pathol. 157:1849–1862. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Carr C, Bild GS, Chang AC, Peer GT,
Palmier MO, Frazier RB, Gustafson ME, Wun TC, Creasey AA and
Hinshaw LB: Recombinant E. coli-derived tissue factor pathway
inhibitor reduces coagulopathic and lethal effects in the baboon
gram-negative model of septic shock. Circ Shock. 44:126–137.
1994.PubMed/NCBI
|
|
67
|
Ware LB and Calfee CS: Biomarkers of ARDS:
what's new? Intensive Care Med. 42:797–799. 2016. View Article : Google Scholar
|